
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking the current limitation of electrochemical CO2 reduction via a silica-hydroxide cycle†
Chulwan
Lim‡
ab,
Sangkuk
Kim‡
ac,
Ji Hwan
Song‡
a,
Man Ho
Han
 a,
Young-Jin
Ko
a,
Kwan-Young
Lee
b,
Jae-Young
Choi
de,
Woong Hee
Lee
*a and
Hyung-Suk
Oh
*ade
a,
Young-Jin
Ko
a,
Kwan-Young
Lee
b,
Jae-Young
Choi
de,
Woong Hee
Lee
*a and
Hyung-Suk
Oh
*ade
aClean Energy Research Center, Korea Institute of Science and Technology, Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: hyung-suk.oh@kist.re.kr; abcabac@kist.re.kr; Tel: +82 (0)2 958 5824
bDepartment of Chemical and Biological Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
cHydrogen and Low-Carbon Energy R&D laboratories, POSCO HOLDINGS, 67 Cheongam-ro, Nam-gu, Pohang-si, Gyeongsangbuk-do 37673, Republic of Korea
dSchool of Advanced Materials Science & Engineering, Sungkyunkwan University (SKKU), Suwon, 16419, Republic of Korea
eKIST-SKKU Carbon-Neutral Research Center, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
First published on 30th April 2024
Abstract
Alkaline local pH during a vigorous electrochemical CO2 reduction reaction (CO2RR) can improve the activity and selectivity of CO2RR. However, it also leads to an alkalinity problem in that hydroxide ions obstruct the mass transfer of CO2 to the active site, thereby limiting the current density. In this study, we introduce a silica-hydroxide cycle, which moderates the local pH by redistributing hydroxide ions, analogous to the carbonate-silicate cycle responsible for the drawdown of atmospheric CO2 on Earth. In the membrane electrode assembly (MEA) of a CO2 electrolyzer, SiO2 undergoes weathering due to the high local pH and consequently consumes OH−, reducing the pH within the MEA. The dissolved silicate ions move to the membrane and are almost regenerated to SiO2 with release of OH−. Geological and spectral observations suggest that the silica-hydroxide cycle reduces the local pH thereby enhancing mass transfer of CO2, breaking the limitation of current density for CO2RR. Our work proposes new chemical approaches to increase current density, mainly improved by physical methods, and contributes valuable insight for improving a variety of electrochemical systems.
Broader contextThe electrochemical CO2 reduction reaction (CO2RR) is viewed as a promising approach to attain carbon neutrality while generating valuable fuels and chemicals. Enhancing the current density in CO2RR presents a significant challenge for the efficient production of chemicals on a meaningful scale, which is essential for economic viability. Recently, various mechanical approaches have enhanced the performance of CO2RR through enhancing the mass transfer of CO2, playing a pivotal role in improving the current density. Nevertheless, in the high current density region, the hydroxide ions created during the reaction form a local alkaline environment, which converts CO2 to carbonate ions, hindering the mass transfer of CO2 to the catalyst surface and limiting the current density for CO2RR. This study delves into the role of SiO2 in the electrochemical CO2RR regarding the silica-hydroxide cycle, which moderates the local pH by redistributing hydroxide ions. This mechanism draws a parallel with the silicate-carbonate cycle, which plays a crucial role in the natural sequestration of atmospheric CO2 on Earth, thus offering a novel avenue for breaking the current density limitation of CO2RR through controlled pH management. |
Introduction
The electrochemical CO2 reduction reaction (CO2RR) is viewed as a promising solution for achieving carbon neutrality while producing valuable fuels and chemicals.1–3 Increasing the current density for CO2RR poses a significant challenge for the mass production of chemicals while ensuring economic feasibility.4,5 Recently, various mechanical approaches, including gas-phase CO2, device design, gas diffusion electrode (GDE), membrane electrode assembly (MEA), pressurized system and nanosized catalyst, have successfully enhanced the performance of CO2RR via enhancing mass transfer of CO2, playing a pivotal role in improving the current density.6–17 Under high current density regime coming from a recently developed system, an intriguing phenomenon, called local pH, clearly appeared for CO2RR. The hydroxide ions generated from the reaction create a local alkaline environment at the electrode, promoting CO2RR via reducing the hydrogen evolution reaction (HER) and modulating binding energies for the intermediates.18–24 Nevertheless, some studies addressing local pH have indicated an alkalinity problem in that hydroxide ions block the mass transfer of CO2 to the catalyst by converting carbonate, limiting the current density for CO2RR.25,26 The alkalinity problem of CO2RR can be validated using alkali cation effects. Numerous experimental and theoretical investigations reveal that a larger size of alkali cation exhibits smaller hydration number at the Helmholtz layer and lower pKa values near the electrode, mitigating the local pH phenomenon.27–29 The current density of CO2RR according to alkali cation was assessed in a CO2 MEA electrolyzer using Ag black sprayed GDE (Fig. 1a and Fig. S1–S3, ESI†). CsHCO3 electrolyte demonstrated a significantly higher maximum current density for CO2RR than KHCO3 electrolyte. To delve into the pH reaction environment of the cathode in the MEA electrolyzer, in situ/operando Raman spectroscopy and gas analysis of the anode were carried out (Fig. 1b and c). In the Raman spectra for the KHCO3 electrolyte, the carbonate peak disappeared at current densities exceeding 400 mA cm−2, while the carbonate peak persisted at a current density of 800 mA cm−2 using CsHCO3 electrolyte. The in situ/operando Raman spectroscopy focused on the catalyst surface. Thus, the disappeared carbonate peak indicates that the catalyst surface possesses high alkalinity with almost no carbonate ions resulting from reaction with OH− and CO2 gas. These observations indicate that the larger size of Cs cations alleviates the local pH phenomenon at high current densities. The gas ratio of CO2 and O2 for the anode side decreased below 2 was observed at 400 mA cm−2 for KHCO3 and 800 mA cm−2 for CsHCO3. The changes of gas ratio by current density are well explained by the schematic illustration in Fig. 1d. The ratio of CO2 and O2 is close 2 in the current density region showing high faradaic efficiency (FE) for CO, indicating the main carbonate ion transfer from cathode to anode.30,31 The decreased CO2 at the anode shows transfer of hydroxide ions from the cathode, suggesting a highly alkaline reaction environment at the cathode sufficient to prevent hydroxide ion contact with CO2. Additionally, as shown in the in situ/operando Raman results in Fig. 1b, the mass transfer of CO2 to the catalyst layer is impeded by high local pH, further limiting the current density for CO2RR. This suggests that, besides physical properties that can be improved by mechanical methods, the chemical reaction environment also plays a significant role in current limitations. Thus, chemical approaches, such as cation effects, are essential for enhancing current density in CO2RR. The key for a chemical strategy is alleviating local pH by rebalancing CO2 and hydroxide ion concentration at the cathode. | ||
| Fig. 1 Alkalinity issue in CO2RR and carbonate-silicate cycle. (a) CO partial current density versus cell voltage in a zero-gap electrolyzer using Ag black with 0.1 M KHCO3 and 0.1 M CsHCO3. (b) In situ/operando Raman spectroscopy results with 1 M KHCO3 and CsHCO3 electrolytes. (c) CO2/O2 ratio at the anode in a zero-gap electrolyzer with 0.1 M KHCO3 and 0.1 CsHCO3. (d) A schematic depiction of the alkalinity problem in a zero-gap CO2 electrolyzer. (e) A schematic representation of the carbonate-silicate geochemical cycle on Earth. | ||
In nature, Earth's CO2 is balanced by various carbon cycles including the carbonate-silicate cycle.32–35 This geochemical process redistributes CO2 in the atmosphere to the oceans and land by rainwater, silicate weathering, carbonate precipitation and sedimentation, resulting in drawdown of atmospheric CO2 (Fig. 1e) The stored CO2 in land is redistributed to the atmosphere by volcanism. In this study, we aim to break the current density limitation for CO2RR using silica oxide via a silica-hydroxide cycle, similar to the carbonate-silicate cycle. The unique silica properties allow distribution of the high OH− concentration at the catalyst surface to the other cathode side, reducing the elevated local pH at the catalyst surface and facilitating mass transfer of CO2 for CO2RR.
Results and discussion
Effects of SiO2 on current density for CO2RR
An Ag–SiO2 electrode was fabricated on a GDE by spraying ink of a mixture of SiO2 nanopowder and commercial Ag black (Fig. S4–S6, ESI†). Fig. 2a illustrates SEM and SEM-EDX mapping images of the Ag–SiO2 electrode, indicating that Ag black and SiO2 nanoparticles are well dispersed on the GDE substrate. The effects of SiO2 for CO2RR performance were studied by using an anion exchange membrane (AEM) zero-gap electrolyzer with 0.1 M CsHCO3 anolyte and humidified CO2 gas (Fig. 2b, d and Fig. S7, ESI†).36,37 At an applied current density below 600 mA cm−2, both the Ag and Ag–SiO2 electrodes show similar CO2RR performance, with a high FECO exceeding 90%. However, above an applied current density of 700 mA cm−2, the FEH2 of the Ag electrode drastically increased with rising current density, resulting in a maximum current density for CO of 549 mA cm−2. On the other hand, the FECO for the Ag–SiO2 electrode remained at over 80% and the selectivity for CO (H2 and CO, SECO) was nearly 100% at an applied current density until 1 A cm−2 while suppressing the HER. The difference of FECO and SECO at a high current density region would come from production of formate (Fig. S8, ESI†). The maximum current density of the Ag–SiO2 electrode for CO reached a value of 806 mA cm−2, implying that additional SiO2 to the Ag black electrode contributed to breaking the current density limitation for CO2RR. Exploring the effects of varying SiO2 ratios indicates the necessity for an optimal amount of SiO2 (Fig. S9, ESI†). To confirm the physical effects, an Ag–TiO2 electrode was fabricated using similar size of TiO2 instead of SiO2 (Fig. S10, ESI†). Ag–TiO2 exhibits a similar limiting current density to Ag for CO2RR. The results of Ag–TiO2 and the comparable ECSA value for both the Ag and Ag–SiO2 electrodes show that the impact of SiO2 does not originate from physical properties (Fig. S11 and S12, ESI†). A similar phenomenon of breaking the maximum current density limit for CO2RR was observed using 0.1 M KHCO3 anolyte at lower current densities (Fig. 2e and Fig. S13, ESI†). To validate the SiO2 effects for high current density, chronopotentiometry tests were conducted at a current density of 700 mA cm−2 where the effect of SiO2 is seen (Fig. 2f and Fig. S14, ESI†). FECO of the Ag electrode was close to 80% and rapidly decreased after 3 h. In contrast, the Ag–SiO2 electrode demonstrated stable cell voltage and consistent FECO above 90% for 12 h, suggesting that the effects of SiO2 are not an instant phenomenon and could serve as a sustained strategy.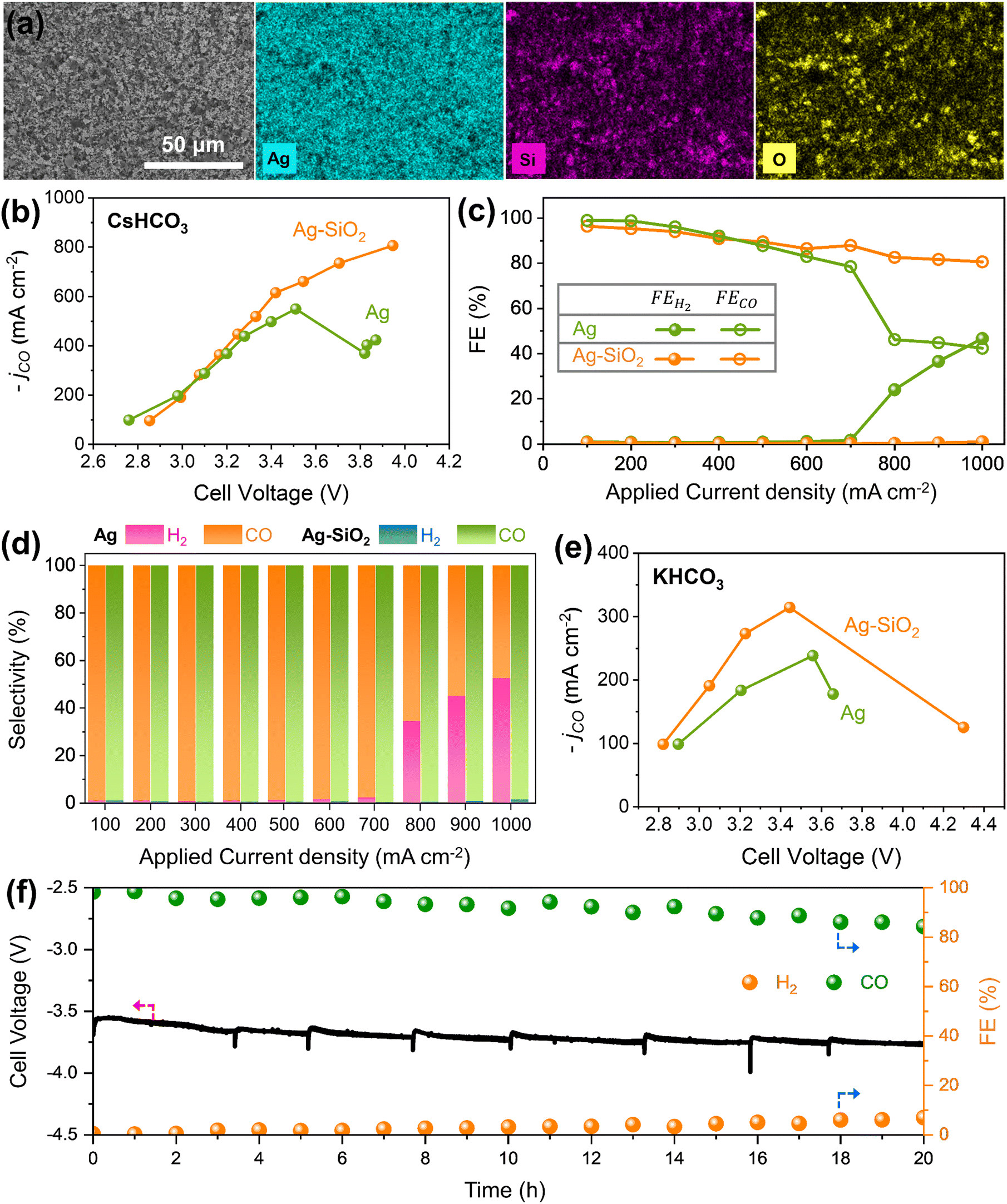 | ||
| Fig. 2 Impact of SiO2 in MEA CO2 electrolyzer, alongside CO2RR performance comparisons between Ag black and Ag–SiO2. (a) SEM image and EDS elemental mapping of the Ag–SiO2 electrode. (b) CO partial current density, (c) H2 and CO faradaic efficiency, and (d) H2 and CO selectivity for both Ag black and Ag–SiO2 using 0.1 M CsHCO3. (e) CO partial current density of Ag black and Ag–SiO2 with 0.1 M KHCO3. (f) Durability test for Ag–SiO2 in the zero-gap electrolyzer using 0.1 M CsHCO3 at 700 mA cm−2 for 12 h. | ||
The origin of SiO2 effects in breaking current density limitation
To elucidate the origins of SiO2 effects that overcome the current density limitation of CO2RR, the cathode reaction environment in the MEA electrolyzer was investigated using gas analysis of the anode and in situ/operando Raman spectroscopy using a GDE. For the Ag–SiO2 electrode, the gas composition ratio of CO2 to O2 remains near 2 within a current density range of 500–1000 mA cm−2. However, for the Ag electrode, this ratio drops to almost 1 when the current density exceeds 800 mA cm−2 (Fig. 3a). This suggests that SiO2 mitigates the local pH effects stemming from high current densities. | ||
| Fig. 3 The analyses of the role of SiO2 in modulating the cathode reaction environment. (a) CO2/O2 ratio at the anode in a zero-gap electrolyzer using Ag–SiO2 with 0.1 M CsHCO3. (b) In situ/operando Raman spectroscopy results for the Ag electrode in 1 M KHCO3 electrolyte. (c) In situ/operando Raman spectroscopy results for the Ag–SiO2 electrode in 1 M KHCO3 electrolyte. (d) In situ/operando Raman spectroscopy results for the Ag electrode in 1 M CsHCO3 electrolyte. (e) In situ/operando Raman spectroscopy results for the Ag–SiO2 electrode in 1 M CsHCO3 electrolyte. (f) Summary of in situ/operando Raman spectroscopy results using carbonate Raman peak shifts and carbonate peak intensity ratios for both Ag and Ag–SiO2 electrodes in KHCO3 and CsHCO3 electrolytes. Reference points for peak shift and intensity: KHCO3, 100 mA cm−2; CsHCO3, 200 mA cm−2. | ||
To more directly observe the reaction environment of the cathode, in situ/operando Raman spectroscopy was carried out using a customized GDE Raman cell (Fig. S15, ESI†).38 To estimate the pH of the cathode according to current density, we constructed two calibration curves for ranges of 9–13 and 13–14 (Fig. S16–S20, ESI†). Even at a low current density of 100 mA cm−2, the bicarbonate peak for all in situ/operando Raman spectra nearly disappears, signifying pronounced local pH effects in the GDE.23 In a KHCO3 electrolyte, the carbonate peak disappears for the Ag electrode at a current density of 600 mA cm−2 (Fig. 3b). Conversely, for the Ag–SiO2 electrode, a minor carbonate peak persists at the same current density, indicating that SiO2 mitigates local pH effects, reminiscent of cation effects (Fig. 3c). The Ag–SiO2 electrode also exhibited a similar trend in CsHCO3 electrolyte. The carbonate peak for the Ag electrode is lower than that for the Ag–SiO2 electrode at a current density of 1 A cm−2 (Fig. 3d and e).
For the Ag–SiO2 electrode, peaks corresponding to Si–ONBO (where NBO is non-bridging oxygen) were observed at a current density between 100 and 400 mA cm−2, hinting at the onset of SiO2 degradation in alkaline conditions (Fig. S21, ESI†).39,40 For intuitive representation of local pH under various conditions, we summarize the in situ/operando Raman results based on two parameters: the log-transformed relative peak area ratio of the carbonate peak and its shift (Fig. 3f). The carbonate peak shift is expected to be related to the Stark effect or cation concentration.41–43 However, since there is no carbonate intermediate in the CO2RR mechanism on the Ag surface, we assumed that this peak shift was caused by cation concentration. The calibration results unequivocally show that carbonate peak area, which is related to carbonate concentration, is correlated to pH and peak shift corresponds to cation concentration (Fig. S22 and S23 and Note S2, ESI†). Based on calibration data, the estimated local pH results from in situ/operando Raman spectra are shown in Fig. S24 and Table S1 (ESI†). While the alkalinity of the Ag electrode increased with current density, the Ag–SiO2 electrode in comparison retained its pH, underscoring the role of SiO2 in the suppression of local pH effects.
Behavior of SiO2 in CO2RR system
In an effort to understand the effects of SiO2, it is necessary to observe the behavior of SiO2 in a medium that simulates the reaction environment for a CO2 MEA electrolyzer. To verify the neutralization of local pH by CO2, we purged the CO2 gas into solutions of various pH and measured the pH every minute (Fig. 4a). As the duration of the CO2 purge increased, the solution's pH decreased, owing to the reaction of OH− ions with CO2, resulting in carbonate ions. The amount of consumed OH− with CO2 is proportional to pH of solution, highlighting the alkalinity problem that blocks the transfer of CO2 to active sites due to the high local pH. When the SiO2 nanopowder was mixed with alkaline solution of pH 12 and 13, SiO2 dissolved, leading to a decrease in the solution's pH. Regardless of the presence of SiO2, the rate of OH− consumption by CO2 was invariably proportional to the pH of the solution, implying that CO2 gas capture by OH− is suppressed by SiO2 (Fig. 4b). Interestingly, suspensions were detected in SiO2-dissolved solution following an extended CO2 purge (Fig. 4c). The analysis of collected solution and precipitate (by centrifugation) after 2 h of CO2 purge (the pH of solution reached 7.5) demonstrates separation of SiO2 and KHCO3 solution,44 suggesting almost reversible behavior of SiO2 (Fig. 4d, e and Fig. S21, S25, ESI†). When the SiO2 nanopowder was mixed with solutions of 0.1 M KHCO3 (pH 8.65) and 1 M KHCO3 (pH 8.69), the pH remained almost unchanged at 8.70 (Fig. S26, ESI†). The presumed reaction involving SiO2 is depicted in Fig. 4f. In alkaline media, SiO2 dissolved and converted to silicate ions by reaction of OH−. These silicate ions then precipitate as SiO2 under neutral conditions with release of OH− due to low solubility.44–46 We hypothesize that this cycle involving SiO2 and hydroxide also takes place in the CO2 MEA electrolyzer. | ||
| Fig. 4 Analysis of SiO2 in CO2RR media. (a) Change in solution pH for various initial pH solutions under CO2 purge, plotted against CO2 purging time (Inset: Quantity of OH− consumed based on current pH). (b) Variation in OH− concentration during CO2 purging, attributed to OH− capture due to the presence of SiO2. (c) Images of pristine SiO2 powder, SiO2-dissolved KOH solution, and an opaque gel obtained within the SiO2-dissolved KOH solution after CO2 purging. (d) Raman spectra comparing 1 M KHCO3 solution (reference), and the CO2-purged, SiO2-dissolved KOH solution (containing the opaque gel). (e) Raman spectra comparing pristine SiO2 powder and centrifuged SiO2 gel obtained from the opaque gel within the CO2-purged solution. (f) A schematic illustrating the reversible behavior of SiO2 species during the reaction. | ||
The silica-hydroxide cycle in MEA CO2 electrolyzer
The mechanism of breaking the current limitation by SiO2 in a CO2 electrolyzer is hypothesized as being due to the reversible cycle of SiO2 between alkaline and neutral media, carrying OH− ions (Fig. 5a). We postulate that this silica-hydroxide cycle decreases the local pH of the electrode surface by increasing hydroxide distribution, thereby enhancing mass transfer of CO2 for CO2RR. This process of silica-hydroxide cycle is similar to the silicate-carbonate cycle that maintains the CO2 level in the atmosphere. When a vigorous CO2RR occurs in the MEA CO2 electrolyzer, the pH of catalyst surface is presumed to be that of an alkaline medium in the range of pH 11–14 by in situ/operando Raman spectroscopy. Concurrently, the pH of the AEM is expected to be neutral owing to abundant transported carbonate ions, indicating a suitable environment for the reversible silica-hydroxide cycle. | ||
| Fig. 5 Geological and spectral observations of silica-hydroxide cycle in MEA CO2 electrolyzer. (a) A schematic representation of the carbonate-silicate geochemical cycle occurring during electrochemical CO2 reduction reaction on Ag–SiO2. SEM images and EDS elemental mapping of the Ag–SiO2 electrode before (b) and after (c)–(e) reaction (c: 900 mA cm−2 for 20 min; (d) and (e): 1 A cm−2 for 20 min) in MEA CO2 electrolyzer. | ||
To demonstrate the silica-hydroxide cycle in the MEA CO2 electrolyzer, silica weathering, evidence of the silica-hydroxide cycle, was monitored using geological and spectral observations with SEM. The pristine Ag–SiO2 electrode clearly delineates the boundary between SiO2 and Ag nanoparticles (Fig. 5b and Fig. S27, ESI†). After CO2RR, this boundary becomes less distinct in SEM images (Fig. 5c and Fig. S28, ESI†), and this blurring intensifies as the current density increases (Fig. S29–S33, ESI†). Nevertheless, SiO2 is well dispersed in the electrode, similar to before the reaction. Notably, no discernible difference in Ag nanoparticles dependent on SiO2 is observed, suggesting that SiO2 has no effect on the chemical state and structure of Ag (Fig. S34–S39, ESI†). These outcomes corroborate the hypothesis of silica weathering under high current densities. To further substantiate the presence of dissolved silicate ions and the regeneration of SiO2, EDS mapping was employed to discern the state of SiO2. When the dissolved silicate ions dried with alkali cations, hydrous alkali silica gel is formed.47 This hydrous alkali silica gel is observed in certain regions of the electrode, signifying the presence of dissolved silicate ions (Fig. 5d and Fig. S40, ESI†). The accumulation of regenerated SiO2 on the Ag electrode, the final step of the silica-hydroxide cycle, is clearly shown in Fig. 5e. SiO2 appears on the Cs carbonate precipitate that is generated during or post-CO2RR, implying regenerated SiO2.
Under high cathodic currents, SiO2 weathering is triggered by high local pH, leading to a decrease in pH by consuming OH− at the catalyst surface. The dissolved silicate ions, not reacted with CO2, move to the AEM side.48,49 The dissolved silicate ions are converted to SiO2 with counterbalancing release of OH− under neutral conditions at the AEM and the converted SiO2 is redistributed on the electrode. However, the silica-hydroxide cycle is not completely reversible in the MEA electrolyzer owing to the AEM, which is highly permeable to anions and cations. SEM-EDS mapping cross-section images after reaction show that the majority of SiO2 remains in the cathode, but a small amount of SiO2 has been moved to the AEM and anode (Fig. S41, ESI†). To eliminate the regeneration step at the AEM, 1 M KOH was used for MEA test instead of 0.1 M CsHCO3 at a current density of 700 mA cm−2. ICP-OES analysis shows that 67.6% of SiO2 is preserved on the cathode with the silica-hydroxide cycle but, without the regeneration step, 5.8% of Si remained on the cathode. These results show that the regeneration step, which is critical for reversibility of the silica-hydroxide cycle, functions in a real MEA electrolyzer. This silica-hydroxide cycle reduces the concentration of OH− that captures CO2, thereby enhancing mass transfer of CO2 to catalyst active sites and breaking the limitation of current density.
Conclusions
Our study introduces a novel strategy to overcome the limitation of the current density for CO2RR by a chemical cycle approach, without the need for physical modification. Hydroxide ions generated from CO2RR elevate the local pH. While this can enhance selectivity, it also hinders the transfer of CO2 by forming carbonate ions, thus imposing a current density limitation for CO2RR. Therefore, controlling the local pH is crucial for achieving a high current density. On Earth, atmospheric CO2 levels are reduced via carbon cycles, such as the silicate-carbonate cycle. This geological carbon cycle redistributes CO2 from the atmosphere to rocks through silicate weathering. Similarly, the silica-hydroxide cycle derived using SiO2 causes a reduction in the local pH of the catalyst surface in the MEA CO2 electrolyzer. SiO2 weathering is triggered by high local pH, and silicate ions transport the hydroxide ions near the AEM without CO2 capture at the catalyst surface and reconvert to SiO2. This cycle of SiO2 enhances the mass transfer of CO2, breaking the current density limitation for CO2RR. Considering commercialization, the future goals of the silica-hydroxide cycle should be focused on enhancing the reversibility in real MEA electrolyzers. This chemical cycle strategy also observed on Earth holds a wide-ranging applicability across various systems, offering fresh insights for augmenting their efficiency and breaking their limitations.Experimental procedures
Materials and chemicals
Silver nanopowder (Alfa Aesar, APS 20–40 nm, 99.9%), silicon dioxide nanopowder (Sigma-Aldrich, 10–20 nm, 99.5% trace metal basis), titanium(IV) oxide nanopowder (Sigma-Aldrich, 21 nm primary particle size (TEM), ≥99.5% trace metals), Nafion 5 wt% ionomer solution (Sigma-Aldrich), isopropanol (DAEJUNG, 99.5%), and iridium(IV) oxide (Alfa Aesar, premion, 99.99%; Ir 84.5% min) were used as received, without further purification. Carbon paper (Fuel Cell Store, Sigracet 39BB) and platinized titanium screen (Fuel Cell Store) were employed as substrates for cathodic and anodic electrodes, respectively. These substrates were cut to the desired size using a homemade punch. KHCO3, which was used as the electrolyte, was procured from Sigma Aldrich (ACS reagent, >99.7%). The AEM used was sourced from Dioxide Materials (Sustainion X37-50 grade RT).Preparation of Ag-based electrode for cathode
The Ag black, Ag–TiO2 and Ag–SiO2 electrodes were fabricated by spraying catalyst ink onto a gas diffusion layer (GDL) at 70 °C. For the Ag–SiO2 catalyst ink, 90 mg of commercial silver nanopowder, 15 mg of silicon dioxide nanopowder, 120 mg of Nafion ionomer solution, and 3.5 mL of isopropanol were ultrasonically mixed. Ag–TiO2 catalyst ink was fabricated with same method as for Ag–SiO2 catalyst ink except for adding TiO2 instead of SiO2. The catalyst ink for Ag black was prepared using the same method as for Ag–SiO2, but without the silicon dioxide nanopowder.Preparation of Ir-based electrode for anode
The electrode for OER, employed as the anode, was fabricated by spraying catalyst ink onto a platinized titanium screen using a 70 °C hot plate. The catalyst ink was prepared by mixing 120 mg of commercial iridium oxide (Alfa Aesar) with 120 mg of 5 wt% Nafion ionomer solution and 4 mL of isopropanol. The electrode had an area of 10 cm2, and the iridium oxide loading was set at 2 mg cm−2.Single-cell electrochemical CO2 reduction reaction test
MEAs were fabricated using the catalyst-coated electrode method for electrochemical reactions, and the geometric electrode area was set at 10 cm2. The fabricated electrodes, along with commercial IrO2-sprayed platinized titanium screen electrodes (Alfa Aesar, with a target loading of 2.0 mg cm−2), were employed as cathodes and anodes in all single-cell tests, respectively. The AEM (Dioxide Materials, X37-50 Grade RT) underwent pretreatment in a 1 M KOH solution for 48 h and was rinsed multiple times with deionized water before use. Following this, a 0.1 M KHCO3 solution, acting as the electrolyte, was introduced to the anode side via a pump. Concurrently, 200 sccm of humidified CO2 gas at 80 °C, heated using a mantle, was introduced to the cathode side. The electrochemical tests were conducted using a VSP potentiostat (BioLogic, VMP3B-20) equipped with a booster up to 20 A. The CO2RR was conducted over 18 min for each applied current density.A gas chromatograph (GC, Agilent 7890A) was employed for product gas analysis at the GC outlet. A water trap was interposed between the GC and the cathode outlet. Argon gas (99.999%) served as the carrier gas. The GC was fitted with a flame ionization detector (FID) to detect hydrocarbons like CO, CH4, and C2H4, and a thermal conductivity detector (TCD) for hydrogen (H2) gas detection. A methanizer was utilized to enhance CO detection before routing to the FID. Measurements of the product gases commenced 9 min after initiation of CO2RR for each current density setting. The FEs of H2 and CO were computed using the following equation:

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1): p = pressure: T = room temperature (298 K): R = ideal gas constant (8.314 J mol−1 K−1).
485 C mol−1): p = pressure: T = room temperature (298 K): R = ideal gas constant (8.314 J mol−1 K−1).
The partial current densities of the products were determined from the volume of a specific product, as indicated by the GC peak.
Physical characterization
The size distribution and microstructure of the Ag black and Ag–SiO2 electrodes were examined using high-resolution transmission electron microscopy (HR-TEM, Titan at 300 kV, FEI Co., USA). HR-TEM images, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images, and energy-dispersive spectroscopy (EDS) mapping were performed with a Talos F200X system (FEI). Raman spectroscopy was employed to characterize various electrodes, powdered samples, and the prepared solutions, and was conducted at room temperature with a 532-nm laser (Renishaw). XPS (X-ray photoelectron spectroscopy, Nexsa, ThermoFisher Scientific) was conducted with a base pressure of 2 × 10−8 mbar and a monochromated Al Kα (1486.6 eV) X-ray source. All XPS spectra were calibrated using the C 1s peak (284.8 eV) as a reference.In situ/operando Raman spectroscopy
In situ/operando SERS (surface-enhanced Raman spectroscopy) was conducted using a standard three-electrode system, a custom-made in situ Raman cell (details provided in Fig. S14, ESI†), and a laser with a 532-nm wavelength. The working electrode was fashioned by spraying catalyst ink (containing Ag and Ag–SiO2 powders) onto a GDL (Sigracet 39BB, SGL Carbon) substrate, which amplified the Raman spectrum signal. A Pt wire and Ag/AgCl (3 M NaCl) served as the counter and reference electrodes, respectively. The electrolyte was either 1 M KHCO3 or 1 M CsHCO3, and gaseous CO2 was purged onto the working electrode at a flow rate controlled with a ball flowmeter. The electrochemical experiments were controlled using a potentiostat (CompactStat, Ivium Technologies, Eindhoven, Netherlands). The thin electrolyte layer enveloping the electrode facilitated Raman spectroscopic observation of the working electrode's surface.Author contributions
C. L., S. K. and J. H. S. designed/conducted the experiments, analyzed the data and wrote the manuscript. M. H. H. performed Raman analysis of Ag black and Ag–SiO2. Y.-J. K. contributed to the electrochemical cell tests. K. Y. L. and J. Y. C. provided an idea for the electrochemical analysis. H.-S. O. and W. H. L. supervised the research and wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by institutional program grants from the Korea Institute of Science and Technology (KIST) and Research Project for ‘Carbon Upcycling Project for Platform Chemicals’ of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (grant number: 2022M3J3A1050053) and ‘Carbon to X Project’ (project no. 2020M3H7A1098229) through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea. This research was also supported by the National Research Council of Science & Technology (NST) grant by the Korean government (MSIT) (no. CAP21011-100) and National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2021R1A6A3A01086623, RS-2023-00256847).References
- W. H. Lee, K. Kim, J. H. Koh, D. K. Lee, D. H. Won, H.-S. Oh, U. Lee and B. K. Min, Nano Energy, 2023, 110, 108373 CrossRef CAS.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, J. Phys.: Energy, 2022, 4, 042003 CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., 2021, 133, 20795–20816 CrossRef.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed.
- B. Endrodi, E. Kecsenovity, A. Samu, F. Darvas, R. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef CAS PubMed.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- D. Kim, Y. Chae, U. Lee, W. Kim and D. H. Won, Curr. Opin. Electrochem., 2023, 39, 101295 CrossRef CAS.
- Y. C. Tan, W. K. Quek, B. Kim, S. Sugiarto, J. Oh and D. Kai, ACS Energy Lett., 2022, 7, 2012–2023 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- W. Choi, Y. Choi, E. Choi, H. Yun, W. Jung, W. H. Lee, H.-S. Oh, D. H. Won, J. Na and Y. J. Hwang, J. Mater. Chem. A, 2022, 10, 10363–10372 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- M. C. O. Monteiro, A. Mirabal, L. Jacobse, K. Doblhoff-Dier, S. C. Barton and M. T. M. Koper, JACS Au, 2021, 1, 1915–1924 CrossRef CAS PubMed.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D.-H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C.-T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- N. Govindarajan, A. Xu and K. Chan, Science, 2022, 375, 379–380 CrossRef CAS PubMed.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager, III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 CrossRef PubMed.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- R. G. Hilton and A. J. West, Nat. Rev. Earth Environ., 2020, 1, 284–299 CrossRef CAS.
- D. E. Penman, J. K. Caves Rugenstein, D. E. Ibarra and M. J. Winnick, Earth-Sci. Rev., 2020, 209, 103298 CrossRef CAS.
- A. Bufe, N. Hovius, R. Emberson, J. K. C. Rugenstein, A. Galy, H. J. Hassenruck-Gudipati and J.-M. Chang, Nat. Geosci., 2021, 14, 211–216 CrossRef CAS.
- C. S. Cockell, T. Bush, C. Bryce, S. Direito, M. Fox-Powell, J. P. Harrison, H. Lammer, H. Landenmark, J. Martin-Torres and N. Nicholson, Astrobiology, 2016, 16, 89–117 CrossRef PubMed.
- M. H. Han, D. Kim, S. Kim, S.-H. Yu, D. H. Won, B. K. Min, K. H. Chae, W. H. Lee and H.-S. Oh, Adv. Energy Mater., 2022, 12, 2201843 CrossRef CAS.
- W. H. Lee, Y.-J. Ko, Y. Choi, S. Y. Lee, C. H. Choi, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Nano Energy, 2020, 76, 105030 CrossRef CAS.
- Y.-J. Ko, J.-Y. Kim, W. H. Lee, M. G. Kim, T.-Y. Seong, J. Park, Y. Jeong, B. K. Min, W.-S. Lee, D. K. Lee and H.-S. Oh, Nat. Commun., 2022, 13, 2205 CrossRef CAS PubMed.
- J.-L. You, G.-C. Jiang, H.-Y. Hou, H. Chen, Y.-Q. Wu and K.-D. Xu, J. Raman Spectrosc., 2005, 36, 237–249 CrossRef CAS.
- A. Osipov, L. Osipova and R. Zainullina, Int. J. Spectrosc., 2015, 2015, 572840 Search PubMed.
- W. W. Rudolph, D. Fischer and G. Irmer, Appl. Spectrosc., 2006, 60, 130–144 CrossRef CAS PubMed.
- Y. Ma, W. Yan, Q. Sun and X. Liu, Geosci. Front., 2021, 12, 1018–1030 CrossRef CAS.
- M. Moradzaman and G. Mul, ChemElectroChem, 2021, 8, 1478–1485 CrossRef CAS.
- J. H. M. Visser, Cem. Concr. Res., 2018, 105, 18–30 CrossRef CAS.
- A. N. Palmer and M. V. Palmer, Speleogenesis and Evolution of Karst Aquifers, 2003, vol. 1, pp. 1–8 Search PubMed.
- B. Lagerblad and J. Trägårdh, Conceptual model for concrete long time degradation in a deep nuclear waste repository, Swedish Nuclear Fuel and Waste Management Co., Sweden, 1994 Search PubMed.
- R. B. Figueira, R. Sousa, L. Coelho, M. Azenha, J. M. de Almeida, P. A. S. Jorge and C. J. R. Silva, Constr. Build. Mater., 2019, 222, 903–931 CrossRef CAS.
- S.-Y. Pan, E. Chang and P.-C. Chiang, Aerosol Air Qual. Res., 2012, 12, 770–791 CrossRef CAS.
- M. Santoro, F. Gorelli, J. Haines, O. Cambon, C. Levelut and G. Garbarino, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7689–7692 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00448e |
| ‡ All authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |