
A concise guide to chemical reactions of atomically precise noble metal nanoclusters
Paulami
Bose
 ,
Krishnadas
Kumaranchira Ramankutty†
,
Papri
Chakraborty‡
,
Esma
Khatun
and
Thalappil
Pradeep
*
,
Krishnadas
Kumaranchira Ramankutty†
,
Papri
Chakraborty‡
,
Esma
Khatun
and
Thalappil
Pradeep
*
DST Unit of Nanoscience & Thematic Unit of Excellence, HSB 148, Indian Institute of Technology Madras, Chennai-600036, Tamil Nadu, India. E-mail: pradeep@iitm.ac.in
First published on 16th November 2023
Abstract
Nanoparticles (NPs) with atomic precision, known as nanoclusters (NCs), are an emerging field in materials science in view of their fascinating structure–property relationships. Ultrasmall noble metal NPs have molecule-like properties that make them fundamentally unique compared with their plasmonic counterparts and bulk materials. In this review, we present a comprehensive account of the chemistry of monolayer-protected atomically precise noble metal nanoclusters with a focus on the chemical reactions, their diversity, associated kinetics, and implications. To begin with, we briefly review the history of the evolution of such precision materials. Then the review explores the diverse chemistry of noble metal nanoclusters, including ligand exchange reactions, ligand-induced structural transformations, and reactions with metal ions, metal thiolates, and halocarbons. Just as molecules do, these precision materials also undergo intercluster reactions in solution. Supramolecular forces between these systems facilitate the creation of well-defined hierarchical assemblies, composites, and hybrid materials. We conclude the review with a future perspective and scope of such chemistry.
1. Introduction
Richard Feynman's historic Caltech address, There's Plenty of Room at the Bottom, in 1959, discussed the concept of nanotechnology which envisioned “maneuvering things atom by atom”.1 Development of atomically precise metal nanoclusters may be viewed as a direction to create materials atom by atom. The term ‘metal cluster’ was originally defined by Cotton in 1964 as “a finite group of metal atoms which are held together mainly or at least to a significant extent, by bonds directly between the metal atoms, even though some nonmetal atoms may also be intimately associated with the cluster” to refer to coordination compounds.2,3 This term was also used in the early literature to refer to plasmonic noble metal particles consisting of several hundreds or a few thousands of atoms, although the term ‘metal nanocluster’ is nowadays used more appropriately to refer to atomically precise, bare, or ligand-protected particles with a precise, molecule-like composition (MxLy; M = metal atom, L = ligands such as thiolate, phosphine, etc.) and well-defined properties. Precision refers to structure as well, both in the molecular form in the gaseous, solution and solid states. Therefore, clusters in the context of this review may be defined as “a finite group of atoms with precise composition and structure, composed of two or more metal atoms held together by chemical bonds between them, and the structure formed stands protected or unprotected with ligands, with well-defined properties.”Electronic confinement of noble metals has been an important subject matter of research in the past few decades.4–6 As the particle size shrinks below ∼3 nm to an intermediate size regime, bridging the dimensions of molecules and condensed matter, molecule-like properties arise in such materials.4,7 Such molecular materials are called atomically precise noble metal nanoclusters (NCs), which have precise composition, structure, and unique properties.7–11 Au102(p-MBA)44 nanocluster (p-MBA=para-mercaptobenzoic acid) was the first reported single-crystal structure in the family of thiolate-protected atomically precise nanoclusters,12 although clusters such as Au11I3[P(C6H4-p-Cl)3]7 (ref. 13) and [Au13(PPhMe2)10C12]3+ (ref. 14) were known since 1970, with other ligands. Since then, more than 250 nanocluster crystal structures have been published.7,11,15 Many more noble metal nanoclusters with proteins,16–21 DNA,22–27 poly(amidoamine)-based dendrimers,28–31 and cyclodextrins32–34 are now known.11 Today, atomic precision in assemblies is attainable in several materials, such as metals, metal oxides, semiconductors, ionic compounds, and even rare gases.35 Carbon clusters, such as Buckminsterfullerene or C60, are some of the most popular clusters investigated so far.36,37 Molecular clusters such as (H2O)2,3,4… and (CH3OH)n(H2O)m, where n, m = 1,2,3,…, and zero-dimensional (0D) particles of perovskites, graphene, etc., are also gaining interest.38,39 In our latest book we presented a comprehensive overview of noble metal nanoclusters and their properties, with a compendium of all reported clusters.15 However, in this review, we will be focusing on the thiolate and phosphine-protected nanoclusters in the context of their chemical reactions.
Noble metal nanoclusters exhibit well-defined physical, chemical, and electronic properties.7,11,12 Unique characteristic properties of nanoclusters include discrete electronic structures,40–47 corresponding HOMO–LUMO transitions,48–53 chemical reactivity,10,54–56,57–59 photoluminescence60–63 and intrinsic magnetism.64–69 Metal nanoclusters have attracted tremendous interest from the scientific community due to their potential applications in optoelectronics,70,71 sensing,63,72–74,75 bioimaging,63,76–79,80 catalysis,8,81–83,84 and others.7,11
Today, the chemistry of well-defined monolayer-protected nanoclusters is an active area of research. We present this review as a mini guide to ligand-protected atomically precise metal nanoclusters and their diverse chemistry (schematically represented in Fig. 1). To begin with, we trace the origin and landmark developments in nanocluster science. The article presents the recent research on ligand-induced chemistry, intercluster & interparticle reactions and their mechanism, thermodynamics, kinetics, and implications. Knowledge of the precise chemical reactivity of such nanoclusters gives a way to control their composition to form alloys and hybrid materials, and also for engineering their properties. Such new materials may find suitable roles in photophysical, catalytic, and optoelectronic applications. Chemical reactions between nanomaterials of various types provide new insights into the dynamics at the nanoscale. Reactivity at the nanoscale is of importance to chemistry in general, and to catalysis, functional materials, photophysics, nanomedicines, sensors, and clean water, in particular.
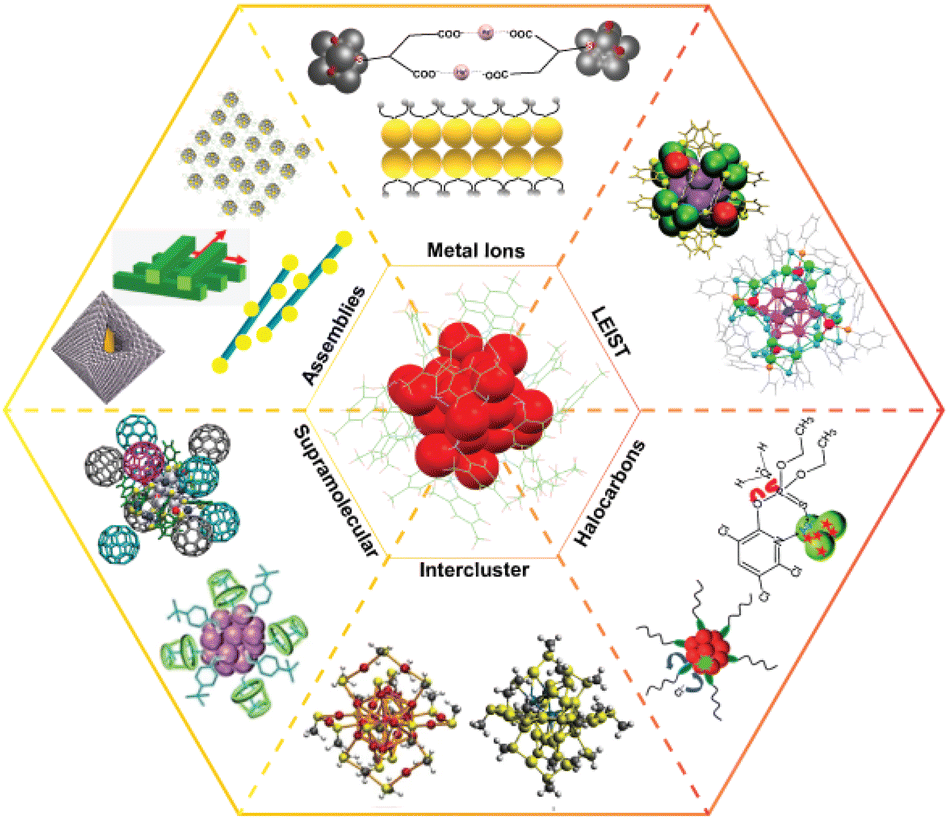 | ||
| Fig. 1 Schematic illustration of the diverse chemistry of the ligand-protected atomically precise noble metal nanoclusters. Images under assemblies are adapted with permission from ref. 275, 281, 278 and 285. Copyright 2014 and 2018 John Wiley and Sons. Copyright 2014 American Chemical Society. Copyright 2020 The Royal Society of Chemistry. Images under halocarbons are adapted from ref. 258 and 257. Copyright 2013 Royal Society of Chemistry. Images under supramolecular are adapted from ref. 259 and 260. Copyright 2014 and 2018 American Chemical Society. Images under intercluster are adapted from ref. 55. Copyright 2016 Springer Nature Group. Images under LEIST are adapted from ref. 218 and 176. Copyright 2018 Royal Society of Chemistry. Copyright 2020 American Chemical Society. | ||
2. Atomically precise noble metal nanoclusters: the evolution
Size-dependent studies of colloidal silver particles in solution using radiolytic and electrochemical methods by Henglein et al. in the 1980s were among the earliest experiments using nanoscale noble metals.85–88 Haruta's discovery, in 1989, of the catalytic activity of finely divided, nanosized gold particles supported on oxide surfaces boosted research on noble metal NPs.89 However, most of the early insights into atomically precise metal nanoclusters were derived from gas-phase investigations.2.1. Insights from gas-phase studies
Gas-phase studies provided the first glimpses into the characteristics of metal nanoclusters. Nanoclusters of alkali, alkaline earth, and noble metals have been studied extensively since the 1980s. Gas-phase investigations of metal nanoclusters made extensive use of techniques like mass spectrometry, ion mobility spectrometry, photoelectron spectroscopy, vibrational spectroscopy, etc.90,91 Using mass spectrometry, Knight et al. characterized sodium Nan (n = 4–100) metal nanoclusters in the gas phase.92 The most abundant peaks observed correspond to the nanoclusters with n = 8, 20, 40, 58, and 92. According to the jellium model, the electrons in these nanoclusters are distributed into discrete electronic shells, just as in atoms. The numbers 8, 20, 40, 58, and 92 correspond to the total number of valence electrons (3s1) in these nanoclusters, analogous to the valence shell electron configurations of noble gases. The nanoclusters whose valence shell electron counts fall in this series of ‘magic’ numbers are referred to as ‘magic clusters’. The abundance of these nanoclusters in the mass spectra is attributed to their stability gained from the completion of electronic shell structures, just like noble gases. This is one of the reasons for the fact that ‘every atom counts’ in the case of atomically precise metal nanoclusters. Magic nanoclusters of metals such as aluminum were also observed. For example, Khanna et al., showed that Al13−, which has a magic number of 40 electrons, exhibits special inertness towards gas-phase etching reactions.93,94 Such nanoclusters are also called ‘superatoms’, a term coined by Khanna et al. in 1995.95 They also introduced the idea of using superatomic nanoclusters as building blocks for nanocluster-assembled materials. The stability of gas-phase metal nanoclusters, especially larger ones, depends on closed geometric shells as well as electronic shells. In the gas phase, the geometry of the nanocluster, rather than the electronic structure, determines the stability. Due to the overlap of electronic bands, there is a negligible change in electronic energy with the addition of each new atom to the nanocluster. For example, positively charged calcium nanoclusters in the gas phase such as Ca561, Ca1412, Ca2865, etc., exhibit mass spectral abundance which is ascribed to the successive addition of layers of atoms to form stable geometries.96 Bare gold nanoclusters in the gas phase have been investigated since the 1980s. Kappes et al. used ion mobility (IM) measurements and trapped ion electron diffraction97–99 in conjunction with density functional theory (DFT) calculations in order to assign structures of Aun− (n < 13) nanoclusters98 and also suggested that planar to three-dimensional transition in these nanoclusters occurs at n = 11.98 The structure of unprotected noble metal nanoclusters deposited on surfaces has also been probed using techniques such as scanning tunneling microscopy.100,1012.2. Atomically precise metal nanoclusters in solution: phosphine- and thiolate-protected metal nanoclusters
Solution-phase nanochemistry of noble metals was accelerated after the discovery of the Brust–Schiffrin method reported in 1994,102 wherein thiolates were used as protecting ligands with limited information on the structure and composition of these particles.The earliest examples of atomically precise metal cluster compounds studied in the solution phase were gold–phosphine coordination complexes, like Au11(PPh3)7(SCN)3 and Au11I3(P(C6H4-p-Cl)3)7. These compounds were synthesized in 1969 and 1970, respectively.13,103 Au11X3[PR3]7 is the first known crystal structure with an incomplete icosahedral core.13 In 1981, Briant et al. reported the first[Au13(PPhMe2)10C12]3+ nanocluster consisting of a perfect icosahedral core.14 Bigger nanoclusters such as [Au39(PPh3)14Cl6]Cl2 consisting of larger, atomically precise Au cores were reported in 1992.104 Schmid et al. synthesized the well-known molecule, Au55[P(C6H5)3]12CI6, in 1981, which attracted significant attention in the community.105 The crystal structure of this nanocluster remained elusive; however, insights into its structure came from later studies.106 Though techniques such as fast atom bombardment mass spectrometry (FABMS) were used to analyze compounds such as [Pt2(AuPPh3)10Ag13Cl7], etc.,107 single-crystal X-ray diffraction was the major tool for probing their compositions and structures. Extensive reviews are available on the single-crystal structure of atomically precise nanoclusters.7,11,108
Even though phosphine-protected noble metal nanoclusters have been known since the 1960s as mentioned above, the big leap in the field of solution-phase nanochemistry of noble metals occurred only after the pioneering efforts of the Whetten and Murray research groups on thiolate-protected noble metal particles. In 1996, Whetten et al. were the first to observe atomically precise compositions for thiolate-protected gold particles using mass spectrometry. Particles with such compositions were referred to as ‘nanocrystal gold molecules’.109 Murray et al. electrochemically observed a molecule-like electronic structure for many such particles. In 2005, Shichibu et al. synthesized glutathione (SG)-protected Au25(SG)18 nanocluster via ligand exchange reaction of preformed Au-phosphine nanoclusters.110–112 In the same year, Tsukuda et al. mass spectrometrically observed a series of glutathione-protected gold nanoclusters with precise and molecule-like compositions, such as Au10(SG)10, Au15(SG)13, Au18(SG)14, Au22(SG)16, Au22(SG)17, Au25(SG)18, Au29(SG)20, Au33(SG)22, and Au39(SG)24 which were separated by polyacrylamide gel electrophoresis (PAGE).113 In 2006, Häkkinen et al. proposed the ‘divide and protect’ structural model wherein these nanoclusters were viewed as consisting of a discrete metal core, protected by well-defined metal–ligand oligomeric units.114 In 2007, Whetten et al. unambiguously assigned the composition of the most popular nanocluster in this family, Au25(PET)18 (PET = 2-phenylethanethiolate) (which was wrongly assigned as Au38(PET)24 in earlier investigations) through electrospray ionization mass spectrometry (ESI MS). In 2007, Shichibu et al. synthesized a biicosahderal nanocluster, [Au25(PPh3)10(SCnH2n+1)5Cl2]2+ (n = 2–18) by the chemical reaction between [Au11(PPh3)8Cl2]+ and n-alkanethiol (CnH2n+1SH, n = 2, 8, 10, 12, 14, 16, and 18).115 This was the first Au25 nanocluster compound whose crystal structure was resolved. Apart from thiolate and phosphinate monolayers, anion templates, such as halides,116 sulfides,117 chalcogenides,118 and polyoxometalates,119,120 are becoming increasingly prominent in the preparation of high-nuclearity atomically precise Ag nanoclusters.121 Mass spectrometry, separation techniques such as electrophoresis, size-exclusion chromatography (SEC), and single-crystal X-ray diffraction have made tremendous contributions to the science of ligand-protected noble metal nanoclusters.91,122,123 Recently, microelectron diffraction has been used to resolve the structures of those nanoclusters for which crystallization was difficult.124,125 Computational methods have made significant contributions to our understanding of the structures and properties of these nanoclusters, complementing the experimental approaches.9,41,47,50,126–129
2.3. Early insights into structures
The identification of atomically precise nanoclusters such as Au25(PET)18 sparked extensive research into numerous nanoclusters and their properties; however, the structures of these early nanoclusters remained unknown for quite some time. Therefore, these particles were called ‘monolayer-protected nanoclusters’ (MPCs); the term was originally used to refer to their larger (consisting of a few hundred metal atoms), plasmonic counterparts wherein the protecting ligands were assumed to be arranged in a uniform, 2D fashion, as in the case of self-assembled monolayers (SAMs)130 of ligands on metals. In 2007, Kornberg et al. were the first to resolve the crystal structure of a thiolate-protected gold nanocluster, namely Au102(MBA)44 (MBA = p-mercaptobenzoic acid).131 Au102(MBA)44 provided significant new insights into the structure of the ligands on the NPs. Au102(MBA)44 consists of an Au79 core protected by nineteen Au(SR)2 and two Au2(SR)3 units, often referred to as staple units. In 2008, the crystal structure of Au25(SR)18, one of the most popular members of this family of nanoclusters,132 was resolved independently by Akola et al. and Heaven et al., which showed that it consisted of an Au13 icosahedron protected by six Au2(SR)3 oligomeric staples.133,134 These findings proved that the structural arrangement of protecting ligands on metal NPs can be completely different from that of SAMs. Recently, atomic precision has been achieved in a larger size regime of nanoclusters wherein plasmonic features start appearing. For example, structures of Au279(SPh-tBu)84,135,136 Au329(SR)84,137,138 [Ag374(SR)113Br2Cl2],139 Au333(SR)79,140 and Au246(SR)80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 141 were reported by different groups. There are also attempts to accurately probe the composition of plasmonic NPs using mass spectrometry.142,143
141 were reported by different groups. There are also attempts to accurately probe the composition of plasmonic NPs using mass spectrometry.142,143
In the few past years, there have been immense advancements in analytic instrumentation, making it possible to study these precision materials in detail.26,91,144–146,147 High-resolution mass spectrometry (HR MS) coupled with soft ionization can precisely determine the composition of the core and ligands as well as the charge states of the nanocluster.55,148 Other advanced mass spectrometric (MS) techniques, like ion mobility MS (IM-MS)97,149–153 and tandem MS (MS/MS),19,154,155 are becoming increasingly powerful to understand the size, shape, and structural evolution. Single-crystal X-ray crystallography has made it possible to resolve the structures of several thiol (–SR)-capped nanoclusters, like Au25(SR)18,134 Au28(SR)20,156 Au38(SR)24,157 Au40(SR)24,158 Au52(SR)32,159 Au40(SR)24,160 Au92(SR)44,159 Au102(SR)44,131 Au133(SR)52,161 Ag44(SR)30,162 Ag25(SR)18,163 Ag29(SR)12,164 and more. Furthermore, with the development of hyphenated techniques, other inherent nanocluster properties, such as electron affinity (EA), ionization energy (IE), electronic transitions, etc., are being studied in greater detail.91 A few of the milestones in the development of noble metal cluster chemistry are briefly presented in Fig. 2.
