 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming passivation through improved mass transport in dense ionic fluids†
Evangelia
Daskalopoulou
 ,
Philip
Hunt
,
Christopher E.
Elgar
,
Minjun
Yang
,
Andrew P.
Abbott
and
Jennifer M.
Hartley
*
,
Philip
Hunt
,
Christopher E.
Elgar
,
Minjun
Yang
,
Andrew P.
Abbott
and
Jennifer M.
Hartley
*
School of Chemistry, University of Leicester, LE1 7RH, UK. E-mail: jmh84@le.ac.uk
First published on 13th March 2024
Abstract
Deep Eutectic Solvents (DESs) have recently been shown to be part of a dense ionic fluid continuum between ionic liquids and concentrated aqueous brines. Charge transport was shown to be governed by fluidity, with no discontinuity between molar conductivity and fluidity irrespective of cation, charge density or ionic radius. By adjusting the activity of water and chloride ions, mass transport, speciation and reactivity can be altered. It has been shown that while brines provide a high chloride content at a lower viscosity than DESs, unlike DESs, brines are unable to prevent metal passivation due to their high water content. This results in the possibility to impart a level of selectivity towards metal dissolution (or passivation) when processing mixed metal materials. Forced convection can be used to avoid the issue of slow mass transport in viscous media, and the use of jets or targeted ultrasound are effective methods for overcoming this issue. High-powered ultrasound was applied to copper, cobalt, and aluminium electrodes undergoing anodic dissolution, and linear sweep voltammetry showed a linear current–voltage response at potentials anodic of the oxidation potential under sonication, with total charge passed being 5 to 134 times greater than under silent conditions. Application of ultrasound to silver and nickel electrodes displayed an initial linear current–voltage response, but the increased water content of the brines resulted in passivation. Mass transport throughout the bulk solution is governed by the forced convection imparted by the ultrasound and ionic species must only migrate across the electrical double layer. It is shown that the anodic dissolution of a range of metals classically expected to passivate, e.g. aluminium, can be significantly accelerated under insonation conditions.
Introduction
Ionic liquids (ILs) and deep eutectic solvents (DESs) have often been described as “tuneable” or “designer solvents” due to the large number of potential cation–anion combinations available.1 This in turn leads to a wide range of physical and chemical properties. However, one of the major downsides of these dry, dense ionic fluids is that they are significantly more viscous than most molecular liquids, leading to a poorer mass transport and conductivity. For example, imidazolium ILs containing chloride generally have viscosity values of >18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (mPa s) at room temperature,2,3 while ILs with weaker coordinating anions such as tetrafluoroborate4,5 or bistriflimide6 have viscosity values of >30 mPa s at room temperature. This viscosity effect is usually proportional to the molecular shape of the cation.7 For example, imidazolium cations with longer alkyl chains form ILs with higher viscosity. Also, cations with six-membered rings form ILs with higher viscosity than those with five-membered rings. The viscosity, and hence conductivity, of DES systems is strongly dependent on the type of hydrogen bond donor (HBD) used. In the case of the common choline chloride-based DESs, viscosity values of 37 mPa s (choline chloride:ethylene glycol), 259 mPa s (choline chloride:glycerol), and 750 mPa s (choline chloride:urea) have been reported at 25 °C.2 However, it should be noted that viscosity will vary depending on the amount of moisture present in the initial DES components, or absorbed from the atmosphere during preparation or use.
000 (mPa s) at room temperature,2,3 while ILs with weaker coordinating anions such as tetrafluoroborate4,5 or bistriflimide6 have viscosity values of >30 mPa s at room temperature. This viscosity effect is usually proportional to the molecular shape of the cation.7 For example, imidazolium cations with longer alkyl chains form ILs with higher viscosity. Also, cations with six-membered rings form ILs with higher viscosity than those with five-membered rings. The viscosity, and hence conductivity, of DES systems is strongly dependent on the type of hydrogen bond donor (HBD) used. In the case of the common choline chloride-based DESs, viscosity values of 37 mPa s (choline chloride:ethylene glycol), 259 mPa s (choline chloride:glycerol), and 750 mPa s (choline chloride:urea) have been reported at 25 °C.2 However, it should be noted that viscosity will vary depending on the amount of moisture present in the initial DES components, or absorbed from the atmosphere during preparation or use.
