
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed diacylmethylation of tyrosine and its peptides with sulfoxonium ylides†
Narendra Dinkar
Kharat
a,
Sushma
Naharwal
a,
Siva S.
Panda
 b,
Kiran
Bajaj
*c and
Rajeev
Sakhuja
*a
b,
Kiran
Bajaj
*c and
Rajeev
Sakhuja
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Rajasthan 333031, India. E-mail: rajeev.sakhuja@pilani.bits-pilani.ac.in
bDepartment of Chemistry and Biochemistry & Department of Biochemistry and Molecular Biology, Augusta University, Augusta, GA 30912, USA
cDepartment of Chemistry, Amity University, Noida, Uttar Pradesh, India. E-mail: kbajaj26@gmail.com
First published on 24th June 2024
Abstract
Pyridyloxy-directed Ir(III)-catalyzed diacylmethylation of protected tyrosines was achieved with alkyl and (hetero)aryl sulfoxonium ylides, furnishing tyrosine-based unnatural amino acids in good yields. Furthermore, the late stage exemplification of the strategy was successfully accomplished in tyrosine-containing dipeptides, tripeptides and tetrapeptides in moderate yields. This methodology is distinguished by its site-selectivity, tolerance of sensitive functional groups, scalability, and retention of the chiral configuration for tyrosine motifs.
Unnatural amino acids (UAAs) prepared by site selective manipulation or conjugation of an amino acid with bioactive molecules have demonstrated remarkable applications in drug optimization and drug delivery studies.1 Such designer amino acids possess unique structural features that foster peptide interactions with biological targets, leading to the discovery of new therapeutic modalities.2 In addition, introducing UAAs at specific sites in a protein can fine-tune its properties and functions as per the desired requirement.3 Thus, the applications of functionalized peptides and peptidomimetics as molecular probes and therapeutics have increased manifold, leading to a growing need for developing novel strategies for chemical modification of amino acids with structural diversity.4
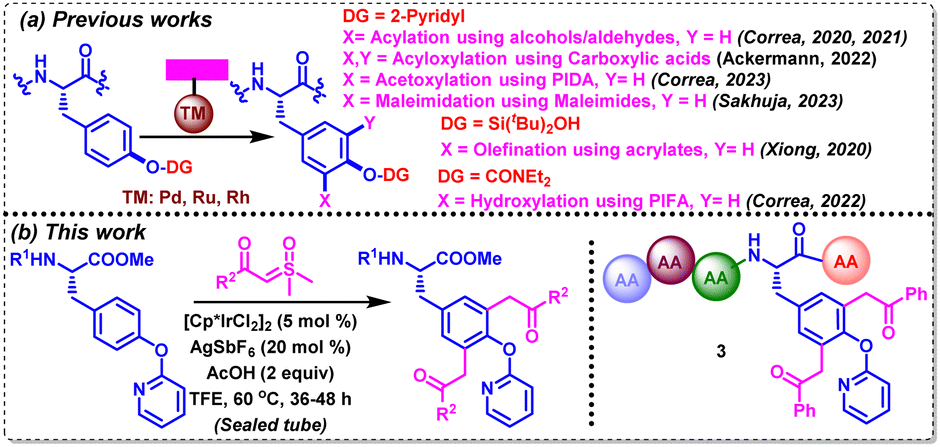
Tyrosine (Tyr) is a proteinogenic amino acid that is ubiquitous in both natural and synthetic bioactive molecules, including endomorphine,5 levothyroxine,6a KN-62,6b OF4949-I,6c and mycocyclosin.6d Due to the presence of an amphiphilic phenolic side chain, it has been involved in varied biochemical processes, exhibiting a significant role in protein–protein and protein–ligand interactions. Its susceptibility to other functionalities by post-translational modifications in the human body further highlight the need for investigating such selectively modified tyrosines in bioactive peptides and proteins. In spite of the low natural abundance of tyrosine, it is found to be embedded in distinct biological environments in most of the proteins.7 Notably, a handful of eye-catching metal-catalyzed and metal-free strategies have evolved over the years, aiming at functionalizing proximal and distal C–H bonds in varied amino acids, including tyrosine.8,9 In this realm, Xiong, Correa and Ackerman have intensified their efforts recently towards the selective functionalization of tyrosine and tyrosine-containing oligopeptides by employing a variety of directing groups (DGs) (Scheme 1a). For example, Xiong's group reported silanol-mediated C3Ar–H olefination in tyrosine and tyrosine-containing peptides with olefins under Pd-catalysis.10a Correa's group achieved pyridyloxy-directed late-stage C3Ar–H acylation and acetoxylation of Tyr-containing oligopeptides with alcohols/aldehydes and (diacetoxyiodo)benzene (PIDA) under Pd- and Ru-catalyzed conditions, respectively.10b–d In addition, the same group achieved C3Ar–H hydroxylation by carbamate-directed Ru-catalyzed activation with PhI-(OCOCF3)2.10e Ackermann's group employed carboxylic acids for the late-stage diacyloxylation of tyrosine-containing peptides under electro-catalyzed conditions.10f Recently, our group has also disclosed a Rh(II)-catalyzed strategy for the C3Ar-maleimidation of tyrosines and tyrosine-containing oligopeptides with maleimides.10g Interestingly, sulfoxonium ylides that are often employed for o-acylmethylation, and annulation of variedly decorated (hetero)arenes, have not been explored for the CAr–H acylmethylation of any aromatic amino acids.11a,b We aimed to functionalize a tyrosine scaffold with such safe carbene precursors via transition-metal-catalyzed C–H bond activation with the aid of an appropriate directing group.
 | ||
| Scheme 1 CAr–H functionalization of Tyr- and Tyr-containing peptides via DG-assisted TM-catalyzed C–H activation. | ||
In the backdrop of the aforementioned discussion, we herein disclose a IrIII-catalyzed pyridyloxy-directed methodology for the synthesis of C3Ar/C5Ar-diacylmethylated tyrosines and tyrosine-containing oligopeptides using a variety of alkyl and (hetero)aryl sulfoxonium ylides in moderate-to-good yields (Scheme 1b).
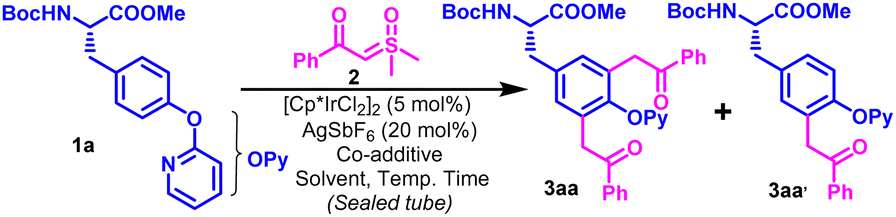
We performed our optimization studies with Boc-L-Tyr(OPy)-OMe (1a) and 2-(dimethyl(oxo)-l6-sulfanylidene)-1-phenylethan-1-one (2a) as model substrates under Ir-catalyzed conditions (Table 1 and Table S1, ESI†). The use of PivOH (2 equiv.) as a co-additive with AgSbF6 (20 mol%) with catalytic amounts of [Cp*IrCl2]2 initiated the stoichiometric reaction between the model substrates in DCE at 60 °C to afford a mixture of mono- and diacylmethyl functionalized products (3aa & 3aa′) in 18% and 12% yields, respectively (Table 1, entry 1). Delightfully, the usage of 3 equivalents of sulfoxonium ylide (2a) dramatically elevated the yield of 3aa to 58% along with a slight increase in the yield of 3aa′ to 22% (Table 1, entry 2). Substitution of PivOH with ADA produced 3aa in 64% yield along with 14% of 3aa′, while its replacement with AcOH furnishes 3aa in 72% yield along with very small (non-isolable) amounts of 3aa′ (Table 1, entries 3–4). Our solvent screening studies suggested that this transformation is highly solvent-dependent (Table S1, ESI†), and the target difunctionalized product could be regioselectivity obtained in 88% yield using TFE as a solvent (Table 1, entry 5). Catalyst loading modulation had no significant effect on the product yield (Table S1, ESI†). It is worth mentioning that increasing the temperature for the aforementioned optimized reaction to 80 °C produced detrimental effects on the yield of 3aa as a few other minor side products were visible on TLC (Table 1, entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Co-additive | Solvent | Yieldb of 3aa (%) | Yieldb of 3aa′ (%) |
| a Reaction conditions: the reactions were carried out with 1a (0.187 mmol) and 2a (0.561 mmol) with [Cp*IrCl2]2 in the presence of additive/Co-additive as indicated in a solvent (2 mL) at 60 °C for 36 h in a sealed tube. b Isolated yields. c 2a (0.187 mmol). d Temperature 80 °C (minor additional spots on TLC were observed). | ||||
| 1c | PivOH (2 equiv.) | DCE | 18 | 12 |
| 2 | PivOH (2 equiv.) | DCE | 58 | 22 |
| 3 | ADA (2 equiv.) | DCE | 64 | 14 |
| 4 | AcOH (2 equiv.) | DCE | 72 | <10 |
| 5 | AcOH (2 equiv.) | TFE | 88 | Traces |
| 6d | AcOH (2 equiv.) | TFE | 72 | Traces |
The scope of this optimized Ir-catalyzed CAr–H diacylmethylation of Boc-protected tyrosine (1a) was next examined with variedly decorated alkyl and (hetero)aryl carbonyl sulfoxonium ylides (2b–k) (Scheme 2). Sulfoxonium ylides substituted with electron-donating groups [2b (R2 = 4-MeC6H4) and 2c (R2 = 4-OMeC6H4)] and strong electron-withdrawing groups [2d (R2 = 4-FC6H4), 2e (R2 = 4-CF3C6H4) and 2f (R2 = 4-NO2C6H4)] reacted smoothly with 1a under the described conditions to furnish their corresponding diacylmethylated products 3ab–3af in 77–84% yields (Scheme 2). Slight variation in the reactivity of para- and meta-substituted chlorophenyl carbonyl sulfoxonium ylides was noticed, producing their respective products (3ag & 3ah) in 76% and 78% yields, respectively. Gratifyingly, the reaction was workable with (hetero)aryl carbonyl sulfoxonium ylides [2i (R2 = 2-thienyl) and 2j (R2 = 2-furyl)] in reasonable reactivity to comfortably accomplish dithienylmethylation and difurylmethylation at the C3/C5 positions in protected tyrosine (1a), affording 3ai and 3aj in 72% and 69% yields, respectively. The C–H diacylmethylation of 1a also proceeded with moderate ease by using alkyl carbonyl sulfoxonium ylide [2k (R2 = nBu)], delivering the desired product 3ak in 65% yield. Interestingly, the presence of other N-protecting groups, such as Ac, and Cbz (substrates 1b–c) was well-tolerated on the N-side of tyrosine esters under the optimized conditions, producing their relevant diacylmethyl functionalized Tyr-based UAAs (3ba–ca) in 73–76% yields. The strategy was scalable on a 1.3 mmol scale to afford 3aa in 78% yield under the standard conditions in 48 h (Scheme 2).
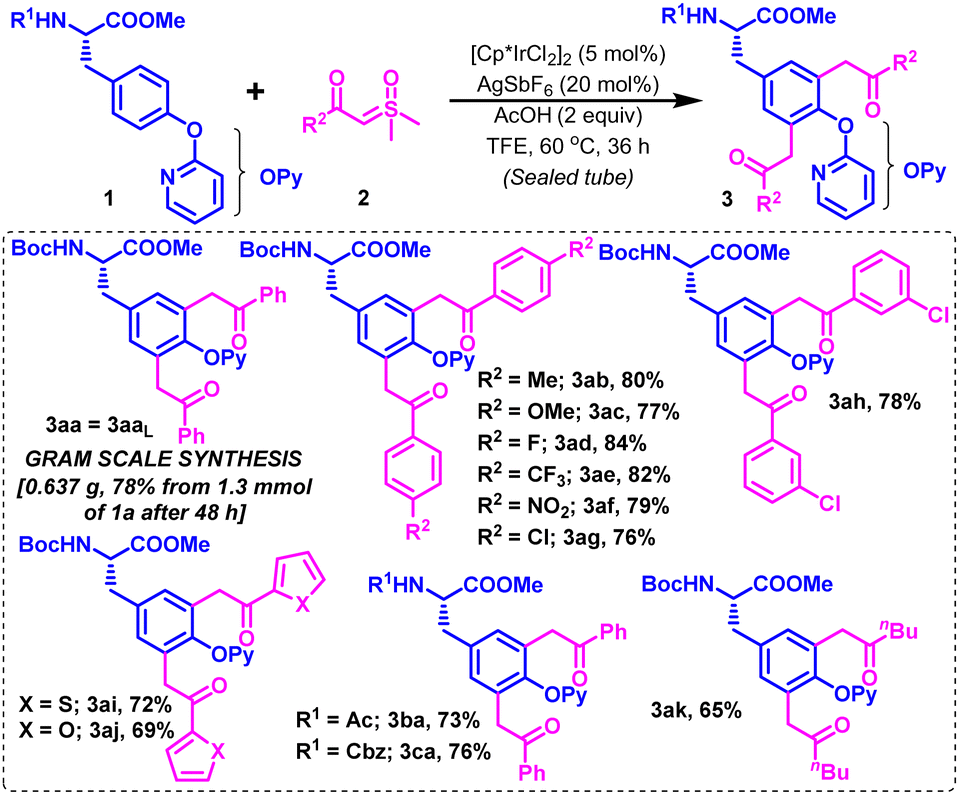 | ||
| Scheme 2 C3Ar/5Ar–H diacylmethylation of tyrosines with sulfoxonium ylides. | ||
Furthermore, we attempted to exemplify the synthetic scope of the strategy towards the late stage functionalization of tyrosine in tyrosine-containing peptides (Scheme 3). Pleasingly, tyrosine-containing peptides underwent functionalization with sulfoxonium ylides in moderate-to-good reactivities. For example, the late stage C3/5Ar-difunctionalization of the dipeptides Cbz-L-Tyr(OPy)-L-Phe-OMe (1d), Cbz-L-Tyr(OPy)-L-Val-OMe (1e), Cbz-L-Val-L-Tyr(OPy)-OMe (1f), Cbz-Gly-L-Tyr(OPy)-OMe (1g), Boc-L-Ala-L-Tyr(OPy)-OMe (1h), Cbz-L-Lys(Nε-Boc)-L-Tyr(OPy)-OMe (1i), Boc-L-Glu(OBn)-L-Tyr(OPy)-OMe (1j), Boc-L-Pro-L-Tyr(OPy)-OMe (1k), Boc-L-Thr-L-Tyr(OPy)-OMe (1l), Boc-L-Asn-L-Tyr(OPy)-OMe (1m), and Boc-L-Trp-L-Tyr(OPy)-OMe (1n), with 2a produced their respective expected dibenzoylmethylated Tyr-containing dipeptides (3da, 3ea, 3fa, 3ga, 3ha, 3ia, 3ja, 3ka, 3la, 3ma, and 3na) in 47–72% yields (Scheme 3). Among these, 3ka was obtained as a mixture of rotamers as usually observed for proline-containing dipeptides. Notably, the strategy is equally workable for o-pyridyl tyrosine-containing dipeptides possessing tyrosine at the C- or N-terminus ends and Cbz or Boc protecting groups at the N-side or at the appended side chain functionalities, albeit in 36–48 h. Gratifyingly, this strategy works reasonably well for the late stage difunctionalization of tyrosine-containing tripeptides Boc-Gly-Gly-L-Tyr(OPy)-OMe (1o) and Boc-Gly-L-Ala-L-Tyr(OPy)-OMe (1p) and tetrapeptides Boc-Gly-Gly-Gly-L-Tyr(OPy)-OMe (1q) and Cbz-L-Val-L-Ala-L-Lys(Nα-Boc)-L-Tyr(OPy)-OMe (1r) with 2a, producing their corresponding bis(benzoylmethyl)-appended Ty-containing tripeptides (3oa, 3pa) and tetrapeptides (3qa, 3ra) in 53–58% and 43–48% yields, respectively.
 | ||
| Scheme 3 Late-stage Tyr-containing peptide diacylmethylation. | ||
The enantiopurity of one of the products 3aa (3aaL) was ascertained by comparing its HPLC profile with that of the corresponding racemic product [3aaDL; obtained in 86% yield from racemic Boc-DL-Tyr(OPy)-OMe (1aDL)]. The high enantiomeric ratio (>98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) observed in the HPLC profile of 3aaL indicates that almost no racemization is observed at the chiral center during the product formation (ESI†). The chemical applicability of the synthesized Tyr-based UAAs was demonstrated by performing the reduction of the two carbonyl groups with sodium borohydride in methanol to afford the corresponding bis(2-hydroxy-2-phenylethyl)-functionalized tyrosine derivative (4aa) in 82% yield as a mixture of diastereomers (Scheme 4i). Furthermore, deprotection of N-Boc and ester groups using TFA/DCM and LiOH/THF:H2O produced their respective N- and C-side deprotected derivatives (5aa & 6aa) in 53% & 63% yields, respectively (Scheme 4ii and iii). It is worth mentioning that attempted deprotection of the O-pyridyl directing group from 3aa (or 3ba/3ca) by using the standard two-step protocol, involving the N-methylation with MeOTf/DCM followed by hydrogenation with Pd(OH)2–C/HCOONH4/MeOH or Na/MeOH could not succeed as messy mixtures were obtained in each case. To gain insights into the reaction mechanism, a few investigations were carried out. A very low H/D scrambling (approx. ∼8%) was observed by performing deuterium labelling experiments on 1a under the standard conditions using 2 equivalents of deuterated acetic acid, suggesting the non-reversible nature of metalation/deutero(proto)demetalation of C3Ar/C5Ar–H bond(s) (Scheme 4iv).
:2) observed in the HPLC profile of 3aaL indicates that almost no racemization is observed at the chiral center during the product formation (ESI†). The chemical applicability of the synthesized Tyr-based UAAs was demonstrated by performing the reduction of the two carbonyl groups with sodium borohydride in methanol to afford the corresponding bis(2-hydroxy-2-phenylethyl)-functionalized tyrosine derivative (4aa) in 82% yield as a mixture of diastereomers (Scheme 4i). Furthermore, deprotection of N-Boc and ester groups using TFA/DCM and LiOH/THF:H2O produced their respective N- and C-side deprotected derivatives (5aa & 6aa) in 53% & 63% yields, respectively (Scheme 4ii and iii). It is worth mentioning that attempted deprotection of the O-pyridyl directing group from 3aa (or 3ba/3ca) by using the standard two-step protocol, involving the N-methylation with MeOTf/DCM followed by hydrogenation with Pd(OH)2–C/HCOONH4/MeOH or Na/MeOH could not succeed as messy mixtures were obtained in each case. To gain insights into the reaction mechanism, a few investigations were carried out. A very low H/D scrambling (approx. ∼8%) was observed by performing deuterium labelling experiments on 1a under the standard conditions using 2 equivalents of deuterated acetic acid, suggesting the non-reversible nature of metalation/deutero(proto)demetalation of C3Ar/C5Ar–H bond(s) (Scheme 4iv).
 | ||
| Scheme 4 Chemical applicability of 3aa and mechanistic studies. | ||
Interestingly, a pale yellow semi-solid (7a) was obtained in high yield by performing a stoichiometric reaction between 1a and [Cp*IrCl2]2 at 60 °C for 18 h; the detailed spectroscopic investigations of 7a suggested that it could possibly be an equimolar mixture of two six-membered iridacyclic intermediates (A′ + B′) (Scheme 4v). Notably, 3aa was obtained in 68% yield by carrying out the standard reaction between 1a and 2a using a catalytic amount of 7a with acetic acid, while traces of 3aa were visible on TLC in the absence of acetic acid (Scheme 4vi). These observations suggested 7a to be a viable intermediate, and a crucial role of acetic acid in the described transformation. Furthermore, real-time standard reaction monitoring through mass spectrometry indicated the presence of C′ and D′ in the reaction mixture (Scheme 4vii).
On the basis of the literature studies12 and our investigation, a plausible mechanism is proposed (Scheme S1, ESI†). The reaction could be believed to proceed by AgSbF6-mediated dissociation of [Cp*IrCl2]2 to monomeric active [Cp*IrCl(SbF6)] species, which undergoes pyridyl-aided C3Ar–H metalation in 1 to furnish a six-membered iridacyclic intermediate BviaA. Subsequently, the coordination of sulfoxonium ylide 2 to the iridium center in B generates Ir–carbene complex C along with the extrusion of DMSO, which is followed by its regioselective migratory insertion between the C3Ar–Ir bond generating a seven-membered iridacyclic intermediate D. Thereafter, protonolysis of the C3Ar-acylmethyl group followed by re-coordination of Ir(III) with C5Ar–H probably via a reversible rollover cyclometalation gives E, which upon repeated coordination of another molecule of sulfoxonium ylide with the iridium center initiates a second catalytic pathway to furnish 3aa, along with the regeneration of the active Ir(III) catalyst for the next catalytic cycle.
In conclusion, we have developed a Ir(III)-catalyzed strategy for C3Ar/C5Ar-difunctionalization of N-protected O-pyridyloxy tyrosines with electron-rich and deficient sulfoxonium ylides to furnish a series of diacylmethylated tyrosine-based unnatural amino acids in moderate-to-good yields. The strategy was successfully employed for the late-stage difunctionalization of tyrosine-containing dipeptides, tripeptides, and tetrapeptides in reasonable reactivity. Isolation of a stable iridacyclic intermediate and the successful deprotection of the C- and N-side protecting groups are the added highlights of the presented work.
The authors acknowledge the Science and Engineering Research Board (SERB), New Delhi, for providing a research grant (CRG/2021/000131). NKD is thankful to BITS Pilani for providing a Senior Research Fellowship. SN is thankful to CSIR, New Delhi for providing a Senior Research Fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed; (b) R. J. Malonis, J. R. Lai and O. Vergnolle, Chem. Rev., 2020, 120, 3210 CrossRef CAS PubMed; (c) E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- (a) L. M. Behera, M. Ghosh and S. Rana, Amino Acids, 2022, 54, 1183–1202 CrossRef CAS PubMed; (b) R. Serfling, C. Lorenz, M. Etzel, G. Schicht, T. Böttke, M. Mörl and I. Coin, Nucleic Acids Res., 2018, 46, 1–10 CrossRef CAS PubMed.
- (a) H. Zhao, W. Ding, J. Zang, Y. Yang, C. Liu, L. Hu, Y. Chen, G. Liu, Y. Fang and Y. Yuan, Nat. Commun., 2021, 12, 7039 CrossRef CAS PubMed; (b) M. Garton, M. Sayadi and P. M. Kim, Plos One, 2017, 12, e0187524 CrossRef PubMed; (c) A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- (a) J. Tang, H. Chen, Y. He, W. Sheng, Q. Bai and H. Wang, Nat. Commun., 2018, 9, 3383 CrossRef PubMed; (b) A. Stevenazzi, M. Marchini, G. Sandrone, B. Vergani and M. Lattanzio, Bioorg. Med. Chem. Lett., 2014, 24, 5349–5356 CrossRef CAS PubMed; (c) A. Mizuno, K. Matsui and S. Shuto, Chem. – Eur. J., 2017, 23, 14394–14409 CrossRef CAS PubMed.
- (a) G. Horvath, Clin. Pharm. Therap., 2000, 88, 437–463 CAS; (b) J. D. Tyndall and R. Sandilya, Med. Chem., 2005, 1, 405–421 CrossRef CAS PubMed; (c) A. Slominski, D. J. Tobin, S. Shibahara and J. Wortsman, Physiol. Rev., 2004, 84, 1155–1228 CrossRef CAS PubMed; (d) A. Leone, G. Noera and A. Bertolini, Curr. Med. Chem., 2013, 20, 735–750 Search PubMed; (e) C. A. Schenck and H. A. Maeda, Phytochem., 2018, 149, 82–102 CrossRef CAS PubMed; (f) J. Fichna, A. Janecka, J. Costentin and J.-C. Do Rego, Pharmacol. Rev., 2007, 59, 88–123 CrossRef CAS PubMed.
- (a) S. Mandel, G. Brent and P. Larsen, Endocrinologist., 1994, 4, 152 CrossRef; (b) J.-H. Park, G.-E. Lee, S.-D. Lee, T. T. Hien, S. Kim, J. W. Yang, J.-H. Cho, H. Ko, S.-C. Lim and Y.-G. Kim, J. Med. Chem., 2015, 58, 2114–2134 CrossRef CAS PubMed; (c) S. Sano, K. Ikai, K. Katayama, K. Takesako, T. Nakamura, A. Obayashi, Y. Ezure and H. Enomoto, J. Antibiot., 1986, 39, 1685–1696 CrossRef CAS PubMed; (d) K. A. Niederer, P. H. Gilmartin and M. C. Kozlowski, ACS Catal., 2020, 10, 14615–14623 CrossRef CAS PubMed.
- (a) G. Rossino, E. Marchese, G. Galli, F. Verde, M. Finizio, M. Serra, P. Linciano and S. Collina, Molecules, 2023, 28, 7165 CrossRef CAS PubMed; (b) T. Schlatzer, J. Kriegesmann, H. Schröder, M. Trobe, C. Lembacher-Fadum, S. Santner, A. V. Kravchuk, C. F. Becker and R. Breinbauer, J. Am. Chem. Soc., 2019, 141, 14931–14937 CrossRef CAS PubMed.
- (a) A. F. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed; (b) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS PubMed; (c) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed; (d) G. Chen, Z. Zhuang, G. C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J. Q. Yu, Angew. Chem., 2017, 129, 1528–1531 CrossRef; (e) A. Correa, Eur. J. Inorg. Chem., 2021, 2928–2941 CrossRef CAS; (f) S. Zhang, L. M. D. L. Rodriguez, F. F. Li, R. Huang, I. K. Leung, P. W. Harris and M. A. Brimble, Chem. Sci., 2022, 13, 2753–2763 RSC; (g) S. Zhang, L. M. D. L. Rodriguez, F. F. Li and M. A. Brimble, Chem. Sci., 2023, 14, 7782–7817 RSC.
- (a) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed; (b) N. D. Kharat, C. K. Mahesha, K. Bajaj and R. Sakhuja, Org. Lett., 2022, 24, 6857–6862 CrossRef CAS PubMed; (c) C. Girón-Elola, I. Sasiain, R. Sánchez-Fernández, E. Pazos and A. Correa, Org. Lett., 2023, 25, 4383–4387 CrossRef PubMed; (d) S. Bhunia, M. Purushotham, G. Karan, B. Paul and M. S. Maji, Synthesis, 2023, 3701–3724 CAS; (e) L. Lukasevics, A. Cizikovs and L. Grigorjeva, Chem. Commun., 2021, 57, 10827–10841 RSC; (f) A. Ville, J. Annibaletto, S. Coufourier, C. Hoarau, R. Tamion, G. Journot, C. Schneider and J. F. Brière, Chem. – Eur. J., 2021, 27, 13961–13965 CrossRef CAS PubMed; (g) P. Singh and S. A. Babu, Eur. J. Org. Chem., 2023, e202300440 CrossRef CAS; (h) S. Shabani, Y. Wu, H. G. Ryan and C. A. Hutton, Chem. Soc. Rev., 2021, 50, 9278–9343 RSC.
- (a) Q.-L. Hu, K.-Q. Hou, J. Li, Y. Ge, Z.-D. Song, A. S. Chan and X.-F. Xiong, Chem. Sci., 2020, 11, 6070–6074 RSC; (b) I. Urruzuno, P. Andrade-Sampedro and A. Correa, Org. Lett., 2021, 23, 7279–7284 CrossRef CAS PubMed; (c) M. San Segundo and A. Correa, Chem. Sci., 2020, 11, 11531–11538 RSC; (d) I. Urruzuno, P. Andrade-Sampedro and A. Correa, Eur. J. Org. Chem., 2023, e202201489 CrossRef CAS; (e) P. Andrade-Sampedro, J. M. Matxain and A. Correa, Adv. Synth. Catal., 2022, 364, 2072–2079 CrossRef CAS; (f) X. Hou, N. Kaplaneris, B. Yuan, J. Frey, T. Ohyama, A. M. Messinis and L. Ackermann, Chem. Sci., 2022, 13, 3461–3467 RSC; (g) N. D. Kharat, S. Naharwal, D. Tank, S. S. Panda, K. Bajaj and R. Sakhuja, Org. Lett., 2023, 25, 7673–7677 CrossRef CAS PubMed.
- (a) A. Kumar, M. S. Sherikar, V. Hanchate and K. R. Prabhu, Tetrahedron, 2021, 101, 132478 CrossRef CAS; (b) P. Bhorali, S. Sultana and S. Gogoi, Asian J. Org. Chem., 2022, 11, 157–176 CrossRef.
- (a) Y. Jia, W. Si, B. Dan, L. Weiwei, C. Yanhui and C. Guolin, Org. Lett., 2019, 21, 6366–6369 CrossRef PubMed; (b) S. Y. Hong, J. Kwak and S. Chang, Chem. Commun., 2016, 52, 3159–3162 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02204a |
| This journal is © The Royal Society of Chemistry 2024 |