
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrostructured silk fiber scaffolds with enhanced stretchability†
Martina
Viola
ab,
Gerardo
Cedillo-Servin
 ac,
Anne Metje
van Genderen
d,
Isabelle
Imhof
a,
Paula
Vena
ace,
Marko
Mihajlovic
b,
Susanna
Piluso
g,
Jos
Malda
af,
Tina
Vermonden
b and
Miguel
Castilho
*ce
ac,
Anne Metje
van Genderen
d,
Isabelle
Imhof
a,
Paula
Vena
ace,
Marko
Mihajlovic
b,
Susanna
Piluso
g,
Jos
Malda
af,
Tina
Vermonden
b and
Miguel
Castilho
*ce
aDepartment of Orthopaedics, University Medical Centre Utrecht, Utrecht, The Netherlands
bDivision of Pharmaceutics, Department of Pharmaceutical Sciences (UIPS), Faculty of Science, Utrecht University, Utrecht, The Netherlands
cDepartment of Biomedical Engineering, Eindhoven University of Technology, Eindhoven, The Netherlands. E-mail: m.dias.castilho@tue.nl
dDivision of Pharmacology, Department of Pharmaceutical Sciences (UIPS), Faculty of Science, Utrecht University, Utrecht, The Netherlands
eInstitute for Complex Molecular Systems, Eindhoven University of Technology, Eindhoven, The Netherlands
fDepartment of Clinical Sciences, Faculty of Veterinary Sciences, Utrecht University, Utrecht, The Netherlands
gSentryX B.V., Utrecht, The Netherlands
First published on 23rd August 2024
Abstract
Despite extensive research, current methods for creating three-dimensional (3D) silk fibroin (SF) scaffolds lack control over molecular rearrangement, particularly in the formation of β-sheet nanocrystals that severely embrittle SF, as well as hierarchical fiber organization at both micro- and macroscale. Here, we introduce a fabrication process based on electrowriting of aqueous SF solutions followed by post-processing using an aqueous solution of sodium dihydrogen phosphate (NaH2PO4). This approach enables gelation of SF chains via controlled β-sheet formation and partial conservation of compliant random coil structures. Moreover, this process allows for precise architecture control in microfiber scaffolds, enabling the creation of 3D flat and tubular macro-geometries with square-based and crosshatch microarchitectures, featuring inter-fiber distances of 400 μm and ∼97% open porosity. Remarkably, the crosslinked printed structures demonstrated a balanced coexistence of β-sheet and random coil conformations, which is uncommon for organic solvent-based crosslinking methods. This synergy of printing and post-processing yielded stable scaffolds with high compliance (modulus = 0.5–15 MPa) and the ability to support elastic cyclic loading up to 20% deformation. Furthermore, the printed constructs supported in vitro adherence and growth of human renal epithelial and endothelial cells with viability above 95%. These cells formed homogeneous monolayers that aligned with the fiber direction and deposited type-IV collagen as a specific marker of healthy extracellular matrix, indicating that both cell types attach, proliferate, and organize their own microenvironment within the SF scaffolds. These findings represent a significant development in fabricating organized stable SF scaffolds with unique microfiber structures and mechanical and biological properties that make them highly promising for tissue engineering applications.
1. Introduction
Natural polymers have become increasingly popular as bio-interactive materials for regenerative medicine (RM). Among these, silk fibroin (SF) stands out due to its biocompatibility, biodegradability, and exceptional mechanical properties,1,2 which stem from its unique chemical composition and hierarchical organization at different length scales. At the nanoscale, SF is composed of stiff β-sheet nanocrystals embedded in a more compliant semi-amorphous phase consisting of random coil chains. These nanostructures further assemble into fibrils at the microscale, which then bundle into fibers or threads at the macroscale (Fig. 1(A)).3,4 Although this complex assembly mechanism has not fully dilucidated, it is thought to occur during silk fiber spinning by spiders or worms, through the formation of a micellar-like pre-assembled silk protein that solidifies upon ejection from the spinning duct.5 This process takes place under ambient conditions and is accompanied by acidification of the surrounding environment, water loss, salt ion gradients (such as sodium and potassium), and exposure to shear and elongation forces, ultimately resulting in the transition of silk micelles to water-insoluble silk fibers composed of sericin-coated SF.5,6 | ||
| Fig. 1 Schematic representation of aqueous electrowriting of organized SF fiber structures and their post-processing for cell culture. (A) Extraction of SF from Bombyx mori cocoons and addition of PEO as rheological modulator. (B) Electrowriting of SF aqueous-based inks, and (C) post-processing treatment with NaH2PO4, to induce controlled formation of secondary β-sheet structures. (D) Assessment of viability and organization of conditionally immortalized proximal tubule epithelial cells (ciPTEC) and conditionally immortalized glomerular endothelial cells (ciGEnC) on flat and tubular SF fiber scaffolds with controlled architectures. | ||
Current methods to create biomimetic SF fibers often involve denaturing conditions during SF extraction from cocoons and employ processing techniques such as wet spinning,7 hand drawing,8 microfluidic systems,9 and electrospinning.10 However, these approaches alone often yield sub-optimal SF conformations that fail to form the native β-sheet crystalline structure, leading to inferior elastic properties compared to native SF fibers.11–13 To enhance β-sheet content and mechanical performance, post-processing of spun fibers is commonly carried out using organic solvent baths based on ammonia,14 hexafluoroisopropanol/methanol,15 hexafluoroacetone hydrate/methanol,16 and formic acid/methanol.12,17 However, these methods lead to the formation of highly compact β-sheets, resulting in either rigid and brittle, or very weak fibers that do not replicate the conformation and elasticity of native SF fibers.12,18–20 Recently, electrowriting—a manufacturing process akin to electrospinning that applies voltage on an extrusion nozzle to generate thin polymer jets that are collected in well-controlled patterns—has been shown as a novel approach to process SF aqueous solutions into microfibers.21 However, the challenge to obtain mechanically robust yet soft crosslinked electrowritten SF fibers has still not been addressed, which is indispensable for cell culture applications. Moreover, the use of organic solvents for tuning the secondary structure and mechanical properties of SF is not compatible with biomedical applications due to potential toxicity for cells. Alternatively, organic solvent-free approaches have been explored to stabilize water-soluble SF, including (a) biomimetic treatments inspired by native spider or silkworm spinning, such as pH changes and treatment in salt solutions (based on Na+, K+, Ca2+ and Cl−),22,23 and (b) treatments that artificially foster the formation of stabilizing covalent or hydrogen bonds, such as sonication, enzymatic crosslinking, and blending with other polymers to create semi-interpenetrating networks.24,25 However, most of these approaches still rely on other support materials or result in soft and brittle hydrogels with time-dependent properties, generally due to predominant β-sheet conformations.22,23
To address these limitations and control the nano/microstructure of SF fibers, a novel processing method is proposed in this study, based on combining aqueous SF electrowriting with post-processing in a sodium dihydrogen phosphate (NaH2PO4) solution, which is then implemented for the first time to generate microfiber scaffolds with well-controlled architectures for a diverse range of cell culture applications (Fig. 1). First, we hypothesized that electrowriting can emulate the shear and elongation process experienced in the spinning ducts of spiders and worms, thereby facilitating the formation of SF microfibers. Additionally, post-processing treatment in a phosphate-rich solution was evaluated as a method to induce controlled conformational changes in SF solutions, specifically for the formation of a mixture of β-sheet and random coil conformations. This was expected to yield more robust stability and mechanical properties that differ from SF hydrogels with a high degree of β-sheet crosslinking, such as those obtained with more common post-processing techniques, e.g. methanol treatment.18,26 Furthermore, SF electrowriting offers the potential to fabricate SF fiber constructs with controlled microgeometries, overcoming limitations of previous silk spinning methods in terms of patterning control, reproducibility, and consequent ability to mimic tissue morphology and biomechanics.21,27 In this work, water-soluble SF inks were developed and optimized, and their suitability for electrowriting of flat and tubular macrostructures with controlled microarchitectures was investigated. The effect of post-printing treatment with NaH2PO4 on controlling the secondary structure of SF was assessed, and the resulting mechanical properties of printed constructs were investigated under both quasi-static and dynamic conditions. Finally, the ability of SF microfiber scaffolds to support the growth and viability of kidney cells, specifically conditionally immortalized proximal tubule epithelial cells (ciPTEC) and conditionally immortalized glomerular endothelial cells (ciGEnC), was evaluated to assess the potential of SF fiber scaffolds for RM applications.
2. Results and discussion
2.1 SF biomaterial ink optimization for aqueous electrowriting
To achieve consistent and durable fibers for manufacturing ordered 3D structures using aqueous electrowriting, the development of high-viscosity inks is crucial.28,29 During degumming, the boiling process causes SF chain breakage, leading to a decrease in molecular weight and the SF solution viscosity. Increasing SF concentration above 20% resulted in inks with high viscosities, it led to electrical instabilities during jet formation due to higher electrical conductivity (Fig. S1†).30 To circumvent this issue, SF concentration was fixed set at 20%, which showed electrical conductivity values below 3 mS cm−1. Ink viscosity was further adjusted by varying the degumming time and incorporating a secondary polymer, poly(ethylene oxide) (PEO). Increasing PEO concentration up to 3% led to an increase in ink viscosity to a value of approximately 3 Pa s, while the highest viscosity was observed for the highest PEO concentration PEO (3%) in combination with the shortest SF degumming time of 5 min (Fig. 2A). The measured viscosity values were consistent with those reported for other natural polymers used in solution-electrospinning processes.31 Among the tested formulations, the ink comprising a 20% SF solution, a short degumming time of 5 min, and 2.5–3% of PEO (SF5DT-3%) exhibited no electrical instabilities during jet formation, which allowed for stable fiber deposition and was therefore selected for further investigations (Fig. 2B).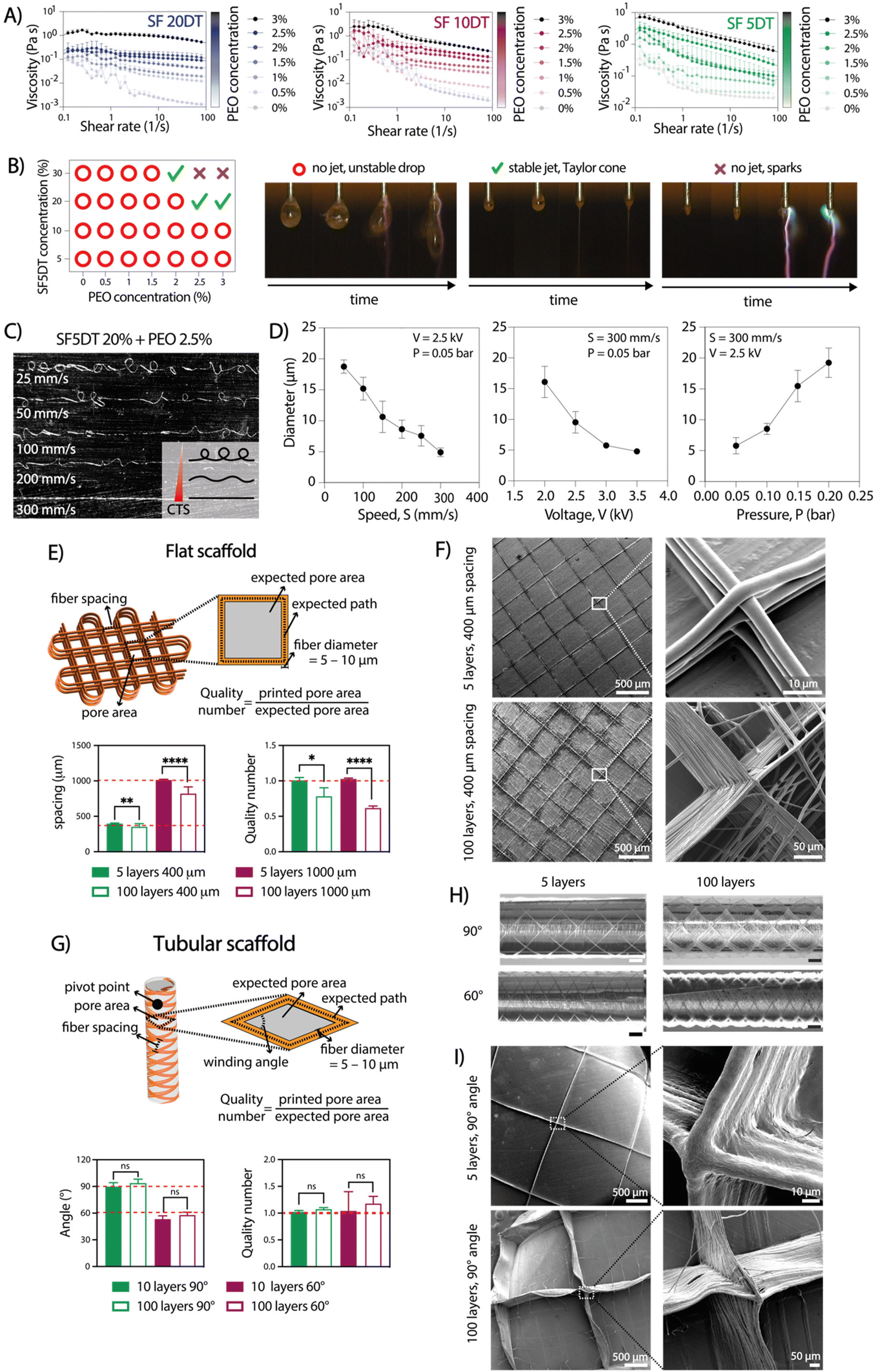 | ||
| Fig. 2 Aqueous electrowriting of rheologically optimized SF inks for controlled fabrication of flat and tubular microfiber scaffolds. (A) Viscosity screening of SF solutions at 20% with increasing degumming time (5, 10 and 20 min DT) and with increasing PEO concentration from 0 to 3% (n = 3). (B) Evaluation of SF ink on jet formation. (C) Effect of printing speed on the morphology of electrowritten microfibers, going from coiled or sinusoidal to straight fibers at or above the critical translation speed (CTS). (D) Effect of key printing parameters on the diameter of electrowritten fibers (n = 3). (E) Scaffold design, measured fiber spacing, and quality number (Q) for flat scaffolds with 5 and 100 layers and with 400 μm and 1000 μm fiber spacing (n = 10). (F) SEM images of flat scaffolds. (G) Scaffold design, measured winding angle, and quality number (Q) for tubular scaffolds with 60° and 90° winding angle and with 10 and 100 layers (n = 10). (H) Light microscope images of tubular scaffolds with 90° and 60° winding angles with 10 pivot points; scale bars: 1 mm. (I) SEM images of tubular scaffolds (unpaired multiple t-tests, *p < 0.05, **p < 0.01, ****p < 0.0001). | ||
The influence of key electrowriting parameters on SF fiber morphology and diameter was investigated. A collector speed of 300 mm s−1 enabled the deposition of straight fibers, as this was found to be above the critical translation speed (CTS) for the voltage and pressure values tested (Fig. 2C). Moreover, by individually varying the collector speed, applied voltage, and pressure, straight fibers with diameters ranging from approximately 5 to 20 μm were obtained (Fig. 2D). Varying the collector distance (Cd) between 8 and 2 mm did not yield a significant effect on the fiber diameter, but a Cd value above 6 mm resulted in jet instabilities and poor control over fiber deposition. A combination of printing parameters, including a collector speed of 300 mm s−1, an applied voltage of 2.5 kV, and pressure of 0.05 bar, was selected for subsequent tests due to its ability to generate a stable jet and produce straight fibers with a consistent diameter throughout the printing process.
2.2 Electrowriting of controlled microfiber shapes
To produce 3D scaffold structures with controlled microarchitectures, we evaluated the ability to stack SF microfibers for both flat and tubular shaped scaffolds. For flat scaffolds, inter-fiber distances of 400 and 1000 μm were evaluated, resulting in pore sizes with theoretical areas of 0.08 mm2 and 1 mm2, respectively (Fig. 2E and F). Optical and SEM imaging analysis confirmed the controlled stacking of SF fibers up to 100 layers. As expected, a lower printing accuracy (as defined by quality number) was achieved for 100 layers scaffolds compared to those with 5 layers. Surprisingly, scaffolds with 400 μm inter-fiber distances resulted in higher quality number (=0.78) than scaffolds with 1000 μm (∼0.62) (Fig. 2E). Additionally, when the fiber spacing was set below 400 μm, unstable jets were observed due to repulsive forces generated between deposited fibers and polymer jet, which is consistent with previous findings.21 Consequently, the observed distances between the fibers were smaller than the theoretical values.32 For tubular scaffolds, winding angles of 60° and 90° were investigated, with theoretical pore areas of 1.82 mm2 and 3.16 mm2, respectively (Fig. 2G–I).33 Optical and SEM analysis confirmed the accurate printing of tubular scaffolds with up to 100 stacked layers for both winding angles. Interestingly, printing accuracy did not lower with an increase in the number of stacked fibers, which is opposite to the observation for flat constructs (Fig. 2G). Overall, the observed instabilities seemed to become more pronounced as the number of layers increased. Similarly to a previous study by Hai et al.,21 we found that jet stability and the printing accuracy were also influenced by environmental factors such as temperature and humidity, which are challenging to control and seem to play a particularly bigger role during electrowriting of aqueous-based materials. Consequently, it may not always be possible to explain the differences in resolution between two theoretically identical scaffolds. Nevertheless, the small standard deviation in the printing accuracy data demonstrates that the process has appropriate reproducibility.2.3 Aqueous electrowriting of SF fibers and post-treatment induce controlled formation of β-sheet crystalline structure
The produced scaffolds exhibited a well-organized and precise 3D structure. However, SF remained soluble in water after electrowriting, causing the printed scaffolds to lose their shape or dissolve within a few minutes upon immersion in aqueous solutions. Previously, ions in solution have been reported to have a crosslinking effect in SF (Table S1†). Thus, to address this challenge, we explored the effect of elevated ion concentrations, taking inspiration from the natural crosslinking process of (spider) silk. However, treatment with potassium, sodium, chloride, and sulfate ions individually resulted in unstable scaffolds that dissolved in water (data not shown). Notably, the treatment with NaH2PO4 led to physical crosslinking and the formation of insoluble SF fiber scaffolds over at least 28 days in PBS at 37 °C (Fig. S2†). Therefore, the treatment of SF with 5 min of degumming time (SF5DT) in NaH2PO4 solution (SF + NaH2PO4) was compared to the standard methanol crosslinking method (SF + MeOH).26 Raman spectroscopy was used to investigate the secondary structure resulting from the different treatments (Fig. 3A). Upon close examination of the spectra, it was observed that in untreated SF, the intensity of the β-sheet bands was relatively low, indicating a predominating random coil secondary structure of the SF protein.25,26 However, both SF + MeOH and SF + NaH2PO4 samples exhibited a more intense, narrower β-sheet band (1200–1300 cm−1), with SF + MeOH showing a higher intensity ratio of β-sheet to coil than SF + NaH2PO4. SF + MeOH also exhibited a shift in the amide I peak from 1666 cm−1 to 1662 cm−1, indicating a strong increase in β-sheet formation, whereas this shift was not observed in the NaH2PO4-treated sample.26 To validate the observation that NaH2PO4 induces different SF conformations than MeOH, small-angle X-ray scattering (SAXS) analysis was performed (Fig. 3B). In the SAXS patterns of all three SF groups and native silk, peaks centered at momentum transfer values of q = 0.65, 1.45, and 1.70 Å−1 were observed, ascribed to the (010) reflection, an overlap of the (020) and (210) reflections, and the (021) reflection, respectively, as reported previously.34 Especially at q = 1.45 Å−1, native silk, untreated dried SF, and SF + MeOH samples all show more prominent peaks and a well-defined scattering profile compared to SF + NaH2PO4, thus indicating that NaH2PO4 treatment leads to a more balanced coexistence of random coils and β-sheets than MeOH-treated or dried SF. These findings are consistent with previous studies suggesting that ionic charges including phosphate ions can facilitate the alignment of silk molecules into β-sheets.35 Importantly, these observations suggest that MeOH has a stronger ability to induce β-sheet formation than NaH2PO4, and thus SF + NaH2PO4 possesses a more balanced mixture of compliant random coils and stiff β-sheets, while MeOH can lead to excessive formation of β-sheet crystallites.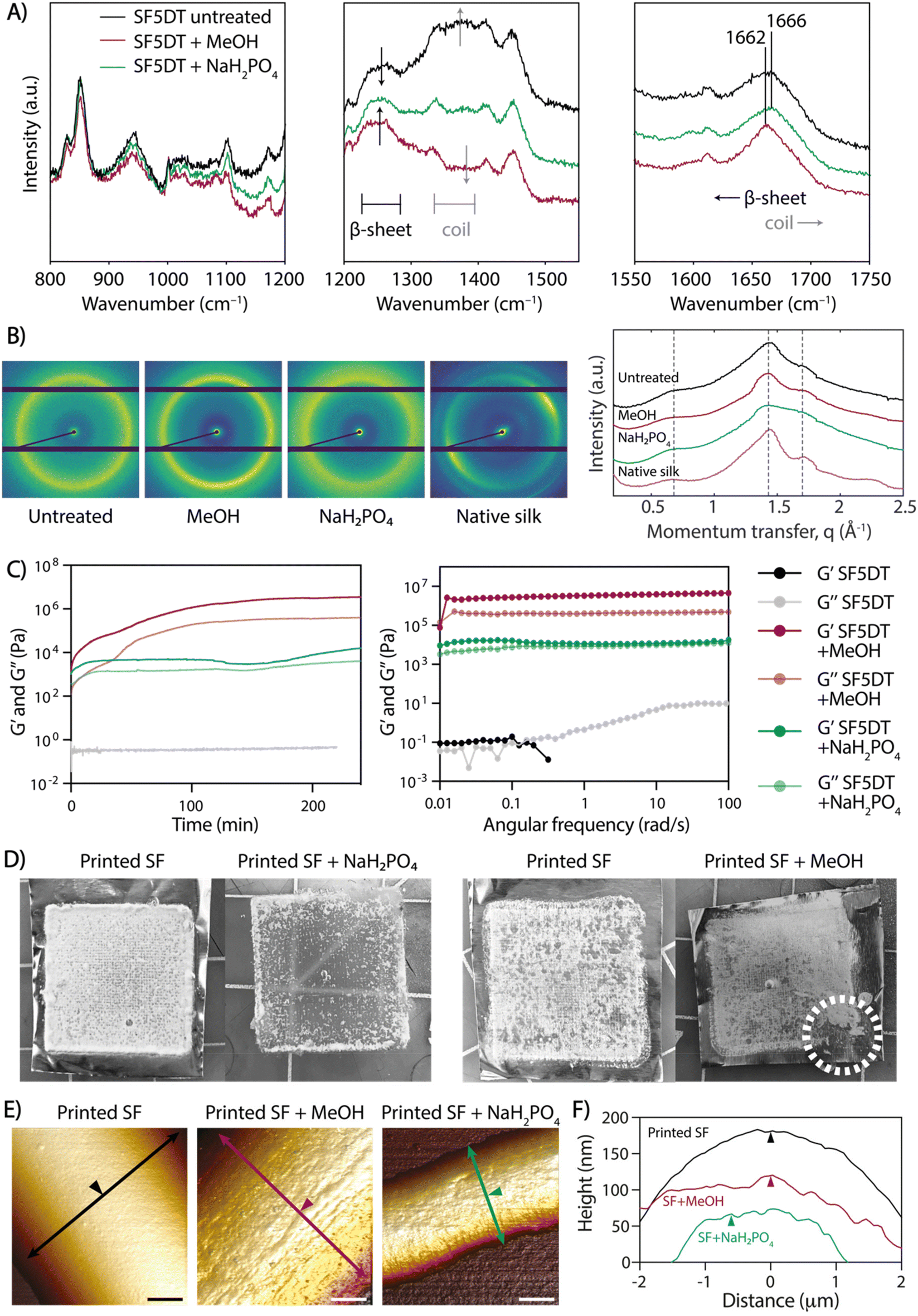 | ||
| Fig. 3 Effect of post-printing treatments on the crosslinking, crystalline formation, and topography of SF microfibers obtained by aqueous electrowriting. (A) Raman spectra of untreated SF, MeOH-treated SF, and NaH2PO4-treated SF, showing absorption bands for random coils and β-sheet crystals. (B) 2D SAXS patterns and 1D profiles of untreated dry and crosslinked SF and native silk fibers, showing characteristic reflections for SF. (C) Rheological analysis of 20% SF + 2.5% PEO crosslinked with MeOH and with NaH2PO4, performed on bulk SF material. (D) Effect of the different crosslinking treatment: stability of printed SF scaffolds in aqueous solutions. White dotted line indicates the breakage in a MeOH-treated SF scaffold. (E) 3D visualization of AFM topography of single fibers in representative images and (F) height profiles of electrowritten SF single fibers before (printed SF) and after crosslinking treatment (printed SF + MeOH and printed SF + NaH2PO4); scale bars: 2 μm. Arrows are perpendicular to fiber axis and show location of height profile; arrowheads indicate equal positions in (E) and (F). | ||
The rheological properties were investigated in SF inks processed with both MeOH and NaH2PO4 crosslinking methods, and time sweep measurements demonstrated that gelation occurred within a few seconds in both cases (Fig. 3C). The shear modulus of SF + NaH2PO4 was lower by almost two orders of magnitude than that of SF + MeOH, which could be attributed to the higher prevalence of stiffer β-sheet structures in SF + MeOH and the higher content of compliant random coils in SF + NaH2PO4 (Fig. 3C). Degumming time was also found to affect the moduli of individual SF inks when exposed to both crosslinking methods. As expected, an increase in degumming time led to a decrease in SF ink modulus (Fig. S3†), thus validating the strong dependence of gel rheology on degumming time.
Additionally, the effect of crosslinking treatment on the stability of SF fibers was evaluated. While both bulk SF + MeOH and SF + NaH2PO4 crosslinked gels are stable in PBS over time (Fig. S4†), printed SF + MeOH microfibers exhibited high brittleness, which hindered their handling. Upon comparing scaffolds before and after crosslinking with NaH2PO4 and MeOH (Fig. 3D), it was observed that upon contact with NaH2PO4 solution, the scaffolds immediately turned opaque, and they eventually detached from the aluminum collecting surface after soaking overnight. In contrast, MeOH-treated scaffolds were brittle to the touch and impossible to remove from the collector surface without breaking them. The ease of handling and mechanical stability of SF + NaH2PO4 scaffolds can be attributed to a lower density of β-sheets, which can be surrounded by random coils that potentially allow for greater fiber deformations and reduce brittleness, as observed in native spider silk.36
To assess the topography of individual SF fibers exposed to different crosslinking conditions, AFM analysis was performed. The surface texture of crosslinked SF fibers exhibited directional nano-striations, which aligned with the fiber's main axis and appeared on the SF fiber surface only after MeOH or NaH2PO4 treatment due to SF protein refolding into secondary structure (Fig. 3E, F and Fig. S5†). The combination of (a) shear-induced chain alignment during the electrowriting process and (b) fiber dehydration upon post-processing treatment contributes to the formation of highly aligned nanofibrils, which were not visible in untreated SF fibers (Fig. 3E and F). This directional topography is in agreement with previous reports of hierarchical mesostructures and spontaneous fibril self-assembly in silk proteins.37,38 Additionally, the 3D microstructure of SF + NaH2PO4 tubular scaffolds was analyzed by micro-computed tomography (μCT; Fig. S6†), revealing a mean strut thickness of 166 ± 48 μm and a scaffold porosity of 97.1%.
2.4 Mechanical analysis of post-processed electrowritten scaffolds
Uniaxial tensile tests were conducted on SF + NaH2PO4 flat scaffolds with fiber spacing of 400 μm and 1 mm, as well as on tubular scaffold with 90° and 60° winding angle (both 100 layers) to investigate the impact of crosslinking on SF scaffold elasticity. After NaH2PO4 treatment, both flat and tubular scaffolds were tested along the main plane and radially, respectively, under uniaxial tension while immersed in PBS to maintain equilibrium swelling of crosslinked SF (Fig. 4A and B). The average elastic modulus of flat scaffolds with 400 μm spacing was higher (1.14 ± 0.27 MPa) than that of the 1 mm spacing (0.752 ± 0.392 MPa), although not significantly higher (Fig. 4A). Tubular scaffolds exhibited a toe region at low strains due to gradual pore elongation along the tensile axis (Fig. 4B). The 90° winding angle scaffolds showed a wider toe region but lower modulus (2.78 ± 1.46 MPa) compared to the 60° winding angle scaffolds (12.0 ± 3.86 MPa), which had fewer elongated pores. Overall, the elastic modulus of SF + NaH2PO4 scaffolds regardless of printing setup was lower than those obtained through common SF processing methods (Table S1†). This is in agreement with the higher stiffness and excessive predominance of β-sheet conformations or covalent crosslinking in recently developed SF spinning or casting approaches,39 whereas the electrowriting process and NaH2PO4 treatment here reported show a considerable presence of random coils that increase SF compliance, while still allowing for mechanical robustness due to the controlled formation of β-sheet crystallites. | ||
| Fig. 4 Mechanical characterization of NaH2PO4-treated electrowritten SF microfiber scaffolds. Uniaxial tensile testing of (A) flat scaffolds and (B) tubular scaffolds: (i) side-view schematics and (ii) top-view photographs of the testing setup; (iii) representative uniaxial tensile curves and (iv) tensile properties (n = 3–5; unpaired t-test with Welch's correction). (C) Cyclical loading and shape recovery of flat scaffolds (400 μm spacing): (i) representative tensile curves, and (ii) representative photographs of scaffold under uniaxial cyclical loading; (iii) snapshot of a flat scaffold delivered through silicone tubing, showing scaffold shape recovery in water. (D) Radial stretching of a tubular scaffold (60° winding angle) using a balloon catheter: (i) schematic of loaded and unloaded states; (ii) snapshots of scaffold stretching and unloading under different pneumatic pressures. | ||
Invasive surgical procedures, as demonstrated in previous studies, are currently utilized for the application of cardiac patches,40 vascular stents,41 and kidney stents.42 However, these procedures are not ideal, and less invasive approaches are preferred. To showcase the versatility of our scaffolds, we tested their shape recovery properties, both for flat and tubular shapes. Two tests were performed: cyclic strain-recovery tests on the scaffold and a catheter analysis to simulate the mechanical environment to which the scaffold may be exposed during microsurgery (Fig. 4C and D). Uniaxial cyclic tests were performed on flat scaffolds (Fig. 4C), involving three sets of 10 cycles with increasing maximum strain with values up to 20% (total of 30 cycles). The scaffold showed complete elastic behavior for strains up to 10%, recovering its original shape between cycles without plastic deformation. However, at strains of 20%, an onset of plastic deformation was observed, resulting in incomplete shape recovery (Fig. 4C(i) and Fig. S7A†). Notably, the SF + NaH2PO4 scaffold exhibited these shape recovery properties, while the SF + MeOH scaffold did not, as it appeared to be more fragile and prone to rupture. Similar behavior was observed for tubular scaffolds, although the uniaxial cyclic tests did not fully replicate the physiological conditions due to the absence of radial deformation, as typically seen in blood vessels and kidney tubules. Flat scaffolds with 400 μm and 1000 μm inter-fiber spacing were found to be optimal for flowing inside silicone catheter tubing (inner diameter = 500 μm), showing no signs of rupture, kink formation, or plastic deformation (Fig. 4C(iii)).
Furthermore, tubular scaffolds were subjected to a balloon catheter test to assess radial shape recovery (Fig. 4D). A deflated balloon catheter was introduced into the tubular scaffold and then inflated to full extension (pressure up to 5 bar). Catheter swelling caused the scaffold fibers to stretch radially without rupture, and subsequent deflation of the catheter enabled the scaffold to restore its original shape and size. The swelling/deswelling of the balloon was applied multiple times (3 times) without any observation of plastic deformation (initial tube diameter = 3 mm; tube diameter after 3 cycles ∼ 3 mm) (Fig. S7B†).
Published studies have focused on maximizing the strength and toughness of SF fibers, given the long-standing historical ambition to replicate the impressive mechanical properties of native spider or worm silk.22 However, in many tissue engineering and cell culture applications, scaffolds mostly operate at low strains, and seeded cells generally experience mechanical properties of the scaffold in equilibrium or at rest. For epithelial and endothelial tissues, for example, low elastic moduli are relevant and even necessary to avoid the introduction of pro-fibrotic cues arising from cell interactions with stiffer substrates.43 Most methods to spin SF lead to fibers with high modulus due to the predominance of β-sheet conformations, and these methods generally overlook the option of a controlled or limited SF refolding into β-sheets, thus allowing for coexistence with random coils and leading to lower moduli.39 Here, electrowriting followed by NaH2PO4 post-processing provided SF fibers with elastic modulus values in the range of native vascular and kidney tissues, while approaching the length scales of features in the native epithelial and endothelial tissue, such as the thickness of kidney proximal tubule and endothelial basement membranes, as well as key components of the vascular ECM such as elastin (Fig. S8†). Moreover, electrowriting provided high control over the deposition of SF fibers, thus enabling the design of scaffolds with complex structures to guide the 3D organization of cells, a characteristic that is often only achievable with 3D printing approaches.
2.5 Flat and tubular scaffolds support cell growth
To demonstrate the suitability of SF scaffolds crosslinked with NaH2PO4 for in vitro cell culture, particularly for renal and vascular tissue engineering, we conducted stability tests over a period of 28 days at 37 °C in PBS. Notably, no visible degradation was observed (Fig. S2†). Subsequently, to investigate whether SF fiber scaffolds could support cell growth, proximal tubular (ciPTEC) and glomerular endothelial (ciGEnC) cells were seeded on both flat and tubular scaffolds. While both cell types were not able to attach to the SF fiber scaffolds without a biofunctionalization step (Fig. S9†), simple L-DOPA coating enabled cell attachment and monolayer formation for both cell types within 7–14 days after initial seeding (Fig. 5). Furthermore, ciPTEC and ciGEnC grown on flat scaffolds showed high viability (>95%), abundant deposition of type-IV collagen and limited production of type-I collagen. F-actin directionality indicated preferential cell alignment along scaffold microstructure, observed with a 90° orientation (Fig. 5). Similarly, ciPTEC and ciGEnC grown in tubular scaffolds formed aligned monolayers and deposited ample type-IV collagen and a limited amount of type-I collagen, indicating that cells began depositing their own tissue-specific extracellular matrix.43,44 Furthermore, the presence of cell-specific ciliary marker α-tubulin (ciPTEC)45 and cell-adhesion molecule CD31 (ciGEnC)46 confirmed cell proliferation within the scaffolds, as well as epithelial monolayer polarization for ciPTEC (Fig. S10†). Remarkably, although the scaffolds showed very high porosity (>97%), both cell types could form continuous monolayers that filled the scaffold pores, suggesting that, even with small amounts of scaffold volume fraction, SF scaffolds provide enough cues to guide cell orientation. Our findings are consistent with previous studies that have explored the influence of various scaffold topographical features, such as scaffold geometry, pore area and pore geometry, on the behaviour of kidney cells.47 For instance, van Genderen et al. demonstrated that the geometry of polycaprolactone-based fiber scaffolds influenced cell alignment, thereby impacting expression of genes with key roles in transport functionality.48 Furthermore, they observed that the maximal scaffold pore size possible to ensure complete pore filling was cell type-dependent, with ciPTECs showing the ability to spread widely and with high adherence over porous scaffolds. Altogether, both flat and tubular scaffolds offer promising substrates for the growth and maturation of ciPTEC and ciGEnC cells. However, further investigation is needed to understand how the elastic properties of the fiber scaffolds influence cell behavior. Besides, it is important to explore whether cell–fiber interactions and the de novo deposition of extracellular matrix impact fiber morphology and, crucially, the mechanical properties of the overall constructs.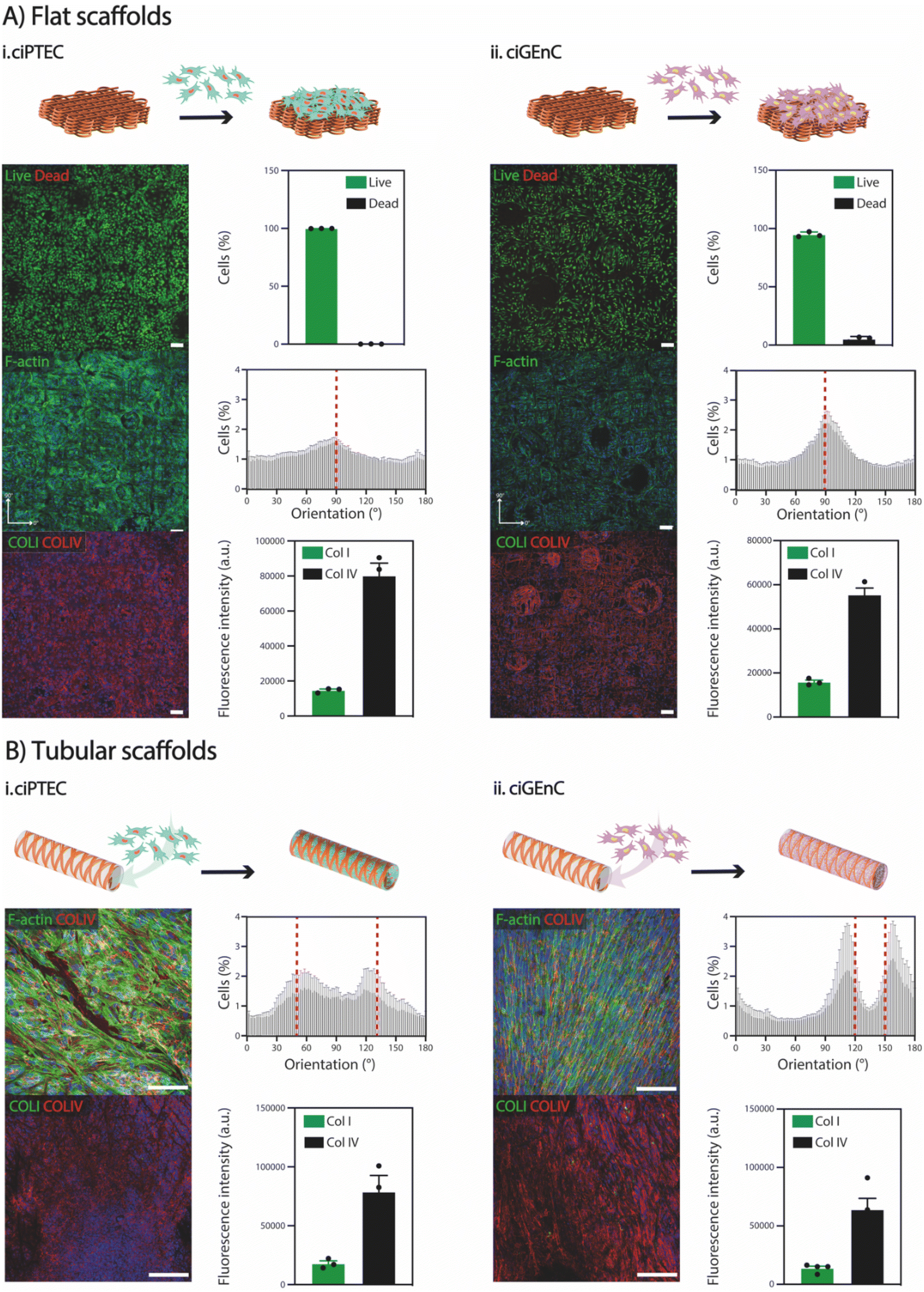 | ||
| Fig. 5 Cell viability, orientation, and growth in (A) flat (400 μm spacing, 100 layers) and (B) tubular (60°, 100 layers) electrowritten SF scaffolds for (i) ciPTEC and (ii) ciGEnC. Scale bars: 100 μm. | ||
3. Conclusions
In this study, an aqueous-based electrowriting technique was developed to create microfiber scaffolds made from silk fibroin with distinct mechanical properties. Our work presents a novel approach to processing silk fibroin that achieves a level of geometric precision and structural robustness while restoring part of the silk fibers’ native elastic behavior, which is not possible with current silk fibroin processing methods. The resulting flat and tubular scaffolds exhibited microfiber sizes with diameters as small as 5 μm, along with well-defined square-based and crosshatch microarchitectures. A crosslinking method based on aqueous NaH2PO4 was also introduced, which induced the controlled formation of β-sheet crystallites in SF while maintaining a considerable amount of compliant random coils, compared to conventional organic-solvent-based crosslinking methods which lead to brittle gels due to excessive formation of stiff β-sheets. While the extraction of SF and use of PEO are well-established practices, our work introduces a novel method for processing silk fibroin that achieves geometric precision and restores part of the silk fibers’ native elastic behavior, which is not possible with current methods. By using NaH2PO4 post-processing, we achieved up to 20% yield strain—significantly higher than the typical 5–10% yield strain, especially in well-defined fiber scaffold geometries. This enhanced elasticity makes the scaffolds reliable for in situ tissue engineering applications, where biological environments are dynamic and the scaffolds need to be easy to handle with surgical instruments. Moreover, when coated with L-DOPA, the scaffolds supported the growth and proliferation of ciPTEC and ciGEnC cells, which formed homogeneous and aligned monolayers and produced type-IV collagen, indicating healthy epithelial and endothelial tissue. Notably, both cell types filled the scaffold pore space despite the low scaffold volume fraction (<5%), indicating a high efficiency of SF scaffolds for directing cell organization with a small amount of material needed. It is important to point out the trade-off between mechanical properties and cellular response. Synthetic materials often have superior mechanical properties, but natural biomaterials like silk fibroin excel in cellular response. Despite their biocompatibility, synthetic materials do not fully replicate the performance of natural ones. Thus, it is crucial to advance biomaterial processing methods that enhance the mechanical properties of natural materials to bridge this gap. Our findings suggest that further optimization of our approach could lead to the production of even more elastic fibers compared to those produced with traditional methods, laying the groundwork for fully reintroducing fibrillar structures in printed silk constructs and achieving the elastic properties of native silk fibers.4. Experimental section
4.1 Materials
SF was extracted from Bombyx mori cocoons (kindly provided by Evrosilk; Czech Republic) as described elsewhere.49 Briefly, the cocoons were degummed at 100 °C in a 0.02 M sodium carbonate (Na2CO3; Sigma Aldrich) solution for 5, 10 or 20 minutes, dissolved in a 9.3 M lithium bromide (LiBr; Acros Organics) solution for 4 h at 60 °C and then dialyzed at 4 °C against pure water for 4 days using cellulose dialysis tubes (MWCO 3.5 kDa, Sigma Aldrich). Subsequently, the diluted solution was concentrated with inverse dialysis at 4 °C using cellulose dialysis tubes (MWCO 3.5 kDa, Sigma Aldrich) against a 40% PEG (6 kDa; Sigma Aldrich) aqueous solution for 16 h to obtain a SF ink concentration between 15 and 20%. Three ink solutions were studied SF5DT, SF10DT and SF20DT, where the number indicates the degumming time (minutes of boiling). Poly(ethylene oxide) (PEO) (10 kDa; Sigma Aldrich) was added prior to printing, in quantities ranging from 0.5 to 3%.4.2 Ink material characterization
The viscosity of 20% SF aqueous solutions with increasing amount of PEO (10 kDa; Sigma Aldrich), was analysed with a rheometer (Discovery HR2, TA Instruments, New Castle, DE, USA) using a cone-plate geometry (20 mm – 1°) performing a flow sweep at 25 °C with shear rate from 0.1 to 1000 s−1. The electrical conductivity of the solution was measured with an electroconductive-meter (Consort C861). Distilled water was used as a standard and calibrated to 1413 μS cm−1. The temperature was set to 25 °C and automatic temperature compensation was applied. The electrodes were washed with DI water before every measurement and afterwards immersed in the SF solution for 1 min (n = 3).4.3 Aqueous electrowriting of SF fibers and fiber scaffolds
Electrowriting was performed using an in-house built set up that allows for both flat and tubular scaffolds manufacturing as described before.40,50 SF5DT aqueous solution (20%) with 2.5% PEO ink was poured in a 3 ml glass syringe (Fortuna Optima Ganzglasspritze, Poulten & Graf GmbH) with a 27 G metal needle (Unimed), connected to a sealed hose delivering pressurized nitrogen (VPPE-3-1-1/8-2-010-E1, Festo). The SF solution was electrified using a high voltage source (Heinzinger, LNC 10000-2neg) and collected either onto a grounded collector plate (x–y) or onto a rotating aluminium mandrel (∅ = 4 mm and 3 mm) mounted on a x–y axis, both covered in aluminium foil and controlled by an advanced motion controller Motion Perfect v5.0.2 (Trio Motion Technology Ltd). The polymer solution processing compatibility was systematically investigated, specifically by tuning the voltage (V), the pressure (p), and the collection distance (Cd). The collector speed (Scol) was studied differently for flat and tubular scaffolds as previously described.40 For both flat and tubular scaffolds, fiber diameter and morphology were investigated by changing the mentioned parameters between the following values: V = [1.5–6] kV, p = [0–0.1] bar, Scol = [20–400] mm s−1, and Cd = [2–6] mm, one parameter at a time. The optimized SF flat scaffolds were manufactured using the following parameters: V = 2.5 kV, p = 0.05 bar, Scol = 300 mm s−1, and Cd = 6 mm. Organized scaffold meshes (40 cm2) with squared microstructures (fibers spacing 400 μm and 1 mm) were fabricated. The SF tubular scaffolds were manufactured using the following parameters: V = 4.8–5.3 kV; p = 0.1 bar; Scol = 100 mm s−1 and Cd = 6 mm. Tubular scaffolds with winding angles 60° and 90° and corresponding to a square or rhomboid microarchitecture with pore sizes of 1.82 mm2 and 3.16 mm2 respectively, were fabricated.4.4 Electrowritten construct imaging before post-processing
Printed SF scaffolds were characterized first with a stereomicroscope (Olympus SZX7) and then with Scanning Electron Microscope (SEM, Quanta3D FEG, Thermo Fisher Scientific, Eindhoven, The Netherlands). For SEM, all scaffolds were coated with Pt (30 s, 30 mA) using a Q150T S high resolution sputter coater (Quorum) and imaged at 5 kV. All the images were analysed with Fiji ImageJ software. The number of fibers used for the diameter measurements was at least n = 10. Fiber spacing, winding angle and quality number were used as references to characterize the accuracy of printed scaffolds. The quality number is identified as the ratio between the experimental and theoretical pore area; in an ideal condition, these two values are identical, and the quality number is 1.4.5 Post-processing treatment
SF scaffolds were immersed in either a solution of methanol (Sigma Aldrich) or 2 M aqueous solution of sodium dihydrogen phosphate (NaH2PO4; Sigma Aldrich) overnight, after which the scaffolds were washed with 10 mL of DI water 5 times and then kept in DI water.4.6 Ink properties after crosslinking and β-sheet formation
To assess SF crosslinked material properties, rheological characterization of SF5DT 20% + PEO 2.5% solution with either MeOH or 2 M NaH2PO4 was performed with a rheometer (Discovery HR2, TA Instruments, New Castle, DE, USA) fitted with a plate-plate geometry (20 mm) with 300 μm gap. A time sweep with angular frequency 10 rad s−1 and strain 1% was conducted at 25 °C to record storage modulus (G′) and loss modulus (G′′) as a function of time. G′ and G′′ were also evaluated as a function of strain (oscillation amplitude sweep, T = 25 °C, angular frequency = 10 rad s−1, strain = 0.1–100%) and as a function of angular frequency (oscillation frequency sweep, T = 25 °C, strain = 1%, angular frequency = 0.1–100 rad s−1). β-sheet formation was evaluated with Raman spectroscopy using a Renishaw InVia Raman microscope with a 785 nm laser. The spectra were collected between 100–3200 cm−1 using a relatively low laser power (0.23 W) to avoid light-induced sample damage with an integration time of 5 minutes and 30 accumulations per spectrum (total acquisition time of 2.5 h).4.7 Small-angle X-ray scattering (SAXS)
20% SF5DT + 2.5% PEO solutions were allowed to gelate under treatment with MeOH or 2 M NaH2PO4 for 12 h, and excess liquid was removed to form a gel film. Untreated 20% SF5DT + 2.5% PEO solution was allowed to air-dry to produce a solid film. Untreated and treated films and unprocessed native silk fibers were mounted on a stage. SAXS measurements were performed on a SAXSLAB GANESHA 300 XL system equipped with a GeniX 3D microfocus sealed tube Cu source producing X-rays (λ = 1.54 Å) at a flux of 1 × 108 ph per s and a Pilatus 300 K silicon detector (487 × 619 pixels, 172 × 172 μm). Momentum transfer (q) was measured at 20 °C to cover a range of 0.0809–0.2087 Å−1, where q = 4π/λ(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ/2). Before each set of measurements, silver behenate was used as a standard to calibrate sample-to-detector distance, beam center, and q-range. The measured profiles are displayed as average intensity I(q) versus q.
θ/2). Before each set of measurements, silver behenate was used as a standard to calibrate sample-to-detector distance, beam center, and q-range. The measured profiles are displayed as average intensity I(q) versus q.
4.8 Atomic force microscopy (AFM)
AFM was used to evaluate the topography of electrowritten SF fibers deposited on glass coverslips. Before imaging, single fibers were immersed in either MeOH or 2 M NaH2PO4 for 12 h and allowed to air-dry. A Multimode Nanoscope IIIA (Veeco) was used to record height and peak force error images. Scans were collected in the ScanAsyst large-amplitude mode at 20 °C in air with silicon tips on nitride levers (ScanAsyst-Air, Veeco, 50–90 kHz, 0.4 N m−1), controlled using Nanoscope 8 (Bruker), and processed using NanoScope Analysis 1.40 (Bruker).4.9 Micro-computed tomography (μCT)
NaH2PO4-crosslinked SF tubular scaffolds (150 layers, 60° winding angle) were immersed in a 1% w v−1 solution of phosphotungstic acid (Sigma Aldrich) as contrast agent for 3 h. The scaffolds were then thoroughly rinsed and imaged in PBS. μCT images were taken on a μCT100 system (Scanco Medical) with the following scanning parameters: voxel size = 36.8 μm, energy level = 70 kV, intensity = 114 μA, and integration time = 200 ms, with no filter, over a region of interest (ROI) of 162 slices. Noise reduction was performed with a constrained Gaussian filter, with a support value of 1.0 and a sigma width of 0.8 voxel. After segmentation, Image Processing Language (IPLFE v2.03, Scanco Medical) was used to analyse the 3D reconstructed models. Strut thickness was estimated as trabecular thickness (Tb.Th) by the distance transformation method,51 while porosity was calculated as P = 1 − (material volume excluding porosity/total volume including porosity).52,534.10 Mechanical characterization
Uniaxial tensile tests were performed at 23 °C in PBS using a BioTester 5000 device (CellScale) with a 1.5 N load cell. Flat scaffolds (width: 10 mm, active length: 10 mm) were tested monotonically at a strain rate of 10% min−1 (n ≥ 3); or under cyclical conditions at a loading-unloading frequency of 0.1 Hz with 10 cycles at 5% max. strain, followed by 10 cycles at 10% max. strain, and 10 cycles at 20% max. strain (n ≥ 3). Tubular scaffolds (axis length: 10 mm, inner diameter: 4 mm) were mounted on custom wire holders to allow for uniform radial deformation and tested monotonically at a strain rate of 10% min−1 (n ≥ 3). Force–displacement curves were recorded using LabJoy (CellScale). Force data were processed with a moving average filter (size: 15 datapoints). Stiffness constant (with units of force) was calculated from least squares fitting of the linear region slope in force–strain curves. Yield strain was set as the upper value of the range used for stiffness calculations. Decrease in peak force was quantified as: ΔFmax = (Fc=1max − Fc=10max)/Fc=1max, where Fc=1max and Fc=10max are the maximal forces during cycle 1 and 10, respectively, of each strain step. For delivery tests, a flat SF scaffold (30 × 30 mm, 100 layers) was passed through a catheter (∅ inner = 500 μm) with a constant flow of PBS. The scaffolds were collected in a Petri dish and imaged. For the balloon catheter test, a tubular scaffold (tube length: 17.41 mm, ∅ = 3 mm, winding angle: 60°, 100 layers) was introduced into a balloon catheter (XTRM Way 3, Blue Medical), with a diameter at maximum pressure of 5 mm. The deflated catheter was inserted into the tubular scaffold, and pressure was gradually increase up to 5 bar. At pressure intervals of 1 bar, images of the scaffold were taken to assess its diameter.4.11 Cell culture
Human conditionally immortalized proximal tubular epithelial cells (ciPTEC, obtained via Cell4Pharma, Nijmegen, The Netherlands) and glomerular endothelial cells ciGEnC were cultured as previously described.53,54 In short, both cell lines were cultured in T75 flasks (Greiner Bio-One, Alphen aan den Rijn, The Netherlands) in their respective culture medium. CiPTEC were cultured in Dulbecco's modified Eagle's medium/HAM's F12 without phenol red (Thermo Fisher Scientific, Paisley, UK), with added insulin, transferrin, selenium (all 5 μg ml−1), hydrocortisone (35 ng ml−1), epidermal growth factor (10 ng ml−1), tri-iodothyronine (40 pg ml−1) (Sigma-Aldrich, Saint Louis, MO, USA), 10% fetal bovine serum (v/v, FBS, Greiner Bio-One), and 1% penicillin/streptomycin (v/v, Gibco, Thermo Fisher Scientific) to prevent infections. CiGEnC were cultured in Endothelial Cell Basal Medium-2 (Lonza) containing Microvascular Endothelial Cell Growth Medium-2 SingleQuots Kit (EGM-2 MV, Lonza). Both cell lines proliferate at the permissive temperature of 33 °C and maturate at 37 °C.4.12 Cell seeding
Tubular scaffolds were sterilized in a laminar flow cabinet by washing 3 times for 15 minutes in 5% penicillin/streptomycin (v/v, Gibco, Thermo Fisher Scientific) in phosphate buffered saline (PBS, Lonza). Thereafter, scaffolds were sterilized by UV exposure in a laminar flow cabinet (365 nm for 30 min). After sterilization, scaffolds were functionalized using 2 mg ml−1L-3,4-dihydroxyphenylalanine (L-DOPA, Sigma-Aldrich) dissolved in 10 mM tris(hydroxyethyl)aminomethane (Tris) pH 8.5 buffer (Sigma Aldrich). After dissolving L-DOPA, it was put at 37 °C for 45 minutes. After 45 minutes L-DOPA was filter-sterilized and used for functionalization. Scaffolds were immersed in L-DOPA solution at 37 °C for a minimum of 4 hours. Tubular scaffolds were washed 3 times using PBS after the coating procedure. CiPTEC or ciGEnC were seeded in the tubular scaffolds (tube length: 17.41 mm, ∅ = 3 mm, winding angle: 60°, 100 layers) at concentrations 15 × 106 cells per ml and 30 × 106 cells per ml respectively, using a positive displacement pipette. Two hours after seeding, scaffolds were turned 180°. Four hours after seeding, the respective culture medium was added. Scaffolds were then cultured until confluency at 33 °C, followed by 7 days of differentiation at 37 °C. Culture medium was refreshed every 2–3 days.4.13 Cell analysis and immunocytochemistry
Live/dead staining was performed by rinsing cells with PBS, followed by incubation with 2 μM calcein-AM and 4 μM ethidium homodimer-1 (Invitrogen) for 30 min to examine viability. For immunocytochemistry, cells were fixed for 10 min with 4 wt% paraformaldehyde (Pierce, Thermo Fisher Scientific) in PBS. Afterwards, cells were permeabilized using 0.3 wt% Triton X-100 (Sigma-Aldrich) in PBS for 30 min and exposed to blocking buffer (2 wt% FBS, 0.5 wt% bovine serum albumin (Sigma Aldrich) and 0.1 wt% Tween-20 (Sigma Aldrich) in PBS) for 60 min at RT. Cells were incubated with primary and secondary antibodies diluted in blocking buffer for 1.5 hours and 1 hour at RT respectively. Table S1† contains a list of primary and secondary antibodies used. Confocal Leica TCS SP8 X microscope and software Leica Application Suite X (Leica) was used to examine immunofluorescence. Images were analysed using ImageJ (National Institutes of Health, USA).4.14 Statistical analysis
All data are shown as mean ± SD, unless otherwise stated. Statistical significance was tested by unpaired t-test with Welch's correction, or two-way ANOVA with Tukey's multiple comparisons test, as noted in each case. All statistical analysis was performed with Prism 9 software (GraphPad).Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Conflicts of interest
S. P. is an employee of SentryX B.V.Acknowledgements
The authors thank Jim de Ruiter (Dept. of Inorganic Chemistry and Catalysis, Faculty of Science, Utrecht University, the Netherlands) for performing the Raman spectroscopy measurements; Marco Hendrix and Prof. Ilja Voets (Dept. of Chemical Engineering & Chemistry, Eindhoven University of Technology, The Netherlands) for the SAXS measurements; and the group of Prof. J. van der Vlag (RadboudUMC, Nijmegen, The Netherlands) and Prof. S. Satchell (Bristol Medical School (THS), Bristol, UK) for providing glomerular endothelial cells. The authors thank the following agencies for their financial support: the European Union's Horizon 2020 research and innovation programme, the Gravitation Program “Materials Driven Regeneration” (024.003.013), the Marie Skłodowska-Curie Actions (RESCUE #801540), and the Netherlands Organization for Scientific Research (NWO) (Reprint project, OCENW.XS5.161). G. C. acknowledges support from the EU Horizon 2020 programme (BRAV3 #874827).References
- C. Lujerdean, G.-M. Baci, A.-A. Cucu and D. S. Dezmirean, Insects, 2022, 13, 286 CrossRef PubMed.
- L.-D. Koh, Y. Cheng, C.-P. Teng, Y.-W. Khin, X.-J. Loh, S.-Y. Tee, M. Low, E. Ye, H.-D. Yu, Y.-W. Zhang and M.-Y. Han, Prog. Polym. Sci., 2015, 46, 86–110 CrossRef CAS.
- S. Tandon, B. Kandasubramanian and S. M. Ibrahim, Ind. Eng. Chem. Res., 2020, 59, 17593–17611 CrossRef CAS.
- C. Belbéoch, J. Lejeune, P. Vroman and F. Salaün, Environ. Chem. Lett., 2021, 19, 1737–1763 CrossRef PubMed.
- T. Asakura, K. Umemura, Y. Nakazawa, H. Hirose, J. Higham and D. Knight, Biomacromolecules, 2006, 8, 175–181 CrossRef PubMed.
- Y. Liu, J. Ren and S. Ling, Compos. Commun., 2019, 13, 85–96 CrossRef.
- J. Yan, G. Zhou, D. P. Knight, Z. Shao and X. Chen, Biomacromolecules, 2009, 11, 1–5 CrossRef PubMed.
- D. E. Chung and I. C. Um, Fibers Polym., 2014, 15, 153–160 CrossRef CAS.
- Q. Peng, H. Shao, X. Hu and Y. Zhang, Macromol. Mater. Eng., 2017, 302, 1700102 CrossRef.
- S. H. Kim, Y. S. Nam, T. S. Lee and W. H. Park, Polym. J., 2003, 35, 185–190 CrossRef CAS.
- M. Wang, H.-J. Jin, D. L. Kaplan and G. C. Rutledge, Macromolecules, 2004, 37, 6856–6864 CrossRef CAS.
- N. Amiraliyan, M. Nouri and M. Haghighat Kish, Polym. Sci., Ser. A, 2010, 52, 407–412 CrossRef.
- V. Catto, S. Farè, I. Cattaneo, M. Figliuzzi, A. Alessandrino, G. Freddi, A. Remuzzi and M. C. Tanzi, Mater. Sci. Eng., C, 2015, 54, 101–111 CrossRef CAS PubMed.
- S. Yazawa, J. Chem. Soc. Jpn., 1960, 63, 1428–1430 Search PubMed.
- O. Liivak, A. Blye, N. Shah and L. W. Jelinski, Macromolecules, 1998, 31, 2947–2951 CrossRef CAS.
- J. Yao, H. Masuda, C. Zhao and T. Asakura, Macromolecules, 2001, 35, 6–9 CrossRef.
- I. C. Um, H. Kweon, Y. H. Park and S. Hudson, Int. J. Biol. Macromol., 2001, 29, 91–97 CrossRef CAS PubMed.
- B. Zuo, L. Liu and Z. Wu, J. Appl. Polym. Sci., 2007, 106, 53–59 CrossRef CAS.
- U.-J. Kim, J. Park, H. Joo Kim, M. Wada and D. L. Kaplan, Biomaterials, 2005, 26, 2775–2785 CrossRef CAS PubMed.
- L. Zhou, X. Chen, Z. Shao, Y. Huang and D. P. Knight, J. Phys. Chem. B, 2005, 109, 16937–16945 CrossRef CAS PubMed.
- A. M. Hai, Z. Yue, S. Beirne and G. Wallace, J. Appl. Polym. Sci., 2023, 140, e53349 CrossRef CAS.
- A. Rising and J. Johansson, Nat. Chem. Biol., 2015, 11, 309–315 CrossRef CAS PubMed.
- C. W. P. Foo, E. Bini, J. Hensman, D. P. Knight, R. V. Lewis and D. L. Kaplan, Appl. Phys. A, 2005, 82, 223–233 CrossRef.
- X. Qu, L. Yan, S. Liu, Y. Tan, J. Xiao, Y. Cao, K. Chen, W. Xiao, B. Li and X. Liao, J. Biomater. Sci., Polym. Ed., 2021, 32, 1635–1653 CrossRef CAS PubMed.
- C. Niu, X. Li, Y. Wang, X. Liu, J. Shi and X. Wang, RSC Adv., 2019, 9, 41074–41082 RSC.
- P. Monti, G. Freddi, A. Bertoluzza, N. Kasai and M. Tsukada, J. Raman Spectrosc., 1998, 29, 297–304 CrossRef CAS.
- H.-J. Jin, S. V. Fridrikh, G. C. Rutledge and D. L. Kaplan, Biomacromolecules, 2002, 3, 1233–1239 CrossRef CAS PubMed.
- H. Jin, Biomaterials, 2004, 25, 1039–1047 CrossRef CAS PubMed.
- L. S. Wray, X. Hu, J. Gallego, I. Georgakoudi, F. G. Omenetto, D. Schmidt and D. L. Kaplan, J. Biomed. Mater. Res., Part B, 2011, 99, 89–101 CrossRef PubMed.
- H. Cao, X. Chen, L. Huang and Z. Shao, Mater. Sci. Eng., C, 2009, 29, 2270–2274 CrossRef.
- M. Rodriguez, J. A. Kluge, D. Smoot, M. A. Kluge, D. F. Schmidt, C. R. Paetsch, P. S. Kim and D. L. Kaplan, Biomaterials, 2020, 230, 119567 CrossRef CAS PubMed.
- T. D. Brown, P. D. Dalton and D. W. Hutmacher, Adv. Mater., 2011, 23, 5651–5657 CrossRef CAS PubMed.
- M. Castilho, R. Levato, P. N. Bernal, M. de Ruijter, C. Y. Sheng, J. van Duijn, S. Piluso, K. Ito and J. Malda, Biomacromolecules, 2021, 22, 855–866 CrossRef CAS PubMed.
- G. Rizzo, V. Petrelli, T. Sibillano, L. De Caro, M. M. Giangregorio, M. Lo Presti, F. G. Omenetto, C. Giannini, P. Mastrorilli and G. M. Farinola, ACS Omega, 2023, 8, 24165–24175 CrossRef CAS PubMed.
- O. Tokareva, M. Jacobsen, M. Buehler, J. Wong and D. L. Kaplan, Acta Biomater., 2014, 10, 1612–1626 CrossRef CAS PubMed.
- S. Rammensee, U. Slotta, T. Scheibel and A. R. Bausch, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6590–6595 CrossRef CAS PubMed.
- M. Stark, S. Grip, A. Rising, M. Hedhammar, W. Engström, G. Hjälm and J. Johansson, Biomacromolecules, 2007, 8, 1695–1701 CrossRef CAS PubMed.
- R. Wu, L. Ma and X. Y. Liu, Adv. Sci., 2022, 9, 2103981 CrossRef CAS PubMed.
- X. Mu, Y. Wang, C. Guo, Y. Li, S. Ling, W. Huang, P. Cebe, H. Hsu, F. De Ferrari, X. Jiang, Q. Xu, A. Balduini, F. G. Omenetto and D. L. Kaplan, Macromol. Biosci., 2020, 20, 1900191 CrossRef CAS PubMed.
- M. Castilho, A. van Mil, M. Maher, C. H. G. Metz, G. Hochleitner, J. Groll, P. A. Doevendans, K. Ito, J. P. G. Sluijter and J. Malda, Adv. Funct. Mater., 2018, 28, 1803151 CrossRef.
- M. Shojaee and C. A. Bashur, Adv. Healthcare Mater., 2017, 6, 1700001 CrossRef PubMed.
- D. J. Wu, K. van Dongen, W. Szymczyk, P. J. Besseling, R. M. Cardinaels, G. Marchioli, M. H. P. van Genderen, C. V. C. Bouten, A. I. P. M. Smits and P. Y. W. Dankers, Front. Mater., 2020, 7 DOI:10.3389/fmats.2020.00220.
- A. M. Genderen, J. Jansen, C. Cheng, T. Vermonden and R. Masereeuw, Adv. Healthcare Mater., 2018, 7, 1800529 CrossRef PubMed.
- M. A. Karsdal, S. H. Nielsen, D. J. Leeming, L. L. Langholm, M. J. Nielsen, T. Manon-Jensen, A. Siebuhr, N. S. Gudmann, S. Rønnow, J. M. Sand, S. J. Daniels, J. H. Mortensen and D. Schuppan, Adv. Drug Delivery Rev., 2017, 121, 43–56 CrossRef CAS PubMed.
- V. Raghavan and O. A. Weisz, Am. J. Physiol.: Renal Physiol., 2016, 310, F1–F5 CrossRef CAS PubMed.
- P. Lertkiatmongkol, D. Liao, H. Mei, Y. Hu and P. J. Newman, Curr. Opin. Hematol., 2016, 23, 253–259 CrossRef CAS PubMed.
- F. Hulshof, C. Schophuizen, M. Mihajlovic, C. Blitterswijk, R. Masereeuw, J. Boer and D. Stamatialis, J. Tissue Eng. Regener. Med., 2018, 12, e817–e827 CAS.
- A. M. van Genderen, K. Jansen, M. Kristen, J. van Duijn, Y. Li, C. C. L. Schuurmans, J. Malda, T. Vermonden, J. Jansen, R. Masereeuw and M. Castilho, Front. Bioeng. Biotechnol., 2021, 8, 617364 CrossRef PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612–1631 CrossRef CAS PubMed.
- M. Castilho, D. Feyen, M. Flandes-Iparraguirre, G. Hochleitner, J. Groll, P. A. F. Doevendans, T. Vermonden, K. Ito, J. P. G. Sluijter and J. Malda, Adv. Healthcare Mater., 2017, 1700311 CrossRef PubMed.
- T. Hildebrand, A. Laib, R. Müller, J. Dequeker and P. Rüegsegger, J. Bone Miner. Res., 1999, 14, 1167–1174 CrossRef CAS PubMed.
- T. L. Conrad and R. K. Roeder, J. Mech. Behav. Biomed. Mater., 2020, 106, 103730 CrossRef CAS PubMed.
- J. Jansen, C. M. S. Schophuizen, M. J. Wilmer, S. H. M. Lahham, H. A. M. Mutsaers, J. F. M. Wetzels, R. A. Bank, L. P. van den Heuvel, J. G. Hoenderop and R. Masereeuw, Exp. Cell Res., 2014, 323, 87–99 CrossRef CAS PubMed.
- S. C. Satchell, C. H. Tasman, A. Singh, L. Ni, J. Geelen, C. J. von Ruhland, M. J. O'Hare, M. A. Saleem, L. P. van den Heuvel and P. W. Mathieson, Kidney Int., 2006, 69, 1633–1640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm00624k |
| This journal is © The Royal Society of Chemistry 2024 |