
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO residence time modulates multi-carbon formation rates in a zero-gap Cu based CO2 electrolyzer†
Siddhartha
Subramanian
 *a,
Jesse
Kok
a,
Pratik
Gholkar
c,
Asvin
Sajeev Kumar
b,
Hugo-Pieter
Iglesias van Montfort
a,
Ruud
Kortlever
b,
Atsushi
Urakawa
c,
Bernard
Dam
a and
Thomas
Burdyny
*a
*a,
Jesse
Kok
a,
Pratik
Gholkar
c,
Asvin
Sajeev Kumar
b,
Hugo-Pieter
Iglesias van Montfort
a,
Ruud
Kortlever
b,
Atsushi
Urakawa
c,
Bernard
Dam
a and
Thomas
Burdyny
*a
aMaterials for Energy Conversion and Storage (MECS), Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, van der Maasweg 9, Delft 2629 HZ, The Netherlands. E-mail: siddhartha.subramanian92@gmail.com; T.E.Burdyny@tudelft.nl
bLarge Scale Energy Storage, Process and Energy Department, Faculty of Mechanical, Maritime and Materials Engineering, Delft University of Technology, Delft 2628 CB, The Netherlands
cCatalysis Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, Delft 2629 HZ, The Netherlands
First published on 1st August 2024
Abstract
Carbon dioxide (CO2) electrolysis on copper (Cu) catalysts has attracted interest due to its direct production of C2+ feedstocks. Using the knowledge that CO2 reduction on copper is primarily a tandem reaction of CO2 to CO and CO to C2+ products, we show that modulating CO concentrations within the liquid catalyst layer allows for a C2+ selectivity of >80% at 200 mA cm−2 under broad conversion conditions. The importance of CO pooling is demonstrated through residence time distribution curves, varying flow fields (serpentine/parallel/interdigitated), and flow rates. While serpentine flow fields require high conversions to limit CO selectivity and maximize C2+ selectivity, the longer CO residence times of parallel flow fields achieve similar selectivity over broad flow rates. Critically, we show that parts of the catalyst area predominantly reduce CO instead of CO2 as supported by CO reduction experiments, transport modelling, and achieving a CO2 utilization efficiency greater than the theoretical limit of 25% for C2+ products.
Broader contextElectrochemical technologies are promising for the reduction of ‘non-electrifiable’ carbon emissions associated with dense chemicals and fuels that are currently derived from fossil fuels. CO2 electrolyzers using the membrane electrode assembly configuration are attractive for producing dense chemicals such as ethylene and ethanol using renewable electricity. While scaling up these systems, research into maximizing CO2 utilization and understanding spatial performance using optimal electrolyzer designs are then required. The work presented here provides insights into the influence of the CO residence time on hydrocarbon production rates from CO2 using modified gas flow field patterns in the copper-based membrane electrode assembly reactor configuration. The variation in CO residence time in the liquid phase of the reactor with varying gas flow field patterns shows design strategies to maximize hydrocarbon production, potentially allowing for new flow field designs to be explored when scaling up CO2 electrolyzers to larger areas which are relevant for industrial operation. |
Introduction
Electrochemical CO2 reduction (ECO2R) using copper (Cu) based catalysts is attractive due to copper's ability to produce hydrocarbons and oxygenates.1–7 As a result, extensive efforts have gone into unravelling the reaction pathways and intermediates in both H-cell and gas diffusion electrode (GDE) cell architectures to understand and improve what enables such broad C1 and C2 product spectra.8–13 These studies have shown that copper's unique ability to moderately bind carbon monoxide (CO) facilitates the coupling of two CO species.14–19 Critically, the coupling of CO-species has been shown to occur through both surface dimerization of two adsorbed CO-species and tandem reactions where CO2 is first converted to CO and desorbs into the aqueous environment as CO(aq). Recent studies have also shown the presence of two distinct active sites on Cu for CO2 to CO conversion and CO to C2+ conversion.20 Copper is then not simply a unique CO2/CO to C2+ catalyst, but an excellent CO2 to CO catalyst.A number of ECO2R experiments performed in fully aqueous reaction environments have sought to understand the effect of CO(aq) on multi-carbon product formation21–23 through a variety of control experiments and modelling. The importance of local CO(aq) concentration has further motivated tandem catalysts and bimetallic systems where one catalyst is included for CO2 to CO conversion, and a copper catalyst further facilitates CO2/CO reduction. A few studies for instance have shown that such spatial variations can be used to tune CO coverage over a Cu catalyst to achieve higher partial current densities of C2+ products.24–26 While this premise is attractive and shows increased oxygenate production rates as a result, these fully tandem systems commonly produce CO in excess amounts except at very precise flow rates and current densities. We conclude that a primary reason for this is that copper itself already reaches excessive CO(aq) concentrations during ECO2R, particularly at elevated current densities. We then posit that the need for local CO regulation can also be met by further understanding and modulating the residence times of CO(aq) in copper-based systems.
Suitable platforms to examine local CO regulation are zero gap membrane electrode assemblies (MEA) using anion exchange membranes due to their elevated reaction rates (>100 mA cm−2), low cell voltages and ability to maintain a favourable alkaline environment around the catalyst surface which reduces by-product H2 production.27–29 A notable consequence of these flow-based systems is the spatial variation in the concentration of reactants (CO2, CO and H2O) and products (CO and C2H4) within the catalyst layer as the flow rate and current density are varied.30 These spatial variations will impact the localized Faradaic efficiency but cannot be resolved through common measurements which give averaged Faradaic efficiency due to external product measurement. More specifically, the effect of the CO2 flow rate on product distribution has been shown to change drastically for a Cu based catalyst with increased C2+ production at lower flow rates. Gabardo et al. showed an increase in CO selectivity and a decrease in ethylene selectivity with an increase in the inlet CO2 flow rate in a Cu based MEA cell at 150 mA cm−2 and attributed the observed differences to favourable CO2/CO coverage and changes in local pH caused by differences in CO2 consumed to hydroxide ions. Tan et al. found similar trends for the CO2 flow rate on C2+ selectivity and postulated that lowering the local CO2 coverage by reducing the local CO2 concentration increases the relative coverage of minority surface species like CO which is beneficial for C2+ production. Crucially, both of these works do not consider how the spatial in-plane reactivity changes along the path length as flow rates are varied.
In this work, we study the influence of CO residence time on C2+ production rates for a Cu catalyst coated on a carbon gas-diffusion layer in an alkaline MEA cell. We use pulse and negative tracer gases to generate residence time distribution curves under various flow rate conditions and flow field patterns (serpentine, parallel and interdigitated) using ex situ mass spectrometry. When contrasting these findings with electrochemical data we are able to infer both local and spatial phenomena related to CO concentrations throughout the MEA cell. We find that C2+ production rates increase at decreased CO2 flow rates because of near-complete CO consumption, achieving a maximum selectivity of 84% at 200 mA cm−2. We further demonstrate how modulating CO concentrations via the choice of flow rate and flow field can enable regions of greater CO reduction instead of CO2 reduction, allowing for elevated CO2 to C2+ utilization efficiency.
Results and discussion
Within a MEA CO2 electrolyzer, CO and other products are produced within the liquid-immersed catalyst layer. These products can then either diffuse across a gas–liquid interface and through the gas diffusion layer (GDL) into the gas channel, or in the case of CO they can further be reduced on the catalyst (Fig. S1, ESI†). To better understand the statistical amount of time that products reside within the liquid layer, we performed residence time distribution experiments of an ex situ CO2 electrolysis cell.As illustrated in Fig. 1, a non-reactive tracer gas of 5% CO and 95% He is injected into an assembled electrolyzer, and a time-resolved output signal of CO is measured using a mass spectrometer (see Fig. S2, ESI†). By using defined input profiles of the tracer gas such as a pulse or negative tracers, the profile and delay of the output signal, known as a residence time distribution (RTD) curve, will give us information about the convective and diffusive properties of the MEA electrolyzer. Inside a CO2 electrolyzer employing a gas diffusion electrode, however, contributions from the gas flow channel, the GDE and the liquid filled catalyst layer all affect the RTD and the dominant transport factor needs to be determined.
 | ||
| Fig. 1 (a) Schematic of residence time distribution (RTD) curve measurements in the zero gap CO2 electrolyzer using carbon monoxide as a tracer gas. (b) Overview of control RTD curve measurement for a 30 s carbon monoxide tracer pulse for two different zero gap configurations at 10 standard cubic centimeters per minute (SCCM). The top configuration uses a non-wetted anion exchange membrane adjacent to the gas-diffusion layer. The bottom configuration uses a wetted Zirfon membrane to provide a water layer next to the gas-diffusion layer. (c) RTD curves for the non-wetted and wetted configurations showing the increase in residence time due to a water layer. | ||
In this regard, previous studies from fuel cells have shown that pulse RTD tests predominantly show the gas flow field channel characteristics as gases pass through the system components (see Fig. 1a center).31–33 In contrast, a negative tracer test saturates the entirety of the system for longer periods of time and provides information on the time required for gases to leave all parts of the cell.34 A negative tracer then gives a good perspective of products that are formed in the liquid phase leaving as the gas channel partial pressure is decreased.35 Here, the eventual release of the tracer is then maximized at the tail.
We began our non-electrochemical RTD tests by first examining the importance of the liquid layer in an MEA system as liquid diffusion and liquid-to-gas diffusion of CO are expected to be the largest transport barriers. For the experiments we used the most common flow field pattern (FFP), a serpentine channel which has a single fluid flow path from the inlet to the outlet, creating a significant pressure drop in the reactor. There are then two scenarios in Fig. 1b. For the non-wetted case, we assembled the MEA cell as usual but without applying a potential, which means that no water wets the anion exchange membrane or is present on the cathode GDL. For the wetted case, we used a porous Zirfon membrane which is wetted by the anolyte flow. When we compare a 30 s pulse RTD test for the two cases, we see a stark difference in the RTD curves (Fig. 1c). For the non-wetted case, we see a CO RTD profile almost identical to the pulse input with a mean residence time of 28 s, whereas the wetted case shows a substantially longer mean residence time of 118 s. This control experiment confirms the impact of the water layer on transport properties and confirms its importance in future tests.
We then performed pulse and negative RTD measurements on the Zirfon wetted system for varied inlet flow rates (Fig. 2a and b). Here a clear difference is observed between a 10 sccm flow (representative of a high CO2 conversion scenario) and 50 sccm (a low CO2 conversion scenario). Consequently, the mean residence time for the tracer at 10 sccm was 118 s, higher than at 50 sccm (110 s). If we relate this finding to a CO2 electrolyzer in operation, it indicates that a CO molecule in either the liquid layer or the gas channel will on average reside there for 8 s longer at 10 sccm than 50 sccm.
 | ||
| Fig. 2 (a) Non-electrochemical pulse RTD results for a serpentine flow field with 30 s CO tracer flow rates of 10 and 50 sccm. (b) Faradaic efficiency of products obtained from ECO2R on the Cu catalyst in the MEA cell at flow rates ranging from 5 sccm to 50 sccm at a geometric current density of 200 mA cm−2. (c) Comparison of CO and C2+ product selectivity at 200 mA cm−2. (d) Single pass conversion of CO2 to C2+ products at the studied inlet flow rates. (e) Fraction of OH− ions consumed to CO2 during the reaction, estimated using carbon balance over the system. Error bars represent the mean and standard deviation of triplicate experiments. | ||
To contrast these non-electrochemical RTD measurements with electrochemical data, we performed CO2 electrolysis experiments in an MEA cell using a Ni foam anode, Cu sputtered carbon GDE (5 cm2) and humidified CO2 as the reactant. The measurement techniques and instrumentation are shown in Fig. S8 (ESI†). For our tests, we used a fixed geometric current density of 200 mA cm−2 with CO2 fed at various flow rates ranging from low to high CO2 conversion conditions.
