
Inhibition of bacterial RNA polymerase function and protein–protein interactions: a promising approach for next-generation antibacterial therapeutics
Jiqing Ye†
 ab,
Cheuk Hei Kan†c,
Xiao Yang*c and
Cong Ma*a
ab,
Cheuk Hei Kan†c,
Xiao Yang*c and
Cong Ma*a
aState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR, China. E-mail: cong.ma@polyu.edu.hk
bSchool of Pharmacy, Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Medical University, Hefei 230032, China
cDepartment of Microbiology, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, Hong Kong SAR, China. E-mail: xiaoyang@cuhk.edu.hk
First published on 26th March 2024
Abstract
The increasing prevalence of multidrug-resistant pathogens necessitates the urgent development of new antimicrobial agents with innovative modes of action for the next generation of antimicrobial therapy. Bacterial transcription has been identified and widely studied as a viable target for antimicrobial development. The main focus of these studies has been the discovery of inhibitors that bind directly to the core enzyme of RNA polymerase (RNAP). Over the past two decades, substantial advancements have been made in understanding the properties of protein–protein interactions (PPIs) and gaining structural insights into bacterial RNAP and its associated factors. This has led to the crucial role of computational methods in aiding the identification of new PPI inhibitors to affect the RNAP function. In this context, bacterial transcriptional PPIs present promising, albeit challenging, targets for the creation of new antimicrobials. This review will succinctly outline the structural foundation of bacterial transcription networks and provide a summary of the known small molecules that target transcription PPIs.
1. Introduction
Bacterial infections have posed significant challenges to human health for centuries, and the discovery of antibiotics revolutionized the field of medicine by providing effective treatments.1 However, the rapid emergence and spread of antibiotic resistance among bacterial pathogens have compromised the efficacy of these life-saving drugs.2–4 Therefore, developing new antibacterial therapeutics with novel mechanisms of action is crucial to addressing the ongoing antibiotic resistance crisis.RNA plays a crucial role in the life cycles of organisms across the three domains of life, and its synthesis is referred to as transcription.5 Generally, RNA is synthesized using DNA as a template, resulting in the production of different types of RNA molecules, such as messenger RNA (mRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA).6 These RNA molecules are then utilized in translation, in which proteins are synthesized in a mRNA-dependent manner.7,8 Notably, some RNA viruses use their RNA genome as a template to synthesize mRNA or make an complementary DNA (cDNA) molecule from the RNA (reverse transcription), subsequently using it to advance their life circle in host cells.9,10
Bacterial transcription is an attractive target for the development of antibacterial agents.11,12 Generally, there are two strategies targeting bacterial transcription. The first involves the direct inhibition of subunits within RNA polymerase (RNAP), while the second is the disruption of the contacts between transcription factors and RNAP core or other factors. Over the past decades, various classes of antibacterial agents have been developed to bind to different sites on bacterial RNAP, including clinically available compounds rifamycin and lipiarmycin.13,14 In recent years, significant progress has been made in elucidating the structures and functions of transcription factors and their contacts with RNAP and other factors. Of note, the identification of critical residues, often referred to as “hotspots”, has facilitated the discovery of protein–protein interaction (PPI) inhibitors with potent antimicrobial activity.
Previously, Ma et al. reviewed the bacterial transcription and inhibitors that bind to different sites on RNAP.11 More recently, the development of modulators of bacterial PPIs and natural products as antimicrobial agents are summarized.15,16 In this perspective, we aim to present the current status of research on bacterial RNAP and transcription factors, especially those that have the potential to be targets for the development of next-generation antibacterial therapeutics. We will emphasize the inhibitors that have been identified thus far and discuss the methods used to screen the hit compounds and their subsequent optimization process.
2. RNA polymerase and clinically used inhibitors
2.1. RNA polymerase
In all three domains of life, multi-subunit RNAPs are the primary enzymes responsible for catalyzing the synthesis of RNA from a DNA template during transcription. Bacteria and archaea utilize only one type of RNAP to transcribe all their genes, whereas eukaryotes utilize four different types of RNAPs (RNAP I, II, III and IV) that transcribe specific subsets of genes without overlapping.17 Amongst, RNAP II is specifically responsible for the synthesis of mRNA.It becomes increasingly clear that all RNAPs are derived from a common ancestor, as evidenced by their amino acid sequence, subunit composition, structures, functions, and molecular mechanisms.18,19 Moreover, the subunit arrangement of all RNAPs shares a common architecture consisting of five subunits, which form the universally conserved core of the RNAP.20 This core structure, which primarily represents the modern bacterial RNAP, accounts for about 75% of the protein mass in archaeal and eukaryotic RNAPs (Fig. 1).21
 | ||
| Fig. 1 Evolution of RNAP structures in the three domains of life from the last universal common ancestor (LUCA). Bacteria: Thermus aquaticus (PDB 1I6V); archaea: Saccharolobus shibatae (PDB 2WAQ); eukaryotes: Saccharomyces cerevisiae RNAP II (PDB 1Y1W). Universally conserved RNAP core subunits are denoted in gray, while subunits unique to the archaeal–eukaryotic lineage are labeled and colored density blue. | ||
Bacterial RNAP is the simplest form of RNAP found among the three domains of life and exists in two forms: the core enzyme and holoenzyme. The core enzyme consists of five subunits: α-dimer (α2), β, β′ and ω.18 The binding of a single protein, the initiation factor σ, to the core enzyme results in the formation of a holoenzyme, which is the active form of RNAP that is essential for transcription initiation.22
Eukaryotic RNAP II, on the other hand, are composed of Rpb1 to Rpb12 subunits, with the numbering based on the subunits in Saccharomyces cerevisiae, whereas the archaeal RNAP subunits are named Rpo1 to Rpo13.23 It is noteworthy that, although archaea and bacteria are both prokaryotes, at the molecular level, archaea RNAP is much more closely related to eukaryotes than bacteria.24 This relationship is supported by molecular characterization and further reinforces the division between the bacterial and archaeal–eukaryotic branches of the universal tree of life. Notably, one of the structural evidence is the presence of a stalk-like protrusion in the core of archaeal and eukaryotic RNAP at a right angle to the transcription direction, which is absent in bacterial RNAP (Fig. 1).25
Structural studies of bacterial RNAP date back to the mid-1960s, and the crystallization of RNAP isolated from Thermus thermophilus was first reported in the late 1970s.26 In 1999, Darst et al. described the X-ray crystal structure of bacterial RNAP core enzyme (PDB 1HQM) (Fig. 2), which was isolated from T. aquaticus.26,27 The core enzyme exhibits a crab claw shape, with a central active site and pincers that bind DNA between them.28 One arm of the claw is primarily the β subunit, and the other is primarily the β′ subunit. The assembly of the β and β′ subunits is aided by the dimer of the α subunit N-terminal domain (αNTD), and the ω subunit completely wraps around the C-terminal tail of β′ subunit.27,28
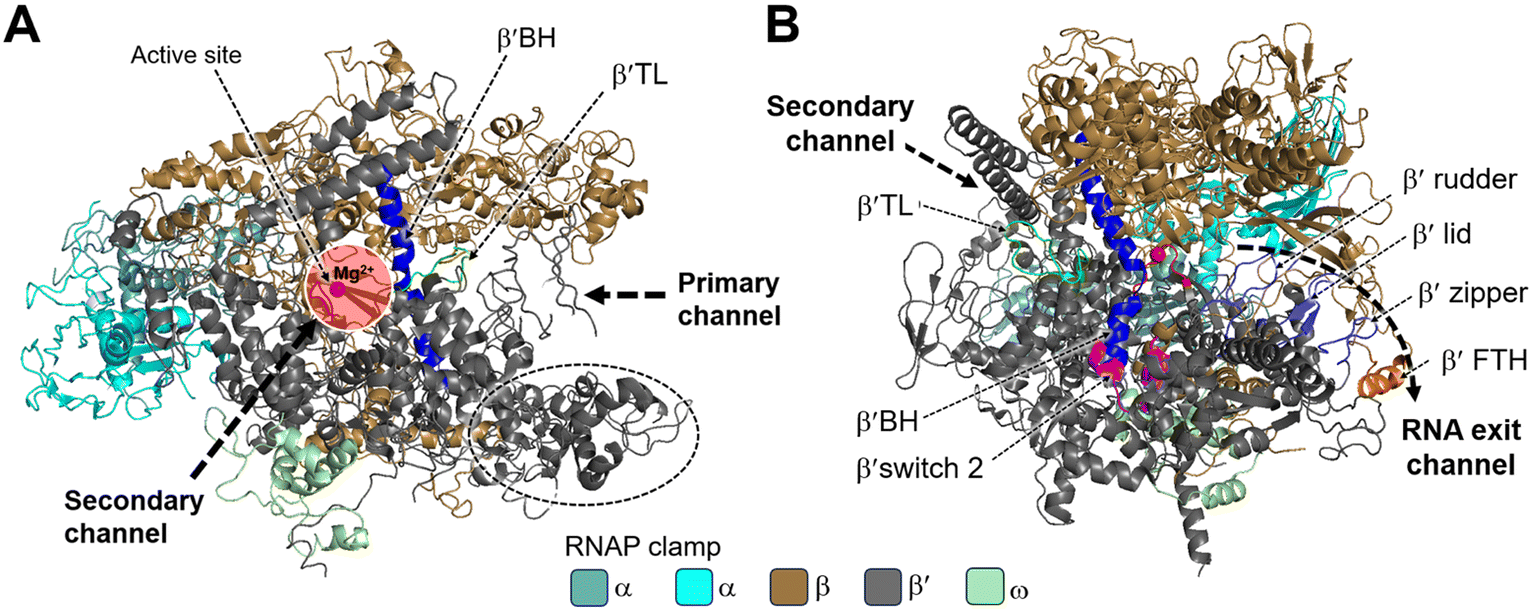 | ||
| Fig. 2 Structural overview of RNAP core and functional motifs from T. aquaticus (PDB 1HQM). A) Secondary channel view; B) primary channel view. The structure is represented as colored ribbons with important domains and structural elements indicated. The directions of primary, secondary and RNA exit channels are indicated by black dashed arrows. | ||
Studies have shown that the active sites of RNAPs are highly conserved across the three domains of life. These active sites are located in the cleft formed by the largest RNAP subunits, β/β′ in bacteria, Rpb1/Rpb2 in eukarya, and Rpo1/Rpo2 in archaea.29,30 In the vicinity of the active site, the α-helical “bridge” (bridge helix, BH) connects the β and β′ clamps, and the two flexible α-helices form a “trigger” loop (TL). In addition to the active site, the cleft contains the primary, secondary, and RNA exit channels (Fig. 2). These structural features form the structural basis for the functions of RNAP.26
During bacterial transcription, the RNAP utilizes several channels to facilitate the process. The primary channel accommodates the double-stranded DNA (dsDNA) template and guides it into the active center cleft of the RNAP. The switch region facilitates conformational changes of the RNAP enzyme, enabling the proper loading of DNA into the active center cleft. Meanwhile, nucleoside triphosphates (NTPs) enter the active center through the secondary channel, serving as building blocks for RNA synthesis. As transcription proceeds, the RNA/DNA hybrid strand is threaded through the active center, and the RNA chain is extruded through the RNA exit channel.31 These channels, hence, play essential roles in the progression of bacterial transcription by facilitating the movement of DNA, NTPs, and the synthesized RNA within the RNAP complex.
2.2. Inhibitors binding to bacterial RNAP directly
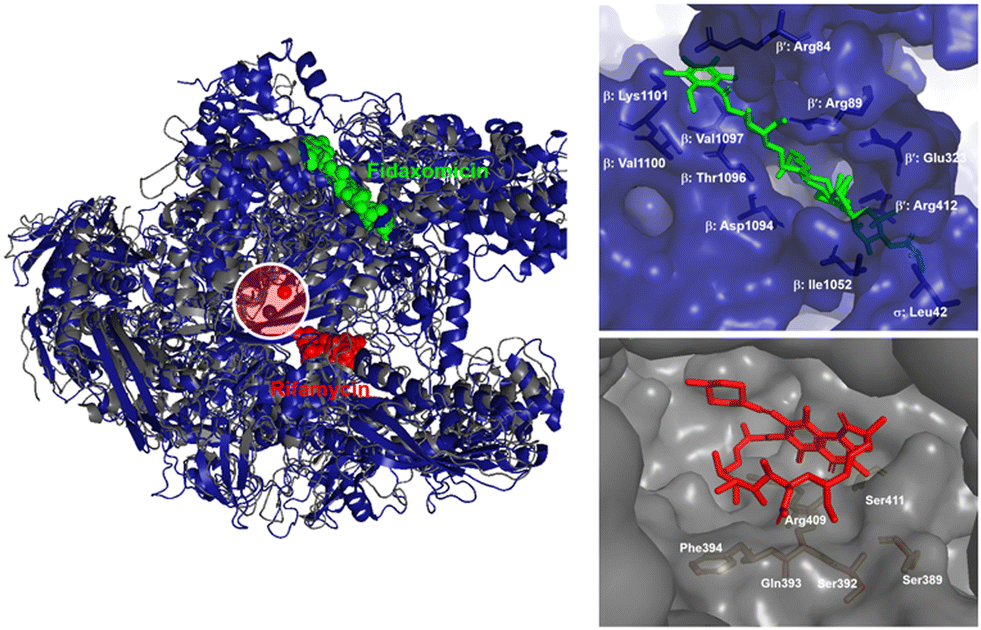
RNAP is an essential enzyme for the viability of organisms and has consequently become a proven target for antibacterial,11,16,32 anticancer33 and antiviral34,35 drug discovery. In the context of bacteria, RNAP is a critical enzyme for gene expression, making it an attractive target for antibacterial therapies. Moreover, RNAP is highly conserved in a wide range of bacterial species, providing a basis for broad-spectrum activity. Together with its low conservation characteristic in eukaryotic RNAPs, which offers a basis for therapeutic selectivity, RNAP is a promising target for antibacterial drug development.36Currently, bacterial RNAP is the target of two classes of antibacterial drugs in clinical use: rifamycin and lipiarmycin (fidaxomicin).22 In addition, several other bacterial RNAP inhibitory agents have been reported, including sorangicin and GE23077 as primary channel inhibitors, streptolydigin, salinamide and CBR703 as inhibitors targeting the mobile element of the primary channel, tagetitoxin and microcin J25 as secondary channel inhibitors, and myxopyronins and squaramides as switch region inhibitors.11,16 The binding sites of these inhibitors are illustrated in Fig. 3. However, despite their potential, none of them enter clinical use as antimicrobials. This is mainly due to their binding sites being located at or near the active site of RNAP, making the development of resistance through point mutations more likely. In addition, most of the molecules are natural products with complex structures which are difficult to prepare.11
 | ||
| Fig. 3 RNAP inhibitors and their corresponding binding sites. | ||
3. Bacterial transcription cycle
During the past few years, exciting progress has been made in understanding the bacterial transcription cycle, especially through the application of single-molecule techniques. These techniques enable us to visualize the real-time movements of RNAP on the DNA template, providing a wealth of quantitative and direct information about transcriptional structures and dynamics.42,43 In general, the transcription cycle can be divided into three phases: initiation, elongation, and termination. While the RNAPs and general transcription cycle are conserved across the three domains of life, details of the regulators in each phase of the cycle can differ among and within different domains of life. In bacteria, the evolution of transcription factors is a good example of this.To ensure transcription in a temporally and spatially coordinated manner during cell growth and development, the enzymatic activity of bacterial RNAP is tightly regulated by a host of transcription factors throughout all stages of the transcription cycle (Fig. 5).44 These factors interact with RNAP through PPIs and directly modify its properties at different stages of transcription. Therefore, these factors and their binding sites on RNAP represent potential targets for developing novel drugs.
 | ||
| Fig. 5 The bacterial transcription cycle. The process contains three general phases: initiation, elongation, and termination. Some of the critical complexes assembled by RNAP and different transcription factors are illustrated. | ||
3.1. Transcription initiation
In bacteria, transcription initiation is a crucial regulatory step in gene expression regulated by σ factors.45 Although the RNAP core is competent for RNA polymerization, it requires a σ factor for promoter recognition and opening.46 Under normal growth conditions, all bacteria possess a primary housekeeping σ factor (such as σ70 in Gram-negative bacteria and σA in Gram-positive bacteria) that controls the transcription of essential genes.47 Furthermore, the σ70 family of sigma factors is further divided into four major groups. Group 1 comprises the primary σ factor, such as σ70 in E. coli. Groups 2 to 4 comprise alternative σ factors that play crucial roles in regulating the expression of specific regulons under stress conditions. These alternative σ factors competitively bind to the core RNAP, thereby directing it to specific promoters and regulating the transcription of genes associated with stress responses or specialized functions.48Progress in structural biochemistry enables the visualization of the structure of σ factor and its interactions with the RNAP core enzyme. The primary σ factor is composed of four conserved regions: σ regions 1.1, 1.2 to 2.4, 3.0 to 3.2, and 4.1 to 4.2, that constitute four domains σ1.1, σ2, σ3 and σ4, respectively, along with a non-conserved region (σNCR) (Fig. 6A). Crystal structures of the RNAP holoenzyme show that the two amino-terminal domains of the σ subunit (σ2) interact with β′ clamp helices (β′CH), while σ4 interacts with β-flap-tip-helix (β-FTH) region of RNAP core, forming a V-shaped structure near the opening of the upstream DNA-binding channel of the active site cleft (Fig. 6A).49 During the core-to-holoenzyme conversion, structural changes of both the core enzyme and the σ factor occur in all the domains of life to facilitate DNA engagement.49 Subsequently, σ70 recognizes the promoter DNA around the −35 and −10 elements to form an open promoter complex through the unwinding of the DNA double helix between the −10 element and the transcription start site (Fig. 6B).50 Although σ forms extensive contacts with the RNAP core, the most conserved interactions exist between σ2.2 and the β′CH region.51
 | ||
| Fig. 6 A) Bacterial RNAP holoenzyme and structural domains and functional motifs of σ factor. σ2.2 interacts with β′CH and σ4 interacts with β-β-FTH. B) The recognition of housekeeping promoter by holoenzyme forms an open promoter complex. σ4 interacts with the discriminator region. σ factor, non-template DNA (ntDNA), template DNA (tDNA), RNA and are colored green, red, black, magenta. Images are generated using PDB file 4YLN. | ||
3.2. Transcription elongation
Upon RNAP binding to the promoter, it melts the dsDNA and starts the polymerization of NTPs to produce an RNA molecule. The initial RNA transcript is typically 13 to 15 nucleotides in length, with 7 to 9 nucleotides forming a pairing with the DNA template strand.52 Subsequently, the initiation complex undergoes promoter clearance and transition into the elongation complex. The elongation phase of transcription can be divided into two distinct phases: active translocation and pausing.53 During active translocation, the polymerase incorporates nucleotides into the growing RNA chain and moves forward along the DNA template.54 In contrast, the paused states represent off-pathway events in the NTP incorporation cycle.55 In these paused states, the polymerase can either remain stationary at a specific position on the DNA or diffuse backwards along the DNA before recovering from the pause. Throughout the elongation phase, diverse factors interact with the polymerase in both the on-pathway (active translocation) and off-pathway (pausing) phases, thereby modulating either the polymerase's activity or its propensity to enter or remain paused. In E. coli, these factors include N-utilizing substance (Nus) factors, RfaH, ribosomal protein S4, Gre-factors (GreA, GreB), Mfd, and RapA (HepA).44The crystal structure of NusA, isolated from M. tuberculosis (PDB 1K0R), shows that NusA is a multidomain protein consisting of an N-terminal domain (NTD) linked with C-terminal modules which have three RNA binding domains: one S1-like domain and two K homology domains (KH1 and KH2)58 (Fig. 7A). Additionally, certain bacterial species, such as E. coli, possess two acidic repeats (AR1, AR2) at the C-terminus of NusA. AR2 contacts with the C-terminal domain (CTD) of the RNAP α subunit (α-CTD), dissociating an auto-inhibitory interaction between AR2 and KH1 and facilitating the binding of RNA to NusA.59 The cryo-EM structure of the E. coli NusA–RNAP complex (PDB 6FLQ) reveals specific contact points between NusA and paused state at histidine pause site (hisPEC). These contact points include: i) RNAP α1-CTD with NusA-AR2, ii) RNAP α2-CTD with NusA-NTD, iii) RNAP ω subunit with NusA-KH1/KH2, and iv) RNAP β-FTH with NusA-NTD60 (Fig. 7B). The S1, KH1, and KH2 domains of NusA are integrated together to form an extensive RNA-binding structure,58 while the NusA-NTD interacts with the β-flap region of the RNA exit channel of RNAP.61 In addition, research has demonstrated that residues F59, R61, E94, and Q96 of the NusA NTD are required for its interaction with conserved residues L895, E899, K900, R903, and E908 in the β-FTH region of RNAP.62
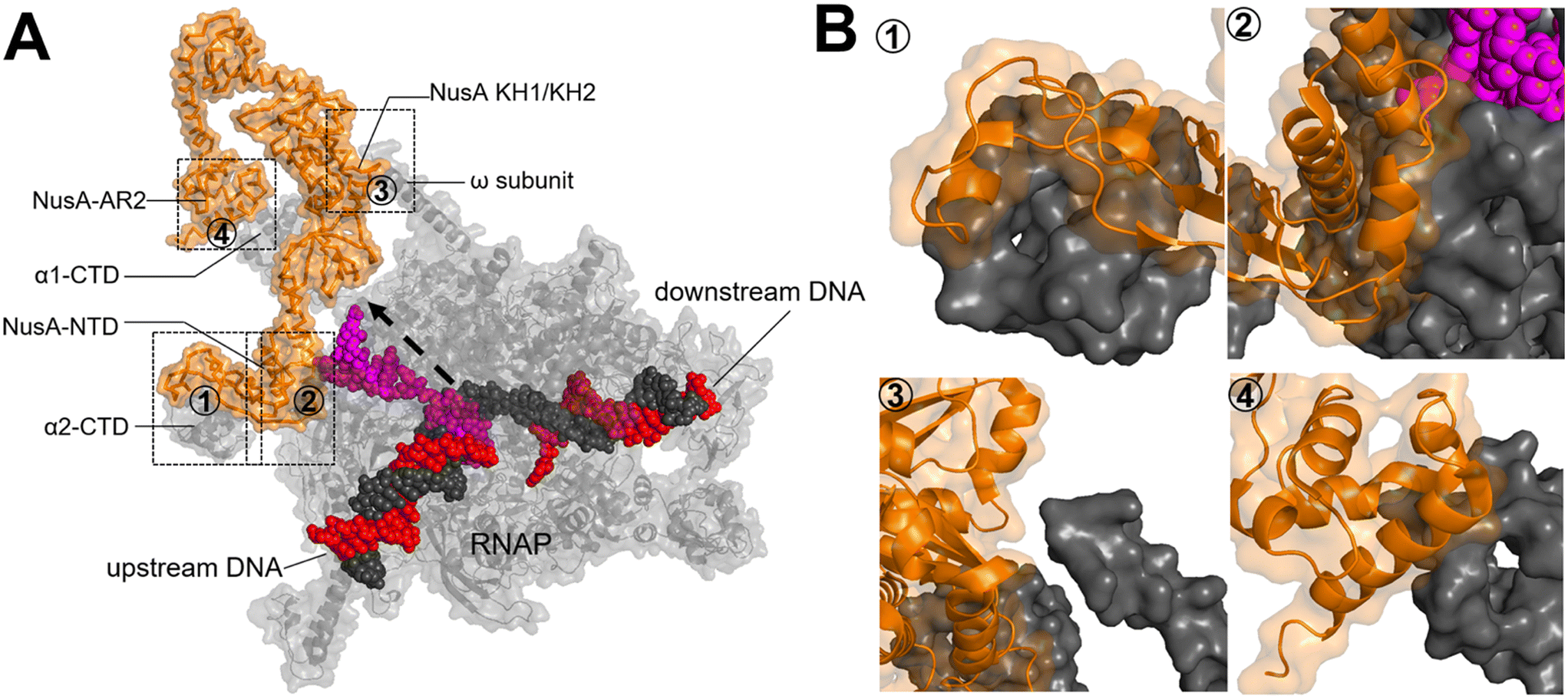 | ||
| Fig. 7 A) Crystal structure of NusA isolated from M. tuberculosis (PDB 1K0R). B) Surface representation of hisPEC–NusA complex. RNAP α1 subunit, α2 subunit, ω subunit, β-flap-tip-helix, NusA, RNA hairpin and upstream and downstream DNA are indicated. The four interaction points are ① RNAP FTH with NusA-NTD; ② RNAP α2-CTD with NusA-NTD; ③ RNAP ω subunit with NusA KH1/KH2; ④ RNAP α1-CTD with NusA-AR2C. (PDB 6FLQ). | ||
NusG is a two-domain protein consisting of the NusG N-terminal domain (NusGN), which binds to RNAP to regulate transcription rates, and the C-terminal (NusGc) Kypride–Onzonis–Woese (KOW) motif, which recruits Rho for termination and NusE (S10) to link transcription and translation.67 Although a previous biochemical study demonstrated that NusG binds to RNAP weakly without nucleic acids, the crystal structure of the NusGN–RNAP complex from E. coli (PDB 5TBZ)67 was assembled in the absence of nucleic acids. This structure reveals that NusG binds to the central cleft of RNAP, which is surrounded by the β′CH, the protrusion, and the lobe domains (Fig. 8A).67 When superimposed on the structure of the σS-transcription initiation complex, NusGN is observed to bind at the upstream fork junction, a position similar to σ2, which may stabilize the upstream junction and thus enhance transcription elongation and RNAP processivity (Fig. 8B).67
 | ||
| Fig. 8 A) Overall structure of the E. coli RNAP–NusG complex (PDB 5TBZ). B) Superimposition of the RNAP–NusG with E. coli σS-transcription initiation complex (PDB 5IPL) via the RNAP. C) Cryo-EM structure of NusG-dependent pausing-transcription complex (PTC) assembled by B. subtilis NusG and M. tuberculosis RNAP (PDB 8EHQ). D) Cryo-EM structure of M. tuberculosis RNAP elongation complex with B. subtilis NusG (PDB 8EOT). RNAP, NusG, σS, tDNA, ntDNA and RNA are colored gray, magenta, green, black, red and blue, respectively. | ||
In contrast, NusG stimulates transcription pausing in Gram-positive bacteria like B. subtilis by identifying a conserved pause-inducing motif, −11TTNTTT−6, located on the ntDNA strand within the transcription bubble.66,68 Structures of B. subtilis NusG and M. tuberculosis RNAP have been assembled to investigate the structural basis of NusG-dependent pausing.69 In the paused transcription complex (PTC), a specific interaction between NusG and ntDNA strand is observed (Fig. 8C). However, in the transcription elongation complex (TEC), the ntDNA passes NusG and RNAP β-lobe domain without any interaction (Fig. 8D). The NusG–ntDNA interaction induces rearrangement of the transcription bubble, inhibiting the folding of the TL and allosterically affecting RNA synthesis.69
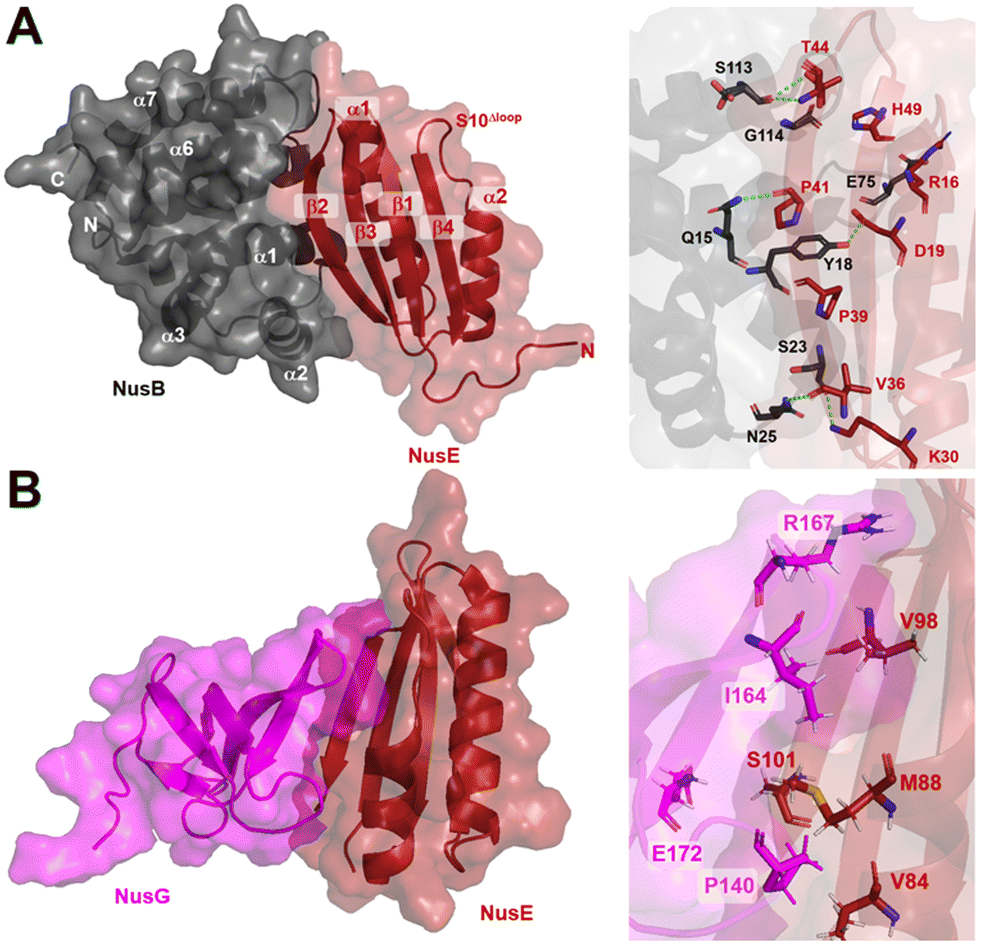 | ||
| Fig. 9 A) Crystal structure of NusB–S10Δloop complex (PDB 3D3B) and details of the interactions between NusB and NusE. B) Crystal structure of NusE–NusGc complex and details of the interactions (PDB 2KVQ). | ||
In addition to its contact with NusB, NusE also binds to the NusGC to anchor the NusB–NusE–boxA RNA complex to RNAP.78 The interaction between NusGC and NusE has been identified through an NMR study (PDB 2KVQ) (Fig. 9B). The structure demonstrates that NusE engages L1 and L2 loops on NusGc, which are also involved in binding to the Rho factor during Rho-dependent termination.79 The competition between NusE and Rho for binding to NusGc provides a plausible explanation for the inability of Rho to terminate transcription, ensuring the stability of the NusB–NusE–boxA RNA complex and preventing premature transcription termination mediated by Rho.
Structurally, both GreA and GreB share a comparable arrangement comprising an coiled-coil NTD and a globular CTD.85,86 The NTD is responsible for readthrough and cleavage stimulatory activities, while the CTD contributes to high-affinity binding with the core of RNAP. Despite their similar architectures, there are notable differences in the activities of the two factors.87,88 GreB exhibits a significantly higher binding affinity to RNAP, approximately 100-fold tighter than that of GreA. Additionally, while GreA only induces cleavage of only 2 to 3 nucleotides of backtracked RNA, GreB is capable of cleaving RNA fragments up to 18 nucleotides. Upon backtracking, RNAP undergoes conformational changes to form a backtracking complex (BC) that allows a Gre factor to access the active site of RNAP. In E. coli, the pre-cleavage complex (PDB 6RIN) of RNAP–GreB reveals that the NTD of GreB extends directly to the active site through the secondary channel of RNAP (Fig. 10A).84 Consequently, two acidic residues located at the tip of the NTD, namely D41 and E44, are positioned in close proximity to the catalytic site of RNAP (Fig. 10B). Outside the RNAP active site channel, the GreB-CTD interacts exclusively with two α helices from the β′ that form a part of the entrance rim of the secondary channel.84 This interaction is critical for the binding of GreB to RNAP. In particular, two residues within this domain, Asp121 and Pro123, have been identified as crucial for the binding affinity of GreB to RNAP through alanine scanning experiment.89
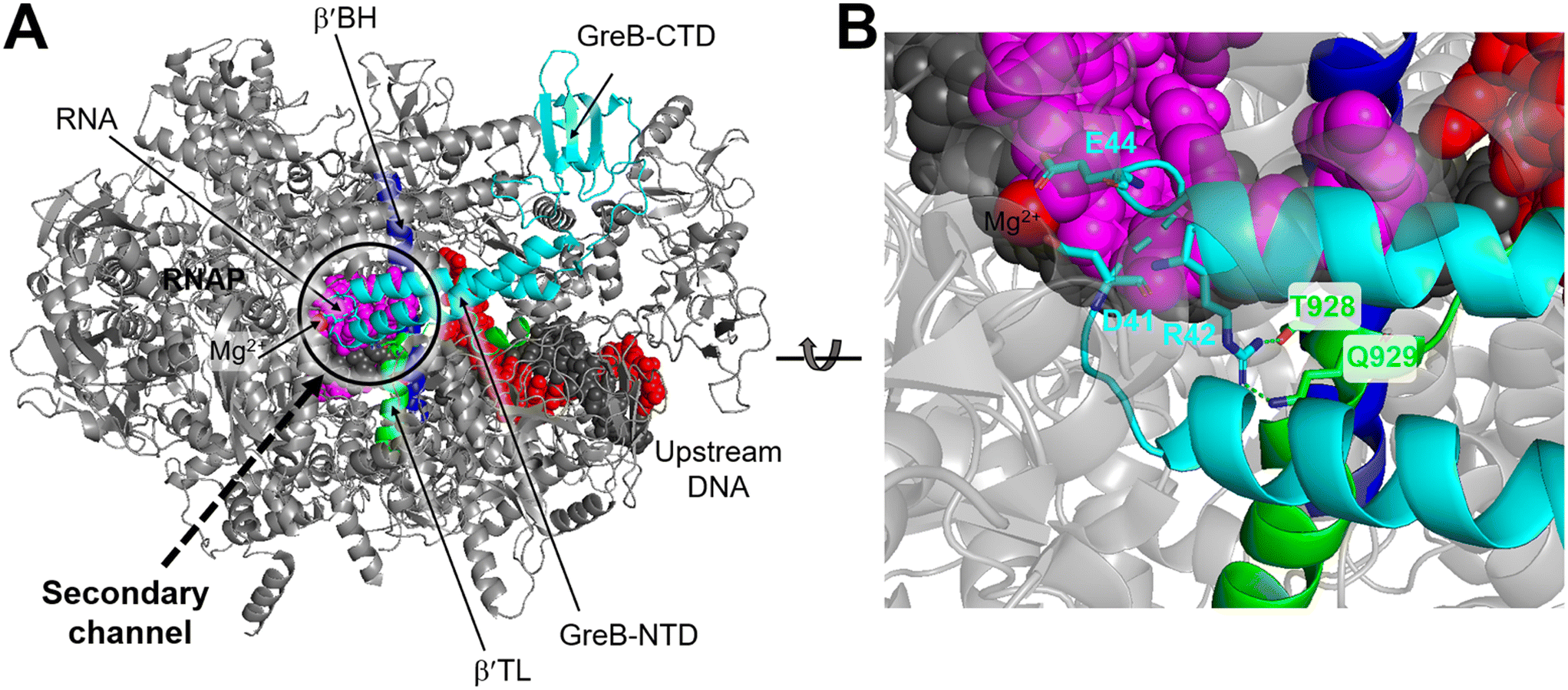 | ||
| Fig. 10 A) Cryo-EM structure of E. coli RNAP backtracked elongation complex bound to GreB transcription factor (PDB 6RIN). B) A closed view of GreB-NTD engaged in RNAP active site. GreB, β′ BH and β′ TL, tDNA, ntDNA and RNA are colored cyan, blue, green, black, red and magenta, respectively. | ||
3.3. Transcription termination
Transcription termination is a critical process that ensures accurate transcription of the DNA and allows for the recycling of RNAP at the end of transcription. Generally, RNAP undergoes programmed transcription termination through two distinct pathways: factor-independent (intrinsic) and factor-dependent termination. Intrinsic termination in bacteria relies on the core enzymatic activity of RNAP and the presence of a terminal uridine-rich segment on the template DNA chain.90 Biochemical and single-molecule studies have provided insights into the sequential intermediate states of intrinsic termination. Initially, RNAP halts at the terminator (referred to as the “pause state”). Afterwards, the nascent RNA forms a partial RNA hairpin structure known as the “hairpin-nucleation state” within RNAP. Finally, the RNA hairpin fully folds in the “hairpin-completion state”, leading to the dissociation of the RNA from the template DNA strand.91 Factor-dependent termination, on the other hand, requires specific termination factors that are critical for genomic stability and RNAP recycling.92 In E. coli there are mainly two distinct termination factors, including Rho and Mfd.93Recently, several structures of the Rho–NusG and Rho–TEC complexes have been reported, providing insights into the molecular interactions involved in the Rho-dependent termination.97–100 One such structure is the pre-termination complex (PTC) assembled by the elongation complex (EC), NusA, NusG and Rho (PDB 6XAV). In this complex, the Rho hexamer interacts with RNAP subunits in an open-ring conformation, and several critical interfaces between Rho and RNAP are determined.99 The structure of the PTC displays that NusG bridges RNAP and Rho factor using its NTD and CTD, while NusA stabilizes PTC and directs the nascent RNA into the central channel of Rho hexamer. This arrangement facilitates the interactions between the components of the complex (Fig. 11A). Also, structures of functional Rho-dependent PTCs with the Rho hexamer in a closed-ring state have been reported.100 In these structures, the Rho hexamer is positioned at the opening of the RNA exit channel of RNAP for direct passage of the RNA from the TEC into and through the Rho hexamer, enabling the uridine-rich SBS ligand to bind to Rho SBS site. On the other hand, a trans-supplied RNA oligomer acts as the PBS ligand and interacts with the Rho PBS site (PDB 8E6X, 8E6V).100 This complex reveals that Rho monomers form three critical protein–protein interfaces with NusG-CTD, the β-subunit flap-tip of RNAP and the ω subunit (Fig. 11B).
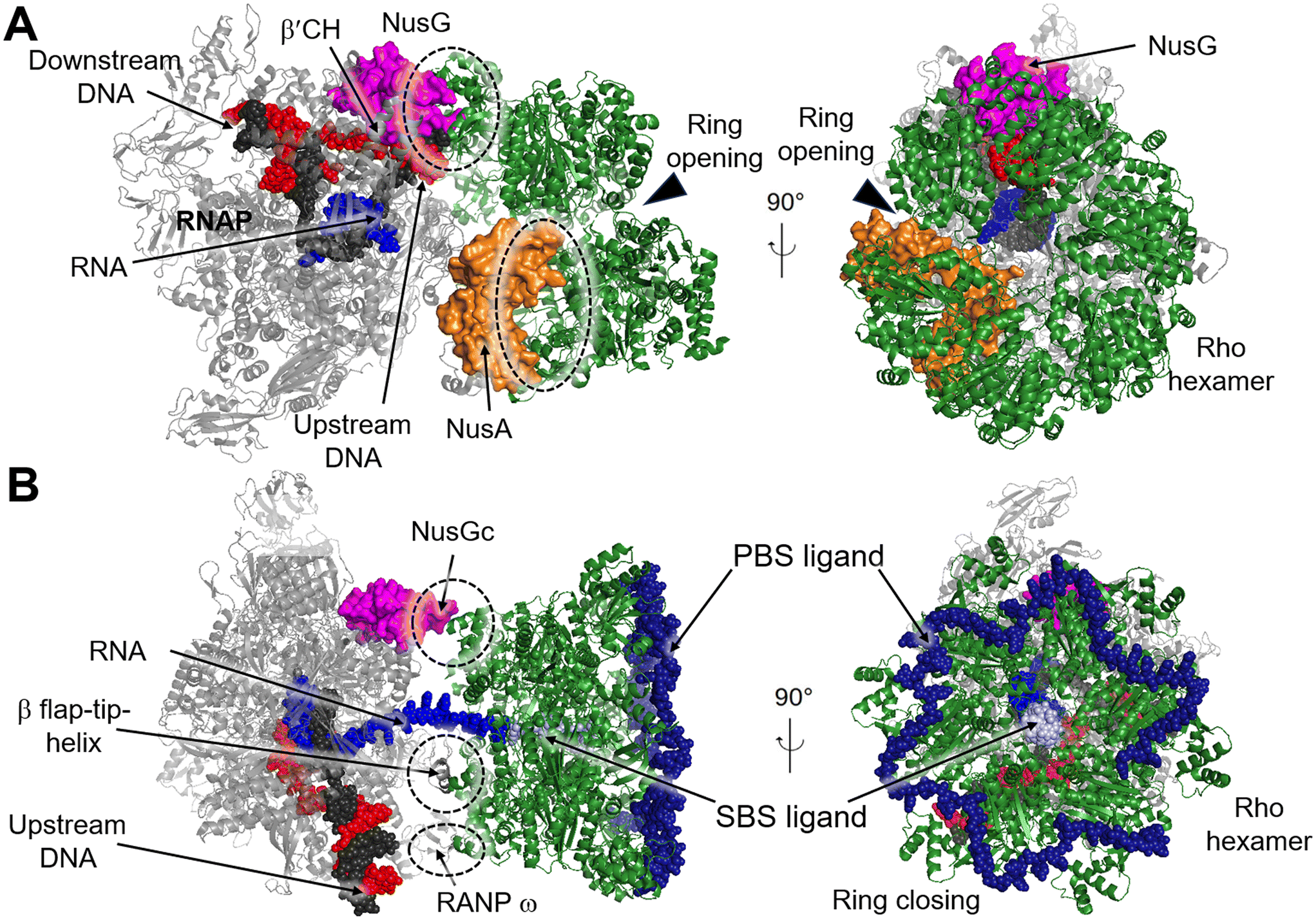 | ||
| Fig. 11 A) Cryo-EM structure of E. coli Rho-dependent transcription pre-termination complex bound with NusG with Rho hexamer in open state (PDB 6XAV). B) E. coli Rho-dependent transcription pre-termination complex (PDB 8E6X and 8E6V) with Rho hexamer in closed state. Rho hexamer, NusG, NusA, tDNA, ntDNA, PBS ligand and SBS are colored forest green, magenta, orange, black, red, pale blue and density blue. Interfaces between Rho and RNAP, NusA or NusG are circled with dashed rings. | ||
4. Development of bacterial transcription inhibitors
The structural studies of RNAP and its interactions with transcription factors provide abundant information for the structure-based drug design. We believe that the interdependent interactions between the RNAP core and transcription factors hold promise as targets for developing next-generation antibacterial agents, as these interactions are critical to gene expression. Moreover, confirmation of the “hotspots” through biological methods or theoretical calculations can accelerate the identification of PPI inhibitors, including those targeting bacterial transcription. In the following section, we would like to introduce some bacterial transcription inhibitors targeting PPI between RNAP and transcription factors.4.1. Inhibitors of RNAP–σ factor interaction
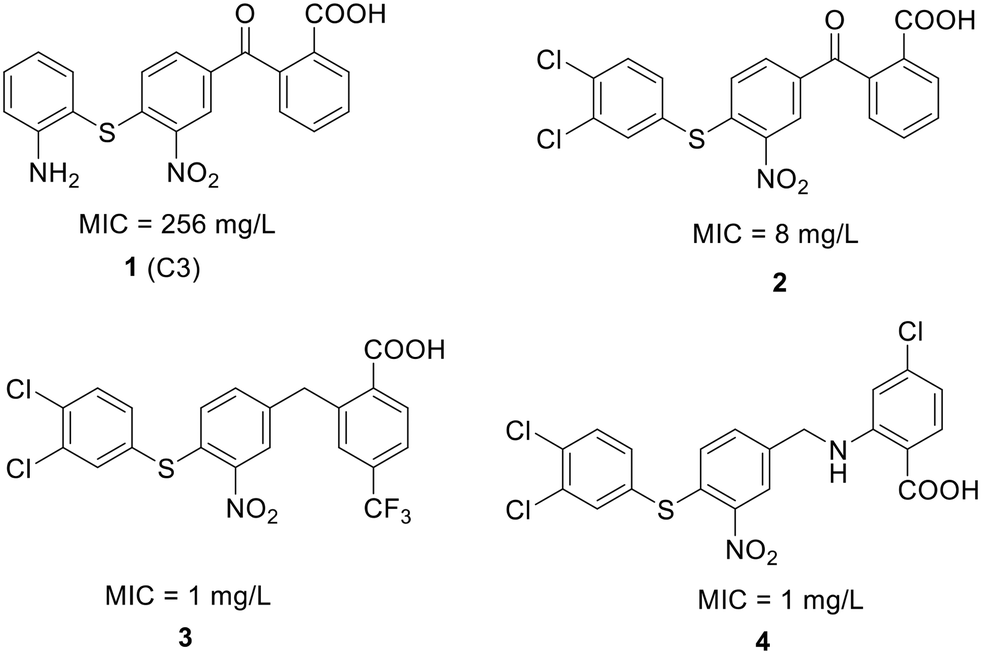 | ||
| Fig. 12 Chemical structure of compound 1 and its derivatives (2 to 4) along with their activity against S. pneumoniae. | ||
To enhance the antibacterial properties of C3, a comprehensive and systematic optimization strategy was employed, resulting in the synthesis of dozens of derivatives, such as compounds 2 to 4 (ref. 107–109) (Fig. 11). Compound C3-005 (2), featuring 3,4-diCl substituents on the left benzene ring instead of the 2-NH2, notably improved the antibacterial activity against S. pneumoniae with a MIC value of 8 μg mL−1.107 Strikingly, compound 3, incorporating a benzyl benzoic acid moiety, demonstrated to be a up-and-coming antibacterial agent with a MIC value of low to 1 μg mL−1.108 Furthermore, compound 4, characterized by an aminomethylene linker instead of a carbonyl linker, proved to be an efficient antimicrobial agent, with a MIC value of 1 μg mL−1 against S. pneumoniae and IC50 value of 6.8 ± 0.7 μM in disrupting the formation of RNAP holoenzyme. This compound also demonstrated time-dependent inhibition of bacterial growth and exerted inhibitory effects on bacterial central metabolism, suggesting its bacteriostatic effect with the reduced release of exotoxin.109 Further sulfonamidyl derivatives of C3 also showed good antimicrobial activity.110 The C3 series of compounds was coined sigmacidins to reflect the inhibitory function by mimicking σ factor and the benzoic acid structure.111
 | ||
| Fig. 13 Chemical structure of compound 5. | ||
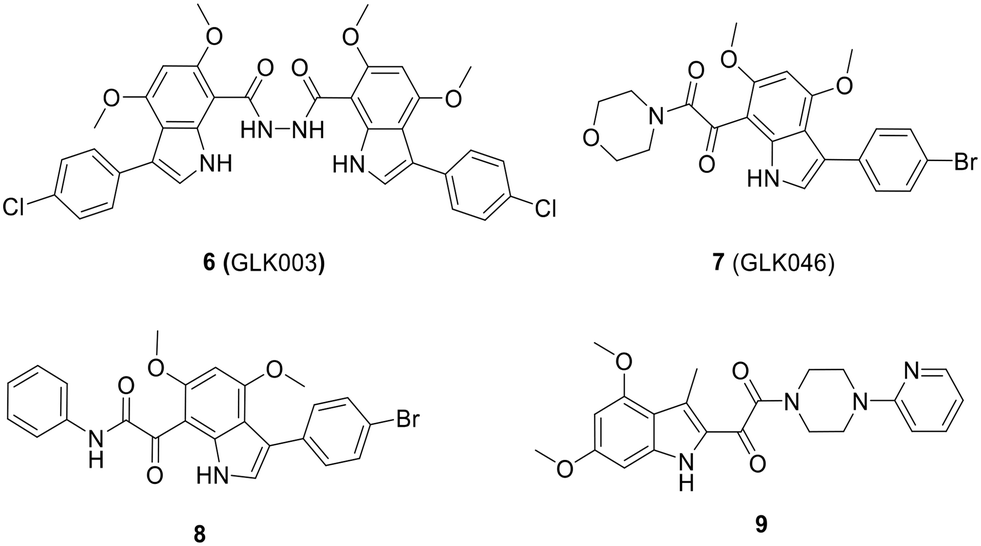 | ||
| Fig. 14 Chemical structure of 6 (GLK003) and its derivatives 7 to 9 with mono-indole scaffold. | ||
The optimization process of GKL003 led to the development of a bis-indole library, focusing on assessing their activity against the interaction between RNAP and σ70/σA.114 In order to develop smaller compounds with improved cell membrane penetration capabilities, the bis-indole scaffold was simplified into mono-indole or related benzofuran scaffolds, resulting in the synthesis of compounds 7 (GLK046) to 9 (Fig. 14). The obtained results showed that some of these derivatives exclusively inhibited E. coli and B. subtilis growth. However, the inhibitory rates of these compounds against β′CH–σ2.2 interaction were relatively modest.115–118
 | ||
| Fig. 15 Chemical structures of compound 9 to 13 and their in vitro activities against E. coli RNAP–σ interaction (IC50 value or percentage inhibition at 200 μM (SD < 40%)). | ||
 | ||
| Fig. 16 Chemical structure of compounds 14 to 16. | ||
4.2. Inhibitors of NusB–NusE interactions
 | ||
| Fig. 17 SAR studies of MC4 and the structures of its derivatives. MIC values represent minimal inhibitory concentrations against S. aureus. | ||
The SAR investigation of MC4 focused on the substitution of the benzyl rings and the bridge connecting them (Fig. 17).123,124,126,127 Some of the derivatives (18 to 22) exhibited a dramatically improved activity against a panel of pathogens, with MICs ranging from 0.25 to 2 μg mL−1 against methicillin- and vancomycin-resistant S. aureus, while demonstrating negligible cytotoxicity. However, the improvement in antibacterial efficacy was not very consistent with the NusB–NusE inhibitory action, which might be due to diverse cell membrane permeability.
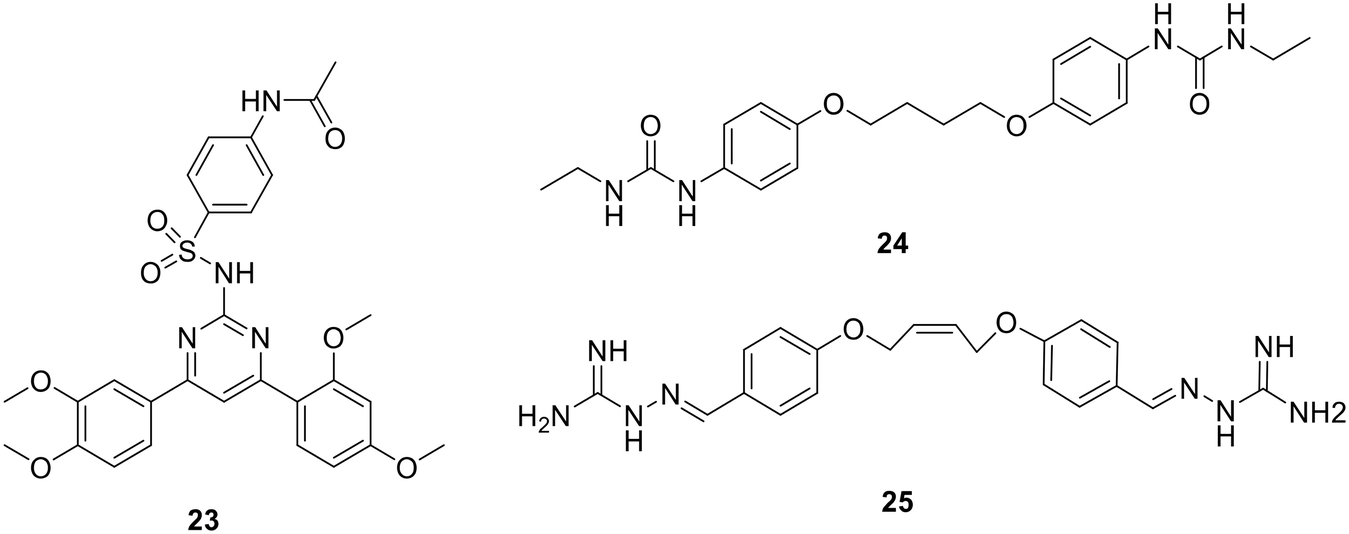 | ||
| Fig. 18 Chemical structures of compounds 23 to 25. | ||
4.3. Inhibitors of Rho factor
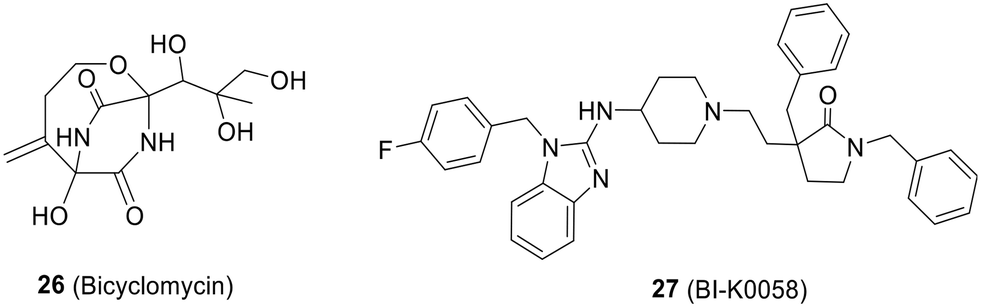 | ||
| Fig. 19 Chemical structures of bicylomycin and BI-K0058. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds. It effectively inhibits the ATPase activity of Rho with an IC50 of 25 μM. Additionally, BI-K0058 exhibited significant antimicrobial activity against two bacterial strains, Neisseria gonorrhoeae and Moraxella catharralis, with MICs ranging from 2.5 to 5 μM (Fig. 19).133
000 compounds. It effectively inhibits the ATPase activity of Rho with an IC50 of 25 μM. Additionally, BI-K0058 exhibited significant antimicrobial activity against two bacterial strains, Neisseria gonorrhoeae and Moraxella catharralis, with MICs ranging from 2.5 to 5 μM (Fig. 19).1335. Discussion and conclusions
The continuing threat of multidrug resistance in bacterial infections, coupled with a lack of innovation in the discovery of new antimicrobial agents, underscores the pressing need for novel therapeutics. However, between 2010 and 2018, the Food and Drug Administration (FDA) only approved eight new antibacterial agents, which were all derived from established drug classes.134 In 2019, the FDA approved lefamulin, a first-in-class semi-synthetic pleuromutilin antibiotic with a unique mechanism of action. Lefamulin inhibits protein synthesis by binding to the peptidyl transferase center of the 50S bacterial ribosome, making it a viable treatment option for community-acquired bacterial pneumonia (CABP) in humans.134,135 However, the potential cost of lefamulin may present a barrier to its widespread use, with intravenous treatment estimated at $205 and oral treatment at $275 per day.136 Therefore, the search for new strategies to discover novel antibiotics remains both valuable and urgent.This review has summarized the effectiveness and efficiency of targeting bacterial RNAP as a strategy for antibacterial therapy, exemplified by clinically utilized drugs, rifamycin and lipiarmycin. This innovative approach disrupts the formation of transcription complex and possess some advantages, in which it has potential to reduce the development of drug resistance. This is primarily because developing resistance to PPI inhibitors typically requires the occurrence of dual mutations, which incurs a higher energy cost.
Structural biology is critical in facilitating drug discovery by providing valuable insights into the three-dimensional structures of macromolecules and their interactions with corresponding partners. Over the past 20 years, a general framework of the structural basis for bacterial transcription cycles has been gradually revealed. By analyzing the interfaces between RNAP and its associated factors, critical domains or hotspots have been identified, providing key clues for structure-based drug design.
Recently, there have been more and more outstanding research efforts towards the inhibition of bacterial transcriptional PPIs, especially some remarkable successes using in silico virtual screening techniques that utilize pharmacophore models constructed on the basis of the structural information. By screening or designing bioactive compounds with a known target, rapid modifications are allowed to enhance the selectivity of the compounds. However, it is worth noting that hit compounds with good activity on purified targets or bacteria may lose potency or become inactive after modifications. This can partially be attributed to the alternations in their interactions with the target or changes in permeability. Despite these challenges, targeting transcriptional PPIs remains a valid but underdeveloped strategy for antibiotic discovery.
Finally, the small molecular antibacterial agents described here are still at a preliminary stage of development and not yet considered clinical drugs. Though some of the most active compounds exhibit very low MIC values, they often require appropriate modifications to optimize their pharmacokinetic properties and enhance their drug-like characteristics, for instance, bioavailability, metabolic stability, and toxicity. Nevertheless, the comprehensive information outlined here serves as a useful foundation for the further development of next-generation antimicrobial agents targeting transcriptional PPIs. By building upon this knowledge and employing strategies to enhance the pharmacokinetic properties of these compounds, it is possible to pave the way for the eventual clinical translation of these promising agents into effective antibacterial therapies.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the financial support from the Research Grants Council of the Hong Kong Special Administrative Region, China (PolyU 15100022 and C5008-19G to C. M., CUHK 14107919 to X. Y.), Hong Kong Polytechnic University (1-ZE2E and the State Key Laboratory of Chemical Biology and Drug Discovery to C. M.), Hong Kong Food and Health Bureau HMRF (19180052 to X. Y.) and the Chinese University of Hong Kong (Faculty of Medicine Faculty Innovation Award FIA2018/A/03, and Passion for Perfection Scheme PFP202210-008 to X. Y.), and Start-up Fund of Anhui Medical University (0601120201 to J. Y.).References
- M. A. Cook and G. D. Wright, Sci. Transl. Med., 2022, 14, eabo7793 CrossRef CAS PubMed.
- R. Aminov, Biochem. Pharmacol., 2017, 133, 4–19 CrossRef CAS PubMed.
- T. M. Uddin, A. J. Chakraborty, A. Khusro, B. R. M. Zidan, S. Mitra, T. B. Emran, K. Dhama, M. K. H. Ripon, M. Gajdács, M. U. K. Sahibzada, M. J. Hossain and N. Koirala, J. Infect. Public Health, 2021, 14, 1750–1766 CrossRef PubMed.
- W. P. J. Smith, B. R. Wucher, C. D. Nadell and K. R. Foster, Nat. Rev. Microbiol., 2023, 21, 519–534 CrossRef CAS PubMed.
- P. G. Higgs and N. Lehman, Nat. Rev. Genet., 2015, 16, 7–17 CrossRef CAS PubMed.
- X. Dai, S. Zhang and K. Zaleta-Rivera, Mol. Biol. Rep., 2020, 47, 1413–1434 CrossRef CAS PubMed.
- G. W. Li and X. S. Xie, Nature, 2011, 475, 308–315 CrossRef CAS PubMed.
- A. Robinson and A. M. Van Oijen, Nat. Rev. Microbiol., 2013, 11, 303–315 CrossRef CAS PubMed.
- M. Wassenegger and G. Krczal, Trends Plant Sci., 2006, 11, 142–151 CrossRef CAS PubMed.
- S. Spiegelman, A. Burny, M. R. Das, J. Keydar, J. Schlom, M. Travnicek and K. Watson, Nature, 1970, 227, 563–567 CrossRef CAS PubMed.
- C. Ma, X. Yang and P. J. Lewis, Microbiol. Mol. Biol. Rev., 2016, 80, 139–160 CrossRef CAS PubMed.
- P. Villain-Guillot, L. Bastide, M. Gualtieri and J. P. Leonetti, Drug Discovery Today, 2007, 12, 200–208 CrossRef CAS PubMed.
- R. A. Adams, G. Leon, N. M. Miller, S. P. Reyes, C. H. Thantrong, A. M. Thokkadam, A. S. Lemma, D. M. Sivaloganathan, X. Wan and M. P. Brynildsen, J. Antibiot., 2021, 74, 786–798 CrossRef CAS PubMed.
- A. Venugopal and S. Johnson, Clin. Infect. Dis., 2012, 54, 568–574 CrossRef PubMed.
- R. Kahan, D. J. Worm, G. V. De Castro, S. Ng and A. Barnard, RSC Chem. Biol., 2021, 2, 387–409 RSC.
- S. H. Kirsch, F. P. J. Haeckl and R. Müller, Nat. Prod. Rep., 2022, 39, 1226–1263 RSC.
- F. Werner, Mol. Microbiol., 2007, 65, 1395–1404 CrossRef CAS PubMed.
- F. Werner and D. Grohmann, Nat. Rev. Microbiol., 2011, 9, 85–98 CrossRef CAS PubMed.
- L. Delaye and A. Becerra, Evol.: Educ. Outreach, 2012, 5, 382–388 Search PubMed.
- M. L. Sokolova, I. Misovetc and K. V. Severinov, Viruses, 2020, 12, 1064 CrossRef CAS PubMed.
- L. Minakhin, S. Bhagat, A. Brunning, E. A. Campbell, S. A. Darst, R. H. Ebright and K. Severinov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 892–897 CrossRef CAS PubMed.
- J. Chen, H. Boyaci and E. A. Campbell, Nat. Rev. Microbiol., 2021, 19, 95–109 CrossRef CAS PubMed.
- F. Werner, Chem. Rev., 2013, 113, 8331–8349 CrossRef CAS.
- C. R. Woese, O. Kandler and M. L. Wheelis, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4576–4579 CrossRef CAS PubMed.
- D. Grohmann and F. Werner, Res. Microbiol., 2011, 162, 10–18 CrossRef CAS PubMed.
- K. S. Murakami, Biomolecules, 2015, 5, 848–864 CrossRef CAS PubMed.
- G. Zhang, E. A. Campbell, L. Minakhin, C. Richter, K. Severinov and S. A. Darst, Cell, 1999, 98, 811–824 CrossRef CAS PubMed.
- C. Sutherland and K. S. Murakami, EcoSal Plus, 2018, 8, 10.1128/ecosalplus.ESP-0004-2018 CrossRef PubMed.
- F. Werner, Trends Microbiol., 2008, 16, 247–250 CrossRef CAS PubMed.
- L. M. Iyer, E. V. Koonin and L. Aravind, BMC Struct. Biol., 2003, 3, 1–23 CrossRef PubMed.
- S. Borukhov and E. Nudler, Trends Microbiol., 2008, 16, 126–134 CrossRef CAS PubMed.
- H. Mosaei and J. Harbottle, Biochem. Soc. Trans., 2019, 47, 339–350 CrossRef CAS PubMed.
- J. Pan, Cancer Lett., 2010, 292, 149–152 CrossRef CAS PubMed.
- Y. Furuta, B. B. Gowen, K. Takahashi, K. Shiraki, D. F. Smee and D. L. Barnard, Antiviral Res., 2013, 100, 446–454 CrossRef CAS PubMed.
- G. Muratore, L. Goracci, B. Mercorelli, A. Foeglein, P. Digard, G. Cruciani, G. Palù and A. Loregian, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6247–6252 CrossRef CAS.
- S. I. Maffioli, Y. Zhang, D. Degen, T. Carzaniga, G. Del Gatto, S. Serina, P. Monciardini, C. Mazzetti, P. Guglierame, G. Candiani, A. I. Chiriac, G. Facchetti, P. Kaltofen, H. G. Sahl, G. Dehò, S. Donadio and R. H. Ebright, Cell, 2017, 169, 1240–1248 CrossRef CAS.
- E. A. Campbell, N. Korzheva, A. Mustaev, K. Murakami, S. Nair, A. Goldfarb and S. A. Darst, Cell, 2001, 104, 901–912 CrossRef CAS PubMed.
- A. Tupin, M. Gualtieri, F. Roquet-Banères, Z. Morichaud, K. Brodolin and J. P. Leonetti, Int. J. Antimicrob. Agents, 2010, 35, 519–523 CrossRef CAS PubMed.
- M. Gualtieri, P. Villain-Guillot, J. Latouche, J. P. Leonetti and L. Bastide, Antimicrob. Agents Chemother., 2006, 50, 401–402 CrossRef CAS PubMed.
- A. Tupin, M. Gualtieri, J. P. Leonetti and K. Brodolin, EMBO J., 2010, 29, 2527–2537 CrossRef CAS PubMed.
- W. Lin, K. Das, D. Degen, A. Mazumder, D. Duchi, D. Wang, Y. W. Ebright, R. Y. Ebright, E. Sineva, M. Gigliotti, A. Srivastava, S. Mandal, Y. Jiang, Y. Liu, R. Yin, Z. Zhang, E. T. Eng, D. Thomas, S. Donadio, H. Zhang, C. Zhang, A. N. Kapanidis and R. H. Ebright, Mol. Cell, 2018, 70, 60–71 CrossRef CAS.
- K. M. Herbert, W. J. Greenleaf and S. M. Block, Annu. Rev. Biochem., 2008, 77, 149–176 CrossRef CAS PubMed.
- J. Griesenbeck, H. Tschochner and D. Grohmann, Macromolecular Protein Complexes: Structure and Function, 2017, pp. 225–270 Search PubMed.
- S. Borukhov, J. Lee and O. Laptenko, Mol. Microbiol., 2005, 55, 1315–1324 CrossRef CAS PubMed.
- C. Mejía-Almonte, S. J. W. Busby, J. T. Wade, J. van Helden, A. P. Arkin, G. D. Stormo, K. Eilbeck, B. O. Palsson, J. E. Galagan and J. Collado-Vides, Nat. Rev. Genet., 2020, 21, 699–714 CrossRef.
- J. D. Helmann, Mol. Microbiol., 2019, 112, 335–347 CrossRef CAS PubMed.
- A. Mazumder and A. N. Kapanidis, J. Mol. Biol., 2019, 431, 3947–3959 CrossRef CAS PubMed.
- M. J. Kazmierczak, M. Wiedmann and K. J. Boor, Microbiol. Mol. Biol. Rev., 2005, 69, 527–543 CrossRef CAS PubMed.
- D. G. Vassylyev, S. I. Sekine, S. Yokoyama and S. Yokoyama, Nature, 2002, 417, 712–719 CrossRef CAS PubMed.
- M. S. B. Paget and J. D. Helmann, Genome Biol., 2003, 4, 1–6 CrossRef PubMed.
- K. S. Murakami and S. A. Darst, Curr. Opin. Struct. Biol., 2003, 13, 31–39 CrossRef CAS PubMed.
- H. Fan, A. B. Conn, P. B. Williams, S. Diggs, J. Hahm, H. B. Gamper Jr, Y.-M. Hou, S. E. O'Leary, Y. Wang and G. M. Blaha, Nucleic Acids Res., 2017, 45, 11043–11055 CrossRef CAS PubMed.
- A. Mustaev, J. Roberts and M. Gottesman, Transcription, 2017, 8, 150–161 CrossRef CAS PubMed.
- G. A. Belogurov and I. Artsimovitch, J. Mol. Biol., 2019, 431, 3975–4006 CrossRef CAS PubMed.
- J. Y. Kang, T. V. Mishanina, R. Landick and S. A. Darst, J. Mol. Biol., 2019, 431, 4007–4029 CrossRef CAS PubMed.
- T. Jayasinghe, Z. F. Mandell, A. V. Yakhnin, M. Kashlev and P. Babitzke, J. Bacteriol., 2022, 204, e00534-21 CrossRef PubMed.
- K. Liu, Y. Zhang, K. Severinov, A. Das and M. M. Hanna, EMBO J., 1996, 15, 150–161 CrossRef CAS PubMed.
- B. Gopal, L. F. Haire, S. J. Gamblin, E. J. Dodson, A. N. Lane, K. G. Papavinasasundaram, M. J. Colston and G. Dodson, J. Mol. Biol., 2001, 314, 1087–1095 CrossRef CAS PubMed.
- K. Schweimer, S. Prasch, P. S. Sujatha, M. Bubunenko, M. E. Gottesman and P. Rösch, Structure, 2011, 19, 945–954 CrossRef CAS PubMed.
- X. Guo, A. G. Myasnikov, J. Chen, C. Crucifix, G. Papai, M. Takacs, P. Schultz and A. Weixlbaumer, Mol. Cell, 2018, 69, 816–827 CrossRef CAS PubMed.
- X. Yang, S. Molimau, G. P. Doherty, E. B. Johnston, J. Marles-Wright, R. Rothnagel, B. Hankamer, R. J. Lewis and P. J. Lewis, EMBO Rep., 2009, 10, 997–1002 CrossRef CAS PubMed.
- C. Ma, M. Mobli, X. Yang, A. N. Keller, G. F. King and P. J. Lewis, Nucleic Acids Res., 2015, 43, 2829–2840 CrossRef CAS PubMed.
- A. V. Yakhnin, M. Kashlev and P. Babitzke, Crit. Rev. Biochem. Mol. Biol., 2020, 55, 716–728 CrossRef CAS PubMed.
- E. Burova, S. C. Hung, V. Sagitov, B. L. Stitt and M. E. Gottesman, J. Bacteriol., 1995, 177, 1388–1392 CrossRef CAS PubMed.
- K. M. Herbert, J. Zhou, R. A. Mooney, A. La Porta, R. Landick and S. M. Block, J. Mol. Biol., 2010, 399, 17–30 CrossRef CAS PubMed.
- A. V. Yakhnin, H. Yakhnin and P. Babitzke, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16131–16136 CrossRef CAS PubMed.
- B. Liu and T. A. Steitz, Nucleic Acids Res., 2017, 45, 968–974 CrossRef CAS PubMed.
- A. V. Yakhnin, P. C. FitzGerald, C. McIntosh, H. Yakhnin, M. Kireeva, J. Turek-Herman, Z. F. Mandell, M. Kashlev and P. Babitzke, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21628–21636 CrossRef CAS PubMed.
- R. K. Vishwakarma, M. Z. Qayyum, P. Babitzke and K. S. Murakami, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218516120 CrossRef CAS PubMed.
- D. L. Court, T. A. Patterson, T. Baker, N. Costantino, X. Mao and D. I. Friedman, J. Bacteriol., 1995, 177, 2589–2591 CrossRef CAS PubMed.
- J. R. Stagno, A. S. Altieri, M. Bubunenko, S. G. Tarasov, J. Li, D. L. Court, R. A. Byrd and X. Ji, Nucleic Acids Res., 2011, 39, 7803–7815 CrossRef CAS PubMed.
- A. S. Altieri, M. J. Mazzulla, D. A. Horita, R. H. Coats, P. T. Wingfield, A. Das, D. L. Court and R. A. Byrd, Nat. Struct. Biol., 2000, 7, 470–474 CrossRef CAS PubMed.
- B. Gopal, L. F. Haire, R. A. Cox, M. J. Colston, S. Major, J. A. Brannigan, S. J. Smerdon and G. Dodson, Nat. Struct. Biol., 2000, 7, 475–478 CrossRef CAS PubMed.
- J. Greenblatt, in Transcriptional Regulation, ed. S. L. McKnight and K. R. Yamamoto, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1992, pp. 203–226 Search PubMed.
- B. T. Wimberly, D. E. Brodersen, W. M. Clemons Jr, R. J. Morgan-Warren, A. P. Carter, C. Vonrheln, T. Hartsch and V. Ramakrishnan, Nature, 2000, 407, 327–339 CrossRef CAS PubMed.
- X. Luo, H. H. Hsiao, M. Bubunenko, G. Weber, D. L. Court, M. E. Gottesman, H. Urlaub and M. C. Wahl, Mol. Cell, 2008, 32, 791–802 CrossRef CAS PubMed.
- R. Das, S. Loss, J. Li, D. S. Waugh, S. Tarasov, P. T. Wingfield, R. A. Byrd and A. S. Altieri, J. Mol. Biol., 2008, 376, 705–720 CrossRef CAS PubMed.
- J. Drögemüller, M. Strauß, K. Schweimer, M. Jurk, P. Rösch and S. H. Knauer, Sci. Rep., 2015, 5, 16428 CrossRef PubMed.
- B. M. Burmann, K. Schweimer, X. Luo, M. C. Wahl, B. L. Stitt, M. E. Gottesman and P. Rösch, Science, 2010, 328, 501–504 CrossRef CAS PubMed.
- M. T. Marr and J. W. Roberts, Mol. Cell, 2000, 6, 1275–1285 CrossRef CAS PubMed.
- A. Lass-Napiorkowska and T. Heyduk, Biochemistry, 2016, 55, 647–658 CrossRef CAS PubMed.
- F. Toulmé, C. Mosrin-Huaman, J. Sparkowski, A. Das, M. Leng and A. R. Rahmouni, EMBO J., 2000, 19, 6853–6859 CrossRef PubMed.
- T. Fouqueau, M. E. Zeller, A. C. Cheung, P. Cramer and M. Thomm, Nucleic Acids Res., 2013, 41, 7048–7059 CrossRef CAS PubMed.
- C. Saint-André, M. Takacs, G. Papai, C. Crucifix, X. Guo, J. Ortiz and A. Weixlbaumer, Mol. Cell, 2019, 75, 298–309 CrossRef PubMed.
- D. Koulich, M. Orlova, A. Malhotra, A. Sali, S. A. Darst and S. Borukhov, J. Biol. Chem., 1997, 272, 7201–7210 CrossRef CAS PubMed.
- R. N. Fish and C. M. Kane, Biochim. Biophys. Acta, Gene Struct. Expression, 2002, 1577, 287–307 CrossRef CAS PubMed.
- N. Opalka, M. Chlenov, P. Chacon, W. J. Rice, W. Wriggers and S. A. Darst, Cell, 2003, 114, 335–345 CrossRef CAS PubMed.
- M. N. Vassylyeva, V. Svetlov, A. D. Dearborn, S. Klyuyev, I. Artsimovitch and D. G. Vassylyev, EMBO Rep., 2007, 8, 1038–1043 CrossRef CAS PubMed.
- N. Loizos and S. A. Darst, J. Biol. Chem., 1999, 274, 23378–23386 CrossRef CAS PubMed.
- A. Ray-Soni, M. J. Bellecourt and R. Landick, Annu. Rev. Biochem., 2016, 85, 319–347 CrossRef CAS PubMed.
- L. You, E. O. Omollo, C. Yu, R. A. Mooney, J. Shi, L. Shen, X. Wu, A. Wen, D. He and Y. Zeng, Nature, 2023, 613, 783–789 CrossRef CAS PubMed.
- P. Mitra, G. Ghosh, M. Hafeezunnisa and R. Sen, Annu. Rev. Microbiol., 2017, 71, 687–709 CrossRef CAS PubMed.
- J. W. Roberts, J. Mol. Biol., 2019, 431, 4030–4039 CrossRef CAS PubMed.
- H. Kohn and W. Widger, Curr. Drug Targets: Infect. Disord., 2005, 5, 273–295 CrossRef CAS PubMed.
- C. Zhu, X. Guo, P. Dumas, M. Takacs, M. M. Abdelkareem, A. Vanden Broeck, C. Saint-André, G. Papai, C. Crucifix and J. Ortiz, Nat. Commun., 2022, 13, 1546 CrossRef CAS PubMed.
- J. M. Peters, R. A. Mooney, J. A. Grass, E. D. Jessen, F. Tran and R. Landick, Genes Dev., 2012, 26, 2621–2633 CrossRef CAS PubMed.
- M. R. Lawson, W. Ma, M. J. Bellecourt, I. Artsimovitch, A. Martin, R. Landick, K. Schulten and J. M. Berger, Mol. Cell, 2018, 71, 911–922 CrossRef CAS PubMed.
- N. Said, T. Hilal, N. D. Sunday, A. Khatri, J. Bürger, T. Mielke, G. A. Belogurov, B. Loll, R. Sen and I. Artsimovitch, Science, 2021, 371, eabd1673 CrossRef CAS PubMed.
- Z. Hao, V. Epshtein, K. H. Kim, S. Proshkin, V. Svetlov, V. Kamarthapu, B. Bharati, A. Mironov, T. Walz and E. Nudler, Mol. Cell, 2021, 81, 281–292.e288 CrossRef CAS PubMed.
- V. Molodtsov, C. Wang, E. Firlar, J. T. Kaelber and R. H. Ebright, Nature, 2023, 614, 367–374 CrossRef CAS PubMed.
- J. Roberts and J.-S. Park, Curr. Opin. Microbiol., 2004, 7, 120–125 CrossRef CAS PubMed.
- A. J. Smith, C. Pernstich and N. J. Savery, Nucleic Acids Res., 2012, 40, 10408–10416 CrossRef CAS PubMed.
- J. Han, O. Sahin, Y.-W. Barton and Q. Zhang, PLoS Pathog., 2008, 4, e1000083 CrossRef PubMed.
- M. N. Ragheb, M. K. Thomason, C. Hsu, P. Nugent, J. Gage, A. N. Samadpour, A. Kariisa, C. N. Merrikh, S. I. Miller and D. R. Sherman, Mol. Cell, 2019, 73, 157–165 CrossRef CAS PubMed.
- H. C. Leyva-Sánchez, N. Villegas-Negrete, K. Abundiz-Yañez, R. E. Yasbin, E. A. Robleto and M. Pedraza-Reyes, J. Bacteriol., 2020, 202, e00807–e00819 CrossRef PubMed.
- C. Ma, X. Yang and P. J. Lewis, ACS Infect. Dis., 2016, 2, 39–46 CrossRef CAS PubMed.
- J. Ye, A. J. Chu, L. Lin, X. Yang and C. Ma, Molecules, 2019, 24, 2902 CrossRef CAS PubMed.
- J. Ye, A. J. Chu, L. Lin, S. T. Chan, R. Harper, M. Xiao, I. Artsimovitch, Z. Zuo, C. Ma and X. Yang, Eur. J. Med. Chem., 2020, 208, 112671 CrossRef CAS PubMed.
- J. Ye, A. J. Chu, R. Harper, S. T. Chan, T. L. Shek, Y. Zhang, M. Ip, M. Sambir, I. Artsimovitch, Z. Zuo, X. Yang and C. Ma, J. Med. Chem., 2020, 63, 7695–7720 CrossRef CAS PubMed.
- J. Ye, C. H. Kan, Y. Zheng, T. F. Tsang, A. J. Chu, K. H. Chan, X. Yang and C. Ma, Bioorg. Chem., 2024, 143, 106983 CrossRef CAS PubMed.
- J. Ye, X. Yang and C. Ma, Int. J. Mol. Sci., 2022, 23, 4085 CrossRef CAS PubMed.
- C. Ma, X. Yang, H. Kandemir, M. Mielczarek, E. B. Johnston, R. Griffith, N. Kumar and P. J. Lewis, ACS Chem. Biol., 2013, 8, 1972–1980 CrossRef CAS PubMed.
- E. B. Johnston, P. J. Lewis and R. Griffith, Protein Sci., 2009, 18, 2287–2297 CrossRef CAS PubMed.
- M. Mielczarek, R. V. Devakaram, C. Ma, X. Yang, H. Kandemir, B. Purwono, D. S. Black, R. Griffith, P. J. Lewis and N. Kumar, Org. Biomol. Chem., 2014, 12, 2882–2894 RSC.
- M. Mielczarek, R. V. Thomas, C. Ma, H. Kandemir, X. Yang, M. Bhadbhade, D. S. Black, R. Griffith, P. J. Lewis and N. Kumar, Bioorg. Med. Chem., 2015, 23, 1763–1775 CrossRef CAS PubMed.
- O. Thach, M. Mielczarek, C. Ma, S. K. Kutty, X. Yang, D. S. Black, R. Griffith, P. J. Lewis and N. Kumar, Bioorg. Med. Chem., 2016, 24, 1171–1182 CrossRef CAS PubMed.
- D. S. Wenholz, M. Zeng, C. Ma, M. Mielczarek, X. Yang, M. Bhadbhade, D. S. C. Black, P. J. Lewis, R. Griffith and N. Kumar, Bioorg. Med. Chem. Lett., 2017, 27, 4302–4308 CrossRef CAS PubMed.
- D. S. Wenholz, M. Miller, C. Dawson, M. Bhadbhade, D. S. Black, R. Griffith, H. Dinh, A. Cain, P. Lewis and N. Kumar, Bioorg. Chem., 2022, 118, 105481 CrossRef CAS PubMed.
- S. Hinsberger, K. Hüsecken, M. Groh, M. Negri, J. Haupenthal and R. W. Hartmann, J. Med. Chem., 2013, 56, 8332–8338 CrossRef CAS PubMed.
- E. André, L. Bastide, P. Villain-Guillot, J. Latouche, J. Rouby and J. P. Leonetti, Assay Drug Dev. Technol., 2004, 2, 629–635 CrossRef PubMed.
- E. André, L. Bastide, S. Michaux-Charachon, A. Gouby, P. Villain-Guillot, J. Latouche, A. Bouchet, M. Gualtiéri and J. P. Leonetti, J. Antimicrob. Chemother., 2006, 57, 245–251 CrossRef PubMed.
- P. Villain-Guillot, M. Gualtieri, L. Bastide, F. Roquet, J. Martinez, M. Amblard, M. Pugniere and J. P. Leonetti, J. Med. Chem., 2007, 50, 4195–4204 CrossRef CAS PubMed.
- J. Ye, X. Yang and C. Ma, Int. J. Mol. Sci., 2022, 24, 339 CrossRef PubMed.
- Y. Qiu, S. T. Chan, L. Lin, T. L. Shek, T. F. Tsang, Y. Zhang, M. Ip, P. K. S. Chan, N. Blanchard, G. Hanquet, Z. Zuo, X. Yang and C. Ma, Bioorg. Chem., 2019, 92, 103203 CrossRef CAS PubMed.
- X. Yang, M. J. Luo, A. C. Yeung, P. J. Lewis, P. K. Chan, M. Ip and C. Ma, Biochemistry, 2017, 56, 5049–5052 CrossRef CAS PubMed.
- Y. Qiu, S. T. Chan, L. Lin, T. L. Shek, T. F. Tsang, N. Barua, Y. Zhang, M. Ip, P. K. S. Chan, N. Blanchard, G. Hanquet, Z. Zuo, X. Yang and C. Ma, Eur. J. Med. Chem., 2019, 178, 214–231 CrossRef CAS PubMed.
- Y. Qiu, A. J. Chu, T. F. Tsang, Y. Zheng, N. M. Lam, K. S. L. Li, M. Ip, X. Yang and C. Ma, Bioorg. Chem., 2022, 124, 105863 CrossRef CAS PubMed.
- P. J. Cossar, C. Ma, C. P. Gordon, J. I. Ambrus, P. J. Lewis and A. McCluskey, Bioorg. Med. Chem. Lett., 2017, 27, 162–167 CrossRef CAS PubMed.
- P. J. Cossar, M. K. Abdel-Hamid, C. Ma, J. A. Sakoff, T. N. Trinh, C. P. Gordon, P. J. Lewis and A. McCluskey, ACS Omega, 2017, 2, 3839–3857 CrossRef CAS PubMed.
- T. Miyoshi, N. Miyairi, H. Aoki, M. Kohsaka, H.-I. Sakai and H. Imanaka, J. Antibiot., 1972, 25, 569–575 CrossRef CAS PubMed.
- A. Zwiefka, H. Kohn and W. R. Widger, Biochemistry, 1993, 32, 3564–3570 CrossRef CAS PubMed.
- A. Magyar, X. Zhang, H. Kohn and W. R. Widger, J. Biol. Chem., 1996, 271, 25369–25374 CrossRef CAS PubMed.
- L. Carrano, P. Alifano, E. Corti, C. Bucci and S. Donadio, Biochem. Biophys. Res. Commun., 2003, 302, 219–225 CrossRef CAS PubMed.
- M. P. Veve and J. L. Wagner, Pharmacotherapy, 2018, 38, 935–946 CrossRef CAS PubMed.
- Y. R. Lee and K. L. Jacobs, Drugs, 2019, 79, 1867–1876 CrossRef CAS PubMed.
- P. N. Malani, JAMA, J. Am. Med. Assoc., 2019, 322, 1671–1672 CrossRef PubMed.
Footnote |
| † Jiqing Ye and Cheuk Hei Kan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |