
Facet-specific ligand dynamics: scaling the descriptors of Cs2AgBiBr6 nanocrystals†
Anamika
Mondal
a,
Manuj
Ahuja
b,
Priya
Johari
 *b and
Sayan
Bhattacharyya
*a
*b and
Sayan
Bhattacharyya
*a
aDepartment of Chemical Sciences and Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur-741246, India. E-mail: sayanb@iiserkol.ac.in
bDepartment of Physics, School of Natural Sciences, Shiv Nadar Institution of Eminence, Gautam Buddha Nagar, Greater Noida, UP 201314, India. E-mail: priya.johari@snu.edu.in
First published on 29th February 2024
Abstract
Facet control by primary amines can bolster the optoelectronic parameters of A2BIB′IIIX6 perovskite nanocrystals (NCs) with large indirect bandgaps. The 18-C amine competitively attaches to the (222) facet of Cs2AgBiBr6 (CABB) NCs, 16-C and 14-C bind to (400) and (440), and 12-C binds to (400). The NCs with only the (400) facet decrease the bandgap and exciton binding energy by 0.26 eV and 15 meV, respectively.
The A2BIB′IIIX6 lead-free double perovskites have a range of structural possibilities with the B-site being occupied by Ag+, Cu+, Au+, Bi3+, Sb3+, or In3+.1 In A2BIB′IIIX6, the octahedra are alternately distributed between the +1 and +3 sites. In the two-interpenetrating face-centered cubic (fcc) sublattices with Fm
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, the cuboctahedral positioning of the monovalent A-site cation remains unaltered. The available elemental choices and compositional flexibility allow them to attain diverse electronic band edges, thereby leading to small or large, direct or indirect bandgaps.2 With a combination of Ag+ and Bi3+ cations at the B-site, CABB has a large indirect bandgap contributed by the 6p and 6s orbitals of Bi to the conduction band minimum (CBM) and valence band maximum (VBM), respectively.3 However, the large indirect bandgap aggravates the optoelectronic characteristics of the CABB NCs, and hence, facet engineering can provide the key to unlocking the potential of these thermodynamically stable nanocrystals (NCs).
m space group, the cuboctahedral positioning of the monovalent A-site cation remains unaltered. The available elemental choices and compositional flexibility allow them to attain diverse electronic band edges, thereby leading to small or large, direct or indirect bandgaps.2 With a combination of Ag+ and Bi3+ cations at the B-site, CABB has a large indirect bandgap contributed by the 6p and 6s orbitals of Bi to the conduction band minimum (CBM) and valence band maximum (VBM), respectively.3 However, the large indirect bandgap aggravates the optoelectronic characteristics of the CABB NCs, and hence, facet engineering can provide the key to unlocking the potential of these thermodynamically stable nanocrystals (NCs).
The binding affinity of organic ligands to specific crystallographic facets is a powerful tool to fine-tune the electronic and structural properties of hybrid perovskite NCs.4–6 Ligand binding depends on the adsorption energy, where higher adsorption energy indicates a stronger facet-specific adherence of the ligand. The dynamic adsorption–desorption of oleylamine (OLAM) and oleic acid (OA) acid–base pair regulates the shape and size of the nanostructures.7–9 With a shorter carbon chain length of the alkylamine and carboxylic acid, the morphology can be tuned with a variety of optoelectronic properties.10,11
In this work, we have employed a series of aliphatic amines, 18-C OLAM, 16-C hexadecylamine (HDA), 14-C tetradecylamine (TDA), and 12-C dodecylamine (DA), to cap the surface of cube-shaped CABB NCs. The reaction temperature and time are kept constant in all cases. The corresponding NCs are abbreviated as CABB-OLAM, CABB-HDA, CABB-TDA, and CABB-DA, respectively. The ligand dynamicity has a particular role in modifying the energy of crystallographic facets through specific adsorption, whereby, only the low-energy facets terminate the NC surface. In our case, the specific ligand binding depends on the carbon chain length. From OLAM to DA, the ligand dynamicity increases due to the feasibility of absorption and desorption of the ligand to and from the binding sites. The adsorption energy calculations show a competing binding affinity to the (hkl) planes. An increasing lattice strain from CABB-OLAM to CABB-DA, decreases the bandgap, exciton binding energy, and trap-state density, which increases the work function and carrier mobility.
