
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the catalytic potential of gold(II) complexes: a comprehensive reassessment
Juan Carlos
Pérez-Sánchez
 ab,
Raquel P.
Herrera
*b and
M.
Concepción Gimeno
*a
ab,
Raquel P.
Herrera
*b and
M.
Concepción Gimeno
*a
aDepartment of Inorganic Chemistry, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: gimeno@unizar.es
bDepartment of Organic Chemistry, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: raquelph@unizar.es
First published on 7th December 2023
Abstract
Gold(II) complexes, unlike their gold(I) and gold(III) counterparts, have been sparsely employed in the field of catalysis. This is primarily due to the challenges associated with isolating and characterising these open-shell species. However, these complexes offer a wide range of possibilities. On one hand, this intermediate oxidation state has proven to be more easily accessible through reduction and oxidation processes compared to the gold(I)/gold(III) redox couple, thereby facilitating potential homo-coupling and cross-coupling reactions. On the other hand, gold(II) exhibits Lewis acid behaviour, bridging the characteristics of the soft acid gold(I) and the hard acid gold(III). In this review, we focus on mono- and dinuclear gold(II) complexes, whether they are isolated and well-studied or proposed as intermediates in cross-coupling reactions induced by the action of oxidants or light. We delve into the unique reactivity and potential applications of these gold(II) species, shedding light on their role in this field. This comprehensive exploration aims to underscore the latent promise of gold(II) complexes in catalysis, offering insights into their structural and mechanistic aspects while highlighting their relevance in contemporary chemical transformations.
 Juan Carlos Pérez-Sánchez | Juan Carlos Pérez-Sánchez was born in 1999 in Zaragoza, Spain. He earned his Bachelor's degree in Chemistry with the highest honors, receiving the "Bachelor Extraordinary Award" from the University of Zaragoza in 2021. In 2023, he achieved a Master's degree in Molecular Chemistry and Homogeneous Catalysis, also with honors and an "Extraordinary Award". Throughout his academic journey, he received prestigious grants, including a Collaboration Fellowship in 2020–2021 and a JAE Intro CSIC Grant in 2021–2022, both under the guidance of Prof. M. Concepción Gimeno. Currently, he is pursuing a Ph.D. in the Department of Inorganic Chemistry at the University of Zaragoza, supported by an FPU grant. He conducts his research under the joint supervision of Dr Raquel P. Herrera and Prof. M. Concepción Gimeno. His research interests primarily revolve around Group 11 Metals & Ferrocene Chemistry, with a particular focus on developing molecular complexity and a diverse array of structures for various applications, including catalysis, synthesis, reactivity, and the exploration of biological properties. |
 Raquel P. Herrera | Raquel P. Herrera was born in Alicante (Spain), in 1977. She received her B.Sc. (1999) and M.Sc. degrees (2000) at the University of Alicante, Spain, and completed her Ph.D. (1999–2003) under the supervision of Prof. Guijarro and Prof. Yus at the same university. Then, she got a European postdoctoral contract with Prof. Ricci (Bologna, Italy) until March 2006, at which time she joined Prof. Lassaletta's and Fernández's group at the IIQ-CSIC (Seville, Spain). She was appointed as a permanent researcher (ARAID program) at the ISQCH-University of Zaragoza in January 2008 and in 2012 she obtained a permanent position as Tenured Scientist of the Spanish Council of Research (CSIC) at the same Institute. Currently, she is Scientific Researcher since 2021. In 2012 she was awarded with the Lilly Prize for the best young scientist less than 40 years, in Spain. In 2021, she received an accessit in the Tercer Milenio 2021 Award in the category “Research and Future”. Her research focuses on asymmetric organocatalysis and its applications. She is the head of the Asymmetric Organocatalysis research group. |
 M. Concepción Gimeno | M. Concepción Gimeno received her PhD from the University of Zaragoza. After completing her postdoctoral work with Prof. Stone at the University of Bristol, she joined the Institute of Chemical Synthesis and Homogeneous Catalysis (ISQCH, CSIC-University of Zaragoza), where she has been a professor since 2008. Her scientific interests are focused on the design, study, and analysis of new group 11 metal compounds with specific catalytic, luminescent, and/or biological properties and potential applications. She has authored more than 300 scientific publications and has received several awards, including the IUPAC 2017 Distinguished Women in Chemistry or Chemical Engineering, the GEQO-Excellence in Organometallic Chemistry Research Award in 2017, the RSEQ-Excellence Research Award in 2018, and the Rafael Uson Medal in 2022, among others. She also is the head of the Gold and Silver Chemistry research group. |
1. Introduction
Gold, a fascinating element, has captivated civilisations throughout history due to its remarkable physical attributes and resplendent golden hue. Nonetheless, from a chemical perspective, gold singularity extends further, it stands apart among transition metals, showing the lowest electrochemical potential across all metals.1,2 Paradoxically, gold has the highest electronegativity among all metals, proving its noble character but rendering its compounds thermodynamically metastable to reduction to elemental gold.3–7 While this chemical inertness might seem limiting, gold role in catalysis has experienced remarkable expansion since 1990 and has emerged as a powerful tool in recent decades.8–15Traditionally, elementary steps in catalytic cycles, such as oxidative addition, reductive elimination, transmetallation, and migratory insertion, were thought to be unfavourable, with gold role often limited to that of a Lewis acid for π-activation. However, recent years have witnessed significant advancements, transforming this perception.2,16,17
The exploration of gold potential in catalysis is significantly augmented by the application of relativistic theory.18 Relativistic effects exert a profound influence on gold chemistry, causing a contraction in atomic and cationic radii. These effects also influence cationic orbital energies, promoting high s-character hybridisation for bonding. This gives rise to a suite of properties unique to gold complexes: an unusually small covalent radius, a distinctive yellow coloration, an inclination towards linear coordination geometry or heightened stability in higher oxidation states.19
Among gold complexes, the prevailing oxidation states are +I and +III, characterised by their distinct coordination geometries. Gold(I) complexes, with a [Xe]5d10 electronic configuration, commonly exhibit linear coordination, although instances of three- and four-coordinated complexes have been reported.20 In contrast, gold(III) complexes, featuring a [Xe]5d8 electronic configuration, favour square-planar coordination.21
On the other hand, the prevalence of gold complexes showing a formal oxidation state +II has exhibited a marked increase, making this oxidation state almost conventional in contemporary gold chemistry. However, the abundance of gold(II) complexes remains limited when compared to their more frequently encountered gold(I) and gold(III) counterparts (Fig. 1).
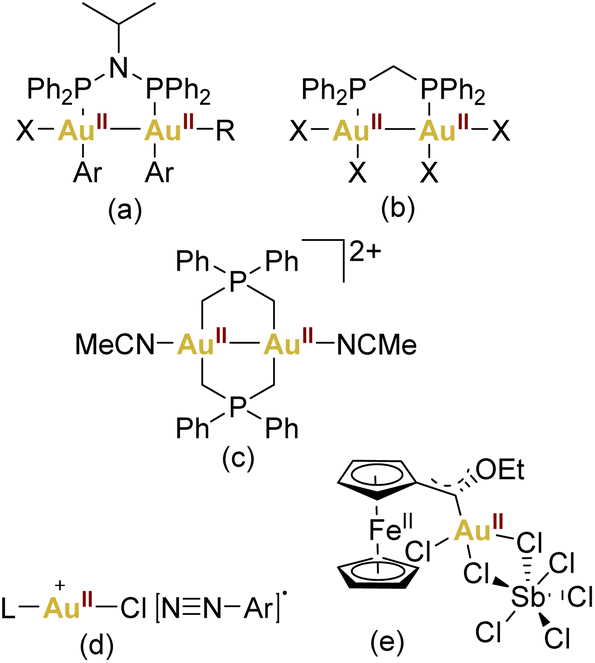 | ||
| Fig. 1 Some selected examples of AuII complexes (a–c) or proposed AuII intermediates (d–e) in catalysis. | ||
The energy required to transition from atomic gold to Au2+ is relatively similar to the energy requirements for Cu2+ and Ag2+ formation. However, gold's tendency towards a +III oxidation state is energetically more favourable than that of Cu or Ag. However, this rationale alone does not entirely explain the relative instability of the +II oxidation state in gold.22
Disproportionation processes favourably generate Au+ and Au3+ species due to the distinctive electronic configuration of d9 metal complexes.23 In these, an odd electron occupies a dx2−y2 orbital, inherently possessing considerably elevated energy compared to copper. This characteristic renders it highly susceptible to ionisation, prompting the favourable formation of gold–gold bonds that enhance compound stability.24
This propensity towards gold–gold bonding is notably exemplified by the formation of Au24+ core species, establishing a robust class of gold(II) dinuclear complexes (Fig. 1c).25,26
Regarding mononuclear gold(II) derivatives, their scarcity is apparent, and a closer examination often reveals instances that can be better described as pseudo-gold(II) complexes. These are exemplified by compounds like the halides “AuX2” or CsAuX3 (Cs2AuIAuIIIX6, X = Cl, Br, I).27–29 This complexity emphasises the challenges associated with isolating mononuclear gold(II) species. Moreover, the interest in low-coordinate gold(II) intermediates has garnered attention, suggesting their plausibility as transient species in both photochemical and redox switchable gold-catalysed reactions.
Notably, Hashmi,30 Toste,31–33 and Glorius,34 have consistently expanded this proposal in the context of photocatalysis. However, a notable void exists in terms of conclusive structural or spectroscopic evidence supporting the presence of gold(II) within photocatalytic cycles (Fig. 1d). Furthermore, in the context of redox switchable catalysis, this field has been represented by Heinze and coworkers by using the ferrocene unit which can undergo a valence isomerisation from FeIII/AuI to FeII/AuII (Fig. 1e).
The direct experimental validation of gold(II) species continues to pose a formidable challenge. This leads to an intriguing disparity wherein gold(II) complexes, in contrast to their gold(I) and gold(III) counterparts, have been relatively unexplored in the context of gold catalysis. Nonetheless, these gold(II) complexes hold latent promise in addressing challenging reactions, and their potential remains underestimated. Therefore, this perspective intends to cover all these pivotal examples regarding gold(II) complexes in catalysis.
2. Gold(II) complexes and their use in catalysis
2.1. Mononuclear gold(II) complexes
Mononuclear gold(II) species featuring a d9 configuration are anticipated to exhibit paramagnetism, manifesting through a distinctive four-line Electron Paramagnetic Resonance (EPR) signal, aligning with the nuclear spin characteristics of 197Au (I = 3/2). This distinctive EPR signature serves as a crucial discriminant between authentic gold(II) complexes and those wherein the unpaired electron resides within the ligand orbitals.The pioneering example of mononuclear gold(II) complexes dates back to 1990 when the Schröder group successfully characterised a gold(II) complex by using a chelating thioether ligand, 1,4,7-trithiacyclononane, encapsulating the gold(II) centre (Fig. 2a).35 This complex, prepared by reduction of HAuCl4·H2O in HBF4/MeOH, exhibits limited delocalisation in the ligand and a configuration that effectively impedes the typical dimerisation and disproportionation pathways observed in highly reactive gold(II) species.
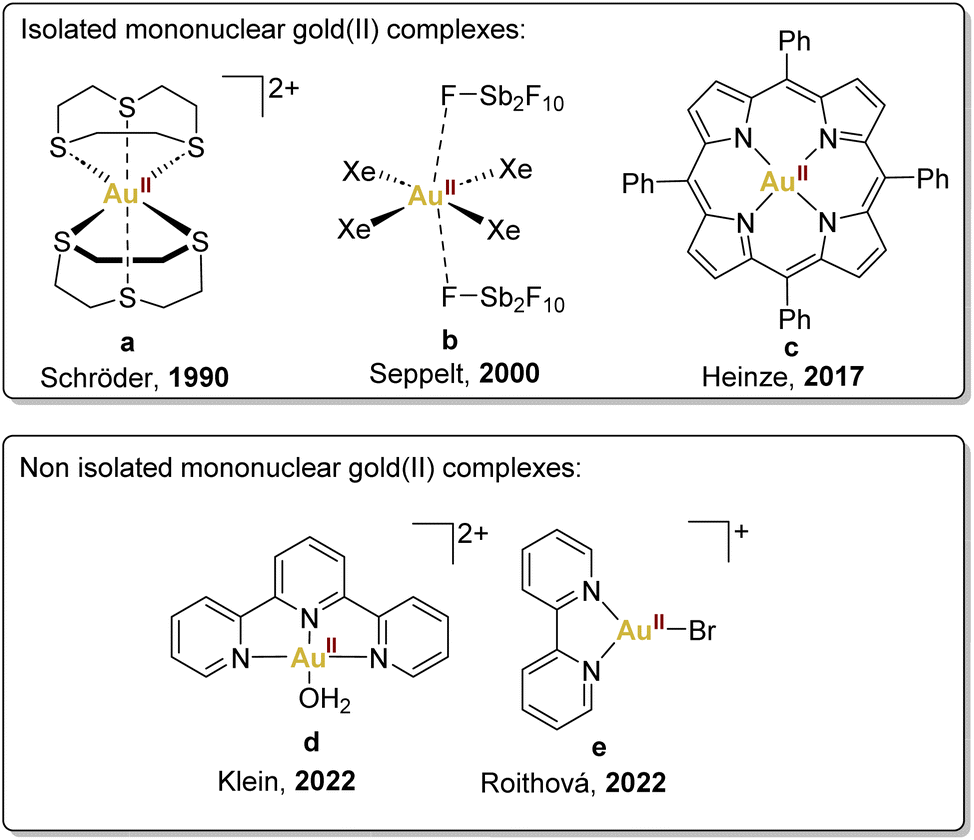 | ||
| Fig. 2 Selected examples of mononuclear gold(II) complexes described previously: Schröder,35 Seppelt,36 Heinze,37 Klein38 and Roithová.39 | ||
Another noteworthy example is [AuIIXe4][Sb2F11]2, synthesised by Seppelt and coworkers by reduction of AuF3 with xenon, contributing significantly to the field (Fig. 2b).36 It is essential to underscore the limiting conditions required for stabilising this xenon complex, achieved within HF/SbF5 mixtures, requiring low temperatures and a xenon atmosphere to prevent decomposition. Unfortunately, these precise conditions restricted the exploration of its reactivity, leaving this aspect largely unexplored.
More recently, in 2017, Heinze's group accomplished the isolation of a neutral mononuclear porphyrinato gold(II) complex, Au(TPP), as a stable and sublimable material (Fig. 2c).37 This was achieved by the reduction of [Au(TPP)][PF6] with cobaltocene or KC8, followed by recrystallisation or sublimation processes. Single-crystal X-ray diffraction analysis of Au(TPP) in the solid-state validated its genuinely mononuclear nature.
In addition, in 2022, Klein's group, employing Density Functional Theory, demonstrated that the gold(III) complex, [AuIII(OH)(terpy)]2+, cleaves C–H and O–H bonds via concerted proton-coupled electron transfer (cPCET).38 This mechanism involves simultaneous yet separate movement of both proton and electron, resulting in the reduction of the gold(III) centre to gold(II) through a single electron transfer, thus generating a [AuII(OH2)(terpy)]2+ (Fig. 2d).
In the same year, Straka, Roithová, and coworkers provided another example of gold(II) complexes with bidentate and tridentate ligands generated in the gas phase (Fig. 2e).39 This exploration delved into the spectroscopic and electronic properties of [AuII(bipy)(X)]+ complexes, revealing intriguing parallels with copper(II) complexes. The analogous electronic spectra of gold(II) and copper(II) complexes underscored the potential for comparative studies in understanding their reactivity.
Despite the existence of mononuclear gold(II) complexes (Fig. 2),35–44 to the best of our knowledge, these species have neither been utilised as catalysts nor suggested as intermediates in catalysis in the literature so far. However, this situation may change in the next future if more stable gold(II) species are reported or if formal mononuclear gold(II) complexes would be used.
2.2. Dinuclear gold(II) complexes or “pseudo-gold(II) complexes”
As previously discussed, the inherent instability of gold(II) complexes can be attributed to the unfavourable energy associated with the odd electron configuration. However, the formation of a metal–metal bond in dinuclear gold(II) complexes introduces an element of additional stability, thereby mitigating decomposition pathways. The use of supporting bridging ligands preorganises the gold centres for oxidative addition, thereby minimising the entropy cost.Notably, to facilitate the valence change in AuI complexes, the involvement of robust external oxidants becomes necessary. Examples of these include Selectfluor™ and hypervalent iodine reagents.45–48 These oxidants play a crucial role in promoting the required valence transformation.
The presence of aurophilic interactions within dinuclear gold(I) complexes manifests in a rich redox chemistry. This encompasses AuI–AuI, AuI–AuIII, and AuII–AuII interactions, which collectively offer exciting prospects for applications in gold(I)-catalysed redox carbon–carbon couplings. The concept of oxidative addition in dinuclear gold complexes was initially explored by Schmidbaur et al. in the 1970s.49,50
The utilisation of stoichiometric gold(I) dinuclear complexes for generating and investigating the reactivity of resulting AuII–AuII species has garnered significant attention. However, the exploration of the potential catalytic activity of these complexes has been somewhat limited. Notably, while the catalytic activity of these complexes in their AuI–AuI form has been extensively studied, it is worth highlighting the emergence of transient gold(II) species as proposed intermediates in catalytic reactions involving dinuclear gold(I) complexes. This intriguing development suggests the exciting prospect of harnessing the catalytic potential of gold(II) complexes through the controlled generation and active involvement of these intermediates in catalysis.
In 2010, Toste’s group introduced an innovative intramolecular amino-arylation reaction employing the [(μ-dppm)(AuBr)2] (dppm = bis(diphenylphosphino)methane) complex 1 as catalyst (Scheme 1a).51
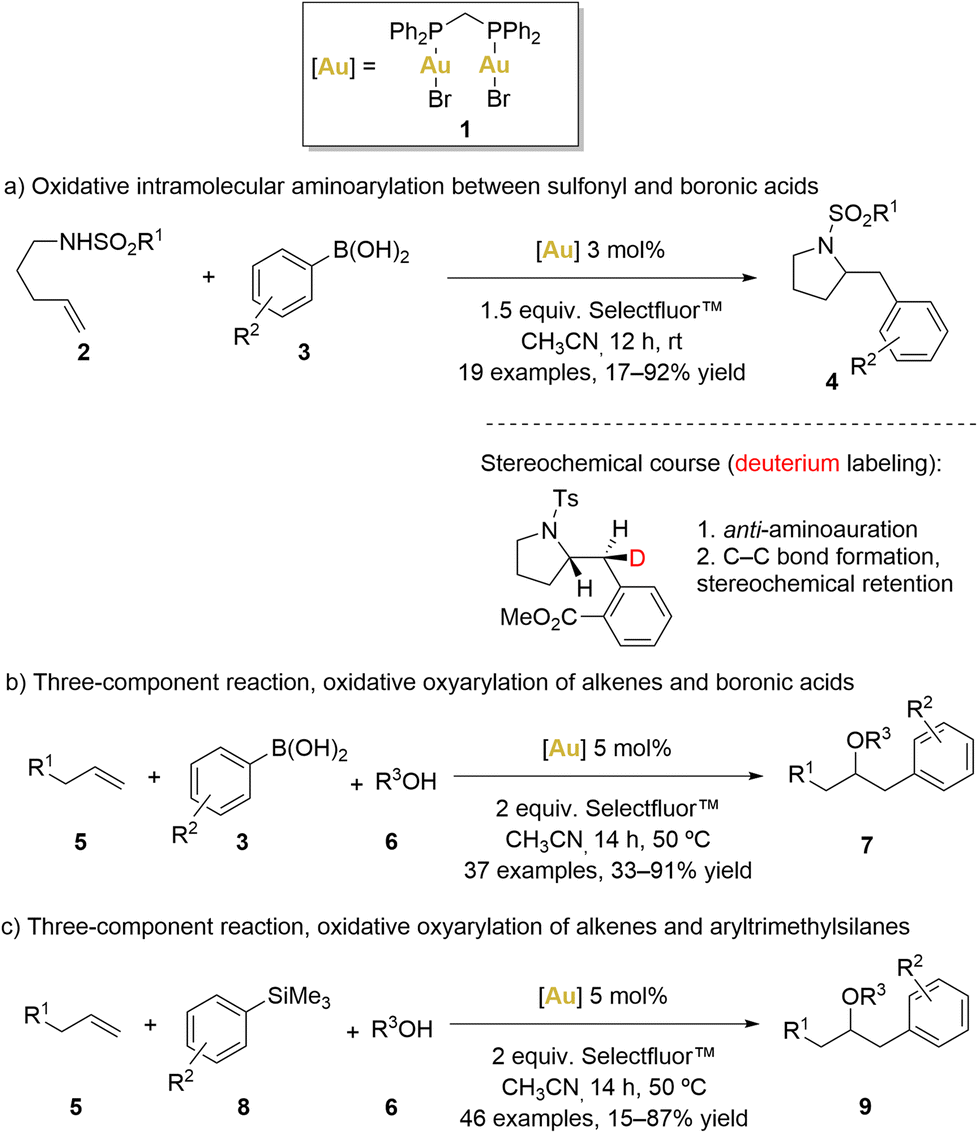 | ||
| Scheme 1 Gold-catalysed oxidative C–C couplings reported by Toste and coworkers. | ||
Initially, a comprehensive catalyst screening involving various monophosphane ([AuPPh3X], X = Cl, OTf, OBz, Br and I) revealed the formation of [Au(PPh3)2]+ species, which are known to be catalytically inert.52 Consequently, a hypothesis was formulated, suggesting that a robust aurophilic interaction between AuI and AuIII centres within complex 1 could disrupt the formation of bisphosphanegold species. As predicted, the use of [(μ-dppm)(AuBr)2] as the catalyst elevated the yield to 81% even at room temperature.53
Motivated by the promising results achieved with this complex, Toste's group expanded their approach in the same year, broadening the catalyst's scope to study additional three-component oxyarylation reactions between alkenes, water or alcohols, and boronic acids (Scheme 1b).54
Similar to previous examples, this strategy involved oxidising the gold(I) species in the presence of Selectfluor™ to generate a gold(II) intermediate. This gold(II) intermediate exhibited sufficient electrophilicity to activate alkenes (5), facilitating a nucleophilic attack by alcohols or water (6) to generate an organo-gold(III) intermediate. Subsequently, a bimolecular reductive elimination process via a 5-membered transition state was proposed, facilitated by the strong B–F bond, leading to the formation of product 7. Importantly, the reluctant behaviour of gold(II) to undergo β-hydride elimination prevented the formation of ketone products. Usually, C–Au bonds exhibit a predilection for protodeauration over β-hydride elimination.2,8,55–57 Remarkably, gold-hydride species are infrequently encountered and pose challenges in terms of accessibility. However, it is only in recent years that gold hydride complexes have been isolated and their reactivity studied.
Notably, Selectfluor™ not only acted as an efficient oxidant but also the strong affinity between boron and fluorine atoms potentially made the AuII–alkyl moiety more electrophilic and the boronic acid component more nucleophilic, facilitating the electrophilic aromatic substitution and the bimolecular reductive elimination process.
In the same year, Toste and coworkers also reported a gold-catalysed oxyarylation of alkenes (5) using aryl silanes (8) and alcohols (6) as coupling partners (Scheme 1c).58 Aryl silanes proved to be efficient coupling partners, avoiding the formation of undesirable homo-coupling by-products. Remarkably, Selectfluor™ played a dual role in this process, serving as both oxidant and activator for the aryl silanes. These functional groups indeed displayed good compatibility when aryl boronic acids were replaced by aryltrimethylsilanes. Furthermore, intramolecular coupling reactions were also found to be accessible with moderate to high yields.
In the preliminary proposals, these coupling reactions were envisioned to proceed through a redox cycle involving the initial oxidation of gold(I) to gold(III) using Selectfluor™ (Scheme 2, 10b), followed by the C–C bond formation through bimolecular reductive elimination. A year later, Toste's group undertook a multifaceted approach, combining density functional theory (DFT) calculations with experimental observations, to unveil a revised oxidative heteroarylation mechanism (Scheme 2).59 This new mechanism would comprise three principal steps: (1) bimetallic oxidation (2-electron, 2-centre), facilitated by Selectfluor™, (2) alkene activation followed by nucleophilic addition, and (3) bimolecular reductive elimination.
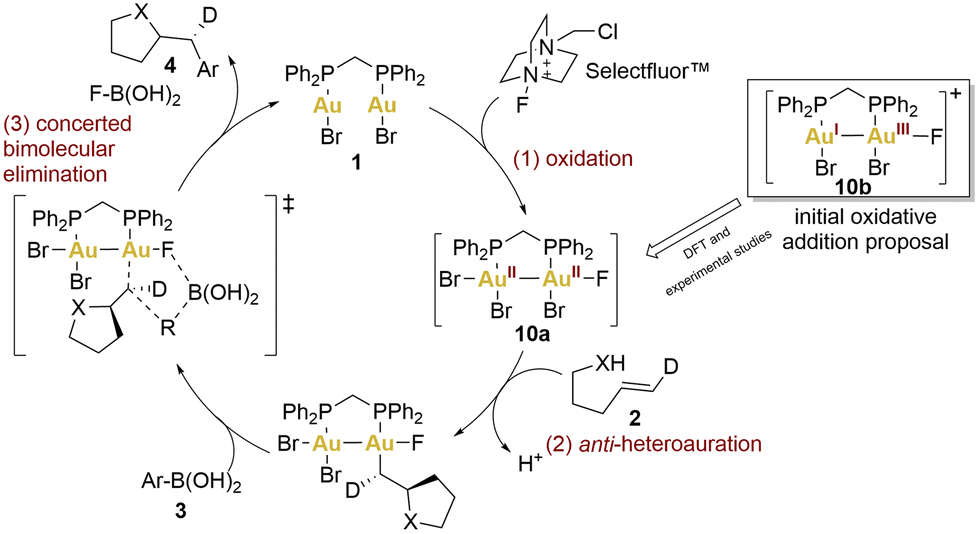 | ||
| Scheme 2 Revised mechanism of the aminoarylation reaction proposed by Toste and coworkers. | ||
According to the authors’ studies, the formation of binuclear Au(II)–Au(II) intermediates lowered the barriers for all depicted steps. Theoretical studies indicated that the bidentate ligand (dppm) facilitated oxidative addition to both gold centres due to minimal entropic costs. This was also supported by cyclic voltammetry (CV). The voltammograms of complex 1 displayed two irreversible, one-electron oxidation waves cathodically shifted by 140 mV compared to benchmark complexes: the mononuclear [Au(BnPPh2)Cl] (Bn = Benzyl) and the dinuclear [(μ-dppm)(AuCl)(AuCl3)] employed as models for a AuI and AuI–AuIII complexes, respectively. These potential shifts could indicate that aurophilic interactions could regulate the redox potential, decreasing the energy barrier. In fact, the facile oxidative addition of the dinuclear complex 1 was driven by AuII–AuII σ-bond formation.
Finally, heteroauration would take place in an anti-sense manner, resulting in the formation of a neutral alkylgold species. Subsequently, a concerted bimolecular reductive elimination with an arylboronic acid succeeds, retaining the stereochemistry and yielding a catalytic process that exhibits an overall inversion of stereochemistry, as has been observed previously.51
In 2014, Levin and Toste introduced a gold-catalysed C–C coupling between arylboronic acids and allyl bromides (Scheme 3).60
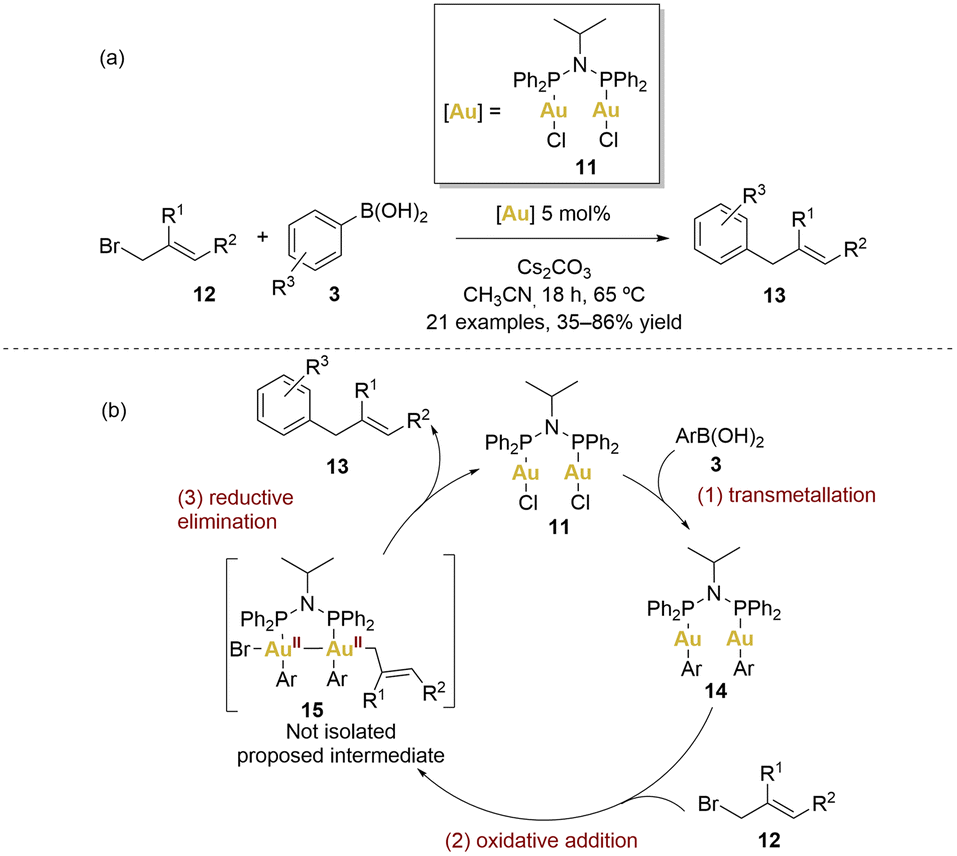 | ||
| Scheme 3 (a) Gold-catalysed cross-coupling reaction between boronic acids and allyl bromides. (b) Proposed catalytic cycle. | ||
This reaction employed [(μ-Ph2PiPrNPPh2)(AuCl)2] 11 as catalyst, working within a net redox-neutral cycle without the need of an external oxidant. This method provided access to diverse sp2–sp3 coupled products under mild conditions (65 °C), exhibiting complete tolerance for air and water. A broad array of electron-donating substituents on the aryl group, as well as iodine substituents and various derivatives of allyl bromides 12 (1-substituted, 2-substitued or cyclic 1,2-disubstituted), were well-tolerated, giving products 13 with yields from 35% to 86%. Remarkably, the reaction demonstrated orthogonal reactivity, high efficiency and chemoselectivity.
A plausible mechanism is depicted in Scheme 3. The initial step involves a transmetallation from the boronic acid 3 generating a gold(I) diaryl complex 14. Based on stoichiometric reactivity of 11 and radical clock experiments (which argue against radical coupling), the authors proposed that transmetallation precedes oxidative addition. The process of transmetallation involving arylboronic acids serves to enhance the electron density around the gold centres, effectively easing the subsequent oxidative addition step with allyl bromides. Achieving two-electron oxidative addition with Au(I) complexes is often a challenging task. Nonetheless, the significance of the AuII–AuII intermediate in this particular transformation cannot be underestimated, as it potentially represents a pivotal factor in the overall mechanism. Regrettably, attempts to directly isolate or detect this intermediate did not yielded fruitful results.
Gold(II) complexes possess the potential to play a significant role, not solely in C–C cross-coupling reactions, but also in enhancing their utility as Lewis acids for activating alkenes and alkynes for nucleophilic additions. In this context, the counterparts of gold(I) and gold(III) have been extensively studied, illustrating distinct reactivity due to their divergent soft/hard Lewis acid behaviour.61,62
In 2016, Wade and coworkers introduced an elegant cascade reaction that involves an intramolecular hydroamination reaction followed by an intermolecular Mukaiyama addition, testing dinuclear gold(II,II) and gold(III,III) phosphorus ylide complexes as catalysts (Scheme 4).63
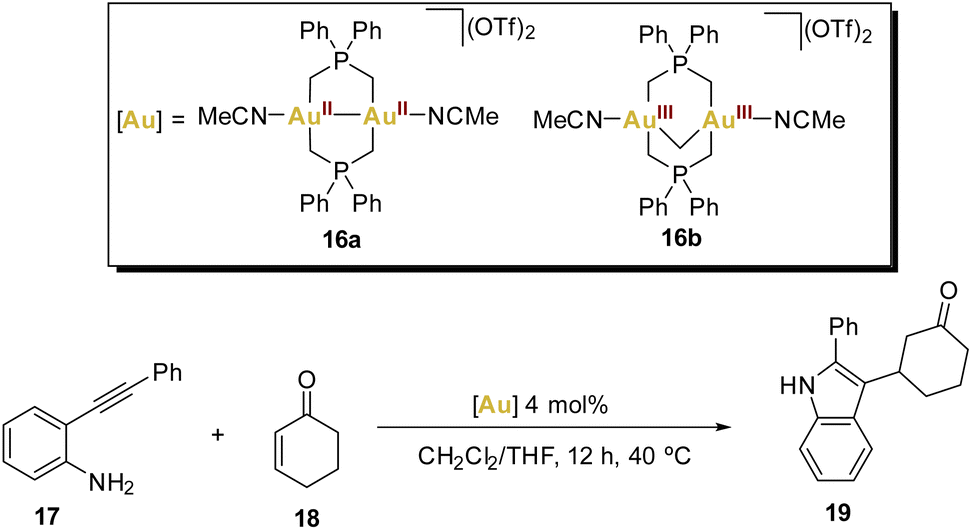 | ||
| Scheme 4 Gold(II)-catalysed tandem reaction. | ||
These bridging ligands with a strong σ-donating character impart remarkable stability across a wide range of oxidation states, prompting the authors to synthesise two acetonitrile solvates of AuII–AuII16a and AuIII–AuIII16b complexes, envisioning their potential as active Lewis acid catalysts.
The crystal structure of both complexes was determined and remarkably both dinuclear complexes showed different behaviour within short Au⋯Au contacts. While for complex 16a the intermetallic distance Au–Au (2.5580(2) Å) was found to be among the shortest observed for a dinuclear gold complex bearing a phosphorus ylide ligand, for complex 16b they found a weak aurophilic interaction (3.0673(10) Å), which was explained as face-to-face pairing of square planar complexes (Fig. 3).
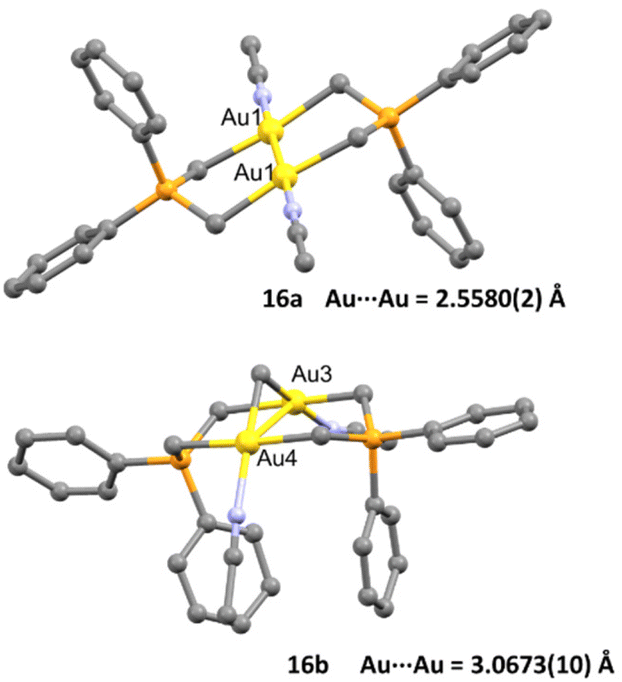 | ||
| Fig. 3 Crystal structure of complexes 16a and 16b. Adapted with permission from ref. 63 Copyright ® American Chemical Society. | ||
With these fully characterised complexes 16a and 16b in hand, authors screened them in a Mukaiyama addition reaction involving a silyl enol ether and either crotonaldehyde or cyclohexenone. The results were compared to those of other gold(I) Lewis acids, such as [Au(OTf)(L)] (L = PPh3 or IPr). Notably, when employing cyclohexenone as the substrate and a catalytic loading of 4 mol%, complex 16a displayed significantly superior activity compared to 16b (88% yield vs. 45% yield). Given that both adjacent gold centres in 16a may participate in substrate activation, the researchers opted to reduce the catalytic loading to 0.5 mol%. This led to complete conversions within 12 hours using complex 16a for the reaction. However, no reaction was observed with [Au(OTf)(PPh3)], likely due to its higher susceptibility to deactivation in contrast to complex 16a.
Indeed, the Gutmann-Becket method64 was employed to assess the relative Lewis acidity of gold complexes, by measuring the variation in the 31P NMR chemical shift between an internal standard like Et3PO and gold-coordinated Et3PO complexes. The results of this analysis indicated that both complexes 16a and 16b demonstrated comparable levels of Lewis acidity. However, complex 16a exhibited faster ligand exchange rate compared to complex 16b. Consequently, it was suggested that the efficiency of catalysing enone additions appeared to correlate more effectively with the rates of ligand exchange than with the apparent Lewis acidity.
Building upon these findings, the authors proceeded to assess the performance of these complexes in a hydroamination reaction involving phenylacetylene. Once again, they observed remarkable results with complex 16a. Interestingly, a trend emerged where the best yields using 16a were obtained with more sterically hindered amines. In contrast, for complex 16b, less hindered amines led to better results. The authors hypothesised that these observed differences could be attributed to increased steric hindrance around the Au centres in complex 16b. This hindrance seemed to block the nucleophilic attack on the activated alkyne, particularly when the amine was more hindered.
Recognising the versatility of both complexes, the researchers eventually applied them in a cascade reaction (Scheme 4) between an o-alkynyl aniline 17 and cyclohexenone 18, involving an intramolecular hydroamination reaction followed by an intermolecular enone addition step, resulting in the generation of a 2,3-substituted indole. Complex 16a facilitated the formation of indole 19 with good yields.
The remarkable ability of the gold(II) complex 16a to act as both a soft carbophilic and hard oxophilic Lewis acid was demonstrated in this cascade reaction sequence. While complex 16a proved to be an effective catalyst for this cascade sequence, complex 16b and the commonly used gold(I) catalysts [Au(OTf)(PPh3)] and [Au(IPr)(OTf)] were found to be ineffective. This further highlights the diverse and promising potential of harnessing gold(II) complexes in various catalytic applications.
In 2019, Xie and coworkers achieved a notable cross-coupling feat by employing dinuclear gold(I) complexes for a challenging reaction between arylsilanes and arylboronates (Scheme 5a).65 The choice of complex 11, influenced by Toste et al., demonstrated superior performance, underscoring the vast versatility and potential scope of dinuclear gold(II) complexes.
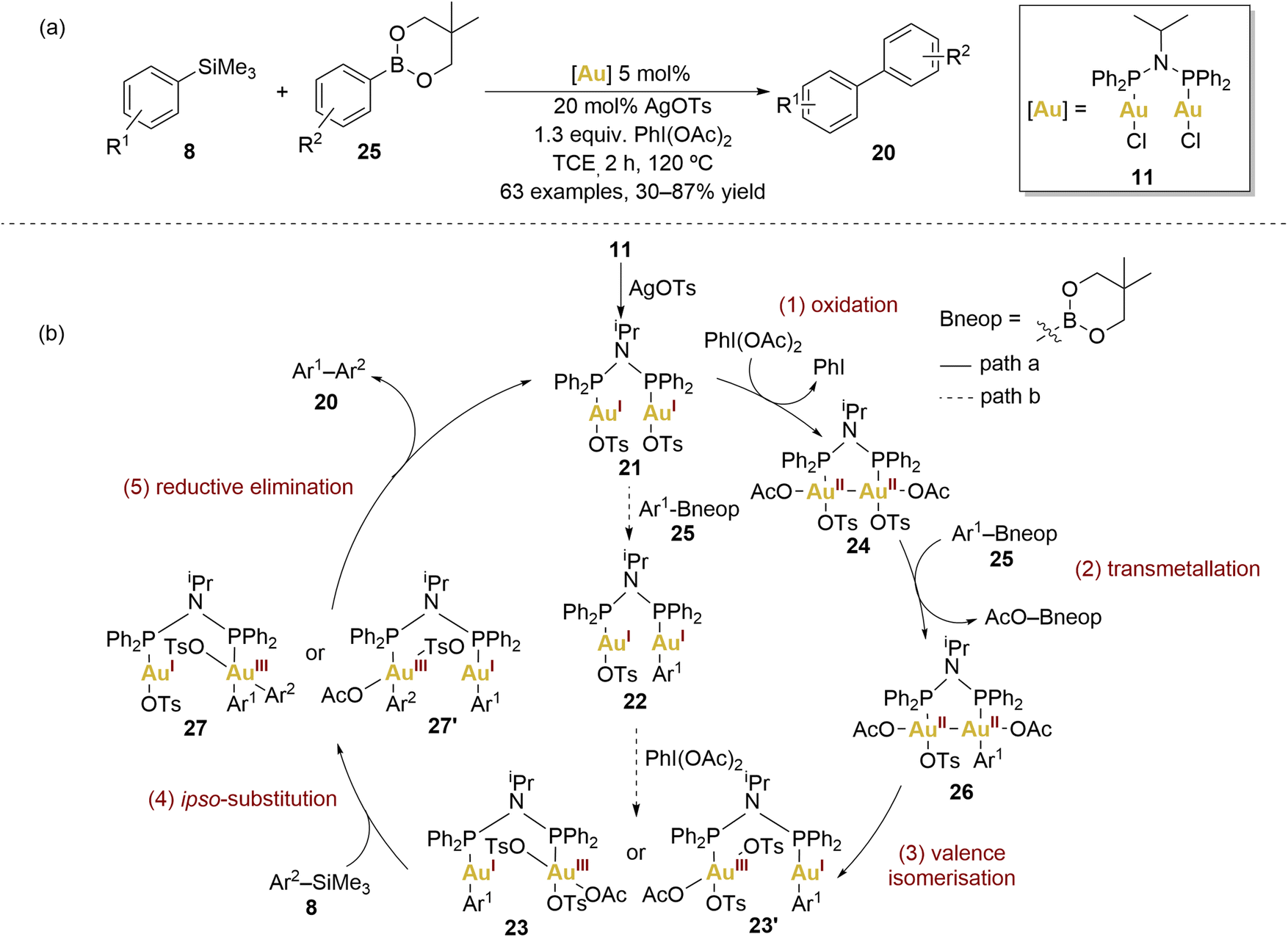 | ||
| Scheme 5 (a) Gold-catalysed biaryl cross-coupling reaction between arylboronates and arylsilanes. (b) Proposed catalytic cycle. | ||
They explored a broad scope of arylsilanes 8 and arylboronates 25 revealing remarkable functional group tolerance and consistently achieving moderate to good yields of the biaryl 17 coupling products. Significantly, the dinuclear gold catalyst effectively suppressed oxidative homocoupling, a challenge persistently encountered in mononuclear gold catalysis. Furthermore, it was highlighted the promising attributes of gold catalysts compared to counterparts based on palladium or nickel.
Xie and coworkers elucidated the plausible mechanism behind this transformation (Scheme 5b, path a). The first step consists of the oxidation of the dinuclear gold(I) complex 21 by PhI(OAc)2, forming a dinuclear AuII–AuII complex 24. Based on their mechanistic experiments, authors gave some insight about the nature of these AuII–AuII intermediates through experimental investigations. The cyclic voltammetry of 11 revealed its tendency to undergo double one-electron oxidation, resulting in the formation of an AuII–AuII intermediate. To confirm this hypothesis, they monitored the reaction between 11, AgOTs, and PhI(OAc)2 in trichloroethylene at room temperature by 31P NMR. They observed the appearance of a new chemical shift within 30 minutes when excess of oxidant and AgOTs were added, suggesting the formation of 24. Notably, oxidation occurred slowly in the absence of silver additives, indicating the importance of the counterion in the oxidation process. Intermediate 24 was further confirmed by high-resolution mass spectrometry but proved to be relatively unstable, decomposing within several minutes at room temperature.
In order to investigate the transmetallation capability of arylsilanes 8, control reactions employing the benchmark complex [AuCl(PPh3)] revealed that arylsilanes undergo exclusive activation in the presence of PhI(OAc)2. In contrast, arylboronates exhibited activation either by the Au(III) complex generated through oxidation with PhI(OAc)2 or the Au(I) complex in the presence of an external base.
To ascertain which coupling partner reacts more rapidly under standard conditions with the dinuclear gold catalyst 11, the authors examined the consumption of both coupling partners (8 and 25) and the yield of the cross-coupling product 20 in the initial 15 minutes. The rate of disappearance of arylboronate 25 significantly outpaced that of arylsilanes 8, with the accumulation of the desired product 20 maintaining a comparable rate to the consumption of arylsilanes. This brief induction period might arise from ligand dissociation, oxidation, or the formation of a highly active gold catalyst.
In view of these results, the second proposed step involved 24 in transmetallation with Ar–Bneop 25, generating another intermediate 26. The loss of symmetry in the dinuclear AuII–AuII complex induced the valence isomerisation of 26 into a dinuclear AuI–AuIII species (23 or 23′). An ipso-substitution on the Au(III) centre was suggested to activate arylsilanes. The final step involved facile reductive elimination from 27 or 27′, yielding the desired biaryl cross-coupling product.
The authors additionally described an alternative, less probable pathway (Scheme 5b, path b), proposing that transmetallation with the arylboronate 25 might precede the subsequent oxidation–isomerisation steps. Nevertheless, experiments involving transmetallation with arylboronates and gold(I) complexes rendered lower yields in comparison to the transmetallation reaction with gold(III) complexes.65
2.3. Gold(II) complexes in photocatalysis
The integration of gold catalysis with photochemical conversions has introduced a novel and synthetically valuable reactivity pattern. This innovative approach involves the fusion of gold catalysis with photoredox catalysis. This strategy has effectively tackled the challenge posed by the AuI/AuIII redox cycle, as it allows for the circumvention of the harsh reaction conditions that were previously required for this type of transformation. Notably, this approach has gained significant interest since Che’ seminal work (Scheme 6a) on light-driven gold catalysis,66,67 with a common theme emerging across various instances: the postulation of gold(II) species as pivotal intermediates in these reactions. However, there is still lack of structural and spectroscopic evidence of gold(II) in the catalytic cycles.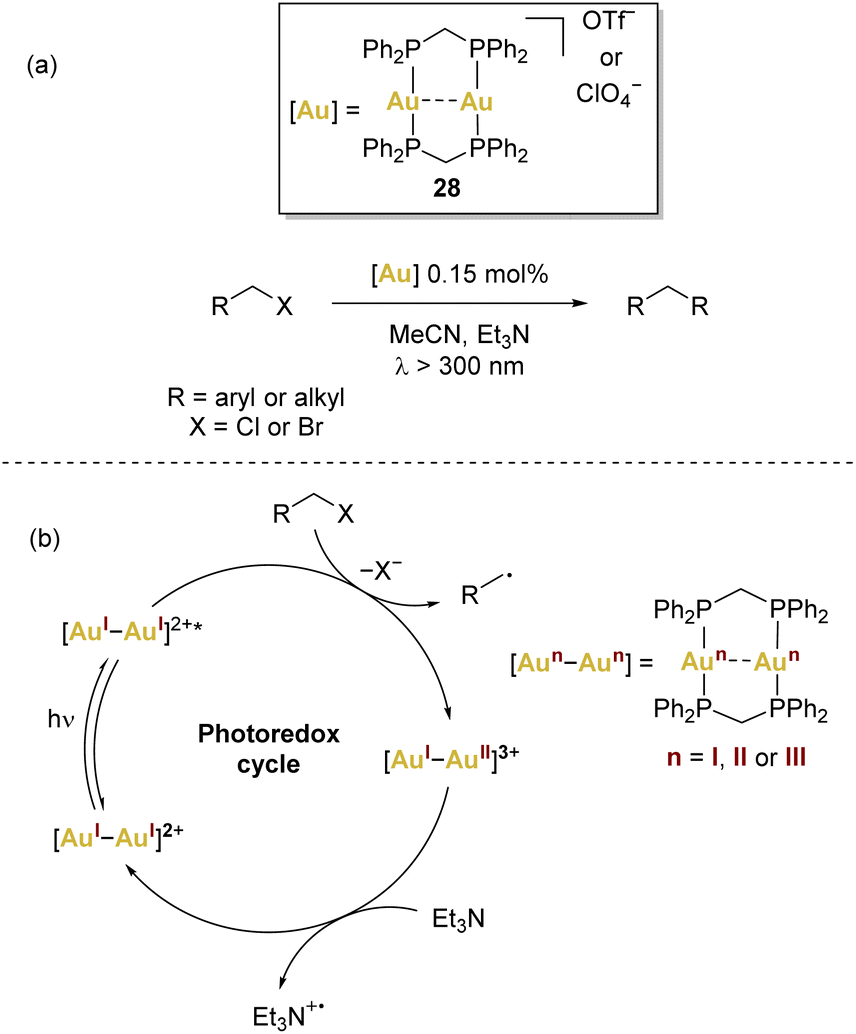 | ||
| Scheme 6 (a) Che's seminal work using [(μ-dppm)2Au2]2+ (dppm = bis(diphenylphosphino)methane), and (b) common proposed mechanism involving complex 28. | ||
An effective approach to categorise gold role in photocatalysis involves dividing it into two distinct groups: the use of mononuclear complexes either in the presence of a cocatalyst or independently (photocatalyst itself), and the utilisation of dinuclear complexes as standalone photocatalysts.
In this context, Glorius and coworkers introduced the combination of gold and photoredox catalysis in 2013,68 showcasing its potential in oxy- and aminoarylation of alkenes using aryldiazonium salts, employing a gold(I) catalyst and a [Ru(bpy)3][PF6]2 complex as photosensitiser. The catalytic cycle (Scheme 7) involves the excitation of the photocatalyst followed by an oxidative quenching by the aryldiazonium salt 29, generating an aryl radical transferred to the gold(I) centre to form gold(II) 32. A single electron transfer (SET) leads to gold(III) 33 and reductive elimination releases the product 35, regenerating the gold(I) catalyst 30.
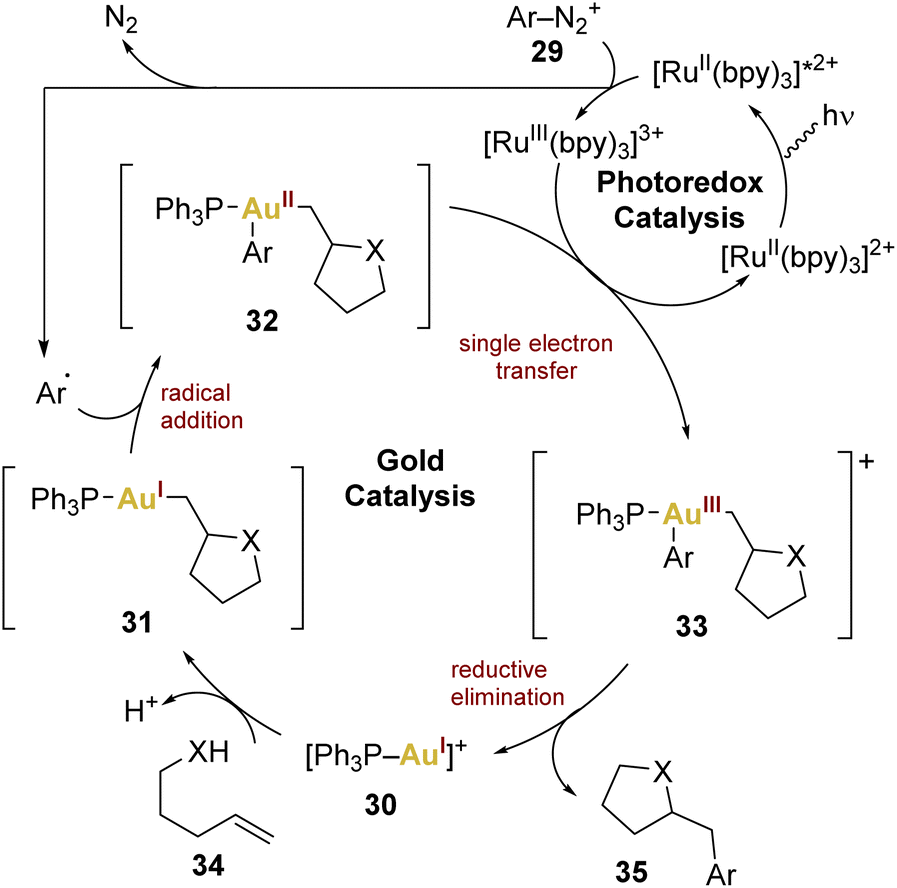 | ||
| Scheme 7 Proposed mechanism employing a mononuclear gold(I) catalyst and a Ru(II) photococatalyst in a oxy- and aminoarylation of alkenes. | ||
This mechanistic trend served as a general insight into understanding dual gold/photoredox catalysis. In 2016, a significant advancement emerged as it was proposed that the interaction between mononuclear gold(I) complexes and aryldiazonium salts could occur without the need for an additional photoredox catalyst.69
Hashmi and collaborators exemplified this concept through the 1,2-difunctionalisation of alkynes, yielding α-arylketones. This pioneering work in photosensitiser-free gold photocatalysis led to propose a mechanism (Scheme 8). This proposal was based on the observation that coordinatively saturated gold(I) catalyst and the aryldiazonium salts do not exhibit absorption of blue LEDs light. Consequently, a significant step was required to initiate the mechanism, specifically, a SET from the gold(I) complex to the aryldiazonium salt, leading to the formation of an aryl diazo radical 37 and a gold(II) complex 36. Subsequent oxidative recombination yielded a gold(III) intermediate 38 (no proof of existence of this intermediate), activating the alkyne 43 for nucleophilic addition giving rise to 39. A reductive elimination from the vinyl intermediate 40 led to the formation of product 42, by hydrolysis of the enol 41, and catalyst regeneration.
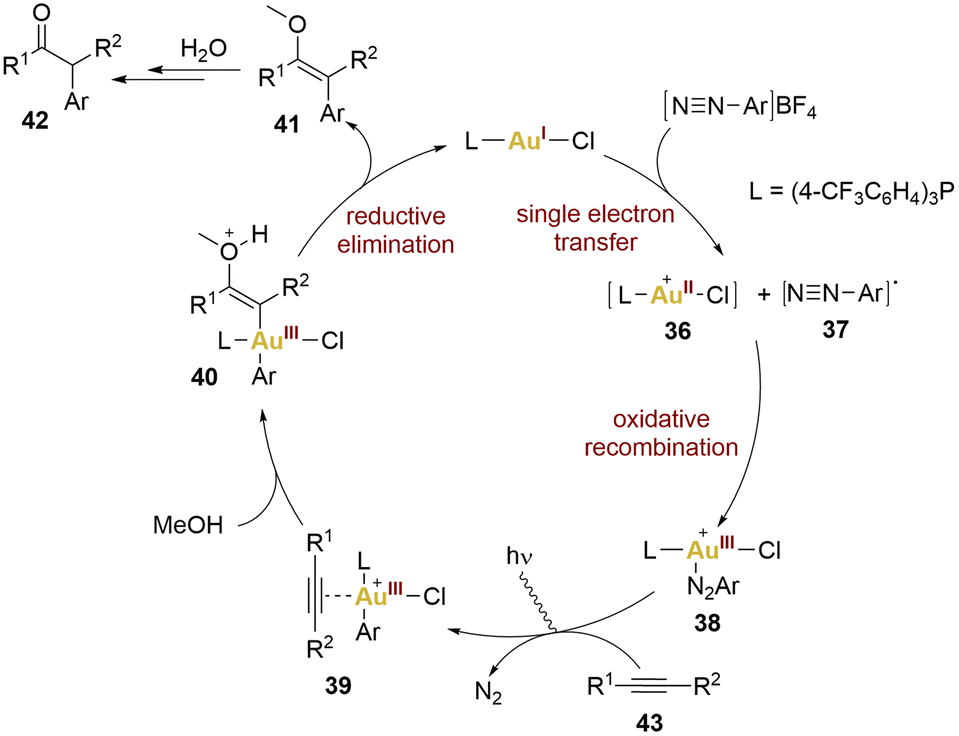 | ||
| Scheme 8 Proposed mechanism for the photosensitiser-free 1,2 defunctionalisation of alkynes. | ||
In the context of dinuclear gold complexes, [(μ-dppm)2Au2]2+28 has garnered significant attention, standing out in a multitude of photoredox transformations. Notably pioneered by Che and coworkers, as far as we are aware, this compound remains the sole dinuclear gold(I) complex employed in photoredox transformations to date. In these reactions, a recurring mechanism involving inner-sphere single electron transfer has been proposed (Scheme 6b), leading to the formation of mixed-valence AuI–AuII species that act as key intermediates.
While a comprehensive discussion is encouraged, it's worth noting that more extensive insights into this field can be found in recent reviews by Xie and Hashmi,70,71 providing a more in-depth exploration of the intricacies surrounding this exciting avenue of research.
2.4. Gold(II) complexes and redox switchable catalysis
In 1995, Wrighton and coworkers introduced the concept of redox-switchable catalysis (RSC),72 which involves modifying the catalytic activity of a transition metal and its complexes by manipulating the electron-donating or electron-withdrawing properties of a coordinated ligand. Ferrocene (Fc), due to its reversible oxidation process, is commonly used as the redox-active component in such catalytic systems.In the context of gold(II) catalysis, Heinze's group made a significant contribution in 2019.73 They synthesised and characterised a ferrocenyl-Fischer carbene gold(I) complex that serves as a precatalyst. This complex can be activated through the oxidation of the ferrocene moiety, leading to a reduction in the donor ability of the ligand and an increase in the catalytic activity of the complex (Scheme 9). Characterisation of the complex was conducted through various methods including NMR, UV/Vis spectroscopy, LIFDI mass spectrometry, and cyclic voltammetry. The CV exhibited a reversible one-electron oxidation at E1/2 = 0.58 V (CH2Cl2), attributed to the ferrocenyl carbene oxidation process. Additionally, small oxidation waves preceding the reversible wave (140 to 310 mV) were tentatively attributed to the oxidation of minority species, dimers of the complex held together by Au⋯Au interactions in both solid and solution states.
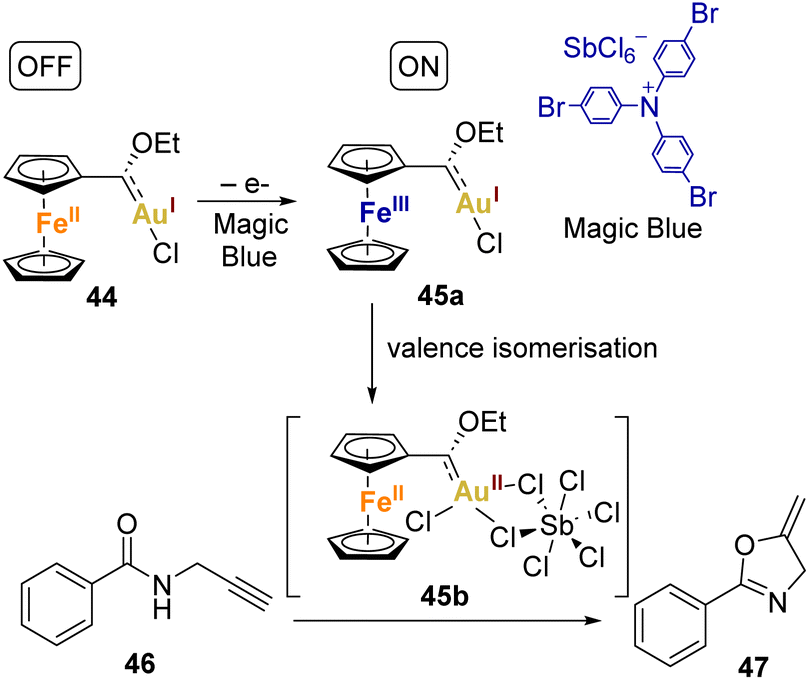 | ||
| Scheme 9 Depiction of the redox switchable catalysis with ferrocenyl gold(II) complexes and its use in a cyclisation reaction. | ||
Heinze and coworkers went further, as they chemically oxidised the complex using Magic Blue ([N(p-C6H4Br)3][SbCl6]) and analysed it through X-band EPR spectroscopy. The EPR studies indicated that Magic Blue initially oxidises the ferrocene unit in the complex 44, resulting in the formation of the cationic FeIIIAuI electromer 45a. Subsequently, the FeIIIAuI electromer 45a gradually transforms into a FeIIAuII electromer 45b, suggesting substantial reorganisation following the initial Fc/Fc+ oxidation. DFT studies supported this concept, indicating that the gold(II) centre needs stabilisation by additional donor ligands (such as counterions or substrates) to prevent aggregation or disproportionation. The Fc/Fc+ couple momentarily stabilises complex via valence isomerisation, preventing aggregation or disproportionation of the unsaturated gold(II) centre in the absence of donor ligands.
To assess the impact of this redox behaviour on catalysis, they applied it to the cyclisation of N-(2-propyn-1-yl)benzamide 46 to 2-phenyl-5-vinylidene-2-oxazoline 47 (Scheme 9). The redox-switchable catalyst displayed a reversible shift from a highly active state (45b), where the coordination of unsaturated gold(II) centres is prominent, to an inactive state characterised by saturated gold(I) centres (44). Interestingly, the addition of extra Ag(I) or Cu(II) salts was unnecessary to activate the catalyst.
Importantly, most of the catalyst remained active during subsequent substrate conversions, maintaining a similar turnover frequency (TOF) and eliminating the need for an induction period after full initial conversion. The addition of decamethylferrocene FeCp2* as a reducing agent halted catalytic turnover, while re-oxidation using Magic Blue re-initiated catalysis. In summary, the catalytic mixture involving catalyst and the oxidant demonstrated the ability to form catalytically active gold(II) species, which could be reversibly turned on and off through reduction and oxidation, respectively.
In 2022, Heinze's group introduced a novel ferrocenyl acyclic diamino carbene.74 This carbene displayed reversible oxidation, evidenced through cyclic and square wave voltammetry. The initial formation of ferrocenium ions was uniquely confirmed using EPR spectroscopy, followed by a slower, potentially intramolecular, electron transfer. This process led to the generation of persistent EPR-active species, possibly with gold(II) character. However, the potential of these species in redox-switchable gold catalysis remains unexplored.
3. Conclusions and perspectives
In this review, our primary focus has been on the role of gold(II) complexes in catalysis, encompassing both mono- and dinuclear gold(II) complexes, whether they are well-studied and isolated or proposed as intermediates in catalytic reactions. While mononuclear gold(II) complexes remain a synthetic challenge, and none have been used in catalysis to date, the potential of gold(II) dinuclear species in the field is significant. Gold(I) dinuclear complexes containing three-atom bridging ligands have shown promise in generating AuII–AuII species upon oxidation. This capability presents an exciting avenue to harness the potential of transient gold(II) species as proposed intermediates in catalytic reactions involving these dinuclear gold(I) complexes. It has been demonstrated that these gold(II) intermediates exhibit sufficient electrophilicity to activate multiple bonds, facilitating nucleophilic attacks, leading to the formation of organo-gold(III) intermediates and subsequent reductive elimination processes. The mechanism has been supported by a combination of density functional theory (DFT) calculations and experimental observations. These studies reveal that the formation of binuclear AuII–AuII intermediates reduces the energy barriers for all the depicted steps, with the bidentate bridging ligand minimising entropic costs during oxidative addition to both gold centres.Additionally, the integration of gold catalysis with photochemical conversions has introduced a novel and valuable reactivity pattern, circumventing the challenges offered by the AuI/AuIII redox cycle and enabling milder reaction conditions. While there is a growing consensus on the importance of gold(II) species as pivotal intermediates in these reactions, there is still a lack of structural and spectroscopic evidence of their presence. Further research in this area holds the potential to provide deeper insights into the mechanisms and applications of gold(II) complexes in photocatalysis.
Lastly, the concept of redox-switchable catalysis, exemplified by the ferrocenyl-substituted Fischer carbene gold(I) complex, demonstrates how the electron-donating or electron-withdrawing properties of ligands can be manipulated to modulate the catalytic activity of gold complexes. This approach offers a versatile tool for fine-tuning the reactivity of gold(II) complexes, opening up exciting possibilities for their use in various catalytic systems.
In summary, this review has highlighted the untapped potential of gold(II) complexes in catalysis, offering a rich field for exploration and innovation. As researchers delve deeper into the structural and mechanistic aspects of these complexes, significant advances in their application across a wide range of chemical transformations can be anticipated. Characterising gold(II) intermediates remains a challenging but achievable task, relying on the careful selection of precatalysts to yield stable intermediates that, if not isolated, can still be characterised through spectroscopic techniques. Ligand design in the coordination sphere of the metal and the selection of appropriate oxidants and organic transformations will be crucial for successful characterisation. Furthermore, redox-active ligands may play a pivotal role in developing redox-active gold complexes with the aim of isolating stable gold(II) species as crucial catalytic intermediates. This approach will enable a feedback loop between stable and isolated species as potential intermediates in catalysis, fostering the development of new catalysts and processes achievable through gold(II) catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Agencia Estatal de Investigación (PID2022-136861NB-I00, PID2020-117455GB-I00 and PID2019-104379RB-C21/AEI/10.13039/501100011033) and Gobierno de Aragón (Research Group E07_23R) for financial support of our research. J. C. Pérez-Sánchez also thanks the Spanish Ministerio de Universidades for a predoctoral grant (FPU21/01888).References
- H. Schmidbaur, Interdiscip. Sci. Rev., 1992, 17, 213–220 CrossRef
.
- R. P. Herrera and M. C. Gimeno, Chem. Rev., 2021, 121, 8311–8363 CrossRef CAS PubMed
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed
- P. Pyykkö, Inorg. Chim. Acta, 2005, 358, 4113–4130 CrossRef
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151–161 CrossRef CAS
- L. F. Pašteka, E. Eliav, A. Borschevsky, U. Kaldor and P. Schwerdtfeger, Phys. Rev. Lett., 2017, 118, 023002 CrossRef PubMed
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed
- Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed
- D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed
- A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed
- S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS PubMed
- A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864–876 CrossRef CAS PubMed
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed
- B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707–720 CrossRef CAS PubMed
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed
- P. Schwerdtfeger, Heteroat. Chem., 2002, 13, 578–584 CrossRef CAS
- M. C. Gimeno and A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS PubMed
-
J. J. Vittal and R. J. Puddephatt, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2011, p. eibc0076 Search PubMed
- A. Laguna and M. Laguna, Coord. Chem. Rev., 1999, 193–195, 837–856 CrossRef CAS
- K. A. Barakat, T. R. Cundari, H. Rabaâ and M. A. Omary, J. Phys. Chem. B, 2006, 110, 14645–14651 CrossRef CAS PubMed
- M. Bardají and A. Laguna, J. Chem. Educ., 1999, 76, 201 CrossRef
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC
- H. Schmidbaur and H. G. Raubenheimer, Angew. Chem., Int. Ed., 2020, 59, 14748–14771 CrossRef CAS PubMed
- D. B. Dell'Amico, F. Calderazzo, F. Marchetti, S. Merlino and G. Perego, J. Chem. Soc., Chem. Commun., 1977, 31–32 RSC
- F. Calderazzo, Pure Appl. Chem., 1978, 50, 49–53 CAS
- J. Strähle, J. Gelinek and M. Kölmel, Z. Anorg. Allg. Chem., 1979, 456, 241–260 CrossRef
- S. Witzel, M. Hoffmann, M. Rudolph, F. Rominger, A. Dreuw and A. S. K. Hashmi, Adv. Synth. Catal., 2022, 364, 581–592 CrossRef CAS
- X. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS PubMed
- M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777–7782 CrossRef CAS PubMed
- S. Kim, J. Rojas-Martin and F. D. Toste, Chem. Sci., 2016, 7, 85–88 RSC
- M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed
- A. J. Blake, J. A. Greig, A. J. Holder, T. I. Hyde, A. Taylor and M. Schröder, Angew. Chem., Int. Ed. Engl., 1990, 29, 197–198 CrossRef
- S. Seidel and K. Seppelt, Science, 2000, 290, 117–118 CrossRef CAS PubMed
- S. Preiß, C. Förster, S. Otto, M. Bauer, P. Müller, D. Hinderberger, H. Hashemi Haeri, L. Carella and K. Heinze, Nat. Chem., 2017, 9, 1249–1255 CrossRef PubMed
- S. Engbers, I. F. Leach, R. W. A. Havenith and J. E. M. N. Klein, Chem. – Eur. J., 2022, 28, e202200599 CrossRef CAS PubMed
- J. Mehara, A. Koovakattil Surendran, T. Van Wieringen, D. Setia, C. Foroutan-Nejad, M. Straka, L. Rulíšek and J. Roithová, Chem. – Eur. J., 2022, 28, e202201794 CrossRef CAS PubMed
- S. H. Elder, G. M. Lucier, F. J. Hollander and N. Bartlett, J. Am. Chem. Soc., 1997, 119, 1020–1026 CrossRef CAS
- T. Drews, S. Seidel and K. Seppelt, Angew. Chem., Int. Ed., 2002, 41, 454–456 CrossRef CAS PubMed
- G. Gencheva, D. Tsekova, G. Gochev, D. Mehandjiev and P. R. Bontchev, Inorg. Chem. Commun., 2003, 6, 325–328 CrossRef CAS
- K. Heinze, Angew. Chem., Int. Ed., 2017, 56, 16126–16134 CrossRef CAS PubMed
- M. Baya, A. Pérez-Bitrián, S. Martínez-Salvador, A. Martín, J. M. Casas, B. Menjón and J. Orduna, Chem. – Eur. J., 2018, 24, 1514–1517 CrossRef CAS PubMed
- A. Kar, N. Mangu, H. M. Kaiser, M. Beller and M. K. Tse, Chem. Commun., 2008, 386–388 RSC
- L. Cui, G. Zhang and L. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 3884–3887 CrossRef CAS PubMed
- G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112–3115 CrossRef CAS PubMed
- T. de Haro and C. Nevado, Angew. Chem., Int. Ed., 2011, 50, 906–910 CrossRef CAS PubMed
- H. Schmidbaur and R. Franke, Inorg. Chim. Acta, 1975, 13, 85–89 CrossRef CAS
- H. Schmidbaur, Acc. Chem. Res., 1975, 8, 62–70 CrossRef CAS
- W. E. Brenzovich Jr., D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard III and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519–5522 CrossRef PubMed
- M. Kumar, J. Jasinski, G. B. Hammond and B. Xu, Chem. – Eur. J., 2014, 20, 3113–3119 CrossRef CAS PubMed
- A. Tamaki, S. A. Magennis and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 6140–6148 CrossRef CAS
- A. D. Melhado, W. E. Brenzovich Jr., A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885–8887 CrossRef CAS PubMed
- B. Alcaide, P. Almendros, T. M. del Campo, E. Soriano and J. L. Marco-Contelles, Chem. – Eur. J., 2009, 15, 9127–9138 CrossRef CAS PubMed
- B. Alcaide, P. Almendros, T. M. Del Campo and I. Fernández, Chem. Commun., 2011, 47, 9054–9056 RSC
- A. S. K. Hashmi, A. M. Schuster, S. Litters, F. Rominger and M. Pernpointner, Chem. – Eur. J., 2011, 17, 5661–5667 CrossRef CAS PubMed
- W. E. Brenzovich, J.-F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728–4731 CrossRef CAS PubMed
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef CAS PubMed
- M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211–6215 CrossRef CAS
- A. W. Sromek, M. Rubina and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 10500–10501 CrossRef CAS PubMed
- N. Morita, A. Yasuda, M. Shibata, S. Ban, Y. Hashimoto, I. Okamoto and O. Tamura, Org. Lett., 2015, 17, 2668–2671 CrossRef CAS PubMed
- B. R. Reiner, M. W. Bezpalko, B. M. Foxman and C. R. Wade, Organometallics, 2016, 35, 2830–2835 CrossRef CAS
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed
- K. Liu, N. Li, Y. Ning, C. Zhu and J. Xie, Chem, 2019, 5, 2718–2730 CAS
- C.-M. Che, H.-L. Kwong, V. W.-W. Yam and K.-C. Cho, J. Chem. Soc., Chem. Commun., 1989, 885–886 RSC
- D. Li, C.-M. Che, H.-L. Kwong and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1992, 3325–3329 RSC
- B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed
- L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808–4813 CrossRef CAS PubMed
- W. Wang, C.-L. Ji, K. Liu, C.-G. Zhao, W. Li and J. Xie, Chem. Soc. Rev., 2021, 50, 1874–1912 RSC
- S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS PubMed
- I. M. Lorkovic, R. R. Duff Jr. and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617–3618 CrossRef CAS
- P. Veit, C. Volkert, C. Förster, V. Ksenofontov, S. Schlicher, M. Bauer and K. Heinze, Chem. Commun., 2019, 55, 4615–4618 RSC
- S. D. Waniek, C. Förster and K. Heinze, Eur. J. Inorg. Chem., 2022, 2022, e202100905 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |