
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-solvation electrolytes for ultra-stable calcium-ion storage†
Junjun
Wang‡
ab,
Ruohan
Yu‡
a,
Yalong
Jiang
a,
Fan
Qiao
a,
Xiaobin
Liao
 a,
Jianxiang
Wang
a,
Meng
Huang
a,
Fangyu
Xiong
a,
Lianmeng
Cui
a,
Yuhang
Dai
a,
Lei
Zhang
a,
Qinyou
An
*ac,
Guanjie
He
*b and
Liqiang
Mai
*ac
a,
Jianxiang
Wang
a,
Meng
Huang
a,
Fangyu
Xiong
a,
Lianmeng
Cui
a,
Yuhang
Dai
a,
Lei
Zhang
a,
Qinyou
An
*ac,
Guanjie
He
*b and
Liqiang
Mai
*ac
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China. E-mail: mlq518@whut.edu.cn; anqinyou86@whut.edu.cn
bChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk
cHubei Longzhong Laboratory, Wuhan University of Technology (Xiangyang Demonstration Zone), Xiangyang 441000, Hubei, China
First published on 30th July 2024
Abstract
Calcium-ion batteries (CIBs) have potential as electrochemical energy storage devices due to the low redox potential of Ca2+/Ca and the abundant reserves of Ca. However, the unsatisfactory calcium storage performance of electrode materials limits the development of CIBs. Here, we propose a design principle of high-solvation electrolytes to achieve ultra-stable calcium-ion storage. In high-solvation electrolytes, the decomposition of TFSI− ions and the formation of a CaF2-rich cathode electrolyte interface with Ca2+ insulation can be suppressed. With this electrolyte, Na2V6O16·2.9H2O shows a high discharge capacity of 240.7 mA h g−1 at 20 mA g−1 and an ultra-long life of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (over 600 days) at 1000 mA g−1. A three-dimensionally reduced graphene oxide aerogel and (NH4)2V6O16·1.5H2O also exhibit a long life of 6000 cycles and 9000 cycles, respectively. These materials have the longest cycle life among reported materials so far in CIBs. This work endows the electrode materials with ultra-stable calcium-ion storage and provides a design principle of electrolytes for cathode materials in CIBs.
000 cycles (over 600 days) at 1000 mA g−1. A three-dimensionally reduced graphene oxide aerogel and (NH4)2V6O16·1.5H2O also exhibit a long life of 6000 cycles and 9000 cycles, respectively. These materials have the longest cycle life among reported materials so far in CIBs. This work endows the electrode materials with ultra-stable calcium-ion storage and provides a design principle of electrolytes for cathode materials in CIBs.
Broader contextThe reserves of raw materials play a crucial role in whether electrochemical energy storage devices can be used for large-scale energy storage. Due to the low redox potential of Ca2+/Ca (−2.87 V vs. SHE close to that of Li+/Li) and the abundant reserves of Ca in the earth's crust, Ca-ion batteries (CIBs) have received widespread attention from researchers as potential electrochemical energy storage devices. However, the unsatisfactory Ca storage performance of electrode materials limits the development of CIBs. In this work, we studied for the first time the correlation between the solvation structure of electrolytes and the performance of cathode materials in CIBs. The results indicated that the decomposition of TFSI− ions and the formation of the CaF2-rich cathode electrolyte interface with Ca2+ insulation can be suppressed in a high-solvation Ca electrolyte. Finally, we proposed a design principle of Ca electrolytes for cathode materials with ultra-stable calcium-ion storage. |
Introduction
The low reserves of raw materials (such as Li, Ni, and Co) for lithium-ion batteries (LIBs) make them unsuitable for large-scale energy storage, which motivates researchers to develop new electrochemical energy storage systems based on abundant materials, such as sodium-ion, potassium-ion, multivalent-ion batteries, etc. Since each multivalent-ion can transfer two or three electrons, multivalent-ion batteries are expected to be energy storage devices with higher energy density and lower cost.1–3 In addition, compared with Li, Na or K metal anodes, Mg and Ca metal anodes are less likely to form dendrites, which can effectively improve the safety of batteries.4 Therefore, multivalent-ion batteries are considered potential candidates for post-LIBs.Among multivalent-ion (Al3+, Zn2+, Mg2+, and Ca2+) batteries, Ca2+/Ca has the lowest redox potential of −2.87 V vs. standard hydrogen electrode (SHE), which is close to that of Li+/Li (−3.04 V vs. SHE). Therefore, calcium-ion batteries (CIBs) can achieve higher working voltage and energy density among these multivalent-ion batteries. In addition, Ca2+ has the lowest charge density, Ca metal tends to be uniformly deposited/stripped, and Ca is the fifth most abundant element, meaning that CIBs have good rate performance, low cost, and high safety. However, the unsatisfactory calcium storage performance of electrode materials, caused by the large radius and divalent nature of Ca2+ and the decomposition of electrolytes, limits the development of CIBs. In the past two decades, considerable efforts have been made to develop electrolytes compatible with Ca metal anodes, and some breakthroughs have been made, such as Ca(BF4)2/ethylene carbonate:propylene carbonate,5 Ca(BH4)2/tetrahydrofuran6 and Ca[B(hfip)4]2/glyme (DME).7 Recently, Zhang et al. realized the compatibility of Ca(TFSI)2-based electrolytes with the Ca metal anode by regulating the solvation structure of Ca2+.8 Unfortunately, these electrolytes are poorly compatible with cathode materials. Ca(TFSI)2, as a commercially available and highly compatible salt, has been used in ∼50% of the reported cathode materials for CIBs. However, most electrode materials exhibited poor cycling performance or low specific capacity (Table S1, ESI†). The electrolyte solvation structure has been shown to have significant impacts on the cathode materials’ performance in monovalent ion (Li+/Na+/K+) and multivalent-ion (Mg2+/Zn2+) batteries.3,9–12 However, the correlation between the solvation structure of electrolytes and the performance of cathode materials in CIBs is unclear. Therefore, it is of great significance to study the correlations between the solvation structure of Ca2+ and the cathode materials’ performance in Ca(TFSI)2-based electrolytes and propose a design principle of Ca electrolytes for ultra-stable calcium-ion storage.
Here, we regulate the solvation structure of Ca2+ in Ca(TFSI)2-based electrolytes by using different solvation power solvents, including DME, diglyme (G2), and tetraglyme (G4). Classical molecular dynamics (CMD) simulation and Raman spectrum data indicate that the solvation degree of Ca2+ increases with the solvent from DME to G2 and G4. With a high-solvation and well-compatible Ca(TFSI)2/G4 electrolyte, Na2V6O16·2.9H2O (NVO) shows an ultra-long life of 20000 cycles at room temperature and 60000 cycles (over 600 days) at 50 °C. A three-dimensional reduced graphene oxide (3D rGO) aerogel and (NH4)2V6O16·1.5H2O (NHVO) also exhibit a long life of 6000 and 9000 cycles, respectively. The formation and composition of the cathode electrolyte interphase (CEI) in CIBs were first studied by ex situ transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). The results demonstrate that the excellent cycling performance of the electrode materials benefits from minimizing the decomposition of the TFSI− ion and preventing the formation of the CaF2-rich CEI in the high-solvation Ca(TFSI)2/G4 electrolyte. In addition, taking NVO as a representative, the differences in the calcium storage mechanism and electrolyte kinetic behavior in different solvation electrolytes were studied through in situ X-ray diffraction (XRD), in situ attenuated total reflectance Fourier-transform infrared (ATR-FTIR), and 3D tomography reconstruction technologies. And, the crystal structure and Ca2+ storage site of NVO were observed for the first time at the atomic scale by double aberration correction TEM.
Results and discussion
Calcium electrolyte design principles
The design principles of Ca(TFSI)2-based electrolytes are summarized in Fig. 1a–c. In low-solvation electrolytes (Fig. 1a), the primary solvation sheath of Ca2+ contains TFSI− ions, and the TFSI− ions are reduced during the discharge process,10 leading to CaF2-rich CEI formation and electrolyte consumption. In addition, CaF2 is unfavorable for the conduction of Ca2+.13 Therefore, electrode materials may exhibit poor cycling performance in low-solvation electrolytes due to the continuous consumption of electrolytes and the formation of the CaF2-rich CEI. In high-solvation electrolytes (Fig. 1b), the TFSI− ions exist as solvent-separated ion pairs (SSIPs) or free ions. The TFSI− ions do not exist in the primary solvation sheath of Ca2+ and do not migrate to the cathode side during discharge. As a result, the reduction of TFSI− ions at the electrode interface is minimized during the discharge process, which prevents the formation of the CaF2-rich CEI and the consumption of electrolyte, further enhancing the cycling performance of electrode materials. As shown in Fig. 1c, the proposed design principle of Ca(TFSI)2-based electrolytes is to inhibit the reduction of TFSI− by utilizing high-solvating power solvents. | ||
| Fig. 1 Design principles and physicochemical characterization of Ca(TFSI)2-based electrolytes. (a) In the low-solvation electrolytes, the reduction of TFSI− ions leads to constant consumption of electrolytes and the formation of CaF2-rich CEI on the electrode surface. (b) In the high-solvation electrolytes, the reduction of TFSI− ions is inhibited. (c) The design principle of Ca(TFSI)2-based electrolyte is to minimize the reduction of TFSI− ions: select high solvating power solvents to increase the solvation degree of electrolytes. (d)–(f) Gaussian calculated electrostatic potentials (φ) of different solvents and the corresponding binding energies between Ca2+ and the solvent: (d) DME, (e) G2, and (f) G4. (g)–(i) CMD simulation of Ca(TFSI)2-based electrolytes: snapshots of CMD simulations of 0.3 M Ca(TFSI)2 in (g) DME, (h) G2 and (i) G4. (j) The coordination numbers of Ca2+ in different electrolytes. (k) Raman spectra of Ca(TFSI)2 salt and Ca(TFSI)2 in different solvents. | ||
To illustrate the correlation between solvents with different solvating powers and the solvation structure of Ca2+ in Ca(TFSI)2-based electrolytes, three ether solvents with different chain lengths (DME, G2, and G4) are considered. Gaussian calculated electrostatic potentials (φ) of different solvents and the corresponding binding energies between Ca2+ and solvents are shown in Fig. 1d–f and Fig. S1 (ESI†). The results indicate that the solvent molecules show high negative charge densities near oxygen atoms, and these oxygen atoms may coordinate with Ca2+ in electrolytes. The binding energies between Ca2+ and solvent oxygen atoms increase with solvent from DME, G2 to G4, and the minimum formation energies are −3.97, −4.52, and −5.6 eV, respectively. The results indicate that G4 has a higher solvating power. CMD simulations were performed to study the solvation structure and the coordinated numbers (CNs) of Ca2+ in Ca(TFSI)2/DME, Ca(TFSI)2/G2 and Ca(TFSI)2/G4 electrolytes (Fig. 1g–j and Fig. S2 and S3, ESI†). The results (Fig. 1g–i and Fig. S2, ESI†) illustrate that the primary solvation sheath of Ca2+ in Ca(TFSI)2/G4 does not contain TFSI−. In addition, we calculated the CNs of Ca2+ in different electrolytes (Fig. 1j) based on the radial distribution functions (RDF) (Fig. S3, ESI†). The CNs of TFSI− decrease as the solvent changed from DME, G2 to G4. Almost no TFSI− are present in the primary solvation sheath of Ca2+ in Ca(TFSI)2/G4, indicating that Ca(TFSI)2/G4 is a high-solvation electrolyte. Raman spectra (Fig. 1k) of different electrolytes are used to analyze TFSI−–Ca2+ interactions due to the TFSI− complex breathing mode located at 735–755 cm−1 and the 740 cm−1 mode corresponding to SSIPs or free TFSI− ions (without Ca2+ coordination).8,14,15 The results suggest that almost no TFSI−–Ca2+ interactions are found in Ca(TSFI)2/G4, which is well consistent with the results of CMD simulations. The Fourier transform infrared (FTIR) spectra of different electrolytes are shown in Fig. S4 (ESI†). The vibrational features in the range of 1300–1400 cm−1 correspond to the asymmetric –SO2 stretching bands in TFSI− ions, which are usually used to observe ion–ion interactions.16–18 In the Ca(TFSI)2/G4 electrolyte, the peak shape of the asymmetric –SO2 stretching bands is consistent with the vibrational peaks of free TFSI− ions in Zn(TFSI)2 and LiTFSI,17,18 indicating the presence of only free TFSI− ions in the Ca(TFSI)2/G4 electrolyte. The analysis of the FTIR spectra further confirms the results of the Raman spectra. Cu//activated carbon cloth (ACC) batteries with different electrolytes were assembled to illustrate the effects of solvent molecules on the decomposition of TFSI−. Cyclic voltammetry (CV) curves of Cu//ACC at 0.1 mV s−1 are shown in Fig. S5 (ESI†). In Ca(TFSI)2/DME and Ca(TFSI)2/G2, the reduction peaks are observed in the range from −0.8 to −0.5 V vs. ACC, corresponding to the decomposition of TFSI−.8 Interestingly, the reduction peak is absent in Ca(TFSI)2/G4. The result indicates that the decomposition of TFSI− can be effectively inhibited in the Ca(TFSI)2/G4 electrolyte. In addition, the results of linear sweep voltammetry (LSV) demonstrate that the Ca(TFSI)2/G4 electrolyte has high oxidation stability (Fig. S6, ESI†).
Calcium storage performance in high-solvation electrolytes
The above results demonstrate that TFSI− is reduced in low-solvation electrolytes during discharge. However, further verification is needed to determine whether the reduction of TFSI− will affect the performance of electrode materials. NVO with large layer spacing and Na+ pillars was synthesized by hydrothermal method (Fig. S7, ESI†) and used as a cathode material to study the effect of the solvation structure of Ca2+ on the Ca2+ storage performance. The XRD pattern of NVO (Fig. 2a) can be well indexed to the PDF card of Na2V6O16·3H2O (PDF#: 00-016-0601), and the scanning electron microscopy (SEM) image shows that NVO is ultra-long nanowires (Fig. S8, ESI†). The high magnification high-angle annular dark-filed (HAADF) scanning transmission electron microscopy (STEM) images of NVO are shown in Fig. 2b and c and Fig. S9 (ESI†). The crystal structure of NVO was observed for the first time at the atomic scale. The bright dots in the HAADF-STEM images are assigned to heavy V atoms, and the position of the V atoms is consistent with the crystal structure of NVO along a and b directions (Fig. 2b and Fig. S9, ESI†). The HAADF-STEM image of NVO further indicates that NVO is a layered structure material with a large layer spacing of 0.79 nm, corresponding to the (001) crystal plane of NVO (Fig. 2c). Some other characterization techniques for NVO are shown in Fig. S10–S12 (ESI†). The results indicate that the valence state of V in NVO is +5 and each NVO contains 2.9 H2O molecules.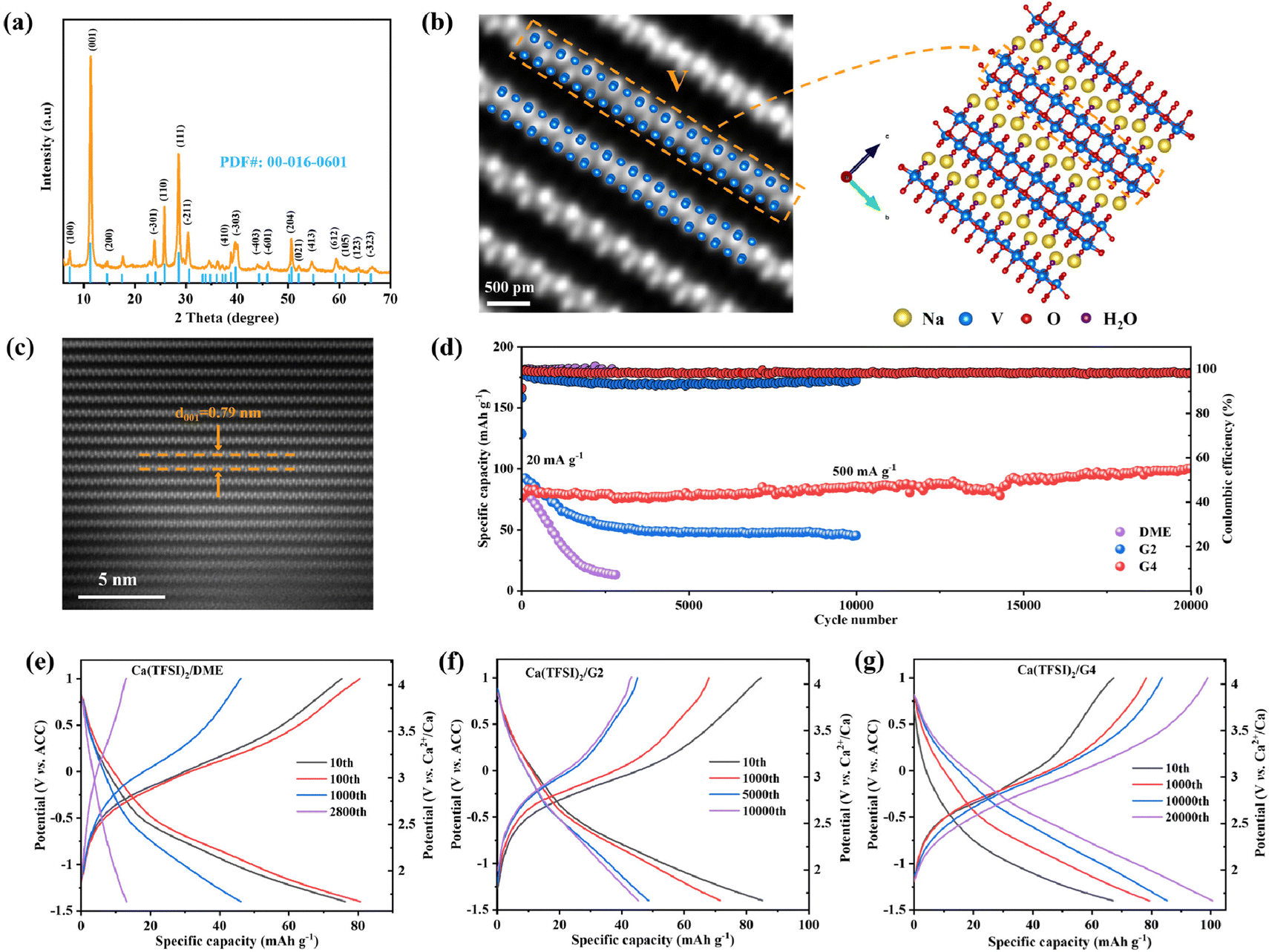 | ||
| Fig. 2 Calcium storage performance of Na2V6O16·2.9H2O in different electrolytes. (a) XRD pattern of NVO. (b) and (c) HAADF-STEM images of NVO and corresponding crystal structures along a direction. (d) Cycling performance of NVO with different electrolytes at 500 mA g−1 and room temperature after five cycles at 20 mA g−1 (the loading of the active material is ∼1.1 mg cm−2). (e)–(g) Charge/discharge profiles as a function of cycle number of NVO using (e) Ca(TFSI)2/DME, (f) Ca(TFSI)2/G2, and (g) Ca(TFSI)2/G4 electrolytes. | ||
Since the Ca(TFSI)2/DME electrolyte is not compatible with the Ca metal,14 ACC is selected as the counter electrode with a stable potential of 3.068 V vs. Ca2+/Ca (Fig. S13, ESI†).19 The Ca2+ storage performance of NVO in three different electrolytes was characterized at room temperature (Fig. 2d). The cell with Ca(TFSI)2/DME shows poor cycling performance, including rapid capacity fading by 84.0% after 2800 cycles and increased cell overpotential during cycling (Fig. 2e). The cell in the Ca(TFSI)2/G2 electrolyte exhibits improved cycling performance, but capacity fading and increased cell overpotential are still observed (Fig. 2f). The capacity retention of NVO is 48.5% after 10000 cycles in the Ca(TFSI)2/G2 electrolyte. In contrast, the cell with a high-solvation Ca(TFSI)2/G4 electrolyte shows a long-life of 20000 cycles and the capacity increases gradually. The overpotential of the cell gradually decreases during 20000 cycles as shown in Fig. 2g with a high Coulombic efficiency (CE). In addition, in the Ca(TFSI)2/G4 electrolyte, NVO can also exhibit a high discharge capacity of 240 mA h g−1 and excellent cycling performance at low current density (Fig. S14, ESI†). The high calcium storage capacity of NVO has also been verified in a three-electrode system (Fig. S15, ESI†). The reasons for the capacity increase and overpotential decrease during the cycle will be discussed in detail later. Combining the CMD simulation, Raman spectroscopy and FTIR spectroscopy analysis results of electrolytes, the electrochemical performance testing indicates that electrolytes with a high proportion of SSIPs or free TFSI− ions are conducive to achieving long cycling lifetimes of electrode materials.
Cathode electrolyte interface components in different solvation electrolytes
The above electrochemical performance tests have shown that NVO has the best cycling performance in high-solvation Ca(TFSI)2/G4 electrolyte, whether this is related to suppressed reduction of TFSI− ions needs to be further studied. The composition and properties of the CEI layer on the cycled NVO electrode surface with different electrolytes were analyzed by XPS and TEM. In low-solvation Ca(TFSI)2/DME and Ca(TFSI)2/G2, the reduction of TFSI− induces the formation of inorganic-rich CEI, including CaF2 and S-based compounds (Fig. 3a and b and Fig. S16a and b, ESI†). In sharp contrast, in high-solvation Ca(TFSI)2/G4, the peak corresponding to CaF2 is very weak even after 210 s Ar+ sputtering (Fig. 3c) and the sulfur-based compounds are not observed (Fig. S16c, ESI†), which indicates that the decomposition of TFSI− ions can be significantly suppressed in a high-solvation electrolyte. | ||
| Fig. 3 Characterization of the cathode electrolyte interface components. (a)–(c) F 1s XPS spectra of the cycled NVO with different sputtering times in (a) Ca(TFSI)2/DME, (b) Ca(TFSI)2/G2, and (c) Ca(TFSI)2/G4 electrolytes. (d) STEM image and (e) HAADF-STEM image with corresponding elemental maps of the cycled NVO in Ca(TFSI)2/DME. (f) EDX spectra corresponding to the selected area in (e). (g) and (h) STEM image of the cycled NVO in (g) Ca(TFSI)2/G2 and (h) Ca(TFSI)2/G4. | ||
The STEM image for the cycled NVO electrode in Ca(TFSI)2/DME (Fig. 3d and Fig. S17, ESI†) demonstrates the presence of a 2.3 nm thick CEI on the surface of NVO. The HAADF-STEM image and elemental maps show that the edge of NVO contains more F, Ca, and C elements (Fig. 3e). The EDX spectra (Fig. 3f) additionally indicate the formation of CaF2-rich CEI in Ca(TFSI)2/DME. The cycled NVO STEM image in Ca(TFSI)2/G2 shows a thinner CEI of 1.3 nm (Fig. 3g). In contrast, in high-solvation Ca(TFSI)2/G4 electrolyte, almost no CEI can be observed on the surface of NVO, which also confirms that the decomposition of TFSI− and the formation of CaF2-rich CEI can be suppressed in the Ca(TFSI)2/G4 electrolyte. In addition, the stable NVO in these electrolytes verifies that the fading of capacity in the low solvation-electrolytes is not caused by the loss of active material (Fig. S18, ESI†). XPS and TEM results show that the cycling performance of NVO is closely related to the solvation structure of Ca2+ and the inhibition of TFSI− decomposition is beneficial for electrode materials to achieve stable calcium-ion storage.
High-temperature performance and compatibility of high-solvation electrolytes
To demonstrate the high-temperature performance and compatibility of the Ca(TFSI)2/G4 electrolyte, the calcium storage performances of NVO and NHVO at 50 °C and the 3D rGO aerogel at room temperature were evaluated. NVO shows a high discharge capacity of 210.7 mA h g−1 without capacity fading after 100 cycles at 100 mA g−1 (Fig. 4a and Fig. S19, ESI†) and exhibits a capacity of 148.0 mA h g−1 at 1000 mA g−1 (Fig. S20, ESI†). It is impressing that NVO can maintain a capacity of 110.9 mA h g−1 with a capacity retention of 75% after 60000 cycles (over 600 days, Fig. 4b). To the best of our knowledge, the 60000 cycles in CIBs are the longest. In addition, when the test temperature turns from 50 °C to 25 °C and back to 50 °C, NVO can still maintain stable cycling performance. Rate performance tests show that NVO has a capacity of 59.1 mA h g−1 at 3000 mA g−1 (Fig. S21, ESI†). The effects of interlayer H2O on the calcium storage performance and the reaction kinetics in Ca(TFSI)2/G4 are additionally investigated (Fig. S22–S23, ESI†). The results demonstrate that the interlayer H2O molecules play an important role in achieving excellent calcium storage performance, and the average diffusion rate Ca2+ in the NVO electrode is 2.8 × 10−11 cm2 s−1.
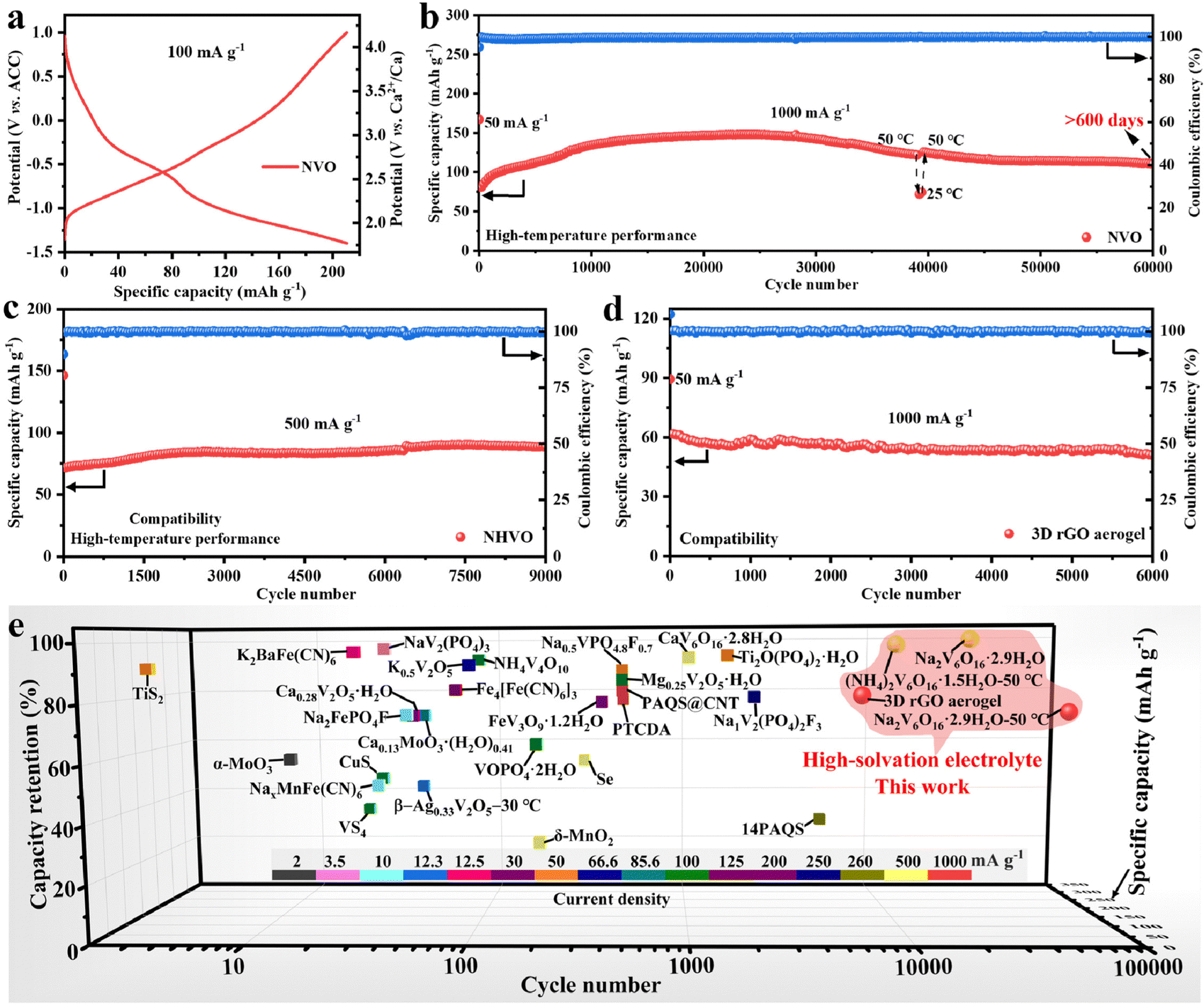 | ||
| Fig. 4 High-temperature performance and compatibility of high-solvation electrolytes. (a) Galvanostatic charge/discharge profiles of NVO at 100 mA g−1 and 50 °C. (b)–(d) Cycling performances of (b) NVO at 1000 mA g−1 and 50 °C, (c) NHVO at 500 mA g−1 and 50 °C and (d) 3D rGO aerogel at 1000 mA g−1. (e) Comparison of the Ca2+ storage performance of the 3D rGO aerogel, NHVO, and NVO with reported cathode materials for CIBs in organic electrolyte.20–45 | ||
In addition, NHVO exhibits a capacity of 71 mA h g−1 and a long-life of 9000 cycles at 500 mA g−1 (Fig. 4c and Fig. S24, ESI†). The 3D rGO aerogel delivers a capacity of 62 mA h g−1 and 6000 cycles with 81.5% capacity retention at 1000 mA g−1 (Fig. 4d and Fig. S25, ESI†). The results indicate that NHVO and 3D rGO aerogel can also exhibit excellent cycling stability in Ca(TFSI)2/G4, meaning that Ca(TFSI)2/G4 has good compatibility with electrode materials. To assess the compatibility of the high-solvation Ca(TFSI)2/G4 electrolyte with the Ca metal anode, the performance of NVO‖Ca and Ca‖Ca cells in the Ca(TFSI)2/G4 electrolyte was evaluated. The observed poor electrochemical performance suggests that the Ca(TFSI)2/G4 electrolyte is not compatible with the Ca metal anode (Fig. S26, ESI†). XPS depth profile analysis indicates that the SEI on the surface of the cycled Ca metal anode contains CaF2 and CaO (Fig. S27, ESI†), which have been shown to have high diffusion barriers for Ca2+ ions.46 The poor compatibility could be attributed to the particularly strong and well-defined G4:Ca2+ interactions.14,47 Finally, the electrochemical performances of 3D rGO aerogel, NHVO, and NVO are compared with the reported cathode materials in organic electrolytes (Fig. 4e). Among the reported cathode materials, 3D rGO aerogel, NHVO, and NVO all exhibit the longest cycle life and high-capacity retention. In particular, NVO exhibits an ultra-long life of 60000 cycles (more than 600 days) and a capacity retention of 75%, which further suggests that high-solvation Ca(TFSI)2-based electrolytes may be the best choice for ultra-stable calcium-ion storage.
A Ca-ion full cell (NVO‖CaxTi3C2) with the Ca(TFSI)2/G4 electrolyte was assembled. The NVO‖CaxTi3C2 full cell has a discharge capacity of 69.1 mA h g−1 (Fig. S28a, ESI†) and 60 cycles life at 20 mA g−1 and room temperature (Fig. S28b, ESI†), which indicates that the potential application of NVO cathode materials and Ca(TFSI)2/G4 electrolyte in CIBs. To further underscore the potential of NVO in Ca–metal batteries, a NVO‖Ca[B(hfip)4]2/DME‖Ca battery was assembled and exhibits a 174.7 mA h g−1 discharge capacity (Fig. S29a, ESI†), affirming the viability of NVO in Ca–metal batteries. However, after 10 cycles, the capacity dwindled to 50.7 mA h g−1 (Fig. S29b, ESI†). This decline is attributed to the insufficient compatibility between Ca[B(hfip)4]2/DME and the Ca–metal anode.48 Therefore, enhancing the compatibility between the electrolyte and the Ca metal is pivotal for advancing CIBs, and it will be a primary focus of our forthcoming endeavors. Moreover, in the Ca-system without using a Ca metal anode, a graphite anode with the low working voltage may be a suitable option to enhance the working voltage of Ca-ion full cells.49 However, the low specific capacity of the graphite anode limits its commercial application. Therefore, it is also crucial to further improve the CE and specific capacity of the graphite anode through the electrolyte solvation structure regulation or material modification in the future work.
Storage and activation mechanism in high-solvation electrolytes
In situ XRD was used to investigate the Ca2+ storage mechanism of NVO. In Ca(TFSI)2/G4, the (001) diffraction peak of NVO shifts to high angle during the discharge process, indicating that the interlayer spacing of NVO decreases (Fig. 5a). Meanwhile, the (−301), (110) and (−211) diffraction peaks of NVO shift to low angle, meaning the increase of a and b. During the charge process, all the diffraction peaks shift reversely and return to the initial position at the end, which indicates that NVO has good structural reversibility and the calcium storage mechanism of NVO is a single-phase solid solution reaction in the Ca(TFSI)2/G4 electrolyte. Interestingly, the shift of the diffraction peaks is not monotonic in the Ca(TFSI)2/G4 electrolyte. In stage I, the (111) and (−211) diffraction peaks shift significantly, while the (−301) and (110) diffraction peaks shift slightly. In stage II, the (111) and (−211) diffraction peaks shift slightly, and the (−301) and (110) diffraction peaks shift significantly. During the charge process, two similar stages are observed. However, the changes of these diffraction peaks in the Ca(TFSI)2/G2 electrolyte are different (Fig. S30, ESI†), which may be due to the presence of solvation co-insertion in high-solvation Ca(TFSI)2/G4 electrolyte. The research on solvation co-insertion will be further discussed later.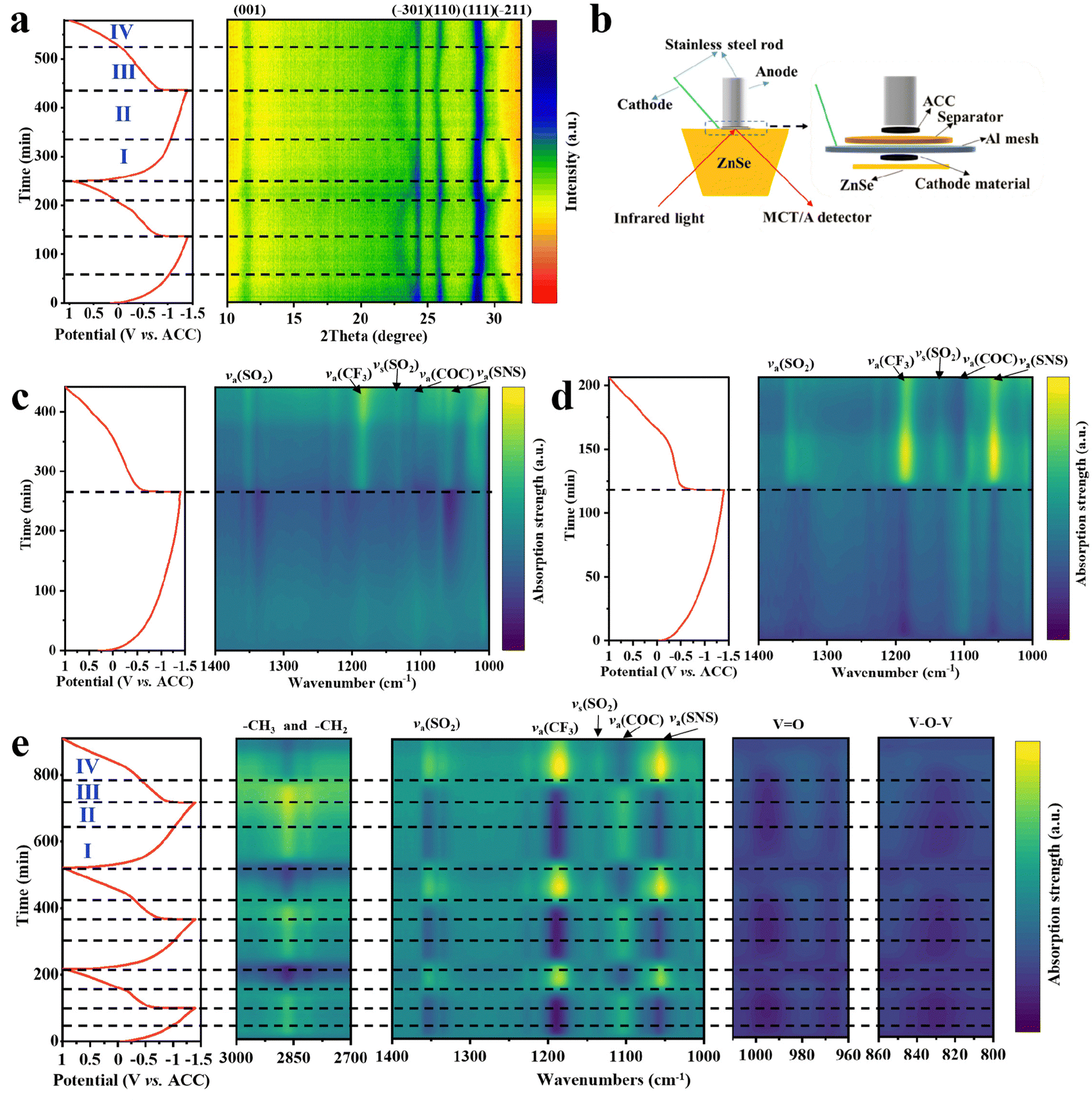 | ||
| Fig. 5 Calcium storage mechanisms of NVO in high-solvation electrolytes. (a) In situ XRD patterns of NVO and the corresponding charge/discharge profiles in the Ca(TFSI)2/G4 electrolyte. (b) The configuration of the cell for in situ ATR-FTIR tests. (c)–(e) In situ ATR-FTIR spectra of NVO and the corresponding charge/discharge curves in (c) Ca(TFSI)2/DME, (d) Ca(TFSI)2/G2 and (e) Ca(TFSI)2/G4 electrolytes. All in situ tests were performed at room temperature. | ||
The dynamic behaviors of electrolytes on NVO electrodes were studied by in situ ATR-FTIR spectroscopy. The configuration of the cell for in situ ATR-FTIR tests is shown in Fig. 5b, and the FTIR spectra of different solvents and electrolytes are shown in Fig. S31 (ESI†). In situ ATR-FTIR spectra of NVO in Ca(TFSI)2/DME (Fig. 5c) indicate that the intensity of the corresponding TFSI− absorption peaks (including va(SO2), va(CF3), vs(SO2) and va(SNS)) gradually weakens50,51 and the intensity of the corresponding DME solvent absorption peak (va(COC))52 increases during discharge, which is due to the migration of TFSI− to the anode and the desolvation of Ca2+ at the interface of the cathode. The reverse peak evolution is observed during charge. In Ca(TFSI)2/G2, similar changes of the absorption peaks were observed, and the more pronounced intensity change is due to the stronger interaction between G2 and Ca2+ (Fig. 5d). In Ca(TFSI)2/G4, the observed absorption peak changes are different from that of other two electrolytes (Fig. 5e). Two distinct stages are observed during discharge and charge, respectively, which are similar to the changes in the in situ XRD pattern (Fig. 5a). It is particularly noteworthy that the intensity of the corresponding G4 solvent absorption peaks, including C–O–C, –CH3 and –CH2, remain unchanged at stage III, which may be caused by G4 solvent co-extraction during charge, corresponding to the presence of solvent co-insertion during discharge. However, in situ ATR-FTIR spectra of NVO for the high wavenumber regions (Fig. S32, ESI†) indicate that there is no solvent co-insertion phenomenon in Ca(TFSI)2/DME and Ca(TFSI)2/G2 electrolytes. In addition, in situ ATR-FTIR spectra reveal the reversible shifts of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and V–O–V absorption bands, further demonstrating the good structural reversibility of NVO. 1D in situ ATR-FTIR spectra of NVO for all 2D plots are additionally shown in Fig. S33–S35 (ESI†).
O and V–O–V absorption bands, further demonstrating the good structural reversibility of NVO. 1D in situ ATR-FTIR spectra of NVO for all 2D plots are additionally shown in Fig. S33–S35 (ESI†).
Ex situ FTIR, inductively coupled plasma atomic emission spectroscopy (ICP-AES), TEM and 3D visualization of tomographic reconstruction techniques were performed to reveal the solvent co-insertion phenomenon and the activation process mechanism during the initial cycles. The ex situ FTIR spectra of G4 solvent and NVO in Ca(TFSI)2/G4 electrolyte show that the absorption peak of G4 is absent at −0.8 V and appears at −1.4 V (Fig. 6a), indicating the solvent co-insertion at low potential. The reversible process can be observed during the charge. The results further confirm the existence of two distinct stages during discharge or charge and solvent co-insertion, consistent with the in situ XRD and in situ ATR-FTIR results. However, the FTIR spectra of the discharged NVO in Ca(TFSI)2/DME and Ca(TFSI)2/G2 have no absorption peaks of solvents (Fig. 6b).
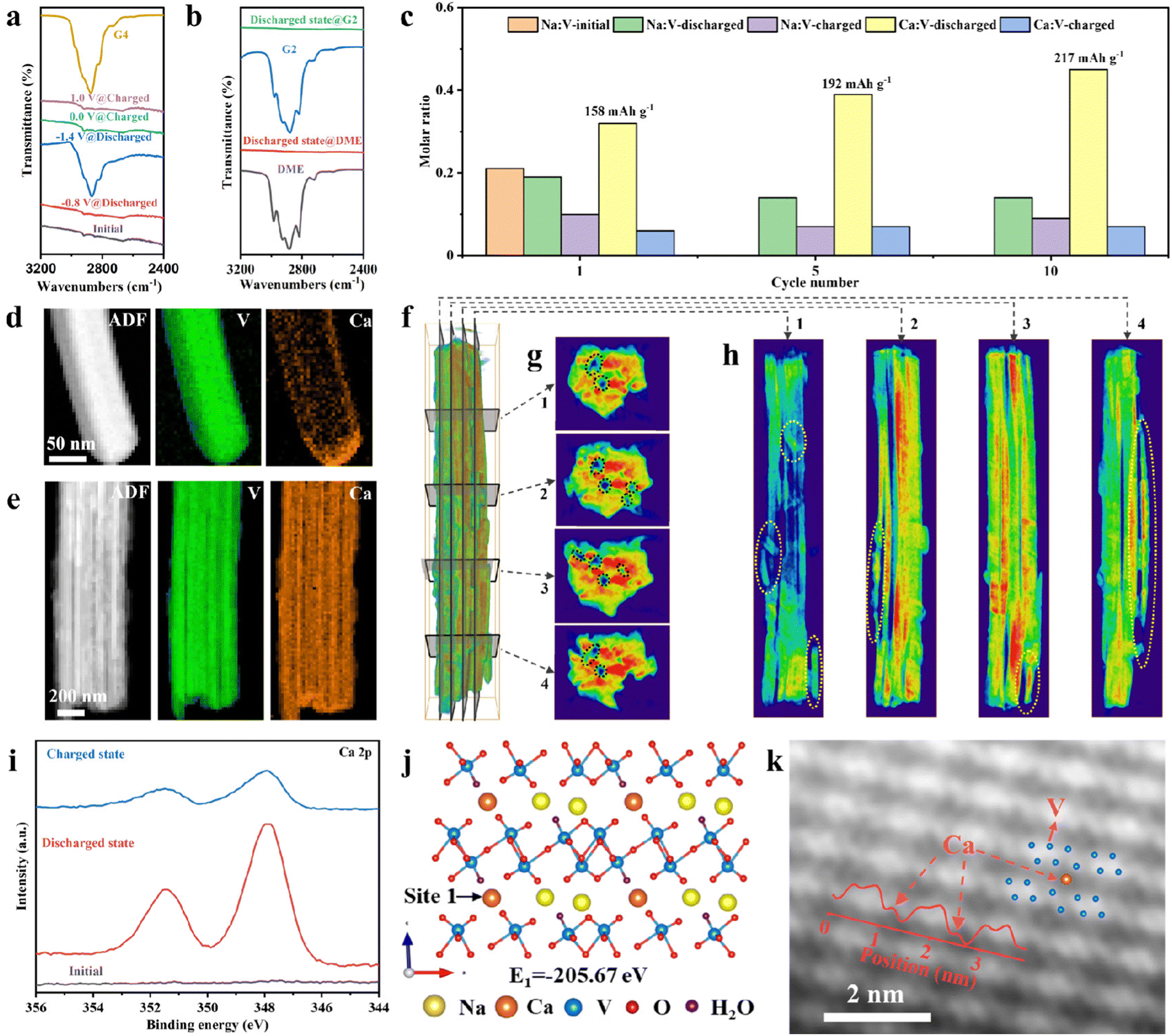 | ||
| Fig. 6 Characterization of solvent co-insertion, the activation mechanisms, and the storage sites of Ca2+ in high-solvation electrolytes. (a) FTIR spectra of G4 solvent and different states NVO in Ca(TFSI)2/G4 electrolyte. (b) FTIR spectra of DME, G2 solvents, discharged NVO in Ca(TFSI)2/DME and Ca(TFSI)2/G2 electrolytes. (c) ICP-AES data of NVO with different states and cycles in Ca(TFSI)2/G4 electrolyte. (d) and (e) ADF image of the fully discharged NVO and EELS mappings of V, Ca at (d) the first cycle and (e) the 10th cycle with Ca(TFSI)2/G4 electrolyte. (f)–(h) 3D visualization of tomographic reconstruction images of NVO after first 10 cycles in Ca(TFSI)2/G4 electrolyte. (i) Ex situ XPS spectra of Ca 2p for NVO at different states. (j) The Ca2+ storage sites (site 1) of NVO determined by DFT calculation. (k) HAADF-STEM images of fully discharged NVO and the inset is the intensity profiles for the corresponding position. | ||
The ICP-AES data of NVO electrodes at different states after different cycles (Fig. 6c) indicate that the amount of Ca2+ ions in NVO increases and a small amount of Na is de-inserted from NVO during the cycling process. At the 10th cycle, the molar ratio of Ca:V is 0.45, corresponding to a capacity of 217 mA h g−1, which is close to the discharge capacity of NVO (Fig. 4a). The ICP-AES data show that the increase in specific capacity is due to the increase in the amount of inserted Ca2+, so the reason why the inserted Ca2+ ions increase during cycling needs to be further revealed. The high spatial and energy resolution of electron energy-loss spectroscopy (EELS) mappings were used to analyze the distribution of Ca in NVO in different states. The ADF image of the discharged NVO and the corresponding EELS mappings of V and Ca (Fig. 6d), at the 1st cycle, reveal that element Ca accumulates at the ends of the nanowires, meaning that Ca2+ ions are inserted into nanowires from the ends. At the 10th cycle, the element Ca is evenly distributed in nanowires (Fig. 6e), further confirming the increase in the amount of inserted Ca2+ and the presence of an activation process. The 3D reconstruction and orthoslicing images of NVO nanowires (Fig. 6f–h and Fig. S36–S38, ESI†) after the first 10 cycles demonstrate that there are some voids (black circles) inside NVO nanowires and some nanowires are broken (yellow circles), which will increase the contact area between NVO and electrolyte. In addition, Ca2+ may be inserted from the end of nanowires, and broken nanowires are beneficial to increase the Ca2+ storage activity of NVO. The electrochemical impedance spectra (EIS) of NVO at different cycles (Fig. S39, ESI†) show a decrease in interfacial charge transfer resistance with cycling, which is also conducive to the insertion of Ca2+ ions. The rotational and orthogonal slicing processes are shown in Movies S1 and S2 (ESI†). Ex situ TEM images with different magnifications after 10 cycles also show that some NVO nanowires are broken (Fig. S40, ESI†). The above results suggest that the activation process is due to the fracture of NVO nanowires. The effect of NVO nanowires fracture on solvents insertion is studied. The EDX spectra of NVO with different states (initial state, 2nd discharged state, and 5th discharged state) indicate a gradual decrease in the atomic ratio of V to O (Fig. S41, ESI†), implying an increase in solvents insertion.49
The insertion/extraction and storage sites of Ca2+ were further investigated by ex situ XPS, density functional theory (DFT) computations, and atomic-resolution TEM. The ex situ Ca 2p XPS spectra of NVO (Fig. 6i) indicate that two strong peaks corresponding to the Ca 2p signal are observed for discharged NVO and the peak intensity of Ca 2p becomes weak for charged NVO, demonstrating the insertion/extraction of Ca2+ during discharge/charge. Some other characterizations, to prove the insertion of Ca2+ and the change of vanadium valence, are shown in Fig. S42–S46 (ESI†), including ex situ XPS, EELS, and TEM. Additionally, the vanadium K-edge X-ray absorption near-edge spectra (XANES) spectra of NVO at the initial state and fully discharged state show that the K-edge of the vanadium of the fully discharged NVO shifts to low energy (Fig. S47a, ESI†), indicating that V5+ is reduced during discharge. The corresponding Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra in R space indicate the V–O distance of initial NVO are 1.06 Å and 1.57 Å (Fig. S47b, ESI†), and the change of the bond length can be observed for the fully discharged NVO, which is due to the insertion of Ca2+ during discharge. DFT computation results show that there are two possible Ca2+ storage sites in NVO (named site 1 and site 2, Fig. 6j and Fig. S48, ESI†) and Ca2+ ions prefer to occupy site 1 to form a more thermodynamically stable structure with a lower formation energy of −205.67 eV (Fig. 6j). Double spherical aberration correction TEM was further used to study the insertion sites of Ca2+ ions. The HAADF-STEM images of fully discharged NVO and the intensity profiles for the corresponding position (Fig. 6k) confirm that Ca2+ ions are inserted into site 1, which is consistent with DFT calculations. The Ca2+ storage site of NVO is determined for the first time at the atomic scale. The feasibility of site 1 accommodating solvent co-insertion is further investigated through DFT calculations. The spatial size of site 1, shown in Fig. S49 (ESI†), indicates that one Ca2+ ion and one G4 molecule can be placed into site 1. After relaxation, the NVO structure with co-inserted Ca2+ ions and G4 solvent can exist stably (Fig. S50, ESI†). These results suggest that G4 solvent co-insertion into site 1 is theoretically possible. However, the extent of solvent insertion may require further experimental and theoretical studies.
Conclusions
In summary, we found that the decomposition of TFSI− ions and the formation of the CaF2-rich CEI can be suppressed in a high-solvation Ca(TFSI)2/G4 electrolyte, which can effectively alleviate the depletion of electrolyte and the irreversible capacity loss, endowing CIBs with excellent stability. With the high-solvation Ca(TFSI)2/G4 electrolyte, NVO as a cathode material for CIBs exhibits a high discharge capacity of 240.7 mA h g−1, an ultra-long life of 20000 cycles at room temperature and 60000 cycles (over 600 days) at 50 °C. In addition, the high-solvation Ca(TFSI)2/G4 electrolyte has good compatibility with other electrode materials. The phenomenon of co-insertion of solvents into the high-solvation Ca(TFSI)2/G4 electrolyte is revealed by in situ ATR-FTIR and ex situ FTIR. In situ XRD and ex situ TEM results show that the calcium storage mechanism of NVO is a single-phase solid solution reaction. The crystal structure of NVO and the Ca2+ storage site of NVO were determined for the first time at the atomic scale by double aberration correction TEM. These findings provide a Ca(TFSI)2-based electrolyte design principle by adjusting the solvent chemistry to enable the electrode materials for CIBs with ultra-long life and are expected to encourage more research to promote the development of CIBs.
Experimental section
Experimental details and characterization are included in the ESI.†Author contributions
L. M., Q. A. and G. H. proposed and supervised the research. J. W. designed the experimental method, performed electrochemical testing and data analysis, and wrote the first draft. F. Q. and J. W. performed material preparation and characterization. R. Y. performed characterization and analysis related to transmission electron microscopy. X. L. performed force-field based classical molecular dynamics simulations. Y. J. performed density functional theory calculations. M. H., F. X., L. C., Y. D., L. Z., G. H., Q. A., and L. M. contributed to the revision of the manuscript.Data availability
The data that support the findings of this study are included in the published article and its ESI.† These data are also available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52172231, 52127816, and 52202290), the National Key Research and Development Program of China (2020YFA0715000), the Engineering and Physical Sciences Research Council (EPSRC, EP/V027433/3), UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding (101077226; EP/Y008707/1), the Natural Science Foundation of Hubei Province (2022CFA087, and 2022CFD090) and the Sanya Science and Education Innovation Park of Wuhan University of Technology (2022KF0011). Junjun Wang is grateful for the funding support from China Scholarship Council/University College London for the joint PhD scholarship (CXXM2110070005). The project is supported by the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology).References
- M. E. Arroyo-de Dompablo, A. Ponrouch, P. Johansson and M. R. Palacin, Chem. Rev., 2020, 120, 6331–6357 CrossRef CAS PubMed
.
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H. M. Cheng, Nat. Chem., 2018, 10, 667–672 CrossRef CAS PubMed
- S. Hou, X. Ji, K. Gaskell, P. F. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed
- S. D. Pu, C. Gong, X. Gao, Z. Ning, S. Yang, J.-J. Marie, B. Liu, R. A. House, G. O. Hartley, J. Luo, P. G. Bruce and A. W. Robertson, ACS Energy Lett., 2020, 5, 2283–2290 CrossRef CAS
- A. Ponrouch, C. Frontera, F. Barde and M. R. Palacin, Nat. Mater., 2016, 15, 169–172 CrossRef CAS PubMed
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2018, 17, 16–20 CrossRef CAS PubMed
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC
- Z. Hou, R. Zhou, Y. Yao, Z. Min, Z. Lu, Y. Zhu, J. M. Tarascon and B. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202214796 CrossRef CAS PubMed
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J.-G. Zhang, Nat. Energy, 2022, 7, 718–725 CrossRef CAS
- P. Bai, X. Ji, J. Zhang, W. Zhang, S. Hou, H. Su, M. Li, T. Deng, L. Cao, S. Liu, X. He, Y. Xu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202202731 CrossRef CAS PubMed
- J. Li, Y. Hu, H. Xie, J. Peng, L. Fan, J. Zhou and B. Lu, Angew. Chem., Int. Ed., 2022, 61, e202208291 CrossRef CAS PubMed
- Q. Ma, R. Gao, Y. Liu, H. Dou, Y. Zheng, T. Or, L. Yang, Q. Li, Q. Cu, R. Feng, Z. Zhang, Y. Nie, B. Ren, D. Luo, X. Wang, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS PubMed
- J. Forero-Saboya, C. Davoisne, R. Dedryvère, I. Yousef, P. Canepa and A. Ponrouch, Energy Environ. Sci., 2020, 13, 3423–3431 RSC
- N. T. Hahn, D. M. Driscoll, Z. Yu, G. E. Sterbinsky, L. Cheng, M. Balasubramanian and K. R. Zavadil, ACS Appl. Energy Mater., 2020, 3, 8437–8447 CrossRef CAS
- D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A1489–A1500 CrossRef CAS
- A. Bakker, J. Lindgren and K. Hermansson, Polymer, 1996, 37, 1871–1878 CrossRef CAS
- Y. Zhang, G. Wan, N. H. C. Lewis, J. Mars, S. E. Bone, H.-G. Steinrück, M. R. Lukatskaya, N. J. Weadock, M. Bajdich, O. Borodin, A. Tokmakoff, M. F. Toney and E. J. Maginn, ACS Energy Lett., 2021, 6, 3458–3463 CrossRef CAS
- N. H. C. Lewis, Y. Zhang, B. Dereka, E. V. Carino, E. J. Maginn and A. Tokmakoff, J. Phys. Chem. C, 2020, 124, 3470–3481 CrossRef CAS
- X. Liu, G. A. Elia and S. Passerini, J. Power Sources Adv., 2020, 2, 100008 CrossRef
- M. S. Chae, H. H. Kwak and S.-T. Hong, ACS Appl. Energy Mater., 2020, 3, 5107–5112 CrossRef CAS
- M. S. Chae, A. Nimkar, N. Shpigel, Y. Gofer and D. Aurbach, ACS Energy Lett., 2021, 6, 2659–2665 CrossRef CAS
- S. Kim, L. Yin, M. H. Lee, P. Parajuli, L. Blanc, T. T. Fister, H. Park, B. J. Kwon, B. J. Ingram, P. Zapol, R. F. Klie, K. Kang, L. F. Nazar, S. H. Lapidus and J. T. Vaughey, ACS Energy Lett., 2020, 5, 3203–3211 CrossRef CAS
- R. Zhou, Z. Hou, Q. Liu, X. Du, J. Huang and B. Zhang, Adv. Funct. Mater., 2022, 32, 2200929 CrossRef CAS
- J. Wang, J. Wang, Y. Jiang, F. Xiong, S. Tan, F. Qiao, J. Chen, Q. An and L. Mai, Adv. Funct. Mater., 2022, 32, 2113030 CrossRef CAS
- C. Zuo, F. Xiong, J. Wang, Y. An, L. Zhang and Q. An, Adv. Funct. Mater., 2022, 32, 2202975 CrossRef CAS
- J. Wang, S. Tan, F. Xiong, R. Yu, P. Wu, L. Cui and Q. An, Chem. Commun., 2020, 56, 3805–3808 RSC
- M. Cabello, F. Nacimiento, R. Alcántara, P. Lavela, C. Pérez Vicente and J. L. Tirado, Chem. Mater., 2018, 30, 5853–5861 CrossRef CAS
- B. Jeon, H. H. Kwak and S.-T. Hong, Chem. Mater., 2022, 34, 1491–1498 CrossRef CAS
- D. S. Tchitchekova, A. Ponrouch, R. Verrelli, T. Broux, C. Frontera, A. Sorrentino, F. Bardé, N. Biskup, M. E. Arroyo-de Dompablo and M. R. Palacín, Chem. Mater., 2018, 30, 847–856 CrossRef CAS
- J. Hyoung, J. W. Heo, B. Jeon and S.-T. Hong, J. Mater. Chem. A, 2021, 9, 20776–20782 RSC
- T. N. Vo, H. Kim, J. Hur, W. Choi and I. T. Kim, J. Mater. Chem., 2018, 6, 22645–22654 RSC
- P. Padigi, G. Goncher, D. Evans and R. Solanki, J. Power Sources, 2015, 273, 460–464 CrossRef CAS
- R. Verrelli, A. Black, R. Dugas, D. Tchitchekova, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2020, 167, 070532 CrossRef CAS
- Z. L. Xu, J. Park, J. Wang, H. Moon, G. Yoon, J. Lim, Y. J. Ko, S. P. Cho, S. Y. Lee and K. Kang, Nat. Commun., 2021, 12, 3369 CrossRef CAS PubMed
- C. Chen, F. Shi, S. Zhang, Y. Su and Z. L. Xu, Small, 2022, 18, e2107853 CrossRef PubMed
- X. Zhang, X. Xu, B. Song, M. Duan, J. Meng, X. Wang, Z. Xiao, L. Xu and L. Mai, Small, 2022, 18, e2107174 CrossRef PubMed
- Z. Li, B. P. Vinayan, P. Jankowski, C. Njel, A. Roy, T. Vegge, J. Maibach, J. M. G. Lastra, M. Fichtner and Z. Zhao-Karger, Angew. Chem., Int. Ed., 2020, 59, 11483–11490 CrossRef CAS PubMed
- J. Bitenc, A. Scafuri, K. Pirnat, M. Lozinsek, I. Jerman, J. Grdadolnik, B. Fraisse, R. Berthelot, L. Stievano and R. Dominko, Batteries Supercaps, 2021, 4, 214–220 CrossRef CAS
- S. Zhang, Y. Zhu, D. Wang, C. Li, Y. Han, Z. Shi and S. Feng, Adv. Sci., 2022, 9, e2200397 CrossRef PubMed
- Z. Zhao-Karger, Y. Xiu, Z. Li, A. Reupert, T. Smok and M. Fichtner, Nat. Commun., 2022, 13, 3849 CrossRef CAS PubMed
- A. L. Lipson, B. Pan, S. H. Lapidus, C. Liao, J. T. Vaughey and B. J. Ingram, Chem. Mater., 2015, 27, 8442–8447 CrossRef CAS
- A. L. Lipson, S. Kim, B. Pan, C. Liao, T. T. Fister and B. J. Ingram, J. Power Sources, 2017, 369, 133–137 CrossRef CAS
- N. Kuperman, P. Padigi, G. Goncher, D. Evans, J. Thiebes and R. Solanki, J. Power Sources, 2017, 342, 414–418 CrossRef CAS
- M. E. Purbarani, J. Hyoung and S.-T. Hong, ACS Appl. Energy Mater., 2021, 4, 7487–7491 CrossRef CAS
- S. J. R. Prabakar, W.-B. Park, J. Y. Seo, S. P. Singh, D. Ahn, K.-S. Sohn and M. Pyo, Energy Storage Mater., 2021, 43, 85–96 CrossRef
- Z. Hou, R. Zhou, Z. W. Min, Z. H. Lu and B. Zhang, ACS Energy Lett., 2022, 8, 274–279 CrossRef
- D. M. Driscoll, N. K. Dandu, N. T. Hahn, T. J. Seguin, K. A. Persson, K. R. Zavadil, L. A. Curtiss and M. Balasubramanian, J. Electrochem. Soc., 2020, 167, 160512 CrossRef CAS
- K. V. Nielson, J. Luo and T. L. Liu, Batteries Supercaps, 2020, 3, 766–772 CrossRef CAS
- S. J. Richard Prabakar, A. B. Ikhe, W. B. Park, K. C. Chung, H. Park, K. J. Kim, D. Ahn, J. S. Kwak, K. S. Sohn and M. Pyo, Adv. Sci., 2019, 6, 1902129 CrossRef CAS PubMed
- A. G. Bishop, D. R. MacFarlane, D. McNaughton and M. Forsyth, J. Phys. Chem., 1996, 100, 2237–2243 CrossRef CAS
- M. Chiku, S. Matsumura, H. Takeda, E. Higuchi and H. Inoue, J. Electrochem. Soc., 2017, 164, A1841–A1844 CrossRef CAS
- F. Shi, P. N. Ross, H. Zhao, G. Liu, G. A. Somorjai and K. Komvopoulos, J. Am. Chem. Soc., 2015, 137, 3181–3184 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02003k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |