
Exploiting natural complexity for substrate controlled regioselectivity and stereoselectivity in tantalum catalysed hydroaminoalkylation†
Cameron H. M.
Zheng
 a,
Ben E.
Nadeau
a,
Heather L.
Trajano
*b and
Laurel L.
Schafer
*a
a,
Ben E.
Nadeau
a,
Heather L.
Trajano
*b and
Laurel L.
Schafer
*a
aDepartment of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: schafer@chem.ubc.ca
bDepartment of Chemical and Biological Engineering, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada
First published on 30th August 2024
Abstract
Naturally occurring and structurally diverse alkene-containing substrates, terpenes, provided a platform for establishing chemo-, regio-, and diastereoselective reactivity in tantalum catalysed hydroaminoalkylation. Naturally derived 1,3-butadienes revealed the unique regio- and diastereoselective (Z)-1,4-addition products accessible from isoprene and β-myrcene by hydroaminoalkylation. Selective terpene functionalisation, within an industrially produced turpentine mixture, demonstrates functionalisation specificity of β-pinene and limonene. Lastly, sesquiterpene functionalisation using β-caryophyllene and humulene provide rare examples of trisubstituted alkene reactivity in hydroaminoalkylation, by leveraging strain-release and stereoelectronic effects to control chemoselectivity. As a result of these reactivity studies using natural substrates, new tools for understanding alkene electronic, strain, and stereoelectronic effects on chemo- and diastereoselectivity outcomes have revealed new mechanistic insights into hydroaminoalkylation.
Introduction
Structurally complex natural products have traditionally served as inspiring synthetic targets for chemists.1–3 Alternatively, natural products, such as terpenes, have been extensively utilised as low-cost enantiopure starting materials for synthesis (Fig. 1).4,5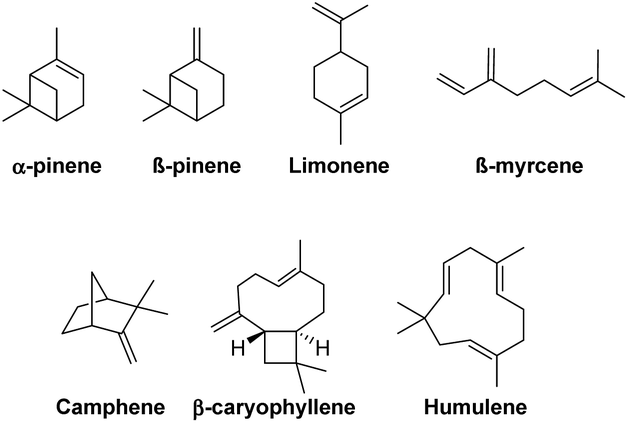 | ||
| Fig. 1 Structural variability in terpene frameworks. | ||
Terpenes themselves also have therapeutic applications ranging from antimicrobial agents to tumour suppressants.6–8 This natural bioactivity has inspired medicinal chemists to use terpenes as templates for the synthesis of terpenoid derivatives in which medicinal properties may be enhanced.9,10 Notably, terpenes contain reactive alkenes in their framework, and may contain other functional groups. The C–C double bonds of the alkene or their adjacent C–H bonds (R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR–CH2–) bonds may be transformed to yield functionalized synthetic terpenoid products bearing new C–C, C–Si, C–O, C–N, and C–P bonds for example.11–20
CR–CH2–) bonds may be transformed to yield functionalized synthetic terpenoid products bearing new C–C, C–Si, C–O, C–N, and C–P bonds for example.11–20
Amines are highly prevalent functional groups in pharmaceuticals and are featured in over 40% of drugs in the FDA's approved small molecule drug database.21 As a result, chemists are highly motivated to develop efficient methods for amination.22,23 Amination of terpene frameworks can result in enhanced bioactivity relative to their unfunctionalized precursors.24 Hydroaminoalkylation is a catalytic atom economic25 reaction which activates the α C–H bond of an amine, followed by addition across an unsaturated C–C bond (Scheme 1).26–28 This reaction produces two possible regioisomers, branched and linear, via 1,2-insertion or 2,1-insertion of the alkene, respectively. This transformation is catalysed by photoredox catalysis,29 early,30–34 and late35–38 transition metal catalysts. Generally, photoredox and late transition metal catalysis favour the linear product, while early transition metal catalysts favour the branched product.
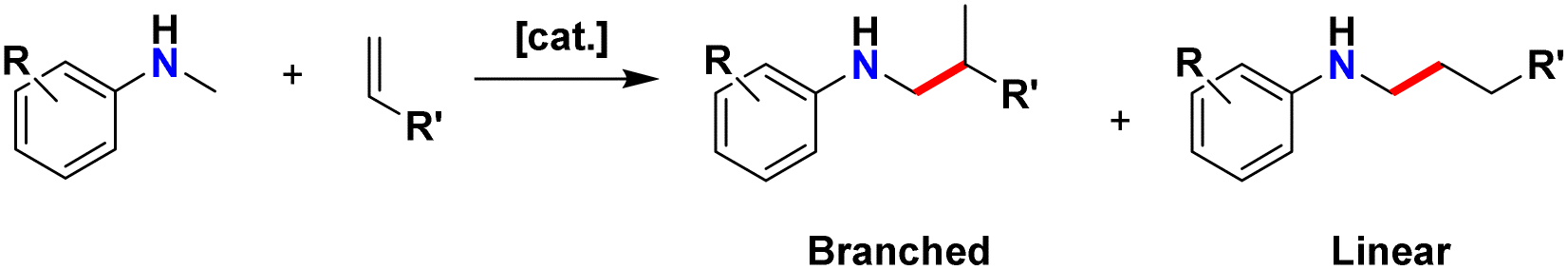 | ||
| Scheme 1 General hydroaminoalkylation reaction scheme. | ||
Our group has focused on the development of 1,3-N,O-chelated early transition metal hydroaminoalkylation catalysts using group 4 and 5 metals.30,39–44 The inclusion of a hemilabile 1,3-N,O-chelating ligand in the design of catalysts has greatly enhanced reactivity such that reaction temperatures and times have decreased from 160 °C to room temperature and from days to minutes.26,30,40,42 Now early transition metal hydroaminoalkylation catalysts bearing 1,3-N,O-chelates can be used with aryl, and alkyl amines,40,44 as well as N-heterocycles,31 for reactions with activated and unactivated, terminal and internal alkenes,40,41,45 as well as alkynes.39,46 Currently, in situ generated catalysts from Ta(CH2SiMe3)3Cl2 with ureate ligands are the state-of-the-art catalysts for the hydroaminoalkylation of challenging unactivated alkenes with excellent selectivity for the branched product.31,40,44,47 Further, these catalysts can be used with challenging macromolecular/polymeric substrates and under neat reaction conditions.45,48,49 Hydroaminoalkylation, as an atom-economic transformation with no by-products, can be readily coupled in synthetic sequences to efficiently assemble complex amine products. For example, we recently reported a telescoped sequence furnishing N-alkyl/arylated indoles, leveraging the unique bond-disconnection enabled by hydroaminoalkylation.50 As such, hydroaminoalkylation represents a Green Advance in the synthesis of amine and N-heterocyclic molecules and materials.
Terpenes, with their inherent structural complexity, are important renewable and abundant biomass-derived starting materials for a range of hydrofunctionalisation reactions.14–17,19,51–56 To date, terpenes have been rarely explored in hydroaminoalkylation reactivity (Scheme 2).47 We previously reported exquisite chemoselectivity for the geminal di-substituted alkene of limonene, over reactivity with the trisubstituted alkene, and diastereospecific reactivity with β-pinene to generate the branched product in both cases. No racemization of the existing stereocentres in either substrate was observed when using our catalyst, in contrast to late transition metal hydrofunctionalisation of terpenes.57
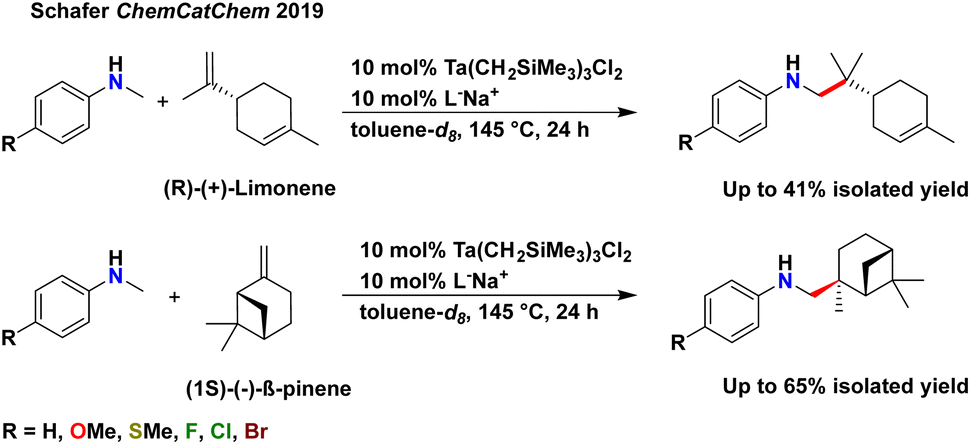 | ||
| Scheme 2 Previously demonstrated limonene and β-pinene functionalisation by hydroaminoalkylation.47 | ||
Here we expand on these results to show that a range of terpenes provide synthetic opportunities to enhance substrate scope and achieving the synthesis of varied synthetic terpenoid alkaloids. These structurally complex starting materials also provide a platform for developing mechanistic insights into chemo-, regio- and stereoselective reactivities in alkene hydroaminoalkylation. This work illustrates how the natural complexity of renewable terpene starting materials, coupled with the principles of Green Chemistry,58 advance critical C–H functionalisation technologies for the direct amination of alkenes.
Results and discussion
Isoprene is a natural product produced by both plants and humans and is one of the most abundant non-methane volatile organic compounds in our atmosphere.59 This abundant natural product contains a 1,3-butadiene moiety which can serve as a source of naturally-found conjugated dienes. The use of 1,3-butadiene as substrates has not been reported previously in tantalum-catalysed hydroaminoalkylation, thus we were highly motivated to test the reactivity of isoprene (Scheme 3). Based upon established reactivity trends with terminal and gem-disubstituted alkenes,40,47 we anticipated the selective formation of branched product, Branched. | ||
| Scheme 3 Synthesis of 1 and 2 by hydroaminoalkylation. Yields were calculated by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. Product ratios were determined by GC. | ||
Here, 10 mol% Ta(CH2SiMe3)3Cl2 and sodium ureate ligand (L−Na+) generated the active tantalum-ureate catalyst in toluene-d8, for the functionalisation of isoprene with N-methylaniline. N-Methylaniline was chosen as amine for exploratory alkene reactivity studies as it is the most reactive amine with our Ta catalyst. Though our tantalum-ureate catalyst is typically capable of achieving high yields using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 amine:alkene ratio, the volatility of isoprene (∼34 °C boiling point) meant that 4 equivalents were needed to ensure substantial amounts of isoprene were not lost to evaporation during reaction set up. 1,3,5-Trimethoxybenzene (TMB) was used as internal standard to determine 1H NMR yields. Integration of the aniline product 1ortho C–H resonances at 6.44 ppm vs. the 6.13 ppm resonance of TMB and compared to the 6.35 ppm resonance of unreacted N-methylaniline allowed for in situ reaction monitoring. The preferred reaction conditions of Scheme 3 showed that the extra equivalents of isoprene also afforded a higher 1H NMR yield of 1 (52%) versus the use of only 1 equivalent of isoprene (ca. 23%).
:1 amine:alkene ratio, the volatility of isoprene (∼34 °C boiling point) meant that 4 equivalents were needed to ensure substantial amounts of isoprene were not lost to evaporation during reaction set up. 1,3,5-Trimethoxybenzene (TMB) was used as internal standard to determine 1H NMR yields. Integration of the aniline product 1ortho C–H resonances at 6.44 ppm vs. the 6.13 ppm resonance of TMB and compared to the 6.35 ppm resonance of unreacted N-methylaniline allowed for in situ reaction monitoring. The preferred reaction conditions of Scheme 3 showed that the extra equivalents of isoprene also afforded a higher 1H NMR yield of 1 (52%) versus the use of only 1 equivalent of isoprene (ca. 23%).
Surprisingly, 2D NMR spectroscopy revealed that the major product 1 resulted from 1,4-addition across the 1,3-butadiene to form the Z-diastereomer. Analysis by Gas Chromatography-Mass Spectrometry (GC-MS) of the reaction mixture prior to purification gave a 15:3:1 product ratio, further demonstrating high selectivity for the 1,4-regioisomer with Z-stereochemistry. This regioselectivity is different from our anticipated product and is different from selective 4,1-addition product obtained when using a zirconium tethered-bisureate catalyst system for the hydroaminoalkylation of isoprene.60
To determine if this regioselectivity was unique to isoprene, we extended the investigation to include the naturally-sourced monoterpene, β-myrcene. This substrate includes not only a 1,3-butadiene but a trisubstituted alkene within its structure. Like isoprene, 10 mol% of Ta(CH2SiMe3)3Cl2 and L−Na+ allowed for functionalisation of β-myrcene with N-methylaniline to access 2 in 50% NMR yield, where TMB served as internal standard (Scheme 3). After product isolation by silica gel chromatography, structural elucidation of major product 2 confirmed the preferred formation of the 1,4-addition product with Z-diastereoselectivity. Further analysis by GC-MS of the reaction mixture after heating revealed a product ratio of 17:1:1. Thus, early-transition metal ureate catalysts favour 1,3-diene isomerization for hydroaminoalkylation, and tantalum-ureates favour 1,4-addition while zirconium-ureates favour 4,1-addition.60
Reported examples of 1,3-diene hydroaminoalkylation using 1-phenyl-1,3-butadiene and titanium metal catalysts afford either a mixture of branched (3,4-addition, major product) and linear products (4,3-addition),61,62 with the 4,1-addition product being observed as a minor regioisomer in one case.33 Ruthenium hydroaminoalkylation catalysts also access 4,1-addition products from isoprene and hydantoins.38 A comparable metallaphotoredox hydroaminoalkylation system featuring an iridium and cobalt catalyst pairing produced the 1,4-addition product as the major product (2:1 ratio vs. the 4,3-addition product) from isoprene and N,N-dimethylaniline in a 2:1 E:Z ratio.29 With β-myrcene using the same metallaphotoredox system, the 4,1-addition product is favoured, rather than 1,4-addition, but with greater diastereoselectivity favouring the E-diastereomer (20:1 E:Z ratio). Our yields of a single product from isoprene and β-myrcene functionalisation are comparable to photoredox-based methodologies. We propose that the relatively high tantalum catalyst loadings, 10 mol%, needed to furnish 1 and 2 in ∼50% yield are due to trace impurities which may be present in our natural product substrates.
Extensive mechanistic investigations into early transition metal catalysed hydroaminoalkylation, both experimental43,63–65 and computational,66–68 have provided key insights that inform a proposed mechanism for the highly regio- and diastereoselective 1,4-addition outcome observed here (Scheme 4). The observation and isolation of related zirconium π-allyl intermediates resulting from 1,3-diene hydroaminoalkylation have also informed this proposal.60 Thus, in summary our proposed mechanism for tantalum catalysed 1,3-diene hydroaminoalkylation invokes readily isomerized tantalum π-allyl intermediates, coupled with a turnover limiting protonolysis step, to result in Curtin–Hammett control over regio- and stereoselectivity. A more detailed analysis is provided below.
 | ||
| Scheme 4 Proposed catalytic cycle for the hydroaminoalkylation of 1,3-butadienes to give the 1,4-addition product with Z-diastereoselectivity. | ||
For early transition metal catalysed hydroaminoalkylation of terminal alkenes, 1,2-insertion of alkene is preferred due to electronic effects which favour the build-up of negative charge on the primary carbon which adds α to metal position of the resultant expanded metallacycle.68,69 As a result, we may have expected an initial 4,3-insertion of 2-substituted-1,3-dienes into the reactive tantallaaziridine (Scheme 4). However, the diene offers a directing group effect, thereby favouring the formation of a π-allyl intermediate, thus facilitating the 2,1-insertion which ultimately produces the 1,4-addition product regioselectivity. In related zirconium catalysed hydroaminoalkylation of 1-phenyl-1,3-dienes, diene insertion was observed to be reversible.60 Thus, here we propose that the observed excellent regioselectivity for the 1,4-addition product with Z-diastereoselectivity can be attributed to the reversible formation of π-allyl intermediates that undergo turnover limiting protonolysis later in the catalytic cycle.69
With regioselective chemistry established with various terpene substrates, we wanted to test the robustness of our catalyst using a raw commercial distillate, turpentine. Turpentine is a fluid distilled at 150 °C from pine tree resin,51 particularly rich in α-pinene, β-pinene and limonene amongst other monoterpenes. Amounts of each of the terpene components can vary depending on tree species and its geographical location, or by the extraction method.51 Turpentine has been used throughout history as medicine, coatings, and as solvent.51,70,71 Advances in the functionalisation of select terpenes within a mixture would forgo the need to purify each terpene from the mixture prior to hydroaminoalkylation. Selective amination would result in the preparation of synthetic terpenoid alkaloids that could be easily separated from unreacted terpene starting materials.
Analysis by GC-MS and NMR spectroscopy of a sample of turpentine72 from our industry partner, Holmen AB, confirms the presence α-pinene (57 mol%), β-pinene (26 mol%), and limonene (9 mol%) as the most abundant monoterpenes. Although α-pinene constitutes 57% of our sample of turpentine, our attempts at hydroaminoalkylation using a commercially pure sample were not successful (see ESI†). This lack of reactivity is presumably due to the steric hinderance associated with this trisubstituted alkene.47 If α-pinene is non-reactive with our catalyst, we wondered if it could act as reaction solvent, thereby allowing us to avoid the addition of further solvent, such as toluene. Thus, turpentine would serve as a source of reactive alkenes, β-pinene and limonene, and the remainder would serve as reaction solvent (Scheme 5).
 | ||
| Scheme 5 Synthesis of 3 and 4 in near turpentine. TMB was used as internal standard for 1H NMR yields & ratios. | ||
Here, N-methylaniline was added to a suspension of 10 mol% Ta(CH2SiMe3)3Cl2 and L−Na+ in turpentine in a 1:1 amine:turpentine ratio. The neat reaction mixture was then heated at 145 °C for 24 hours. High vacuum was next applied after the reaction to remove all volatiles, and the resulting crude product mixture was dissolved in toluene-d8, and filtered over a Celite™-packed Pasteur pipette for 1H NMR yield quantification. A yield of 38% of 3 and 70% of 4 were measured by 1H NMR spectroscopy, respectively. Product formation was confirmed by the appearance of new signals corresponding to the product methyl C–H resonances at 0.99 and 0.72 ppm for 3 and 4, respectively, after hydrofunctionalisation (Scheme 5). Product formation was further confirmed by GC (see ESI†). Notably, selective reactivity with the geminally substituted alkene of limonene was observed and there was no reactivity with the endocyclic tri-substituted alkene. Finally, purification via filtration through a silica gel plug provided a 1.7:1 mixture of 3:4 in 57% isolated yield relative to the β-pinene and limonene starting material present in the turpentine mixture. This represents a total isolated yield of 20% of aminated material from the initial turpentine mixture. When toluene was added as reaction solvent, 3 and 4 were furnished in 33% and 60% NMR yields, respectively, further demonstrating the efficiency of the neat turpentine reaction (see ESI†). Thus, the hydroaminoalkylation of a turpentine mixture serves to demonstrate the capability our tantalum-ureate catalyst to react with select terpenes within a complex biomass-sourced alkene mixture containing at least 6 monoterpenes (see ESI† for monoterpene species).
Next, we sought to study the reactivity of our catalyst with structurally complex sesquiterpenes containing multiple alkenes within their frameworks. β-Caryophyllene and its isomer humulene are examples of sesquiterpenes and are notable for providing the ‘hoppy’ aroma in beer (Fig. 1).73,74 Both sesquiterpenes contain unique structural features such as trisubstituted alkenes as well as endo- or exocyclic disubstituted alkenes. These different unsaturated Csp2–Csp2 bonds create opportunities to explore factors controlling alkene chemoselectivity in hydroaminoalkylation.
We first tested β-caryophyllene for reactivity in the presence of our catalyst and N-methylaniline (Scheme 6). β-Caryophyllene contains an exocyclic geminal disubstituted alkene and an E-endocyclic trisubstituted alkene within its framework. Based upon reactivity trends with limonene and β-pinene we anticipated selective functionalization of geminal disubstituted alkene of β-caryophyllene.
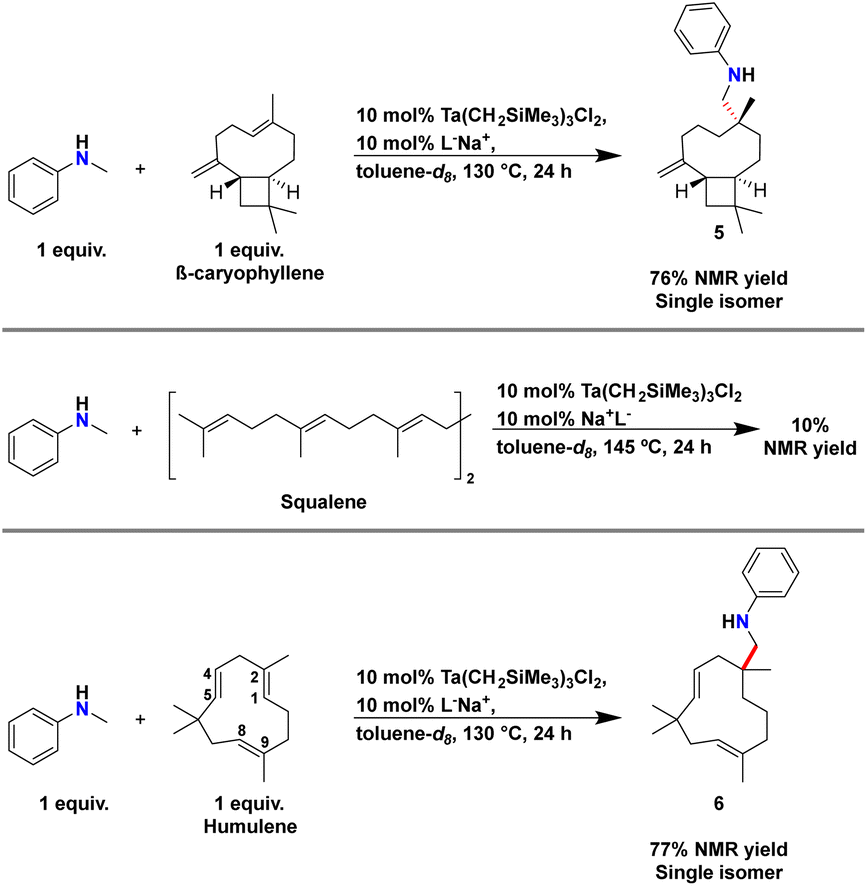 | ||
| Scheme 6 Synthesis of 5 and 6 by hydroaminoalkylation and attempt at squalene functionalisation. Yields were calculated by 1H NMR spectroscopy using TMB as internal standard. | ||
On NMR tube scale, 1 equivalent of N-methylaniline and 1 equivalent of β-caryophyllene were sequentially added to a solution of 10 mol% of Ta(CH2SiMe3)3Cl2 and L−Na+ in toluene-d8. The reaction mixture was heated to 130 °C for 24 hours.751H NMR spectroscopy was used to monitor the disappearance of the ortho C–H resonances of N-methylaniline and a new signal at 6.50 ppm for product formation (Scheme 6). A 1H NMR yield of 76% could be calculated for the new product by using TMB as internal standard. GC-MS analysis of the reaction mixture after heating confirmed that a single monoaminated product was formed.
Remarkably, structural analysis of this product by 2D NMR spectroscopy revealed the selective formation of 5, which results from selective hydroaminoalkylation of the endocyclic trisubstituted alkene while the geminal disubstituted alkene remained unreacted. Our structural assignment was particularly aided by diagnostic HSQC and HMBC NMR (see ESI† for discussion). The relative diastereoselectivity and connectivity were further confirmed by X-ray crystallography by generating an ammonium chloride salt from 5 (see ESI†). This remarkably chemoselective and diastereospecific reactivity at a sterically hindered site represents a dramatic departure from previously reported reactivity trends in hydroaminoalkylation.
Such reactivity with the trisubstituted alkene was completely unexpected. As a result, we questioned the driving force inherent to our terpene substrate that must be at play to unlock such unprecedented reactivity. It is established that E-endocyclic alkenes carry considerably more strain energy relative to their Z-endocyclic counterparts, therefore we propose ‘strain release’ within a cyclic framework could be a major driving force in this observed reactivity.76
To probe ‘strain release’ effects with our tantalum catalyst, the reactivity of squalane, a triterpene containing an acyclic series of non-conjugated E-trisubstituted alkenes, was tested with N-methylaniline (Scheme 6) using comparable reaction conditions. After heating at 145 °C for 24 hours, a 1H NMR yield of 10% was measured, which is consistent with no catalytic turnover. These results suggest that the strain of E-trisubstituted endocyclic alkenes provide a crucial driving force for unlocking challenging alkene functionalization. To confirm this hypothesis, we turned our attention next to humulene which also bears 2 E-endocyclic trisubstituted alkenes as well as an E-endocyclic disubstituted alkene in its framework (Scheme 6).
Similar to the synthesis of 5, 1 equivalent of each N-methylaniline and humulene were added to a solution of 10 mol% of Ta(CH2SiMe3)3Cl2 and L−Na+ in toluene-d8. The reaction mixture was then heated at 130 °C for 24 hours. 1H NMR spectroscopy was used to monitor the appearance of a new set of signals for the ortho C–H resonances of the aniline moiety at 6.51 ppm and once again, 2D NMR spectroscopic analysis indicated the formation of 6, a product from trisubstituted alkene functionalisation. A 1H NMR yield of 77% was determined and GC-MS analysis of the reaction mixture after heating confirmed that a single monoaminated product was produced.
Specifically, trisubstituted alkene C1–C2 was selectively functionalised according to structural analysis by 2D NMR spectroscopy of 6. HSQC and HMBC correlations also suggest that C4–C5 and C8–C9 remain untransformed (see ESI† for discussion). It is interesting that the E-disubstituted alkene remains unreacted, even though we have reported the successful functionalization of similar alkenes with tantalum-ureate catalysts.40 This remarkable chemoselectivity in hydroaminoalkylation is consistent with substrate strain-release being a critical driving force for controlling hydroaminoalkylation reactivity. However, the exquisite selectivity for C1–C2 over C8–C9 in hydroaminoalkylation required further analysis.
To further support the structure for 6, as assigned by NMR spectroscopy, we next sought to obtain a solid-state molecular structure. An ammonium chloride salt of 6 was generated from the addition of HCl to 6, an oil, in DCM. After 5 minutes of stirring, high vacuum was applied which furnished a crystalline powder (see ESI†). Next, crystals suitable for X-Ray Diffraction (XRD) were grown by Et2O diffusion into a saturated DCM solution of the hydrochloride salt (6·HCl) followed by slow solvent evaporation. The solid-state molecular structure of 6·HCl shows C1–C2 alkene functionalisation by hydroaminoalkylation and the hydrochlorination of C8–C9 (formed upon treatment with HCl) while the disubstituted alkene C4–C5 remains unfunctionalized. This result provides more evidence of the profound electronic effect of C1–C2 which dictates complete chemoselective reactivity in hydroaminoalkylation.
The selective formation of 5 and 6 show that strain release is dictating reactivity in hydroaminoalkylation. Such strain factors were previously demonstrated to be critical for high epoxidation reactivity as isocaryophyllene (a β-caryophyllene isomer with a Z-endocyclic trisubstituted alkene) was reported to be a magnitude less reactive than β-caryophyllene and humulene under the same reaction conditions.77 Furthermore, calculations and conformational analyses (bond lengths and angles) via X-ray crystal structures of β-caryophyllene, humulene, and their derivatives were used to rationalize the preferred reactivity of the same trisubstituted alkenes in epoxidation.12,77–81 Previous reports have attributed the selectivity observed with humulene to be due to the nearly perpendicular dihedral angle between the plane of the alkene and its allylic σC–C bonds, creating a strong hyperconjugative effect that maximizes electron density onto C1–C2 over C8–C9.80 We suggest that a combination of strain release as well as hyperconjugation within natural substrates work synergistically to overcome steric inhibition to realize selective trisubstituted alkene hydroaminoalkylation.
Previous computational results supporting hydroaminoalkylation reaction outcome analysis have asserted electronic features as being critical for regioselective reactivity of terminal alkenes.68 Recent results reported for the tantalum-ureate catalyst system used here have shown that substrate electronic effects in substituted styrene derivatives also impact regioselectivity.31 We attribute the high regioselectivity observed in β-caryophyllene and humulene to the preferred orientation of the least sterically hindered carbon of the alkene α-to tantalum in the formation of the aza-metallacyclopentane intermediate from alkene insertion. Like terminal alkenes, the less substituted carbon is appropriate for the accumulation of greater negative charge, resulting in preferred metallacycle formation with the electrophilic metal centre.68 Overall, this work has unlocked new strategies in hydroaminoalkylation reactivity through substrate-controlled alkene strain and stereoelectronics effects.
Conclusions
In summary, we report the use of naturally sourced terpenes as an abundant starting-material pool to install nitrogen functionality with high regio- and diastereoselectivity via tantalum catalysed hydroaminoalkylation. Naturally sourced 1,3-butadienes produced selectively the (Z)-1,4-addition product, a new achievement in early-metal catalysed hydroaminoalkylation. As well, we were able to demonstrate the selectivity of our catalyst to achieve select terpene functionalisation in a neat turpentine mixture to produce aminated β-pinene and limonene derivatives. New synthetic reactivity was explored with β-caryophyllene and humulene. These complex substrates revealed that electron rich, strained trisubstituted alkenes can undergo chemoselective functionalisation. We propose that the selectivity is dictated by stereoelectronic and strain release effects to yield unique aminated sesquiterpenoids. Overall, this work featuring terpene substrates has revealed new tools in hydroaminoalkylation for predicting chemoselective, diastereoselective, and regioselective outcomes in diene and alkene hydroaminoalkylation. These insights expand the understanding and substrate scope in the direct catalytic addition of amines to alkenes in a 100% atom economic transformation.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Author contributions
L. L. S. and H. L. T. directed this project and revised the manuscript. C. H. M. Z conducted all catalytic experiments and syntheses. B. E. N. collected XRD data and solved X-ray structures.Conflicts of interest
The authors declare the following competing financial interest(s): The Ta catalyst used in this work is patented (PCT/CA2018/050619).Acknowledgements
We thank Dr M. Ezhova of UBC NMR facility for assistance in characterization of product structures, and B. Herring of UBC Shared Instrument Facility for assisting in monoterpene identification in turpentine by GC.As well, we thank NSERC, Canada Research Chairs program, UBC Energy Reduction in Mechanical Pulping, and UBC for funding. We also thank Holmen AB for providing a sample of turpentine. B. E. N. and C. H. M. Z thank NSERC for scholarships.
This work was conducted as part of the Energy Reduction in Mechanical Pulping program, funded by a Collaborative Research and Development Grant provided by NSERC (Grant No. CRDPJ 538628-19) and the following partners: AB Enzymes, Alberta Newsprint Company, BC Hydro, BC Institute of Technology, Canfor, Paper Excellence, FPInnovations, Holmen Paper, McMaster University, Millar Western, The University of British Columbia Pulp and Paper Centre, University of Victoria, West Fraser, and Valmet, who we greatly thank for their continuous support.
References
- R. B. Woodward and W. E. Doering, J. Am. Chem. Soc., 1944, 66, 849 Search PubMed.
- A. Hinman and J. Du Bois, J. Am. Chem. Soc., 2003, 125, 11510–11511 CrossRef CAS PubMed.
- W. L. Scott and D. A. Evans, J. Am. Chem. Soc., 1972, 94, 4779–4780 CrossRef CAS PubMed.
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed.
- R. J. Nyamwihura and I. V. Ogungbe, RSC Adv., 2022, 12, 11346–11375 RSC.
- D. Cox-Georgian, N. Ramadoss, C. Dona and C. Basu, Med. Plants From Farm to Pharm, 2019, pp. 333–359 Search PubMed.
- S. J. Greay and K. A. Hammer, Phytochem. Rev., 2011, 14, 1–6 CrossRef.
- M. Zielińska-Błajet and J. Feder-Kubis, Int. J. Mol. Sci., 2020, 21, 7078 CrossRef PubMed.
- S. R. Ferrarini, C. S. Graebin, J. Limberger, R. F. Canto, D. O. Dias, R. G. da Rosa, M. de F. Madeira and V. L. Eifler-Lima, Mem. Inst. Oswaldo Cruz, 2008, 103, 773–777 CrossRef CAS PubMed.
- S. Liao, S. Shang, M. Shen, X. Rao, H. Si, J. Song and Z. Song, Bioorg. Med. Chem. Lett., 2016, 26, 1512–1515 CrossRef CAS PubMed.
- W. B. Cunningham, J. D. Tibbetts, M. Hutchby, K. A. Maltby, M. G. Davidson, U. Hintermair, P. Plucinski and S. D. Bull, Green Chem., 2020, 22, 513–524 RSC.
- C. Lescot, B. Darses, F. Collet, P. Retailleau and P. Dauban, J. Org. Chem., 2012, 77, 7232–7240 CrossRef CAS PubMed.
- M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed.
- Q. Wang, H. Jung, D. Kim and S. Chang, J. Am. Chem. Soc., 2023, 145, 24940–24951 CAS.
- C. S. Kuai, D. W. Ji, C. Y. Zhao, H. Liu, Y. C. Hu and Q. A. Chen, Angew. Chem., Int. Ed., 2020, 59, 19115–19120 CrossRef CAS PubMed.
- L. Bareille, S. Becht, J. L. Cui, P. Le Gendre and C. Moïse, Organometallics, 2005, 24, 5802–5806 CrossRef CAS.
- A. Perrier, V. Comte, C. Moïse and P. Le Gendre, Chem. – Eur. J., 2010, 16, 64–67 CrossRef CAS PubMed.
- A. Çelik, S. L. Flitsch and N. J. Turner, Org. Biomol. Chem., 2005, 3, 2930–2934 RSC.
- C. G. Vieira, M. C. De Freitas, K. C. B. De Oliveira, A. De Camargo Faria, E. N. Dos Santos and E. V. Gusevskaya, Catal. Sci. Technol., 2015, 5, 960–966 RSC.
- T. S. Chamblee, R. R. Weikel, S. A. Nolen, C. L. Liotta and C. A. Eckert, Green Chem., 2004, 6, 382–386 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- J. Li, J. S. Cisar, C. Y. Zhou, B. Vera, H. Williams, A. D. Rodríguez, B. F. Cravatt and D. Romo, Nat. Chem., 2013, 5, 510–517 CrossRef CAS PubMed.
- B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- R. C. DiPucchio, S.-C. Rosca and L. L. Schafer, J. Am. Chem. Soc., 2022, 144, 11459–11481 CrossRef CAS PubMed.
- P. M. Edwards and L. L. Schafer, Chem. Commun., 2018, 54, 12543–12560 RSC.
- M. Manßen and L. L. Schafer, Trends Chem., 2021, 3, 428–429 CrossRef.
- S. M. Thullen and T. Rovis, J. Am. Chem. Soc., 2017, 139, 15504–15508 CrossRef CAS PubMed.
- M. Manßen, D. Deng, C. H. M. Zheng, R. C. DiPucchio, D. Chen and L. L. Schafer, ACS Catal., 2021, 11, 4550–4560 CrossRef.
- R. C. DiPucchio, K. E. Lenzen, P. Daneshmand, M. B. Ezhova and L. L. Schafer, J. Am. Chem. Soc., 2021, 143, 11243–11250 CrossRef CAS PubMed.
- A. E. Nako, J. Oyamada, M. Nishiura and Z. Hou, Chem. Sci., 2016, 7, 6429–6434 RSC.
- J. Dörfler, T. Preuß, A. Schischko, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2014, 53, 7918–7922 CrossRef PubMed.
- T. Kaper, M. Fischer, H. Thye, D. Geik, M. Schmidtmann, R. Beckhaus and S. Doye, Chem. – Eur. J., 2021, 27, 6899–6903 CrossRef CAS PubMed.
- P. Verma, J. M. Richter, N. Chekshin, J. X. Qiao and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 5117–5125 CrossRef CAS PubMed.
- G. Lahm and T. Opatz, Org. Lett., 2014, 16, 4201–4203 CrossRef CAS PubMed.
- M. Schinkel, L. Wang, K. Bielefeld and L. Ackermann, Org. Lett., 2014, 16, 1876–1879 CrossRef CAS PubMed.
- D. C. Schmitt, J. Lee, A.-M. R. Dechert-Schmitt, E. Yamaguchi and M. J. Krische, Chem. Commun., 2013, 49, 6096–6098 RSC.
- E. N. Bahena, S. E. Griffin and L. L. Schafer, J. Am. Chem. Soc., 2020, 142, 20566–20571 CrossRef CAS PubMed.
- R. C. DiPucchio, S.-C. Roşca and L. L. Schafer, Angew. Chem., Int. Ed., 2018, 57, 3469–3472 CrossRef CAS PubMed.
- E. Chong, J. W. Brandt and L. L. Schafer, J. Am. Chem. Soc., 2014, 136, 10898–10901 CrossRef CAS PubMed.
- P. Garcia, Y. Y. Lau, M. R. Perry and L. L. Schafer, Angew. Chem., Int. Ed., 2013, 52, 9144–9148 CrossRef CAS PubMed.
- P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Angew. Chem., Int. Ed., 2009, 48, 8361–8365 CrossRef CAS PubMed.
- P. Daneshmand, S. C. Roşca, R. Dalhoff, K. Yin, R. C. Dipucchio, R. A. Ivanovich, D. E. Polat, A. M. Beauchemin and L. L. Schafer, J. Am. Chem. Soc., 2020, 142, 15740–15750 CrossRef CAS PubMed.
- M. Manßen, S. S. Scott, D. Deng, C. H. M. Zheng and L. L. Schafer, Green Chem., 2023, 25, 2629–2639 RSC.
- E. Nuñez Bahena, S. A. Sirohey and L. L. Schafer, Organometallics, 2023, 42, 1291–1299 CrossRef.
- R. C. DiPucchio, S. Rosca, G. Athavan and L. L. Schafer, ChemCatChem, 2019, 11, 3871–3876 CrossRef CAS.
- S. S. Scott, S. C. Roşca, D. J. Gilmour, P. Brant and L. L. Schafer, ACS Macro Lett., 2021, 10, 1266–1272 CrossRef CAS PubMed.
- S. S. Scott, B. Kaur, C. H. M. Zheng, P. Brant, D. J. Gilmour and L. L. Schafer, J. Am. Chem. Soc., 2023, 145, 22871–22877 CrossRef CAS PubMed.
- C. H. M. Zheng, D. A. Balatsky, R. C. Dipucchio and L. L. Schafer, Org. Lett., 2022, 24, 6571–6575 CrossRef CAS PubMed.
- M. Golets, S. Ajaikumar and J. P. Mikkola, Chem. Rev., 2015, 115, 3141–3169 CrossRef CAS PubMed.
- A. Perrier, M. Ferreira, J. N. H. Reek and J. I. Van Der Vlugt, Catal. Sci. Technol., 2013, 3, 1375–1379 RSC.
- H. C. Brown and N. N. Joshi, J. Org. Chem., 1988, 53, 4059–4062 CrossRef CAS.
- F. Xianjie, P. Yu and B. Morandi, Science, 2016, 351, 832–836 CrossRef PubMed.
- E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 382–411 CrossRef CAS.
- J. Zhang, C. G. Yang and C. He, J. Am. Chem. Soc., 2006, 128, 1798–1799 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. Lewandowski, M. Jaoui, J. H. Offenberg, J. D. Krug and T. E. Kleindienst, Atmos. Chem. Phys., 2015, 15, 3773–3783 CrossRef CAS.
- E. Nuñez Bahena, K. Hosseini, S. Galván Curto and L. L. Schafer, Chem. Sci., 2024 10.1039/D4SC00636D.
- T. Preuß, W. Saak and S. Doye, Chem. – Eur. J., 2013, 19, 3833–3837 CrossRef PubMed.
- J. Dörfler, T. Preuß, C. Brahms, D. Scheuer and S. Doye, Dalton Trans., 2015, 44, 12149–12168 RSC.
- J. M. Lauzon, P. Eisenberger, S.-C. Roşca and L. L. Schafer, ACS Catal., 2017, 7, 5921–5931 CrossRef CAS.
- M. Manßen, N. Lauterbach, J. Dörfler, M. Schmidtmann, W. Saak, S. Doye and R. Beckhaus, Angew. Chemie Int. Ed., 2015, 54, 4383–4387 CrossRef PubMed.
- A. L. Reznichenko and K. C. Hultzsch, J. Am. Chem. Soc., 2012, 134, 3300–3311 CrossRef CAS PubMed.
- G. Luo, F. Liu, Y. Luo, G. Zhou, X. Kang, Z. Hou and L. Luo, Organometallics, 2019, 38, 1887–1896 CrossRef CAS.
- N. Thoben, T. Kaper, S. de Graaff, L. Gerhards, M. Schmidtmann, T. Klüner, R. Beckhaus and S. Doye, ChemPhysChem, 2023, 24, e202300370 CrossRef CAS PubMed.
- F. Liu, G. Luo, Z. Hou and Y. Luo, Organometallics, 2017, 36, 1557–1565 CrossRef CAS.
- D. J. Gilmour, J. M. P. Lauzon, E. Clot and L. L. Schafer, Organometallics, 2018, 37, 4387–4394 CrossRef CAS.
- Z. Schelz, J. Molnar and J. Hohmann, Fitoterapia, 2006, 77, 279–285 CrossRef CAS PubMed.
- B. Mercier, J. Prost and M. Prost, Int. J. Occup. Med. Environ. Health, 2009, vol. 22, pp. 331–342 Search PubMed.
- Our sample of turpentine was extracted via steam recovery.
- S. T. Katsiotis, C. R. Langezaal and J. J. C. Scheffer, Planta Med., 1989, 55, 634–634 CrossRef.
- F. Francomano, A. Caruso, A. Barbarossa, A. Fazio, C. La Torre, J. Ceramella, R. Mallamaci, C. Saturnino, D. Iacopetta and M. S. Sinicropi, Appl. Sci., 2019, 9, 5420 CrossRef CAS.
- Increasing reaction temperature results in lower product yields.
- K. J. Shea and J. S. Kim, J. Am. Chem. Soc., 1992, 114, 3044–3051 CrossRef CAS.
- B. Steenackers, A. Neirinckx, L. De Cooman, I. Hermans and D. De Vos, ChemPhysChem, 2014, 15, 966–973 CrossRef CAS PubMed.
- P. J. Li, G. Dräger and A. Kirschning, Org. Lett., 2019, 21, 998–1001 CrossRef CAS PubMed.
- E. Grau and S. Mecking, Green Chem., 2013, 15, 1112–1115 RSC.
- U. Neuenschwander, B. Czarniecki and I. Hermans, J. Org. Chem., 2012, 77, 2865–2869 CrossRef CAS PubMed.
- S. Wang, Y. Li, H. Zhou, L. Wang and R. Wang, J. Org. Chem., 2022, 87, 8648–8655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2238920 and 2344466. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc01614a |
| This journal is © The Royal Society of Chemistry 2024 |