
Molecular structural engineering of donor–acceptor-based porous organic polymers for sulfide photooxidation in water: a sustainable approach†
Neha
Saini
,
Kirti
Dhingra
,
Amit
Kumar
and
Kamalakannan
Kailasam
 *
*
Institute of Nano Science and Nanotechnology, Knowledge city, Sector-81, Manauli, SAS Nagar, 140306 Mohali, Punjab, India. E-mail: kamal@inst.ac.in
First published on 11th September 2024
Abstract
The utilization of mild and environment-friendly reaction conditions for the photocatalytic oxidation of sulfides to highly valuable sulfoxides represents a sustainable approach that is highly desirable yet quite challenging. Herein, we present a novel approach to enhance the photocatalytic oxidation of sulfides in water by molecular structural engineering of donor–acceptor (D–A) based polymeric networks. By incorporating electron-deficient heptazine or triazine units as acceptors and electron-rich 2,5-diamino fluorene as donors, a synergistic effect is achieved, promoting efficient charge separation upon light absorption. The two polymeric networks namely HEP-FL and TZ-FL efficiently carried out selective oxidation of sulfides to sulfoxide with 100% conversion within 1.3 h and 3.5 h, respectively under blue light irradiation. Through advanced spectroscopic and electrochemical measurements, the correlation between molecular structures and optoelectronic properties is elucidated, unveiling tunable band structures and exciton binding energies. Notably, the heptazine-containing polymeric network (HEP-FL) exhibited superior charge separation efficiency and enhanced catalytic activity, attributed to improved electron delocalization and reduced exciton binding energy. Additionally, we have performed green metrics calculations for the synthesis of sulfoxide using HEP-FL as a photocatalyst to prove the sustainability of the reaction system. These findings underscore the significant prospects of donor–acceptor-based polymeric networks as highly effective photocatalysts for selective oxidation reactions, highlighting their potential to advance environmentally conscious practices in organic synthesis and industrial applications.
Introduction
Photocatalysis is considered as one of the most promising strategies for developing the catalytic process for a sustainable future.1,2 Utilizing solar energy to produce value-added chemicals and fuels has witnessed unprecedented advancements in the past few decades. Researchers have made substantial strides in developing advanced functional materials geared toward efficient solar energy utilization for water splitting, CO2 reduction, photocatalytic degradation, biomass valorization, and various other organic transformation reactions.3–5 Among these, photocatalytic oxidation reactions represent a paradigm that has attracted significant interest as they provide a sustainable pathway for mild and green synthesis.6 Aerobic oxidation reactions are extremely important reactions, especially at industrial levels, but they suffer a great challenge of unselectively and over-oxidation due to lack of rational control of the reactive species generated from photogenerated electrons and holes.7–9The oxidation of sulfides to sulfoxides emerges as a compelling and consequential oxidation reaction, finding wide utility in organic synthesis and serving as active components in the pharmaceutical and agrochemical sectors.10–13 Many conventional methods for oxidizing sulfides to sulfoxides rely on oxidants such as m-CPBA, TBHP, UHP, IBX, or Oxone. These methods often involve excessive amounts of oxidants, catalysts, or high temperatures, leading to the formation of undesired byproducts. To mitigate these challenges and advance environmentally conscious practices, ongoing research endeavors focus on the development of novel catalysts and sustainable strategies.
In this regard, oxygen is considered a green and mild oxidant, which offers several advantages including its ambient nature, abundant availability, and harmlessness at ambient pressure.14,15 Consequently, the use of molecular oxygen as an oxidant can be considered the most environmentally conscious approach for oxidation reactions, aligning with the principles of sustainability and green chemistry. However, its activation is a challenging task due to the substantial kinetic barriers involved.
In the pursuit of efficient photocatalytic systems for sulfide oxidation to sulfoxides, inorganic photocatalytic materials like Bi4O5Br216 and Pt/BiVO417 have been investigated but face limitations such as low activity under visible light and poor selectivity. To address these issues, researchers have explored molecular coordination complexes, organic–inorganic hybrid18 photocatalysts (Table S1†), including dye-TiO2 assemblies,19 CdS/C3N4,20 metal–organic frameworks21,22 such as Zr6-Irphen,23 and composites like C60@PCN-222.24 However, most of these systems often require the incorporation of a crucial redox mediator like TEMPO.25,26 Despite these efforts, the photocatalytic activity of these systems remains relatively low for practical applications.
In recent years, there has been a growing interest in exploring porous organic polymer (POPs) based photocatalysts due to their inherent advantages, such as cost-effectiveness, high stability, facile separation and recovery processes.27,28 These materials have emerged as a prominent class of heterogeneous photocatalysts for selective transformations involving organic molecules in the presence of molecular oxygen.29–32 However, it remains challenging as the photocatalytic performance of most of the reported POPs is still unsatisfactory mainly due to the limited light utilization, fast recombination of charge carriers, inefficient charge generation, and charge transfer within the organic semiconductors. Addressing these limitations necessitates concerted efforts towards refining the molecular architecture of POPs to enhance photon absorption and exciton dissociation efficiencies. In this regard a novel strategy was conceived for the incorporation of donor–acceptor (D–A) units within the polymeric networks POPs.33–35
Herein, two polymeric photocatalysts with fluorene as donor unit and heptazine or triazine as the acceptor unit (named as HEP-FL and TZ-FL, respectively) were designed and fabricated using nucleophilic substitution reaction between 2,5-diamino fluorene and heptazine chloride or triazine chloride aiming at increased charge separation and high photocatalytic performance. Heptazine and triazine are highly electron-deficient units and act as excellent acceptors. They are the main repetitive units and the primary active centres within polymeric graphitic carbon nitride (g-C3N4), possess notable characteristics including high nitrogen density, which contributes to the photocatalytic activity of g-C3N4 by providing a greater number of reactive sites for chemical transformations.36 Additionally, heptazine and triazine possess π-conjugated systems leading to their interesting electronic and optical properties.37,38 By varying the acceptor unit of the polymeric photocatalysts, the electron affinities can be readily modulated, leading to tunable band structures and exciton binding energies. The correlation between molecular structures and optoelectronic properties of organic semiconductors is unveiled by a series of advanced spectroscopic and electrochemical measurements. Both of these were employed for the chemo-selective photocatalytic oxidation of sulfides to sulfoxide under blue light irradiation using molecular oxygen as an oxidant and water as solvent. The optimal sample, heptazine-containing photocatalyst HEP-FL exhibits efficient charge separation efficiency, thanks to improved electron delocalization and reduced exciton binding energy, resulting in superior photocatalytic activity of HEP-FL over TZ-FL. To the best of our knowledge, this is the first study that features the heterogeneous metal-free polymeric network for the photocatalytic selective aerobic oxidation of sulfides to sulfoxides using water as solvent. We anticipate that these findings will facilitate new possibilities for the development and implementation of environmentally conscious and sustainable protocols for selective organic transformations.
Experimental
Materials
All the chemicals unless specifically mentioned were commercially obtained and used without any further purification. 2,7 diamino fluorene, triazine chloride, N,N-diisopropylethylamine (DIPEA), dimethyl pyridine N-oxide (DMPO), methyl phenyl sulfide (thioanisole) and few of its substituted derivatives were purchased from TCI chemicals. Other substituted methyl phenyl sulfides were bought from BLD Pharma. Phosphorus(V) oxychloride, phosphorus pentachloride, and other common organic solvents were obtained from CDH. Melamine, 2,2,6,6-tetramethyl piperidine (TEMP) silver nitrate, deuterated chloroform, and potassium iodide (KI) was purchased from Sigma Aldrich.Synthesis of heptazine/triazine and fluorene-based POPs (HEP-FL/TZ-FL)
In a flame-dried 100 mL two-necked round bottom flask equipped with a magnetic stir bar, 2,7-diamino fluorene (1.5 eq.) was dissolved into anhydrous 1,4-dioxane (40 mL) followed by the addition of N,N-diisopropylamine (DIPEA; 2.064 mmol). The reaction mixture was purged with N2 for 20 min and then cooled to 15 °C. A solution of heptazine chloride/triazine chloride (1 eq.) in 10 mL anhydrous 1,4-dioxane was added dropwise to the above reaction mixture and stirred at 15 °C for 1 h followed by stirring for the next 1 h at room temperature. Finally, the reaction mixture was refluxed at 110 °C for 3 days. After completion of the reaction, the precipitates were filtered out and washed thoroughly with THF, MeOH, water, ethyl acetate, acetone, DMSO, and 1,4-dioxane followed by soxhlet purification with THF/MeOH for 48 h. The precipitates were dried overnight in a vacuum oven at 120 °C (reaction yield-84% for HEP-FL and 77% for TZ-FL).Results and discussion
Heptazine chloride and triazine chloride were specifically chosen as the monomeric unit for the synthesis of the polymeric networks due to their strong acceptor properties and possession of a highly labile C–Cl bond, which readily undergoes nucleophilic substitution reactions. So, herein we successfully synthesized donor acceptor-based polymeric networks namely, HEP-FL and TZ-FL through nucleophilic substitution reactions of heptazine chloride and triazine chloride (acceptor units) using 2,7-diamino fluorene which acts as donor units (Scheme 1). Notably, both resulting polymeric networks exhibited insolubility in a majority of organic solvents and aqueous solutions thus exhibiting their remarkable stability in different solvents.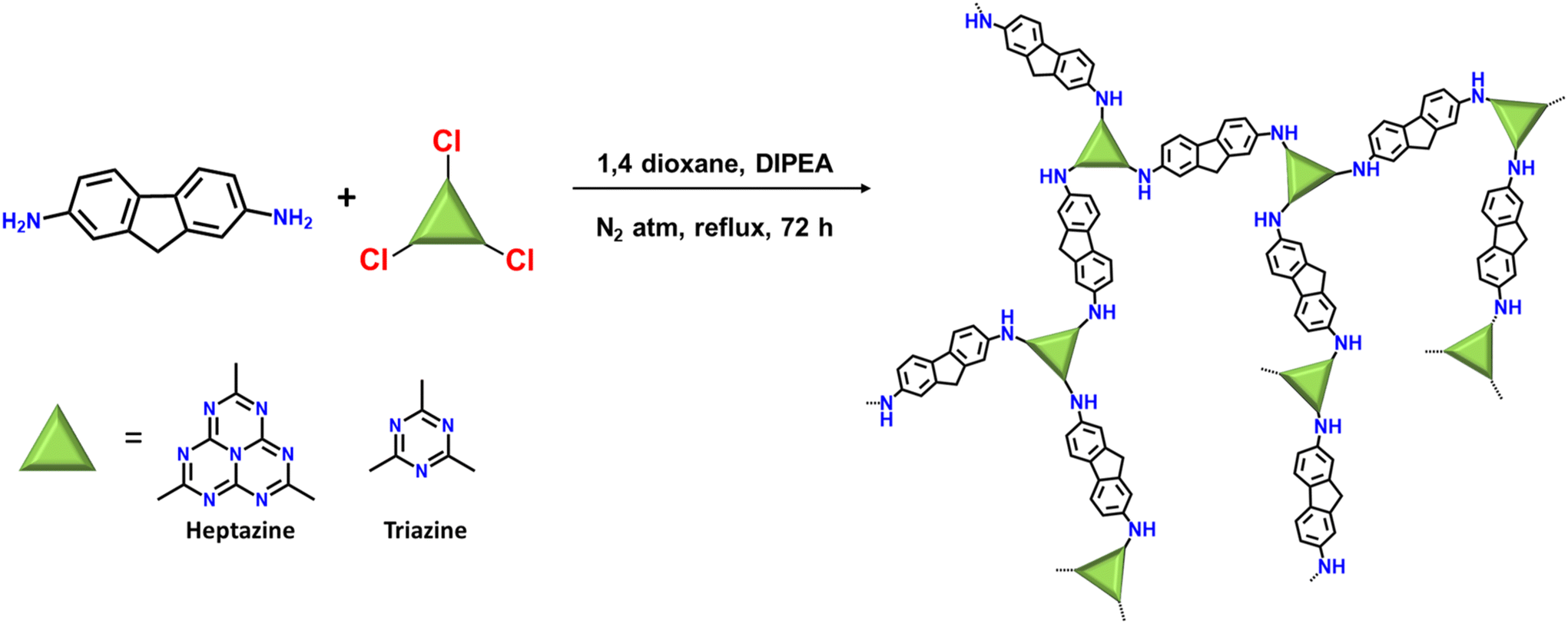 | ||
| Scheme 1 Synthesis scheme of HEP-FL and TZ-FL. | ||
The chemical structure of these polymeric networks was initially scrutinized using solid-state 13C CP/MAS NMR, confirming the successful integration of heptazine and triazine units in the polymeric network. The peaks observed at 166 and 168 ppm correspond to the heptazine and triazine carbons of HEP-FL and TZ-FL, respectively where the chlorine atoms are substituted by fluorene moiety forming a new C–N bond (Fig. 1a).39,40 The peak of heptazine carbon bonded to three nitrogen atoms, appeared at 156 ppm. The aromatic carbons of fluorene moiety gave rise to peaks in the range of 120–150 ppm. The peak at 37 ppm indicated the methylene carbons of fluorene.
 | ||
| Fig. 1 (a) 13C SS-NMR, (b) FTIR spectra, (c) and (d) high-resolution C 1s and N 1s of HEP-FL, (e) and (f) high-resolution C 1s and N 1s of HEP-FL, (g) and (h) FESEM images of HEP-FL and TZ-FL respectively. | ||
Further, the successful synthesis of HEP-FL and TZ-FL was corroborated using the FTIR technique (Fig. 1b). The unambiguous indication of the presence of the heptazine and triazine moiety in the polymeric networks was demonstrated by breathing mode vibration at 800 cm−1 and 796 cm−1, respectively. In contrast, the absence of the C–Cl stretching vibration at 942 cm−1 and 850 cm−1 in the case of HEP-FL and TZ-FL, respectively served as evidence that heptazine chloride and triazine chloride efficiently underwent substitution.40 Moreover, the peaks at 1620 cm−1 corresponded to the characteristic peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the heterocycles. This along with the aromatic vibrations of FL between 1470 to 1600 cm−1 validated the presence of both the precursors in their respective polymeric network. Additionally, the high-resolution C 1s spectrum of HEP-FL showed peaks at 284.4 eV and 288.0 eV and TZ-FL showed peaks at 284.4 eV and 287.6 eV corresponding to C–C/CC and CN linkages, respectively. N 1s high-resolution XPS spectrum of HEP-FL was fitted into three peaks at 398.4 eV (CN of heptazine), 399.7 eV (C3–N), and 400.5 eV (C–N) (Fig. 1d). For TZ-FL, the N 1s XPS spectrum was deconvoluted into three peaks at 398.1 eV (CN triazine), 399.5 eV (C–N), and 400.2 eV (N–H) (Fig. 1f). The thermal and chemical stabilities of the synthesized polymers were then characterized by thermogravimetric analysis (TGA) and FTIR measurements. The thermogravimetric analysis (TGA) carried out under the N2 atmosphere showed that HEP-FL is thermally stable up to 540 °C, whereas the TF-FL is stable up to 300 °C (Fig. S3†). To check their chemical stabilities, the as-synthesized polymeric networks were immersed under different solvents for 3 days and then recorded the FTIR spectra of the collected powder (Fig. S4 and S5†). The main peaks in FTIR spectra of all the samples soaked in THF, dioxane, hexane, acetonitrile (ACN), methanol (MeOH), ethanol (EtOH), dichloromethane (DCM), dimethyl sulfoxide (DMSO), acetone, water, 6 N HCl and 6 N NaOH respectively, appear at the same wavenumber indicating that both HEP-FL and TZ-FL possess high chemical stability in wide organic solvents (Fig. S6 and S7†). The morphologies of the synthesized polymeric network were investigated using FESEM (Fig. 1g and h). They both demonstrate aggregated particle-like morphology with irregularity in the structure and absence of long-range order. Further, the broad peak obtained in the PXRD spectra revealed that both HEP-FL and TZ-FL are amorphous in nature (Fig. S8†). The disorderly growth of the polymeric networks observed could be attributed to the irreversible kinetic control exhibited during the polymerization process.39
N in the heterocycles. This along with the aromatic vibrations of FL between 1470 to 1600 cm−1 validated the presence of both the precursors in their respective polymeric network. Additionally, the high-resolution C 1s spectrum of HEP-FL showed peaks at 284.4 eV and 288.0 eV and TZ-FL showed peaks at 284.4 eV and 287.6 eV corresponding to C–C/CC and CN linkages, respectively. N 1s high-resolution XPS spectrum of HEP-FL was fitted into three peaks at 398.4 eV (CN of heptazine), 399.7 eV (C3–N), and 400.5 eV (C–N) (Fig. 1d). For TZ-FL, the N 1s XPS spectrum was deconvoluted into three peaks at 398.1 eV (CN triazine), 399.5 eV (C–N), and 400.2 eV (N–H) (Fig. 1f). The thermal and chemical stabilities of the synthesized polymers were then characterized by thermogravimetric analysis (TGA) and FTIR measurements. The thermogravimetric analysis (TGA) carried out under the N2 atmosphere showed that HEP-FL is thermally stable up to 540 °C, whereas the TF-FL is stable up to 300 °C (Fig. S3†). To check their chemical stabilities, the as-synthesized polymeric networks were immersed under different solvents for 3 days and then recorded the FTIR spectra of the collected powder (Fig. S4 and S5†). The main peaks in FTIR spectra of all the samples soaked in THF, dioxane, hexane, acetonitrile (ACN), methanol (MeOH), ethanol (EtOH), dichloromethane (DCM), dimethyl sulfoxide (DMSO), acetone, water, 6 N HCl and 6 N NaOH respectively, appear at the same wavenumber indicating that both HEP-FL and TZ-FL possess high chemical stability in wide organic solvents (Fig. S6 and S7†). The morphologies of the synthesized polymeric network were investigated using FESEM (Fig. 1g and h). They both demonstrate aggregated particle-like morphology with irregularity in the structure and absence of long-range order. Further, the broad peak obtained in the PXRD spectra revealed that both HEP-FL and TZ-FL are amorphous in nature (Fig. S8†). The disorderly growth of the polymeric networks observed could be attributed to the irreversible kinetic control exhibited during the polymerization process.39
Next, the porous characteristics of these polymeric networks were analyzed by N2 physisorption measurements at 77 K. Both HEP-FL and TZ-FL showed characteristic type-IV isotherm indicating the mesoporous nature of the polymeric networks (Fig. S9†). The Brunauer–Emmett–Teller (BET) specific surface areas of HEP-FL and TZ-FL were found to be 72 m2 g−1 and 58 m2 g−1, respectively (Fig. S10†). The larger surface area of HEP-FL compared to TZ-FL could be attributed to the rigid structure of heptazine moiety which is composed of three fused triazine rings.40
Further, to examine their light-harvesting ability, the DR UV-vis technique was utilized. It was found that both of these polymeric networks absorb well in the visible range with an absorption maximum of 420 nm for HEP-FL and 460 nm for TZ-FL (Fig. 2a inset). Using the Tauc plot band gaps (Eg) of HEP-FL and TZ-FL were determined to be 2.47 eV and 1.72 eV, respectively (Fig. 2a). To determine the band potentials, Mott–Schottky analysis was carried out. The positive slope of the curve indicated that both the polymeric networks possess characteristics of n-type semiconductors (Fig. 2b and c). The X-intercept of the Mott–Schottky plot gives the value of flat band potential which is considered to lie 0.1 V below the conduction band (CB) minimum for an n-type semiconductor.41 Therefore, the CB potential (ECB) for HEP-FL and TZ-FL was found to be −0.79 V and −0.69 V vs. Ag/AgCl, respectively. These were converted into −0.59 V and −0.49 V vs. normal hydrogen electrode (NHE) respectively using the Nernst equation (ENHE = EAg/AgCl + 0.197, at pH = 7).42,43 Using the band energy equation, EVB = ECB + Eg, the valence band potential (EVB) was calculated to be 1.88 V and 1.20 V vs. NHE at pH = 7 (Fig. 2d). Both of these have the suitable band potential to reduce molecular oxygen to superoxide radical anion (O2/O2˙− −0.33 V vs. NHE at pH = 7).
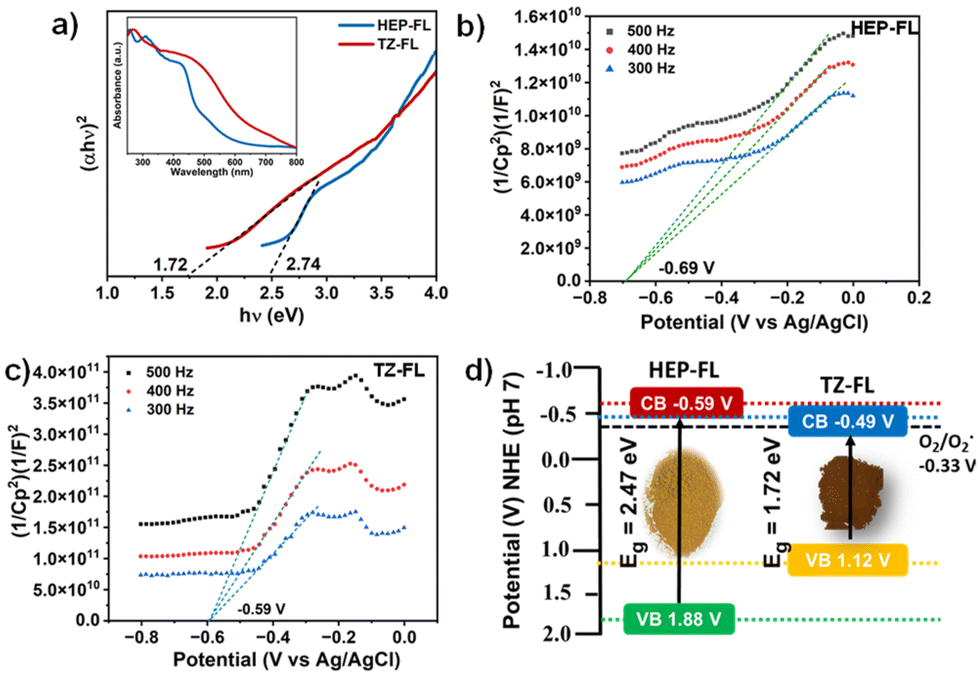 | ||
| Fig. 2 (a) Tauc plot (inset DR UV-vis spectrum), Mott–Schottky plot of (b) HEP-FL and (c) TZ-FL, and (d) band energy diagram. | ||
Photocatalytic oxidation of sulfides
Selective sulfide oxidation to sulfoxides is quite challenging yet highly demanding as these sulfoxides are the key intermediates of many pharmaceutically and industrially important compounds. Therefore, considering the excellent optoelectronic properties including visible light absorption and remarkable stability of these polymeric networks, they were eventually utilized for highly selective photocatalytic sulfide oxidation. Encouragingly, these networks exhibited impressive photocatalytic results for the oxidation of methyl phenyl sulfide under blue light.Firstly, a suitable solvent was scrutinized for the photooxidation of sulfides. Evidently, it can be inferred from Table 1 that solvent had a significant effect on the photocatalytic performance of the polymeric catalysts. No reaction is observed in THF and chloroform as solvent (Table 1, entries 1–4). Acetonitrile (ACN) which is one of the most commonly used solvents for photocatalytic reactions gave 8% and 23% conversion of sulfide with TZ-FL and HEP-FL (Table 1, entries 5 and 6). On replacing ACN with MeOH, the conversion was found to be increased. This could be attributed to the stabilization of the persulfoxide intermediate in polar protic solvent (Table 1, entries 7 and 8).44 By adding water to MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the conversion was further enhanced (Table 1, entries 9 and 10). This indicates the important role of water in accelerating the sulfide oxidation reaction. So, this encouraged carrying out the reaction in water which is a stronger polar protic solvent and green universal solvent. The reaction was highly efficient and indeed 100% conversion of methyl phenyl sulfide was observed in just 1.3 h using HEP-FL with selective formation of methyl phenyl sulfoxide (Table 1, entry 12). No over-oxidized product i.e., methyl phenyl sulfone was formed. Similar results were found using TZ-FL as photocatalyst, however, in this case it took a longer time for >99% sulfide conversion i.e., 3.5 h (Table 1, entry 11). Fig. 3a and b show that the methyl phenyl sulfide is linearly oxidized to sulfoxide by using HEP-FL and TZ-FL respectively, while maintaining the >99% product selectivity. No over-oxidized product i.e., methyl phenyl sulfone was formed even after longer irradiation hours.
:1), the conversion was further enhanced (Table 1, entries 9 and 10). This indicates the important role of water in accelerating the sulfide oxidation reaction. So, this encouraged carrying out the reaction in water which is a stronger polar protic solvent and green universal solvent. The reaction was highly efficient and indeed 100% conversion of methyl phenyl sulfide was observed in just 1.3 h using HEP-FL with selective formation of methyl phenyl sulfoxide (Table 1, entry 12). No over-oxidized product i.e., methyl phenyl sulfone was formed. Similar results were found using TZ-FL as photocatalyst, however, in this case it took a longer time for >99% sulfide conversion i.e., 3.5 h (Table 1, entry 11). Fig. 3a and b show that the methyl phenyl sulfide is linearly oxidized to sulfoxide by using HEP-FL and TZ-FL respectively, while maintaining the >99% product selectivity. No over-oxidized product i.e., methyl phenyl sulfone was formed even after longer irradiation hours.
 | ||
| Fig. 3 Time study of photocatalytic sulfide oxidation using photocatalyst (a) HEP-FL and (b) TZ-FL; water contact angle of (c) HEP-FL and (d) TZ-FL. | ||
| S. No. | Catalyst | Solvent | Time | Conv.a (%) |
|---|---|---|---|---|
| Reaction conditions: 0.1 mmol of methyl phenyl sulfide, 10 mg photocatalyst, 3 mL solvent, O2 atmosphere, temperature 27 ± 0.5 °C, under 20 W blue LED light irradiation (λ = 450 nm).a Conv. (conversion) determined using 1H NMR; n.d. = not detected. | ||||
| 1 | HEP-FL | THF | 4 h | n.d. |
| 2 | TZ-FL | THF | 4 h | n.d. |
| 3 | TZ-FL | CHCl3 | 4 h | n.d. |
| 4 | HEP-FL | CHCl3 | 4 h | n.d. |
| 5 | TZ-FL | ACN | 4 h | 8% |
| 6 | HEP-FL | ACN | 4 h | 23% |
| 7 | TZ-FL | MeOH | 4 h | 17% |
| 8 | HEP-FL | MeOH | 4 h | 35% |
| 9 | TZ-FL | H2O:MeOH (1:2) |
4 h | 34% |
| 10 | HEP-FL | H2O:MeOH (1:2) |
4 h | 79% |
| 11 | TZ-FL | H2O | 3.5 h | >99% |
| 12 | HEP-FL | H2O | 1.3 h | >99% |
To get deeper insights into the structure-photocatalytic activity relationship and investigate the underlying reason for the better photocatalytic performance of HEP-FL compared to TZ-FL, various techniques were used. The interaction between the polymeric network and solvent (water) is an important parameter for determining the reaction kinetics and for this purpose, water contact angle measurements were carried out. As shown in Fig. 3c and d, the water contact angles of HEP-FL and TZ-FL were found to be 24.9° and 117°, respectively. A lower contact angle with water is an indicator of hydrophilic nature. The increased hydrophilicity of HEP-FL results in better interaction between catalyst and substrate leading to efficient charge transport.
The charge separation and transfer ability of any photocatalyst determines the fate of its photocatalytic performance. The steady-state photoluminescence (PL) spectra of HEP-FL and TZ-FL showed their emission maxima at 610 nm and 640 nm respectively (Fig. 4a). HEP-FL was found to have much lower PL intensity as compared to TZ-FL. Additionally, the lifetime of charge carriers was determined using time-resolved fluorescence lifetime decay spectroscopy (Fig. 4b). The decay spectra of HEP-FL and TZ-FL were fitted biexponentially, resulting in an average fluorescence lifetime of 3.41 ns and 2.71 ns, respectively. The two distinct time constants, namely, τ1 and τ2 represents the instantaneous relaxation (or the thermal relaxation) which corresponds to the non-radiative recombination and slow decay component which is associated with radiative recombination. Since the relaxation lifetime is inversely proportional to the recombination efficiency, so the longer relaxation time corresponds to the lesser recombination. It is evident from the TRPL data that there is minimised non-radiative and radiative recombination in HEP-FL than TZ-FL. Hence, there will be a greater number of charge carriers available in HEP-FL to carry out the photocatalytic process. This decrease in the PL intensity and longer fluorescence lifetime of HEP-FL as compared to TZ-FL implies the enhanced charge separation and lower recombination of charge carriers in HEP-FL. This could be explained based on the fact that heptazine has more electron-withdrawing ability in comparison to triazine. Therefore, heptazine being a better acceptor than triazine forms a superior donor–acceptor-based polymer with increased charge separation.
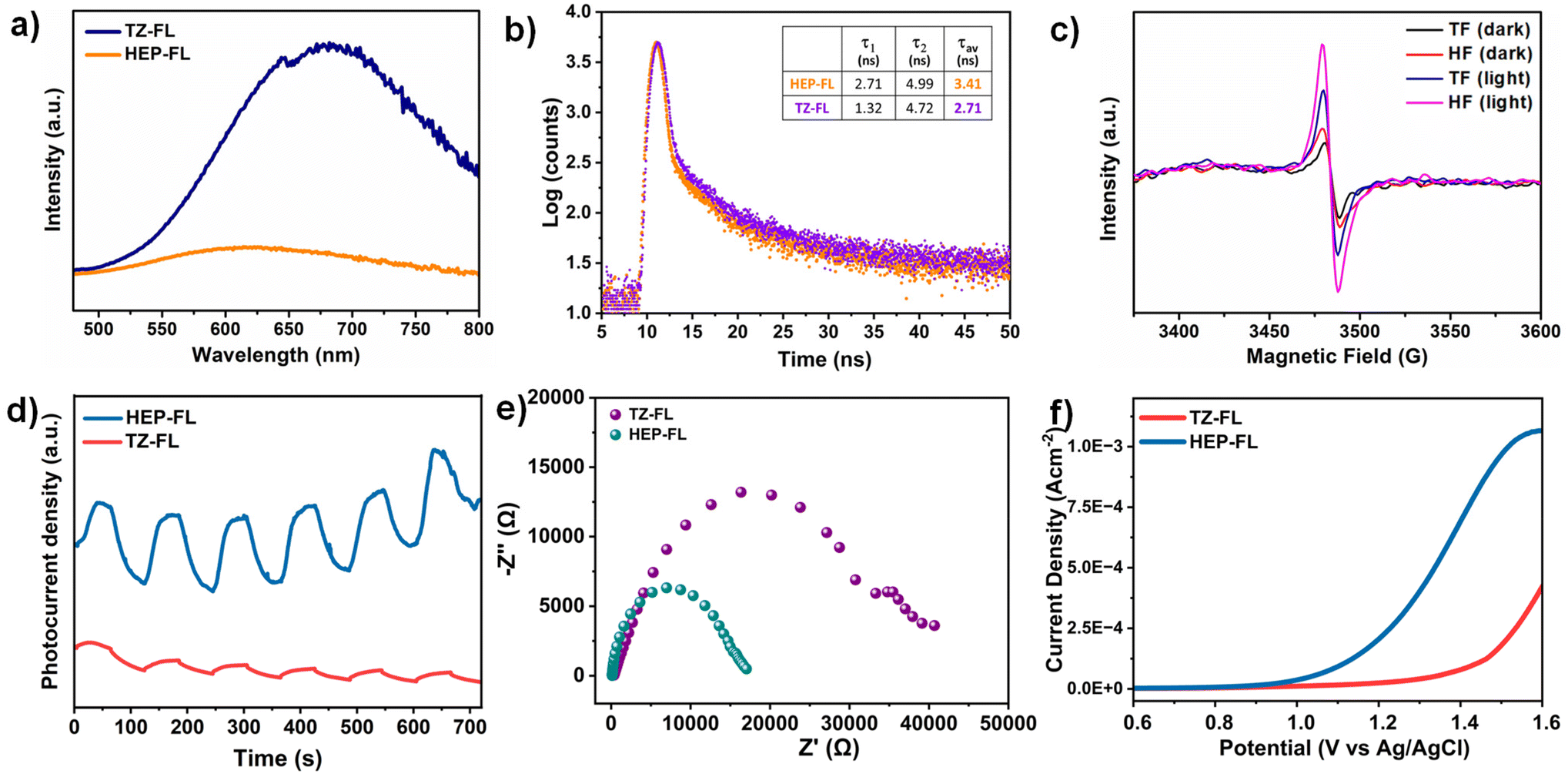 | ||
| Fig. 4 (a) Photoluminescence spectra, (b) fluorescence lifetime decay spectra, (c) EPR spectra (in dark and light), (d) transient photocurrent response, (e) Nyquist plot, and (f) LSV curve (in presence of methyl phenyl sulfide) of HEP-FL and TZ-FL. | ||
Further, to investigate the effect of acceptor units on the generation of charge carriers, charge transfer dynamics, and optoelectronic properties, electron paramagnetic resonance (EPR), transient photocurrent and (photo)electrochemical studies, respectively were employed. Upon visible light irradiation, HEP-FL displayed a more intense EPR signal in contrast to TZ-FL which demonstrates its superior ability to photogenerated electrons generation (Fig. 4c). HEP-FL exhibited a higher photocurrent response than TZ-FL under several cycles of intermittent on–off light experiments (Fig. 4d). The photocurrent response is positively related to enhanced generation and separation of photogenerated charge carriers in HEP-FL. Additionally, the reaction dynamics were analyzed using electrochemical impedance spectroscopy (EIS). As shown in the Nyquist plot, the arc radius for HEP-FL is much smaller than that of TZ-FL which indicates the lesser resistance to the charge carriers and thereby enhanced charge mobility in the case of HEP-FL (Fig. 4e).45 Moreover, we performed the LSV experiment in the presence of thioanisole to look into the charge transfer dynamics. As shown in Fig. 4f, HEP-FL shows increased anodic current as compared to TZ-FL which indicates HEP-FL facilitates better charge transfer for sulfide oxidation. All these experimental results unveil the crucial impact of monomer engineering in structuring the polymeric networks. Replacing the triazine with heptazine unit into the donor–acceptor-based polymeric network promotes electron delocalization and boosts charge separation which ultimately leads to better photocatalytic performance.
Substrate scope
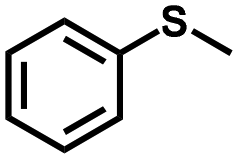
The convincingly established high photocatalytic efficacy of HEP-FL over TZ-FL, prompted an extension of the substrate exploration to encompass diverse sulfides, thereby investigating the universality of HEP-FL for such transformations. A variety of substituted sulfides underwent transformation into their respective sulfoxides, exhibiting exceptional selectivity and underscoring the broad applicability of HEP-FL in the context of photocatalytic selective oxidation. It is noteworthy that the conversion outcomes were contingent upon the structural attributes of the organic sulfides, as elucidated in Table 2. In the scenario of halogen substitution at the para-position of sulfides, the conversion rates were observed to align with the electronegativity of the respective halides. Electronic considerations elucidated that the contribution of the halogen atom to the reaction kinetics increased proportionally with its electronegativity, manifesting a hierarchy as F > Cl > Br. Additionally, a notable impact was observed when evaluating the different substituted positions, underscoring the significant influence of the chosen substitution position on the results. The ortho-substituted halides resulted in a decrease in the activity as compared to the para-substituted one, which could be attributed to the combined effects of steric hindrance and electronic effects. Moreover, 4-nitromethyl phenyl sulfide and 4-(methylthio) benzaldehyde, with strong electron-withdrawing groups were also converted into corresponding sulfoxides with good yields by extending the reaction time. In the case of electron-donating groups, –CH3 substituent showed better results as compared to –OCH3 substitution at para-position. In addition, aliphatic sulfide substrates were also converted into corresponding sulfoxides with >99% selectivity thus broadening the substrate scope of our photocatalyst.| S. No. | Substrate | Product | Time (h) | %Conv.a (%sel.) |
|---|---|---|---|---|
| Reaction conditions: 0.1 mmol of sulfide, 10 mg photocatalyst (HEP-FL), 3 mL H2O, O2 atmosphere, temperature 27 ± 0.5 °C, under 20 W blue LED light irradiation (λ = 450 nm).a Conv. (conversion) and selectivity (sel.) determined using 1H NMR. | ||||
| 1 |
|
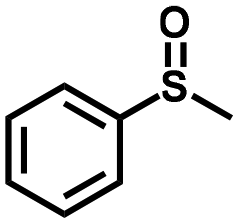
|
1.3 | >99 (>99) |
| 2 |
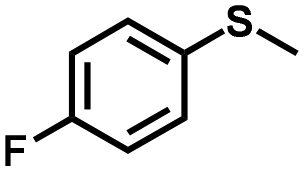
|
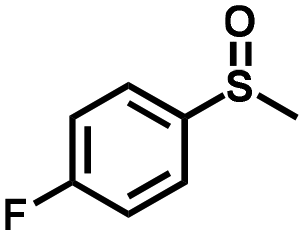
|
1.0 | >99 (99) |
| 3 |
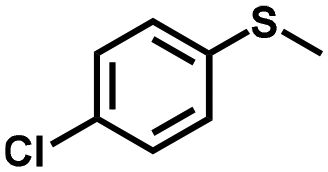
|
|
1.3 | >99 (99) |
| 4 |
|
|
1.8 | >99 (>99) |
| 5 |
|
|
2.0 | 99 (99) |
| 6 |
|
|
3.0 | 98 (>99) |
| 7 |

|
|
4.0 | 94 (>99) |
| 8 |
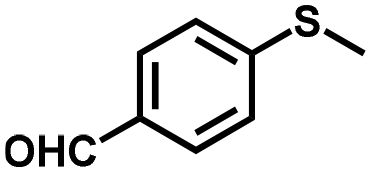
|
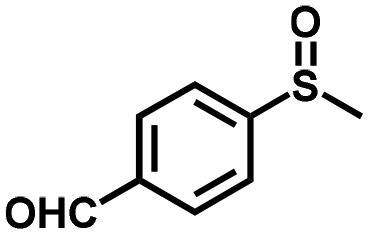
|
5.0 | 89 (>99) |
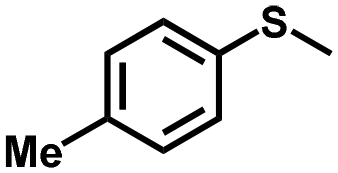
| 9 |
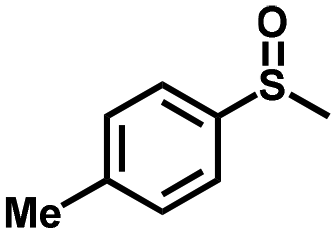
|
|
2.5 | 99 (98) |
| 10 |
|
|
2.0 | >99 (98) |
| 11 |
|
|
1.0 | >99 (>99) |
| 12 |

|
|
0.8 | >99 (>99) |
In addition, various green metrics parameters were calculated for the model reaction which are provided in Table S2.† To our delight, promising values of the green metric parameters were obtained which proves the environment-friendliness and sustainability of this photocatalytic reaction. Moreover, the green metrics for some of the reported catalysts were calculated and compared with those of our catalyst (Table S3†). Clearly, our catalyst outperformed most of these catalysts in terms of green metrics values and reaction conditions thus representing a significant step forward in developing greener catalytic processes.
Mechanistic insights
To unveil the underlying mechanism of photocatalytic studies, several controlled studies were carried out. These studies reveal that the presence of light and photocatalyst is essential for the progress of the reaction (Table 3, entries 1 and 2). On replacing oxygen with atmospheric air, there was no significant effect on the photocatalytic activity of HEP-FL (Table 3, entry 3). For any oxidation reaction, the holes play a significant role in oxidizing the substrate. This was further confirmed in the present reaction system when the conversion was drastically decreased by adding KI as a hole scavenger (Table 3, entry 4). To understand the mechanism deeply, it is important to determine the role of reactive oxygen species generated during the reaction. Generally, superoxide radicals (O2˙−) and singlet oxygen (1O2) have been reported in the literature as the key reactive intermediates in photocatalytic sulfoxidation reactions. In the presence of p-benzoquinone which is known to scavenge superoxide O2˙− radicals, the sulfide conversion was found to be 65% (Table 3, entry 5). For scavenging 1O2, sodium azide (NaN3) was used and the conversion decreased up to 48% (Table 3, entry 6). This shows that both O2˙− and 1O2 participate in photocatalytic sulfide oxidation reaction. Further, to rule out the possibility of hydroxyl radical OH˙ participation, IPA was used as OH˙ radical scavenger (Table 3, entry 7). No suppression for the sulfide conversion was observed which confirmed that OH˙ radical is not the reactive intermediate in this reaction. This is also supported from the band energy diagram (Fig. 2d), as the valence band potential of HEP-FL is less positive than the potential required for generating OH˙ radical (E° (H2O/OH˙) = 1.97 eV).| S. No. | Conditions | Conva (%) |
|---|---|---|
| Reaction conditions: 0.1 mmol of methyl phenyl sulfide, 10 mg photocatalyst (HEP-FL), 3 mL H2O, O2atmosphere, temperature 27 ± 0.5 °C, under 20 W blue LED light irradiation (λ = 450 nm).a Conv. (conversion) determined using 1H NMR; n.d. = not detected. | ||
| 1 | Dark | n.d. |
| 2 | Without catalyst | n.d. |
| 3 | Under air | 94% |
| 4 | KI (h+ scavanger) | 13% |
| 5 | p-BQ (O2˙− scavanger) | 65% |
| 6 | NaN3 (1O2 scavanger) | 48% |
| 7 | IPA (OH˙ scavanger) | >99% |
Furthermore, the generation of reactive oxygen species i.e., O2˙− and 1O2 was confirmed by in situ EPR experimental studies. DMPO was utilized to trap the superoxide radical.46 Under dark conditions, no signal was observed in the EPR spectra (Fig. 5a). On irradiation of light for 10 min, a quadruple signal was indeed observed corresponding to adduct formed between DMPO and O2˙−.46 For trapping singlet oxygen, TEMP was used. No signal was observed in the dark, but upon light irradiation, a triplet signal was observed corresponding to TEMPO which is formed by trapping of 1O2 by TEMP (Fig. 5b).46 Since the reaction was carried out using water as the solvent which itself can act as a source of oxygen and lead to simultaneous production of H2O2. To investigate this, the iodometric technique using UV-vis spectroscopy was used which confirmed the presence of H2O2 in the reaction mixture (Fig. S11, experimental details in ESI†) thereby demonstrating that water is also acting as a source of the oxygen atom for generation of sulfoxide.31 To validate this further the experiment was carried out in the absence of oxygen i.e., under Argon atmosphere where 42% conversion of methyl phenyl sulfide to corresponding sulfoxide (selectivity >99%) was observed in 1.3 h using HEP-FL as photocatalyst. This confirms that water also acts as the source of oxygen for sulfide oxidation. The protons of water were reduced to produce H2 which was detected using gas chromatography (GC) technique.
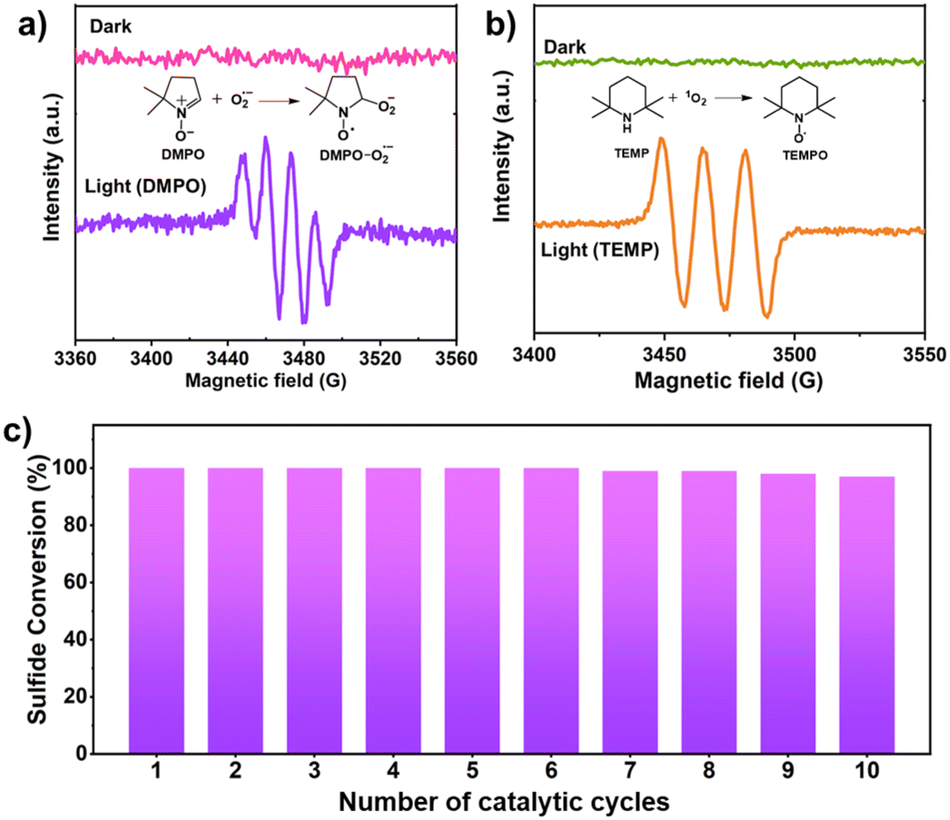 | ||
| Fig. 5 EPR detection of in situ generated (a) O2˙− with DMPO, (b) 1O2 with TEMP over HEP-FL, and (c) recyclability studies of HEP-FL. | ||
Based on the preceding investigations, the plausible mechanism for photocatalytic sulfide oxidation is delineated in Scheme 2. Upon blue light irradiation, the photocatalyst is excited to produce the photogenerated electrons and holes. The holes oxidize sulfide (I) to sulfide radical cation (II). The electrons on the other hand by single electron transfer (SET) to molecular oxygen (3O2) produce superoxide radical anion (O2˙−). The oxygen for the formation of sulfoxide can come from two sources: (i) Molecular oxygen or (ii) water. Based on these two possible pathways have been proposed (pathway 1 and pathway 2). In pathway 1 in which molecular oxygen acts as oxygen source for sulfoxide formation, the O2˙− radical reacts with (I) to produce persulfoxide anion (III) intermediate.26 Moreover, singlet oxygen (1O2) which is the ROS species generated by energy transfer, can combine with sulfide (I) to directly produce intermediate (III) which on subsequent proton abstraction gives peroxide cation intermediate (IV) (R2S+–OOH) and finally the desired sulfoxide.47 In pathway 2, the water acts as a source of oxygen for sulfoxide formation which was confirmed by the simultaneous production of H2O2 during the photocatalytic reaction (Fig. S11†).31 Herein, the sulfide radical cation (I) is hydrolyzed to form intermediate (V).31 Superoxide ion which is formed by the reduction of oxygen, on subsequent abstraction of a proton (H+) and H˙ radical from intermediate (V and VI), produces H2O2 and desired sulfoxide.
 | ||
| Scheme 2 Plausible mechanism for the photocatalytic sulfide oxidation under blue light irradiation. | ||
Recyclability
The investigation of robustness and reusability of the photocatalyst which are the critical parameters for its practical utility was conducted through recycling experiments. HEP-FL showed excellent performance even up to ten photocatalytic cycles without any significant loss of activity (Fig. 5c). The recovered catalyst, HEP-FL, underwent FTIR and FESEM characterization post ten catalytic runs, exhibiting consistent spectroscopic and structural profiles (Fig. S12 and 13†). Additionally, porosity analysis using N2 physisorption corroborated its physical stability (Fig. S14†). These findings demonstrate HEP-FL's robustness and suitability for sustained photocatalytic sulfide oxidation.Conclusions
In conclusion, we have successfully synthesized metal free-heptazine and triazine-based-polymeric networks which were eventually exploited as heterogeneous photocatalysts for highly selective photocatalytic sulfide oxidation under environment-friendly and ambient conditions. The mechanistic investigations revealed the important role of water not just as a solvent but also as the source of oxygen for sulfoxide formation. Controlled experiments and in situ EPR studies reveal the activation of molecular oxygen via both electron transfer (O2˙−) and energy transfer (1O2) to form sulfoxide. In comparison to TZ-FL, HEP-FL showed better photocatalytic performance which could be attributed to better charge mobility and lower recombination rate in the case of HEP-FL as confirmed by various experiments. Remarkably, the recycled photocatalyst HEP-FL could endure multiple catalytic cycles without any loss in the catalytic activity. Thus, the present work foreshadows the use of molecular-engineered organic networks as the green and sustainable approach for the upgradation of chemicals via photocatalytic organic transformation reactions.Author contributions
K. Kailasam and N. Saini conceptualized the idea for this manuscript. N. Saini designed and performed the experiments and analyzed the data. K. Dhingra and A. Kumar helped in characterization and data analysis. N. Saini prepared the manuscript draft with the help of K. Dhingra and A. Kumar.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
N. Saini thanks CSIR, New Delhi for the fellowship under grant 09/1129(0031)/2020-EMR-I. K. Kailasam thanks INST Mohali, for the fundings and instrumental facilities. The authors acknowledge Central Research Facility, IIT Delhi (Sonepat campus) for the EPR facility. K. Kailasam thanks Indo-French Centre for the Promotion of Advanced Research, CEFIPRA CSRP Project no. 70T10-2 for the financial support.References
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- R. Ballini, Eco-friendly Synthesis of Fine Chemicals, Royal Society of Chemistry, 2009 Search PubMed.
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC.
- C. Rosso, G. Filippini, A. Criado, M. Melchionna, P. Fornasiero and M. Prato, ACS Nano, 2021, 15, 3621–3630 CrossRef.
- N. Sharma, D. K. Chauhan, N. Saini and K. Kailasam, ACS Appl. Polym. Mater., 2023, 5, 4333–4341 CrossRef CAS.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- A. Rezaeifard, R. Haddad, M. Jafarpour and M. Hakimi, ACS Sustainable Chem. Eng., 2014, 2, 942–950 CrossRef CAS.
- C. O. Kinen, L. I. Rossi and R. H. de Rossi, J. Org. Chem., 2009, 74, 7132–7139 CrossRef CAS PubMed.
- K. Liu, J. Meng and X. Jiang, Org. Process Res. Dev., 2023, 27, 1198–1202 CrossRef CAS.
- Y. Uozumi and R. Niimi, Synfacts, 2022, 18, 897 CrossRef.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- Q. Fan, L. Zhu, X. Li, H. Ren, G. Wu, H. Zhu and W. Sun, Green Chem., 2021, 23, 7945–7949 RSC.
- Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898–2910 CrossRef CAS PubMed.
- N. L. Reed and T. P. Yoon, Chem. Soc. Rev., 2021, 50, 2954 RSC.
- W. Zhao, C. Yang, J. Huang, X. Jin, Y. Deng, L. Wang, F. Su, H. Xie, P. K. Wong and L. Ye, Green Chem., 2020, 22, 4884–4889 RSC.
- B. Zhang, J. Li, B. Zhang, R. Chong, R. Li, B. Yuan, S.-M. Lu and C. Li, J. Catal., 2015, 332, 95–100 CrossRef.
- Y. Zheng, H. Zhu, X. Xie, L. Yang, Q. Fan, Z. Le and Z. Xie, Green Chem., 2024, 26, 7031–7037 RSC.
- F. Huang, H. Hao, W. Sheng and X. Lang, Chem. Eng. J., 2021, 423, 129419 CrossRef.
- Y. Xu, Z.-C. Fu, S. Cao, Y. Chen and W.-F. Fu, Catal. Sci. Technol., 2017, 7, 587–595 RSC.
- H. Wei, Z. Guo, X. Liang, P. Chen, H. Liu and H. Xing, ACS Appl. Mater. Interfaces, 2019, 11, 3016–3023 CrossRef PubMed.
- X.-N. Zou, D. Zhang, T.-X. Luan, Q. Li, L. Li, P.-Z. Li and Y. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 20137–20144 CrossRef PubMed.
- L.-Q. Wei and B.-H. Ye, ACS Appl. Mater. Interfaces, 2019, 11, 41448–41457 CrossRef PubMed.
- D.-Y. Zheng, E.-X. Chen, C.-R. Ye and X.-C. Huang, J. Mater. Chem. A, 2019, 7, 22084–22091 RSC.
- B. Zeng, W. Sheng, F. Huang, K. Zhang, K. Xiong and X. Lang, Chem. Eng. J., 2023, 474, 145559 CrossRef.
- X. Dong, F. Zhang, Y. Wang, F. Huang and X. Lang, Appl. Catal., B, 2024, 345, 123660 CrossRef CAS.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- F. Zhang, H. Hao, X. Dong, X. Li and X. Lang, Appl. Catal., B, 2022, 305, 121027 CrossRef CAS.
- Q. Li, X. Lan, G. An, L. Ricardez-Sandoval, Z. Wang and G. Bai, ACS Catal., 2020, 10, 6664–6675 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef.
- Y. Li, T.-X. Luan, K. Cheng, D. Zhang, W. Fan, P.-Z. Li and Y. Zhao, ACS Mater. Lett., 2022, 4, 1160–1167 CrossRef.
- M. Deng, L. Wang, Z. Wen, J. Chakraborty, J. Sun, G. Wang and P. V. D. Voort, Green Chem., 2024, 26, 3239–3248 RSC.
- J. Li, S.-Y. Gao, J. Liu, S. Ye, Y. Feng, D.-H. Si and R. Cao, Adv. Funct. Mater., 2023, 33, 2305735 CrossRef.
- L. Wang and W. Zhu, Adv. Sci., 2024, 11, 2307227 CrossRef PubMed.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- S. Kumar, N. Sharma and K. Kailasam, J. Mater. Chem. A, 2018, 6, 21719–21728 RSC.
- P. Puthiaraj, Y.-R. Lee, S. Zhang and W.-S. Ahn, J. Mater. Chem. A, 2016, 4, 16288–16311 RSC.
- N. Sharma, S. Kumar, V. R. Battula, A. Kumari, A. Giri, A. Patra and K. Kailasam, Chem. – Eur. J., 2021, 27, 10649–10656 CrossRef CAS.
- D. Abdullatif, A. Khosropour, A. Khojastegi, I. Mosleh, L. Khazdooz, A. Zarei and A. Abbaspourrad, ACS Appl. Polym. Mater., 2023, 5, 412–419 CrossRef CAS.
- D. K. Chauhan, A. Verma, A. Jain, N. Saini, P. K. Prajapati, C. Bera and K. Kailasam, J. Mater. Chem. A, 2023, 11, 18672–18678 RSC.
- S. Behera, S. T. Aziz, N. Singla and B. Mondal, Chem. Commun., 2023, 59, 11528–11531 RSC.
- Z. Liu, J. Zhang, X. Li, R. Cui, J. Ma and R. Sun, iScience, 2023, 26(8), 107416 CrossRef CAS PubMed.
- L. Ding, Y. Zhao, Y. Li, C. Ge, L. Lv, H. Ji, W. Song, C. Chen and J. Zhao, J. Catal., 2023, 426, 328–335 CrossRef CAS.
- J. Cheng, Y. Wu, W. Zhang, J. Zhang, L. Wang, M. Zhou, F. Fan, X. Wu and H. Xu, Adv. Mater., 2024, 36, 2305313 CrossRef.
- X. Lan, J. Wang, Q. Li, A. Wang, Y. Zhang, X. Yang and G. Bai, ChemSusChem, 2022, 15, e202102455 CrossRef PubMed.
- J. Jiang, Z. Liang, X. Xiong, X. Zhou and H. Ji, ChemCatChem, 2020, 12, 3523–3529 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03255a |
| This journal is © The Royal Society of Chemistry 2024 |