While one of the major benefits of carrying out (electro)chemical reactions using ILs and DESs is the absence of water and (oxy)hydroxide chemistry, many authors have used the addition of small amounts of water to improve mass transport and conductivity without significantly altering the metal ion speciation in solution.8–14 Above a water content of ca. 40 wt% the water-in-DES matrix is inverted to become DES-components-distributed-in-water.15 Chloride-based DESs containing water have been shown to be part of a continuum with concentrated aqueous chloride brines (NaCl, LiCl, KCl, CaCl2etc.). The conductivity of DES, and various chloride brines, was shown to vary linearly with the fluidity of the medium on a log–log scale, within a salt concentration range of 0.05 to 10.8 mol per kg of solvent, irrespective of the species, charge density or ionic radius of the cation.16 A line of best fit for the 18 data sets described on the log–log scale has the function y = 0.82x − 0.80. However, this can only be considered a general approximation due to the variation in measurement methods. By adjusting the activity of water and chloride ions, mass transport, speciation and reactivity can be altered. Concentrated brines provide a high chloride content at a lower viscosity than DESs, but unlike dry DESs, brines are unable to prevent metal passivation due to their high water content. This behaviour has been utilised to impart a level of selectivity towards metal dissolution (or metal passivation) when processing mixed metal materials such as printed circuit boards.16,17
An alternative way to improve the mass-transport of DESs without introducing the undesired water chemistry as discussed above is to utilise forced convections. Specifically, forced convection solves the issue of slow mass transport kinetics in viscous media, without risking a change in metal ion speciation or chemical properties of the solvent. The use of jets or targeted ultrasound are effective methods of obtaining fast reaction kinetics in viscous liquids. High-powered ultrasound has been applied to copper and nickel electrodes undergoing anodic dissolution, and linear sweep voltammetry showed a linear current–voltage response at potentials anodic of the oxidation potential under sonication, with dissolution currents roughly 14 times greater than under silent conditions.18 Application of ultrasound to a solution will cause bubble formation. Bubbles are generated from dissolved gases in solution. As the sound waves oscillate between high and low pressure phases, so the bubbles expand and compress. Upon reaching a critical size, bubbles will collapse, sending shockwaves through the solution, causing micromixing.19 Bubble propagation next to a surface can cause asymmetric collapse to occur, with the bubble sending a powerful jet into the surface.20,21 Typically, sonication experiments are carried out in an aqueous environment due to the low cost and ability to generate reactive oxygen species under intense thermal environments seen during sonication.22 Sonication run in different liquids can result in very different bubble profiles due to the increase in viscosity.19,23 For example, cavitation in glycerol can produce bubble halos rather than the stream of bubbles seen more typically in water,23 with this effect also being seen in ChCl:2EG at certain powers.24 The use of ultrasound in combination with hydrometallurgy to improve leaching rates has been studied by Bao et al. in a recent review.25 Ultrasound can enhance the recovery of metals from ores or waste streams in a variety of different lixiviants via three main pathways. Firstly, an increase in mass transport means more collisions between the lixiviant and the metallic particles. Secondly, cavitation can cause deformation of surfaces, increasing surface area or the exfoliation of passivation layers, and finally when run in aqueous conditions the cavitation can generate reactive oxygen species which can help oxidise and solubilise metallic species.
In this work, the anodic dissolution behaviour of a range of metals in a choline chloride-based DES and two aqueous choline chloride brines of different chloride concentrations is explored, under both silent and ultrasonic conditions. The DES and the choline chloride brine (1:6.85 molar ratio) have the same chloride content (ca. 3.79 mol kg−1) but the brine has a tenth of the viscosity of the DES. The metals studied include those classically expected to passivate, such as aluminium and titanium, along with the technology critical metals copper, nickel, silver, and cobalt. It is shown that all of the target metals except for titanium have enhanced dissolution under ultrasonic conditions, providing insights into how the nature of passivation layers formed on the native metal surface or during dissolution affects electrochemical dissolution.
Experimental
Chemicals
The DES ChCl:2EG was prepared from choline chloride (ChCl) (Sigma-Aldrich, >98%) and ethylene glycol (EG) (Sigma-Aldrich, 98%), by mixing the two components in a 1:2 molar ratio on a hotplate stirrer at 80 °C until a homogeneous clear liquid had formed. This was stored in a storage bottle in a 50 °C oven until needed, in order to prevent crystallisation of the ChCl in cold lab conditions. The chloride concentration was equivalent to 3.79 mol per kg solvent. The brines were prepared by mixing ChCl and deionised water (18.2 Ω, Elga Purelab Option apparatus) in a 1:3 or 1:6.85 molar ratio at 80 °C until fully dissolved, equivalent to 5.16 and 3.79 mol chloride per kg total solvent respectively. Solvents were prepared in bulk to avoid variation in chloride concentration across the different experiments from multiple preparations, all brines were stored at room temperature as crystallisation is not a problem faced by these solvents.
Voltammetry
Electrochemical testing was carried out using a Metrohm Autolab potentiostat/galvanostat (PGStat302N) with the compatible Nova 2.1 software, with a 3-electrode configuration. The working electrode (WE) was a disc electrode made from the target metal of interest, i.e. nickel, copper, cobalt, silver, aluminium, or titanium (see Table S1†), embedded in a glass electrode housing. The diameter of the disc was between 0.5 and 3 mm, depending on the metal. The counter electrode was an iridium oxide-coated titanium mesh. The quasi-reference electrode (QRE) was a wire of the same material as the WE in direct contact with the bulk solution to ensure that the same oxidation overpotential was applied to each metal. This QRE was housed in an open-ended glass pipette to minimise disturbances in reference potential due to solution turbulence. The scan rates were 10, 50, and 100 mV s−1. The working electrodes were polished using SiC abrasive papers (Metprep Ltd, UK) with an increasing grit size of 320, 600, 1200 and 2500. In each condition (silent, 53 W cm−2, and 132 W cm−2 ultrasonic power density), three sequential sweeps are performed at three separate scan rates (10, 50, and 100 mV s−1), polishing only before the first sweep of each. The total charge passed during the electrodissolution experiments was determined via integration of the LSV curves between 0 and +1.5 V vs. the quasi-reference wire using the Origin 2021 graphing program.Ultrasonics
The sonicated experiments were conducted using a Model CL-334 Q700 sonicator and compatible FisherBrand controller, with an operating frequency of 20 kHz and power output variable up to 700 W (527 W cm−2). The sonotrode (dia. = 13 mm) was placed 4 mm away from the working electrode. Ultrasound was applied for the full duration of the LSV scan when required.Viscosity & conductivity
The viscosities of the different solvents were measured at 25 °C using a Seiko EG&G QCM922A Quartz Crystal Microbalance (QCM) using the previously reported procedure,26 and their densities were estimated by weighing 10 mL of solvent at 25 °C in a 10 mL volumetric flask, using a 4 d.p. analytical balance (Mettler Toledo). Reported densities are average of at least three repeat measurements. Conductivities were recorded using a Mettler Toledo SevenDirect SD30 conductivity meter with Automatic Temperature Compensation (ATC). The cell constant for the commercial conductivity probe was 5.2 ± 0.5 cm−1, and conductivity standards of 12.88 mS cm−1 and 1413 μS cm−1 (Mettler Toledo, values certified at 19 °C) were used as received in order to perform calibrations. The reported conductivity values are an average of at least three repeat measurements.Results and discussion
Effect of ultrasound on metal dissolution in a DES
Under silent conditions, the electrochemical etching of metals is usually hindered by the presence of passivation layers. These can be oxides formed on the surface of the metal through contact with the atmosphere, or precipitates formed during electrochemical dissolution, salty slurries, or highly saturated solutions of metal ions. It has previously been shown that all 9 common metals studied in the ChCl:2EG liquid passivated during anodic oxidation.27 Depending on the identity of this passivation layer, further oxidation may be possible as charge migrates across or through it, or the passivation layer may be removed by sufficiently strong agitation. High powered ultrasound has been shown to be effective at removing passivation layers formed during electrochemical oxidation of copper and nickel in a DES formed from ChCl:2EG.18Linear sweep voltammetry (LSV) of Cu, Ni, Ag, and Co under silent conditions in ChCl:2EG shows the development of passivation layers on all four metals (Fig. 1a). The presence of passivation layers can be identified by the decrease in current density from a maximum. Depending on the metal investigated, these passivation layers could be poorly soluble insulating metal salts, resulting in almost zero current flow, metal salts where further oxidation of the metal can take place, resulting in steady state conditions, or a concentrated solution of metal complexes, where current flow is dependent on diffusion of ions to and from the electrode surface. In ChCl:2EG, these species are known to be CuCl27 on the copper electrode, and nickel glycolates28 or hydroxylates29 on the nickel electrode, where the insoluble species are formed from reaction of nickel ions with dissolved water/oxygen, or the solvent itself. Visual observations of the cobalt disc during electrodissolution under silent conditions show a blue liquid flowing off the electrode surface (Fig. 2), indicative of the [CoCl4]2– species.30 At the surface of the silver electrode, a layer of AgCl is expected to form which passivates the anodic dissolution of silver in ChCl:2EG. Note that ChCl:2EG contains 3.79 mol of the Cl− anion per kg of solution and although the solubility of AgCl(s) in ChCl:2EG is relatively high (approximately 0.2 mol dm−3),31 the dissolution of AgCl is kinetically slow. Neither Al nor Ti show appreciable electrochemical dissolution under silent conditions.
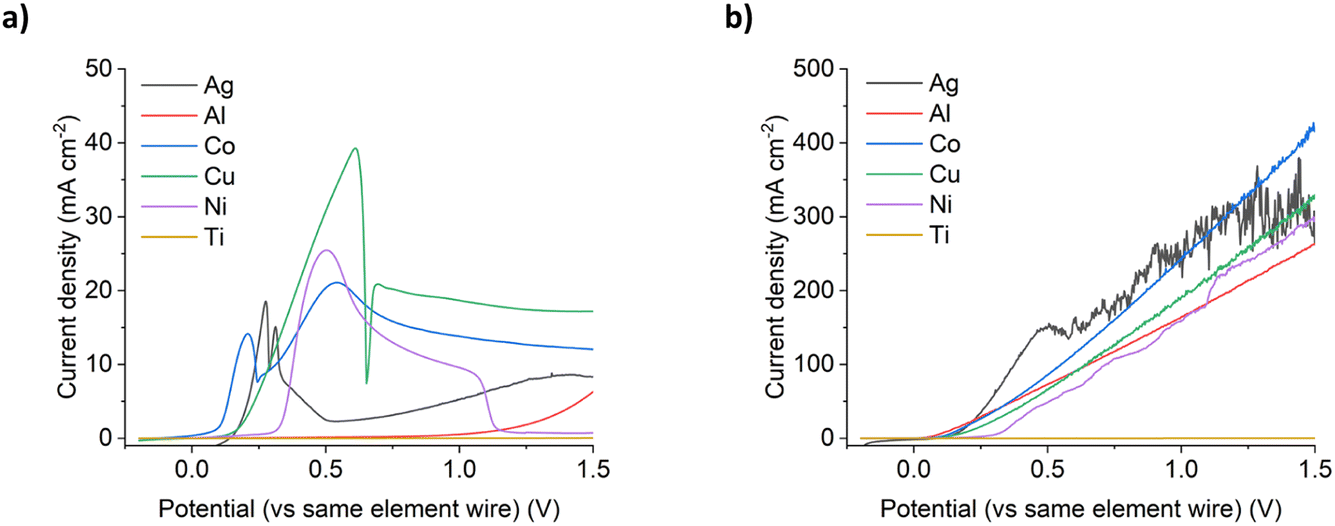 | ||
| Fig. 1 LSVs of the six metals investigated in ChCl:2EG, under (a) silent, and (b) 132 W cm−2 ultrasound. The scan rate was 10 mV s−1, with a quasi-reference electrode made from the same metal as the metal under investigation. Note 10× increase in magnitude of Y axis in (b). | ||
 | ||
| Fig. 2 Cobalt disc working electrode after linear sweep voltammetry in ChCl:2EG under silent conditions. The reference electrode was the same metal as the working electrode, and the scan rate was 10 mV s−1. The blue colour is characteristic of [CoCl4]2–. | ||
With the application of 132 W cm−2 ultrasound, a significant difference in the voltammetric response is observed. With ultrasound, a linear increase in current density with increasing electrode potential was observed, up to a maximum of ca. 300–450 mA cm−2 for all metals except titanium, with similar line gradients (Fig. 1b). Depending on the metal, this is equivalent to an 8 to 134 times increase in total charge passed across the potential range investigated, and hence an increase in metal etching rates. It should be noted that the metals that benefited most from the application of ultrasound were nickel (a 48 times increase in total charge passed), and aluminium (a 134 times increase in total charge passed).
This observed behaviour suggests that ultrasound prevents the metals from passivating at higher over-potentials as fast mass transport removes the metal complexes from the transport layer close to the electrode surface and means that the concentration never exceeds the solubility limit which is what happens during passivation. This enables very high current densities and fast dissolution to be achieved without the formation of insulating layers at the electrode solution interface.
Comparison of metal dissolution in DESs and ChCl brines
The results for the DES tested above were compared with two ChCl brines. The first had the same chloride concentration as the ChCl:2EG system (3.79 mol chloride per kg of solvent) but a tenth of the viscosity (Table S2†). The second ChCl brine contained 5.16 mol chloride per kg of solvent, but at only ca. half the viscosity of the ChCl:2EG system. These systems would permit the effect of chloride concentration to be probed as well as the effect of viscosity, and hence solvent conductivity. Voltammetry was recorded for the same metals in two different concentrated ChCl brines.For the silent LSVs (Fig. 3 and S2†) the currents are highest for the ChCl:6.85H2O brines which have the lowest viscosities of the liquids studied, and lowest currents for ChCl:2EG which are the most viscous. The exception is for aluminium in ChCl:2EG where the metal is relatively passive due to the low water activity and the inability to break down the oxide. The static corrosion of aluminium has been previously studied in DESs and shown to be very slow due primarily to the slowness of the cathodic reaction resulting from the low activity of water. For the insonated systems no passivation is observed despite the high water content of some of the brines. The slopes of the LSVs, i.e. rate of metal dissolution as a function of potential, vary for each metal, but the overall magnitudes are similar within a solvent.
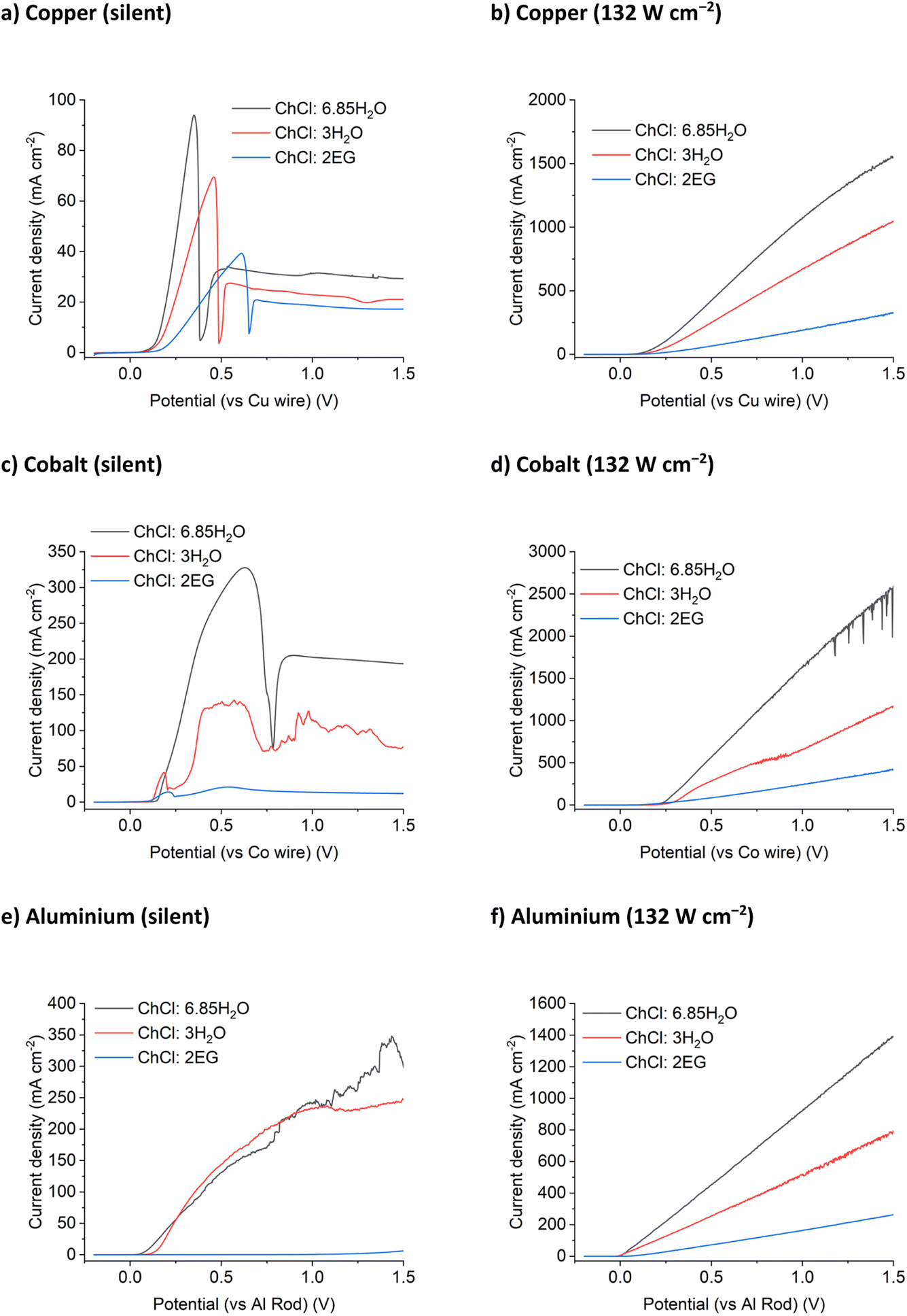 | ||
| Fig. 3 Linear sweep voltammograms of (a and b) copper, (c and d) cobalt, and (e and f) aluminium disc working electrodes in ChCl:2EG and two different ChCl brines, under (a, c and e) silent, and (b, d and f) 132 W cm−2 ultrasound. The quasi-reference electrode was the same metal as the working electrode, and the scan rate was 10 mV s−1. First scans presented only. | ||
Effect of transport layer structure on dissolution rate in dense ionic fluids
In typical electrochemical experiments, a significantly high concentration of electrolyte (typically 0.1 mol dm−3) is added to a molecular solvent such that a double layer forms close to the electrode surface and most of the surface charge is neutralised close to (<0.5 nm) the electrode surface ensuring that mass transport is diffusion controlled. The diffusion layer thickness is typically 20 to 200 μm. In most dense ionic fluids, the double layer structure is different with alternating layers of anions and cation32 and the thicknesses of the double layer are significantly thicker due to the large ionic radii. The diffusion layer thickness is proportional to D1/2 so it should become longer in viscous dense ionic fluids compared to electrolytes in molecular fluids. The effect of ultrasound is to significantly decrease the thickness of the diffusion layer to 1–10 μm.33It has previously been suggested that the linear current vs. voltage responses for the LSVs in sonicated solutions are due to a different mass transport mechanism occurring in dense ionic fluids under forced mass transport conditions.18 It was proposed that a much thinner diffusion layer existed with a different structure to the double layer from that observed in dilute electrolyte solutions. This would enable migration to take over from diffusion as the main mass transport mechanism. If that was the case then the slope of the LSVs in Fig. 3 should be a measure of the solution conductivity. This was shown to be the case for copper dissolution in a range of brines at two different ultrasonic powers.
Fig. 4a shows the effect of conductivity on the slope of the LSVs for the six different metals under 132 W cm−2 ultrasound. It could be expected that the migration of metal complexes should be relatively similar in the same medium, and while that is the case in the ChCl:2EG DES, there is significant deviation in the different brines. For Al, Co and Cu the LSV slopes increase roughly linearly with solution conductivity and it can be seen that an increase in conductivity results in increased oxidation rates for Cu, Co, and Al, rather than chloride content alone. If migration is controlling the mobility of ions in dense ionic fluids under ultrasound then the molar ionic conductivity, Λ, should be described by the Stokes–Einstein equation:34
| Λ = z2Fe/6πη(R+ + R−) | (1) |
:5 for Co2+,36,37 whereas for Ni2+, only the hexaaqua complex is present.38
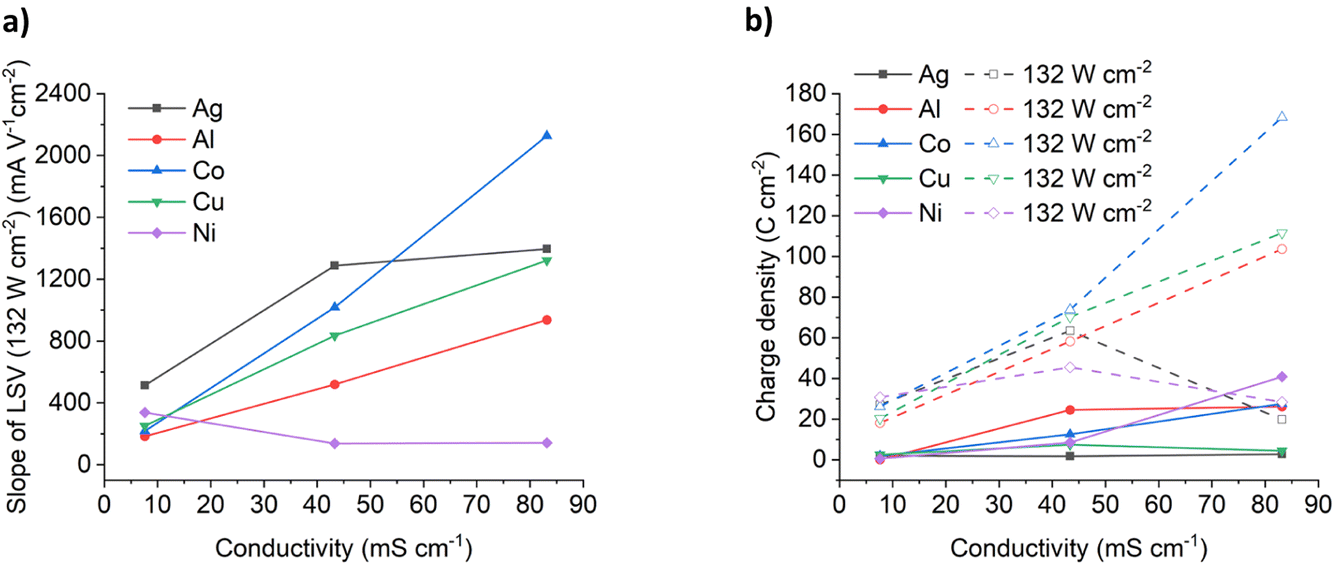 | ||
| Fig. 4 Effect of conductivity on (a) the slope of the insonated LSV curves for metal etching in ChCl:2EG, ChCl:3H2O and ChCl:6.85H2O, and (b) the total charge passed during etching for both silent and insonated conditions. | ||
 | ||
| Fig. 5 Photos of solutions containing 0.1 mol dm−3 of (a) CuCl2·2H2O, (b) NiCl2·6H2O, and (c) CoCl2·6H2O dissolved in the three chloride solvents. For each metal salt, the solutions are: left – ChCl:2EG, middle – ChCl:3H2O, right – ChCl:6.85H2O. | ||
Electroanalysis of copper passivation under silent conditions
Fig. S1† shows the anodic stripping LSVs for a macro-sized copper electrode in ChCl:2EG DES and ChCl:H2O (1:3 and 1:6.85 molar ratio) at voltage scan rates of 10, 50, and 100 mV s−1. Interestingly, the magnitude of the peak current is seen to increase with the voltage scan rate before the anodic stripping of copper is passivated by the electrogenerated product. It is known that oxidation of Cu0(s) to Cu+ in DES, where the concentration of Cl− is approximately 3.8 mol kg−1 (Table S1†), leads to CuCl precipitates. Therefore, the upper maximum concentration of Cu+ that can feasibly form at the electrode interface is determined by the amount of chloride anions available to solvate the copper ions formed at the interface. Electroanalysis of the LSV of copper shows that the ratio of [Cu+]:[Cl−]bulk at the electrode interface prior to the onset of passivation is close to unity.
The measured current increases linearly with the applied potential before a peak is seen around ca. +0.6 V versus a copper pseudo reference wire. Note that the working electrode is a macro-sized copper disc and over the course of the LSVs the anodic dissolution of the copper metal disc is negligible compared to the overall geometry of the electrode. It is evident that the drop in the current during the LSVs as seen in Fig. S1† is due to the formation of a passivation layer prohibiting further anodic stripping of Cu0, not because the copper working electrode is completely dissolved during the experiment.
At sufficiently low overpotential with respect to the copper reference electrode, copper is known to undergo one-electron oxidation in ChCl DES/brine medium to form CuCl(s)
| Cu0(s) − e− + Cl−(aq) ⇌ CuCl(s) | (2) |
| CuCl(s) − e− + Cl−(aq) ⇌ [CuCl4]2−(aq) | (3) |
:3 ChCl brine and 1:6.85 ChCl brine solutions are approximately 4.25, 5.70, and 4.02 mol dm−3, respectively. The current density of a macro-electrode is described by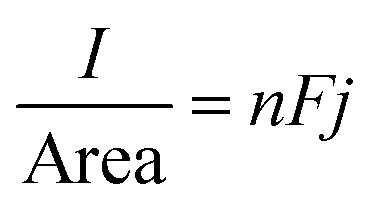 | (4) |
 | (5) |
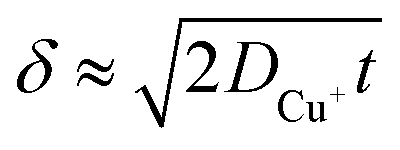 , where t in the case of LSV of copper is the time taken to scan from the onset potential of copper oxidation (Eox) to the onset of electrode passivation (Epass). The peak potential is used for the latter since a delay in the faradaic current in this case is indicative of passivation effects. Substituting the definition of δ into the above two equations results in the following expression
, where t in the case of LSV of copper is the time taken to scan from the onset potential of copper oxidation (Eox) to the onset of electrode passivation (Epass). The peak potential is used for the latter since a delay in the faradaic current in this case is indicative of passivation effects. Substituting the definition of δ into the above two equations results in the following expression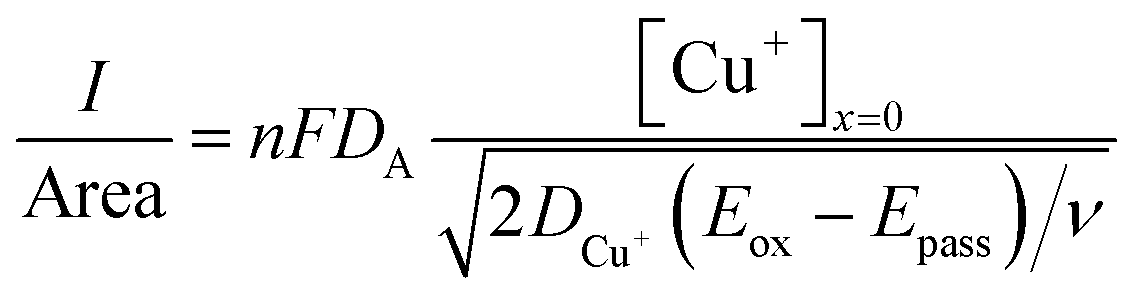 | (6) |
As can be seen, the concentration ratio of Cu:Cl−bulk at the electrode interface is approximately 1:1 prior to passivation. This indicates that the chemical identity of the passivation layer is likely to be CuCl(s). Note that the ratio of Cu:Cl−bulk for ChCl:2EG DES is a little higher than unity, this is likely because the local concentration of chloride anions is likely higher than that of the bulk value during the dynamic anodic potential sweep. Under insonation conditions, as shown in Fig. 4b, no evidence of passivation is observed and the total charge passed increases by a factor of 10 for the 5.16 mol kg−1 brine and ChCl:2EG, and by a factor of 25 in the 3.79 mol kg−1 brine, as compared to silent conditions.
The trend of fluidity controlling the maximum current density is continued for the anodic dissolution of a cobalt electrode in concentrated ChCl systems. Blue passivation layers formed of CoCl2 or solution-based [CoCl4]2– develop at the electrode surface during anodic dissolution for all three ChCl systems. This results in the noisy LSV response shown in Fig. 3c, as the current density increases temporarily when the layer of concentrated metal ions flows off the electrode surface, before being reformed. As was observed for copper, the application of 132 W cm−2 of ultrasound to the dissolution of a cobalt electrode results in a maximum current density increase of up to 10 times (Fig. 3d). While the diffusion coefficients of the Co2+ species are not currently known in these systems, it could reasonably be anticipated that the limiting concentration of CoII at the electrode surface at the point of passivation is likely to be half that of the chloride concentration, assuming a solid CoCl2 species is initially present.
Aluminium is notoriously difficult to anodically dissolve due to the highly inert passivation layer formed in contact with the atmosphere. Previous research into the anodic dissolution of aluminium in DES media has used significant overpotentials to achieve dissolution.39,40 Upon application of ultrasound, aluminium dissolution follows the same trend of increased dissolution with increased fluidity and conductivity (Fig. 3f). White precipitates were observed to form in all of the ChCl systems after dissolution, most likely an aluminium (oxy)hydroxide or glycolate species.
In contrast to Cu, Co and Al, Ag and Ni display a decrease in etching rate in the most conducting solution (ChCl:6.85H2O). Silver ions are highly sensitive to the presence of chloride in water and the resulting AgCl has poor dissolution kinetics in these chloride brines in comparison to the production of Ag+ ions. This behaviour can be seen in the silent scans as well, where both the ChCl brines display similar current maximum values before passivation, suggesting that the rate of AgCl formation is more important than solvent fluidity for dissolution under silent conditions (Fig. S2a†). Nickel will passivate in contact with water, even in the presence of a strong oxidising agent (iodine) and >3 mol kg−1 of chloride (10 wt% water in ChCl:2EG).17
Unlike the dissolution of aluminium under ultrasonic conditions, titanium does not dissolve electrochemically under a sonic horn. This is the case regardless of the chloride content or fluidity of the solvent, even under high-powered ultrasound (Fig. 1a and b). In ChCl:2EG, titanium and its ions are expected to react with any residual water content (ca. 1–4 wt%) in the DES,41 limiting the formation of soluble titanium chloride species. In aqueous systems, and adjusting the titanium quasi reference for a NHE, Pourbaix diagrams indicate that the most stable present species would be TiO2, and aqueous phase diagrams suggest potential TiOCl2 derivatives at [Cl]− molalities greater than 0.1 mol dm−3 of which all 3 systems are.42 These species could be preferentially forming instead of a soluble titanium chloride species, suggesting why the current densities remain lower than other metals in the same conditions. Research has been carried out into the anodic dissolution of titanium in non-aqueous systems such as DESs, but large overpotentials and long subjection times are still required to overcome the natural passivation layer that is formed under normal atmospheric conditions.43
Conclusions
Concentrated ionic fluids where the non-ionic component is water or an organic compound are shown to be part of a continuum where conductivity is controlled solely by fluidity and this appears to be relatively independent of the cation/anion types. The dissolution of metal through electrooxidation is limited in all cases by the formation of concentrated layers of metal ions close to the electrode surface which result from slow mass transport. The use of enhanced mass transport in the form of jets, or in the case of this study, ultrasound, enables passivation to be circumvented. This results in enhanced mass transport and very rapid metal dissolution rates.Depending on the metal, etching can be dependent on solvent fluidity, the chloride:water ratios and/or the solubility of the species formed in the solvent. For example, copper, cobalt, and aluminium all showed a direct correlation of improved etching under ultrasound as a function of improved solvent fluidity. Application of ultrasound means that etching rate is no longer dependent on metal ion and ligand diffusion, but rather migration dominates the mass transport process. Dissolution can be tuned using brines and this study showed that nickel and silver etching could be hindered by increasing the water activity in the liquid.
Enhanced mass transport is not a universal solution for overcoming passivation as it depends on the solubility of the oxidised species and the activity of any ligands which render the metal ions less soluble e.g. H2O or OH−. While dissolution rates were similar for most metals using ultrasound, there are some metals which are unaffected due to the stability of the initial passive layer on the electrode surface. This was most notable for titanium although it was also observed for metals such as Pt, W and Mo. From an electrochemical perspective, the change in the dominant mass transport factor from diffusion to migration can also be changed depending on the rate at which the overpotential is scanned as a function of time.
This study has importantly shown that the generally invoked maxim that reaction rates are slow in ionic liquids because they are viscous can be overcome in some cases using forced mass transport such as ultrasound.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Faraday Institution (Faraday Institution grant code FIRG027, project website https://relib.org.uk), the UKRI Interdisciplinary Circular Economy Centre for Technology Metals, Met4Tech project (EP/V011855/1), NERC CENTA2 grant NE/S007350/1, the EPSRC SonoCat project (EP/W018632/1), and Johnson Matthey for funding this work.References
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC
.
- C. D’Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC
-
K. R. Seddon, A. Stark and M.-J. Torres, in Clean Solvents, 2002, vol. 819, pp. 34–49 Search PubMed
- J. Fuller, R. T. Carlin and R. A. Osteryoung, J. Electrochem. Soc., 1997, 144, 3881–3886 CrossRef CAS
- T. Nishida, Y. Tashiro and M. Yamamoto, J. Fluorine Chem., 2003, 120, 135–141 CrossRef CAS
- P. Bonhote, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef CAS PubMed
- S. Seki, T. Kobayashi, Y. Kobayashi, K. Takei, H. Miyashiro, K. Hayamizu, S. Tsuzuki, T. Mitsugi and Y. Umebayashi, J. Mol. Liq., 2010, 152, 9–13 CrossRef CAS
- A. Y. M. Al-Murshedi, J. M. Hartley, A. P. Abbott and K. S. Ryder, Trans. IMF, 2019, 97, 321–329 CrossRef CAS
- P. E. Valverde, T. A. Green and S. Roy, J. Appl. Electrochem., 2020, 50, 699–712 CrossRef CAS
- V. S. Protsenko, A. A. Kityk, D. A. Shaiderov and F. I. Danilov, J. Mol. Liq., 2015, 212, 716–722 CrossRef CAS
- L. S. Bobrova, F. I. Danilov and V. S. Protsenko, J. Mol. Liq., 2016, 223, 48–53 CrossRef CAS
- M. Lukaczynska, E. A. Mernissi Cherigui, A. Ceglia, K. Van Den Bergh, J. De Strycker, H. Terryn and J. Ustarroz, Electrochim. Acta, 2019, 319, 690–704 CrossRef CAS
- Z. Liu, S. Z. E. Abedin and F. Endres, Electrochim. Acta, 2013, 89, 635–643 CrossRef CAS
- C. Du, H. Yang, X.-B. Chen, L. Wang, H. Dong, Y. Ning, Y. Lai, J. Jia and B. Zhao, J. Mater. Sci., 2018, 53, 10758–10771 CrossRef CAS
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed
- G. Zante, C. E. Elgar, K. George, A. P. Abbott and J. M. Hartley, Angew. Chem., Int. Ed., 2023, 62, e202311140 CrossRef CAS PubMed
- J. M. Hartley, S. Scott, R. Marin Rivera, P. Hunt, A. J. Lucio, P. Bird, R. Harris, G. R. T. Jenkin and A. P. Abbott, RSC Sustainability, 2023, 1, 107–116 RSC
- C. E. Elgar, S. Ravenhill, P. Hunt, B. Jacobson, A. Feeney, P. Prentice, K. S. Ryder and A. P. Abbott, Electrochim. Acta, 2024, 476, 143707 CrossRef CAS
- I. Tzanakis, G. S. Lebon, D. G. Eskin and K. A. Pericleous, Ultrason. Sonochem., 2017, 34, 651–662 CrossRef CAS PubMed
- J. R. Blake and D. C. Gibson, Annu. Rev. Fluid Mech., 1987, 19, 99–123 CrossRef
- T. B. Benjamin and A. T. Ellis, Colloids Surf., A, 1966, 260, 221–240 Search PubMed
- K. Yasui, Molecules, 2022, 27, 4788 CrossRef CAS PubMed
- R. W. Time and A. H. Rabenjafimanantsoa, Annu. Trans. Nord. Rheol. Soc., 2011, 19, 1–12 Search PubMed
- B. Jacobson, S. Li, R. Marin Rivera, P. Daly, C. E. Elgar, D. M. Mulvihill, A. P. Abbott, A. Feeney and P. Prentice, Ultrason. Sonochem., 2023, 101, 106701 CrossRef CAS PubMed
- S. Bao, B. Chen, Y. Zhang, L. Ren, C. Xin, W. Ding, S. Yang and W. Zhang, Ultrason. Sonochem., 2023, 98, 106525 CrossRef CAS PubMed
- A. J. Lucio, I. Efimov, O. N. Efimov, C. J. Zaleski, S. Viles, B. B. Ignatiuk, A. P. Abbott, A. R. Hillman and K. S. Ryder, Chem. Commun., 2021, 57, 9834–9837 RSC
- A. P. Abbott, G. Frisch, J. Hartley, W. O. Karim and K. S. Ryder, Prog. Nat. Sci., 2015, 25, 595–602 CrossRef CAS
-
S. Spathariotis, Recovery of Metals Using Deep Eutectic Solvents, PhD thesis, University of Leicester, 2019 Search PubMed
- P. Sebastian, M. I. Giannotti, E. Gómez and J. M. Feliu, ACS Appl. Energy Mater., 2018, 1, 1016–1028 CrossRef CAS
- J. M. Hartley, C. M. Ip, G. C. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed
- A. P. Abbott, G. Frisch, H. Garrett and J. Hartley, Chem. Commun., 2011, 47, 11876–11878 RSC
- O. K. Coskun, M. Munoz, S. Dongare, W. Dean and B. E. Gurkan, Langmuir, 2024, 40, 3283–3300 CAS
- F. Marken, R. P. Akkermans and R. G. Compton, J. Electroanal. Chem., 1996, 415, 55–63 CrossRef
- A. P. Abbott, R. C. Harris and K. S. Ryder, J. Phys. Chem. B, 2007, 111, 4910–4913 CrossRef CAS PubMed
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972–4981 CrossRef CAS PubMed
- W. Liu, S. J. Borg, D. Testemale, B. Etschmann, J.-L. Hazemann and J. Brugger, Geochim. Cosmochim. Acta, 2011, 75, 1227–1248 CrossRef CAS
- P. Pan and N. J. Susak, Geochim. Cosmochim. Acta, 1989, 53, 327–341 CrossRef CAS
- Y. Tian, B. Etschmann, W. Liu, S. Borg, Y. Mei, D. Testemale, B. O’Neill, N. Rae, D. M. Sherman, Y. Ngothai, B. Johannessen, C. Glover and J. Brugger, Chem. Geol., 2012, 334, 345–363 CrossRef CAS
- T. M. Abdel-Fattah and J. D. Loftis, Molecules, 2020, 25, 5712 CrossRef CAS PubMed
- A. A. Kityk, V. S. Protsenko, F. I. Danilov, O. V. Kun and S. A. Korniy, Surf. Coat. Technol., 2019, 375, 143–149 CrossRef CAS
- J. M. Hartley, J. Allen, J. Meierl, A. Schmidt, I. Krossing and A. P. Abbott, Electrochim. Acta, 2022, 402, 139560 CrossRef CAS
- M. L. P. Reddy and J. Saji, Miner.Process. Extr. Metall. Rev., 2002, 23, 199–227 CrossRef CAS
- A. Kityk, V. Protsenko, F. Danilov, V. Pavlik, M. Hnatko and J. Šoltýs, Colloids Surf., A, 2021, 613, 126125 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00030g |
| This journal is © The Royal Society of Chemistry 2024 |