
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hypervalent iodine chemistry with a mechanochemical twist†
Sayad
Doobary
 *a,
Miguel M.
de Vries Ibáñez
a and
Berit
Olofsson
*ab
*a,
Miguel M.
de Vries Ibáñez
a and
Berit
Olofsson
*ab
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, 106 91 Stockholm, Sweden. E-mail: Berit.Olofsson@su.se
bStockholm University Center for Circular and Sustainable Systems (SUCCeSS), Stockholm University, 106 91 Stockholm, Sweden
First published on 30th October 2024
Abstract
The combination of mechanochemistry and hypervalent iodine chemistry has rarely been reported, despite the numerous advantages offered by this enabling technology. With this in mind, this study addresses the key issue of transforming hypervalent iodine-mediated, solution-based reactions into the mechanochemical realm, accompanied by benchmarking and sustainability studies of the different types of reactions. Interestingly, several reagents displayed quite different reactivity and regioselectivity under mechanochemical conditions.
Mechanochemistry provides a unique avenue to explore organic reactivity due to its inherent solvent-free nature. The mechanical impacts created by ball-milling, pestle and mortar or extruders impart mechanical energy to activate a reaction in a similar way to thermal or irradiation activation.1 Thus, mechanochemistry has emerged as a useful enabling technology in organic chemistry.2 Whilst the solvent-free nature can lead to chemical advantages such as increased selectivity, faster reaction times and novel reactivity,3 there can also be potential sustainability benefits like less waste produced and decreased use of energy.4
Hypervalent iodine reagents have in recent years been demonstrated as powerful electrophilic reagents that can be utilized to give chemo- and stereoselective reactions under transition metal-free conditions.5 The vast majority of iodine(III) reactions are reported in solution, but there are some examples of solvent-free reactions, such as the microwave-assisted acetoxylation of oxo-benzoxazines6 or transamidation reactions.7 While mechanochemical activation has been employed in iodine(III)-mediated oxidations8 and in halogenation of imidazo[1,2-α]pyridines (Scheme 1A),9 very few reports concentrate on their use as group transfer reagents. Prominent examples include a metal-catalysed indole alkynylation with ethynylbenziodoxolone (EBX, Scheme 1B),10 a fluorocyclisation to form fluorinated heterocycles,11 and a copper-catalysed N-arylation of diaryliodonium salts.12
 | ||
| Scheme 1 The use of hypervalent iodine reagents in mechanochemistry as (A) an oxidant; (B) as a group transfer agent. (C) This work. | ||
Further investigations are needed to properly assess the power of combining mechanochemistry with hypervalent iodine chemistry, and discover novel applications of this emerging technology. Hence, we have conducted a study on a multitude of hypervalent iodine-mediated transformations, which are reported in solution, to understand how to convert solution-phase to solid-phase, compare yields, and evaluate the sustainability of mechanochemical vs. solution reactions. Herein, we report the optimisation and scope of mechanochemical S-, O- and C-arylations,13S- and C-vinylations, as well as a catalytic C-tosyloxylation (Scheme 1C). The important parameters and variables are highlighted during the work and comparisons to solution-based methods elucidate the superiority of mechanochemistry in many transformations.
In order to assess the key parameters governing the transformation of a solution-phase reaction to mechanochemical conditions, we first evaluated the O-arylation of phenols to give diaryl ethers, a transformation that proceeds well under solution-phase conditions.14 The optimisation was performed with unsymmetric diaryliodonium salt 1a to chemoselectively give diaryl ether 3a (Table 1).15 Initial ball milling reactions were performed at 25 Hz for 30 min, using a 1.5 mL stainless steel vessel with a 5 mm stainless steel ball. Very low yields were observed using sodium t-butoxide, sodium hydroxide or sodium carbonate as base (entries 1–3). A screen of frequencies showed that 35 Hz gave higher yield, which is in line with previous reports (entries 3–5).16 The effect of liquid-assisted grinding (LAG) was evaluated next,1c,17 and the presence of a small amount of LAG (20 μL = 0.33 μL mg−1) increased the yield markedly (entries 6–9). The green solvent EtOAc18 was selected as LAG for further studies, and the optimum amount of LAG proved to be 20 μL since a smaller amount resulted in a lower yield (entries 9–11). Potassium bases often work well with iodine(III) reagents,14e,19 and the combination of increased reaction time (3 h) and the use of potassium carbonate indeed improved the yield substantially (entries 12–14). Whilst the use of two stainless steel balls was detrimental (entry 15), a larger ball-milling vessel (5 mL) produced similar results (entry 16). Finally, it was found that pre-milling phenol 2a with base for 15 min, followed by 30 min of milling with diaryliodonium salt 1a delivered diaryl ether 3a in 91% isolated yield (entry 17). Interestingly, when the reaction was carried out in a microwave vial with a stirring bar under the same conditions, only 28% yield was garnered, indicating the importance of milling for the reaction to occur (entry 18). This optimisation serves as an example of the process of switching from solution- to solid-phase, and accounts for the most important factors, such as choice of base and LAG, amount of LAG, frequency and reaction setup.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Frequency (Hz) | Base | LAG | Time (min) | Yieldb (%) |
| a Standard setup: 1.5 mL stainless steel vessel with a 5 mm stainless steel ball. b NMR yield using 1,3,5-trimethoxybenzene (TMB) as internal standard. c With 2 balls. d With a 5 mL vessel and a 10 mm ball. e Pre-milling of 2a with base for 15 min before addition of 1a, isolated yield. f Reaction carried out in a microwave vial. | |||||
| 1 | 25 | NaOtBu | — | 60 | 5 |
| 2 | 25 | NaOH | — | 60 | 5 |
| 3 | 25 | Na2CO3 | — | 60 | 8 |
| 4 | 30 | Na2CO3 | — | 60 | 10 |
| 5 | 35 | Na2CO3 | — | 60 | 15 |
| 6 | 35 | Na2CO3 | THF | 60 | 23 |
| 7 | 35 | Na2CO3 | MeOH | 60 | 39 |
| 8 | 35 | Na2CO3 | Toluene | 60 | 40 |
| 9 | 35 | Na2CO3 | EtOAc | 60 | 40 |
| 10 | 35 | Na2CO3 | EtOAc (10 μL) | 60 | 33 |
| 11 | 35 | Na2CO3 | EtOAc (30 μL) | 60 | 41 |
| 12 | 35 | Na2CO3 | EtOAc | 180 | 60 |
| 13 | 35 | KOtBu | EtOAc | 180 | 69 |
| 14 | 35 | K2CO3 | EtOAc | 180 | 88 |
| 15c | 35 | K2CO3 | EtOAc | 180 | 63 |
| 16d | 35 | K2CO3 | EtOAc | 180 | 90 |
| 17e | 35 | K2CO3 | EtOAc | 15 + 30 | (91) |
| 18f | — | K2CO3 | EtOAc | 15 + 30 | 28 |
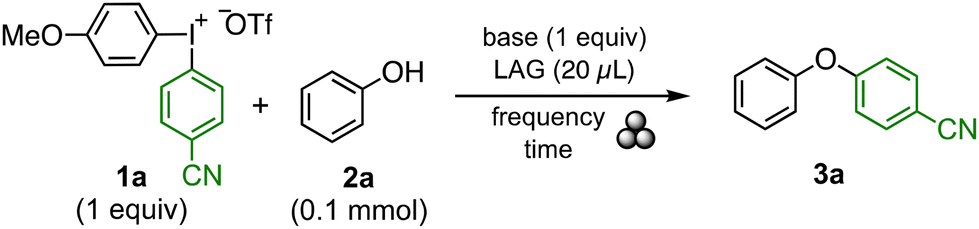
The scope of the diaryl ether synthesis is depicted in Scheme 2A. A range of phenols with varying electronic properties were well tolerated, including halogen, nitro, t-butyl and methoxy substituents (3a–3h). Steric hindrance was well accepted, as demonstrated by ortho-substituted products 3f–3h. Finally, the functional group tolerance was briefly evaluated with allyl and pyridyl groups (3h, 3i). The diaryliodonium structure was varied next, keeping the anisyl group as “dummy” ligand to ensure chemoselective transfer of the green aryl group.20 Reactions of phenol 2a with varied diaryliodonium salts 1 produced 3j–3m in good to excellent yields. Next, 2-methoxyphenol was used as nucleophile to evaluate a sterically demanding environment. Arylation with a variety of electron-deficient aryl groups, including halides, cyano and ester substituents (3n–3q) was achieved upon extending the reaction time to 2 h to ensure complete conversion with less reactive salts 1. To the contrary, transfer of electron rich aryl groups was limited to a tolyl group (3r, 3s), whereas a heteroaryl group was successfully transferred (3t). The method was subsequently evaluated for arylation of a variety of other nucleophiles, which are reported in solution. Indeed, aliphatic alcohols21 could be arylated in good yields (3u–3y) using acetonitrile as LAG.
 | ||
Scheme 2 Mechanochemical arylation scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3.0 mmol scale; b15 min pre-mill with base, then 2 h milling time with 1, c2 h reaction time, MeCN as LAG, dEtOAc as LAG. 3.0 mmol scale; b15 min pre-mill with base, then 2 h milling time with 1, c2 h reaction time, MeCN as LAG, dEtOAc as LAG. | ||
Next, the S-arylation of thiols22 to yield diaryl sulfides 4 was investigated (Scheme 2B). An optimisation15 showed that cyclopentanone, which is a more sustainable LAG than EtOAc,18 was equally efficient (see Table S5†). Similar to the O-arylation, varied nucleophiles could be used (4a–4e). While the reaction was limited to incorporation of electron deficient aryl rings (4f–4k) and a pyridyl group (4j), the arylation of mercaptobenzoxazole and aliphatic thiols was feasible (4k–4m). The chemoselectivity of the reaction is demonstrated by the selective S-arylation to product 4l, which has a free amino group.
Finally, the C-arylation of nitroalkanes23 to give products 5 was investigated under mechanochemical conditions (Scheme 2C).15 The reaction was found to proceed well in the absence of a LAG, and was high-yielding for both cyclic and acyclic nitroalkanes (5a–5h), including transfer of the electron-rich anisyl group (5d) and a pyridyl group (5f). While a nitroester reacted sluggishly (5i), the C-arylation to β-keto ester 5j proceeded smoothly.
We have recently reported metal-free, regio- and stereoselective vinylations using vinylbenziodoxolones (VBX).24 Due to the increased stability of the benziodoxolone core, reactions with VBX often give enhanced selectivity compared to vinyl(aryl)iodonium salts.24b,d,25 The mechanochemical vinylation efficiency of VBX and vinyliodonium salts was interesting to study, and the solution-phase S-vinylation of thiols with VBX and KOtBu was examined first.24a We were pleased to discover that the reaction proceeded well with VBX 6a and the milder base K2CO3 in the ball mill at 30 Hz (Scheme 3). Similar to the original report, the mechanochemical yields of vinyl aryl sulfides 7 were quite high, and the products were formed with complete regioselectivity and generally with E/Z > 20:1.26 The thiol scope was wide, and tolerant of free amino groups and heteroaromatics (7a–7f). Additionally, reactions with VBX reagents 6b, 6c provided 7g, 7h in good yields. Importantly, since the synthesis of 7g and 7h was higher-yielding using the mechanochemical methodology than in solution, isolation could be completed using a chromatography- free protocol, which considerably improves the sustainability of the transformation.
 | ||
| Scheme 3 Mechanochemical vinylation scope. Products 7 formed with VBX 6a–6c and K2CO3, products 8 formed with salt 6d and KOtBu. aTHF as LAG, 135 min; b10 min; c3.0 mmol scale; d30 min. | ||
Finally, the C-vinylation of nitroalkanes24b was investigated under mechanochemical conditions. The optimisation uncovered that the acyclic styryl(phenyl)iodonium tetrafluoroborate27 (6d) produced higher yields than VBX, delivering C-vinylated products 8a, 8b. Remarkably, this mechanochemical protocol can be finished in as little as 10 min and the products were formed with excellent regioselectivity (ratio of internal/terminal alkene >20:1). In solution, the same reaction required 18 h reaction time with 6d, and produced a measly internal/terminal ratio of 4:1, whereas solution reactions with VBX gave the terminal product in 14:1 ratio.24bC-vinylation to give β-keto ester 8c was also feasible.
To evaluate the scalability of the developed mechanochemical protocols, the scale was increased from 0.3 mmol to 3 mmol for products 3h, 4a, and 8a. A 5 mL vessel and a 10 mm stainless steel ball were utilised to ensure sufficient space for milling to occur. Good yields were still obtained in these reactions, illustrating that upscaling is indeed possible.
Organic oxidative transformations utilising catalytic iodine(III) reagents are ubiquitous in the literature owing to the readily accessible redox couple iodine(I)/(III).5a,28 However, a literature search showed a complete lack of mechanochemical reactions utilizing catalytic iodine(III) reagents.29 The tosyloxylation of C-nucleophiles has been profusely studied in solution,30 and served as a good model reaction to evaluate mechanochemically. The optimisation showed that it was indeed possible to form the iodine(III) oxidant in situ, using a catalytic amount of 2,4-dimethylphenyl iodide (9) together with stoichiometric mCPBA and tosic acid (Scheme 4). In this fashion, several classes of substrates were tosyloxylated to give products 10 in high yields.
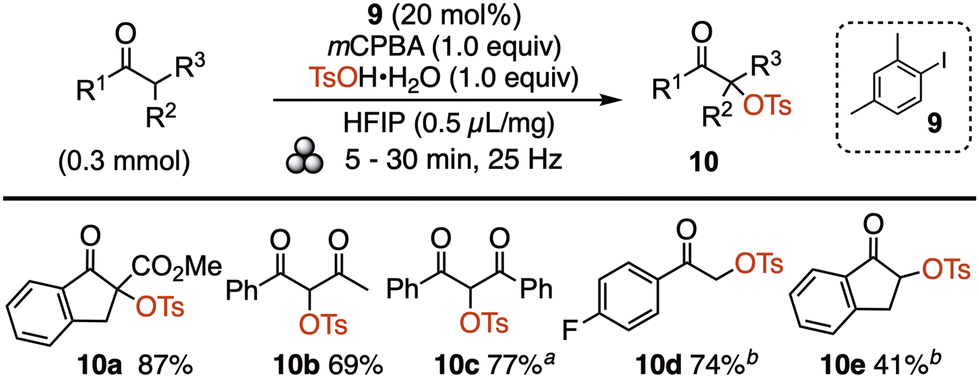 | ||
| Scheme 4 Catalytic tosyloxylation of C-nucleophiles. aNo HFIP added; b1.5 equiv. mCPBA and tosic acid, preheated vessel. mCPBA = m-chloroperbenzoic acid; TsOH = p-toluenesulfonic acid; HFIP = hexafluoroisopropanol. | ||
To evaluate the reasons for the observed reaction efficiency, we carried out the respective solution reactions in concentrations similar to that of a LAG, which usually resulted in a stark drop in yield.15 The same was true when our standard mechanochemical reactions were carried out in microwave vials with stirring bars as opposed to ball milling conditions.15 Together these results show that the mechanochemical activation is indeed required for high yields.
Having demonstrated the facile conversion of known solution-phase transformations to mechanochemical conditions, we wanted to evaluate the efficiency and sustainability of the developed mechanochemical methods. The E-factor, which is defined as the mass ratio of waste to desired product, is a very useful sustainability benchmarking method.4,31 To highlight the differences between mechanochemical and solution-phase reactions, we decided to calculate the E-factors excluding the waste created via purification (which was expected to be rather similar for both methods). Chart 1 details the comparison of yields and E-factors for selected products15 using the arylation, vinylation and tosyloxylation methodologies. The mechanochemical arylations sometimes produced markedly higher yields (3h, 3q, 4g) than the reactions in solution.21,22e,23 A similar trend was seen for S-vinylations, where the high yields obtained mechanochemically enabled column-free purification and hence higher E/Z ratios (7g).24a For the C-vinylations, the mechanochemical protocol provides higher yields and better regioselectivity (internal/terminal alkene) than in solution.
 | ||
| Chart 1 Comparison of yields and E-factors for mechanical and solution conditions. a1 (1 equiv.), KOtBu (1.2 equiv.), THF (0.2 M), 40 °C, 1 h;14ab1 (1 equiv.), DBU (1.1 equiv.), MeCN (0.1 M), 80 °C for 1.5 h;22acYields taken from the literature; 5f,237g,24a10c,30ad6d (1.1 equiv.), KOtBu (1.1 equiv.), THF (0.024 M), rt, 18 h.24a | ||
The comparison of E-factors highlights that the mechanochemical methods produce far less waste than the corresponding solution-phase reactions; particularly striking are the results for 7g and 8c, which are low-yielding in solution and have high E-factors. The difference is especially interesting for products with higher yields using solution-phase methodologies (3q, 10c), as the solution-phase still has much higher E-factors.
Conclusions
In conclusion, we have successfully transferred known hypervalent iodine-mediated arylations, vinylations and tosyloxylations from solution- to solid-phase. Furthermore, the first catalytic hypervalent iodine protocol utilising mechanochemistry was developed. The scope studies revealed several examples where the mechanochemical methods were superior, and a comparison of E-factors showed that the novel protocols are far more sustainable. The results establish the value of combining mechanochemistry with hypervalent iodine chemistry, and the discovered key parameters for methodology transfer from solution are likely also applicable when developing other mechanochemical hypervalent iodine reactions.Author contributions
Sayad Doobary: conceptualization, investigation, methodology, supervision, writing – original draft, writing – review & editing; Miguel M. de Vries Ibáñez: investigation, methodology; Berit Olofsson: funding acquisition, validation, project administration, resources, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Raw data can be found online at: https://doi.org/10.5281/zenodo.13991537.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Carl Trygger Foundation (20:316) and Olle Engkvist Foundation (224-0058) are kindly acknowledged for financial support.References
-
(a) B. R. Naidu, T. Sruthi, R. Mitty and K. Venkateswarlu, Green Chem., 2023, 25, 6120–6148 RSC
; (b) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
-
(a) J.-L. Do and T. Friščić, Synlett, 2017, 2066–2092 Search PubMed
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC
-
(a) N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC
-
(a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2024, 124, 11108–11186 CrossRef CAS PubMed
- R. Prajapati, A. K. Gola, A. Kumar, S. Jaiswal and N. Tadigoppula, New J. Chem., 2022, 46, 5719–5724 RSC
- R. Vanjari, B. Kumar Allam and K. Nand Singh, RSC Adv., 2013, 3, 1691–1694 RSC
-
(a) M. S. Yusubov and T. Wirth, Org. Lett., 2005, 7, 519–521 CrossRef CAS
- D. R. Indukuri, G. R. Potuganti and M. Alla, Synlett, 2019, 1573–1579 CAS
- G. N. Hermann, M. T. Unruh, S.-H. Jung, M. Krings and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 10723–10727 CrossRef CAS PubMed
- W. Riley, A. C. Jones, K. Singh, D. L. Browne and A. M. Stuart, Chem. Commun., 2021, 57, 7406–7409 RSC
- J. Jiang and J. Li, ChemistrySelect, 2020, 5, 542–548 CrossRef CAS
- Mechanochemical N-arylations were not investigated as they are already reported, see ref. 12.
-
(a) N. Jalalian, E. E. Ishikawa, L. F. Silva and B. Olofsson, Org. Lett., 2011, 13, 1552–1555 CrossRef CAS
- See the ESI† for details.
- S. Ni, M. Hribersek, S. K. Baddigam, F. J. L. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2021, 60, 6660–6666 CrossRef PubMed
-
(a) L. E. Wenger and T. P. Hanusa, Chem. Commun., 2023, 59, 14210–14222 RSC
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC
-
(a) F. Gang, Y. Che, Z. Du, H. Feng and Y. Fu, Synlett, 2017, 1624–1629 CAS
-
(a) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334–10342 CrossRef CAS PubMed
- R. Ghosh, E. Lindstedt, N. Jalalian and B. Olofsson, ChemistryOpen, 2014, 3, 54–57 CrossRef CAS PubMed
-
(a) S. Sarkar, N. Wojciechowska, A. A. Rajkiewicz and M. Kalek, Eur. J. Org. Chem., 2022, e202101408 CrossRef CAS
- C. Dey, E. Lindstedt and B. Olofsson, Org. Lett., 2015, 17, 4554–4557 CrossRef
-
(a) L. Castoldi, E. M. Di Tommaso, M. Reitti, B. Gräfen and B. Olofsson, Angew. Chem., Int. Ed., 2020, 59, 15512–15516 CrossRef PubMed
- A. Yoshimura, A. Saito and V. V. Zhdankin, Adv. Synth. Catal., 2023, 365, 2653–2675 CrossRef CAS
- In solution, the S-vinylation provided product 7e with an E/Z ratio of only 5:1 (see ref. 24a). The E-product is formed through a stereospecific ligand coupling mechanism (see ref. 24e), but some products are prone to isomerization during purification on silica.
- M. Ochiai, T. Shu, T. Nagaoka and Y. Kitagawa, J. Org. Chem., 1997, 62, 2130–2138 CrossRef CAS PubMed
- F. V. Singh, S. E. Shetgaonkar, M. Krishnan and T. Wirth, Chem. Soc. Rev., 2022, 51, 8102–8139 RSC
- Ref. 8d demonstrates in situ-formation of iodine(III) utilizing 50 mol% of ArI combined with 1.5 equiv. mCPBA under mechanochemical conditions. Since the product yield is 30%, the reaction is not catalytic.
-
(a) A. Tanaka and H. Togo, Synlett, 2009, 3360–3364 CAS
- R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04903a |
| This journal is © The Royal Society of Chemistry 2024 |