As shown in Fig. 2b and c, we find that the product distribution varies as CO2 flow rates decrease from 50 sccm to 5 sccm. Notably, we see a decrease in the Faradaic efficiency (FE) of CO from 12.6% at 50 sccm to just 3% at 10 sccm. In contrast, the FE of ethanol increases substantially from 20.3% at 50 sccm to 30.8% at 10 sccm. The shift is even more stark when observing the oxygenate (ethanol + acetate) trend in Fig. 2c. In particular, the product spectrum where oxygenates outcompete ethylene FE is more indicative of ECOR on copper than that of ECO2R.36,37 When coupled to the non-electrochemical RTD data it implies that the production rate of CO in both the 10 and 50 sccm cases may actually be similar (see Table S2, ESI†), but the CO produced in the 10 sccm case remains longer in the gas channel and liquid layer such that it is further reduced, providing a more oxygenated product spectrum. Interestingly, rates of ethylene production remain unchanged (FE ∼ 40%) across all flow rates, implying that any decrease in CO(aq) predominantly contributed to ethanol product pathways (eqn (S38) in ESI†).38 When the flow rates were further decreased (5 sccm), competing HER took over (FEH2 of 40%) due to mass transport limitations of CO2 reaching the entire 5 cm2 Cu GDE (Fig. 2c). Here, however, the oxygenate to ethylene trend continues.
A single pass conversion efficiency of 24% (Fig. 2d) and a maximum C2+ selectivity of 84% (jC2+ of 168 mA cm−2) are then achieved at 10 sccm, due to the higher residence time of CO in the reactor. A higher residence time ensures that there is sufficient time for the dimerization of two *CO molecules, thereby achieving a CO utilization of 87.6% for C2+ production at 10 sccm (see eqn (S28)–(S30) in ESI† and Table S2). Notably, then we can say that the highest combined Faradaic efficiency and single-pass conversion at a fixed current density both occur at low flow rates for a serpentine channel. At a higher flow rate of 50 sccm however, the C2+ selectivity drops to 65% with a single-pass conversion of only 4%. Additionally, a very low stoichiometric CO2 excess of 1.13 (see Table S2, ESI†) is obtained at 10 sccm, which is beneficial for achieving product rich gas streams and reducing downstream gas separation costs as shown in a recent study.39
Lastly, for the serpentine results, we found during our experiments that we lost less CO2 to OH− interactions than expected at 10 and 20 sccm. In particular, only 75% of OH− ions generated at the cathode are converted to CO32−/HCO3− due to buffer reactions with CO2 at 10 sccm, whereas all OH− is reacted above 30 sccm (Fig. 2e). Assuming that these 75% of OH− ions reacting with CO2 are converted to CO32− ions at these higher reaction rates (local pH > 12), this would mean that the ions transported across the AEM (towards the anode) is a mixture of CO32− and OH− ions (see eqn (S15)–(S23), ESI†). Typically, we would expect such a result only in CO2 depleted cases where high H2 FE's are seen. However, the total ECO2R FE are 92% and H2 FE is only 7%. This finding implies that we have regions in the reactor where reactions can proceed to C2+ products without parasitic reactions of CO2 and OH− ions. We then conclude that regions of our 5 cm2 electrode are CO2 depleted but not CO depleted. A portion of the catalyst performing CO electrolysis then also explains the constant ethylene selectivity observed across various flow rates (Fig. 2c) since selectivity shift to oxygenates rather than ethylene occurs when moving from CO2 to CO electrolysis.40,41
Using the above findings, we can then predict the dominant electrochemical reaction occurring along the flow channel length for low, moderate, and high CO2 conversion cases (Fig. 3a). In green regions the primary reactant is CO2, whereas in purple regions, CO is more in abundance than CO2. As discussed elsewhere,42 the maximum CO2 utilization efficiency to ethylene and ethanol products for the green region is 25% (Fig. 3b). However, the purple region performing primarily ECOR has no such limitation as CO does not react with OH− ions to form (bi) carbonates (Fig. 3c). Indeed, when we calculate the CO2 utilization efficiency across various flow rates (eqn (S26), ESI†), we reach a value of 31% (Fig. 3d), breaking the limit of 25% obtained for pure C2+ product formation, where CO32− ions act as the sole charge carrier.
