
Augmentation of Pd-catalysed oxidative C–H/C–H carbonylation through alternating current electrosynthesis†
Haoran
Li‡
a,
Jiaqi
Peng‡
a,
Li
Zeng
*a,
Linpu
Zhou
a,
Muhammad
Shabbir
a,
Feiran
Xiao
b,
Jiaxin
Yuan
b,
Hong
Yi
 *a and
Aiwen
Lei
*ab
*a and
Aiwen
Lei
*ab
aCollege of Chemistry and Molecular Sciences, The Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China. E-mail: zengli123@whu.edu.cn; hong.yi@whu.edu.cn; aiwenlei@whu.edu.cn
bState Key Laboratory of Power Grid Environmental Protection, School of Electrical Engineering and Automation, Wuhan University, P. R. China
First published on 16th October 2024
Abstract
In light of the burgeoning biological applications associated with xanthones, the development of highly efficient synthetic methodologies for their production has emerged as a pivotal objective of chemical research. Amidst the array of available protocols, the oxidative carbonylation of diaryl ethers with carbon monoxide (CO) stands out as a notably uncomplicated route, often necessitating stoichiometric oxidants. Herein, we present a feasible approach employing unsymmetrical-waveform alternating current (AC) electrolysis to facilitate Pd-catalysed oxidative C–H/C–H carbonylation. Leveraging a straightforward catalytic system, we demonstrate the conversion of diverse diaryl ethers into xanthones with moderate to commendable yields. Our mechanistic investigations illuminate the indispensable role played by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in the electrochemical system, particularly its ability to recycle heterogeneous palladium species within the solution.
Xanthones play a pivotal role within the realms of medicine, biology, and pharmacology, attributable to their distinctive heterocyclic architectures (Fig. 1a).1–3 This particular structural motif is recognized as a “privileged structure”, endowed with the capacity to engage with a diverse spectrum of biomolecules.4,5 Consequently, substantial attention has been devoted to the synthesis of this structural moiety.6 Traditional methods for the assembly of xanthone frameworks encompass nucleophilic cyclization of 2,2′-dioxygenated benzophenones,7 electrophilic cycloacylation of 2-aryl oxybenzoic acids,8 and cyclization of 2-arylcarboxaldehydes or 2-arylbenzaldimines through C–H activation.9,10 In contrast to these methodologies, which necessitate rigorous conditions and result in the generation of chemical waste, the oxidative carbonylation utilizing carbon monoxide (CO) emerges as one of the most straightforward approaches for carbonyl compound construction.11–14 Within the context of our ongoing exploration into transition-metal-catalysed oxidative carbonylation, we have pioneered the inaugural palladium-catalysed oxidative carbonylation of diaryl ethers, facilitating the synthesis of xanthones (Fig. 1b).15 However, the submillimole reaction scale of this carbonylative strategy restricts its application because heterogeneous palladium species – “Pd black” – are difficult to convert to the active catalyst under stoichiometric oxidant conditions.16,17 Exploiting the efficient recycling of heterogeneous palladium species will be much more appealing.
 | ||
| Fig. 1 AC-promoted Pd catalysed C–H/C–H carbonylation (a) Bioactive molecules with xanthone skeletons. (b) Known synthetic methods of xanthones. (c) Unsymmetrical AC electrolysis recycles and activates heterogeneous palladium species. | ||
In light of the advancements in electrosynthesis and Pd-catalysed oxidative coupling reactions,18–26 we anticipate that substituting external stoichiometric oxidants with anodic oxidation presents a more environmentally sustainable protocol.27–30 Nonetheless, during the course of the direct current (DC) electrolysis, the generation of low oxidation state palladium, stemming from reductive elimination or cathodic reduction, may lead to the formation of substantial-sized particles.31,32 Such Pd-containing particles prove to be resistant to direct oxidation by electrodes.33,34 Therefore, the catalytic process necessitates the presence of an appropriate mediator and an efficient electrolytic system capable of re-oxidizing Pd(0) species (Fig. 1c, left). The realm of alternating current (AC) electrosynthesis has emerged as a burgeoning area within the domain of organic synthesis.35,36 In contrast to the conventional DC electrolysis, alternating current electrolysis introduces novel parameters, including waveform, frequency, and the duty ratio (i.e., the proportion of the positive half-period to the entire period) (Fig. 1c, right).37 These parameters endow researchers with the prospect to tune the course of the reaction precisely. Furthermore, the reductive metal foil present on the electrode's surface undergoes re-oxidation when the direction of the current changes.38–40 Consequently, both electrodes become available for oxidation and reduction, effectively doubling the active surface area of the electrodes and thereby enhancing reactivity.41 In this work, we present an AC electrolytic strategy for the attainment of oxidative C–H/C–H carbonylation under mild conditions, minimizing the formation of heterogeneous palladium species.
Results and discussion
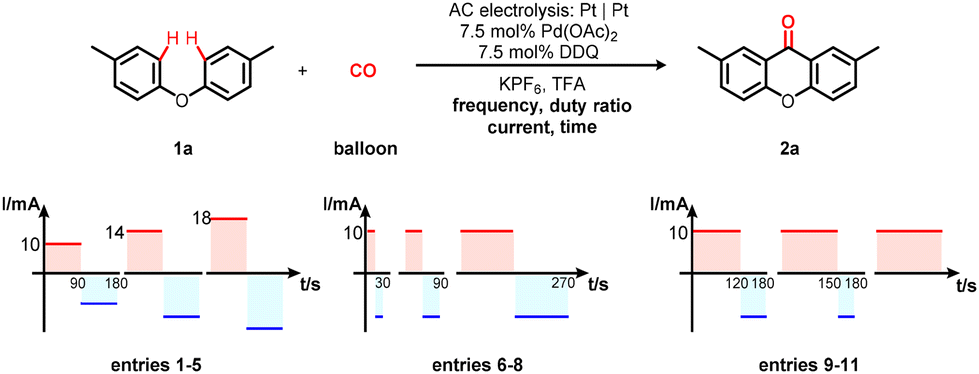
We started our research by applying 4-tolyl ether (1a) as a model substrate for palladium-catalysed electrochemical oxidative carbonylation reaction. By utilizing PdCl2 as the catalyst and BQ as the mediator, 2,7-dimethyl-9H-xanthen-9-one 2a was obtained with alternating current supply (square wave, 10 mA, 1/180 Hz and 50% duty ratio) in TFA at 50 °C for 2.0 h (Table 1, entry 1). Various palladium catalyst precursors were tested to improve reaction efficiency. Pd(PPh3)2Cl2 was not suitable for this transformation (Table 1, entry 2). Pd(OAc)2 was found to be the most suitable catalyst (Table 1, entries 3 and 4). In the next step, optimization of mediators was carried out (Table 1, entries 5–7). Reaction results showed that DDQ was an appropriate mediator, which furnished 2a in 64% yield (Table 1, entry 7). Investigation of supporting electrolytes was also carried out (Table 1, entries 8–10), indicating that LiClO4, NH4PF6, and Et4NPF6 were ineffective to promote the transformation. Control experiments revealed that both the palladium catalyst and the mediator were indispensable to afford the product (Table 1, entries 11 and 12).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | [Pd] | Mediator | Electrolyte | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a 1a (0.30 mmol), [Pd] (7.5 mol%), mediator (7.5 mol%), electrolyte (0.20 mmol) and TFA (1.5 mL) in an undivided cell with two Pt plates as electrodes, CO balloon, 1/180 Hz, D = 50%, 10 mA, 2.0 h, 50 °C. b The yield of 2a was determined by GC analysis with biphenyl as internal standard. n.d.: not detected, TFA: trifluoroacetic acid, BQ: p-benzoquinone, TEMPO: 2,2,6,6-tetramethylpiperidinyl-1-oxide. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | PdCl 2 | BQ | KPF6 | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Pd (PPh 3 ) 2 Cl 2 | BQ | KPF6 | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Pd(TFA) 2 | BQ | KPF6 | 54 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Pd(OAc) 2 | BQ | KPF6 | 57 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Pd(OAc)2 | KI | KPF6 | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Pd(OAc)2 | TEMPO | KPF6 | 41 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Pd(OAc)2 | DDQ | KPF 6 | 64 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Pd(OAc)2 | DDQ | NH 4 PF 6 | 11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Pd(OAc)2 | DDQ | Et 4 NPF 6 | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Pd(OAc)2 | DDQ | LiClO 4 | n.d. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | None | DDQ | KPF6 | n.d. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | Pd(OAc)2 | None | KPF6 | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Intrigued by the impact of AC parameters on the electrolytic process, initial endeavors concentrated on optimizing current and duration (Table 2, entries 1–5). These investigations revealed that a current of 14 mA and a duration of 1.5 hours formed a optimal combination, yielding compound 2a in a noteworthy 67% yield (Table 2, entry 5). Subsequently, various frequencies were explored to enhance reactivity; however, these efforts failed to yield favorable outcomes (Table 2, entries 6–8). It was imperative to emphasize that the adjustment of the duty ratio from 50% to 67% elicited a notable improvement, elevating the isolated yield of the desired product to an impressive 82% (Table 2, entries 9 and 10). It was pertinent to mention that reasonably respectable yields were also achievable via room-temperature AC electrolysis (Table 2, entry 12). Furthermore, AC electrolysis displayed significantly superior efficiency compared to its DC counterpart (Table 2, entry 11). Control experiments unequivocally established the indispensable role of electricity in effecting this transformation (Table 2, entry 13).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Current, time | Frequency | Duty ratio | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a 1a (0.30 mmol), Pd(OAc)2 (7.5 mol%), DDQ (7.5 mol%), KPF6 (0.20 mmol) and TFA (1.5 mL) in an undivided cell with two Pt plates as electrodes, 1 atm. CO, X Hz, D = Y%, Z mA, W h, 50 °C. b The yield of 2a was determined by GC analysis with biphenyl as the internal standard. c Isolated yield. d Room temperature. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 10 mA, 2 h | 1/180 Hz | 50% | 64 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 14 mA, 2 h | 1/180 Hz | 50% | 69 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 18 mA, 2 h | 1/180 Hz | 50% | 52 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 14 mA, 1 h | 1/180 Hz | 50% | 44 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 14 mA, 1.5 h | 1/180 Hz | 50% | 67 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 14 mA, 1.5 h | 1/30 Hz | 50% | 48 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 14 mA, 1.5 h | 1/90 Hz | 50% | 59 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 14 mA, 1.5 h | 1/270 Hz | 50% | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 14 mA, 1.5 h | 1/180 Hz | 67% | 78 (82) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 14 mA, 1.5 h | 1/180 Hz | 83% | 50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 14 mA, 1.5 h | 0 Hz (DC) | 100% | 21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 14 mA, 1.5 h | 1/180 Hz | 67% | 57d | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | Turn-off, 1.5 h | — | — | 14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To gain a deeper insight into the reaction mechanism, a series of experiments were conducted. The yield comparison of the desired product 2a and the gravimetric comparison of Pd species between AC, AC (w/o DDQ), and DC conditions were shown in Fig. 2a. The absence of DDQ and the DC condition both resulted in remarkably decreased yield of xanthone 2a. Gravimetric experiments demonstrated that the content of homogeneous palladium in the reaction solvent under AC conditions was much higher than the others (Fig. 2a, ns/nw), indicating that both the presence of DDQ and the AC electrolysis were indispensable to minimize the reduction or agglomerative deposition of the palladium catalyst. Moreover, it was observed that palladium species was deposited on the electrodes under DC and AC (w/o DDQ) conditions. The contents of palladium in the precipitation after reaction were also relatively higher than the standard AC condition (Fig. 2a, nd/nw), which agreed with the observed phenomena.
 | ||
| Fig. 2 Mechanistic insight. (a) The yield and Pd species comparison between DC and AC electrolysis. ns/nw, the ratio of Pd species in solution (ns; mole) to whole Pd catalyst (nw; mole); nd/nw, the ratio of Pd deposition (existed on the electrodes and sink to the bottom (nd; mole)) to whole Pd catalyst. (b) CV for 5.0 mM Pd(OAc)2, 5.0 mM DDQ and their mixture, scan range: 1.5–0.5–1.5 V, scan rate: 100 mV s−1. (c) Current variations under the constant-potential AC electrolysis with 0.4, 1.0 V working potentials and their product yields. | ||
To verify the role of AC electricity in the reaction system, cyclic voltammetry (CV) experiments were meticulously conducted to shed light on the mechanistic intricacies of this carbonylation reaction, as delineated in Fig. 2b. Initially, an exhaustive examination encompassed DDQ, Pd(OAc)2 and their mixture. Investigations were focused on the oxidation potentials (Eoxi) of the existent species. Pd(OAc)2 displayed a distinct oxidation peak during positive scan within the −0.5 V to 1.5 V potential window (Fig. 2b, 0.24 V vs. Ag/AgCl ref., red line). Notably, an inconspicuous peak at 1.03 V manifested for DDQ (Fig. 2b, vs. Ag/AgCl ref., blue line). It is noteworthy that the premixing of Pd(OAc)2 with DDQ showed two distinct peaks at 0.13 V and 0.72 V, respectively (Fig. 2b, green line). This potential shift upon the amalgamation of Pd(OAc)2 and DDQ prompted a series of constant-potential AC electrolytic experiments conducted under various applied working potentials. As shown in Fig. 2c, 0.4 V was selected as a working potential to drive the electrochemical carbonylation since it was greater than Eoxi(Pd0/PdII). Only trace amount of desired product 2a could be obtained by GC analysis. This result indicated that catalytic Pd species was difficult to be formed by direct anodic oxidation (red line). Notably, as the working potential was elevated to 1.0 V, surpassing the Eoxi of the Pd(OAc)2 and DDQ mixture, product 2a materialized with a 33% yield (blue line). These findings collectively signified that the catalytically active Pd(II) species predominantly emanated from the mediation of DDQ. It is noteworthy that the application of AC electricity served a multifaceted purpose: it facilitated the re-oxidation of Pd foil on the electrode surface, recycled the mediator DDQ, and concurrently liberated hydrogen.
Encouraged by these results, the oxidative carbonylation of an array of diaryl ethers was systematically examined (Fig. 3). For 4,4′-substituted diaryl ethers, halogen moieties, namely F, Cl, and Br, were all well-tolerated in the formation of xanthones, thereby affording enticing prospects for late-stage functionalization (Fig. 3, 2b–2d, 2s). Likewise, diaryl ethers bearing diverse alkyl and aryl substituents exhibited compatibility with this AC electrolysis, yielding the coveted products in yields ranging from 82% to 49% (Fig. 3, 2a, 2e, 2f, 2i, 2m). Treatment of ethers featuring electron-donating or electron-withdrawing groups yielded the desired products in yields between 49% and 31% (Fig. 3, 2g, 2h). Furthermore, multi-substituted ethers displayed facile reactivity toward CO, as evidenced by the formation of products 2j and 2l (Fig. 3). 1-Phenoxy naphthalene also underwent carbonylation, furnishing the corresponding product with a commendable 69% yield (Fig. 3, 2k). In an effort to comprehensively investigate the breadth of the reaction's scope, a range of mono-substituted diaryl ethers were subjected to scrutiny. Substrates bearing substituents such as methyl, ethyl, tert-butyl, and methoxyl groups demonstrated a harmonious coalescence with the AC-promoted methodology, yielding the target products with distinction (Fig. 3, 2n–2r). Even non-substituted diphenyl ether yielded a xanthone product, albeit with a modest yield of 31% (Fig. 3, 2t). Two regioselective products were isolated in 35% and 24% yields when the diaryl ether had a substituent at the meta-position of phenyl ring (2u and 2u′). Compared to standard conditions using CO balloon as CO source, blowing CO could further increase yield by 17%. In a deliberate expansion of the synthetic domain, a 5 mmol scale electrolysis was undertaken to underscore the practical utility of this electrochemical approach. Notably, under the 50 mA conditions (2.6 F mol−1), a reaction involving 1a yielded the corresponding xanthone (2a) with a noteworthy 65% yield. Moreover, this AC-promoted Pd catalytic system could also be extended to intermolecular oxidative C(sp2)–H/C(sp3)–H cross-coupling and C(sp2)–H/C(sp2)–H reactions, and targeted products could be obtained in moderate to good yields but in only poor efficiencies using DC and AC (w/o mediator) electrolysis (4a–4b).42,43
 | ||
Fig. 3 Substrate scopes and reaction expansions. Reaction conditions: diphenyl ethers (0.3 mmol), Pd(OAc)2 (7.5 mol%), DDQ (7.5 mol%), KPF6 (0.2 mmol), TFA (1.5 mL) in an undivided cell with two Pt plates as electrodes, 1/180 Hz, D = 67%, 14 mA, 50 °C, 90 min, (2.6 F mol−1), isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.2 mL DCE and 1.3 mL TFA. b2.5 h (4.4 F mol−1). cGram-scale conditions: 1a (5.0 mmol), Pd(OAc)2 (7.5 mol%), DDQ (7.5 mol%), KPF6 (0.4 mmol), TFA (30.0 mL), Pt and Pt electrode couples, 1/180 Hz, D = 67%, 50 mA, 50 °C, 7 h (2.6 F mol−1). dDetailed conditions are described in ESI Fig. S11, S12 and S15.† 0.2 mL DCE and 1.3 mL TFA. b2.5 h (4.4 F mol−1). cGram-scale conditions: 1a (5.0 mmol), Pd(OAc)2 (7.5 mol%), DDQ (7.5 mol%), KPF6 (0.4 mmol), TFA (30.0 mL), Pt and Pt electrode couples, 1/180 Hz, D = 67%, 50 mA, 50 °C, 7 h (2.6 F mol−1). dDetailed conditions are described in ESI Fig. S11, S12 and S15.† | ||
Conclusions
In summary, we have pioneered a transformative approach in the form of palladium-catalysed electrochemical oxidative carbonylation, specifically targeting diaryl ethers for the synthesis of xanthones through AC electrolysis. Leveraging a suitabe catalytic system, we achieved the conversion of a diverse spectrum of diaryl ethers into xanthones. Notably, this method facilitates the construction of xanthones under mild conditions, obviating the necessity for external oxidants, and thus rendering a substantial contribution to the overall sustainability. The implementation of this AC-driven methodology highlights the substantial benefits conferred by Pd-catalysed conversions in comparison to traditional DC electrolysis, thereby emphasizing the pragmatic virtues inherent in our approach.Author contributions
L. Zeng., H. Y. and A. L. conceived the project. H. L., J. P., L. Zeng, L. Zhou, M. S., F. X. and J. Y. performed the experiments, analysed the data, and discussed the results. H. L., L. Zeng, M. S., H. Y. and A. L. wrote the paper, supplementary methods, and related materials.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the National Key R&D Program of China (No. 2022YFA1505100 and 2021YFA1500100), National Natural Science Foundation of China (22031008, 22401221), the Fundamental Research Funds for the Central Universities 2042022rc0030, the China Postdoctoral Science Foundation (2021M702520, BX2021225) and the Postdoctoral Innovation Research Foundation of Hubei Province (211000163).References
- R. H. El-Seedi, A. M. El-Barbary, M. H. D. El-Ghorab, L. Bohlin, A.-K. Borg-Karlson, U. Goransson and R. Verpoorte, Curr. Med. Chem., 2010, 17, 854–901 CrossRef.
- B. Lesch and S. Bräse, Angew. Chem., Int. Ed., 2004, 43, 115–118 CrossRef PubMed.
- M. M. M. Pinto, E. M. Sousa and S. J. M. Nascimento, Curr. Med. Chem., 2005, 12, 2517–2538 CrossRef CAS PubMed.
- K.-S. Masters and S. Bräse, Chem. Rev., 2012, 112, 3717–3776 CrossRef CAS PubMed.
- M. M. L. Vieira and A. Kijjoa, Curr. Med. Chem., 2005, 12, 2413–2446 CrossRef.
- E. M. Sousa and M. M. M. Pinto, Curr. Med. Chem., 2005, 12, 2447–2479 CrossRef.
- A. J. Quillinan and F. Scheinmann, J. Chem. Soc., Perkin Trans. 1, 1973, 1329–1337 RSC.
- W. T. Jackson, R. J. Boyd, L. L. Froelich, D. M. Gapinski, B. E. Mallett and J. S. Sawyer, J. Med. Chem., 1993, 36, 1726–1734 CrossRef CAS.
- H. Rao, X. Ma, Q. Liu, Z. Li, S. Cao and C.-J. Li, Adv. Synth. Catal., 2013, 355, 2191–2196 CrossRef CAS.
- X. Yu and X. Lu, Tetrahedron Lett., 2011, 52, 2076–2079 CrossRef CAS.
- Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed.
- C. Shen and X.-F. Wu, Chem. – Eur. J., 2017, 23, 2973–2987 CrossRef CAS.
- D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851–857 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229–241 CrossRef CAS PubMed.
- H. Zhang, R. Shi, P. Gan, C. Liu, A. Ding, Q. Wang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5204–5207 CrossRef CAS PubMed.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- L. Zeng, H. Li, J. Hu, D. Zhang, J. Hu, P. Peng, S. Wang, R. Shi, J. Peng, C.-W. Pao, J.-L. Chen, J.-F. Lee, H. Zhang, Y.-H. Chen and A. Lei, Nat. Catal., 2020, 3, 438–445 CrossRef CAS.
- J. Chen, S. Lv and S. Tian, ChemSusChem, 2019, 12, 115–132 CrossRef CAS.
- S. Torabi, M. Jamshidi, P. Amooshahi, M. Mehrdadian and S. Khazalpour, New J. Chem., 2020, 44, 15321–15336 RSC.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 Search PubMed.
- J.-S. Zhong, Y. Yu, Z. Shi and K.-Y. Ye, Org. Chem. Front., 2021, 8, 1315–1328 Search PubMed.
- F. Kakiuchi and T. Kochi, Isr. J. Chem., 2017, 57, 953–963 CrossRef CAS.
- T. Wirtanen, T. Prenzel, J.-P. Tessonnier and S. R. Waldvogel, Chem. Rev., 2021, 121, 10241–10270 CrossRef CAS.
- F. Kakiuchi and T. Kochi, Chem. Rec., 2021, 21, 2320–2331 CrossRef CAS.
- C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS.
- M. Jamshidi, C. Fastie and G. Hilt, Synthesis, 2022, 54, 4661–4672 CrossRef CAS.
- S. Rodrigo, D. Gunasekera, J. P. Mahajan and L. Luo, Curr. Opin. Electrochem., 2021, 28, 100712 CrossRef CAS.
- L. Zeng, J. Wang, D. Wang, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2023, 62, e202309620 Search PubMed.
- C. Schotten, C. J. Taylor, R. A. Bourne, T. W. Chamberlain, B. N. Nguyen, N. Kapur and C. E. Willans, React. Chem. Eng., 2021, 6, 147–151 RSC.
- L. Zeng, Y. Jiao, W. Yan, Y. Wu, S. Wang, P. Wang, D. Wang, Q. Yang, J. Wang, H. Zhang and A. Lei, Nat. Synth., 2023, 2, 172–181 Search PubMed.
- L. Zeng, Q. Yang, J. Wang, X. Wang, P. Wang, S. Wang, S. Lv, S. Muhammad, Y. Liu, H. Yi and A. Lei, Science, 2024, 385, 216–223 CrossRef CAS.
- E. O. Bortnikov and S. N. Semenov, Curr. Opin. Electrochem., 2022, 35, 101050 CrossRef CAS.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750–5753 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04569f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |