
Solar driven CO2 reduction: from materials to devices
Lili
Wan
,
Rong
Chen
,
Daniel Wun Fung
Cheung
 ,
Linxiao
Wu
and
Jingshan
Luo
*
,
Linxiao
Wu
and
Jingshan
Luo
*
Institute of Photoelectronic Thin Film Devices and Technology, Solar Energy Research Center, Key Laboratory of Photoelectronic Thin Film Devices and Technology of Tianjin, Ministry of Education Engineering Research Center of Thin Film Photoelectronic Technology, Renewable Energy Conversion and Storage Center, Nankai University, Tianjin 300350, China. E-mail: jingshan.luo@nankai.edu.cn
First published on 28th February 2023
Abstract
Solar driven CO2 reduction for the production of fuels and chemicals is a promising technology for achieving carbon neutrality. Photocatalytic CO2 reduction, photoelectrochemical CO2 reduction and photovoltaic-electrochemical CO2 reduction, are three main approaches for solar driven CO2 reduction that have attracted a lot of interest in both the academic and industrial communities. However, in spite of the decades of work that have been devoted to this research area, low solar to fuel efficiency, poor product selectivity and unsatisfactory stability continue to impede the application of these three technologies. Herein, we summarize the recent advances in photo-absorbers, catalysts and device designs for solar driven CO2 reduction and have identified the following requirements that are essential for realizing a highly efficient solar driven CO2 reduction system: optimized photo-absorbers, tailored catalysts, well-designed devices and the synergistic operation of these three parts. In addition, we provide perspectives for the future development of the solar driven CO2 reduction field.
 Lili Wan | Lili Wan received her bachelors degree in Environmental Science from Inner Mongolia University in 2011 and obtained her masters and doctorate degrees in Environmental Science from Nankai University in 2015 and 2019, respectively. During her PhD study, she visited Geoffrey A. Ozin's group at the University of Toronto as an exchange doctoral student for one and a half years, where she focused on photocatalytic CO2. She is currently a Postdoc at the Institute of Photoelectronic Thin Film Devices and Technology of Nankai University. Her research interests primarily focus on electrochemical and photochemical CO2 reduction. |
 Jingshan Luo | Jingshan Luo is a full professor and vice director of the Institute of Photoelectronic Thin-Film Devices and Technology at Nankai University. He received his BSc degree from Jilin University in 2010 and his PhD degree from Nanyang Technological University in 2014. After that, he went to École Polytechnique Fédérale de Lausanne (EPFL) for postdoctoral research with Professor Michael Grätzel. In 2018, he joined Nankai University. He has authored/coauthored more than 120 peer-reviewed publications, which have been cited more than 23 |
10th Anniversary StatementCongratulations on the 10th anniversary of Journal of Materials Chemistry A! The journal was launched in the middle of my PhD study, and its theme fitted well with my research topic. It has accompanied me through my scientific career from a PhD student, postdoc, to my current position as a full professor. I have witnessed the fast growth and expanding impact of the journal, and I am a frequent reader, author, and active reviewer of the journal. Since 2020, I have been an advisory board member of the journal. It is my great pleasure to contribute a review article as the corresponding author to this special issue. As the world's demand for energy and sustainability is continuously increasing, the journal will continue to excel and contribute to the common benefits of human kind. |
1. Introduction
1.1 Motivation for CO2 recycling
The atmospheric concentration of CO2 has increased by around 100 ppm over the past century due to human activity,1 mainly the excessive combustion of fossil fuels, which has disrupted the natural carbon cycle and caused severe environmental and ecological problems.2 Without efficient carbon emission reduction strategies, the concentration of CO2 in the atmosphere would rise up to 570 ppm by the year 2100, which would lead to an increase of approximately 1.9 °C in the mean global temperature.3 One of the ideal strategies of CO2 reduction is converting CO2 into value-added chemicals and carbonaceous fuels using renewable energy, such as solar energy. It has been estimated that 10% of the solar energy received by the Earth via sunlight irradiating 0.3% of the Earth's surface would be sufficient to fulfill the global energy needs for a year.4 In 1978, Halmann discovered the photo-induced reduction of CO2 into carbonaceous products on semiconductors,5 which led to large amount of research focusing on solar driven CO2 conversion.1.2 Solar driven CO2 reduction
Solar driven CO2 reduction utilizes the energy from sunlight to reduce CO2 into value-added carbonaceous chemicals, such as CO, CH4, HCOO−, alkenes and alcohols. Solar driven CO2 reduction is typically divided into photocatalytic CO2 reduction (PC CO2R), photoelectrochemical CO2 reduction (PEC CO2R) and photovoltaic-electrochemical CO2 reduction (PV-EC CO2R) approaches.In the PC CO2R system, CO2 reduction and oxidation reactions (most commonly the water oxidation reaction) both occur on the surface of semiconductors under light irradiation. Three crucial steps are involved in this process (Fig. 1A).6–9 In the first step, the semiconductor absorbs incident light with energy equal to or higher than its bandgap and photo-induced electron–hole pairs are subsequently generated. The photo-induced electrons are excited to the conduction band (CB) and the holes remain in the valence band (VB). In the second step, the photo-induced electrons and holes migrate to the surface of the semiconductor, but a large proportion of the electrons and holes may be consumed by bulk recombination and surface recombination. Photo-induced electrons which successfully migrate to the surface reduce CO2 into fuels and H2O into H2 as the by-product, whilst holes on the surface of the semiconductors oxidize H2O into O2. The hydrogen evolution reaction (HER) accompanies the CO2 reduction due to the similar equilibrium potentials of these half-reactions (Fig. 1A).10 At the semiconductor–liquid interface, the difference between the Fermi level (EF) of the semiconductor and the redox potential level of the liquid (electrolyte) (ER) induces band bending at the semiconductor/liquid interface, which facilitates the separation of photo-generated electron–hole pairs during light irradiation.11EF is close to VB in p-type semiconductors, and it is typically lower than ER, which results in a tendency for electrons flowing from the liquid to the semiconductor, building up an electric field in the semiconductor near the interface. This electric field causes band bending at the semiconductor–liquid interface. Therefore, the CB and VB of a p-type semiconductor bend downward, from the semiconductor to the liquid (Fig. 1B), enhancing electron–hole pair separation and facilitating electron transport to the semiconductor–liquid interface, which is critical for improving the photo to electron conversion efficiency.
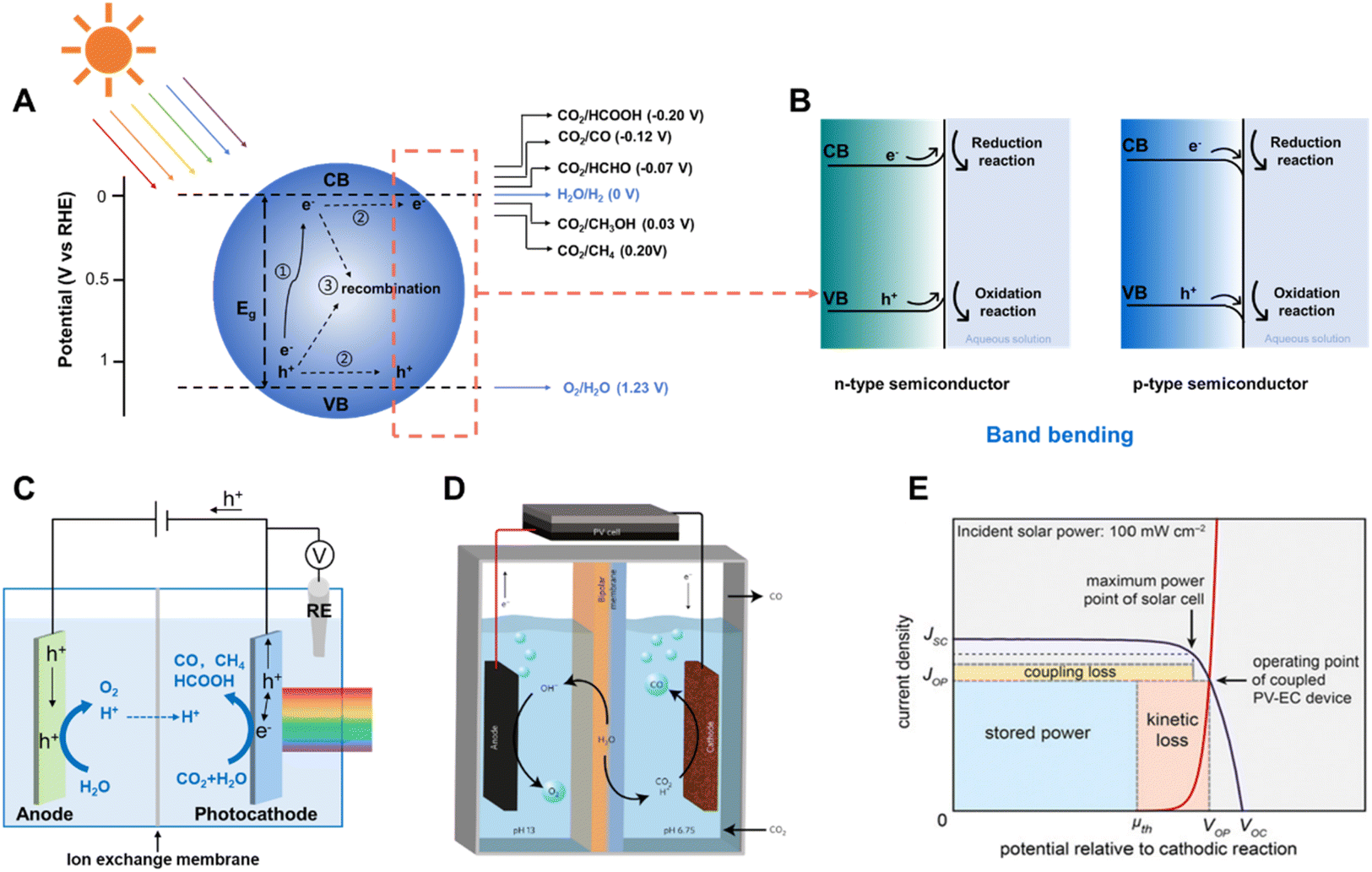 | ||
| Fig. 1 (A) Schematic of photocatalytic CO2 reduction, illustrating the three main steps of the process, and (B) band bending of an n-type and a p-type semiconductor at the semiconductor/liquid interface.12 Copyright 2015, Springer Nature. (C) Schematic of a typical photoelectrochemical CO2 reduction device. (D) Schematic of the PV-EC CO2 reduction device.13 Copyright 2017, Springer Nature. (E) General current density–voltage diagram for a coupled PV-EC system.14 Copyright 2013, Proceedings of the National Academy of Sciences. | ||
The PEC CO2 reduction is generally performed in a cell with two compartments (H-type cell), which mostly consists of a photoelectrode, a counter/dark counter electrode, reference electrode and a membrane (proton- or anion-exchange membrane, for separating the electrolyte of two half-reactions, transferring the protons or the donor of protons). As illustrated in Fig. 1C, photo-induced electrons are generated on the photocathode under light irradiation conditions, transferred to the electrode/electrolyte interface and reduce CO2 or H2O into carbonaceous products or H2, whilst photo-induced holes are transferred from the external circuit to the anode and oxidize H2O into O2. Similar to the situation of a semiconductor in contact with an aqueous solution, semiconductors on an electrode exhibit band bending due to equilibration of the semiconductor Fermi level with the redox potential of the electrolyte, which creates a space charge region providing an electric field at the interface of the electrode/electrolyte.15 The “downward” band bending in the space charge region of the semiconductor enables efficient separation of photo-induced electron–hole pairs, directing electrons toward the electrolyte solution to drive the reduction reaction. The PEC system is different from the PC system because it separates the photo-induced electrons and holes from the surface of one particle to two different electrodes which can be placed in two chambers, allowing further separation of the redox products. The directional flow of the photo-induced electrons and holes reduces the amount of recombination of electrons and holes in the PEC system. In addition, the separation of photo-induced electrons and holes can be further enhanced by applying an external bias. However, the ultimate goal of the PEC CO2 reduction system is to construct a bias-free two-electrode cell, which is driven by the solar irradiation.16 It is challenging to construct photoelectrodes offering sufficient photo-voltage and with suitable conduction and valence band edges.
