
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of moisture on the stability and degradation of perovskites in solar cells
Bhushan P.
Kore
,
Mahboubeh
Jamshidi
 and
James M.
Gardner
*
and
James M.
Gardner
*
Department of Chemistry, Division of Applied Physical Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: jgardner@kth.se
First published on 12th February 2024
Abstract
Efficiency and stability are the two most important factors in commercially scalable solar cells. In spite the high-power conversion efficiencies (PCE), the commercialization of perovskite solar cells (PSC) has been limited due to their low stability under ambient conditions. Environmental factors like moisture, heat, and light can all adversely affect PSC performance and limit device lifetime. In this review, we refer to the literature addressing the moisture induced stability issue of perovskite based solar cells. We present an overview on the moisture stability of the perovskite solar cells and clarify the effect of moisture on different layers in perovskite solar cells and the corresponding degradation process. Then we extend the discussion highlighting the strategies to prevent the moisture induced degradation in hybrid perovskite solar cells. The methods include composition engineering (cation and halide engineering) and interfacial layer engineering/surface passivation. We further summarize the utilization of doping techniques and use of organic/Inorganic passivators. We address methods of producing moisture stable 2D perovskites and admixtures of 2D and 3D perovskites. Lastly, the review highlights research directions focused on improving perovskite stability without compromising power conversion efficiency.
1. Introduction
According to international standards, commercially available solar cells for outdoor use should have a lifespan of 25 years and should survive temperature fluctuations from −40 °C to 85 °C.1 While there are challenges that need to be addressed, there are number of reasons to think that organic inorganic metal halide perovskite solar cell (PSC) panels can exceed present international standards. The organic inorganic metal halide perovskite materials with ABX3 formula (where A-CH3NH3+, NH2CHNH2+, Cs+; B–Pb2+, Sn2+; and X–Cl−, Br−, I−) will be referred to solely as perovskites unless otherwise stated. Perovskites readily react with water/moisture, that causes degradation into their precursor materials or formation of hydrated phases and results in lower power output and short circuit in the solar cells.2–4 Perovskites are susceptible to chemical decomposition from moisture due to the presence of hygroscopic ammonium and Pb(II) salts. Moisture instability is a persistent concern for perovskite solar cells and comprehensive strategies are required to overcome this problem.4–8 While impressive progress has been made on increasing PSC efficiency, stability under ambient conditions remains a major concern. Thermal degradation, and ion migration are also challenges to perovskite stability; however, significant improvements have been made and these effects have been largely suppressed.9–14From commercialization point of view, external encapsulation technique can be a straightforward process to prevent the infiltration of moisture and oxygen,15,16 but two factors have impeded the progress toward enhancing the efficiency and stability of PSCs. Firstly, the presence of interfacial and bulk vacancy defects creates a primary pathway for the diffusion or migration of halide ions, consequently leading to the degradation of devices due to light exposure. Secondly, the surplus PbI2 within perovskite absorbers undergoes decomposition when subjected to light, resulting in compromised illumination stability. Additionally, it is important to building shields within the PSCs to boost their own internal resistance to water and oxygen.17,18
If we look at the PV triangle (efficiency, cost, stability) requirements for commercialization, then it is only long-term stability that remains a challenge for perovskite photovoltaic (PV) technology. The ionic crystal structure, and weakly bonded organic cations make perovskite vulnerable to moisture, heat, and oxygen. In this review, we summarize the stabilization strategies against moisture for PSCs and highlighted the challenges/outlook for the future research. Our perspective in writing this review is to give readers an overview of major advances in PSCs, which either resulted in significant improvement in the solar cell stability or shows promise for commercializing this technology.
2. Device architecture of hybrid perovskite device
PSCs emerged from dye sensitized solar cells (DSSC) when the light absorbing dye was replaced with perovskite nanocrystals material by Kojima and co-workers.19 The rapid reaction of perovskite with the liquid electrolyte promoted its replacement with the solid-state hole conducting material (Spiro-OMeTAD). This advancement created a tremendous interest in the photovoltaic community and drew the attention of experts from various other scientific communities.Inspired by CIGS and CdTe architectures, monolithic devices were developed in which the light absorbing perovskite material was sandwiched between electron transporting material (ETM) and hole transporting material (HTM), as shown in Fig. 1(a)–(c). Mesoscopic and planar are the two basic structures that are used in the fabrication of PSCs. The mesoscopic structure has n–i–p configuration with compact ETM/mesoscopic ETM/perovskite/HTM/electrode stacking, as shown in Fig. 1a. The planar configuration is further divided into two categories n–i–p planar and p–i–n planar, as shown in Fig. 1b and c. Usually in n–i–p structure, the perovskite material is deposited onto transparent substrates covered with compact TiO2 and then mesoporous TiO2 scaffold layer, whereas in p–i–n structure the perovskite layer is deposited onto transparent electrodes covered with HTM such as poly(3,4-ethylene dioxythiophene):polystyrene sulfonic acid (PEDOT:PSS). Both mesoporous and planer structures are known to deliver high efficiency and stable solar cells, however, the comparison of stability of two different structures is still under debate7,20 though there are several reports published in literature claiming the advantage of one structure over other.21,22
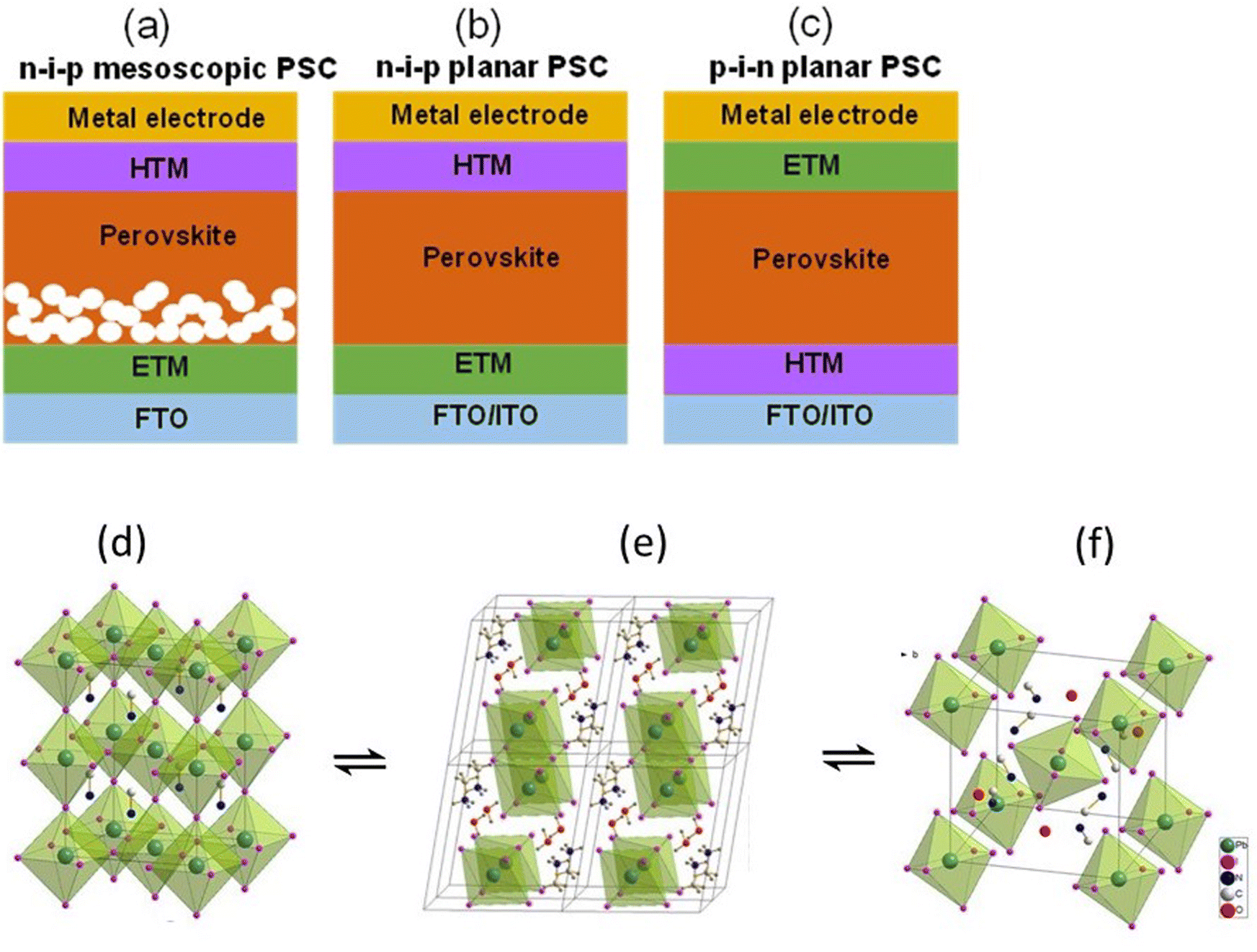 | ||
| Fig. 1 Device structures of (a) n–i–p mesoscopic, (b) n–i–p planar, and (c) p–i–n planar PSCs. If the incident light goes through ETM then it is known as n–i–p (regular) structure and if the light goes through the HTM then it is known as p–i–n (inverted) structure. (d) shows the structure of MAPI3 in its cubic phase, (e) shows the structure of the monohydrate phase, CH3NH3PbI3·H2O, and (f) displays the structure of the dihydrate, (CH3NH3)4PbI6·2H2O. The position of the hydrogens on the (CH3NH3)+ ions and the water is not assigned in panels (d) and (f) Reprinted with permission from ref. 2. Copyright 2015 American Chemical Society. | ||
2.1. Mesoporous structure
A certified 16.2% efficiency was achieved by depositing MAPbI3 perovskite film on TiO2 mesoporous layer using the ‘antisolvent’ method.23 Later this has become one of the most preferred methods of perovskite deposition. Furthermore, efficiencies more than 20% were achieved by incorporating multiple (organic/inorganic) cations into the perovskite like Cs/FA/MA.24–26 The substitution of multiple cations was found to help in stabilizing the perovskite structure. The inclusion of mesoporous scaffold in the PSCs greatly facilitates the charge collection particularly in perovskite absorbers where light absorption length is longer than the carrier diffusion length. As an example, tin halide perovskites with mesoscopic scaffolds show external quantum efficiency (EQE) of ∼80% whereas in planer structure configuration this ratio is quite low.27,28 Thus, the advantage of mesoscopic configuration is that it greatly facilitates the collection of charge carriers and enhance the performance of PSCs.292.2. Planar structure
The planar n–i–p architecture of PSC came into existence when the mesoporous layer was completely removed from the solar cell configuration.30 The inverted p–i–n planar structured solar cells initially exhibited lower PCE values of 1.6% which was further improved by Snaith et al., to 9.8% with the help of mixed halide composition of perovskite and replacing the HTL by PC61BM.31 Solar cell efficiencies exceeding 15% were reported by Tong et al., using NiO nanocrystals as HTL and PCBM/BCP as ETL.32 The PCE over 20% was achieved by Yang et al., in 2018.33 They proposed Cs doped FAPbI3 perovskite with EDTA complex tin oxide (SnO2) as ETM and Spiro-OMeTAD as HTM to achieve high performance solar cells. The PSCs attained record PCE of 21.6% and the unsealed devices show drop of only 8% PCE when exposed to ambient condition for 2880 h.3. Effect of moisture on different layers in perovskite solar cells
3.1. Effect of moisture on light absorbing perovskite material
Among the various hybrid perovskite materials, MAPbI3 (CH3NH3PbI3) has been most thoroughly studied and extensively used in the solar cells as a light absorbing material owing to its appropriate band gap for solar cell material and ideal optical and electrical properties. However, it has been now well established that MAPbI3 undergoes degradation when exposed to moisture or water. The perovskite MAPbI3 visibly degrades: its black color fades and transforms into the yellow PbI2 and gases CH3NH2 and HI as given in eqn (1).2,34–36 The impact of water on the perovskite layer can be understood through the following reactions.| CH3NH3PbI3 (s) ↔ PbI2 (s) + CH3NH3I (aq.) ↔ PbI2 (s) + CH3NH2 (aq.) + HI (aq.) | (1) |
| 4CH3NH3PbI3 + 4H2O ↔ 4[CH3NH3PbI3·H2O] ↔ (CH3NH3)4PbI6·2H2O + 3PbI2 + 2H2O | (2) |
| (CH3NH3)4PbI6·2H2O ↔ 4 CH3NH3I + PbI2 + 2H2O | (3) |
| CH3NH3I ↔ CH3NH2 + HI | (4) |
The perovskites are soluble in polar solvents, like water, thus when the perovskites come in contact with water/moisture they rapidly degrade.37 The hydrogen bond between organic and inorganic units defines the structural stability of perovskite and it is strongly affected by the moisture.38 However, an appropriate percentage of moisture is known to be favorable for the crystallization of perovskite layer, it further helps in improving the thin film quality and grain growth/size distribution,2,39–43 on the other hand excess amount of water damages the crystal structure of the perovskite and disintegrate into the starting compounds and hydrated phases.
Mainly the water molecule interacts with the perovskite by the hydrogen bond between the organic cation and inorganic units and then forming new bonds with perovskite giving rise to hydrated phases.3,44 For example by reacting with H2O, solid MAPbI3 will first decompose through hydrolysis reaction into CH3NH3I solution (aq.) and solid (s) PbI2. Furthermore, in the next step MAI (aq.) gets decompose into volatile compounds like CH3NH2 (aq.) and HI (aq.). Thus, in the presence of moisture there is high probability that MAI, PbI2, CH3NH2 and HI will coexist within the thin film. Furthermore, the presence of crystalline defects in perovskite act as a channel for infiltration of water molecules this enables more severe accelerated degradation by breaking the interactions of alkylamine cations with other ions.6
A similar explanation has been proposed by Walsh et al., by considering a simple acid base reaction.3 According to their study, in case of water exposure, the H2O molecules can interact with MAPbI3 and can take away one proton from the organic cation (ammonium) this will form an intermediate phase/compound [(CH3NH3+)n−1(CH3NH2)nPbI3][H3O]. The formed intermediate then subsequently decompose into the volatile CH3NH2, HI and solid PbI2.3,45
The hydrated structure of perovskite can be fully reverted back to the perovskite phase (without H2O) by keeping the hydrated perovskite sample in a dry air for 48 h, as shown in Fig. 1d–f.2 The incorporation of H2O molecule significantly deforms the crystal structure by separating the [PbI6]4− octahedra, as shown in Fig. 1e and f. This makes the perovskite structure transform from a 3D network of [PbI6]4− octahedra to a 1D and 0D chain of octahedra for monohydrated and dehydrated phase, respectively.6,46
The moisture induced degradation of perovskite can be further accelerated by UV light and O2.34,47,48 The degradation of perovskite under moisture along with UV light and oxygen can be expressed as follows,
| CH3NH3PbI3 H2O ↔ CH3NH3I (aq.) + PbI2 (s) | (5) |
| CH3NH3I (aq.) ↔ CH3NH2 (aq.) + HI (aq.) | (6) |
| 4HI (aq.) + O2 ↔ 2I2 (s) + 2H2O | (7) |
| 2HI (aq.) hv ↔ H2(g) + I2 (s) | (8) |
It is noteworthy to mention that oxygen gas and water vapor can influence the energetics for degradation by significantly changing the energy levels in organometal halide perovskite films, such as CH3NH3PbI3 and CH3NH3PbI3−xCl shown by J. Yang et al.49 Exposure to oxygen gas causes a surface dipole-induced shift in perovskite films, leading to an upward shift in the vacuum level (work function increase). This shift saturates due to the surface dipole effect. The valence band remains unchanged, resulting in an increase in ionization potential (IP) that corresponds to the work function increase. Additionally, oxygen exposure triggers a minor conversion from CH3NH3PbI3 to PbI2 within the films. Water vapor has distinct effects on perovskite films as compared to oxygen gas. Specifically, exposure to water vapor leads to a downward shift in the vacuum level (resulting in a decrease of the work function), while simultaneously causing an increase in the binding energy of the valence band in relation to the Fermi level. This combined effect ensures that the ionization potential (IP) of the perovskite films remains unaltered.
3.2. Effect of moisture on other layers
Not only perovskite but the presence of moisture can also degrade the HTMs which also leads to further drop in the solar cell performance. The 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (Spiro-OMeTAD) is the most commonly used HTM in the regular n–i–p structured PSCs. The pure form of Spiro-OMeTAD shows poor electrical properties owing to its incompletely conjugated structure and intermolecular spacing.50 In order to improve the electrical properties of Spiro-OMeTAD it is doped with mild oxidants and salts such as lithium-bis-(trifluoromethane)sulfonimide (Li-TFSI).51 Spiro-OMeTAD doped with Li-TFSI shows improved hole mobility and conductivity. However, Li-TFSI is hygroscopic in nature which induces moisture and degrades the device/HTM.52,53 Furthermore, the presence of Li+ ions in Li-TFSI initiates Li+ migration into the perovskite and ETM. This acts as a channel for the moisture penetration and accelerates the degradation of HTM and perovskite layer leading to the drop in the device performance.6 It is therefore equally important to select a moisture resistive HTM, which can not only act as an efficient HTM but also help in encapsulating perovskite and further improving the stability of the PSC devices.Compared to HTM the ETM are less susceptible towards moisture. The most commonly used ETM in regular n–i–p structure TiO2 is hydrophilic in nature whereas [6,6]-phenyl C61 butyric acid methyl ester (PCBM) used in inverted PSCs can absorb moisture.54 In regular structure of PSCs, mesoporous TiO2 layer adsorb atmospheric water that is later retained in the final device structure. In inverted structure PSCs, most commonly PCBM is used which has higher water contact angle compared to TiO2 but still undergoes water induced degradation. Though PCBM helps in reducing the trap states it cannot efficiently passivate the undercoordinated Pb2+ ion defects.55 This causes degradation of PSC like in the case of TiO2, moreover the poor morphology of the PCBM creates passage for the water permeation.6
4. Strategies to enhance perovskite solar cells’ stability against moisture
In this section we will discuss the different strategies to enhance the overall stability of the perovskite materials.4.1. Composition engineering
Though hybrid perovskite materials have shown great potential as a light absorbing layer their stability remains an important concern before the industry. Since the fabrication of the first PSC, there have been significant improvements in the stability of the light absorbing perovskite through multiple cation substitution, mixed halide substitution, and doping of inorganic/organic cations has been observed. The partial or complete substitution of cations/halides significantly improves the stability of the perovskite layer and boosts the efficiency of the PSCs. Composition tuning of perovskite alters the lattice parameters and bonding environment, which helps in retaining the perovskite structure and improves their moisture stability.56 | (9) |
For obtaining the perovskite structure the value of ‘t’ should be between 0.8 and 1, if this value is lower than 0.8 and greater than 1 then the formation of non-perovskite structure can be expected.60 There are very few organic/inorganic cations available for the substitution at A site. In most of the solar cell MA cation-based perovskite were explored but due to the lower stability of MAPbI3 under moist conditions people started exploring partial substitution of other perovskites such as FAPbI3 and CsPbI3. The FAPbI3 and CsPbI3 perovskites are not stable in the cubic α-phase and relax to the orthorhombic phase at room temperature. A site cation substitution with other cations such as FA+, Cs+, Rb+, and K+ led to enhancement in the device performance and stability. The A site cation engineering also helps in tuning the band gap or absorption edge. Number of studies on A site cation engineering have been reported in the literature for enhancing the moisture stability of the PSCs.56,58,61
In 2022, F. Cao et al. investigated the degradation mechanism of FA-based perovskites and proposed the existence of FABr–H2O.62 They implemented an approach where MABr powder was introduced into a PbI2 precursor solution, creating a seed solution through a two-step preparation method. This technique was employed to boost the efficiency and durability of FA-based perovskite solar cells (PSCs). As a result, the degradation rate of the resulting perovskite film exhibited a notable reduction when subjected to high humidity, in comparison to films prepared using a two-step method without the application of a seed solution. The resulting device demonstrated an impressive power conversion efficiency of 23.22%, and even after being exposed to air for 900 hours, the efficiency of this device remained unchanged.
The improved photovoltaic properties of the mixed cation MAxFA1−xPbI3 perovskite have attracted many researchers into the fundamental investigation and further development of mixed cation PSCs and large number of studies on A-site cation engineering have been carried out to improve the moisture stability of the perovskites.26,63–66 There are various systematic investigations reported in literature where several combinations of possible cations such as MA+/FA+, CS+/MA+, Cs+/FA+, Cs+/FA+/MA+ are explored.26,59,64,66–68 Based on the first principle calculations, Zhu et al., reported that alloying of FAPbI3 and CsPbI3 can effectively tune the tolerance factor and enhance the stability of photoactive FA1−xCsxPbI3 α phase.59 Further, they experimentally confirmed that alloyed FA0.85Cs0.15PbI3 demonstrate improved stability under high humidity environments, in contrast to FAPbI3 which degrades to δ phase under humid conditions. Due to the structural stabilization the alloyed FA0.85Cs0.15PbI3 solar cells showed improved performance and better device stability in comparison with FAPbI3 perovskite. These results confirmed that Cs alloying at A site could significantly improve the stability of perovskite under humid conditions.
Despite efforts to exclude all moisture from the triple cation perovskite solar cells, a minimal quantity of moisture within the perovskite films was found to enhance the growth of triple cation perovskite (Cs0.05FA0.85MA0.15Pb(I0.85Br0.15)3) crystals and amplify the difference in potential energy between individual crystals and their grain boundaries (Fig. 2a). This, in turn, aids the movement and capture of charge carriers across these boundaries.69,70
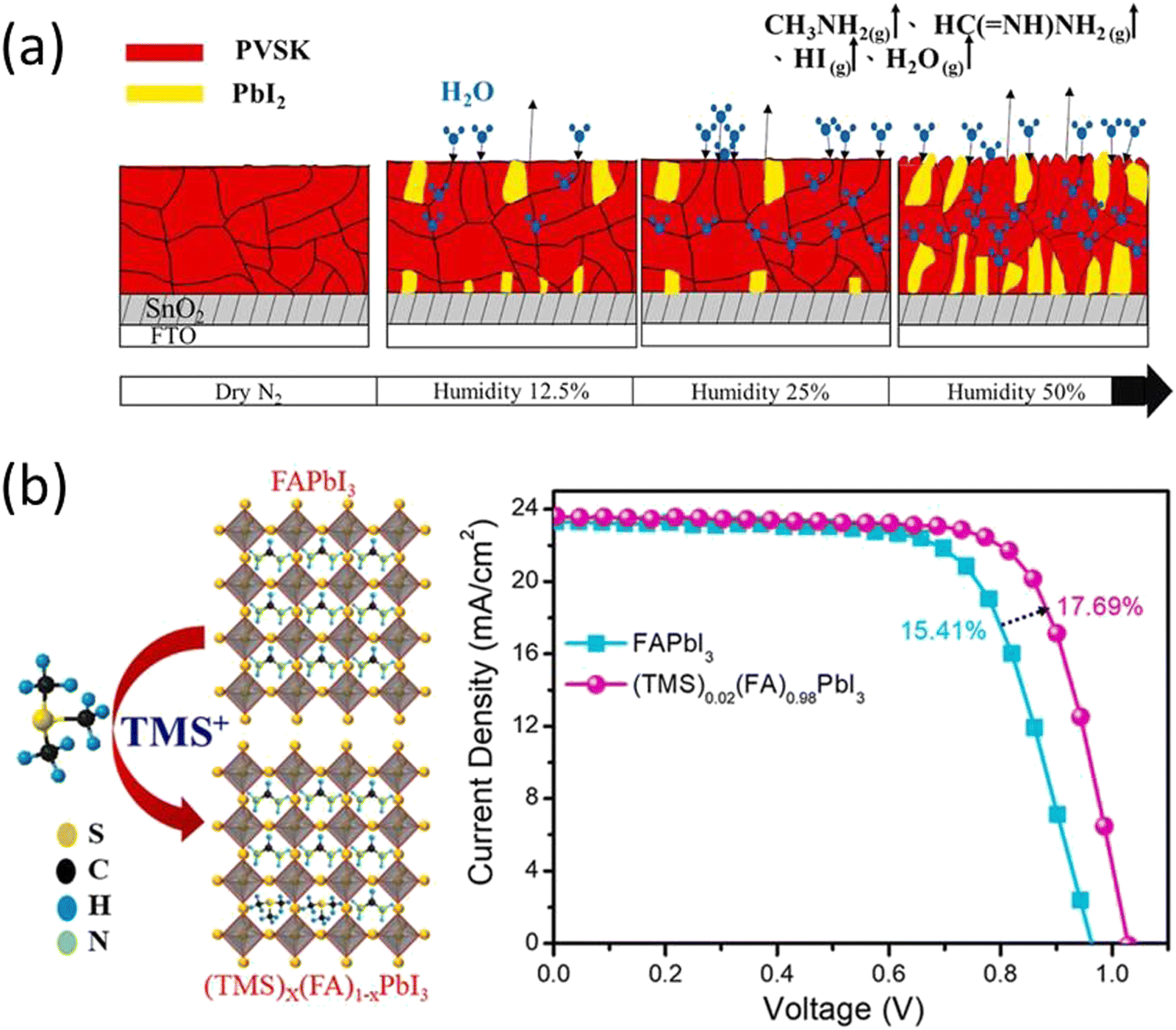 | ||
Fig. 2 (a) Water molecules in contact with (Cs0.05 FA0.85MA0.15Pb(I0.85Br0.15)3) perovskite crystal grain boundaries form a hydrated phase; PVSK in the figure. Continuous heating during the annealing process returns partially hydrated phases to the original perovskite phase, while a portion of the samples underwent degradation to form CsI, CH3NH2, HC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NH2, HI, PbI2, PbBr2, and H2O. Increasing the humidity levels to 50% accelerated degradation, resulting in the formation of large quantities of CH3NH2, HC(NH)NH2, and PbI2. Reproduced from ref. 69 with permission from John Wiley and Sons, copyright 2022. (b) Schematic crystal structure and JV plots of FAPbI3 before and after TMS+ incorporation. Reproduced from ref. 73 with permission from Elsevier, copyright 2022. NH)NH2, HI, PbI2, PbBr2, and H2O. Increasing the humidity levels to 50% accelerated degradation, resulting in the formation of large quantities of CH3NH2, HC(NH)NH2, and PbI2. Reproduced from ref. 69 with permission from John Wiley and Sons, copyright 2022. (b) Schematic crystal structure and JV plots of FAPbI3 before and after TMS+ incorporation. Reproduced from ref. 73 with permission from Elsevier, copyright 2022. | ||
Apart from small cation doping in perovskite there are also reports of substituting organic cations at A site present. PEA was reported to partially substitute the FAPbI3 using phenylethylammonium iodide (PEAI) to form FAxPEA1–xPbI3. The substitution of PEA alleviates both the phase stability and moisture degradation of FAPbI3 as well as the performance of the PSCs.61 A similar study of substitution of acetamidinium at MA+ sites led to improved perovskite crystal structure and significant enhancement in the stability of the PSCs.71 Seok et al., stabilized the α-FAPbI3 phase by doping methylenediammonium dichloride (MDACl2) into perovskite. This doping facilitates the stable PCE of 23.7% after 600 hours of operation. Moreover, the unencapsulated devices show high thermal stability and retained more than 90% efficiency under continuous annealing at 150 °C in air for 20 hours.72 In 2022, S. Sandhu et al. introduced trimethylsulfonium (TMS+), as a moisture-stable and aprotic organosulfonium cation to form a mixed-cation (TMS)x(FA)1−xPbI3 perovskite with enhanced moisture stability with PCE of 17.69% (Fig. 2b).73
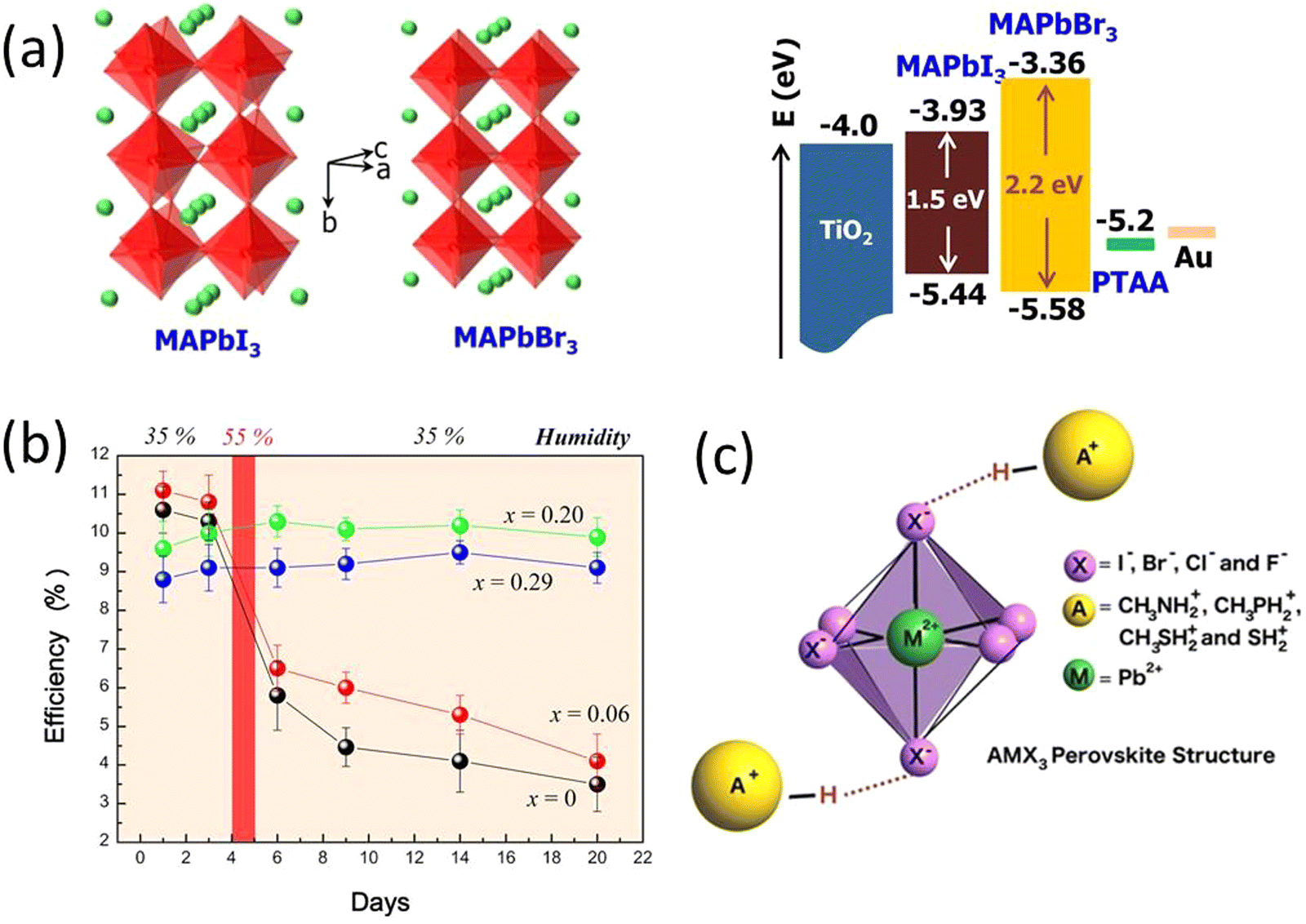 | ||
| Fig. 3 (a) Crystal structures and energy levels of MAPbI3 and MAPbBr3, and CBM and VBM of MAPbI3, MAPbBr3, and TiO2 are represented in eV. (b) Power conversion efficiency variation of the heterojunction solar cells based on MAPb(I1–xBrx)3 (x = 0, 0.06, 0.20, 0.29) with time stored in air at room temperature without encapsulation. Reprinted with permission from ref. 74. Copyright 2013 American Chemical Society. (c) Replacing the methylammonium cation with alternative protonated cations allows an increase in the stability of the perovskite by forming strong hydrogen bonds with the halide anions. Reproduced from ref. 75 with permission from John Wiley and Sons, copyright 2016. | ||
An early breakthrough work by Snaith et al., suggested that the stability of MAPbI3−xClx was remarkably enhanced compared with MAPbI3, when both processed under ambient conditions.30 Similarly, other studies have found that the iodide-chloride mixed-halide perovskite CH3NH3PbI3−xClx performed well when the cells were fabricated under ambient conditions.30,76,86 The small amount of chloride substitution leads to a smoother and more crystalline surface morphology of the thin films.87,88 Grätzel et al., reported the introduction of small amount of methylammonium chloride (MACl) to the perovskite precursor solution facilitate improved photovoltaic performance, suppresses the formation of bulk and surface defects with high quality and larger grain size perovskite thin films.87 In a similar study Kim et al., demonstrated addition of MACl into the FAPbI3 perovskite induces an intermediate to the pure FAPbI3 α-phase without annealing with increased gain size and phase crystallinity.88 A successful use of blade coating technique was reported by Zhu et al., for realization of high efficiency large area PSCs having comparable efficiencies with small area solar cells fabricated through spin coating technique.89 They used excess MACl to achieve a highly crystalline film with a very wide thermal processing window and achieved a similar perovskite morphology and device efficiency for both spin coated and blade coated devices.
Partial substitution of acetate ions (OAc−, CH3COO−) in place of I− ions also significantly enhance the device efficiency and stability. The introduction of saturated Pb(OAc)2 solution on the top of the MAPbI3 perovskite precursor restructures the lattice to form MAPbI3−x(OAc)x resulting in perovskite film with high crystallinity, large grain size and uniform film morphology. Moreover, the OAc− ions enable bond with uncoordinated Pb2+ resulting in reduction of uncoordinated Pb2+, leading to stable perovskite film.90 In another study Pan et al., substituted super halide BH4− to simultaneously immobilize methylammonium and substitute iodide vacancy based on the dihydrogen bonding interactions for achieving high-performance PSCs. The incorporation of BH4− anion inhibits the decomposition of CH3NH3+ cation by forming CH3NH3PbI3−x(BH4−)x perovskite.91
4.2. Interfacial layer engineering/surface passivation
Most of the PSC devices are fabricated with sandwich like structure, where light absorbing perovskite layer is sandwiched between ETM and HTM. To effectively transport and extract the photogenerated charge carriers in the device, the band alignment of the interfaces must be optimized in order to reduce the potential barrier and charge trapping. In general, the performance of a PSC relies on efficient charge transportation, charge extraction, and lastly collection of charge carriers. The processing of hybrid perovskites via low temperature solution methods and use of near ambient temperatures induces the formation of polycrystalline films with vacancies at the interfaces and this significantly impacts the PSC performance.92 Based on the defect formation energy, the defects are categorised as shallow and deep trap defects. Vacancies are known to be shallow defect whereas interstitials and uncoordinated ions as deep trap defects, which act as a charge trapping/recombination center enabling the loss of the photogenerated charges.Grain boundaries, interfaces, crystallographic defect, and surface imperfections within traditional polycrystalline perovskites can function as reactive entry points for moisture and oxygen into the cell. Single crystal perovskites have lower densities of defects and grain boundaries that act as susceptible areas for deterioration. Further, single-crystalline perovskites have a reduced trap density, extended charge carrier diffusion lengths, longer carrier lifetimes, and improved carrier mobility.93
4.3. Doping
Prolonging the stability of the light absorbing perovskite material is the highest priority for achieving the high stability and high efficiency solar cells devices. As discussed in the above sections, the effect of mixing of multiple cations and anions is encouraging and promising. In addition to this method, dopant engineering in perovskites has proven to further stabilize the perovskite and ultimately enhance their performance in devices.Grätzel's group in 2015 used a one step solution processing strategy using phosphonic ammonium additives that result in efficient PSCs with enhanced moisture stability.94 They used butylphosphonic acid 4-ammonium chloride ((4-AB2PACl)) as additive into the perovskite precursor solution and achieved a uniform perovskite surface. As illustrated in Fig. 4a, the additives act as a cross linker between neighboring grains in the perovskite film through hydrogen bonding of PO(OH)2 and NH3+ terminal groups. The PSCs show an increase in photovoltaic performance from 8.8% to 16.7% and robust air stability over 1000 h after the addition of (4-ABPACl). Use of a self healing polymer scaffold architecture has been demonstrated by Zhao et al., to prevent the moisture induced degradation of perovskite.95 The use of polyethylene glycol (PEG) helps in achieving uniform perovskite film, humidity resistance and self healing perovskite crystals. Though they found that light has a strong destructive influence on the stability of PSC, due to the light accelerated decomposition of perovskite with the aid of hydrolysis by water molecules under illumination. Moreover, in moisture stability tests the devices retain PCE for up to 300 h under highly humid environment (RH 70%). Air stable and efficient PSCs have been reported by Bi et al., by adding aliphatic fluorinated amphiphilic additive, 1,1,1-trifluoro ethyl ammonium iodide (FEAI), to the perovskite precursor solution.96 The FEAI treated PSCs imparts greater moisture tolerance when compared to pristine devices after 4 months under ambient conditions. Most importantly, the moisture tolerance is enhanced without formation of 2D perovskite due to the presence of CF3-terminal group on the surface of the 3D perovskite which prevents the direct chemical reaction between MAPbI3 and water molecules. In another study, Priya et al., proposed a formation of highly stable MAPbI3 perovskite through a “green” self assembly.97 In this study poly(methyl methacrylate) (PMMA) was added to the 2D layered MAPbI3 perovskite intermediates. This gives rise to 3D perovskite with grain boundaries covered with PMMA which effectively blocks the moisture from intrusion, of boundary coated perovskites through the self-assembly process. The PMMA treated PSCs exhibit no degradation under high humid conditions (70% RH) for over a month and excellent device performance.
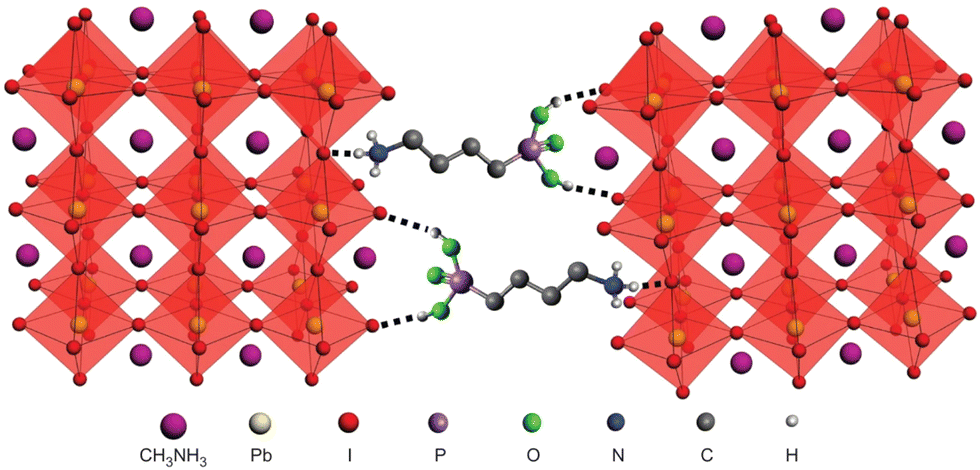 | ||
| Fig. 4 Schematic illustration of two neighboring grain structures (in which the methyl ammonium groups are shown as one sphere for clarity) and the PbI64− octahedra (shown in red) are crosslinked by butylphosphonic acid 4-ammonium chloride (4-ABPACl) hydrogen-bonding interactions (OH⋯I and N–H⋯I) of the iodide from the iodoplumbate complex with the phosphonic acid (–PO(OH)2) and the ammonium (–NH3+) end groups of the 4-ABPACl species. Reproduced from ref. 94 with permission from Springer Nature, copyright 2015. | ||
Recently, Bakr et al., reported a methodology to reduce the efficiency gap between inverted PSCs and regular PSC by using a trace amount of surface anchoring alkylammonium ligands (AAL) with varying length as grain and interface modifiers/passivators.82 The addition of long chain AAL to the precursor solution supresses non-radiative recombination, trap-state formation, and improves the optoelectronic properties of the perovskite. The devices with AAL showed PCE of 22.3% and improved operational stability of over 1000 h at maximum power point (MPP) without any loss of efficiency under N2 atmosphere. Unfortunately, the study does not report the moisture stability of the devices. Ding et al., adopted a cost effective and widely available water soluble additive polyvinyl alcohol (PVA) to improve the quality of the perovskite film and enhance the moisture stability of PSCs without compromising the efficiency.98 The incorporation of PVA have been found to enhance the efficiency by nearly 12% compared to those without additives. Moreover, the devices with PVA additive show retention of more than 90% efficiency even after 30 days of storage under high humidity environments (around 90% RH).
Moreover, some other additives such as alkylamines,82 1,3,7-trimethylxanthine (caffeine)99 2-aminoterephthalic acid,100 thioctic acid,101 have also been used and successfully implemented in developing highly efficient and stable PSCs. Overall, the use of additives suppresses the non-radiative losses, decreases the trap density, permits self healing of the perovskite, and stabilizes the surface of the perovskite film by terminating the grain boundaries with moisture resistant groups. There are several fullerene derivatives and organic/polymer materials such as phenyl-C61-butyric acid methyl ester (PCBM),102–105 a fullerene derivative (α-bis-PCBM),54 cross-linkable [6,6]-phenyl-C61-butyric styryl dendron ester (PCBSD),106 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ),107,108 graphitic carbon nitride (g-C3N4),109 have been explored and found to reduce the ion migration, excellent photo and thermal stability, improved solar cell performance, growth of perovskite crystallite size, enhanced charge transfer efficiency, and most importantly enhanced moisture resistance by hindering moisture from infiltrating the perovskite layer.95–97
4.4. Organic passivator/inorganic passivators
The organic cations or organic salts have been widely explored for passivating the perovskite surface.110,111 Along with passivation of perovskite surface the use of organic salts make the surface of the perovskite more hydrophobic and prevents the intrusion of water molecules into the perovskite layer.In most of the studies, ammonium halide salts have been explored for passivating the perovskite film. Zhu et al., used two different passivators phenylethylammonium iodide (PEAI) and 4-tert-butyl-benzylammonium iodide (tBBAI) in PSCs and compared the passivation effects.112 They observed that the surface treatment with tBBAI significantly accelerates the charge extraction from perovskite to HTM and suppresses the non-radiative carrier recombination. The PSCs retain over 95% of their initial PCE (23.5%) after 500 h of continuous operation at maximum power point (MPP) and over 90% of their initial PCE (23.5%) after 55 days of storage under ambient conditions with RH 50–70%. Yang et al., demonstrated the constructive molecular configurations for surface defect passivation of PSC.113 The chemical environment of a functional group was systematically studied with theophylline, caffeine and theobromine treatment, through hydrogen bond formation (between N–H and I) assisted CO binding with anti-site Pb defects. Notably, the theophylline treated PSCs maintained 95% of their original PCE value when stored under ambient conditions (with RH 20–30%) for 60 days. Yang et al., successfully fabricated stable PSCs using moisture tolerant molecules on the surface of the perovskite. They used some hydrophobic tertiary and quaternary alkyl ammonium cations on the perovskite surface by facile surface functionalization technique. They demonstrated that these molecules can protect the perovskite film under high relative humidity (90 ± 5%) for about a month.114 Furthermore, Guo et al.,115 reported that the use of Cs oleate in the interface between perovskite and HTM has the similar effect.
The use of polymer as an insulating tunnelling layer in PSCs was proposed by Wang et al.116 They used cross linked fluoro-silane polymer as the tunnelling layer between perovskite and ETL, this layer can significantly improves the device performance by suppressing the carrier recombination at cathode and prevents the penetration of water or moisture into the perovskite film. Niu et al., demonstrated the use of ultrathin Al2O3 layer on the top of the perovskite to fabricate moisture stable PSCs.34 Later a similar strategy with ALD deposited ultrathin Al2O3 layer on top of perovskite and on top of HTL was investigated to prolong the lifetime of the PSCs.117 In both these studies efficiencies were retained after several days of storage in air. Poly(3-hexylthiophene) (P3HT) is an organic, polymer with p-type conductivity. P3HT is thermally stable and hydrophobic. When applied in PSC, P3HT modifies the interface between the perovskite and electrode, which boosts moisture stability and device efficiency.118–122
In 2022, T. H. Han et al. synthesized polyurea (PU) with polydimethylsiloxane (PDMS) elastomer blocks and and polyurea with diisocyanate linkages and introduced the molecules into the perovskite precursor.123 The inclusion of the polymers in the perovskite solar cells regulated crystal growth, mitigated defect formation, safeguarded against moisture, increased structural integrity, and demonstrated a power conversion efficiency of 23.25%. This is attributed to the robust covalent interaction between the polymer and the perovskite material. Significantly, the incorporation of polymers led to a substantial enhancement in the mechanical durability of PSCs through thermal-induced healing effect. The inclusion of the PDMS blocks resulted in an elastic response to mechanical stress, coupled with the hydrophobic encapsulation of the MHP thin film. Furthermore, interchain hydrogen bonding helped dissipate strain energy when stretching the MHP thin film. Subsequently, the reconfiguration of the hydrogen bonding network following mechanical damage contributed to the development of the self-repairing feature (Fig. 5a). The devices without encapsulation remained stable for over 1000 hours.
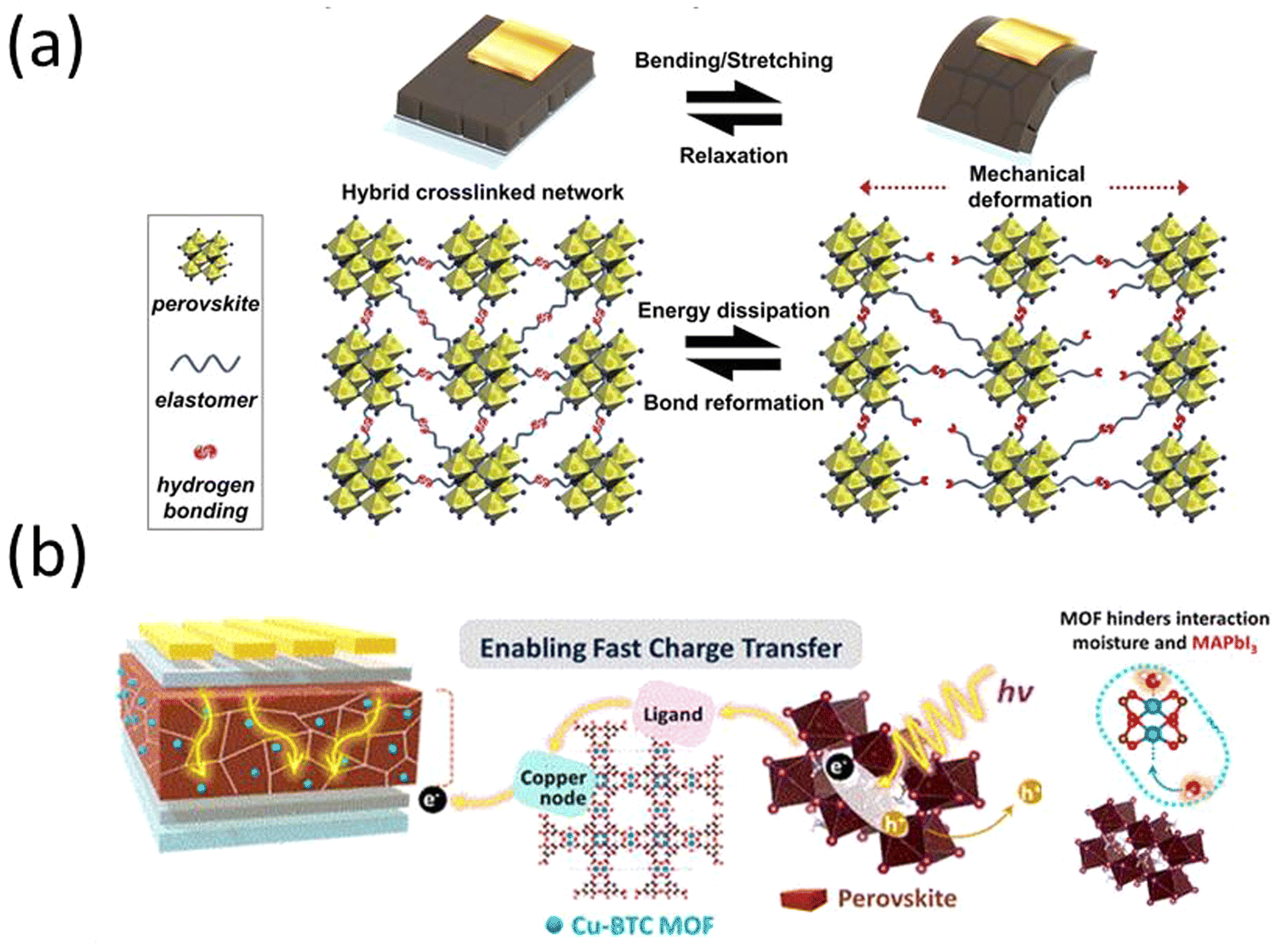 | ||
| Fig. 5 (a) Schematic illustration of hybrid cross-linked network and its energy-dissipation and self-recovery mechanism. Reproduced from ref. 123 with permission from John Wiley and Sons, copyright 2022. (b) Schematic design of a MOF/perovskite hybrid layer to facilitate charge transfer and increase moisture stability in a PSC. Reprinted with permission from ref. 124. Copyright 2022 American Chemical Society. | ||
An organic/inorganic hybrid approach was introduced by J. Lee et al., wherein a perovskite-MOF hybrid was synthesized.124 The MOF was a hygroscopic copper(II) benzene-1,3,5-tricarboxylate metal−organic framework (Cu-BTC MOFs) that bound up moisture instead of the perovskite, which lead to larger perovskite crystal grains. Additionally, the MOF facilitated charge transfer when exposed to light by transferring photoexcited electrons from the perovskite to TiO2 (Fig. 5b).
Recently, J. Sun et al. fabricated PSCs with 4,4′-dinonyl-2,2′-dipyridine (DN-DP) instead of hygroscopic DMSO.125 DN-DP exhibits a stronger ability to coordinate with Pb2+ ions compared to the SO group present in DMSO. This heightened coordination effect is essential for eliminating adducts derived from DMSO and minimizing the formation of empty spaces. Through the electron-donating characteristics of pyridine, the presence of DN-DP within the perovskite film can effectively address defects and fine-tune the energy level alignment of the perovskite structure. As a result of incorporating DN-DP, the open-circuit voltage (Voc) of the resulting perovskite solar cell (PSC) is enhanced from 1.107 V (in the control device) to 1.153 V, leading to a notable increase in power conversion efficiency (PCE) to 24.02% (Fig. 6). Furthermore, owing to the moisture resistance conferred by the hydrophobic nonyl group, the PCE maintains 90.4% of its initial performance after enduring 1000 hours of storage in ambient conditions.
 | ||
| Fig. 6 The influence of DN-DP on the formation of the perovskite film. First, DN-DP exchange with DMSO to forms a new adduct of DN-DP–PbI2 following the extension of the PbI2 lattice. Then FA ions were inserted into the layered PbI2 and exchanged with DN-DP.126 The coordination between DN-DP and PbI2 can retard the fast reaction between organic ammonium halides and PbI2, thus slowing down the crystallization rate and ensuring large grain growth. DN-DP reduces the residual DMSO in perovskite film, optimizes the contact of the buried interface, passivates the defects and changes the energy level, and finally improves the performance of the device. Reproduced from ref. 125 with permission from Elsevier, copyright 2023. | ||
4.5. 3D-2D perovskites
Over the past few years, tremendous efforts have been made to improve the photovoltaic performance of perovskite solar cells by passivating the interfaces and minimizing the surface trap defects by means of a long chain organic cation coating or 2D perovskite coating on the top of the light absorbing perovskite layer.84,94,112,114,127–136 The enhanced stability of devices against moisture in 2D layered perovskites is attributed to the presence of a large organic cation, rendering the 2D perovskite hydrophobic.137,138 Additionally, the stacking of organic–inorganic layers creates a natural quantum well structure, where inorganic layers act as potential wells and organic layers serve as potential barriers.139,140 This quantum well structure allows for easy tuning of dielectric and excitonic properties by adjusting halide anions and varying the thickness of the inorganic layer.141,142 Coating 2D layer on the top of 3D perovskite is one of the most commonly used approach these days for preventing the 3D perovskite from the moisture.143 There are several 2D perovskite materials have been explored using this approach and significant improvement in the device stability was achieved but very few of them showed higher photovoltaic efficiencies.130,144,145Wang et al., demonstrated the introduction of n-butylammonium (BA, C4H9NH3) cations in the mixed cation mixed halide 3D perovskite to realize efficient and ambient air stable solar cells (Fig. 7a).143 The incorporation of BA into the system led to formation of thin films with increased crystallinity and preferentially oriented growth of grains and 2D-3D perovskite heterostructure with 3D and 2D perovskite domains and 2D perovskite platelets interspersed between 3D perovskite grains. The PSCs with optimal amount of BA exhibit average efficiency of 17.5% with 1.61 eV band gap perovskite. The PSCs with BA cations sustain more than 80% of their efficiency after 1000 h in air. In a similar study, Chen et al.,146 proposed the reaction of 1,4-butanediamine iodide (BEAI2) vapor with 3D perovskite facilitate crystallization of large grain perovskite films to obtain stable and efficient PSCs. The BEAI2 reacts with PbI2 from 3D perovskite at 110 °C to form 2D BEAPbI4 perovskite on the surface of the 3D perovskite. The PSCs kept at 55% RH show drop of only 13% of its initial PCE when monitored for 1000 h.
 | ||
| Fig. 7 (a) Schematic illustration of the proposed self-assembled 2D-3D perovskite film structure. Reproduced from ref. 143 with permission from Springer nature, copyright 2017. (b) Schematic illustration of channels for iodide ion diffusion provided by the vacancy defects inside the device and the effect of the internal encapsulation. Reproduced from ref. 147 with permission from John Wiley and Sons, copyright 2022. | ||
In 2022, Y. Ge et al. developed an internal encapsulation strategy involving the incorporation of NbCl5 at the concealed junction between the perovskite layers.147 Simultaneously, a layer of BABr was applied atop the perovskite through spin coating. The NbCl5 treatment leads to a notable decrease in oxygen vacancies within the SnO2 layer. This, in turn, yields a more uniform and well-crystallized perovskite layer, along with a diminished presence of PbI2 on the surface of the perovskite. Following the deposition of BABr onto the perovskite layer, the formation of 2D perovskites occurs (Fig. 7b). This process effectively addresses defects on the surface, leading to their passivation, and additionally results in a further reduction of PbI2 content. A PCE of 24.01% and an excellent stability of 88% remaining PCE after 1000 h were achieved.
Formation of phase pure α-FAPbI3 was observed by Lee et al. after incorporation of small percentage of PEA2PbI4 2D perovskite to 3D perovskite precursor solution.131 The formation of 2D perovskite at the grain boundaries of FAPbI3 helps in preventing the moisture induced degradation of 3D perovskite and also helps in reducing ion migration, as illustrated in Fig. 8. The nonpolar aromatic rings and longer alkyl chains of the 2D perovskite protects the grain boundaries of 3D perovskite and significantly enhanced the moisture stability of the film. They achieved certified PCE close to 20% in 2D perovskite incorporated devices and these devices show less than 20% drop in the PCE when kept at 40 °C and 50% RH for about 400 h under 1 sun illumination. During this time 3D only, devices decreased more than 60% in the PCE values under similar circumstances. In a similar study, stabilization of all inorganic α-CsPbI3 phase has been achieved, by adding small amount of EDAPbI4 perovskite.148 The efficiency of the devices drops from 13% to 10% within a month while retaining the α-CsPbI3 phase, when stored in a dry box (in dark) without any encapsulation.
 | ||
| Fig. 8 Schematics of the device incorporating polycrystalline 3D perovskite film with 2D perovskite at grain boundaries and band structure of each layer in device. Reproduced from ref. 131 with permission from Springer nature, copyright 2018. | ||
In situ incorporation of an ultrathin (5-AVA)2PbI4 (5-AVA = 5 ammoniumvaleric acid) 2D perovskite passivation layer at the interface between the perovskite and hole transporting layer (CuSCN) is demonstrated.149 This passivation layer improves both moisture stability and photostability of the PSC. The unencapsulated devices retain 98% of initial PCE after 60 days of storage in dark with relatively low humidity of 10%. A PSC device stable for 1-year was achieved by Grancini et al., by engineering an ultra-stable 2D/3D (HOOC(CH2)4NH3)2PbI4/MAPbI3 perovskite interface.130 The 2D/3D multidimensional interface yields ∼15% PCE in standard mesoporous solar cell structure. They also demonstrated fabrication of large area fully printable PSCs with 11.2% efficiency without any drop in the performance for >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h.
000 h.
In one of our recent studies, we demonstrated the use of long chain alkylammonium chain cation based 2D perovskite (C14H29NH3)2PbI4, (C16H33NH3)2PbI4, and (C18H37NH3)2PbI4 to prevent the moisture induced degradation of 3D perovskite.127 The 2D perovskites were selected based on previous literature indicating that as a powders these perovskites were stable in water.150 We observed that 2D perovskite coating not only encapsulate the 3D perovskite but also suppress PbI2 impurity and aids improvement in the Voc by passivating the surface trap states. The 2D perovskite incorporated PSCs achieved ∼17% efficiency, which is slightly lower than its 3D counterpart (18.19%). In stability test the 2D incorporated PSCs retained about 80% of their PCE after more than 200 days of storage under ambient conditions with RH 25–80%, within this period 3D only devices show more than 60% loss in PCE (Fig. 9a). More importantly when the 3D@2D film was dipped in water it remains unaffected at least for few minutes before the degradation started indicating 2D perovskite strongly prevent the penetration of water molecules into the 3D perovskite, see Fig. 9b.
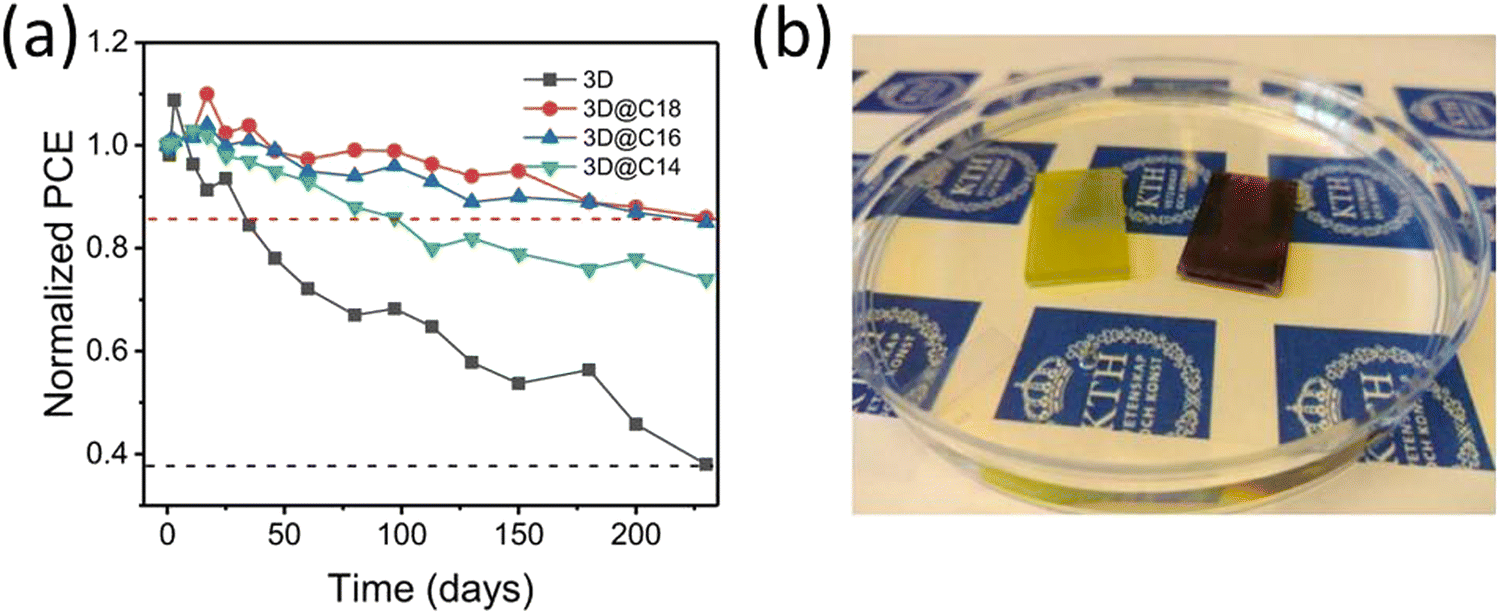 | ||
| Fig. 9 (a) Variation in PCE of nonencapsulated 3D-only PSCs and (C14H29NH3)2PbI4 (C14), (C16H33NH3)2PbI4 (C16), and (C18H37NH3)2PbI4 (C18) perovskite-coated PSCs kept under ambient atmosphere (RH 25–80%, 10–25 °C, in the dark). (b) After 3 minutes of direct immersion in water, the purely 3D perovskite has reacted with water to form PbI2 (left film) while same 3D perovskite coated with (C18H37NH3)2PbI4 perovskite (right film) is unaffected. Reproduced from ref. 127 with permission from Springer nature, copyright 2021. | ||
Bismuth based 2D perovskites like MA3Bi2I9 have also been explored in PSCs owing to their strong moisture stability. Hu et al., demonstrated the coating of vertically aligned MA3Bi2I9 platelets on the top of MAPbI3 grains, exhibits excellent photoelectric properties and robust tolerance against moisture and oxygen.145 They achieved PCE of 18.97% with significantly reduced hysteresis and remarkably enhanced air stability. There are several other studies, which successfully demonstrate the use of 2D perovskites for encapsulating 3D perovskites and achieved improved stability and photovoltaic performance.45,128,149,151,152
By adjusting the dimensional components of room temperature-formed two-dimensional perovskite layers using oleylammonium iodide molecules, R. Azmi et al., created PSCs that are resistant to damp heat.153 These layers serve to passivate the perovskite surface at the electron-selective contact. As a result, the inverted PSCs achieved an impressive 24.3% power conversion efficiency (PCE). Even under rigorous damp-heat testing for over 1000 hours, these cells maintained over 95% of their initial efficiency. This accomplishment fulfills a crucial industrial stability benchmark for photovoltaic modules.
4.6. 2D perovskite only
Lower dimensional materials, especially 2D perovskite materials, are found to be the most suitable materials for passivating the interface between HTM and perovskite. Both 2D perovskite and organic cations offer excellent surface passivation, suppression of defects, and improved grain size and film quality, while making the surface of perovskite films more hydrophobic and thus delivering moisture stable devices. Researchers took motivation from the above findings and started exploring 2D only PSCs.154–156The early pioneering work on 2D perovskite based solar cells delivered an efficiency of up to 4.73% but with greater moisture resistance compared to 3D perovskite based PSCs.157 Later, after significant investigations higher efficiencies up to ∼13% have been achieved by fabricating near single crystal quality thin films out of layered 2D Ruddlesden–Popper (RP) perovskites.158 Even higher certified PCE (17.8%) have recently been reported in bulky 2-(methylthio)ethylamine hydrochloride (MTEACl) based 2D RP PSCs.159 Apart from the improved moisture stability 2D perovskite capping layers offer reduced ion migration and ion accumulation resulting in reduced hysteresis and increased Voc compared to 3D only PSCs.58,149 Tsai et al., reported (BA)2(MA)2Pb3I10 (n = 3) and (BA)2(MA)3Pb4I13 (n = 4) 2D perovskites, which were derived from the parent 3D perovskite (MAPbI3) using a hot casting technique.158 In stability tests, for over 2250 h under constant illumination and relative humidity of 65%, the encapsulated PSC devices showed negligible degradation compared to 3D PSCs that lost 90% in PCE. In another study Ma et al., reported excellent moisture and thermal stability in propane 1,3 diammonium (PDA) cation based 2D perovskite.129 PDA based 2D perovskites can retain over 90% of their efficiency upon storage for over 1000 h under high humidity conditions (RH 85%).
Recently Ren et al., demonstrated use of bulky alkylammonium MTEA cations in 2D perovskite framework.159 The (MTEA)2(MA)4Pb5I16 2D perovskite based solar cell devices were fabricated and the photovoltaic parameters were compared with that of (BA)2(MA)4Pb5I16. The MTEA cation based 2D PSCs show better photovoltaic properties than the BA based 2D perovskites, with highest PCE of 18.06% for MTEA based PSCs and 15.94% for BA based PSCs. Furthermore, in stability tests MTEA-based PSCs retain more than 85% of initial PCE after continuous power output at MPP for 1000 h and better moisture tolerance of up to 1512 h (under 70% RH) for (MTEA)2(MA)4Pb5I16 thin films.
4.7. Charge-transporting materials
As discussed above, the performance of the PSCs is not solely hampered due to the degradation of perovskites but also the degradation of charge-transporting layers is responsible for decreasing PSC performance. While this is beyond the scope of this review, we summarize and cite some of the important reports on them. As an example, a solution processed strategy to stabilize the interface between perovskites and graphene oxide charge-transporting heterostructures was proposed by Wang et al.160 Strong bonding between Pb-rich perovskite and graphene oxide (GO) favors the formation of stable heterostructures based on ionic Pb–Cl and Pb–O bonds. As a demonstration of the stabilized structure, the PCSs with perovskite/Cl–GO heterostructures retained >90% of their initial efficiency after 1000 h of continuous light soaking at 60 °C. The use of hydrophobic HTM or carbon-based electrodes help in improving the device longevity; however, these materials have suppressed the efficiency of the PSCs.161–164 Following the initial exploration of carbon based perovskite solar cells introduced by Chang et al. in 2016,165 a range of strategies has been suggested to enhance device performance and reaching to 15.35% PCE.166 These strategies encompass optimizing fabrication techniques, engineering solvents, adjusting compositions, refining interfaces, optimizing charge transport layers, and more.167–171 Carbon-based electrodes were developed to address the challenges associated with metal electrodes in perovskite solar cells—such as high-temperature and low pressure deposition conditions, sensitivity to humidity, limited scalability, and cost concerns. Carbon materials offer numerous advantages over metal electrodes, including a reduced environmental impact, compatibility with flexible materials, hydrophobicity, high surface area, and porosity. Utilizing carbon as both the HTM and the top electrode can safeguard the device from moisture, leading to the development of an air-stable solar cell even in the absence of encapsulation.172–174 To resolve the stability issue in carbon-based electrode, the inorganic HTMs such as NiO,175–179 Al2O3,180 CuSCN,181–185 and Cu2O186 have been investigated.Apart from HTM, the interface contact between the electron transport layer (ETL) specifically TiO2, SnO2, and ZnO-based ETLs and perovskite affects the stability of PSCs.187–189 As an example, the impact of UV-induced O2− desorption in TiO2 accelerates PSC degradation. Q. Ye et al.190 investigated NaTaO3, a perovskite oxide, as a novel ETL for PSCs. It effectively shields against UV damage, suppresses perovskite layer degradation, and enhances overall PSC stability. PSCs incorporating NaTaO3 demonstrated a power conversion efficiency (PCE) of 21.07%, maintaining over 80% of the initial PCE after 240 minutes of UV irradiation in air. In contrast, the reference device, with a PCE of 20.16%, retained only approximately 53% of its initial PCE under the same testing conditions. In 2023, J. Liu et al.191 presented SnO2 ETL embedded with carbon quantum dots (CQDs). The power conversion efficiency (PCE) saw a substantial increase from 21.62% to 24.05%, attributed to improved charge extraction. Notably, perovskite solar cells with the CQD-SnO2 ETL retained over 84% of their initial PCE after 1000 hours of continuous irradiation under 1-sun illumination, effectively suppressing perovskite degradation from the bottom contact. Moreover, utilizing a TiO2/SnO2 double ETL192 and surface modification of ZnO by anchoring fullerene material,193 and interlayer modifiers between oxide ETLs and perovskite can improve stability in PSCs.194
5. Conclusion and outlook
The hybrid perovskites have evolved as low cost, solution processable material for third generation solar cells owing to their large absorption coefficient, high charge carrier mobility, longer diffusion lengths and high-power conversion efficiency. The fabrication of PSCs through facile and cost-effective screen printing, spin coating, and roll-to-roll printing methods makes these materials an easy technology for commercialization. However, the instability of the perovskite material under ambient conditions presently limits the prospects for commercialization of this technology.In this review, we discussed the major advancements to improve the stability of the perovskites under ambient air through, mixed cation-mixed anion substitution, doping of organic/inorganic materials into perovskite, interface engineering, and using moisture resistant 2D perovskite/organic cations. However, modification of perovskites using the strategies above complicates the understanding of degradation and charge transport processes and there is an urgent need to obtain a simple and sustainable solution to overcome these complications. To achieve stable working PSC the HTM, ETM, and electrodes need further stabilization. This opens a wide scope for exploration of dopant-free HTM and inorganic HTM to increase device stability.
The PSC lifetime is limited by two major factors, the first is intrinsic instability of the halide perovskite absorber and second is poor resistance to environmental factors such as heat and moisture. An in-depth knowledge of intrinsic and extrinsic degradation mechanism in materials and devices is prerequisite to achieve stable and robust solar cell devices. The moisture induced degradation of MAPbI3 is very well understood, but in most of the high performing PSCs mixed cation (Cs/MA/FA) and mixed anion (I/Br) composition of perovskites are used and moisture induced degradation of mixed cation-mixed halide perovskites is not investigated in detail and therefore, it is equally important to understand the moisture induced degradation of these high performing perovskites.
The lifetime of PSCs has been significantly prolonged from few hours to ten thousand of hours by employing interface engineering and mixed cation–anion substitution but still the stability is far away from commercialization requirements. Furthermore, the future work should be focused on prolonging the stability of PSC up to 5 to 10 years or even longer. The inclusion of 2D perovskite in PSCs along with 3D perovskite has provided promising results with significantly enhanced stability under humid conditions and at elevated temperatures. However, the stability tracking over several months is lacking in the literature, which hinders the actual use of 2D perovskite as long term encapsulation. Moreover, the crystallization mechanism of perovskite after molecular passivation/2D perovskite have not been fully understood.
External encapsulation techniques can be straightforward processes to prevent the infiltration of moisture and oxygen; however, internal encapsulation is still needed. In this regard It is important to select a moisture resistive HTM, or organic/inorganic compounds as passivators in perovskite layers. Moreover, we anticipate that the combination of 3D/2D heterostructure and appropriate internal encapsulation techniques can proficiently alleviate perovskite instability in the presence of humidity. Appropriate encapsulation strategies need to be developed in order to achieve high performance and stable PSC within a universally acceptable strategy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support from the Swedish government through the research initiative “STandUP for ENERGY”, the Swedish Foundation for Strategic Research (SSF: RMA15-0130), and the Swedish Energy Agency (Energimyndigheten; 49278-1).References
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. Liu, Y.-L. Loo, J. M. Luther, C.-Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N.-G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- J. Huang, S. Tan, P. D. Lund and H. Zhou, Energy Environ. Sci., 2017, 10, 2284–2311 RSC.
- D. Wang, M. Wright, N. K. Elumalai and A. Uddin, Sol. Energy Mater. Sol. Cells, 2016, 147, 255–275 CrossRef CAS.
- W. Chi and S. K. Banerjee, Chem. Mater., 2021, 33, 4269–4303 CrossRef CAS.
- R. Wang, M. Mujahid, Y. Duan, Z.-K. Wang, J. Xue and Y. Yang, Adv. Funct. Mater., 2019, 29, 1808843 CrossRef CAS.
- B. Chen, S. Wang, Y. Song, C. Li and F. Hao, Chem. Eng. J., 2022, 430, 132701 CrossRef CAS.
- Y. Lin, Y. Bai, Y. Fang, Z. Chen, S. Yang, X. Zheng, S. Tang, Y. Liu, J. Zhao and J. Huang, J. Phys. Chem. Lett., 2018, 9, 654–658 CrossRef CAS PubMed.
- Y. Wu, F. Xie, H. Chen, X. Yang, H. Su, M. Cai, Z. Zhou, T. Noda and L. Han, Adv. Mater., 2017, 29, 1701073 CrossRef PubMed.
- Y. Zhao, W. Zhou, Z. Han, D. Yu and Q. Zhao, Phys. Chem. Chem. Phys., 2021, 23, 94–106 RSC.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS PubMed.
- Y. Ye, Y. Yin, Y. Chen, S. Li, L. Li and Y. Yamauchi, Small, 2023, 19, 2208119 CrossRef CAS PubMed.
- Y. Yin, M. Wang, V. Malgras and Y. Yamauchi, ACS Appl. Energy Mater., 2020, 3, 10447–10452 CrossRef CAS.
- Y. Jiang, L. Qiu, E. J. Juarez-Perez, L. K. Ono, Z. Hu, Z. Liu, Z. Wu, L. Meng, Q. Wang and Y. Qi, Nat. Energy, 2019, 4, 585–593 CrossRef CAS.
- Q.-Q. Chu, Z. Sun, D. Wang, B. Cheng, H. Wang, C.-P. Wong and B. Fang, Matter, 2023, 6, 3838–3863 CrossRef CAS.
- A. Uddin, M. B. Upama, H. Yi and L. Duan, Coatings, 2019, 9, 65 CrossRef.
- S. Zhang, Z. Liu, W. Zhang, Z. Jiang, W. Chen, R. Chen, Y. Huang, Z. Yang, Y. Zhang, L. Han and W. Chen, Adv. Energy Mater., 2020, 10, 2001610 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- A. F. Akbulatov, L. A. Frolova, M. P. Griffin, I. R. Gearba, A. Dolocan, D. A. Vanden Bout, S. Tsarev, E. A. Katz, A. F. Shestakov, K. J. Stevenson and P. A. Troshin, Adv. Energy Mater., 2017, 7, 1700476 CrossRef.
- W. Chen, H. Sun, Q. Hu, A. B. Djurišić, T. P. Russell, X. Guo and Z. He, ACS Energy Lett., 2019, 4, 2535–2536 CrossRef CAS.
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2020, 5, 44–60 CrossRef CAS.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- S. Yang Woon, H. Noh Jun, J. Jeon Nam, C. Kim Young, S. Ryu, J. Seo and I. Seok Sang, Science, 2015, 348, 1234–1237 CrossRef PubMed.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, M. Zakeeruddin Shaik, K. Nazeeruddin Mohammad, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed.
- N. K. Noel, S. D. Stranks, A. Abate, C. Wehrenfennig, S. Guarnera, A.-A. Haghighirad, A. Sadhanala, G. E. Eperon, S. K. Pathak, M. B. Johnston, A. Petrozza, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 3061–3068 RSC.
- M. H. Kumar, S. Dharani, W. L. Leong, P. P. Boix, R. R. Prabhakar, T. Baikie, C. Shi, H. Ding, R. Ramesh, M. Asta, M. Graetzel, S. G. Mhaisalkar and N. Mathews, Adv. Mater., 2014, 26, 7122–7127 CrossRef CAS PubMed.
- N.-G. Park, M. Grätzel, T. Miyasaka, K. Zhu and K. Emery, Nat. Energy, 2016, 1, 16152 CrossRef CAS.
- M. Lee Michael, J. Teuscher, T. Miyasaka, N. Murakami Takurou and J. Snaith Henry, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- P. Docampo, J. M. Ball, M. Darwich, G. E. Eperon and H. J. Snaith, Nat. Commun., 2013, 4, 2761 CrossRef PubMed.
- J. Tang, D. Jiao, L. Zhang, X. Zhang, X. Xu, C. Yao, J. Wu and Z. Lan, Sol. Energy, 2018, 161, 100–108 CrossRef CAS.
- D. Yang, R. Yang, K. Wang, C. Wu, X. Zhu, J. Feng, X. Ren, G. Fang, S. Priya and S. Liu, Nat. Commun., 2018, 9, 3239 CrossRef PubMed.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705–710 RSC.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- P. Raval, M. A. Akhavan Kazemi, J. Ruellou, J. Trébosc, O. Lafon, L. Delevoye, F. Sauvage and G. N. Manjunatha Reddy, Chem. Mater., 2023, 35, 2904–2917 CrossRef CAS.
- S. N. Habisreutinger, D. P. McMeekin, H. J. Snaith and R. J. Nicholas, APL Mater., 2016, 4, 091503 CrossRef.
- J. Liu, Y. Wu, C. Qin, X. Yang, T. Yasuda, A. Islam, K. Zhang, W. Peng, W. Chen and L. Han, Energy Environ. Sci., 2014, 7, 2963–2967 RSC.
- J. You, Y. Yang, Z. Hong, T.-B. Song, L. Meng, Y. Liu, C. Jiang, H. Zhou, W.-H. Chang, G. Li and Y. Yang, Appl. Phys. Lett., 2014, 105, 183902 CrossRef.
- C.-H. Chiang, M. K. Nazeeruddin, M. Grätzel and C.-G. Wu, Energy Environ. Sci., 2017, 10, 808–817 RSC.
- K. K. Bass, R. E. McAnally, S. Zhou, P. I. Djurovich, M. E. Thompson and B. C. Melot, Chem. Commun., 2014, 50, 15819–15822 RSC.
- G. E. Eperon, S. N. Habisreutinger, T. Leijtens, B. J. Bruijnaers, J. J. van Franeker, D. W. deQuilettes, S. Pathak, R. J. Sutton, G. Grancini, D. S. Ginger, R. A. J. Janssen, A. Petrozza and H. J. Snaith, ACS Nano, 2015, 9, 9380–9393 CrossRef CAS PubMed.
- P. Docampo and T. Bein, Acc. Chem. Res., 2016, 49, 339–346 CrossRef CAS PubMed.
- Z. Zhu, V. G. Hadjiev, Y. Rong, R. Guo, B. Cao, Z. Tang, F. Qin, Y. Li, Y. Wang, F. Hao, S. Venkatesan, W. Li, S. Baldelli, A. M. Guloy, H. Fang, Y. Hu, Y. Yao, Z. Wang and J. Bao, Chem. Mater., 2016, 28, 7385–7393 CrossRef CAS.
- N. H. Tiep, Z. Ku and H. J. Fan, Adv. Energy Mater., 2016, 6, 1501420 CrossRef.
- S. Ma, G. Yuan, Y. Zhang, N. Yang, Y. Li and Q. Chen, Energy Environ. Sci., 2022, 15, 13–55 RSC.
- X. Yu, Y. Qin and Q. Peng, J. Phys. Chem. A, 2017, 121, 1169–1174 CrossRef CAS PubMed.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- J. Yang, Z. Yuan, X. Liu, S. Braun, Y. Li, J. Tang, F. Gao, C. Duan, M. Fahlman and Q. Bao, ACS Appl. Mater. Interfaces, 2018, 10, 16225–16230 CrossRef CAS PubMed.
- L. Caliò, M. Salado, S. Kazim and S. Ahmad, Joule, 2018, 2, 1800–1815 CrossRef.
- S. Luo and W. A. Daoud, J. Mater. Chem. A, 2015, 3, 8992–9010 RSC.
- J. Luo, J. Xia, H. Yang, L. Chen, Z. Wan, F. Han, H. A. Malik, X. Zhu and C. Jia, Energy Environ. Sci., 2018, 11, 2035–2045 RSC.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- F. Zhang, W. Shi, J. Luo, N. Pellet, C. Yi, X. Li, X. Zhao, T. J. S. Dennis, X. Li, S. Wang, Y. Xiao, S. M. Zakeeruddin, D. Bi and M. Grätzel, Adv. Mater., 2017, 29, 1606806 CrossRef PubMed.
- S. Wang, H. Chen, J. Zhang, G. Xu, W. Chen, R. Xue, M. Zhang, Y. Li and Y. Li, Adv. Mater., 2019, 31, 1903691 CrossRef CAS PubMed.
- Z. Wang, Z. Shi, T. Li, Y. Chen and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 1190–1212 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- B. Kim and S. I. Seok, Energy Environ. Sci., 2020, 13, 805–820 RSC.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- N. Li, Z. Zhu, C.-C. Chueh, H. Liu, B. Peng, A. Petrone, X. Li, L. Wang and A. K. Y. Jen, Adv. Energy Mater., 2017, 7, 1601307 CrossRef.
- F. Cao, P. Zhang, H. Sun, M. Wang and L. Li, Nano Res., 2022, 15, 8955–8961 CrossRef CAS.
- Y. Jiang, M. R. Leyden, L. Qiu, S. Wang, L. K. Ono, Z. Wu, E. J. Juarez-Perez and Y. Qi, Adv. Funct. Mater., 2018, 28, 1703835 CrossRef.
- F. Xu, T. Zhang, G. Li and Y. Zhao, J. Mater. Chem. A, 2017, 5, 11450–11461 RSC.
- L. Gil-Escrig, C. Momblona, M.-G. La-Placa, P. P. Boix, M. Sessolo and H. J. Bolink, Adv. Energy Mater., 2018, 8, 1703506 CrossRef.
- P. McMeekin David, G. Sadoughi, W. Rehman, E. Eperon Giles, M. Saliba, T. Hörantner Maximilian, A. Haghighirad, N. Sakai, L. Korte, B. Rech, B. Johnston Michael, M. Herz Laura and J. Snaith Henry, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- G. Niu, W. Li, J. Li, X. Liang and L. Wang, RSC Adv., 2017, 7, 17473–17479 RSC.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- R.-Y. Hsu, Y.-J. Liang, Y.-J. Hung and Y.-C. Lin, Mater. Sci. Semicond. Process., 2022, 152, 107100 CrossRef CAS.
- B. P. Dhamaniya, P. Chhillar, B. Roose, V. Dutta and S. K. Pathak, ACS Appl. Mater. Interfaces, 2019, 11, 22228–22239 CrossRef CAS PubMed.
- P. Singh, R. Mukherjee and S. Avasthi, ACS Appl. Mater. Interfaces, 2020, 12, 13982–13987 CrossRef CAS PubMed.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, H. Lee Jun and I. Seok Sang, Science, 2019, 366, 749–753 CrossRef CAS PubMed.
- S. Sandhu, M. M. Rahman, M. Senthilkumar, B. Yadagiri, J. Park, K. Yoo and J.-J. Lee, J. Power Sources, 2022, 551, 232207 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- F. El-Mellouhi, A. Marzouk, E. T. Bentria, S. N. Rashkeev, S. Kais and F. H. Alharbi, ChemSusChem, 2016, 9, 2648–2655 CrossRef CAS PubMed.
- Z. Liu, J. Chang, Z. Lin, L. Zhou, Z. Yang, D. Chen, C. Zhang, S. Liu and Y. Hao, Adv. Energy Mater., 2018, 8, 1703432 CrossRef.
- K. A. Bush, K. Frohna, R. Prasanna, R. E. Beal, T. Leijtens, S. A. Swifter and M. D. McGehee, ACS Energy Lett., 2018, 3, 428–435 CrossRef CAS.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T.-Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS PubMed.
- E. Jung, K. Budzinauskas, S. Öz, F. Ünlü, H. Kuhn, J. Wagner, D. Grabowski, B. Klingebiel, M. Cherasse, J. Dong, P. Aversa, P. Vivo, T. Kirchartz, T. Miyasaka, P. H. M. van Loosdrecht, L. Perfetti and S. Mathur, ACS Energy Lett., 2020, 5, 785–792 CrossRef CAS.
- S. Wu, Z. Li, J. Zhang, X. Wu, X. Deng, Y. Liu, J. Zhou, C. Zhi, X. Yu, W. C. H. Choy, Z. Zhu and A. K. Y. Jen, Adv. Mater., 2021, 33, 2105539 CrossRef CAS PubMed.
- Y. Chen, Z. Yang, X. Jia, Y. Wu, N. Yuan, J. Ding, W.-H. Zhang and S. Liu, Nano Energy, 2019, 61, 148–157 CrossRef CAS.
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D.-J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z.-H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS.
- Q. Dong, C. Zhu, M. Chen, C. Jiang, J. Guo, Y. Feng, Z. Dai, S. K. Yadavalli, M. Hu, X. Cao, Y. Li, Y. Huang, Z. Liu, Y. Shi, L. Wang, N. P. Padture and Y. Zhou, Nat. Commun., 2021, 12, 973 CrossRef CAS PubMed.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- J. Wang, J. Li, Y. Zhou, C. Yu, Y. Hua, Y. Yu, R. Li, X. Lin, R. Chen, H. Wu, H. Xia and H.-L. Wang, J. Am. Chem. Soc., 2021, 143, 7759–7768 CrossRef CAS PubMed.
- S. Colella, E. Mosconi, P. Fedeli, A. Listorti, F. Gazza, F. Orlandi, P. Ferro, T. Besagni, A. Rizzo, G. Calestani, G. Gigli, F. De Angelis and R. Mosca, Chem. Mater., 2013, 25, 4613–4618 CrossRef CAS.
- M. M. Tavakoli, M. Saliba, P. Yadav, P. Holzhey, A. Hagfeldt, S. M. Zakeeruddin and M. Grätzel, Adv. Energy Mater., 2019, 9, 1802646 CrossRef.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- M. Yang, Z. Li, M. O. Reese, O. G. Reid, D. H. Kim, S. Siol, T. R. Klein, Y. Yan, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Energy, 2017, 2, 17038 CrossRef CAS.
- Y. Zhang, Y. Ma, I. Shin, Y. K. Jung, B. R. Lee, S. Wu, J. H. Jeong, B. H. Lee, J. H. Kim, K. H. Kim and S. H. Park, ACS Appl. Mater. Interfaces, 2020, 12, 7186–7197 CrossRef CAS PubMed.
- S. Xu, G. Liu, H. Zheng, X. Xu, L. Zhang, H. Xu, L. Zhu, F. Kong, Y. Li and X. Pan, ACS Appl. Mater. Interfaces, 2020, 12, 8249–8259 CrossRef CAS PubMed.
- J. Shi, X. Xu, D. Li and Q. Meng, Small, 2015, 11, 2472–2486 CrossRef CAS PubMed.
- R. K. Battula, C. Sudakar, P. Bhyrappa, G. Veerappan and E. Ramasamy, Cryst. Growth Des., 2022, 22, 6338–6362 CrossRef CAS.
- X. Li, M. Ibrahim Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Grätzel, Nat. Chem., 2015, 7, 703–711 CrossRef CAS PubMed.
- Y. Zhao, J. Wei, H. Li, Y. Yan, W. Zhou, D. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- D. Bi, P. Gao, R. Scopelliti, E. Oveisi, J. Luo, M. Grätzel, A. Hagfeldt and M. K. Nazeeruddin, Adv. Mater., 2016, 28, 2910–2915 CrossRef CAS PubMed.
- C. Wu, H. Li, Y. Yan, B. Chi, K. M. Felice, R. B. Moore, B. A. Magill, R. R. H. H. Mudiyanselage, G. A. Khodaparast, M. Sanghadasa and S. Priya, Sol. RRL, 2018, 2, 1800052 CrossRef.
- Y. Sun, Y. Wu, X. Fang, L. Xu, Z. Ma, Y. Lu, W.-H. Zhang, Q. Yu, N. Yuan and J. Ding, J. Mater. Chem. A, 2017, 5, 1374–1379 RSC.
- R. Wang, J. Xue, L. Meng, J.-W. Lee, Z. Zhao, P. Sun, L. Cai, T. Huang, Z. Wang, Z.-K. Wang, Y. Duan, J. L. Yang, S. Tan, Y. Yuan, Y. Huang and Y. Yang, Joule, 2019, 3, 1464–1477 CrossRef CAS.
- Z. Liu, F. Cao, M. Wang, M. Wang and L. Li, Angew. Chem., Int. Ed., 2020, 59, 4161–4167 CrossRef CAS PubMed.
- H. Chen, T. Liu, P. Zhou, S. Li, J. Ren, H. He, J. Wang, N. Wang and S. Guo, Adv. Mater., 2020, 32, 1905661 CrossRef CAS PubMed.
- C. Park, H. Ko, D. H. Sin, K. C. Song and K. Cho, Adv. Funct. Mater., 2017, 27, 1703546 CrossRef.
- J. Xu, A. Buin, A. H. Ip, W. Li, O. Voznyy, R. Comin, M. Yuan, S. Jeon, Z. Ning, J. J. McDowell, P. Kanjanaboos, J.-P. Sun, X. Lan, L. N. Quan, D. H. Kim, I. G. Hill, P. Maksymovych and E. H. Sargent, Nat. Commun., 2015, 6, 7081 CrossRef CAS PubMed.
- C.-H. Chiang and C.-G. Wu, Nat. Photonics, 2016, 10, 196–200 CrossRef CAS.
- Y. Fang, C. Bi, D. Wang and J. Huang, ACS Energy Lett., 2017, 2, 782–794 CrossRef CAS.
- M. Li, Y.-H. Chao, T. Kang, Z.-K. Wang, Y.-G. Yang, S.-L. Feng, Y. Hu, X.-Y. Gao, L.-S. Liao and C.-S. Hsu, J. Mater. Chem. A, 2016, 4, 15088–15094 RSC.
- W.-Q. Wu, Q. Wang, Y. Fang, Y. Shao, S. Tang, Y. Deng, H. Lu, Y. Liu, T. Li, Z. Yang, A. Gruverman and J. Huang, Nat. Commun., 2018, 9, 1625 CrossRef PubMed.
- C. Liu, Z. Huang, X. Hu, X. Meng, L. Huang, J. Xiong, L. Tan and Y. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 1909–1916 CrossRef CAS PubMed.
- L.-L. Jiang, Z.-K. Wang, M. Li, C.-C. Zhang, Q.-Q. Ye, K.-H. Hu, D.-Z. Lu, P.-F. Fang and L.-S. Liao, Adv. Funct. Mater., 2018, 28, 1705875 CrossRef.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- F. Zhang, Q. Huang, J. Song, Y. Zhang, C. Ding, F. Liu, D. Liu, X. Li, H. Yasuda, K. Yoshida, J. Qu, S. Hayase, T. Toyoda, T. Minemoto and Q. Shen, Sol. RRL, 2020, 4, 1900243 CrossRef CAS.
- H. Zhu, Y. Liu, F. T. Eickemeyer, L. Pan, D. Ren, M. A. Ruiz-Preciado, B. Carlsen, B. Yang, X. Dong, Z. Wang, H. Liu, S. Wang, S. M. Zakeeruddin, A. Hagfeldt, M. I. Dar, X. Li and M. Grätzel, Adv. Mater., 2020, 32, 1907757 CrossRef CAS PubMed.
- R. Wang, J. Xue, K.-L. Wang, Z.-K. Wang, Y. Luo, D. Fenning, G. Xu, S. Nuryyeva, T. Huang, Y. Zhao, L. Yang Jonathan, J. Zhu, M. Wang, S. Tan, I. Yavuz, N. Houk Kendall and Y. Yang, Science, 2019, 366, 1509–1513 CrossRef CAS PubMed.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef CAS.
- X. Guo, T. M. Koh, B. Febriansyah, G. Han, S. Bhaumik, J. Li, N. F. Jamaludin, B. Ghosh, X. Chen, S. Mhaisalkar and N. Mathews, ACS Appl. Mater. Interfaces, 2019, 11, 27882–27889 CrossRef CAS PubMed.
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed.
- X. Dong, X. Fang, M. Lv, B. Lin, S. Zhang, J. Ding and N. Yuan, J. Mater. Chem. A, 2015, 3, 5360–5367 RSC.
- G. Wang, W. Dong, A. Gurung, K. Chen, F. Wu, Q. He, R. Pathak and Q. Qiao, J. Power Sources, 2019, 432, 48–54 CrossRef CAS.
- G. Chen, Z. Gong, X. Bin and S. Agbolaghi, Polym.-Plast. Technol. Mater., 2023, 62, 162–176 CAS.
- Y. Zhang, W. Liu, F. Tan and Y. Gu, J. Power Sources, 2015, 274, 1224–1230 CrossRef CAS.
- D. Sharma, R. Mehra and B. Raj, J. Mater. Sci., 2022, 57, 21172–21191 CrossRef CAS.
- Y. Wang, Y. Xiong, H. Wang, X. Wu, J. Sha, Y. Shang, Y. Zhang, W. Li and S. Wang, Curr. Appl. Phys., 2023, 47, 54–59 CrossRef.
- T.-H. Han, Y. Zhao, J. Yoon, J. Y. Woo, E.-H. Cho, W. D. Kim, C. Lee, J.-W. Lee, J.-M. Choi, J. Han, J.-S. Nam, K. Wang, S. Priya, M. Balaban, I. Jeon and Y. Yang, Adv. Funct. Mater., 2022, 32, 2207142 CrossRef CAS.
- J. Lee, N. Tsvetkov, S. R. Shin and J. K. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 35495–35503 CrossRef CAS PubMed.
- J. Sun, W. Chen, Y. Ren, Y. Niu, Z. Yang, L. E. Mo, Y. Huang, Z. Li, H. Zhang and L. Hu, J. Energy Chem., 2023, 82, 219–227 CrossRef CAS.
- W. Hui, L. Chao, H. Lu, F. Xia, Q. Wei, Z. Su, T. Niu, L. Tao, B. Du, D. Li, Y. Wang, H. Dong, S. Zuo, B. Li, W. Shi, X. Ran, P. Li, H. Zhang, Z. Wu, C. Ran, L. Song, G. Xing, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Science, 2021, 371, 1359–1364 CrossRef CAS PubMed.
- B. P. Kore, W. Zhang, B. W. Hoogendoorn, M. Safdari and J. M. Gardner, Commun. Mater., 2021, 2, 100 CrossRef CAS.
- C. Ma, C. Leng, Y. Ji, X. Wei, K. Sun, L. Tang, J. Yang, W. Luo, C. Li, Y. Deng, S. Feng, J. Shen, S. Lu, C. Du and H. Shi, Nanoscale, 2016, 8, 18309–18314 RSC.
- C. Ma, D. Shen, T.-W. Ng, M.-F. Lo and C.-S. Lee, Adv. Mater., 2018, 30, 1800710 CrossRef PubMed.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- J.-W. Lee, Z. Dai, T.-H. Han, C. Choi, S.-Y. Chang, S.-J. Lee, N. De Marco, H. Zhao, P. Sun, Y. Huang and Y. Yang, Nat. Commun., 2018, 9, 3021 CrossRef PubMed.
- F. Zhang, H. Lu, J. Tong, J. J. Berry, M. C. Beard and K. Zhu, Energy Environ. Sci., 2020, 13, 1154–1186 RSC.
- Y. Liu, S. Akin, L. Pan, R. Uchida, N. Arora, V. Milić Jovana, A. Hinderhofer, F. Schreiber, R. Uhl Alexander, M. Zakeeruddin Shaik, A. Hagfeldt, M. I. Dar and M. Grätzel, Sci. Adv., 2019, 5, eaaw2543 CrossRef CAS PubMed.
- T. Zhou, H. Lai, T. Liu, D. Lu, X. Wan, X. Zhang, Y. Liu and Y. Chen, Adv. Mater., 2019, 31, 1901242 CrossRef PubMed.
- H. Kim, S.-U. Lee, D. Y. Lee, M. J. Paik, H. Na, J. Lee and S. I. Seok, Adv. Energy Mater., 2019, 9, 1902740 CrossRef CAS.
- G. Wu, R. Liang, M. Ge, G. Sun, Y. Zhang and G. Xing, Adv. Mater., 2022, 34, 2105635 CrossRef CAS PubMed.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2019, 4, 4–22 CrossRef CAS.
- W. Fu, H. Liu, X. Shi, L. Zuo, X. Li and A. K.-Y. Jen, Adv. Funct. Mater., 2019, 29, 1900221 CrossRef.
- B. Traore, L. Pedesseau, L. Assam, X. Che, J.-C. Blancon, H. Tsai, W. Nie, C. C. Stoumpos, M. G. Kanatzidis, S. Tretiak, A. D. Mohite, J. Even, M. Kepenekian and C. Katan, ACS Nano, 2018, 12, 3321–3332 CrossRef CAS PubMed.
- J. C. Blancon, A. V. Stier, H. Tsai, W. Nie, C. C. Stoumpos, B. Traoré, L. Pedesseau, M. Kepenekian, F. Katsutani, G. T. Noe, J. Kono, S. Tretiak, S. A. Crooker, C. Katan, M. G. Kanatzidis, J. J. Crochet, J. Even and A. D. Mohite, Nat. Commun., 2018, 9, 2254 CrossRef PubMed.
- C. M. Mauck and W. A. Tisdale, Trends Chem., 2019, 1, 380–393 CrossRef CAS.
- D. B. Straus and C. R. Kagan, J. Phys. Chem. Lett., 2018, 9, 1434–1447 CrossRef CAS PubMed.
- Z. Wang, Q. Lin, F. P. Chmiel, N. Sakai, L. M. Herz and H. J. Snaith, Nat. Energy, 2017, 2, 17135 CrossRef CAS.
- Y. Liu, S. Akin, L. Pan, R. Uchida, N. Arora, J. V. Milić, A. Hinderhofer, F. Schreiber, A. R. Uhl, S. M. Zakeeruddin, A. Hagfeldt, M. I. Dar and M. Grätzel, Sci. Adv., 2019, 5, eaaw2543 CrossRef CAS PubMed.
- Y. Hu, T. Qiu, F. Bai, W. Ruan and S. Zhang, Adv. Energy Mater., 2018, 8, 1703620 CrossRef.
- M. Chen, P. Li, C. Liang, H. Gu, W. Tong, S. Cheng, W. Li, G. Zhao and G. Shao, J. Energy Chem., 2020, 45, 103–109 CrossRef.
- Y. Ge, F. Ye, M. Xiao, H. Wang, C. Wang, J. Liang, X. Hu, H. Guan, H. Cui, W. Ke, C. Tao and G. Fang, Adv. Energy Mater., 2022, 12, 2200361 CrossRef CAS.
- T. Zhang, M. I. Dar, G. Li, F. Xu, N. Guo, M. Grätzel and Y. Zhao, Sci. Adv., 2017, 3, e1700841 CrossRef PubMed.
- J. Chen, J.-Y. Seo and N.-G. Park, Adv. Energy Mater., 2018, 8, 1702714 CrossRef.
- B. P. Kore and J. M. Gardner, Mater. Adv., 2020, 1, 2395–2400 RSC.
- Y. Hu, J. Schlipf, M. Wussler, M. L. Petrus, W. Jaegermann, T. Bein, P. Müller-Buschbaum and P. Docampo, ACS Nano, 2016, 10, 5999–6007 CrossRef CAS PubMed.
- Y. Bai, S. Xiao, C. Hu, T. Zhang, X. Meng, H. Lin, Y. Yang and S. Yang, Adv. Energy Mater., 2017, 7, 1701038 CrossRef.
- R. Azmi, E. Ugur, A. Seitkhan, F. Aljamaan, A. S. Subbiah, J. Liu, G. T. Harrison, M. I. Nugraha, M. K. Eswaran, M. Babics, Y. Chen, F. Xu, T. G. Allen, A. U. Rehman, C.-L. Wang, T. D. Anthopoulos, U. Schwingenschlögl, M. De Bastiani, E. Aydin and S. De Wolf, Science, 2022, 376, 73–77 CrossRef CAS PubMed.
- X. Zhang, G. Wu, W. Fu, M. Qin, W. Yang, J. Yan, Z. Zhang, X. Lu and H. Chen, Adv. Energy Mater., 2018, 8, 1702498 CrossRef.
- W. Fu, J. Wang, L. Zuo, K. Gao, F. Liu, D. S. Ginger and A. K. Y. Jen, ACS Energy Lett., 2018, 3, 2086–2093 CrossRef CAS.
- F. Rehman, I. H. Syed, S. Khanam, S. Ijaz, H. Mehmood, M. Zubair, Y. Massoud and M. Q. Mehmood, Energy Adv., 2023, 2, 1239–1262 RSC.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- H. Ren, S. Yu, L. Chao, Y. Xia, Y. Sun, S. Zuo, F. Li, T. Niu, Y. Yang, H. Ju, B. Li, H. Du, X. Gao, J. Zhang, J. Wang, L. Zhang, Y. Chen and W. Huang, Nat. Photonics, 2020, 14, 154–163 CrossRef CAS.
- Y. Wang, T. Wu, J. Barbaud, W. Kong, D. Cui, H. Chen, X. Yang and L. Han, Science, 2019, 365, 687–691 CrossRef CAS PubMed.
- W. Wei, B. Hu, F. Jin, Z. Jing, Y. Li, A. A. García Blanco, D. J. Stacchiola and Y. H. Hu, J. Mater. Chem. A, 2017, 5, 7749–7752 RSC.
- H. Zhou, Y. Shi, Q. Dong, H. Zhang, Y. Xing, K. Wang, Y. Du and T. Ma, J. Phys. Chem. Lett., 2014, 5, 3241–3246 CrossRef CAS PubMed.
- Y. Zhu, S. Poddar, L. Shu, Y. Fu and Z. Fan, Adv. Mater. Interfaces, 2020, 7, 2000118 CrossRef CAS.
- K. Patel, D. Prochowicz, S. Akin, A. Kalam, M. M. Tavakoli and P. Yadav, Energy Technol., 2023, 11, 2300228 CrossRef CAS.
- X. Chang, W. Li, L. Zhu, H. Liu, H. Geng, S. Xiang, J. Liu and H. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 33649–33655 CrossRef CAS PubMed.
- H. Wang, H. Liu, Z. Dong, X. Wei, Y. Song, W. Li, L. Zhu, Y. Bai and H. Chen, Nano Energy, 2022, 94, 106925 CrossRef CAS.
- X. Zhang, Z. Yu, D. Zhang, Q. Tai and X.-Z. Zhao, Adv. Energy Mater., 2023, 13, 2201320 CrossRef CAS.
- S. Pandey, M. Karakoti, D. Bhardwaj, G. Tatrari, R. Sharma, L. Pandey, M.-J. Lee and N. G. Sahoo, Nanoscale Adv., 2023, 5, 1492–1526 RSC.
- M. Stefanelli, L. Vesce and A. Di Carlo, Nanomaterials, 2023, 13, 313 CrossRef CAS PubMed.
- D. Bogachuk, B. Yang, J. Suo, D. Martineau, A. Verma, S. Narbey, M. Anaya, K. Frohna, T. Doherty, D. Müller, J. P. Herterich, S. Zouhair, A. Hagfeldt, S. D. Stranks, U. Würfel, A. Hinsch and L. Wagner, Adv. Energy Mater., 2022, 12, 2103128 CrossRef CAS.
- M. Que, B. Zhang, J. Chen, X. Yin and S. Yun, Mater. Adv., 2021, 2, 5560–5579 RSC.
- P. Pradid, K. Sanglee, N. Thongprong and S. Chuangchote, Materials, 2021, 14, 5989 CrossRef CAS PubMed.
- D. Pourjafari, N. G. García-Peña, W. Y. Padrón-Hernández, D. Peralta-Domínguez, A. M. Castro-Chong, M. Nabil, R. C. Avilés-Betanzos and G. Oskam, Materials, 2023, 16, 3917 CrossRef CAS PubMed.
- P. Xie, G. Zhang, Z. Yang, Z. Pan, Y. Fang, H. Rao and X. Zhong, Sol. RRL, 2020, 4, 2000431 CrossRef CAS.
- W. Zhang, X. Zhang, T. Wu, W. Sun, J. Wu and Z. Lan, Electrochim. Acta, 2019, 293, 211–219 CrossRef CAS.
- S. Pang, C. Zhang, H. Dong, D. Chen, W. Zhu, H. Xi, J. Chang, Z. Lin, J. Zhang and Y. Hao, ACS Appl. Energy Mater., 2019, 2, 4700–4707 CrossRef CAS.
- S. S. Mali, H. Kim, H. H. Kim, S. E. Shim and C. K. Hong, Mater. Today, 2018, 21, 483–500 CrossRef CAS.
- L. Pan, C. Liu, H. Zhu, M. Wan, Y. Li and Y. Mai, Sol. Energy, 2020, 196, 521–529 CrossRef CAS.
- X. Guo, G. Luo, J. Liu, C. Liao, G. Wang and S. Li, IEEE J. Photovolt., 2018, 8, 1039–1043 Search PubMed.
- D. Koushik, W. J. H. Verhees, Y. Kuang, S. Veenstra, D. Zhang, M. A. Verheijen, M. Creatore and R. E. I. Schropp, Energy Environ. Sci., 2017, 10, 91–100 RSC.
- N. Arora, M. I. Dar, A. Hinderhofer, N. Pellet, F. Schreiber, M. Zakeeruddin Shaik and M. Grätzel, Science, 2017, 358, 768–771 CrossRef CAS PubMed.
- M. Jung, Y. C. Kim, N. J. Jeon, W. S. Yang, J. Seo, J. H. Noh and S. Il Seok, ChemSusChem, 2016, 9, 2592–2596 CrossRef CAS PubMed.
- K. Zhao, R. Munir, B. Yan, Y. Yang, T. Kim and A. Amassian, J. Mater. Chem. A, 2015, 3, 20554–20559 RSC.
- D. Sharma, R. Mehra and B. Raj, Opt. Mater., 2022, 126, 112221 CrossRef CAS.
- T. Wang, G.-J. Xiao, R. Sun, L.-B. Luo and M.-X. Yi, Chin. Phys. B, 2022, 31, 018801 CrossRef.
- L. Lin, L. Jiang, P. Li, B. Fan and Y. Qiu, J. Phys. Chem. Solids, 2019, 124, 205–211 CrossRef CAS.
- S. Huang, P. Li, J. Wang, J. C.-C. Huang, Q. Xue and N. Fu, Chem. Eng. J., 2022, 439, 135687 CrossRef CAS.
- T. Singh, J. Singh and T. Miyasaka, ChemSusChem, 2016, 9, 2559–2566 CrossRef CAS PubMed.
- M. Shahiduzzaman, S. Fukaya, E. Y. Muslih, L. Wang, M. Nakano, M. Akhtaruzzaman, M. Karakawa, K. Takahashi, J.-M. Nunzi and T. Taima, Materials, 2020, 13, 2207 CrossRef CAS PubMed.
- Q.-Q. Ye, M. Li, X.-B. Shi, M.-P. Zhuo, K.-L. Wang, F. Igbari, Z.-K. Wang and L.-S. Liao, ACS Appl. Mater. Interfaces, 2020, 12, 21772–21778 CrossRef CAS PubMed.
- J. Liu, Y. Yin, B. He, P. Wang, M. Wang, W. Cai, Y. Han, Z. Su, J. Guo, R. Cai, S. Jin, X. Gao, J. Bian and Y. Shi, Mater. Today Phys., 2023, 33, 101041 CrossRef CAS.
- C. Li, H. Xu, C. Zhi, Z. Wan and Z. Li, Chin. Phys. B, 2022, 31, 118802 CrossRef.
- K. Yao, S. Leng, Z. Liu, L. Fei, Y. Chen, S. Li, N. Zhou, J. Zhang, Y.-X. Xu, L. Zhou, H. Huang and A. K. Y. Jen, Joule, 2019, 3, 417–431 CrossRef CAS.
- Y. Rui, T. Li, B. Li, Y. Wang and P. Müller-Buschbaum, J. Mater. Chem. C, 2022, 10, 12392–12401 RSC.
| This journal is © The Royal Society of Chemistry 2024 |