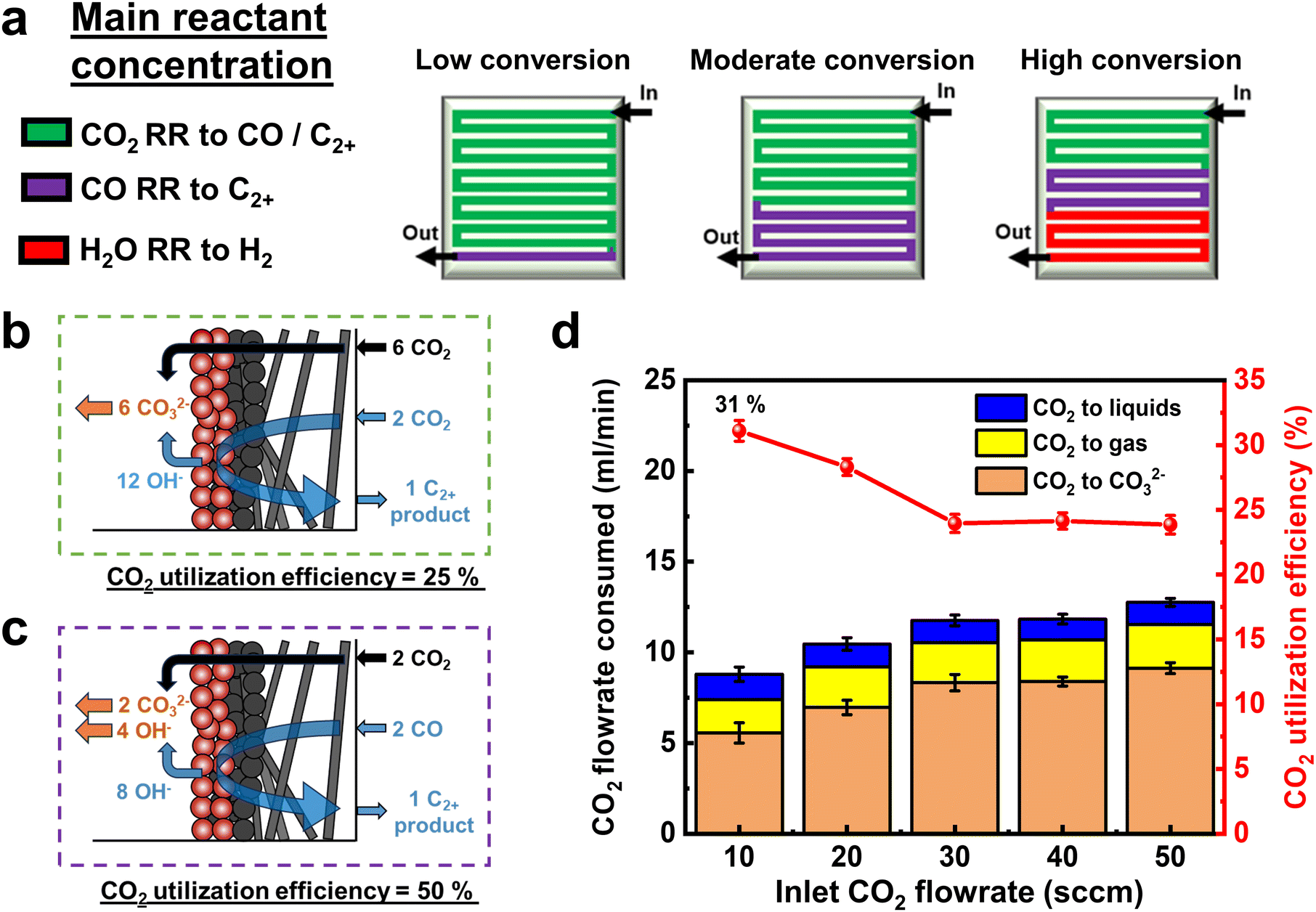 | ||
| Fig. 3 (a) Schematic of the predicted dominant reactions occurring within the catalyst layer along the serpentine flow field during ECO2R under varying CO2 conversion regimes. (b) Schematic depicting the theoretical CO2 utilization efficiency of 25% for 100% Faradaic production of ethylene and ethanol from CO2. (c) Schematic depicting how CO2 depletes regions with access to by-product CO can exceed the theoretical CO2 utilization efficiency. Shown here is an example when it can reach 50% (d) A carbon balance of the consumed input CO2 in the serpentine MEA tests system and CO2 utilization efficiencies at the studied inlet flow rates. | ||
To further test the above conclusion regarding CO residence times, we performed further RTD and electrochemical tests on parallel flow fields. In contrast to serpentine channels, a parallel FFP has channels divided into parallel paths with a very low pressure drop between the inlet and outlet.43 Mass transport through the GDE is then dominated predominantly by diffusion. Due to fundamental differences in GDE transport between FFP's, we performed negative tracer RTD measurements to compare the release of gases from each system (Fig. 4a and Fig. S6, ESI†). Again, all components of our standard electrochemical MEA cell were assembled except with a pretreated Zirfon membrane pressed against the carbon GDL to mimic the wetted catalyst in real ECO2R conditions (see ESI† for details).
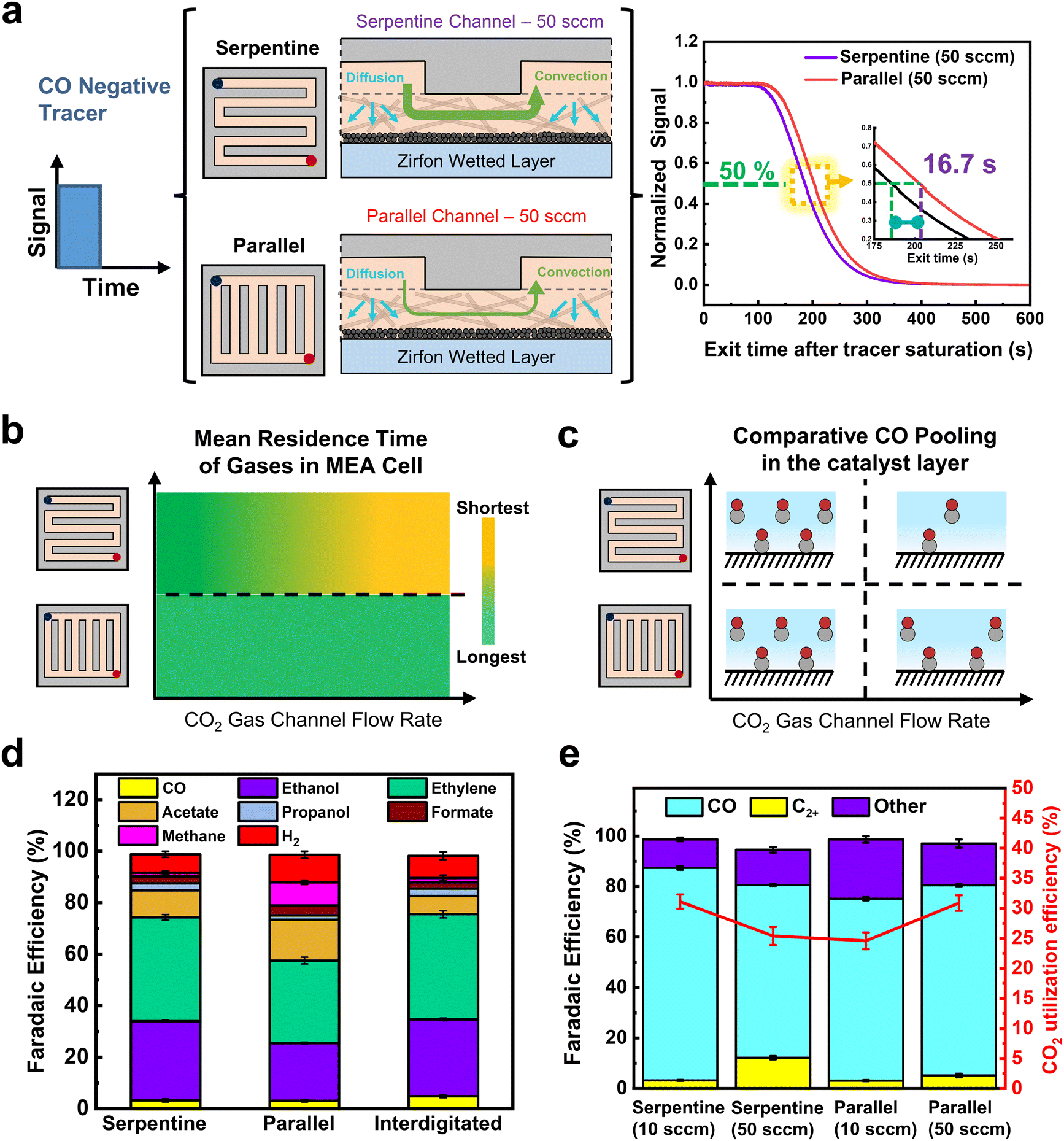 | ||
| Fig. 4 (a) Non-electrochemical negative tracer RTD results for serpentine and parallel flow fields at tracer flow rates of 50 sccm showing longer residence times for parallel flow fields as a result of lower convective forces. (b) A qualitative comparison of the RTD results as a function of flow rate and flow field. (c) A qualitative assessment of the CO pooling inside the catalyst layer during CO2 electrolysis as a function of flow rate and flow field. (d) Faradaic efficiency of products obtained from ECO2R in an MEA cell at 10 sccm and 200 mA cm−2 for serpentine, parallel and interdigitated flow fields. (e) Comparable CO and C2+ product selectivity for varied flow rates and flow fields. The inset (red line) shows the calculated CO2 utilization efficiencies. Error bars represent the mean and standard deviation of triplicate experiments. | ||
Observing the normalized RTD curves in (Fig. 4a), the negative tracer experiments show a large difference in the serpentine and parallel FFP curves at 50 sccm. Despite being at identical flow rates, there is 16.7 s delay for the tracer to exit the reactor using a parallel FFP in comparison to the serpentine FFP, illustrating an increased residence time of the tracer gas inside the reactor. Additionally, using a Cu GDE at 200 mA cm−2, a 5 s delay was observed for the tracer to exit the reactor using a parallel FFP, showing the residence time effect is attributed to the flow channel design used (see Fig. S9, ESI†). The higher residence time shows that the use of a parallel FFP creates the likelihood of higher reactant pooling in the wetted regions of the GDL surface. These results can be anticipated as the parallel flow field has lower channel velocities than the serpentine channel (Table S4, ESI†), which impacts concentration gradients between the gas channel and liquid layer, thus slowing gas removal from the liquid.
By combining the flow rate and flow field RTD data together we can compose the qualitative graph in Fig. 4b. Here we see that the serpentine channel can have long or short residence times depending on the flow rate inputted. Conversely, the parallel channel has a lower sensitivity to flow rate as the fluid velocity is always at a substantially lower value than the serpentine channel at equivalent volumetric rates. These conclusions then lead to a representative image of CO pooling during electrochemical CO2 reduction for each of the different cases as shown in Fig. 4c.
We then performed ECO2R using varied gas flow field patterns (FFPs) at the cathode. Fig. 4d shows the product distribution using all three different FFPs at 200 mA cm−2 and a CO2 inlet at a flow rate of 10 sccm. While all three FFPs show a similar selectivity of CO (3–4%), there were differences in the individual C2+ product distribution. For both the serpentine and interdigitated FFPs, FE of ethylene (40%) and oxygenates (40%) remain quite similar, achieving a C2+ selectivity of 82–84% with a low CH4 (FE ∼1%) and H2 (FE ∼8%). However, when a parallel FFP is used at the cathode, the FE of acetate doubles to 16% and CH4 increases to 9%. This comes at the expense of decreased ethylene (FE ∼32%) and ethanol (FE ∼22%), leading to a drop in the total C2+ selectivity of 72% (Fig. 3b). The selectivity switch from ethylene/ethanol to acetate for the parallel FFP suggests that higher local alkalinity around the catalyst is more likely, due to CO2 depletion within the GDE.44 We also see in Fig. 3e that the total FE is <100% which is possibly due to the volatility of products such as ethanol to enter the gas phase. While it is possible that this ethanol entering the gas phase increases with an increase in flow rates, the difference in total FE between 10 and 50 sccm is only 3.9%, and hence the overall CO and C2+ trends would remain similar.
The higher CH4 production also shows that an increased *H coverage (from *H2O) occurs within the catalyst layer due to depleted CO2 in some parts of the catalyst layer. An increased *H coverage is plausible since CH4 formation is well known to occur through surface recombination of *CO and *H via a Langmuir–Hinshelwood mechanism.45 Overall, the use of a parallel FFP on the cathode side at 10 sccm results in decreased CO2 access at some portions of the Cu catalyst layer and a subsequent increase in local alkalinity, producing higher CH4 and acetate, respectively. Previous studies in CO reduction on Cu have also attributed the increased acetate production to the abundance of OH− ions, which leads to a higher local pH around the catalyst surface.46,47
Furthermore, this depletion in CO2 access for a parallel FFP, suggests that a significant portion of the catalyst surface is predominantly used for the electrolysis of CO (produced from CO2) to C1 (methane) and C2+ products. Supporting this hypothesis are the results from our empirical numerical transport model which shows that about 18% of GDE has no CO2 access when a parallel FFP is used at the cathode (Fig. S12, ESI†). The total C2+ selectivity is however lower (72%) for the parallel FFP due to depleted CO2 and increased *H coverage as is evident from the increased CH4 selectivity. This would then imply that, if excess CO2 is fed into the system to ensure no mass transport limitations, the parallel FFP should maximize C2+ production due to the increased residence time of CO within the GDE as shown earlier (Fig. 4a).
To assess some of the above statements, we operated the serpentine and parallel FFPs at 200 mA cm−2 and at an excess CO2 flow rate of 50 sccm to ensure that sufficient CO2 is available for both cases to prevent CO2-depleted regions. Interestingly, we find a switch in the product distribution, with a significantly higher C2+ selectivity for the parallel FFP (75.2%), compared to the serpentine case (68%). As shown in Fig. 4e, the FE of CO was then more than twice lower (5%) for the parallel FFP compared to the serpentine case (FE 12.6%). This increased CO utilization to C2+ products for the parallel FFP shows the benefit of an increased residence time within the GDE for the dimerization of two CO molecules. A modified FFP taking the benefit of both the parallel FFP to achieve a higher CO residence time and a serpentine FFP to increase under-rib convection might then be attractive to achieve both higher C2+ production and higher single pass conversion efficiencies. Such considerations, however, are beyond the scope of this study. In addition, while these observations of residence time distribution effects are shown in our work at a geometric current density of 200 mA cm−2, the design rules can be generalized to other Cu based catalysts as well in MEA cells using ECSA normalized current densities of the Cu catalysts (see Fig. S21, ESI†).
The calculated CO utilization rate towards C2+ products then reached 83.6% for the parallel FFP (see Table S2, ESI†) at 50 sccm, significantly higher than the 65% obtained for the serpentine case. Operating electrolyzers using a parallel FFP is then beneficial at higher flow rates but comes at a cost of lower single pass conversion of CO2 fed into the reactor. Recent studies have however shown that operating at lower single pass conversion efficiencies (5–10%) is sufficient since the energy required for gas separation is 100 times lower than the actual electrolyzer energy requirements.48 Considering this aspect, a parallel FFP might be beneficial at higher and a broader range of flow rates due to its inherent ability to increase the reactant residence time inside the liquid filled catalyst layer. In addition, a parallel FFP also benefits from a very low pressure drop in the reactor (Table S4, ESI†), which might be beneficial as CO2 electrolyzers are scaled to larger areas (>100 cm2). Trade-offs associated with single pass conversion efficiency due to gas separation costs and energy efficiency due to higher full cell voltages will ultimately exist and careful consideration of all these parameters is essential for the selection of the right flow field design for ECO2R on Cu based catalysts.
ECOR experiments
While much of the work here showcased the influence of residence time of CO on C2+ production, the Faradaic efficiency results of individual C2 products (ethylene, ethanol and acetate) also showed distinct trends. For instance, the use of a parallel FFP at the cathode produced the highest acetate (FE 15–16%) at both 10 and 50 sccm inlet flow rates, which was twice higher than the serpentine and interdigitated FFPs. We hypothesized that the local catalyst microenvironment, specifically the local alkalinity as a result of differences in CO2 availability might be altered due to the FFP used, which may explain selectivity differences. We then performed CO electrolysis at 200 mA cm−2.Fig. 5a shows the product distribution obtained from electrochemical CO reduction (ECOR) for the three FFPs. As the reactant feed is switched from CO2 to CO, we see a clear selectivity switch from ethylene/ethanol to acetate for all three FFPs. Acetate production under these conditions is similar to the existing ECOR literature but it is interesting to see that the differences we observed in ECO2R have mostly been removed here. When we consider that most of the ECO2R differences for serpentine vs. parallel channels are explained to be a result of CO pooling and tandem reactions, it then makes sense that we do not see a stark serpentine-parallel difference for CO electrolysis in Fig. 5a where no products can be further reduced. Another interesting trend we observe from both ECO2R and ECOR tests is that the interdigitated flow field pattern produces the lowest acetate and lowest cell voltage during electrolysis. This variation in acetate production and cell voltage could be attributed to the lower CO residence time and hence lower CO concentration in the liquid phase for the interdigitated case (see Fig. S23, ESI†), due to the higher degree of under-rib convection through the GDL. Higher reactant flux through the GDE can result in lower mass transport overpotential as has been shown in some previous studies.49,50 However, this is speculative and further research on the reasons behind low acetate formation rates using interdigitated flow fields are required.
 | ||
| Fig. 5 (a) Product distribution from CO electroreduction for the three flow field patterns at a geometric current density of 200 mA cm−2. (b) Cell voltages obtained ECOR for the three FFP at 200 mA cm−2. | ||
The higher acetate production observed during this reactant switch from CO2 to CO for all FFPs also suggests a stronger dependence of product distribution on the local alkalinity around the catalyst layer. The concentration of local OH− ions during ECOR is more than one order of magnitude higher than for ECO2R,44 where neutralization by buffering reactions with CO2 occurs. It has been shown before that these abundant hydroxide ions react with the CH2CO intermediates (eqn (S40) in ESI†) relevant for ethylene and ethanol, leading to a switch in product towards acetate.51,52 A moderate interfacial pH, observed during ECO2R (pH < 12–13),53 is then beneficial for avoiding this switch from ethylene/ethanol to acetate. However, modulating the interfacial pH in these catholyte free MEA cells at higher current densities is quite challenging, as this would require modifications either in the type of the ion exchange membrane used or ionomers54 within the catalyst coated GDL. Additionally, although our combined results indicate that low flow rates and parallel FFPs contain higher acetate FEs due to local pH differences, further research is needed to fully determine the factors to promote or suppress acetate further.
This interplay of product formation rates highlight important implications for CO electrolysis in zero gap MEA electrolyzers. Although ECOR is beneficial due to the absence of carbonate crossover and lower full cell voltages (Fig. 5b), the findings here show that these advantages come at the expense of lower ethylene and ethanol formation rates. In addition, the calculated full cell energy efficiency (Table S7, ESI†) for a combined ethylene and ethanol production from ECOR is similar (23.4%) to ECO2R (29%), highlighting that the main benefit of COER lies in the long-term operational stability, due to the absence of carbonate formation at the cathode. Overall, this shows that CO2 electrolysis still has the potential for producing high rates of ethylene and ethanol if stability issues due to (bi) carbonate formation and Cu catalyst stability can be addressed as shown in recent studies.55,56
Finally, the major challenge that hinders commercialization of CO2 electrolyzers using Cu based catalysts lies in its inability to selectively produce ethylene or ethanol with high selectivity (>70%). Most studies have however shown that a combined 70–80% selectivity towards ethylene and ethanol can be obtained at industrially relevant current densities. While these branching pathways towards ethylene and ethanol cannot be well controlled as shown in a recent study,57 we posit here that researchers must look into a combined ethylene + ethanol selectivity as a performance metric. This is because ethanol as a liquid product can be separated from the MEA reactor and further be used as a starting material to produce ethylene through a catalytic dehydration reaction.58
Importantly, the energy requirements for this ethanol dehydration reaction to ethylene (45 kJ mol−1 of ethylene) are two orders of magnitude lower than a CO2 electrolyzer producing ethylene (2900 KJ mol−1 of CO2).59 We then argue here that the research community should look into integrating catalytic dehydration of ethanol to ethylene as an additional process step to CO2 electrolysis in order to make it energy efficient and industrially viable.
Conclusion
In conclusion, the residence time of CO in the liquid catalyst region greatly impacts product distribution from CO2 electrolysis. Here, we show that modulating the CO residence time is possible with varying inlet flow rates and flow field patterns and is an important consideration for both catalyst and system studies. We show that while both the interdigitated and serpentine flow patterns require higher single pass conversions to limit CO selectivity, a parallel flow pattern shows the highest C2+ selectivity at larger and a broader range of flow rates. Under lower flow conditions, we also show that the electrolyzers begin to split into CO2 and CO dominated regions, which has implications for selectivity, CO2 utilization efficiencies, and local catalytic and component effects.Author contributions
Conceptualization: SS and TB. Experimental investigation: SS, JK, PG, HPIvM, and ASK. Transport model and simulations: SS. Supervision: TB. Writing – original draft: SS and TB. Writing – review & editing: SS, TB, JK, ASK, HPIvM, JM, PG, BD, RK, and AU.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Thomas Burdyny and Siddhartha Subramanian would like to acknowledge the co-financing provided by Shell and a PPP allowance from Top Consortia for Knowledge and Innovation (TKI) of the Ministry of Economic Affairs and Climate in the context of the TU Delft e-Refinery Institute. In addition, the authors thank Joost Middelkoop for technical support with flow field design fabrication and Herman Schreuders for technical support with the magnetron sputter deposition technique.References
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, Selective Formation of C2 Compounds from Electrochemical Reduction of CO2 at a Series of Copper Single Crystal Electrodes, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C2 Products, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311 RSC.
- G. Mangione, J. Huang, R. Buonsanti and C. Corminboeuf, Dual-Facet Mechanism in Copper Nanocubes for Electrochemical CO 2 Reduction into Ethylene, J. Phys. Chem. Lett., 2019, 10, 4259–4265 CrossRef CAS PubMed.
- Y. Zheng, et al., Understanding the Roadmap for Electrochemical Reduction of CO2 to Multi-Carbon Oxygenates and Hydrocarbons on Copper-Based Catalysts, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, Mechanistic Understanding of CO2 Reduction Reaction (CO2RR) Toward Multicarbon Products by Heterogeneous Copper-Based Catalysts, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- K. Rossi and R. Buonsanti, Shaping Copper Nanocatalysts to Steer Selectivity in the Electrochemical CO2 Reduction Reaction, Acc. Chem. Res., 2022, 55, 629–637 CrossRef CAS PubMed.
- G. L. De Gregorio, et al., Facet-Dependent Selectivity of Cu Catalysts in Electrochemical CO2 Reduction at Commercially Viable Current Densities, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- C. Zhan, et al., Revealing the CO Coverage-Driven C–C Coupling Mechanism for Electrochemical CO2 Reduction on Cu 2 O Nanocubes via Operando Raman Spectroscopy, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- C.-T. Dinh, et al., CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- F. P. García De Arquer, et al., CO2 electrolysis to multicarbon products at activities gareater than 1 A cm−2, Science, 2020, 367, 661–666 CrossRef PubMed.
- S. Nitopi, et al., Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 Reduction: A Classification Problem, Chem. Phys. Chem., 2017, 18, 3266–3273 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, The influence of pH on the reduction of CO and CO2 to hydrocarbons on copper electrodes, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- X. Wang, et al., Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2–CO co-feeds on Cu and Cu-tandem electrocatalysts, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- W. Gao, Y. Xu, L. Fu, X. Chang and B. Xu, Experimental evidence of distinct sites for CO2-to-CO and CO conversion on Cu in the electrochemical CO2 reduction reaction, Nat. Catal., 2023, 6, 885–894 CrossRef CAS.
- S. Louisia, et al., The presence and role of the intermediary CO reservoir in heterogeneous electroreduction of CO2, Proc. Natl. Acad. Sci., 2022, 119, e2201922119 CrossRef CAS PubMed.
- C. G. Morales-Guio, et al., Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- Y. Lum and J. W. Ager, Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- T. Zhang, Z. Li, J. Zhang and J. Wu, Enhance CO2-to-C2+ products yield through spatial management of CO transport in Cu/ZnO tandem electrodes, J. Catal., 2020, 387, 163–169 CrossRef CAS.
- C. Chen, et al., Cu-Ag Tandem Catalysts for High-Rate CO2 Electrolysis toward Multicarbons, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- T. Zhang, et al., Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, A systematic analysis of Cu-based membrane-electrode assemblies for CO2 reduction through multiphysics simulation, Energy Environ. Sci., 2020, 13, 3592–3606 RSC.
- C. M. Gabardo, et al., Continuous Carbon Dioxide Electroreduction to Concentrated Multi-carbon Products Using a Membrane Electrode Assembly, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Gas diffusion electrodes and membranes for CO2 reduction electrolysers, Nat. Rev. Mater., 2021, 7, 55–64 CrossRef.
- H. Simonson, et al., Direct Measurement of Electrochemical Selectivity Gradients over a 25 cm2 Copper Gas Diffusion Electrode, ACS Energy Lett., 2023, 8, 3811–3819 CrossRef CAS.
- U. Krewer, et al., Direct methanol fuel cell (DMFC): analysis of residence time behaviour of anodic flow bed, Chem. Eng. Sci., 2004, 59, 119–130 CrossRef CAS.
- F. Barreras, A. Lozano, L. Valiño, C. Marín and A. Pascau, Flow distribution in a bipolar plate of a proton exchange membrane fuel cell: experiments and numerical simulation studies, J. Power Sources, 2005, 144, 54–66 CrossRef CAS.
- F. Trachsel, A. Günther, S. Khan and K. F. Jensen, Measurement of residence time distribution in microfluidic systems, Chem. Eng. Sci., 2005, 60, 5729–5737 CrossRef CAS.
- J. Diep, D. Kiel, J. St-Pierre and A. Wong, Development of a residence time distribution method for proton exchange membrane fuel cell evaluation, Chem. Eng. Sci., 2007, 62, 846–857 CrossRef CAS.
- N. T. Nesbitt, et al., Liquid–Solid Boundaries Dominate Activity of CO2 Reduction on Gas-Diffusion Electrodes, ACS Catal., 2020, 10, 14093–14106 CrossRef CAS.
- P. Wei, et al., Coverage-driven selectivity switch from ethylene to acetate in high-rate CO2/CO electrolysis, Nat. Nanotechnol., 2023, 18, 299–306 CrossRef CAS PubMed.
- D. S. Ripatti, T. R. Veltman and M. W. Kanan, Carbon Monoxide Gas Diffusion Electrolysis that Produces Concentrated C2 Products with High Single-Pass Conversion, Joule, 2019, 3, 240–256 CrossRef CAS.
- J. Li, et al., Enhanced multi-carbon alcohol electroproduction from CO via modulated hydrogen adsorption, Nat. Commun., 2020, 11, 3685 CrossRef CAS PubMed.
- S. Brückner, et al., Design and Diagnosis of High-Performance CO2-to-CO Electrolyzer Cells, Nat. Chem. Eng., 2024, 1, 229–239 CrossRef.
- E. Bertheussen, et al., Acetaldehyde as an Intermediate in the Electroreduction of Carbon Monoxide to Ethanol on Oxide-Derived Copper, Angew. Chem., Int. Ed., 2016, 55, 1450–1454 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, High-rate electroreduction of carbon monoxide to multi-carbon products, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- M. Ma, et al., Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- S. Subramanian, et al., Geometric Catalyst Utilization in Zero-Gap CO2 Electrolyzers, ACS Energy Lett., 2023, 8, 222–229 CrossRef CAS PubMed.
- J. Li, et al., Constraining CO coverage on copper promotes high-efficiency ethylene electroproduction, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- M. Schreier, Y. Yoon, M. N. Jackson and Y. Surendranath, Competition between H and CO for active sites governs copper-mediated electrosynthesis of hydrocarbon fuels, Angew. Chem., Int. Ed., 2018, 130(32), 10378–10382 CrossRef.
- W. Luc, et al., Two-dimensional copper nanosheets for electrochemical reduction of carbon monoxide to acetate, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- L. Wang, et al., Electrochemical Carbon Monoxide Reduction on Polycrystalline Copper: Effects of Potential, Pressure, and pH on Selectivity toward Multicarbon and Oxygenated Products, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- S. C. Da Cunha and J. Resasco, Maximizing single-pass conversion does not result in practical readiness for CO2 reduction electrolyzers, Nat. Commun., 2023, 14, 5513 CrossRef CAS PubMed.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Enhancing carbon dioxide gas-diffusion electrolysis by creating a hydrophobic catalyst microenvironment, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- Z. Zhang, et al., Porous metal electrodes enable efficient electrolysis of carbon capture solutions, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- H. H. Heenen, et al., The mechanism for acetate formation in electrochemical CO (2) reduction on Cu: selectivity with potential, pH, and nanostructuring, Energy Environ. Sci., 2022, 15, 3978–3990 RSC.
- S. Guo, et al., Promoting Electrolysis of Carbon Monoxide toward Acetate and 1-Propanol in Flow Electrolyzer, ACS Energy Lett., 2023, 8, 935–942 CrossRef CAS.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- C. Kim, et al., Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- B. Endrődi, et al., Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolysers, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- T. N. Nguyen, et al., Catalyst Regeneration via Chemical Oxidation Enables Long-Term Electrochemical Carbon Dioxide Reduction, J. Am. Chem. Soc., 2022, 144, 13254–13265 CrossRef CAS PubMed.
- G. Kastlunger, H. H. Heenen and N. Govindarajan, Combining First-Principles Kinetics and Experimental Data to Establish Guidelines for Product Selectivity in Electrochemical CO2 Reduction, ACS Catal., 2023, 13, 5062–5072 CrossRef CAS.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, Bioethylene Production from Ethanol: A Review and Techno-economical Evaluation, ChemBioEng Rev., 2017, 4, 75–91 CrossRef.
- T. Moore, et al., Electrolyzer energy dominates separation costs in state-of-the-art CO2 electrolyzers: Implications for single-pass CO2 utilization, Joule, 2023, 7, 782–796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02004a |
| This journal is © The Royal Society of Chemistry 2024 |