Photovoltaic (PV) driven electrochemical (EC) CO2 reduction, as another key strategy for converting solar energy into fuels and chemicals, employs photo-electrons provided by PV to reduce CO2 at the cathode in the EC cell (Fig. 1D).17 It combines the benefit of photovoltaics that it is a proven technique for photoelectric conversion with high efficiency and the advantage of electrocatalysis that it is a relatively mature technology close to practical application. The coupling of two distinct systems, both of which can be optimized independently, makes it an attractive integrated system.18 Collaborative coupling of the PV cell and EC is essential to achieve a highly efficient integrated system. It is better to operate the PV-EC system close to the maximum power point of the PV device, which is located at the intersection point of the electrocatalytic J–V curve of the electrocatalyst and PV cell close to the maximum power point of the PV device (Fig. 1E). Otherwise, the impedance mismatch significantly curtails the power available to run the EC cell.8,14
1.3 Strategies for improving the efficiency of solar driven CO2 reduction systems
Although PC, PEC and PV-EC systems are all driven by solar energy, they are intrinsically different from each other. The major difference between the PC system and the PEC system is the directional flow of the photo-induced electrons and holes in the PEC system and that the direction of the built-in electric field can be interchanged through the adjustment of the applied bias voltage. The Fermi level of semiconductors will move up or down when an external potential is applied and the band bending will therefore invert to form an accumulation region instead of a depletion region. This increases the band bending and enhances the electron–hole pair separation and interfacial charge transfer.8 Different from PC and PEC systems, the PV-EC system decouples the processes of solar energy conversion into electricity and redox reactions driven by electricity, which makes it easier to optimize the system efficiency by improving the efficiency of the two parts, PV and EC cell, separately.However, apart from their differences, the three solar driven CO2 reduction systems also share some common key elements. They all require photo-absorbers to convert solar energy, catalysts to accelerate the reaction kinetics and a well-designed device for realizing a highly efficient system. The first requirement for a highly efficient solar driven system (except for the PV-EC system) is a befitting photo-absorber with suitable band gap and band positions, which decide the absorbed light spectrum and the redox potential of the photo-induced electrons and holes, respectively. This is not a limitation for the PV-EC system because the photon to electron conversion is decoupled from the electrons involved catalysis. The active components, most commonly cocatalysts, are used to accelerate the reaction kinetics, improve product selectivity, and enhance the surface charge carrier transport.19 A well designed device with suitable characteristics also promotes the efficient utilization and conversion of solar energy. Therefore, materials and device designs are the two key aspects for realizing an efficient solar driven CO2 reduction system.
2. Materials for solar driven CO2 reduction
2.1 Photo-absorbers
The photo-absorbers are fundamental materials for a solar driven CO2 reduction system, which undertake the most important step of photo-absorption and conversion. Two basic properties are required for the photo-absorbers in PC and PEC systems: (i) the band gap of the semiconductors is better to be located in the range of 1.6 eV to 1.7 eV, (ii) the position of the CB and VB should be thermodynamically more negative and positive to the equilibrium potentials of CO2 reduction and water oxidation, respectively (Fig. 2A). For the single absorber system, the semiconductors must inherently satisfy the above properties concurrently. The multi-junction photo-absorber, such as the Z-scheme structure, type II structure and p–n junction can be adopted in PC and PEC systems for CO2 reduction when one of the semiconductors has a suitable CB and the other has an appropriate VB (Fig. 2B–D). Transition metal oxides, Si, III–V and II–VI photo-absorbers, perovskite materials, carbon nitrides, metal–organic frameworks and metal complexes are generally employed as photo-absorbers in PC and PEC systems.20 Most semiconductors in PC systems can also be adopted as the photo-absorbers in PEC systems. The photovoltaic materials functioning as a photovoltaic cell in the PV-EC system were also introduced into PC and PEC systems as photo-absorbers in recent research studies.21 Here, we discuss some representative semiconductors to demonstrate the importance of photo-absorbers in a highly efficient solar driven CO2 reduction system. | ||
| Fig. 2 (A) Conduction band and valence band potentials of several typical semiconductors relative to the standard redox potentials of CO2 reduction in water at pH 7.22 Copyright 2018, Science China Press and Springer-Verlag GmbH Germany. (B) Type II heterostructure semiconductors, (C) Z-scheme heterostructure semiconductors, (D) semiconductors constructed as a p–n junction.23 Copyright 2020, Wiley. | ||
Cuprous oxide (Cu2O) is a p-type semiconductor with a bandgap of 2.0–2.2 eV and a suitable conduction band for CO2 reduction (Fig. 2A), which makes it an attractive semiconductor for solar driven CO2 reduction.35 In 1989, Tennakone et al. found that methanol was produced from CO2 in a PC system on Cu2O powders under full spectrum illumination from a mercury lamp.36 However, Cu2O is unstable under aqueous conditions and light illumination due to the oxidation or reduction of Cu2O into CuO or Cu, respectively. The efficient strategies of preventing the photo-corrosion of Cu2O are constructing heterojunction structures, employing co-catalysts to improve the separation and transfer of photo-excited charge carriers or employing a protection layer to prevent direct contact with the electrolyte. Grela et al. synthesized octahedral Cu2O covered with TiO2 nanoparticles (Cu2O/TiO2) and investigated its photocatalytic CO2 activity in humidified CO2.37 As illustrated in Fig. 3A, the p–n heterojunction consisted of Cu2O and TiO2, preventing the photo-corrosion of Cu2O. A higher stability and CO production rate were achieved when using the Cu2O/TiO2 p–n heterojunction compared to the individual component materials (Fig. 3B). A ternary Ag–Cu2O/ZnO nanorod (NR) hybrid structure was also reported by Xu and co-workers. The Z-scheme formed between Cu2O and ZnO facilitated the photogenerated charge separation and the Ag nanoparticles on Cu2O promoted the electron transfer, leading to a higher photocatalytic CO2 reduction activity.38 Similar strategies were also widely employed in PEC systems. A buried p–n heterojunction structured photocathode (Fig. 3C) composed of Cu2O/AZO/TiO2 with a rhenium bipyridyl co-catalyst was investigated by Grätzel and co-workers.39 The Cu2O and AZO were combined to create a p–n junction which promoted the separation of photo-excited charge carriers and the TiO2 further prevented the contact of Cu2O with the electrolyte. The Cu2O photocathode exhibited an almost 100% faradaic efficiency (FE) of CO and 5.5 hour stability in anhydrous MeCN with 2 mM Re(tBu-bipy)(CO)3Cl and 7.5 M MeOH for CO2 reduction (Fig. 3D).
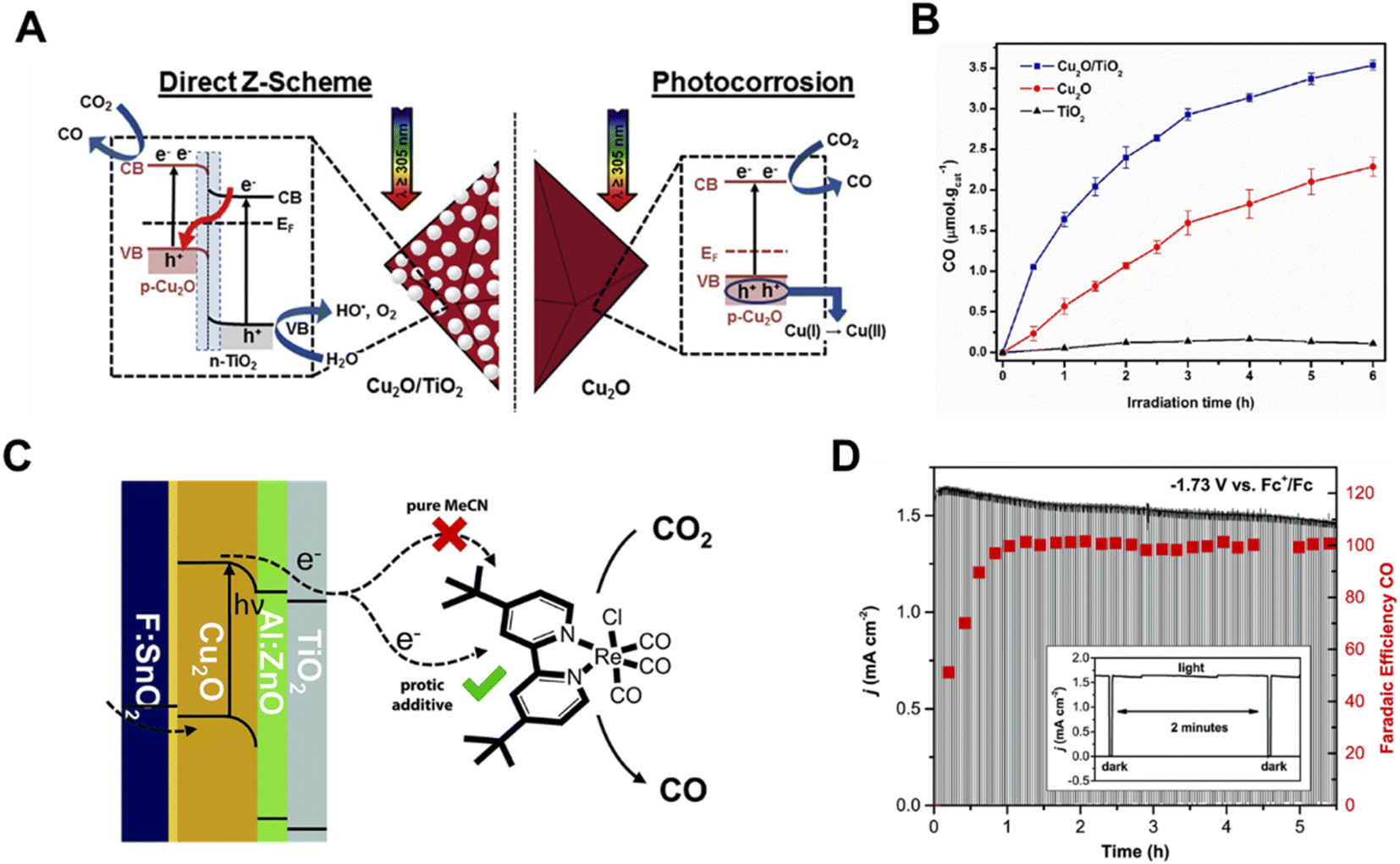 | ||
| Fig. 3 (A) Schematic illustration of a direct Z-scheme consisting of Cu2O and TiO2, (B) photocatalytic CO2 reduction performance of TiO2, Cu2O and Cu2O/TiO2.37 Copyright 2017, Elsevier. (C) Schematic of the photoelectrochemical CO2 reduction process involving TiO2 protected Cu2O photocathodes with a Re-based molecular co-catalyst, (D) cathodic current density and FE of CO on a Cu2O photocathode under chopped light illumination at a constant potential of −1.73 V vs. Fc+/Fc in MeCN with 2 mM Re(tBu-bipy)(CO)3Cl and 7.5 M MeOH.39 Copyright 2015, Royal Society of Chemistry. | ||
In a recent study, Toma et al. investigated the degradation mechanisms of the Cu2O photoelectrode when used for CO2 reduction.40 The transformation of Cu2O into Cu2+–OH in aqueous solution under illumination, which was contributed by both the photo-excited electrons and holes, prevented the transfer of photo-excited electrons from Cu2O to the catalysts (Fig. 4A and B). Therefore, the authors replaced the aqueous electrolyte with 2 mM Re(tBu-bipy)(CO)3Cl and 7.5 M MeOH, and a heterostructure system consisting of a WO3/Fe2O3 bilayer as the hole transfer layer and Cu2O as the photo-absorber was constructed (Fig. 4C) to facilitate the transfer of photo-excited holes. A FE of C2H4 of 60% over 3 h was achieved using this system (Fig. 4D).
 | ||
| Fig. 4 (A) Schematic of the charge transfer between Cu2O and the catalysts, in the presence of Cu2+–OH (aq) species. (B) Schematic of the charge transfer between Cu2O and the catalysts, in the absence of Cu2+–OH (aq) species. (C) The band diagram of suitable candidates for constructing the Z-scheme system. (D) faradaic efficiencies on the Ag/Cu2O/ZS/FTO photocathode for CO2R over time at −1.2 V versus Fc+/Fc under AM 1.5G simulated sunlight (100 mW cm−2) using 0.1 M Bu4NPF6 as the electrolyte and 0.1 M TEOA as the proton donor in AcCN solution (CO2 saturated).40 Copyright 2021, Springer Nature. | ||
Cu2O, as an attractive semiconductor with a near ideal band structure for the CO2 reduction reaction, still has a lot of potential for wider application,35 and future efforts can be focused on improving its stability by introducing newly developed protection layers, and improving its efficiency by constructing optimized p–n junctions and nanostructures.
Delafossite materials of the general stoichiometry ABO2 are a new class of promising photocatalysts for solar driven CO2 reduction. Symmetry breaking in these materials, by chemical substitution, modifies the band structure of the solid. Therefore, the photocatalytic performance of delafossites can be enhanced through engineering by adjusting the alignment of its band edges. Bocarsly et al. found that the addition of a Mg2+ dopant increases the conductivity of CuFeO2. Mg-doped CuFeO2 was demonstrated to have a conduction band edge at −1.1 V vs. SCE (pH = 6.8) and the ability to reduce CO2 to formate with an underpotential of 400 mV.41 In 2019, Park and co-workers mixed copper and iron oxide (CuO/CuFeO2; CFO) to form bulk heterojunction films, which were capable of converting CO2 and water into C1–C6 aliphatic acid anions and O2 with a solar-to chemical energy conversion (STC) efficiency close to 3% under simulated sunlight in the absence of any sacrificial chemicals or electrical biases.42
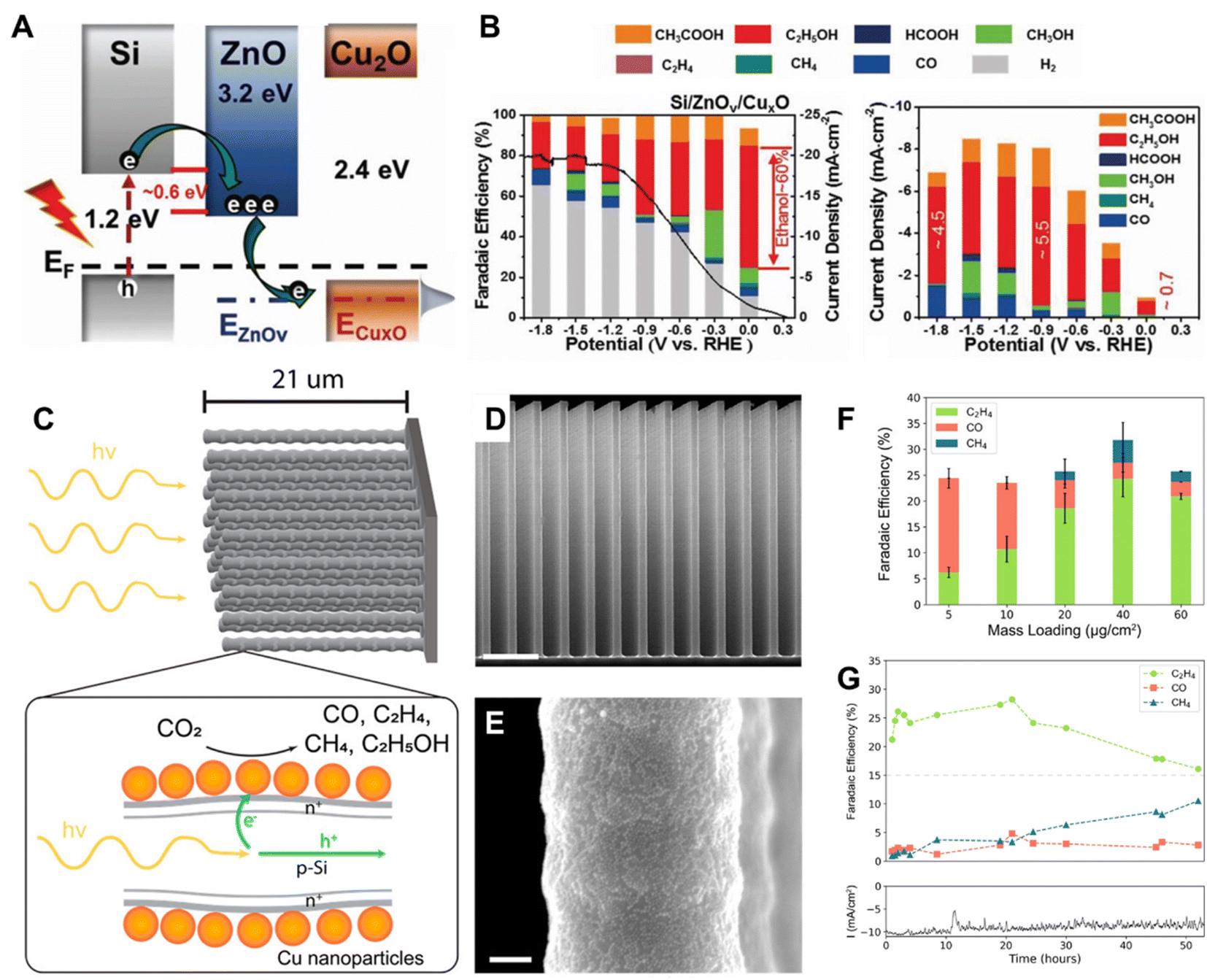 | ||
| Fig. 5 (A) A schematic illustration of the band structure of Si/ZnOv/CuxO, (B) FE of photoelectrochemical CO2 reduction on Si/ZnOv/CuxO in CO2-dissolved 0.1 M KHCO3 under airmass 1.5G (left), partial photocurrent densities and products obtained on Si/ZnOv/CuxO (right).45 Copyright 2022, Wiley. (C) A schematic of the CuNPs/Si nanowire arrays, illustrating the dopant layer and the charge separation processes occurring within the arrays, (D) SEM image of the Si NW array (scale bar 4 μm), (E) the corresponding SEM image with greater magnification (scale bar 100 nm), (F) FE of CO2 reduction products as a function of mass loading of Cu NPs at −0.50 V vs. RHE. (G) Stability test of the CuNP/SiNW photocathode operating at −0.50 V vs. RHE at 1 Sun using a loading mass of Cu NPs of 40 μg cm−2.46 Copyright 2022, American Chemical Society. | ||
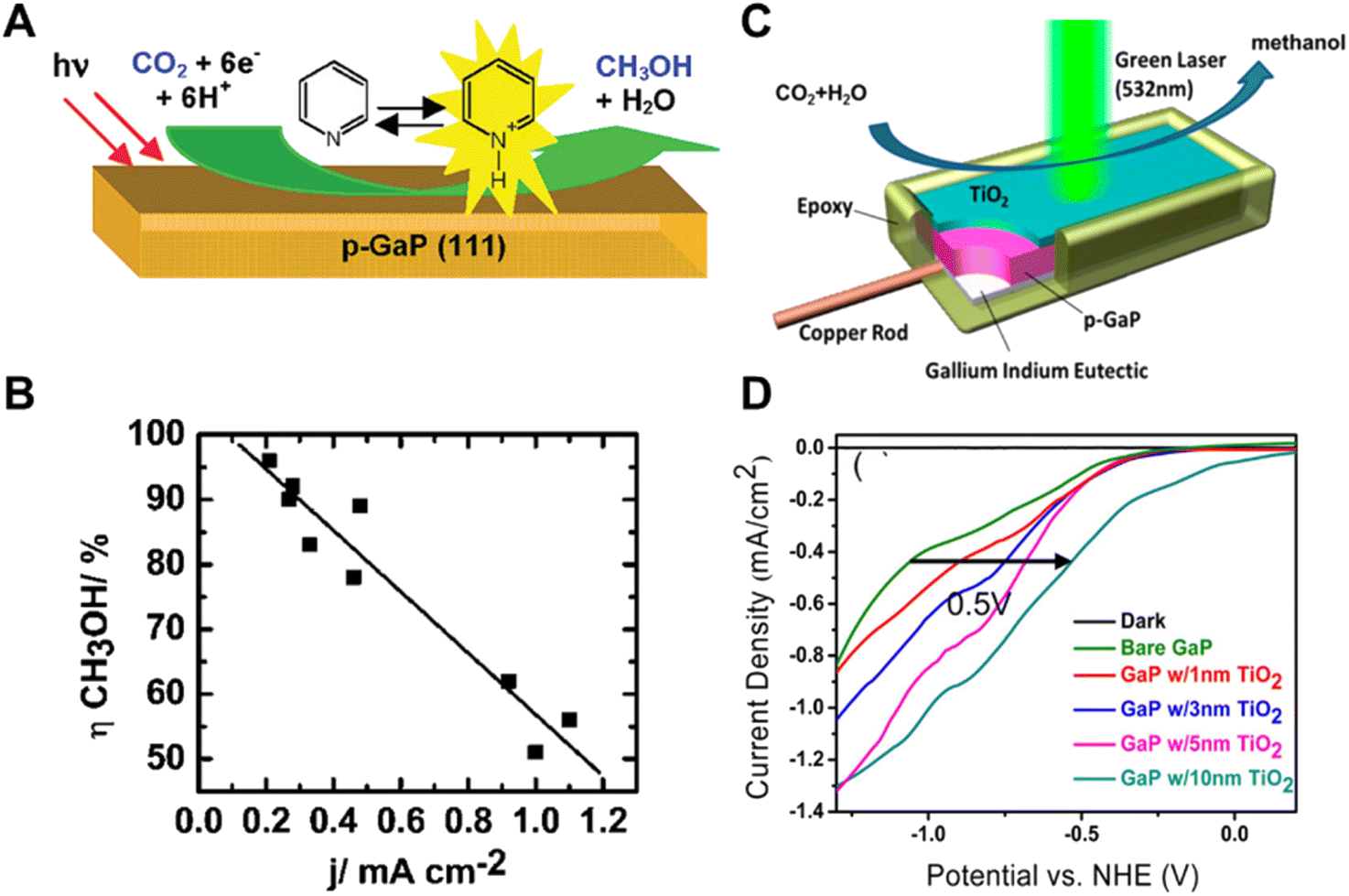 | ||
| Fig. 6 (A) Schematic of pyridinium ion catalyst decorated p-GaP for CO2 reduction, (B) relationship of increasing methanol yield with decreasing current density.49 Copyright 2008, American Chemical Society. (C) Schematic of TiO2 passivated GaP photocathodes in photocatalytic CO2 reduction under 532 nm wavelength laser illumination, (D) photocatalytic current–potential curves of GaP photocatalysts with different TiO2 thicknesses in a 0.5 M NaCl, 10 mM pyridine solution under 532 nm wavelength laser illumination.50 Copyright 2014, American Chemical Society. | ||
 | ||
| Fig. 7 (A) Schematic illustration of the device combining photovoltaics with an electrochemical cell. (B) Generalized energy diagram for converting CO2 into CO with three perovskite solar cells connected in series, (C) J–V curves of three series-connected perovskite cells under simulated AM 1.5G 1 Sun solar irradiation and in the dark, overlaid with the matched J–V characteristic of the CO2-reduction and oxygen-evolution electrodes. The maximum power point of the photovoltaics is indicated by the circled red dot on the curve.57 Copyright 2015, Springer Nature. (D) Schematic diagram of CO2 photoreduction over the MAPbI3@PCN-221(Fe0.2) photocatalyst, photocatalytic performance: yield of the CO2 reduction to CH4 and CO in the CO2-saturated ethyl acetate/water solution after (E) 25 h and (F) 80 h.58 Copyright 2019, Wiley. | ||
Apart from their use as a PV cell, the lead halide perovskites are also attractive semiconductor compounds for photocatalytic CO2 reduction due to their suitable bandgap, conduction and valence band levels.59 In 2017, Kuang's group first reported the artificial photosynthesis based on halide perovskite QDs for CO2 reduction in non-aqueous media.60 Under AM 1.5G simulated illumination, the CsPbBr3 QD/graphene oxide (CsPbBr3 QD/GO) composite catalyzed CO2 reduction into CO and CH4 with a selectivity over 99.1%. Afterwards, they further investigated a Pt decorated CsPbBr3 perovskite for photocatalytic CO2 reduction in non-aqueous solvents.61 The previous studies on perovskites as photocatalysts to convert CO2 into solar fuels were commonly carried out in non-aqueous media due to the water sensitivity of perovskites. To improve the water stability of perovskites, strategies for preventing the contact of perovskites with water were commonly adopted. Lu et al. encapsulated low-cost CH3NH3PbI3 (MAPbI3) perovskite QDs in the pores of Fe porphyrin MOF (PCN-221(Fex)) and obtained an enhanced catalytic performance and stability with water as the electron donor for photocatalytic CO2 reduction (Fig. 7D–F).58 Coating a thin graphdiyne layer on CsPbBr3 nanocrystals also delivered a significant improvement in stability for CO2 reduction in an aqueous system.62 It is worth noting that an isotopic labeling test using 13CO2 must be carried out for experiments involving organic media as the solvent. This is to confirm that the CO2 reduction products are indeed derived from CO2. To increase the application of perovskite materials, more practical hydrophobic treatments and encapsulation techniques are required. The relationship between photocatalytic activity and crystal structure, and the behavior of the photoelectrons at the surface should be further investigated to develop more efficient perovskite materials. In addition, new water stable perovskites are demanded.
On the other hand, PCN materials have emerged as promising cheap and benign semiconductors for PEC cells during the last decade, owing to their stability under harsh conditions and suitable energy band edges for water-splitting and other chemical transformations.69 A thin film composed of g-C3N4 doped with or without boron atoms was fabricated using an electrophoresis deposition method, and it was found that the BCN3.0 exhibited a higher CO2 reduction activity owing to its suitable band edges.70 Furthermore, cocatalysts, including Ag, Rh and Au were decorated on the surface of g-C3N4 and B-doped g-C3N4. The photocurrent of Rh–BCN3.0 was approximately 10 times larger than that of the original g-C3N4, and the Au–BCN3.0 produced more ethanol compared to the other electrodes. A type-II g-C3N4/ZnTe heterojunction was constructed by Wang and co-workers for photoelectrochemical CO2 reduction and it was proven to possess an enhanced photo-generated electron–hole separation and efficient electron transfer from g-C3N4 to ZnTe.71 The g-C3N4/ZnTe photocathode yielded an impressive ethanol generation rate of 17.1 μmol cm−2 h−1 at −1.1 V (vs. Ag/AgCl) in CO2-saturated 0.1 M KHCO3 aqueous solution under AM 1.5G illumination.
PCNs are promising photo-absorber materials due to their unique electronic structure and high stability against high temperature, acids, bases, and organic solvents. Further exploration into functionalized g-C3N4 with specific chemical groups and elemental doping may be practical approaches for extending their absorption in visible light range. Furthermore, it is important for the application of g-C3N4 as a photoelectrode by exploring thin film-formation methods and developing protection layers.
Re(bpydc) (CO)3Cl complexes have been incorporated into UiO-67 MOF for photocatalytic CO2 reduction, leading to high catalytic activity for the light-driven reduction of CO2 to CO (Fig. 8A).78 Inspired by this work, intensive research has been conducted on constructing photoactive MOFs for visible light-driven CO2 reduction. Li et al. reported an amine-functionalized MOF, NH2-MIL-125(Ti), which presented a broad absorption band and enhanced photocatalytic activity of CO2 to HCOO− compared with MIL-125(Ti) (Fig. 8B).79 In this titanium-based MOF, NH2-BDC linkers absorbed light and generated photoelectrons, which were then transferred to the Ti centers (active sites for CO2 reduction) by the ligand to metal charge transfer (LMCT) process. Porphyrin-incorporated MOF (PCN-222) was also proven to be a highly efficient photocatalyst for CO2 reduction under visible-light irradiation.80 PCN-222 was found to contain deep electron trap states, which inhibited electron−hole recombination and prolonged the lifetime of photo-excited electrons.
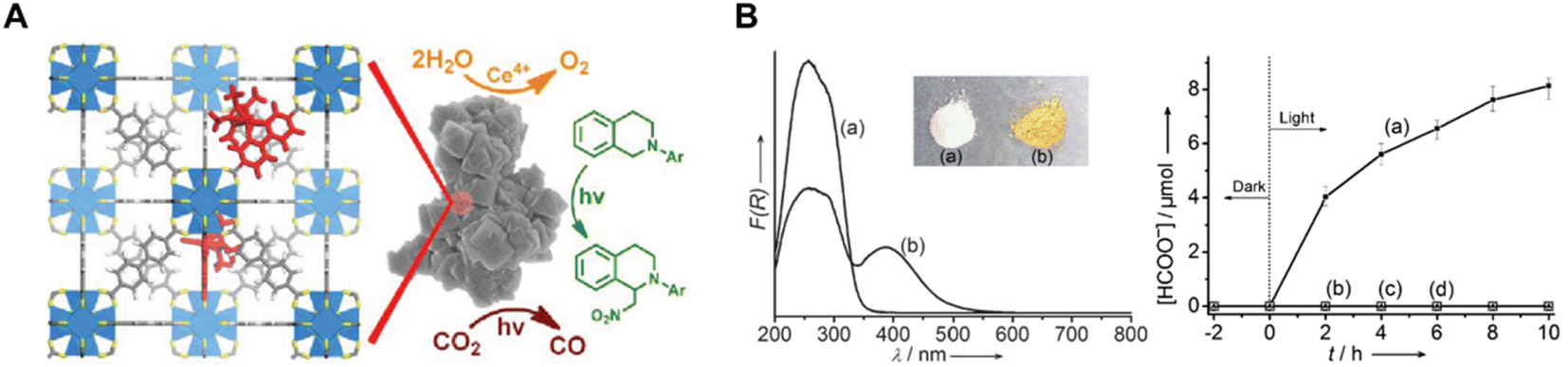 | ||
| Fig. 8 (A) Schematic illustration of the MOF [ReI(CO)3(bpydc)Cl] for photocatalytic CO2 reduction.78 Copyright 2011, American Chemical Society. (B) (left) UV/Vis spectra (a) MIL-125(Ti) and (b) NH2-MIL-125(Ti) and the inset shows a photo of the samples of the corresponding samples and (right) the amount of HCOO− produced as a function of the irradiation time.79 Copyright 2012, Wiley. | ||
By far, plenty of strategies have been adopted to achieve highly active photo-driven CO2 reduction using MOF-based materials.81 The methods of designing highly active MOF composites include improving the visible-light absorption and the charge-carrier separation.82 Adjusting organic ligands and functionalization of metal centers are common strategies for enhancing visible-light absorption and charge-carrier separation.82,83
2.2 Catalysts
In PC, PEC and PV-EC CO2 reduction systems, catalysts are generally needed.84,85 In the PC or PEC system, the photo-absorber may also act as the catalytically active ingredient, but the discussion in this part mainly focuses on the catalysts, which play a key role in improving the performance of photochemical CO2 reduction systems. The catalysts can promote photochemical CO2 reduction in the following ways: (i) by lowering the activation energy or overpotential required for CO2 reduction, (ii) by promoting the separation and transport of photo-excited electron–hole pairs, (iii) by improving the stability of the photoelectrodes or photo-absorbers through rapidly transferring the surface charge carriers and (iv) by increasing the product selectivity through tuning the adsorption strengths of key reaction intermediates.4,86Recently, there have been many well-designed electrocatalysts for electrocatalytic CO2 reduction that have been adopted in solar driven CO2 reduction systems. Driven by electricity, CO2 is converted into carbonaceous products on the electrocatalysts. As shown in Table 1, the production of general C1–C3 products: CO/HCOOH, HCHO, CH3OH, CH4/CH3COOH, CH3CHO, C2H5OH/C2H4, C2H6, C2H5CHO and C3H7OH requires the consumption of 2, 4, 6, 8, 10, 12, 14, 16 and 18 electrons, respectively.87 Although the equilibrium potentials of CO2 reduction are all close to 0 V vs. RHE, the overpotentials required are still high due to the reaction barriers. In addition, the similar equilibrium potentials of the various half-reactions of CO2 reduction and the competing hydrogen evolution reaction (HER) lead to a low target product selectivity of CO2 reduction. Hence, catalysts that facilitate the reaction kinetics and enhance the product selectivity are required. The most investigated materials are metal-based catalysts, which are widely studied from the aspect of geometric structures, compositional survey, mixing patterns of constituent metals, oxidation states, coordination structure and defect engineering to improve catalytic activity and product selectivity. Metal-free and hybrid catalysts, like bacteria and the hybrid materials combining bacteria and metals have also been used for electrocatalytic CO2 reduction. Over the past few decades, CO2 reduction electrocatalysts have been well studied and milestones have been achieved. Catalysts that convert CO2 into C1 products with FE close to 100% were reported in recent years.88–92 In another study, a FE of 85% for CH4 was also achieved on a single-atom Zn catalyst.93 Huang et al. reported copper nanowires with rich surface steps which converted CO2 into C2H4 with a FE of more than 70% over 200 h.94 Recently, long-chain C3 to C6 hydrocarbons were produced with a sustained FE of up to 6.5% using polarized nickel catalysts.95
| Reaction | Product | Number of electrons transferred | Equilibrium potentials (V vs. RHE) |
|---|---|---|---|
| CO2R | CO (g) | 2 | −0.10 |
| HCOOH (aq) | 2 | −0.12 | |
| HCHO | 4 | −0.09 | |
| CH3OH (aq) | 6 | 0.03 | |
| CH4 (g) | 8 | 0.17 | |
| CH3COOH (aq) | 8 | 0.11 | |
| CH3CHO (aq) | 10 | 0.06 | |
| C2H5OH (aq) | 12 | 0.09 | |
| C2H4 (g) | 12 | 0.08 | |
| C2H6 (g) | 14 | 0.14 | |
| C2H5CHO (aq) | 16 | 0.09 | |
| C3H7OH (aq) | 18 | 0.10 | |
| HER | H2 | 2 | 0 |
The well-designed CO2 reduction electrocatalysts promote the rapid development of catalysts for solar driven CO2 reduction. Metal-based catalysts (including mono-metals,96 bimetals,89,97 metal oxides,98 metal sulfides,99 metal carbides100) and biomimicking catalysts101 are commonly studied catalysts.
Li et al. prepared a Si/Ag photocathode by controllable chemical etching on a Si wafer by Ag+ ions (Fig. 9A).105 The resultant photocathode exhibited a large photocurrent density of −10 mA cm−2 under 0.5 Sun illumination (Fig. 9B), an excellent CO faradaic efficiency of 90% at −0.5 V versus the reversible hydrogen electrode (Fig. 9C) and an operational stability exceeding 12 h. Photoelectrochemical CO2 reduction to methanol, over a TiO2-passivated InP nanopillar photocathode, was greatly enhanced after the deposition of Cu NPs (Fig. 9D and E).106 To combine the catalytic effect and plasmonic effect of Au, Atwater et al. fabricated a gold/p-type gallium nitride (Au/p-GaN) Schottky junction tailored for photoelectrochemical studies involving plasmon-induced hot-hole capture and conversion.107 On the plasmonic Au/p-GaN photocathodes, hot holes from Au nanoparticles were injected into p-GaN upon plasmon excitation, since the vast majority of hot holes generated via interband transitions in Au were sufficiently hot to inject above the 1.1 eV interfacial Schottky barrier at the Au/p-GaN heterojunction (Fig. 9F).
 | ||
| Fig. 9 (A) Schematic illustration of how a Si/Ag photocathode is prepared by controllable chemical etching on a Si wafer by Ag+ ions (top) and SEM images of the Si/Ag photocathode preparation over time (bottom), (B) photocurrent density of the Si/Ag photocathode under 0.5 Sun illumination, (C) FE of the Si/Ag photocathode under 0.5 Sun illumination.105 Copyright 2018, Royal Society of Chemistry. (D) Schematic illustration of a TiO2-passivated InP nanopillar photocathode decorated with Cu NPs, (E) FE of a TiO2-passivated InP nanopillar photocathode and control samples.106 Copyright 2015, American Chemical Society. (F) Schematic illustration of gold/p-type gallium nitride (Au/p-GaN) Schottky junctions tailored for photoelectrochemical studies of plasmon-induced hot-hole capture and conversion.107 Copyright 2017, American Chemical Society. (G) Schematic diagram of the CO2 photoreduction mechanism by using Pt–TiO2 nanostructured films, (H) CO and CH4 yields of commercially available TiO2 powders (P25), pristine TiO2 columnar films (TiO2 film), and Pt–TiO2 films with different Pt deposition times.109 Copyright 2012, American Chemical Society. | ||
In PC CO2 reduction systems, metal co-catalysts, including Pt,108–110 Ag,111,112 Pd,113,114 Ru,115 Au116 and others, have been more widely studied. A platinized titanium dioxide (Pt–TiO2) nanostructured film was constructed based on unique one-dimensional (1D) columnar TiO2 single crystals coated with ultrafine 0.5–2 nm Pt nanoparticles (Fig. 9G).109 The Pt NPs improved the electron–hole pair separation and the electron-transfer rate in TiO2 single crystals, which enhanced the amount of photocatalytic CO2 reduction to CH4 with a maximum yield of 1361 μmol gcat−1 h−1 (Fig. 9H). Therefore, in some cases metals functioning as catalysts in photochemical CO2 reduction systems do not always display the same product selectivity as when they are used for electrochemical CO2 reduction,109 the mechanism of which needs further investigation.
It is important to note that bimetallic catalysts sometimes exhibit better catalytic activity for CO2 reduction than mono-metals,19 and the cost of the noble metal catalysts can be reduced by alloying noble metals with non-noble metals.86 Yang et al. created an integrated photoelectrode by directly assembling Au3Cu alloy NPs on TiO2-protected n+p-Si NW arrays (Fig. 10A).117 Well-dispersed Au3Cu NPs decorated Si NW arrays served as an effective CO2 reduction photoelectrode, exhibiting a high CO2-to-CO selectivity close to 80% at −0.20 V vs. RHE and remained stable for up to 18 h (Fig. 10B and C). A high-rate sunlight-driven conversion of diluted CO2 to hydrocarbons was achieved by constructing a Cu–Pt inner coating and modulated-diameter TiO2 nanotube photocatalyst.118 As illustrated in Fig. 10D and E, the core–shell nanotube arrays with the inner walls of the double-walled TiO2 nanotubes coated with bimetallic Cu0.33–Pt0.67 generated light hydrocarbons from low concentration CO2 at room temperature.
 | ||
| Fig. 10 (A) Schematic illustration of Au3Cu alloy NPs on TiO2-protected n + p-Si NW arrays, (B) and (C) FE and current density of hybrid Si NW arrays, respectively.117 Copyright 2016, American Chemical Society. (D) SEM image and XPS spectra of core–shell nanotubes with bimetallic Cu0.33–Pt0.67 coating on the inner walls of the double-walled TiO2 nanotube arrays, (E) CO2 conversion performance of core–shell nanotubes with bimetallic Cu0.33–Pt0.67 coating on the inner walls of the double-walled TiO2 nanotube arrays.118 Copyright 2012, Wiley. | ||
In addition to monometals and bimetals, metal oxides, metal sulfides and metal carbides have also been utilized as co-catalysts in solar driven CO2 reduction systems. A mixed-phase material consisting of CuFeO2 and CuO was prepared by cathodic deposition on FTO substrates (Fig. 11A), and was employed as the photoelectrocatalyst in a PEC CO2 reduction system.119 The selectivity of the CO2 reduction products could be adjusted from primarily acetate to primarily formate by varying the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu atomic ratio from 1.3 to 0.1 (Fig. 11B and C). Purging with H2S gas led to a spontaneous transformation of Cu to CuS, which presented a high FE of HCOOH.120 Inspired by this work, Mi et al. prepared a Cu/GaN/Si photocathode,121 and converted it into CuS/GaN/Si by exposing Cu/GaN/Si to an industrial CO2 gas environment (contaminated with H2S) during CO2 reduction. The CuS-decorated GaN/Si photocathode exhibited a superior FE of 70.2% for formate and partial current density of 7.07 mA cm −2 at −1.0 V vs. RHE under AM 1.5G illumination (Fig. 11D). In PC CO2 reduction systems, metal oxides, metal sulfides and metal carbides were studied more extensively as co-catalysts. RuO2 loaded mesoporous ZnGa2O4 exhibited a high photocatalytic activity for converting CO2 into CH4 under light irradiation.122 The two-dimensional MoS2–TiO2 hybrid nanosheet (Fig. 11E) performed an enhanced photocatalytic reduction of CO2 to methanol.123 As demonstrated by the photoluminescence (PL) emission spectra and normalized time-resolved photoluminescence (TRPL) decay traces (Fig. 11F and G), the MoS2-loaded TiO2 nanosheets promoted the electron transfer from TiO2 to MoS2 and minimized charge carrier recombination, improving the conversion efficiency of the photoreduction of CO2 to CH3OH. However, the real active sites during the catalytic reaction on the co-catalysts of metal oxides and metal sulfides should be studied by in situ and operando physical and chemical characterizations, since most of the metal oxides and metal sulfides are not stable under reduction conditions.
:Cu atomic ratio from 1.3 to 0.1 (Fig. 11B and C). Purging with H2S gas led to a spontaneous transformation of Cu to CuS, which presented a high FE of HCOOH.120 Inspired by this work, Mi et al. prepared a Cu/GaN/Si photocathode,121 and converted it into CuS/GaN/Si by exposing Cu/GaN/Si to an industrial CO2 gas environment (contaminated with H2S) during CO2 reduction. The CuS-decorated GaN/Si photocathode exhibited a superior FE of 70.2% for formate and partial current density of 7.07 mA cm −2 at −1.0 V vs. RHE under AM 1.5G illumination (Fig. 11D). In PC CO2 reduction systems, metal oxides, metal sulfides and metal carbides were studied more extensively as co-catalysts. RuO2 loaded mesoporous ZnGa2O4 exhibited a high photocatalytic activity for converting CO2 into CH4 under light irradiation.122 The two-dimensional MoS2–TiO2 hybrid nanosheet (Fig. 11E) performed an enhanced photocatalytic reduction of CO2 to methanol.123 As demonstrated by the photoluminescence (PL) emission spectra and normalized time-resolved photoluminescence (TRPL) decay traces (Fig. 11F and G), the MoS2-loaded TiO2 nanosheets promoted the electron transfer from TiO2 to MoS2 and minimized charge carrier recombination, improving the conversion efficiency of the photoreduction of CO2 to CH3OH. However, the real active sites during the catalytic reaction on the co-catalysts of metal oxides and metal sulfides should be studied by in situ and operando physical and chemical characterizations, since most of the metal oxides and metal sulfides are not stable under reduction conditions.
 | ||
| Fig. 11 (A) Schematic illustration of a mixed-phase material consisting of CuFeO2 and CuO for CO2 conversion, (B) and (C) CO2 reduction performance according to the ratio of Fe and Cu.119 Copyright 2017, American Chemical Society. (D) Schematic illustration of the CuS-decorated GaN/Si photocathode for CO2 reduction.121 Copyright 2021, American Chemical Society. (E) Schematic illustration of two-dimensional MoS2–TiO2 hybrid nanosheets, (F) PL emission spectra of MoS2–TiO2 hybrid nanosheets, (G) normalized TRPL decay traces of MoS2–TiO2 hybrid nanosheets.123 Copyright 2017, Royal Society of Chemistry. | ||
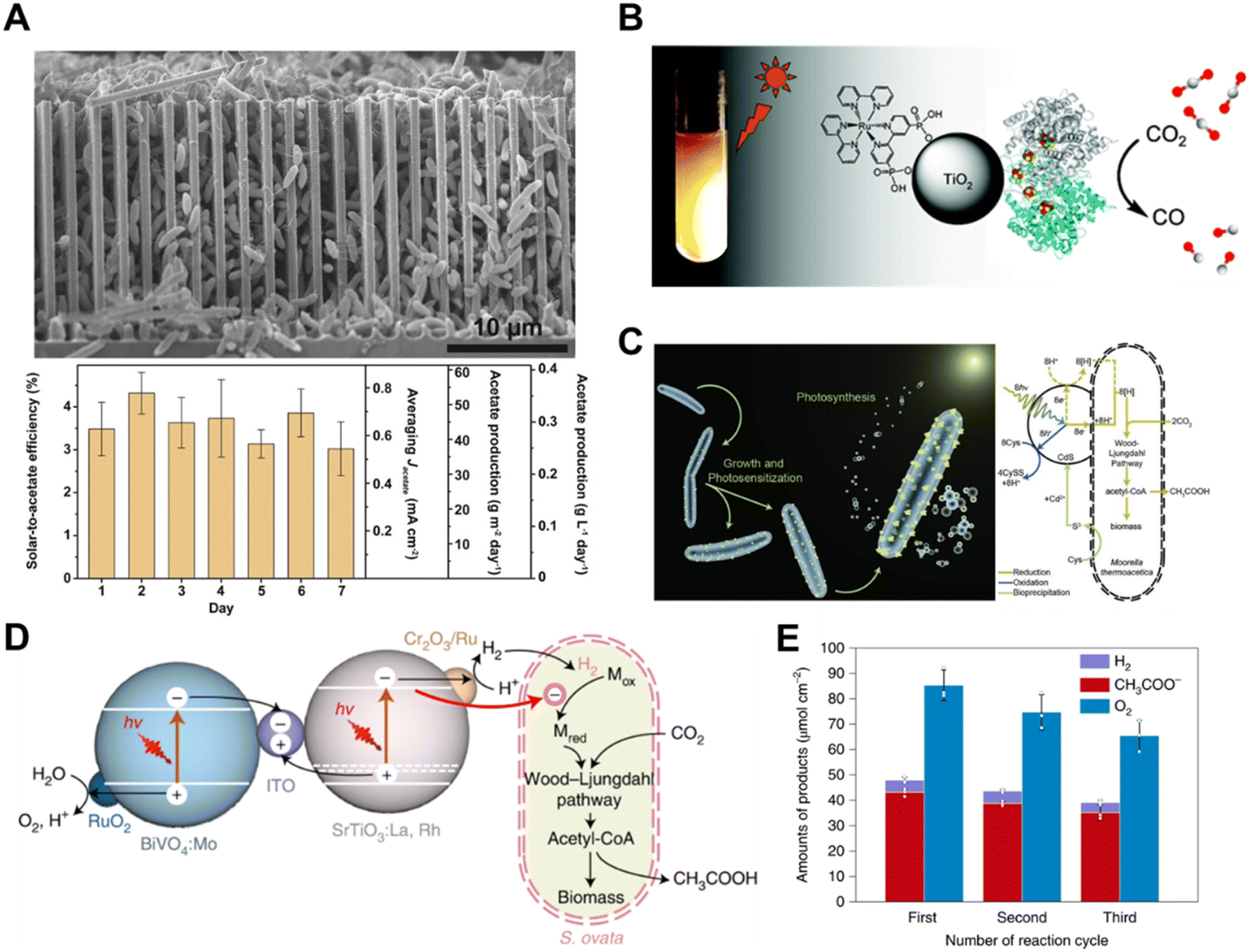 | ||
| Fig. 12 (A) SEM image of the silicon nanowire/Sporomusa ovata system.126 Copyright 2020, Elsevier. (B) Schematic illustration of a hybrid enzyme–nanoparticle system for CO2 conversion.124 Copyright 2010, American Chemical Society. (C) Schematic illustration of a non-photosynthetic bacterium integrated with cadmium sulfide nanoparticles for CO2 conversion. Bacteria–photocatalyst hybrid system for CO2 reduction.101 Copyright 2016, American Association for the Advancement of Science. (D) Schematic illustration of the mechanistic pathway of photosynthetic CO2-to-acetate conversion coupled with water oxidation over S. ovata|Cr2O3/Ru–SrTiO3:La,Rh|ITO|RuO2–BiVO4:Mo, and (E) reduction products accumulation for three runs of 15 h, with reloading of Cr2O3 for every cycle.127 Copyright 2022, Springer Nature. | ||
The diverse electrocatalysts available for use in the EC system greatly promote the development of the PV-EC system, since almost all the electrocatalysts can also be utilized in the PV-EC system. Except for the co-catalysts mentioned above, Cu based catalysts have unparalleled selectivity for hydrocarbons and multi-carbon products and single atom catalysts show high selectivity towards C1 products. However, the selectivity of reduction products in the PV-EC system is also affected by the operating voltage of PV. To achieve a highly selective PV-EC system, the optimum output voltage of the PV needs to be compatible with the electrocatalysts.
3. Devices: progress and challenges
Highly efficient solar driven CO2 reduction requires not only active materials for CO2 conversion, but also a rationally designed device. A higher CO2 conversion efficiency is achievable when the device has enhanced light absorption, improved mass transfer, optimal gaseous/liquid flow field management and the heat management from device engineering aspects.128,1293.1 Recent progress of PC devices
The PC devices that are commonly in use are slurry photoreactors and fixed bed photoreactors.129–131 According to the type of illumination method employed, the slurry photoreactors are further divided into externally illuminated and internally illuminated reactors.132,133 The catalytic reactions carried out in slurry photoreactors involve a slow mass transfer of gas–liquid–solid triple-phase and the solubility of CO2 in liquid is low, which limits the catalytic reaction rate. To simplify this process and improve the concentration of CO2 in the catalyst surroundings, fixed bed photoreactors involving a two-phase, gas–solid catalytic reaction were developed.134 In fixed bed photoreactors, the catalysts are fixed on various supports, such as glass,135 monoliths136 and packed beds.137In a recent study, Cu2O decorated reduced titania (RT) nanoparticles were packed to form a fixed bed in a continuous flow reactor (Fig. 13A).137 In this system, the optimized RT-Cu2O photocatalyst enabled CH4 evolution with a yield of 462 nmol g−1 (Fig. 13B). The reaction was conducted using highly diluted CO2 in water vapor under illumination for 6 h, and it exhibited excellent stability over seven testing cycles (42 h). The direct Z-scheme charge transfer mechanism taking place across a well-designed interface inhibited the photo-corrosion of Cu2O (Fig. 13C). This coupled with the packed bed flow reactor synergistically enabled the excellent stability. Zhang et al. set up a photoreactor which enabled a triple-phase photocatalytic CO2RR reaching a high CO2 reduction rate of 305.7 μmol g−1 h−1, which was approximately 8 times higher than that of the bi-phase system.138 The Ag decorated TiO2 supported on a gas diffusion layer (GDL) formed a triple-phase at the gas–water boundary (Fig. 13D), which greatly promoted the mass transfer of CO2 (Fig. 13E). Therefore, the triple-phase photocatalytic CO2RR system exhibited a high CO2 reduction performance.
 | ||
| Fig. 13 (A) Schematic illustration of the experimental setup for photocatalytic CO2 reduction involving a fixed bed of Cu2O decorated RT nanoparticles, (B) CH4 evolution rate over seven sequential 6 h tests of the optimized RT-Cu0.75 sample, (C) energy band alignment (in V vs. NHE at pH = 7), charge transfer and reaction mechanism for the described RT-Cu2O Z-scheme showing CO2 + H2O into CH4 transformation.137 Copyright 2020, Elsevier. (D) Schematic illustration of the tri-phase photocatalytic CO2RR system based on Ag–TiO2 supported at the gas–water boundary, (E) time-dependent CO2RR over Ag–TiO2 NPs in the water phase (left) and supported at the gas–water boundary (right). Insets schematically illustrate the photocatalytic processes of the two systems.138 Copyright 2022, Wiley. | ||
3.2 Recent progress of PEC devices
The H-type photo-electrolyzer has received the most practical lab use in the PEC system and the batch mode is the most common operation mode, in which the products can be accumulated for detection (Fig. 14A). However, the mass transfer limitation and the low CO2 solubility in aqueous solution promote the upgrading of the H-type photo-electrolyzer. Therefore, continuous flow PEC reactor (CFPR) devices have been developed for PEC CO2 reduction.139 With a hybrid p-type CuO/Cu2O semiconductor nanorod array as the photocathode, the flow system exhibited a high activity for producing alcohols (ethanol and isopropanol) with a liquid alcohol production rate of 0.22 ml m−2 h−1, which was approximately 6 times higher than the production rate of a batch mode electrolyzer design. The photocurrent density was significantly enhanced in the CFPR, benefiting from its high surface area-to-volume ratio provided by the narrow reaction channels (Fig. 14B). To highlight the significance of the reactor design, Andresen et al. constructed a continuous flow microfluidic PEC reactor with an α-Fe2O3/CuO thin film as the photocathode.140 The sandwich-type planar microfluidic photo-reactor was a one chamber reactor constituting an α-Fe2O3/CuO/FTO photocathode, Pt/FTO anode, laser cut PMMA parts and Surlyn gaskets without any separator between the electrodes (Fig. 14C). Under backside AM 1.5 solar irradiation, the microfluidic system converted CO2 to formate with an STF efficiency of 0.2% after 12 h illumination. Compared with the batch reactor, the continuous flow microfluidic PEC reactor promoted the formation of methanol.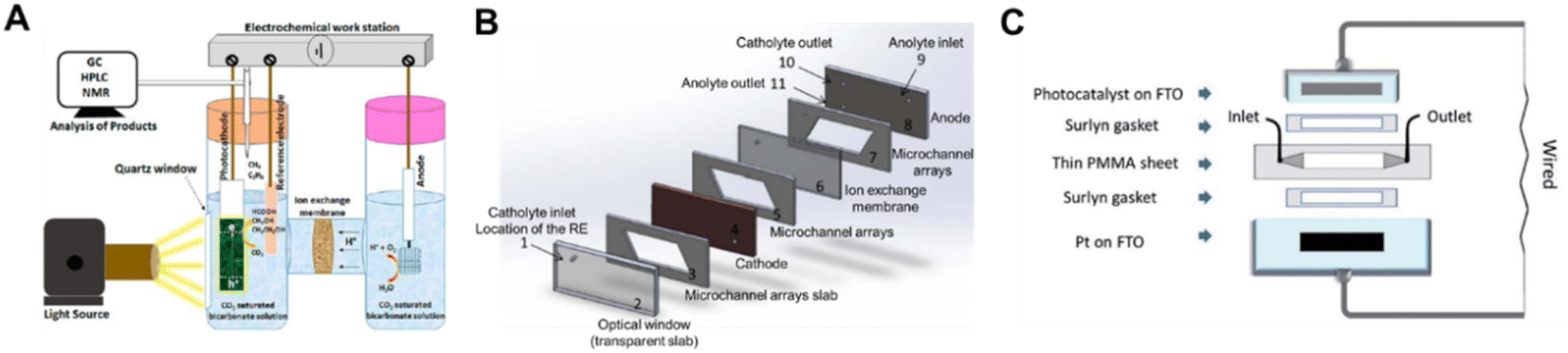 | ||
| Fig. 14 Schematic diagrams of PEC devices for CO2 conversion. (A) H-type PEC reactor.141 Copyright 2020, American Chemical Society. (B) Continuous flow PEC reactor.139 Copyright 2015, IOP Publishing. (C) Wired sandwich-type microfluidic PEC cell.140 Copyright 2019, Royal Society of Chemistry. | ||
In recent years, Reisner et al. constructed a series of highly integrated PV-PEC systems for CO2 reduction. By immobilizing a cobalt porphyrin catalyst on carbon nanotubes with triple-cation mixed halide perovskite and BiVO4 photoabsorbers, they demonstrated a highly tunable perovskite-BiVO4 tandem PV-PEC syngas production system (Fig. 15A).21 The tandem photocathodes exhibited a different syngas production ratio under different light intensities (Fig. 15B) for aqueous CO2 reduction and maintained one day continuous syngas production at a light intensity of 0.1 Sun (Fig. 15C). They further fabricated a lightweight perovskite-BiVO4 device with flexible substrates and carbonaceous protection layers.142 A wired floating Ti|BiVO4|TiCo-fPVK|GE|CoMTPP@CNT device was constructed by attaching a flexible perovskite photocathode (fPVK) with CoMTPP@CNT as the CO2 reduction catalyst. The BiVO4 photoanode was spin-coated with TiCoOx (TiCo) mixed with a contact adhesive, as the O2 evolution catalyst (Fig. 15D). With an active light irradiation area of 1.7 cm2, the Ti|BiVO4|TiCo-fPVK|GE|CoMTPP@CNT device achieved a STF efficiency of 0.053 ± 0.006% (CO) and 0.021 ± 0.004% (H2), as demonstrated in Fig. 15E. The impressive scalable integrated PV-PC device opens a new prospect for the application of solar driven CO2 reduction.
 | ||
| Fig. 15 (A) Schematic illustration showing the architecture of the standalone perovskite–BiVO4 PEC tandem device for bias-free syngas production, (B) amounts of products produced and CO:H2 selectivity after 4 h irradiation with different light intensities, (C) stability test under 0.1 Sun irradiation and the corresponding product amounts.21 Copyright 2020, Springer Nature. (D) Photo of the actual lightweight floating perovskite-BiVO4 device (top) schematic illustration of the floating perovskite-BiVO4 device showing the concurrent water-splitting and CO2 reduction reactions taking place (bottom), (E) photoelectrochemical performance of the floating perovskite-BiVO4 devices: CVs of small-scale individual perovskite and BiVO4 photoelectrodes assembled in a 1.7cm2 wired PEC device (left), chronoamperometric trace and amounts of products obtained for a 1.7 cm2 wired perovskite-BiVO4 device with a CoMTPP@CNT catalyst over 24 h at zero applied bias voltage (right).142 Copyright 2022, Springer Nature. | ||
As one of the key components, ion exchange membranes also play a key role in improving the performance of the photo-electrolyzer. Compared with anion exchange membranes, proton exchange membranes (PEMs) are preferred in H-type PEC devices with 0.1 M KHCO3 as the electrolyte, owing to their ability to allow proton transportation from the anode to the cathode.143 Recently, bipolar membranes (BPMs), which combine the characteristics of cation and anion exchange membranes, have gained great attention. Their high pH sensitivity enables the selective transport of OH− to the anode and H+ to the cathode under different electrolyte conditions.143–145 Xiang et al. constructed a solar driven CO2 reduction system with a TiO2 protected III−V tandem photoanode in conjunction with a bipolar membrane and a Pd/C cathode.145 Adopting two electrolytes with different bulk pHs, the bipolar membrane maintained a steady-state operation of the system, and the system achieved a CO2 to HCOOH FE > 94% (8.5 mA cm−2) and a ∼10% solar-to-fuel conversion efficiency.
3.3 Recent progress of PV-EC devices
The key aspects to make an efficient PV-EC CO2 reduction device are decreasing the reaction energy barriers, minimizing the electrical energy losses and tuning the selectivity of the desired products.146 The research on PV-EC for CO2 reduction has demonstrated higher STF efficiencies than PC and PEC devices,18,147–149 suggesting that it is a promising technology for direct CO2 conversion using solar energy.Morikawa et al. reported a monolithic tablet-shaped device with a solar-to-chemical energy conversion efficiency of 4.6% for CO2 photoreduction to formate by utilizing water as an electron donor under simulated solar light irradiation.150 In the monolithic tablet-shaped PV-EC device, a triple-junction of amorphous silicon–germanium (SiGe-jn) functionalizing as the PV, converted solar energy into electricity and powered the catalytic reaction. Concurrently, the porous ruthenium complex polymer (p-RuCP) as the CO2 reduction catalyst and iridium oxide (IrOx) as the water oxidation catalyst reduced CO2 into formate and oxidized water into O2, respectively (Fig. 16A). Different from the monolithic device, the Ager group used a four-terminal III–V/Si tandem solar cell configuration coupled with a nanostructured Cu–Ag bimetallic cathode (Fig. 16B), which yielded a solar conversion efficiency to hydrocarbons and oxygenates exceeding 5% under 1 Sun illumination (Fig. 16C).151 The Gratzel group constructed a more efficient device with an atomic layer deposited SnO2 on CuO nanowires as the cathode and a GaInP/GaInAs/Ge cell as the photovoltaic (Fig. 16D), producing a peak solar-to-CO conversion efficiency of 13.4% (Fig. 16E).13 In that work, the energy loss was minimized by matching the current density of the photovoltaic with the cathode catalyst. Recently, they also reported a solar driven CO2 reduction system which coupled a flow cell composed of a tin oxide surface dominant copper/tin-oxide catalyst with a III–V triple junction solar cell, reaching a solar-to chemical energy conversion efficiency approaching 20%.152 To further improve the STF efficiency of the PV-EC device, Salehi-Khojin et al. constructed a one compartment electrochemical flow cell consisting of a MoS2 nanoflakes (NFs)/GDL cathode and an IrO2 nanopowder/GDL anode.153 By modifying the catalyst and adjusting the electrolyte, the PV-EC device composed of the flow cell coupled to a triple-junction photovoltaic (TJ-PV) cell showed a CO2 to CO STF efficiency of 23% (Fig. 16F).
 | ||
| Fig. 16 (A) Schematic illustration of the monolithic tablet-shaped PV-EC device.150 Copyright 2015, Royal Society of Chemistry. (B) Solar driven CO2 RR measurements performed in a two-electrode configuration with a CuAg nanocoral cathode and an IrO2 nanotube anode in tandem with two series-connected solar cells and a maximum power point (MPP) tracker, (C) measured current density (left axis, red) and voltage (right axis, blue) of PV-EC (PV was two tandem cells connected in parallel) as a function of electrolyte concentration at 1 Sun illumination. Inset depicts the circuit diagram.151 Copyright 2017, Royal Society of Chemistry. (D) Schematic illustration of the device combining series-connected photovoltaics with an electrochemical cell, (E) photovoltaic and electrocatalytic J–V behaviours of the series-connected photovoltaic-electrochemical cell.13 Copyright 2017, Springer Nature. (F) Schematic illustration of the flow cell using solar energy to drive CO2 reduction reaction and photovoltaic and electrocatalytic J–V curves.153 Copyright 2019, Wiley. (G) Schematic illustration of large-sized cell with stacked electrode catalysts, (H) photographs of the large cell for solar driven CO2 conversion with stacked electrode catalysts, (I) LSV curves of the large cells equipped with the stacked FTO/Ag grid cathodes and Ti cathodes, and current (I)–voltage (V) characteristics of the six series-connected c-Si-PV cells under the AM 1.5G 1 Sun solar irradiation.47 Copyright 2021, Elsevier. | ||
Recently, a large-sized PV-EC system with a 1000 cm2 irradiation area composed of five stacked electrodes (electrically parallel connected) and six series-connected single-crystalline Si PV cells was constructed by Kato and co-workers.47 As illustrated in Fig. 16G and H, the anodes and cathodes (∼1000 cm2 in size) faced each other with a small distance of 1 cm in a transparent plastic housing without membrane separation and are directly connected to c-Si PV cells with similar sizes, which solves the proton diffusion resistance problem and avoids the use of a converter. The I–V curves of 6-series-connected single-crystalline Si PV cells and five stacked electrodes showed a good match between the EC reactor and PV cell (Fig. 16I), achieving a solar to formate conversion efficiency of 7.2%.
4. Conclusion and perspectives
At present, the highest achieved STF efficiencies for PC, PEC and PV-EC CO2 reduction systems are ∼1%,154 ∼10% (ref. 145) and ∼20%,152,153 respectively. The PC, PEC systems are still confronted with the problems of low STF efficiency and poor stability. Only the PV-EC CO2 reduction system showed an STF efficiency that meets industrial operation requirements. However, its high cost, poor stability and low selectivity for desired products limit its large-scale application. The photo-absorber, catalyst and device design, as three fundamental factors, collectively determine the solar-to-fuel efficiency of the system. It is essential to optimize the solar driven CO2 reduction systems from a three-pronged approach and construct a system with these three elements optimized synergistically.At present, the photo-absorbers suffer from low photo conversion efficiency and low stability. To improve the solar harvesting and conversion efficiency of the photo-absorbers, frequently used strategies are constructing heterojunction structures, elemental doping, nano-morphology control, defect engineering, amongst others. In addition, we can also explore new chemical functionalization, discover new semiconductors suitable for CO2 reduction and construct composite photo-absorbers with multiple materials, such as organic semiconductors combined with inorganic semiconductors, biomimetic materials/biomaterials combined with inorganic semiconductors, or other newly developed photo-absorbers.
The catalysts, which promote CO2 conversion, also require further development for reducing the reaction energy barriers and improving the product selectivity and stability. Although recent studies on the catalyst morphology control, bimetal or multi-metal construction, molecular catalyst modification, surface basic sites and the introduction of surface defects proved to be effective, there is still much work to be done. For example, we can construct a suitable reaction micro-environment that aims to achieve an appropriate local pH and surrounding ions for CO2 reduction by chemical modification of the catalyst or specified nano-structure design of the catalysts using 3D printing or other techniques. To solve the problem of low CO2 solubility in aqueous PC and PEC systems, materials with high CO2 adsorption capacity can be applied to the catalysts. Furthermore, the synergetic operation of photo-absorbers and catalysts requires further in-depth exploration. With a clear mechanism of the interaction of photo-absorbers and catalysts during photochemical CO2 reduction, a highly effective combination of photo-absorber and catalyst can be developed and the product selectivity can also be controlled by adjusting the combination strategy and mode.
The PC, PEC and PV-EC devices have been updated for several generations from the aspects of improving light absorption, enhancing the mass transfer and reducing the gas/liquid transfer resistance. As a result, the efficiency of CO2 conversion has gradually increased. However, an optimized design for the devices remains challenging and demands suitable solutions to address key scientific and engineering problems. Accordingly, the following factors should be considered for the design of the device: the light source illumination way (built-in light, fiber lamp, etc.), mode of operation (batch, continuous flow, etc.), parameters of the chamber (volume, thickness, geometrical configuration, the in/out flow pathway of reactants, etc.) and membrane characteristics (for PEC and PV-EC systems). Meanwhile, we can also improve the reaction rate of product yields by designing a high pressure device for PC, PEC and PV-EC systems, since the solubility of CO2 in aqueous media is a rate-limiting step in CO2 reduction.
Finally, the synergetic operation of each component of the system is important for achieving high efficiency. The three key components, an efficient photo-absorber, a catalyst with high product selectivity and an optimized device design, working together synergistically will greatly improve the solar to fuel efficiency. The effect of how the photo-absorber and catalysts are combined, the light illumination method, device integration, and the supply of reactants on system efficiency requires further and systematic investigation.
Besides the scientific research of solar driven CO2 reduction systems, great efforts should also be made on facilitating the industrial application of these technologies. The scale-up of existing high-performance systems is required in spite of the challenges faced with the optimization of the system parameters. For the scale-up of PC and PEC devices, it is necessary to design the devices in flow mode with multi-cells connected in series or parallels, and the problems of the increased resistance of aqueous solution and flowing gases need to be resolved. Furthermore, the efficient utilization of solar light must be considered when designing stacked devices. To improve the product yield rate of solar driven CO2 reduction systems in practical use, high intensity light can be adopted. However, the low solubility of CO2 in aqueous solution might hinder the reaction rate in this mode. We can refer to the flow cell design used for EC CO2 reduction that separates the CO2 gas from the aqueous solution and constructs a solid–liquid–gas triple-phase boundary for sufficient CO2 supply. Due to the rapid development of PV cells and EC cells, there is a strong drive for PV-EC devices to reach maturity and be employed in industrial application. Nevertheless, it is essential to further improve the STF efficiency, optimize the selectivity, reduce the cost and improve the stability before industrial scale implementation. With the advantage of efficient thermal and flow field management, the integrated stacked PV-EC device will be a promising approach in the future. Inspired by the positive effect of external fields and inner micro-environments on the performance of EC devices,91,155–159 the field or reaction environment effects should be thoroughly considered when designing the solar driven CO2 reduction systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. L. acknowledges the funding support from the National Key Research and Development Program of China (2019YFE0123400 and 2022YFE0114800), the Excellent Young Scholar Fund from the National Natural Science Foundation of China (22122903), and the Tianjin Distinguished Young Scholar Fund (20JCJQJC00260). L. W. acknowledges the funding support from the Natural Science Foundation of China (grant no. 22209081) and the fellowship of China Postdoctoral Science Foundation (grant no. 2021M690082). J. L. and L. W. acknowledge the funding support from the Major Science and Technology Project of Anhui Province (202203f07020007).References
- H. Balat and C. Öz, Energy Explor. Exploit., 2007, 25, 357–392 CrossRef CAS
.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 CrossRef CAS PubMed
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS
- W. J. Ong, L. K. Putri and A. R. Mohamed, Chem.–Eur. J., 2020, 26, 9710–9748 CrossRef CAS PubMed
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu and Y. Yan, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558–2568 CrossRef CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- S. Xu and E. A. Carter, Chem. Rev., 2018, 119, 6631–6669 CrossRef PubMed
- M. Kibria, F. Chowdhury, S. Zhao, B. AlOtaibi, M. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 1–8 Search PubMed
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 1–9 Search PubMed
- M. T. Winkler, C. R. Cox, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 1076–1082 Search PubMed
- C. E. Creissen and M. Fontecave, Adv. Energy Mater., 2021, 11, 2002652 CrossRef CAS
- D. Li, K. Yang, J. Lian, J. Yan and S. Liu, Adv. Energy Mater., 2022, 12, 2201070 CrossRef CAS
- T. N. Huan, D. A. Dalla Corte, S. Lamaison, D. Karapinar, L. Lutz, N. Menguy, M. Foldyna, S. H. Turren-Cruz, A. Hagfeldt and F. Bella, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 9735–9740 CrossRef CAS PubMed
- J. L. White, J. T. Herb, J. J. Kaczur, P. W. Majsztrik and A. B. Bocarsly, J. CO2 Util., 2014, 7, 1–5 CrossRef CAS
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed
- Q. Zhou, S. Ma and S. Zhan, Appl. Catal. B Environ., 2018, 224, 27–37 CrossRef CAS
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS PubMed
- N. Zhang, R. Long, C. Gao and Y. Xiong, Sci. China Mater., 2018, 61, 771–805 CrossRef CAS
- L. Liu, Y. Zhang and H. Huang, Solar RRL, 2021, 5, 2000430 CrossRef CAS
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC
- Z. Xiong, Z. Lei, Y. Li, L. Dong, Y. Zhao and J. Zhang, J. Photochem. Photobiol., C, 2018, 36, 24–47 CrossRef CAS
- Y. Zhang, Z. Zhao, J. Chen, L. Cheng, J. Chang, W. Sheng, C. Hu and S. Cao, Appl. Catal. B Environ., 2015, 165, 715–722 CrossRef CAS
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed
- A. Meng, B. Cheng, H. Tan, J. Fan, C. Su and J. Yu, Appl. Catal. B Environ., 2021, 289, 120039 CrossRef CAS
- A. Ahmad Beigi, S. Fatemi and Z. Salehi, J. CO2 Util., 2014, 7, 23–29 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS
- Y. Xu, Y. Jia, Y. Zhang, R. Nie, Z. Zhu, J. Wang and H. Jing, Appl. Catal. B Environ., 2017, 205, 254–261 CrossRef CAS
- S. K. Kuk, J. Jang, J. Kim, Y. Lee, Y. S. Kim, B. Koo, Y. W. Lee, J. W. Ko, B. Shin and J. K. Lee, ChemSusChem, 2020, 13, 2940–2944 CrossRef CAS PubMed
- J. Cheng, L. Wu and J. Luo, Chem. Phys. Rev., 2022, 3, 031306 CrossRef CAS
- K. Tennakone, A. Jayatissa and S. Punchihewa, J. Photochem. Photobiol., A, 1989, 49, 369–375 CrossRef CAS
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal. B Environ., 2017, 217, 485–493 CrossRef CAS
- F. Zhang, Y. H. Li, M. Y. Qi, Z. R. Tang and Y. J. Xu, Appl. Catal. B Environ., 2020, 268, 118380 CrossRef CAS
- M. Schreier, P. Gao, M. T. Mayer, J. Luo, T. Moehl, M. K. Nazeeruddin and S. D. T. andM. Grätzel, Energy Environ. Sci., 2015, 8, 855–861 RSC
- G. Liu, F. Zheng, J. Li, G. Zeng, Y. Ye, D. M. Larson, J. Yano, E. J. Crumlin, J. W. Ager, L. W. Wang and F. M. Toma, Nat. Energy, 2021, 6, 1124–1132 CrossRef CAS
- J. Gu, A. Wuttig, J. W. Krizan, Y. Hu, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Phys. Chem. C, 2013, 117, 12415–12422 CrossRef CAS
- U. Kang, S. H. Yoon, D. S. Han and H. Park, ACS Energy Lett., 2019, 4, 2075–2080 CrossRef CAS
- R. Hinogami, Y. Nakamura, S. Yae and Y. Nakato, J. Phys. Chem. B, 1998, 102, 974–980 CrossRef CAS
- S. K. Choi, U. Kang, S. Lee, D. J. Ham, S. M. Ji and H. Park, Adv. Energy Mater., 2014, 4, 1301614 CrossRef
- M. Kan, C. Yang, Q. Wang, Q. Zhang, Y. Yan, K. Liu, A. Guan and G. Zheng, Adv. Energy Mater., 2022, 26, 2201134 CrossRef
- I. Roh, S. Yu, C.-K. Lin, S. Louisia, S. Cestellos-Blanco and P. Yang, J. Am. Chem. Soc., 2022, 144, 8002–8006 CrossRef CAS PubMed
- N. Kato, S. Mizuno, M. Shiozawa, N. Nojiri, Y. Kawai, K. Fukumoto, T. Morikawa and Y. Takeda, Joule, 2021, 5, 687–705 CrossRef CAS
- J. Kim, S. Jeong, M. Beak, J. Park and K. Kwon, Chem. Eng. J., 2022, 428, 130259 CrossRef CAS
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS PubMed
- G. Zeng, J. Qiu, Z. Li, P. Pavaskar and S. B. Cronin, ACS Catal., 2014, 4, 3512–3516 CrossRef CAS
- M. Lessio and E. A. Carter, J. Am. Chem. Soc., 2015, 137, 13248–13251 CrossRef CAS PubMed
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC
- K. Yang, Z. Yang, C. Zhang, Y. Gu, J. Wei, Z. Li, C. Ma, X. Yang, K. Song and Y. Li, Chem. Eng. J., 2021, 418, 129344 CrossRef CAS
- J. Low, B. Dai, T. Tong, C. Jiang and J. Yu, Adv. Mater., 2019, 31, 1802981 CrossRef PubMed
- Z. Zhu, J. Qin, M. Jiang, Z. Ding and Y. Hou, Appl. Surf. Sci., 2017, 391, 572–579 CrossRef CAS
-
J. Luo, M. T. Mayer and M. Grätzel, in Organic-Inorganic Halide Perovskite Photovoltaics: From Fundamentals to Device Architectures, ed. N.-G. Park, M. Grätzel and T. Miyasaka, Springer International Publishing, Cham, 2016, pp. 285–305 Search PubMed
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 1–6 Search PubMed
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed
- R. Chen, G. Gao and J. Luo, Nano Res., 2022, 15, 10084–10089 CrossRef CAS
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed
- Y. X. Chen, Y. F. Xu, X. D. Wang, H. Y. Chen and D. B. Kuang, Sustain. Energy Fuels, 2020, 4, 2249–2255 RSC
- K. Su, G. X. Dong, W. Zhang, Z. L. Liu, M. Zhang and T. B. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 50464–50471 CrossRef CAS PubMed
- H. Li, J. Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 122–138 CrossRef CAS PubMed
- T. D. Nguyen, C. T. Dinh and T. O. Do, Chem. Commun., 2015, 51, 624–635 RSC
- M. Irfan, M. Sevim, Y. Koçak, M. Balci, Ö. Metin and E. Ozensoy, Appl. Catal. B Environ., 2019, 249, 126–137 CrossRef CAS
- G. Dong and L. Zhang, J. Mater. Chem., 2012, 22, 1160–1166 RSC
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal. B Environ., 2015, 176–177, 44–52 CAS
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS PubMed
- N. Sagara, S. Kamimura, T. Tsubota and T. Ohno, Appl. Catal. B Environ., 2016, 192, 193–198 CrossRef CAS
- Q. Wang, X. Wang, Z. Yu, X. Jiang, J. Chen, L. Tao, M. Wang and Y. Shen, Nano Energy, 2019, 60, 827–835 CrossRef CAS
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 86 CrossRef
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 1–14 CrossRef
- Q. Wang, Y. Zhang, H. Lin and J. Zhu, Chem.–Eur. J., 2019, 25, 14026–14035 CrossRef CAS PubMed
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed
- Y. C. Hao, L. W. Chen, J. Li, Y. Guo, X. Su, M. Shu, Q. Zhang, W. Y. Gao, S. Li and Z. L. Yu, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed
- I. I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali and G. Palmisano, Catal. Today, 2020, 340, 209–224 CrossRef CAS
- X. Li and Q. L. Zhu, EnergyChem, 2020, 2, 100033 CrossRef
- Z. Qin, J. Wu, B. Li, T. Su and H. Ji, Acta Phys.-Chim. Sin., 2021, 37, 2005027 Search PubMed
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS PubMed
- X. Chang, T. Wang, P. Yang, G. Zhang and J. Gong, Adv. Mater., 2019, 31, 1804710 CrossRef PubMed
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Small, 2020, 16, 2001847 CrossRef CAS PubMed
- Q. Yang, Q. Wu, Y. Liu, S. Luo, X. Wu, X. Zhao, H. Zou, B. Long, W. Chen and Y. Liao, Adv. Mater., 2020, 32, 2002822 CrossRef CAS PubMed
- L. Wan, X. Zhang, J. Cheng, R. Chen, L. Wu, J. Shi and J. Luo, ACS Catal., 2022, 12, 2741–2748 CrossRef CAS
- Q. Chen, K. Liu, Y. Zhou, X. Wang, K. Wu, H. Li, E. Pensa, J. Fu, M. Miyauchi, E. Cortés and M. Liu, Nano Lett., 2022, 22, 6276–6284 CrossRef CAS PubMed
- C. Cai, B. Liu, K. Liu, P. Li, J. Fu, Y. Wang, W. Li, C. Tian, Y. Kang and A. Stefancu, Angew. Chem., Int. Ed., 2022, 61, e202212640 CAS
- Q. Wang, K. Liu, K. Hu, C. Cai, H. Li, H. Li, M. Herran, Y.-R. Lu, T.-S. Chan, C. Ma, J. Fu, S. Zhang, Y. Liang, E. Cortés and M. Liu, Nat. Commun., 2022, 13, 6082 CrossRef CAS PubMed
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Nat. Catal., 2022, 5, 545–554 CrossRef CAS
- J. T. Song, H. Ryoo, M. Cho, J. Kim, J. G. Kim, S. Y. Chung and J. Oh, Adv. Energy Mater., 2017, 7, 1601103 CrossRef
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Catal. B Environ., 2017, 204, 548–560 CrossRef CAS
- J. Qu, X. Zhang, Y. Wang and C. Xie, Electrochim. Acta, 2005, 50, 3576–3580 CrossRef CAS
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed
- J. Low, L. Zhang, T. Tong, B. Shen and J. Yu, J. Catal., 2018, 361, 255–266 CrossRef CAS
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc. Faraday Trans. I, 1989, 85, 2309–2326 RSC
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed
- Y. Zhou, Y. Liang, J. Fu, K. Liu, Q. Chen, X. Wang, H. Li, L. Zhu, J. Hu, H. Pan, M. Miyauchi, L. Jiang, E. Cortés and M. Liu, Nano Lett., 2022, 22, 1963–1970 CrossRef CAS PubMed
- Y. Hu, F. Chen, P. Ding, H. Yang, J. Chen, C. Zha and Y. Li, J. Mater. Chem. A, 2018, 6, 21906–21912 RSC
- J. Qiu, G. Zeng, M.-A. Ha, M. Ge, Y. Lin, M. Hettick, B. Hou, A. N. Alexandrova, A. Javey and S. B. Cronin, Nano Lett., 2015, 15, 6177–6181 CrossRef CAS PubMed
- J. S. DuChene, G. Tagliabue, A. J. Welch, W.-H. Cheng and H. A. Atwater, Nano Lett., 2018, 18, 2545–2550 CrossRef CAS PubMed
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS
- W. N. Wang, W. J. An, B. Ramalingam, S. Mukherjee, D. M. Niedzwiedzki, S. Gangopadhyay and P. Biswas, J. Am. Chem. Soc., 2012, 134, 11276–11281 CrossRef CAS PubMed
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, J. Mater. Chem. A, 2015, 3, 11313–11319 RSC
- W. Kim, T. Seok and W. Choi, Energy Environ. Sci., 2012, 5, 6066–6070 RSC
- S. Bai, X. Wang, C. Hu, M. Xie, J. Jiang and Y. Xiong, Chem. Commun., 2014, 50, 6094–6097 RSC
- N. Sasirekha, S. J. S. Basha and K. Shanthi, Appl. Catal. B Environ., 2006, 62, 169–180 CrossRef CAS
- L. Cao, S. Sahu, P. Anilkumar, C. E. Bunker, J. Xu, K. S. Fernando, P. Wang, E. A. Guliants, K. N. Tackett and Y.-P. Sun, J. Am. Chem. Soc., 2011, 133, 4754–4757 CrossRef CAS PubMed
- Q. Kong, D. Kim, C. Liu, Y. Yu, Y. Su, Y. Li and P. Yang, Nano Lett., 2016, 16, 5675–5680 CrossRef CAS PubMed
- X. Zhang, F. Han, B. Shi, S. Farsinezhad, G. P. Dechaine and K. Shankar, Angew. Chem., Int. Ed., 2012, 51, 12732–12735 CrossRef CAS PubMed
- X. Yang, E. A. Fugate, Y. Mueanngern and L. R. Baker, ACS Catal., 2017, 7, 177–180 CrossRef CAS
- P. Shao, S. Ci, L. Yi, P. Cai, P. Huang, C. Cao and Z. Wen, ChemElectroChem, 2017, 4, 2593–2598 CrossRef CAS
- W. J. Dong, I. A. Navid, Y. Xiao, J. W. Lim, J. L. Lee and Z. Mi, J. Am. Chem. Soc., 2021, 143, 10099–10107 CrossRef CAS PubMed
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye and Y. Zhou, Angew. Chem., 2010, 122, 6544–6548 CrossRef
- W. Tu, Y. Li, L. Kuai, Y. Zhou, Q. Xu, H. Li, X. Wang, M. Xiao and Z. Zou, Nanoscale, 2017, 9, 9065–9070 RSC
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Solar RRL, 2020, 4, 1900546 CrossRef CAS
- Y. Su, S. Cestellos-Blanco, J. M. Kim, Y.-x. Shen, Q. Kong, D. Lu, C. Liu, H. Zhang, Y. Cao and P. Yang, Joule, 2020, 4, 800–811 CrossRef CAS
- Q. Wang, S. Kalathil, C. Pornrungroj, C. D. Sahm and E. Reisner, Nat. Catal., 2022, 5, 633–641 CrossRef CAS
-
P. Yue, in Photoelectrochemistry, Photocatalysis and Photoreactors, Springer, 1985, pp. 527–547 Search PubMed
- A. A. Khan and M. Tahir, J. CO2 Util., 2019, 29, 205–239 CrossRef CAS
- L. Wan, Q. Zhou, X. Wang, T. E. Wood, L. Wang, P. N. Duchesne, J. Guo, X. Yan, M. Xia, Y. F. Li, A. A. Jelle, U. Ulmer, J. Jia, T. Li, W. Sun and G. A. Ozin, Nat. Catal., 2019, 2, 889–898 CrossRef CAS
- S. Bai, H. Qiu, M. Song, G. He, F. Wang, Y. Liu and L. Guo, eScience, 2022, 2, 428–437 CrossRef
- I. H. Tseng, W. C. Chang and J. C. Wu, Appl. Catal. B Environ., 2002, 37, 37–48 CrossRef CAS
- P. Zhang, S. Wang, B. Y. Guan and X. W. D. Lou, Energy Environ. Sci., 2019, 12, 164–168 RSC
- M. Tahir and N. S. Amin, Appl. Catal. B Environ., 2013, 142, 512–522 CrossRef
- E. G. Look and H. D. Gafney, J. Phys. Chem. A, 2013, 117, 12268–12279 CrossRef CAS PubMed
- M. Tahir and N. S. Amin, Appl. Catal., A, 2013, 467, 483–496 CrossRef CAS
- S. Ali, J. Lee, H. Kim, Y. Hwang, A. Razzaq, J. W. Jung, C. H. Cho and S. I. In, Appl. Catal. B Environ., 2020, 279, 119344 CrossRef CAS
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Angew. Chem., 2022, 134, e202200802 Search PubMed
- H. Homayoni, W. Chanmanee, N. R. de Tacconi, B. H. Dennis and K. Rajeshwar, J. Electrochem. Soc., 2015, 162, E115 CrossRef CAS
- E. Kalamaras, M. Belekoukia, J. Z. Tan, J. Xuan, M. M. Maroto-Valer and J. M. Andresen, Faraday Discuss, 2019, 215, 329–344 RSC
- V. Kumaravel, J. Bartlett and S. C. Pillai, ACS Energy Lett., 2020, 5, 486–519 CrossRef CAS
- V. Andrei, G. M. Ucoski, C. Pornrungroj, C. Uswachoke, Q. Wang, D. S. Achilleos, H. Kasap, K. P. Sokol, R. A. Jagt, H. Lu, T. Lawson, A. Wagner, S. D. Pike, D. S. Wright, R. L. Z. Hoye, J. L. MacManus-Driscoll, H. J. Joyce, R. H. Friend and E. Reisner, Nature, 2022, 608, 518–522 CrossRef CAS PubMed
- S. Castro, J. Albo and A. Irabien, ACS Sustainable Chem. Eng., 2018, 6, 15877–15894 CrossRef CAS
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS
- S. Sultan, J. H. Kim, S. Kim, Y. Kwon and J. S. Lee, J. Energy Chem., 2021, 60, 410–416 CrossRef CAS
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS
- T. Sekimoto, S. Shinagawa, Y. Uetake, K. Noda, M. Deguchi, S. Yotsuhashi and K. Ohkawa, Appl. Phys. Lett., 2015, 106, 073902 CrossRef
- D. Ren, N. W. X. Loo, L. Gong and B. S. Yeo, ACS Sustain. Chem. Eng., 2017, 5, 9191–9199 CrossRef CAS
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC
- J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC
- J. Gao, J. Li, Y. Liu, M. Xia, Y. Z. Finfrock, S. M. Zakeeruddin, D. Ren and M. Grätzel, Nat. Commun., 2022, 13, 5898 CrossRef CAS PubMed
- M. Asadi, M. H. Motevaselian, A. Moradzadeh, L. Majidi, M. Esmaeilirad, T. V. Sun, C. Liu, R. Bose, P. Abbasi and P. Zapol, Adv. Energy Mater., 2019, 9, 1803536 CrossRef
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S. I. In, Energy Environ. Sci., 2018, 11, 3183–3193 RSC
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortés, S. A. Maier and M. Liu, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef CAS PubMed
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O'Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Sustain., 2022, 5, 563–573 CrossRef
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, Nature, 2020, 581, 178–183 CrossRef CAS PubMed
- K. Xie, R. K. Miao, A. Ozden, S. Liu, Z. Chen, C.-T. Dinh, J. E. Huang, Q. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O'Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |