
Annulative coupling of α-substituted acrylic acids and sulfoxonium ylides: easy access to bioactive γ-butyrolactones†
Naveen
Kumar
and
Satyendra Kumar
Pandey
 *
*
Department of Chemistry, Institute of Science, Banaras Hindu University, Varanasi 221005, India. E-mail: skpandey.chem@bhu.ac.in
First published on 25th July 2024
Abstract
We present a straightforward, catalyst- and additive-free method for synthesizing keto γ-butyrolactones using readily available β-keto sulfoxonium ylides and acrylic acids. This robust approach demonstrates exceptional compatibility with various functional groups on β-keto sulfoxonium ylides and α-substituted acrylic acids, resulting in good to high yields of the anticipated products. Moreover, the practicality of this approach was validated through large-scale reactions and the successful conversion of some synthesized derivatives into bioactive natural products, including L-factor, muricatacin, and cytosporanone A.
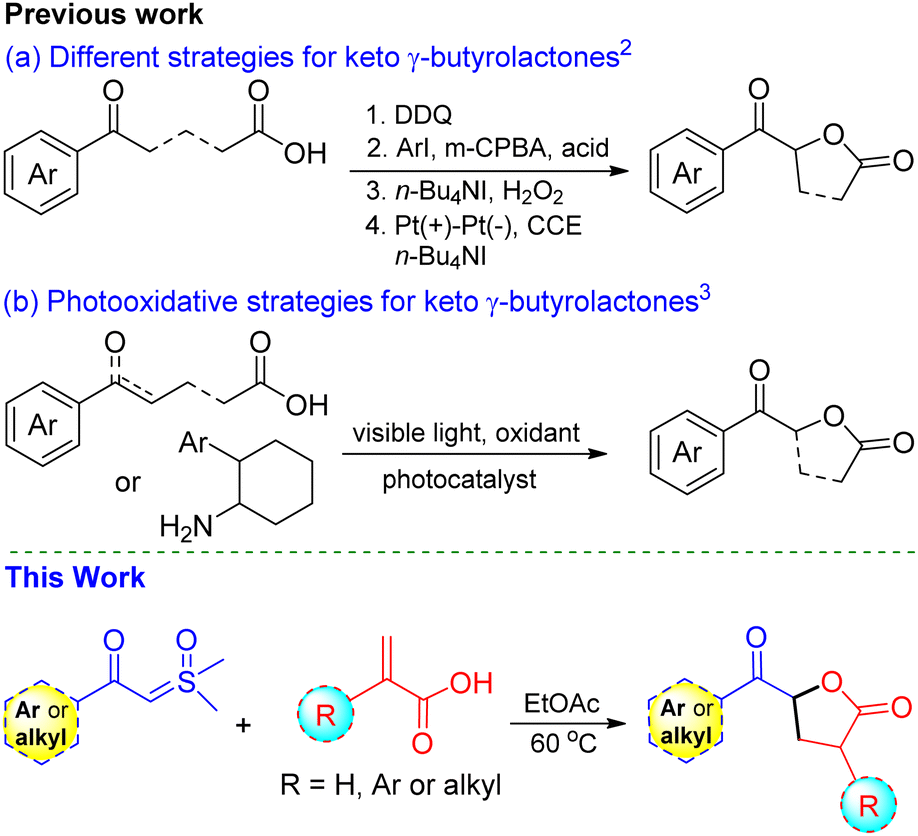
The functionalized γ-butyrolactone structural motif is highly regarded as a privileged structure in medicinal chemistry, found in numerous synthetic compounds and natural products with diverse biological activities (Fig. 1).1 Due to its array of applications and biological activities, the synthesis of keto γ-butyrolactone has recently garnered significant interest in the literature, following Ochiai's pioneering work.2 Zhang and co-workers described an alternative approach employing DDQ as the catalyst.2a Notably, Ishihara and co-workers significantly advanced the synthesis of keto γ-butyrolactones through their remarkable research work on iodine- and hypervalent iodine-catalyzed lactonization (Scheme 1a).2b,c Photoredox-catalyzed photooxidative methods have also been successfully utilized to synthesize keto γ-butyrolactones from a variety of substrates (Scheme 1b).3 Recently, Zhang, Xu, and co-workers disclosed an elegant approach employing electrocatalytic dehydrogenative esterification of aryl-substituted γ-keto butanoic acids, using n-Bu4NI as a catalyst, for the synthesis of keto γ-butyrolactones.4 Despite advancements in synthesizing keto γ-butyrolactones most existing methods are still constrained by expensive reagents, restricted raw material availability, limited substrate scopes and stoichiometric waste. Therefore, it remains highly important to develop a simple, versatile, and metal-free procedure for access to these scaffolds, which are essential for research in the pharmaceutical industry and academia in their search for bioactive compounds. To our knowledge, the synthesis of keto γ-butyrolactone derivatives from α-substituted acrylic acids and sulfoxonium ylides has not been previously documented.
 | ||
| Fig. 1 Representative natural products with γ-butyrolactone skeleton. | ||
 | ||
| Scheme 1 Synthetic strategies for keto γ-butyrolactones. | ||
Sulfoxonium ylides have recently gained popularity as diazo surrogates in various chemical reactions owing to their ease of use, bench stability, and extended reactive profiles.5 They have been widely employed as C1 or C2 synthons in the synthesis of diverse heterocyclic compounds, including furan, pyrrole, pyrimidine, quinolone, and indole, using transition metal catalysts such as Rh, Pd, Ru, Co, and Ir.6 In our ongoing efforts to develop novel synthetic approaches employing sulfoxonium ylides under metal-free conditions,7 we present a new method for synthesizing highly functionalized keto γ-butyrolactone derivatives using α-substituted acrylic acids and sulfoxonium ylides under mild conditions, without any catalysts or additives.
We hypothesized that the acidic proton of acrylic acid could protonate the α-carbon of sulfoxonium ylides, resulting in the in situ formation of a carboxylate ion. This carboxylate ion would then react with the α-carbon of the ylides to form an α-oxycarbonyl-β-ketone intermediate, releasing DMSO in the process. This intermediate would subsequently undergo tautomerization and intramolecular Michael addition reaction to afford keto γ-butyrolactone. To investigate this hypothesis, we performed experiments at room temperature with the model substrates β-ketosulfoxonium ylide 1a (0.5 mmol) and acrylic acid 2a (0.75 mmol) in a toluene (Table 1). Unfortunately, the desired product was not obtained, and the initial substrates remained unreacted (entry 1). However, we were pleased to observed that increasing the temperature to 60 °C, the model reaction, successfully afforded the anticipated keto γ-butyrolactone 3a with a 65% yield (entry 2). We further explored a variety of solvents to enhance the reaction conditions and observed that ethyl acetate yielded the best results among aprotic solvents such as such as CH3CN, DCM, CHCl3, DMSO, DMF, and DCE (Table 1, entry 9 vs. entries 2–8). We also tested protic solvents such as MeOH, EtOH, and H2O, but the results were not promising (entries 13–15). Finally, we established the optimal conditions, resulting in product 3a with high purity and yield (Table 1, entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time (h) | Temp. (°C) | Yield of 3ab (%) |
| a General conditions: β-keto sulfoxonium ylides 1a (0.50 mmol) and acrylic acids 2a (0.75 mmol) were mixed to a solvent (1.0 mL) and heated to 60 °C, unless otherwise noted. b Isolated yield. c No reaction. Acrylic acid 2a. d 1.0 mmol (entry 10). e 1.25 mmol (entry 11). f 1.50 mmol (entry 12). | ||||
| 1 | Toluene | 48 | rt | nrc |
| 2 | Toluene | 36 | 60 | 65 |
| 3 | CH3CN | 36 | 60 | 45 |
| 4 | DCM | 36 | 60 | 25 |
| 5 | CHCl3 | 36 | 60 | 32 |
| 6 | DMSO | 36 | 60 | 15 |
| 7 | DMF | 36 | 60 | 24 |
| 8 | DCE | 36 | 60 | 60 |
| 9 | EtOAc | 30 | 60 | 88 |
| 10 | EtOAcd | 24 | 60 | 90 |
| 11 | EtOAce | 22 | 60 | 95 |
| 12 | EtOAcf | 22 | 60 | 91 |
| 13 | EtOH | 36 | 60 | 22 |
| 14 | MeOH | 36 | 60 | 35 |
| 15 | H2O | 36 | 60 | nrc |

The optimized conditions provided a foundation for synthesizing keto γ-butyrolactone derivatives and facilitated their application to various other α-substituted acrylic acids and aryl/alkyl ylide derivatives. A range of ylides 1 were effectively transformed into their corresponding keto γ-butyrolactones 3, as shown in Scheme 2 (entries 3a–3t, 85–96%). Initially, employing acrylic acid 2a, the effects and generality of aryl/alkyl-containing sulfoxonium ylides 1 were assessed. We observed that β-keto sulfoxonium ylides with electron-donating groups, including methyl and methoxy, at various positions on the aromatic ring, were well accommodated and yielded the anticipated compounds in good yields (entries 3b–3e, 93–96%). When the aromatic ring of the ylides is decorated with electron-withdrawing substituents, such as bromo, fluoro, trifluoromethyl, and nitro groups at various positions, the reactions proceeded smoothly, yielding the estimated products in high yields (entries 3f–3k, 90–94%). In addition, some heterocyclic compounds, including thiophene and furan containing sulfoxonium ylides and naphthalene sulfoxonium ylides, were tested with acrylic acid 2a, resulting in the corresponding keto γ-butyrolactone derivatives in good yields (Scheme 2, entries 3l, 3m, and 3n: 88%, 86%, and 90%, respectively). Moreover, under optimized conditions, some aliphatic β-keto sulfoxonium ylides were assessed, showing good compatibility and resulting in moderate to high yields of their respective products (entries 3o–3t, 85–90%). Sulfoxonium ylides with other electron-withdrawing groups, such as –CO2Et and –CO2Bn, were also well tolerated and resulted in the anticipated products 3u and 3v, with yields of 85% and 88%, respectively. To demonstrate the practical applicability of our approach, we conducted a gram-scale reaction employing β-keto sulfoxonium ylide 1a (1.00 g, 5.10 mmol) and acrylic acid 2a (0.90 g, 12.75 mmol), which afforded the keto γ-butyrolactone derivative 3a in 92% yield (0.90 g).
 | ||
| Scheme 2 Substrate scope. Reaction conditions: β-keto sulfoxonium ylides 1 (0.5 mmol) and acrylic acids 2a (1.25 mmol) were reacted in EtOAc (1.0 mL) in a sealed tube at 60 °C. | ||
The influence of α-substituted acrylic acids 2 was then evaluated using sulfoxonium ylide 1a, as illustrated in Scheme 3. The reactions between sulfoxonium ylide 1a and various α-substituted acrylic acids 2 were well-tolerated, resulting in the anticipated products as single diastereomers (>99/1 dr) with anti-configuration in moderate to good yields (entries 3w–3ae, 75–85%).8 Both electron-donating and electron-withdrawing groups attached to the α-position of acrylic acids 2 afforded keto γ-butyrolactone derivatives 3 in good to high yields (entries 3w–3ab, 76–81%). The reaction of sulfoxonium ylide 1a with naphthyl-substituted acrylic acid 2ac was found to be highly compatible, yielding the respective compound 3ac in a 75% yield. Acrylic acids with α-substitution by alkyl functional groups, such as methyl and benzyl, were also compatible under the optimized conditions and afforded the keto γ-butyrolactone derivatives in good to high yields (85% and 75% for 3ad and 3ae, respectively). Substitutions at the β-position of acrylic acid, such as methyl (crotonic acid) and phenyl (cinnamic acid), were incompatible with the existing reaction conditions and failed to afford the anticipated products.
 | ||
| Scheme 3 Substrate scope. Reaction conditions: β-keto sulfoxonium ylide 1a (0.5 mmol) and acrylic acids 2 (1.25 mmol) were reacted in EtOAc (1.0 mL) in a sealed tube at 60 °C. | ||
Inspired by the aforementioned methods, we conducted several application experiments to enhance the usefulness and versatility of our developed approach, as illustrated in Scheme 4. Notably, under optimized conditions, we synthesized the keto γ-butyrolactone derivative 3s on a gram-scale using ylide 1s (1.20 g, 6.3 mmol) and acrylic acid 2a (1.08 g, 15.75 mmol) with a yield of 86% (1.00 g) (Scheme 4a). Similarly, the reaction of ylide 1t (1.8 g, 6.3 mmol) and acrylic acid 2a (1.08 g, 15.75 mmol) afforded keto γ-butyrolactone derivative 3t with a 84% (1.5 g) yield. The large-scale synthesis of compounds 3t and 3s demonstrates the prospective of the established method, and these derivatives were then used to synthesize a few bioactive natural products. For example, diastereoselective reduction of keto γ-butyrolactone derivative 3s with BH3. NH3 complex afforded the natural products L-factor94 and muricatacin105 with >99/1 dr in 75% and 78% yields, respectively (Scheme 4b). Muricatacin, an acetogenin extracted from the seeds of the tropical fruit Annona muricata L., has been identified as a quasi-racemic compound (ca. 25% ee).1g It has shown cytotoxic effects on human tumor cell lines and served as a precursor for synthesizing higher acetogenin natural products.11 Additionally, the diastereoselective reduction of keto γ-butyrolactone derivative 3a furnished the natural product cytosporanone A 61b,d (>99/1 dr)4 with 80% yield (Scheme 4c).
 | ||
| Scheme 4 Synthetic applications. | ||
Following a notable demonstration of the practical synthesis of keto γ-butyrolactone derivatives from sulfoxonium ylides and α-substituted acrylic acids, a probable reaction mechanism isillustrated in Scheme 5. Initially, the α-carbon of β-keto sulfoxonium ylide 1a engages with the acidic proton of the acrylic acid, forming intermediate I. Subsequently, a nucleophilic attack at the α-carbon position of intermediate I by the in situ generated carboxylate ion produces α-acyloxy ketone intermediate II,12 releasing DMSO. Finally, the α-acyloxy ketone intermediate II tautomerizes to II′, which undergoes intramolecular cyclization via a Michael addition reaction, resulting in the formation of the keto γ-butyrolactone derivative 3a.
 | ||
| Scheme 5 Plausible reaction mechanism. | ||
In conclusion, we have established a robust, catalyst- and additive-free synthetic strategy for the synthesis of keto γ-butyrolactones from α-substituted acrylic acids and β-keto sulfoxonium ylides. These β-keto sulfoxonium ylides and α-substituted acrylic acids, featuring various functional groups, showed exceptional compatibility, yielding the desired products with good yields. The practicality of this strategy was further validated through a large-scale reaction, the synthesis of numerous keto γ-butyrolactone derivatives, and their conversion into some bioactive molecules such as L-factor, muricatacin, and cytosporanone A.
N. K. acknowledges the CSIR, New Delhi, for the SRF (Senior Research Fellowship). S. K. P. expresses gratitude to the SERB, New Delhi, for the project grant no. CRG/2020/005562. The authors are grateful to the BHU IoE incentive grant and SATHI-BHU for NMR analysis.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. F. Yan, S.-T. Fang, W.-Z. Li, S.-J. Liu, J.-H. Wang and C.-H. Xia, Nat. Prod. Res., 2015, 29, 2013 CrossRef CAS PubMed; (b) Y.-H. Yi, S. Lu, W. Zhang, L. Li, P. Sun, B.-S. Liu and H. Tang, Patent, CN101880266A, 2010; (c) T. Fukuda, A. Matsumoto, Y. Takahashi, H. Tomoda and S. Omura, J. Antibiot., 2005, 58, 252 CrossRef CAS; (d) J. Hargreaves, J. Park, E. L. Ghisalberti, K. Sivasithamparam, B. W. Skelton and A. H. White, Aust. J. Chem., 2002, 55, 625 CrossRef CAS; (e) F. Q. Alali, X. X. Liu and J. L. McLaughlin, J. Nat. Prod., 1999, 62, 504 CrossRef CAS PubMed; (f) M. C. Zafra-Polo, B. Figadere, T. Gallardo, J. R. Tormo and D. Cortes, Phytochemistry, 1998, 48, 1087 CrossRef CAS; (g) M. J. Rieser, J. F. Kozlowski, K. V. Wood and J. L. McLaughlin, Tetrahedron Lett., 1991, 32, 1137 CrossRef CAS.
- For selected examples, see: (a) Y. Ding, Z.-J. Huang, J. Yin, Y.-S. Lai, S.-B. Zhang, Z.-G. Zhang, L. Fang, S.-X. Peng and Y.-H. Zhang, Chem. Commun., 2011, 47, 9495 RSC; (b) M. Uyanik, D. Suzuki, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2011, 50, 5331 CrossRef CAS PubMed; (c) M. Uyanik, T. Yasui and K. Ishihara, Bioorg. Med. Chem. Lett., 2009, 19, 3848 CrossRef CAS PubMed; (d) M. Ochiai, Y. Takeuchi, T. Katayama, T. Sueda and K. Miyamoto, J. Am. Chem. Soc., 2005, 127, 12244 CrossRef CAS PubMed.
- For photooxidative lactonization, see: (a) X.-K. Cheng, B. Yang, X.-G. Hu, Q. Xu and Z. Lu, Chem. – Eur. J., 2016, 22, 17566 CrossRef CAS PubMed; (b) Q.-B. Zhang, Y.-L. Ban, D.-G. Zhou, P.-P. Zhou, L.-Z. Wu and Q. Liu, Org. Lett., 2016, 18, 5256 CrossRef CAS PubMed; (c) N. Tada, L. Cui, T. Ishigami, K. Ban, T. Miura, B. Uno and A. Itoh, Green Chem., 2012, 14, 3007 RSC.
- S. Zhang, F. Lian, M.-Y. Xue, T.-T. Qin, L.-J. Li, X. Zhang and K. Xu, Org. Lett., 2017, 19, 6622 CrossRef CAS.
- For selected examples, see: (a) H. Jiang, H. Zhang, W. Xiong, C. Qi, W. Wu, L. Wang and R. Cheng, Org. Lett., 2019, 21, 1125 CrossRef CAS; (b) C. Li, M. Li, W. Zhong, Y. Jin, J. Li, W. Wu and H. Jiang, Org. Lett., 2019, 21, 872 CrossRef CAS; (c) J. Vaitla and A. Bayer, Synthesis, 2019, 612 CrossRef CAS; (d) X. Wu, S. Sun, J.-T. Yu and J. Cheng, Synlett, 2019, 21 CAS; (e) A. G. Talero, B. S. Martins and A. C. B. Burtoloso, Org. Lett., 2018, 20, 7206 CrossRef CAS PubMed; (f) J. Vaitla, A. Bayer and K. H. Hopmann, Angew. Chem., 2017, 129, 4341 CrossRef; (g) A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, Eur. J. Org. Chem., 2013, 5005 CrossRef CAS; (h) C. Molinaro, P. G. Bulger, E. E. Lee, B. Kosjek, S. Lau, D. Gauvreau, M. E. Howard, D. J. Wallace and P. D. O’Shea, J. Org. Chem., 2012, 77, 229 CrossRef.
- For recent reviews see: (a) P. Bhorali, S. Sultana and S. Gogoi, Asian J. Org. Chem., 2022, 11, e202100754 CrossRef CAS; (b) S. Kumar, S. Nunewar, S. Oluguttula, S. Nanduri and V. Kanchupalli, Org. Biomol. Chem., 2021, 19, 1438 RSC; (c) A. Kumar, M. S. Sherikar, V. Hanchate and K. R. Prabhu, Tetrahedron, 2021, 101, 132478 CrossRef CAS.
- (a) A. Sharma and S. K. Pandey, Chem. Commun., 2023, 59, 1509 RSC; (b) A. K. Gola, A. Sharma and S. K. Pandey, Org. Lett., 2023, 25, 1214 CrossRef CAS PubMed; (c) T. N. Chaubey, P. J. Borpatra and S. K. Pandey, Org. Lett., 2023, 25, 5329 CrossRef CAS PubMed.
- The diastereomeric ratio (dr) was determined from 1H-NMR and 13C-NMR data, while the relative configuration was established using NOESY NMR data.
- U. Gräfe, G. Reinhardt, G. Schade, D. Krebs, I. Eritt, W. F. Fleck, E. Heinrich and L. Radics, J. Antibiot., 1982, 35, 609 CrossRef PubMed.
- M. C. Murcia, C. Navarro, C. Moreno and A. G. Csaky, Curr. Org. Chem., 2010, 14, 1510 CrossRef.
- (a) R. A. Fernandes, A. Bhowmik and P. Choudhary Muricatacin, Chem. − Asian J., 2020, 15, 3660 CrossRef CAS PubMed; (b) R. A. Fernandes, A. J. Gangani, A. Kumari and P. A. Kumar, Eur. J. Org. Chem., 2020, 6845 CrossRef CAS.
- Intermediate II was detected in the HRMS data of the crude reaction mixture taken after 1 hour.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and the NMR spectra data. See DOI: https://doi.org/10.1039/d4cc03187c |
| This journal is © The Royal Society of Chemistry 2024 |