The CABB NCs were synthesized by the hot-injection method through the dissolution of AgNO3 and BiBr3 in a high-boiling organic solvent, octadecene (ODE), followed by HBr addition (Fig. S1a, ESI†). In the case of CABB-OLAM NCs, OLAM and OA were used as surface passivation agents to dissolve the metal precursors at 160 °C, turning the solution from white to transparent yellow. The nucleation originated within 5 s after injecting Cs-oleate into the metal oleate precursor solution at 200 °C. The NC growth occurred only after ice quenching. For all four amines, OLAM, HDA, TDA, and DA, identical reaction conditions (200 °C and 5 s) were maintained. A precise addition of HBr is crucial to prevent the formation of side products, such as AgBr. Therefore, the HBr content was varied for the different amines because when the basicity decreases for a shorter chain amine, the required HBr amount also decreases.
Rietveld refinement of the powder X-ray diffraction (XRD) patterns demonstrates the phase purity of the NCs (Fig. S1b, S2a–d, and Table S1, ESI†). The refined crystal structures match the elpasolite crystal structure of the K2NaAlF6 family (Fig. 1 and Fig, S1c, ESI†). The AgBr65− and BiBr63− octahedra are alternatively placed, and Cs+ sits in the octahedral void. From OLAM to DA, the overall unit cell length and volume increase due to the elongation of the Ag–Br and Bi–Br bond lengths (Fig. S1d, ESI†). The lattice strain calculated from Williamson–Hall (W–H) plots rose from 6.4 × 10−4 for CABB-OLAM NCs to 20.9 × 10−4 for CABB-DA NCs (Fig. S2e–h, ESI†). Transmission electron microscopy (TEM) shows cubic NCs (Fig. S3a–d, ESI†) with uniform elemental distribution (Fig. S4, ESI†). The average edge length of CABB-OLAM NCs is 10.6 ± 0.1 nm, and with the reduction in the ligand chain length from 18-C to 12-C, the edge length increases to 11.8 ± 0.1 nm (Fig. S3e, ESI†). The binding of OLAM induces a higher nucleation rate than DA. In addition, as the nuclei have to compete more with the remaining seats for growth, the NC growth is suppressed, and therefore, the size of CABB-OLAM NCs is smaller than that of the CABB-DA NCs by 1.2 nm. The average inter-NC distance, calculated over 40 NCs is reduced as expected, when the NCs are capped with shorter ligands, decreasing from 2.0 ± 0.1 nm for CABB-OLAM to 1.2 ± 0.1 nm for CABB-DA (Fig. S3f, ESI†). Due to an intricate curling of the carbon chain, the observed inter-NC distance is found to be less than the calculated chain length of the amine molecules, 2.15 nm for OLAM, 2.01 nm for HDA, 1.76 nm for TDA, and 1.51 nm for DA.
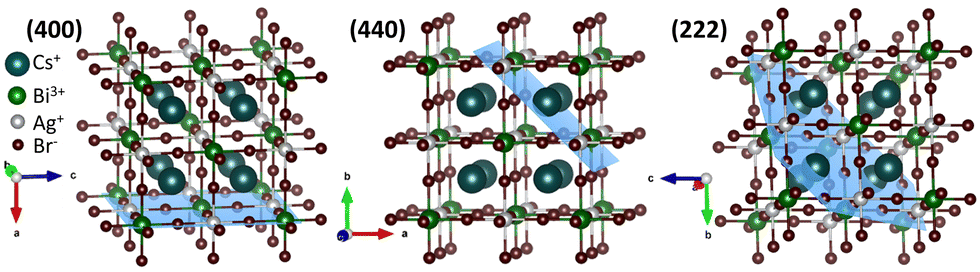 | ||
| Fig. 1 The (400), (440), and (222) facets in the crystal structure obtained from Rietveld refined XRD patterns for CABB-DA NCs. | ||
The ligand binding interaction is understood from the X-ray photoelectron spectral (XPS) analysis of the Ag 3d, Bi 4f, and Br 3d core levels of the NCs and N 1s core level of the aliphatic amine (Fig. S5 and Table S2, ESI†). Br 3d5/2 and 3d3/2 core levels at 69.4 eV and 70.5 eV are slightly downshifted from CABB-OLAM to CABB-DA NCs (Fig. S3g, ESI†). This shift is complemented by the higher binding energy shift of the N 1s core level from 401.4 to 401.8 eV for CABB-OLAM to CABB-DA. Firstly, the binding energy range of the N 1s level indicates the presence of protonated amine groups (NH3+) to bind electrostatically to the Br− on the NC surface. Secondly, the decreasing basicity from OLAM to DA increases the acidity of the NH3+ group, which also increases the electron-withdrawing tendency of NH3+. The charge transfer between the NC surface and the amine ligands is also evident from the Ag 3d3/2, Ag 3d5/2, Bi 4f5/2, and Bi 4f7/2 core-level spectra. The infrared spectra of all the NCs show intense symmetric and asymmetric stretching vibrations of the CH2 and CH3 groups of the amine ligands at 2840–2960 cm−1 (Fig. S6, ESI†). Due to the charge transfer from Br− to NH3+, these vibrational bands shift to lower frequencies relative to the bare ligands.
The experimental evidence for the facet-specific ligand dynamics is obtained from the TEM analyses of individual NCs (Fig. 2 and Fig. S7–S9, ESI†). The CABB-OLAM NC is populated by the (222) facet with interplanar d-spacing of 0.33 nm (Fig. 2a), along with the less abundant (444) and (440) planes having interplanar spacings of 0.16 and 0.20 nm, respectively. CABB-HDA NC surface features facets of 0.29 nm (400) and 0.20 nm (440) (Fig. 2b), in addition to the less frequent (220) and (600) planes. CABB-TDA NCs also show the (400) and (440) facets, along with the (220) and (800) planes (Fig. 2c). The CABB-DA NC surface is primarily composed of the (400) facet with 0.29 nm spacing, along with (800) facet with 0.14 nm spacing (Fig. 2d). Overall, CABB-HDA and CABB-TDA have (400) and (440) as the significant facets, while CABB-OLAM and CABB-DA are primarily exposed with a single facet of either (222) or (400), respectively (Fig. S10, ESI†). (400) is the most populated with Ag+, Bi3+, and Br−, followed by (440) with only Cs+, and (222) with Cs+ and Br− ions (Fig. 1).
 | ||
| Fig. 2 TEM images of (a) CABB-OLAM, (b) CABB-HDA, (c) CABB-TDA, and (d) CABB-DA NCs, showing two regions (i) and (ii). The results from the analyses of each region are shown by the mask-filtered TEM image obtained by masking the diffraction spot corresponding to a particular lattice plane. | ||
Computationally, the amines were placed over various locations on the fully relaxed 2 × 2 × 1 supercell of (100), (110), and (111) surfaces (Fig. 3a and Fig. S11a, ESI†). The ligand adsorption leads to alterations in the bond lengths of Ag–Br (Bi–Br) in the pristine (100), (110), and (111) surfaces as 2.79 Å (2.85 Å), 2.90 Å (2.94 Å), and 3.00 Å (2.79 Å), respectively (Fig. S12a and b, ESI†). The calculated XRD patterns by VESTA agree well with the experimental XRD data (Fig. S12c, ESI†). DFT calculations show that the ligand interaction introduces an out-of-plane strain, whose trend is also in sync with the W–H lattice strain (Fig. S11b, ESI†). The adsorption energy was calculated on the (400), (440), and (222) surfaces. The (400) facet is the most favoured with maximum adsorption energy (Fig. 3b). While DA binds most strongly with an adsorption energy of −4.27 eV on the (400) facet, OLAM is loosely bound (−3.71 eV). As compared to (400), HDA and TDA are more strongly affixed on the (440) plane than DA. For the CABB-DA NCs, (440) has comparatively lower adsorption energy, which makes it the primary facet. Since DA, TDA, and HDA have higher adsorption energies on the (400) and (440) facets, these planes are primarily stabilized. For CABB-OLAM NCs, the lower adsorption energies of OLAM on (400) and (440) make it prone to attach to (222) through competing ligand binding.
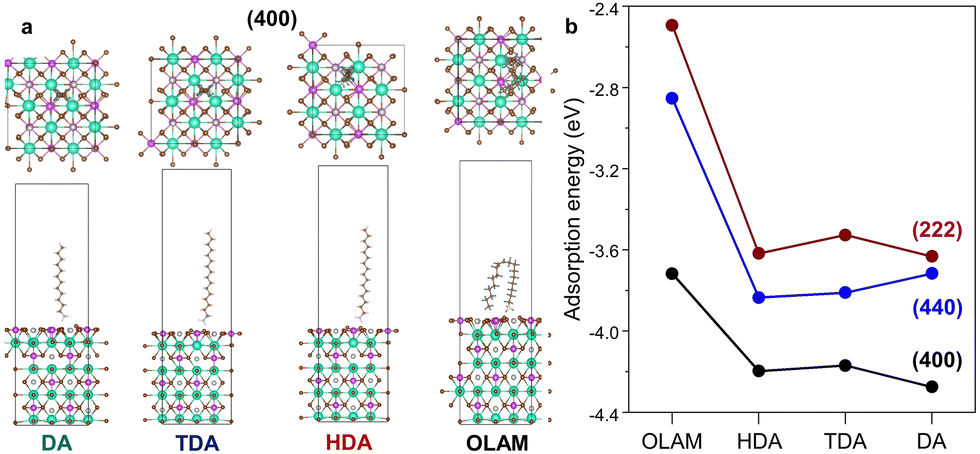 | ||
| Fig. 3 (a) Side and top-views of the optimized geometries after amine ligand adsorption on the (400) facet. (b) Amine-dependent variation of the adsorption energy on (400), (222), and (440) facets. | ||
All the NCs show sharp optical absorption at a wavelength of 440 nm (Fig. S13a, ESI†), a likely result of the excitation of the defect-bound excitons at the Ag+ vacancies, which act as hole-localized states.12 The valence band maxima (VBM) increases from −5.73 for CABB-OLAM NCs to −5.41 eV for CABB-DA NCs, whereas the conduction band minima (CBM) decreases from −3.69 to −3.71 eV (Fig. S13b, ESI†). Since the NC size increases from CABB-OLAM to CABB-DA, the indirect bandgap decreases from 2.04 ± 0.02 to 1.76 ± 0.01 eV (Fig. S14a and b, ESI†). The photoluminescence (PL) spectra recorded with an excitation wavelength (λex) of 365 nm displays a PL band maximum, which is red-shifted from 640 nm for CABB-OLAM to 650 nm for CABB-DA NCs (Fig. S13c, ESI†). With increased λex from 320 to 390 nm, the PL intensity gradually enhances without changing the PL maxima (Fig. S13d, ESI†). From 300 to 80 K, the PL intensity is enhanced due to the reduction in non-radiative recombination (Fig. S15, ESI†), and the PL bandwidth becomes narrower (Fig. S13e, ESI†).
The local lattice deformations and the increasing cell volume from the CABB-OLAM to CABB-DA NC lattice may lead to the development of additional energy states between the VBM and CBM. Also, the spatial separation of the adjacent BiBr63− and AgBr65− octahedra localizes the charge carriers to generate self-trapped excitons (STE).13 The broadness of the PL bands with high Stokes shift is related to the recombination of indirectly bound excitons in these mid-gap states.14 The increasing cell volume impacts the STE emission by red-shifting the PL band. The thermal energy activates the charge carriers at higher temperatures to overcome the energy barrier and reach the deeper STE (RT) states (Fig. S13f, ESI†). These states are stabilized at reduced temperatures to create the STE (LT) states, thereby partially clearing the mid-gap region and blue-shifting the PL bands from 300 to 80 K.15,16
The exciton binding energy (Eb) decreases with a decrease in the chain length of the amine ligand as follows: 74.0 ± 1.4, 72.0 ± 0.6, 73.0 ± 0.2, and 59.0 ± 0.2 meV for CABB-OLAM, CABB-HDA, and CABB-TDA to CABB-DA NCs, respectively (Fig. S14c, ESI†). As a smaller Eb facilitates carrier mobility, the reduced Eb of CABB-DA NCs is sufficient for a facile charge carrier generation in any optoelectronic and photovoltaic device application.17 The surface potential distribution due to the facet-specific ligand binding was investigated by Kelvin probe force microscopy (KPFM) on the FTO/c-TiO2/CABB-NC films (Fig. S16a–d, ESI†). The RMS roughness of the films is 151.2 ± 51.1, 137.5 ± 43.7, 56.2 ± 4.9, and 24.6 ± 5.6 nm for CABB-OLAM, CABB-HDA, CABB-TDA, and CABB-DA NCs, respectively. This reflects the ligand binding effect, i.e., the shorter the amine chain length, the more compact is the film due to shorter NC distances. The contact potential difference (CPD) decreases from 252 ± 28 mV for CABB-OLAM to 195 ± 50, 62 ± 12, and 16 ± 12 mV for CABB-HDA, CABB-TDA, and CABB-DA NCs, respectively (Fig. S16e–h, ESI†). The better film quality with the NCs bound by shorter chain amines leads to a lesser variation of surface potential. With decreasing CPD, the work function increases to 4.75 ± 0.03, 4.81 ± 0.04, 4.94 ± 0.01, and 4.98 ± 0.01 eV for CABB-OLAM, CABB-HDA, CABB-TDA, and CABB-DA NCs respectively. With the reduction in CPD, the work function increases, altering the band alignment and the Fermi energy and making the charge carrier transport more facile (Fig. S17, ESI†). From KPFM and energy band alignment, CABB-DA NCs have a greater tendency to facilitate the flow of charge carriers when this layer is sandwiched between TiO2 and Au.
The electron-only devices were fabricated with the FTO/c-TiO2/CABB-NC/Au configuration, and the current–voltage (J–V) curves were measured between 0–3 V under both light and dark conditions (Fig. S18, ESI†). The current increases non-linearly with the increment of the potential bias (J ∝ Vn) (Fig. S19a–d, ESI†). The trap state density (n) and charge carrier mobility (μ) were analyzed in the dark by the space charge limited current (SCLC) method.18 The μ values vary as (1.50 ± 0.08) × 10−2, (1.26 ± 0.01) × 10−2, (1.30 ± 0.00) × 10−2, and (2.00 ± 0.07) × 10−2 cm2 V−1 s−1 for CABB-OLAM, CABB-HDA, CABB-TDA to CABB-DA NCs, respectively (Fig. 4a and Fig. S19e–h, ESI†). The electron mobility of the 10−2 order of magnitude is at par or better than the reported CABB NCs (Table S3, ESI†),8,19–23 but lower than the CABB single crystals (Table S4, ESI†).24–26 The increase in mobility is due to a steady decrease in n, which decreases to (46.3 ± 0.5) × 1015, (44.9 ± 0.5) × 1015, (43.0 ± 0.9) × 1015, and (41.7 ± 0.2) × 1015 cm−3 for CABB-OLAM, CABB-HDA, CABB-TDA, and CABB-DA NCs, respectively (Fig. 4b). For CABB-OLAM to CABB-DA NCs, the trap-filled limited voltage decreases from 1.00 ± 0.01 to 0.90 ± 0.02 V. While all the NCs show decent light sensitivity, the CABB-DA NCs with their (400) facet populated with Ag+, Bi3+ and Br− show the highest carrier mobility and photocurrent (Fig. S18, ESI†).
 | ||
| Fig. 4 Amine chain length-dependent variation of (a) charge carrier mobility (μ) and (b) trap-state density (n). The inset of panel a shows the digital image of a typical electron-only device. | ||
In summary, the dynamics of the facet-specific primary amine ligands with chain lengths of 18-C to 12-C in the CABB NCs could stabilize the crystallographic facets of (222) for CABB-OLAM NCs, (400) and (440) for CABB-HDA and CABB-TDA NCs, and (400) for CABB-DA NCs. The calculated adsorption energy of the amine molecules corroborates the TEM results. The PL bands originate from the localized defect states of the bound exciton. From CABB-OLAM to CABB-DA NCs, the differently exposed facets increase the NC size and lattice strain, while lowering the bandgap from 2.04 to 1.76 eV, decreasing the binding energy from 74 to 59 meV, and increasing the electron mobility from 1.5 × 10−2 to 2 × 10−2 cm2 V−1 s−1. In particular, the exposed (400) facets with the B-site (Ag+, Bi3+) and Br− ions allow the CABB-DA NCs to have the highest carrier mobility and work function, making them more suitable for optoelectronic devices. Our study shows that facet engineering of double perovskite NCs can make them suitable for optoelectronic and photovoltaic applications, beyond the CsPbX3 (X = Br/I) systems.
A. M. acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi. M.A. acknowledges the Shiv Nadar Foundation for financial aid. P. J. and M. A. acknowledge the high-performance computing facility “Magus” and workstations at the School of Natural Sciences, Shiv Nadar Institution of Eminence. S. B. acknowledges the financial support from SERB (CRG/2020/000084 and STR/2021/000001).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. S. Lamba, P. Basera, S. Singh, S. Bhattacharya and S. Sapra, J. Phys. Chem. C, 2021, 125, 1954–1962 CrossRef CAS.
- A. H. Slavney, B. A. Connor, L. Leppert and H. I. Karunadasa, Chem. Sci., 2019, 10, 11041–11053 RSC.
- K.-Z. Du, W. Meng, X. Wang, Y. Yan and D. B. Mitzi, Angew. Chem., Int. Ed., 2017, 56, 8158–8162 CrossRef CAS PubMed.
- S. Banerjee, S. Bera and N. Pradhan, ACS Nano, 2023, 17, 678–686 CrossRef CAS PubMed.
- D. Ghosh, Md. Y. Ali, A. Ghosh, A. Mandal and S. Bhattacharyya, J. Phys. Chem. C, 2021, 125, 5485–5493 CrossRef CAS.
- S. P. Chaudhary, S. Bhattacharjee, V. Hazra, S. Shyamal, N. Pradhan and S. Bhattacharyya, Nanoscale, 2022, 14, 4281–4291 RSC.
- A. Stelmakh, M. Aebli, A. Baumketner and M. V. Kovalenko, Chem. Mater., 2021, 33, 5962–5973 CrossRef CAS PubMed.
- A. Mandal, A. Ghosh, D. Ghosh and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2021, 13, 43104–43114 CrossRef CAS PubMed.
- V. Hazra, S. Mondal, P. Pattanayak and S. Bhattacharyya, Small, 2023, 2304920 Search PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- A. Mandal, A. Ghosh, S. P. Senanayak, R. H. Friend and S. Bhattacharyya, J. Phys. Chem. Lett., 2021, 12, 1560–1566 CrossRef CAS PubMed.
- S. J. Zelewski, J. M. Urban, A. Surrente, D. K. Maude, A. Kuc, L. Schade, R. D. Johnson, M. Dollmann, P. K. Nayak, H. J. Snaith, P. Radaelli, R. Kudrawiec, R. J. Nicholas, R. J. P. Plochocka and M. Baranowski, J. Mater. Chem. C, 2019, 7, 8350–8356 RSC.
- A. D. Wright, L. R. V. Buizza, K. J. Savill, G. Longo, H. J. Snaith, M. B. Johnston and L. M. Herz, J. Phys. Chem. Lett., 2021, 12, 3352–3360 CrossRef CAS PubMed.
- A. Dey, A. F. Richter, T. Debnath, H. Huang, L. Polavarapu and J. Feldmann, ACS Nano, 2020, 14, 5855–5861 CrossRef CAS PubMed.
- A. D. Wright, C. Verdi, R. L. Milot, G. E. Eperon, M. A. Pérez-Osorio, H. J. Snaith, G. Feliciano, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2017, 27, 1700860 CrossRef.
- A. Mandal, S. Roy, A. Mondal, S. Gupta, B. Pal and S. Bhattacharyya, J. Phys. Chem. Lett., 2022, 13, 9103–9113 CrossRef CAS PubMed.
- A. Mandal, S. K. Khuntia, D. Mondal, P. Mahadevan and S. Bhattacharyya, J. Am. Chem. Soc., 2023, 145, 24990–25002 CrossRef CAS PubMed.
- V. M. L. Corre, E. A. Duijnstee, O. E. Tambouli, J. M. Ball, H. J. Snaith, J. Lim and L. J. A. Koster, ACS Energy Lett., 2021, 6, 1087–1094 CrossRef PubMed.
- Z. X. Zhang, C. Li, Y. Lu, X. W. Tong, F. X. Liang, X. Y. Zhao, D. Wu, C. Xie and L. B. Luo, J. Phys. Chem. Lett., 2019, 10, 5343–5350 CrossRef CAS PubMed.
- Y. Zhang, M. Sun, N. Zhou, B. Huang and H. Zhou, J. Phys. Chem. Lett., 2020, 11, 7610–7616 CrossRef CAS PubMed.
- S. Kumar and C. J. Shih, J. Appl. Phys., 2020, 128, 120901 CrossRef CAS.
- B. Maynard, Q. Long, E. A. Schiff, M. Yang, K. Zhu, R. Kottokkaran, H. Abbas and V. L. Dalal, Appl. Phys. Lett., 2016, 108, 173505 CrossRef.
- A. Kumar, S. K. Swami, S. S. Rawat, V. N. Singh, O. P. Sinha and R. Srivastava, Int. J. Energy Res., 2021, 45, 16769–16780 CrossRef CAS.
- Y. Dang, G. Tong, W. Song, Z. Liu, L. Qiu, L. K. Ono and Y. Qi, J. Mater. Chem. C, 2019, 8, 276–284 RSC.
- Y. Yin, W. Tian, J. Leng, J. Bian and S. Jin, Phys. Chem. Lett., 2020, 11, 6956–6963 CrossRef CAS PubMed.
- K. M. McCall, Z. Liu, G. Trimarchi, C. C. Stoumpos, W. Lin, Y. He, I. Hadar, M. G. Kanatzidis and B. W. Wessels, ACS Photonics, 2018, 5, 3748–3762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental Section; Rietveld refined XRD patterns; W–H plots; XPS; IR spectra; HAADF-STEM maps; TEM and FFT analyses; Computational parameters; Tauc plots; Bandgap; KPFM; Optical properties; Work function; Current–voltage plots; Carrier mobility and trap state density. See DOI: https://doi.org/10.1039/d4cc00270a |
| This journal is © The Royal Society of Chemistry 2024 |