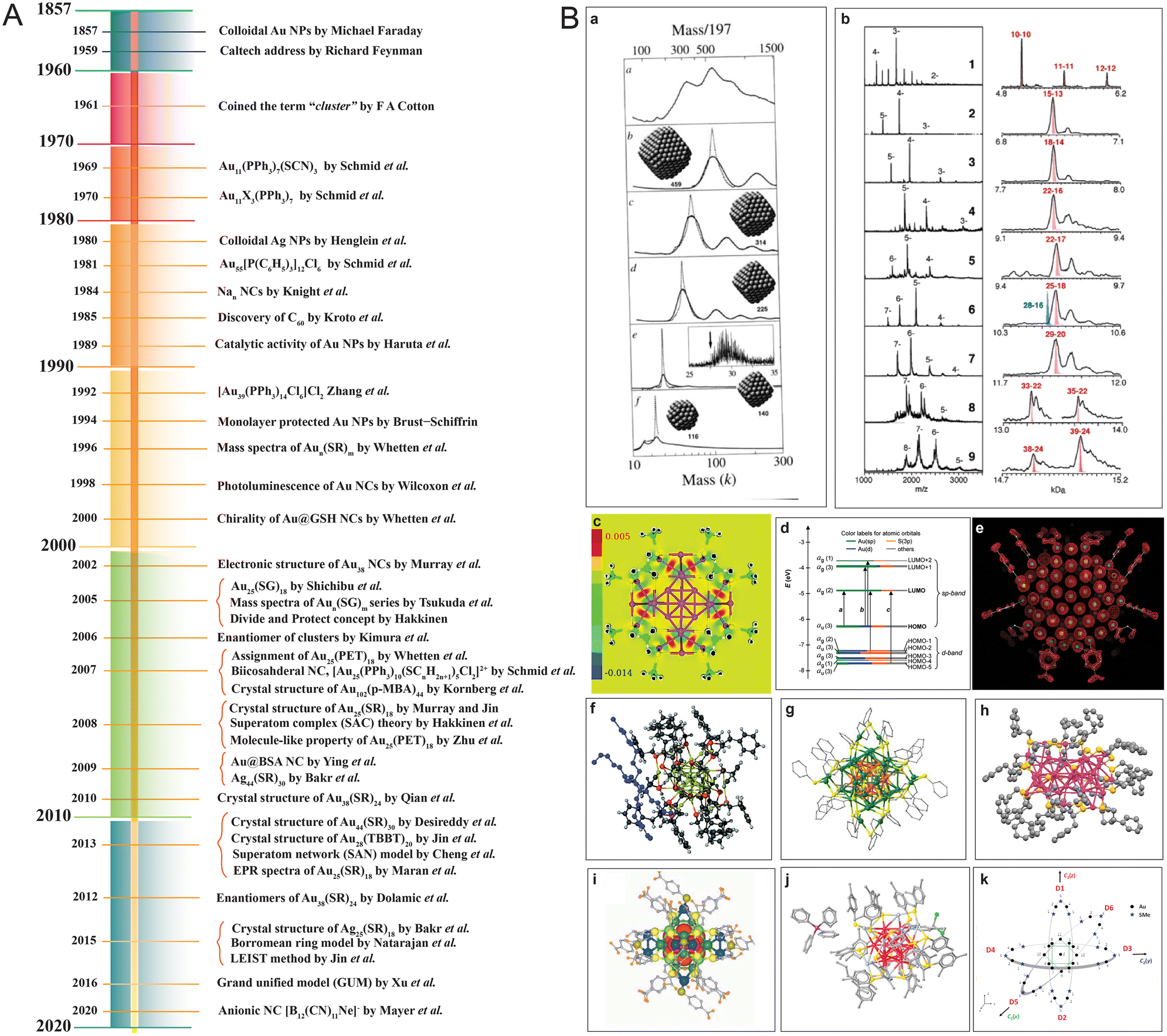 | ||
| Fig. 2 (A) A representative timeline of the evolution of nanocluster science. (B) A few notable mentions from the nanocluster timeline, namely (a) mass spectra of Aun(SR)m, (b) mass spectra of Aun(SG)m series, (c) divide and protect concept as visualized from the difference in electron density due to bonds, (d) Kohn–Sham (KS) orbital energy level diagram of Au25(SH)18−, crystal structures of (e) Au102(p-MBA)44, (f) [N(C8H17)4][Au25(SH)18], (g) Ag44(4-FTP)30(PPh4)4, (h) Au38(SR)24, (i) Na4Ag44(MBA)30, and (j) Ag25(DMBT)18, and (k) Borromean rings diagram of Au25(SMe)18 nanocluster. Adapted from ref. a,109 b,113 c,114 d50,126 e,131 f,134 g,291 h,292 i,162 j,163 and k.166 Copyright 1996 John Wiley and Sons. Copyright 2005, 2006, 2008, 2010, and 2015 American Chemical Society. Copyright 2007 American Association for the Advancement of Science. Copyright 2013 Springer Nature Group. | ||
2.4. Structural models
Various models have been proposed to understand the structure and stability of these nanoclusters.9,41,129 When the ‘divide and protect model’,114 which was one of the earliest, was put forward, no crystal structures were known for thiolate-protected noble metal nanoclusters. In 2008, Häkkinen et al. proposed the superatom complex (SAC), which was an extension of the superatom theory and was used in the case of gas-phase metal nanoclusters, to understand the stability of ligand-protected noble metal nanoclusters.130 In 2013, Cheng et al. proposed the superatom network (SAN) model for some of the thiolate-protected noble metal nanoclusters.165 They also proposed the superatom valence bond (SVB) model for non-spherical nanoclusters such as Au38(SR)24 for which the ordinary SAC model cannot be used for explaining the stability. In 2015, Natarajan et al. proposed a new structural model, namely the Borromean ring model,166 for thiolate-protected noble metal nanoclusters wherein these nanoclusters are viewed as a single structural unit, i.e., interlocked oligomeric metal–ligand rings in contrast to the ‘divide and protect model’ wherein nanoclusters possess a discrete core and staple units. According to this model, Au25(SR)18, for example, is viewed as three interlocked Au8(SR)6 rings surrounding the central Au atom. For the first time, a systematic method of precise naming of alloy nanoclusters and mixed-ligand nanoclusters based on this model was proposed. In 2016, Xu et al. proposed another structural model, namely the grand unified model (GUM) that successfully comprehended the structures of all the gold nanoclusters known by then.167 Here, these gold nanoclusters are viewed as built up from triangular and tetrahedral elementary building blocks. An interesting suggestion based on this model is that the evolution of the gold cores in these nanoclusters cannot be viewed simply as the addition of an Au atom alone, but rather as built from these elementary building blocks.2.5. Properties of metal nanoclusters
Soon after the crystal structures of Au102(SR)44 and Au25(SR)18 were resolved, attempts to understand the structure–property relations in these nanoclusters also commenced. For example, distinct electronic absorption bands of Au25(SR)18 were assigned to various electronic transitions within the molecular orbitals derived from the metal atoms and the ligands.50,126 Several groups have studied the electrochemistry of various metal nanoclusters168 and their alloys,169–171 such as Au25(SR)18, Au38(SR)24, Au67(SR)35, Au102(SR)44, Au144(SR)60, Au333(SR)79 AgxAu25−x(SR)18 (x = 1–5), M2Au36(SR)24 (M = Pd, Pt), etc., further establishing the molecule-like electronic structures of these nanoclusters.172,173 In 2008, distinct charge states of Au25(SR)18 were observed by a few research groups.66,130 Electron paramagnetic resonance (EPR) spectroscopy of Au25(SR)18 was also reported subsequently.66 Photoluminescence in gold nanoclusters was first reported by Wilcoxon et al. in 1998174 and subsequently by many other groups.50,111,126,128 Photoluminescence was observed from protein-protected noble metal nanoclusters as well.19 Nuclear magnetic resonance (NMR) spectra of Au25(SR)18 were reported by different groups.67,175,176 In 2010, the structure of Au38(SR)24, another popular member of the thiolate-protected gold nanoclusters, was theoretically predicted by Lopez-Acevedo et al.,44 and its crystal structure was revealed in the same year by Qian et al.157 Chirality in thiolate-protected noble metal nanoclusters was first reported by Schaaff and Whetten in 2000.177 In 2012, the first separation of enantiomers of Au38(SR)24 was achieved using chiral HPLC by Dolamic et al.,178 which was a significant step toward understanding the chirality of nanoclusters. In 2015, the structural isomerism in Au38(SR)24 was observed by Tian et al.179 Infrared and Raman spectroscopy of these nanoclusters were reported by Dolamic et al. and Varnholt et al., in 2013 and 2014, respectively, revealing the distinct vibrational features of the staples of these nanoclusters.180,181 Whereas most of these advancements were centered around gold nanoclusters, the search for an atomically precise silver nanocluster was fruitful only in 2013 when Desireddy et al. reported the structure of Ag44(SR)30.162 Ag25(SR)18, which is structurally and compositionally analogous to Au25(SR)18, was discovered in 2015 by Joshi et al.163 Clusters of other metals and alloy nanoclusters composed of two, three and four elements have been reported.10,182 Apart from thiolates, a wide variety of ligands, such as selenolates,183–187 tellurolates, alkynes,188,189 carbenes,190etc., have also been used as protecting ligands for noble metal nanoclusters. For a comprehensive summary of the advancements in the field of thiolate-protected noble metal nanoclusters, please consult several additional references.7,8,10–12,41,57,91,173,191–194
3. Chemical reactivity of ligand-protected atomically precise metal clusters
The molecule-like nature of the physical properties of these nanoclusters is evident from the structural and spectroscopic studies discussed above. Recently, we have shown that these nanoclusters exhibit molecule-like chemical reactivity as well. In the following sections, we discuss different aspects of their chemistry in detail.3.1. Ligand exchange and ligand-induced transformations
Post-synthetic modification in particles is a versatile approach for the transformation in atomically precise nanoclusters in terms of compositional, morphological, and structural changes. Reactions of nanoclusters with structurally different ligands are another way to synthesize new nanoclusters. Substitution or exchange of ligands is one of the earliest reactions of such nanoclusters.195Over the past two decades, monolayer-protected metal nanoclusters reacting with various ligands have produced nanoclusters with novel physical and chemical properties.196 Murray et al. performed the first of such ligand-exchange reactions with thiol-protected gold nanoclusters.197 Murray et al. also studied the mechanism of these ligand-exchange reactions in detail using mass spectrometry, NMR spectroscopy, and electrochemistry.195,198 The rate of such reactions depends on the concentrations of both the nanocluster and the foreign ligand. The electron-donating and withdrawing nature of the functional groups on the ligands also governs the reaction rates. Using electrochemistry, Parker et al. showed that the electron-withdrawing ligands accelerate the exchange rate relative to electron-donating ligands.199 Murray et al. observed that the ligand exchange is a second-order reaction.200 Rate of reaction is determined by the bonding of incoming and outgoing ligands with metal, much like an associative mechanism. However, the rate is independent of the size of the nanoclusters. For example, both Au38(PET)24 and Au140(PET)53 showed similar rate constants during ligand exchange using various p-substituted aryl thiols.201 The understanding of site selectivity and specificity of ligands in exchange reactions significantly improved with the availability of single-crystal structures of nanoclusters.
When noble metal nanoclusters react with foreign ligands, they undergo a transformation that leads to three types of ligand-exchange products. These products can result in the nanocluster with (i) retention of its structure and composition upon exchange, (ii) alteration in geometry while retaining its composition, or (iii) alteration in both structure and composition.
The initial reports on ligand-exchange reactions indicate that structure and composition of the nanocluster remain unaltered in the process. Murray et al. extensively studied the ligand-exchange reactions on Au25(PET)18 with different SR (where, R = Ph–CH3, Ph–F, etc.), which resulted in Au25(SR)18−x(SR′)x series (x = 1–12).200,202,203 Partial ligand exchange was observed during the reaction of p-BBT (BBT = bromobenzenethiol) with Au102(p-MBA)44 and Au25(PET)18 which led to the formation of Au102(p-MBA)40(p-BBT)4 and Au25(PET)16(p-BBT)2, respectively.204,205 In 2014, Abdulhalim et al. reported the ligand exchange on Ag44(4-FTP)30 (FTP = fluorothiophenol) with various other aryl thiols such as MNBA (5-mercapto-2-nitrobenzoic acid), 4-NTP (NTP = nitrothiophenol), and 2-NT (NT = naphthalenethiol) which resulted in the formation of Ag44(SR)30 (where SR = MNBA/4-NTP/2-NT).206 A second type of ligand exchange, referred to as ligand-induced isomerization, was reported in 2016 by Jin et al. upon the reaction of Au28(CHT)20 (CHT = cyclohexanethiol) with TBBT ligand (4-tert-butylbenzenethiol), in which the structure of the nanocluster changed while the composition remained unchanged.207
In 2008, Shibu et al. came up with the first report on post-synthetic modification of atomically precise Au25 nanoclusters via ligand exchange reaction.195 Performing a ligand exchange with functionalized glutathione on the Au25(SG)18 nanocluster altered its optical and photoluminescence properties. These reactions have since found widespread application for modifying the chemical and other properties of nanoclusters through the introduction of new ligands to the parent clusters. Recently, ligand-exchange-induced structure transformation (LEIST) has become a rapidly developing technique in nanoclusters. In such reactions, when a foreign ligand is introduced, it can cause significant distortion of the core, resulting in both structural and compositional changes within the nanocluster. In 2013, Jin et al. introduced the term LEIST when they observed the transformation of Au38(SR)24 to Au36(SR′)24 nanocluster through a ligand-exchange reaction. Jin et al. performed the ligand-exchange reaction on Au38(PET)24 with excess TBBT (TBBT = 4-tert-butylbenzenethiol) under thermal conditions, resulting in molecularly pure Au36(TBBT)24 in excellent yield.208 The process of ligand exchange brings about a change in the structure of biicosahedral Au38(PET)24, transforming it into a truncated tetrahedral Au36(TBBT)24 with an FCC kernel. Interestingly, one of the first examples of an FCC-structured Aun(SR)m nanocluster is Au36(TBBT)24. Zeng et al. established the universality of the LEIST method by reacting Au25(PET)18 with TBBT under thermal conditions, which led to the formation of Au28(TBBT)20.156 The Jin group's discovery of the LEIST method led to the synthesis of numerous new nanoclusters.209 Following this work, a large number of transformations was studied by different groups, such as conversions of Au11(PPh3)7Cl3 to [Au25(SR)5(PPh3)10X2]2+,210 Au15(SG)13 to Au16(S-Adm)12,211 Au18(S-c-C6H11)14 to Au21(S-Adm)15,212etc., which proved the method to be versatile for making new structures. Furthermore, similar structural changes were observed in silver nanoclusters, and the mechanisms underlying these changes were investigated in detail. Bakr et al. showed the reversible conversion between Ag25(2,4-DMBT)18 (DMBT = dimethylbenzenethiol) and Ag44(4-FTP)30 (FTP = fluorothiophenol).213 Upon reaction with 2,4-DMBT, Ag44(4-FTP)30 underwent a disproportionation reaction to form smaller sized Ag25(4-FTP)1(2,4-DMBT)17 and bigger sized Ag46–50(4-FTP)4–9(2,4-DMBT)21–26. After complete ligand exchange, other less stable nanoclusters transformed to more stable Ag25(2,4-DMBT)18. On the other hand, the conversion of Ag25(2,4-DMBT)18 to Ag44(4-FTP)30 occurred via dimerization of Ag25(2,4-DMBT)18 followed by a rearrangement pathway. A similar mechanism was observed during the conversion of Ag35(SG)18 to Ag44(4-FTP)30.214 Khatun et al. showed a distinctly different mechanistic pathway during the transformation of Ag59(2,5-DCBT)32 (DCBT = dichlorobenzenethiol) to Ag44(2,4-DCBT)30, Ag25(2,4-DMBT)18 and Ag29(1,3-BDT)12(PPh3)4 (BDT = benzenedithiol) upon reaction with 2,4-DCBT, 2,4-DMBT and 1,3-BDT/PPh3, respectively (Fig. 3).215 In the presence of incoming thiol ligands, Ag59(2,5-DCBT)32 dissociated completely into smaller nanoclusters and thiolates instead of ligand exchange. Then, these smaller nanoclusters and thiolates recombined and rearranged to form the final product. The nature of the thiolate ligand plays an important role in determining the structure and composition of the product nanoclusters. Khatun et al. also reported the synthesis of MAg28(1,3-BDT)12(PPh3)4 from MAg24(2,4-DMBT)18via the LEIST method. Recently, a phosphine-protected nanocluster Ag18(PPh3)10H16 synthesized by Bakr et al. was observed as a very good precursor for the LEIST reaction (Fig. 4).216 Bodiuzzaman et al. synthesized two new nanoclusters, Ag46(DMBT)24(PPh3)8 and Ag40(DMBT)24(PPh3)8via the LEIST method, using Ag18(PPh3)10H16.217 Manju et al. synthesized NIR-emitting [Ag34S3(SBB)20(CF3COO)6]2+ nanocluster from Ag18(PPh3)10H16 upon reacting it with tertiary-butylbenzylthiol (SBB).218 Upon reacting Ag18(PPh3)10H16 with 2-pyrene imine thiol (2-PIT), Jana et al. synthesized a new dual-emitting nanocluster, [Ag35(2-PIT)7(PPh3)7@(H2O)]3+.219 Kang and Zhu published an extensive review on the evolution of the LEIST methodology and its application.220
 | ||
| Fig. 3 Schematic representation of ligand exchange-induced conversion of Ag59(2,5-DCBT)32 to Ag44(2,4-DCBT)30, Ag25(2,4-DMBT)18 and Ag29(1,3-BDT)12(PPh3)4 after the reaction with 2,4-DCBT, 2,4-DMBT and 1,3-BDT/PPh3, respectively, under ambient conditions. Adapted from ref. 215. Copyright 2017 Royal Society of Chemistry. | ||
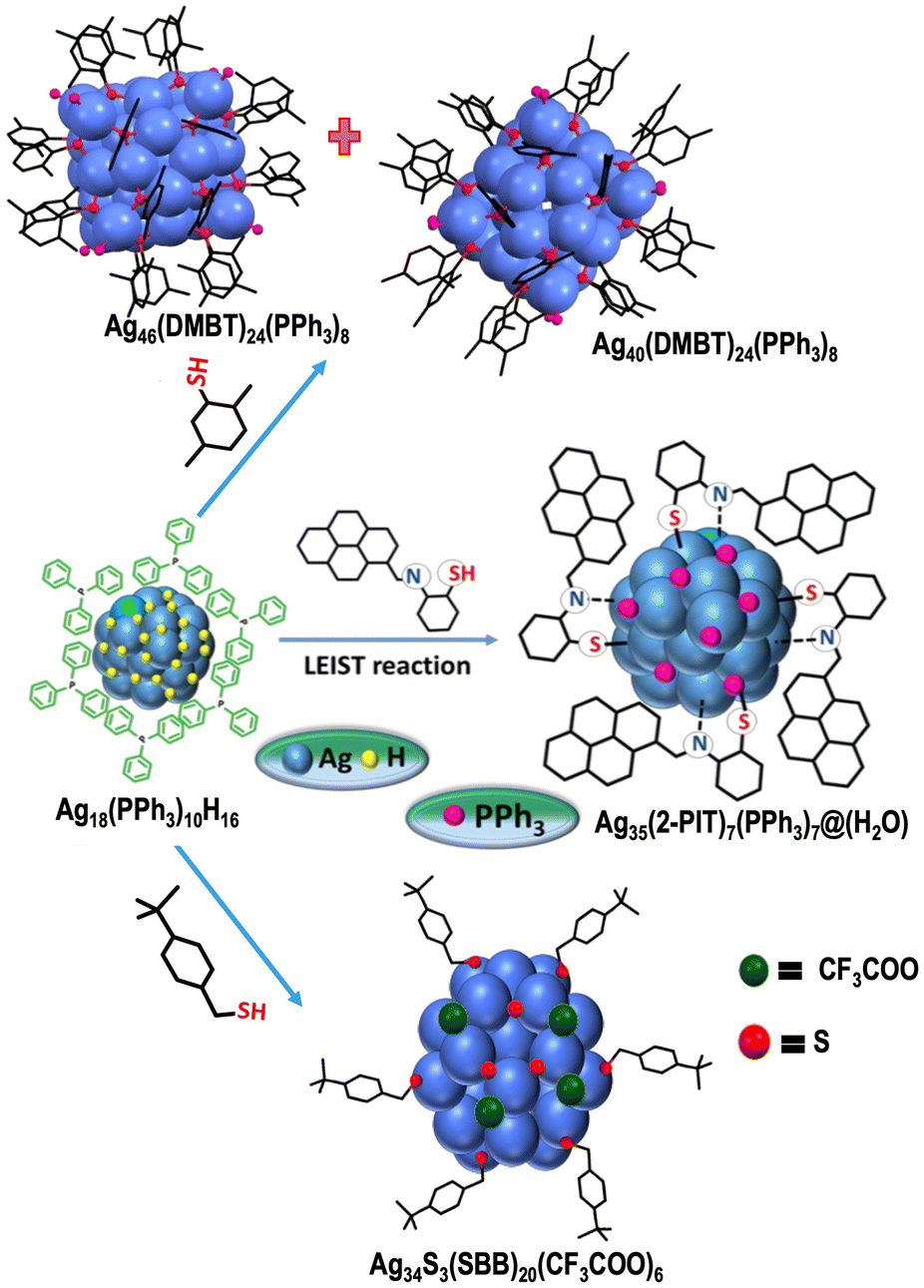 | ||
| Fig. 4 Formation of [Ag34S3(SBB)20(CF3COO)6]2+, [Ag35(2-PIT)7(PPh3)7@(H2O)]3+, Ag46(DMBT)24(PPh3)8 and Ag40(DMBT)24(PPh3)8 from Ag18(PPh3)10H16via LEIST method. Adapted from ref. 217. Copyright 2019 John Wiley and Sons. | ||
Ligand exchange is an effective strategy to improve the physical and chemical properties of nanoclusters. The method has largely been used to enhance the emission quantum yield (QY) of several non-luminescent or feebly luminescent nanoclusters. Jin et al. showed an improvement in photoluminescence (PL) intensity while preserving the composition of Au25(SR)18.221 They found higher QY for Au25(PET)18 (PET = 2-phenylethanethiol) than for Au25(DDT)18 (DDT = dodecanethiol) and Au25(HT)18 (HT = hexanethiol). Later on, the PL intensity of Au25(PET)18 enhanced 6.5 fold on using NAP (NAP = 2-(naphthalen-2-yl)ethanethiolate) ligand instead of PET.222 Kim et al. showed enhancement in PL intensity of Au36(TBBT)24 (TBBT = tert-butylbenzenethiol) by partial ligand exchange using CPT (CPT = cyclopentanethiol) ligand.223 Similar to gold nanoclusters, ligand engineering in silver nanoclusters also led to the enhancement of the PL QY. Khatun et al. found structure-conserved ligand exchange in Ag29(BDT)12(PPh3)4 (BDT = 1,3-benzenedithiol) using various diphosphine ligands such as DPPM (1,1-bis(diphosphino)methane), DPPE (1,2-bis(diphosphino)ethane) and DPPP (1,3-bis(diphosphino)propane) which resulted in the increment of PL QY, as shown in Fig. 5.224 Among these nanoclusters, Ag29(BDT)12(DPPP)4 exhibited highest PL QY which is 30 fold higher than that of Ag29(BDT)12(PPh3)4. The PL intensity can also be modified by the structure, transformed due to ligands. Such an example is the conversion of Pt1Ag24(SR)18 to Pt1Ag28(SAdm)18(PPh3)4 (SAdm = adamentanethiol) which displayed 50-fold higher PL QY than the parent nanocluster.225 Other properties such as chirality is introduced in nanoclusters using the ligand exchange method.226,227 Bürgi et al. introduced chirality in Au38(PET)24 nanocluster by partial ligand exchange using chiral bidentate thiol, BINAS (BINAS = 1,1′-binaphthyl-2,2-dithiol).228
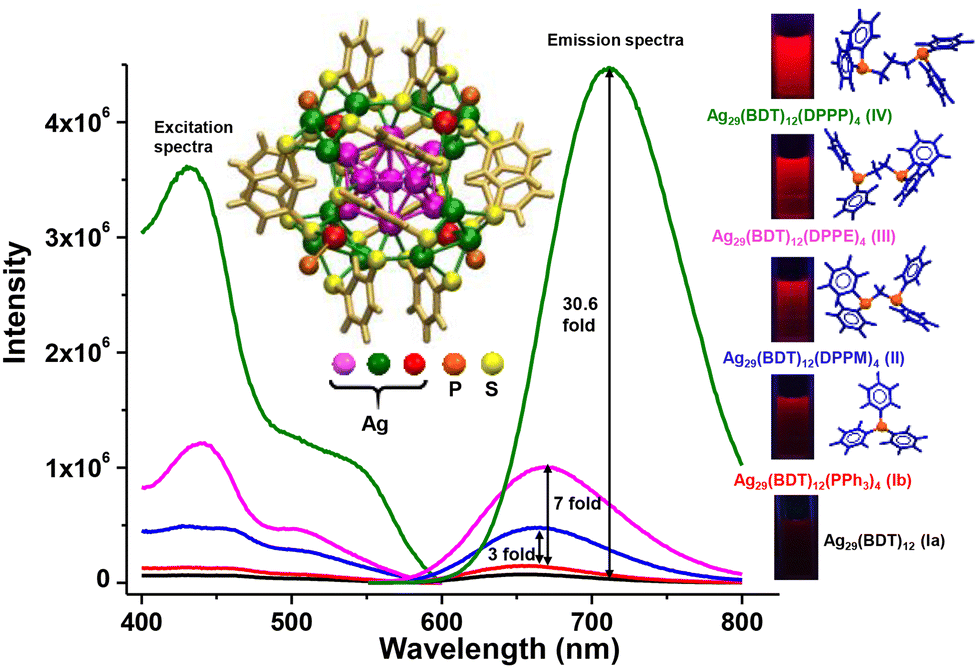 | ||
| Fig. 5 Excitation and emission spectra of Ag29(BDT)12 and Ag29(BDT)12(P)4 where P = PPh3, DPPM, DPPE, DPPP. A systematic enhancement of PL intensity is observed. Photographs of nanoclusters under UV light are also shown. Reproduced from ref. 224. Copyright 2018 Royal Society of Chemistry. | ||
3.2. Reactions with metal ions
The interaction of a metal ion with the noble metal nanocluster is reflected in the changes observed in the absorption and emission spectra of the nanocluster. Some metal nanoclusters are known to be highly fluorescent compared to their bulk counterparts. Also, the post-synthetic metal-exchange reactions with noble metal nanoclusters are an important method for the preparation of alloy nanoclusters. Of all the known metal-exchange methods, galvanic reduction is one of the most efficient approaches for the preparation of multimetallic alloy nanoclusters.Metal ion-induced alteration in the fluorescence of nanoclusters is one of the most used strategies for sensing heavy metal ions, like Hg2+, Cu2+, As3+, Cr3+, Pb2+, etc.75 In 2007, Habeeb et al. were the first to report the reactivity of Au25SG18 nanocluster to AuCl4−.229 The Au25SG18 nanocluster underwent an instantaneous decomposition in the presence of AuCl4− ions to form an insoluble gold–glutathione coordination polymer (AunSGm). The characteristic absorption features of Au25SG18 nanocluster get quenched immediately after addition AuCl4− ions due to the formation of Au(I) – glutathione complex. Upon reaction with other metal ions, such as Ag+, Fe3+, Cu2+, Ni2+, Cd2+, Zn2+ and Sr2+, the Au25SG18 nanocluster decomposes but at a slower rate. The net reaction of Au25SG18 nanocluster – Au3+ ion can be represented as an electron-transfer process where the electrons from the nanocluster core reduce AuCl4− to AuCl2− ions.
Mercury (Hg) is one of the most toxic heavy metals. Bootharaju and Pradeep reported that the Ag7,8(MSA)7,8 nanocluster (MSA = mercaptosuccinic acid) can act as a Hg and other heavy metal scavenger.230 In the reaction medium, the silver nanocluster interacts preferentially through its core and the carboxylate group of mercaptosuccinic acid (MSA) ligand depending upon the concentration of Hg2+ ion. The Ag7,8(MSA)7,8 nanocluster undergoes luminescence quenching as it interacts with Hg2+. To understand the scavenging property of the Ag7,8(MSA)7,8 nanocluster, it is important to note that the redox potentials of Ag1+/Ag0 decrease with the particle size compared with the bulk metal, which is +0.79 V, whereas it is +0.8 V for the interacting metal ion (Hg2+/Hg0). The net cell electromotive force (emf) for the reduction of Hg2+ by the silver nanocluster is positive. The alumina-loaded Ag7,8(MSA)7,8@Al2O3 nanocluster can be used quantitatively for Hg2+ removal from contaminated water. Later on, from the same group, Chakraborty et al. went ahead to report the selective reaction of Ag25SG18 nanocluster with Hg2+ ion. The chemical interaction of Ag25SG18 nanocluster and Hg2+ ion resulted in the formation of Ag3Hg2 alloy (paraschachnerite with an orthorhombic crystal structure), as observed with the appearance of new blue-shifted features in the optical absorption spectra (Fig. 6). XPS studies show that the Ag25SG18 nanocluster – Hg2+ ion reaction is a redox process which involves oxidation of Ag0 to Ag+ and reduction of Hg2+ to Hg0. The luminescent Ag25SG18 nanocluster can act as a sensing material, as it undergoes fluorescence quenching upon interaction with Hg2+ ions with a limit of detection of 1 ppb.
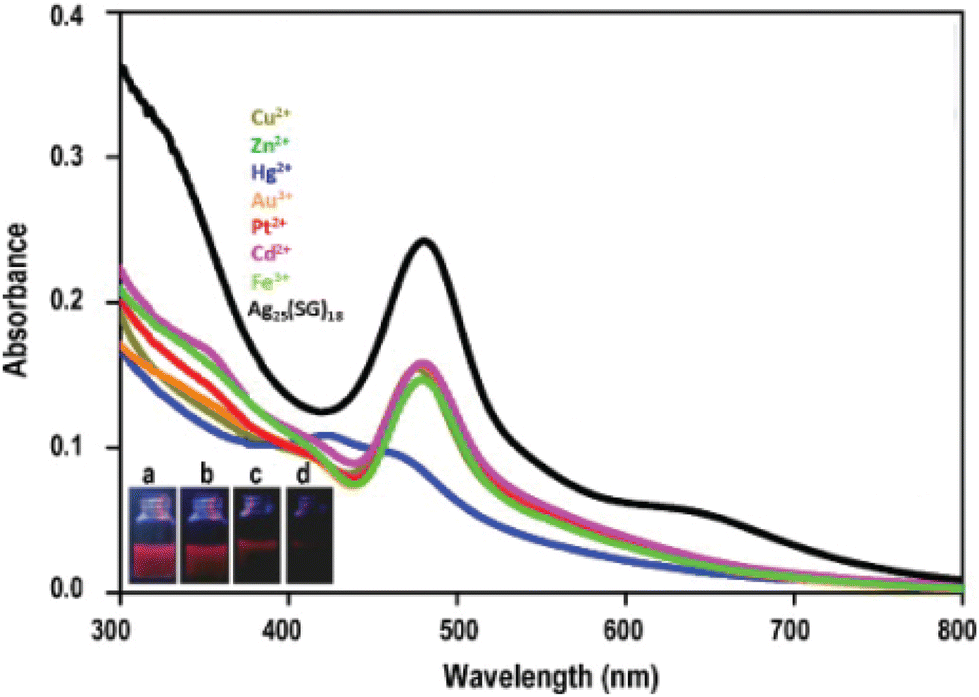 | ||
| Fig. 6 Optical absorption spectra of the Ag25SG18 nanocluster solution on addition of different metal ions. Under UV excitation, images corresponding to fluorescence quenching of (a) phase transferred nanocluster and on the addition of (b) 1 ppm, (c) 2 ppm, and (d) 10 ppm of Hg2+. Adapted from ref. 290. Copyright 2012 Elsevier. | ||
Another heavy metal present in drinking water is Cu2+, which has a permissible limit of 1.3 ppm in drinking water as set by the U.S. Environmental Protection Agency (EPA). Glutathione (GSH) is a natural tripeptide in amino acids, such as cysteine, glutamic acid, and glycine. GS-protected gold nanoclusters are biocompatible and have strong near-IR fluorescence emission compared with other non-aqueous Aun(SR)m, making them a popular candidate for developing biological and heavy metal sensors. Back in 2009, fluorescent Au@GS NPs made a debut as a highly selective Cu2+ sensor, which is a classic example of aggregation-induced fluorescence quenching.231 Later on, efforts were undertaken by George et al. for sensing Cu2+ ions using thiolate-protected gold nanoclusters.34 This work involved a GS-protected Au15 nanocluster encapsulated in cyclodextrin (CD) cavities (denoted by Au15@CD). The sensor material is prepared by loading the Au15@CD nanocluster onto a freestanding film of chitosan. This nanocluster composite material is a bright luminescent film under UV light. Upon exposing the nanocluster composite material to Cu2+ ions, luminescence quenching happens which is selective to Cu2+ ion concentration. A change in emission maximum was observed in the PL spectra of the material before and after its exposure to Cu2+ ions. The sensing specificity of the nanocluster composite material towards Cu2+ ion was studied using XPS analysis, which suggested a reduction of Cu2+ to Cu1+/Cu0 by the glutathione ligand or the Au15 core. The reported limit of detection of the GS-protected-Au15@CD nanocluster composite material is 1 ppm of Cu2+ ion present in the medium. Zhang et al. reported the application of water-soluble GS-protected gold nanoclusters in Cu2+ sensing with a limit of detection of 86 nM.72 The quenching of the fluorescence is attributed to the carboxylic group in GSH-ligand, which is a chelating agent with a high affinity and selectively towards Cu2+ ion over other metal ions, like Hg2+ and Pb2+, present in the medium. Krishnadas et al. reported a highly luminescent MSA-protected Ag–Au bimetallic nanocluster (denoted as AgAu@MSA) material as a Cu2+ sensor. Initially, the preparation of the AgAu@MSA nanocluster involved a galvanic reduction of polydispersed Ag NPs by AuI-MSA thiolates. A methanolic solution of the AgAu@MSA nanocluster undergoes immediate luminescence quenching selectively upon interaction with Cu2+ even in the presence of other metal ions. The mechanism of metal-induced fluorescence quenching of the nanocluster was investigated using XPS, and it was concluded that the Cu2+ ion interacts with the AgAu metal core of the nanocluster. The nanocluster–metal ion interaction is a redox process; the AgAu metal core reduces the Cu2+ ion to Cu1+/Cu0, while it gets oxidized in the process.
In the following part, we will focus on the metal-exchange reactions of noble metal nanoclusters resulting in alloy nanoclusters. As per the galvanic theory, the metal ion with a higher reduction potential replaces another metal with a lower reduction potential, and subsequently, it gets reduced (Fig. 7A). The reduction potential of metals in decreasing order is Fe2+ > Cd2+ > Co2+ > Ni2+ > [Au NCs] > Cu2+ > Hg2+ > Ag1+ > Pd2+ > Pt2+ > Au1+.232,233 Using the galvanic replacement method, various multimetallic noble metal alloy nanoclusters are formed. One such example is the Au-incorporated Ag24Au(DMBT)18 nanocluster derived from Ag25(DMBT)18 studied by Bootharaju et al., where the reduction potentials of Ag+/Ag and Au+/Au are 0.080 and 1.69 V, respectively (Fig. 7C(a)).234 The structure of the bimetallic Ag24Au(DMBT)18 nanocluster is analogous to its monometallic counterpart with enhanced stability and photoluminescence. The reaction of PdAg24(SR)18 and Au-salt was found to proceed via trimetallic PdAuAg23(SR)18 nanocluster intermediate and finally resulting in a replacement product, the AuAg24(SR)18 nanocluster. But the Au ionic reaction with the structurally equivalent PtAg24(SR)18 nanocluster led to the formation of Au2PdAg22(SR)18 nanocluster. Later, Kang et al. reported dopant-dependent shape-controlled galvanic exchange reactions.235 The Au-doping in a PtAg24(DMBT)18 nanocluster with the precursors Au-DMBT and AuBrPPh3 resulted in the formation of trimetallic nanoclusters with shape-unaltered PtAuxAg24−x(DMBT)18 and altered Pt2Au10Ag13(PPh3)10Br7 nanoclusters, respectively (Fig. 7C(b)). Similarly, multimetallic PtCuxAg28−x(BDT)12(PPh3)4 and Pt1Ag12Cu12Au4(S-Adm)18(PPh3)4 nanoclusters were prepared using the galvanic exchange method.236 Bootharaju et al. studied the metal-exchange reaction between MAg24(SR)18 (M = Pd/Pt) and AuPPh3Cl salt, using mass spectrometry (Fig. 7C(c)).237 Likewise, using the templated galvanic metal-exchange route, a highly stable Au5.34Ag44.66(Dppm)6(TBBM)30 (Dppm = bis(diphenylphosphino)methane and TBBM = 4-tert-butylbenzyl mercaptan) nanocluster was prepared from Ag50(Dppm)6(TBBM)30 (Fig. 7C(d)).238
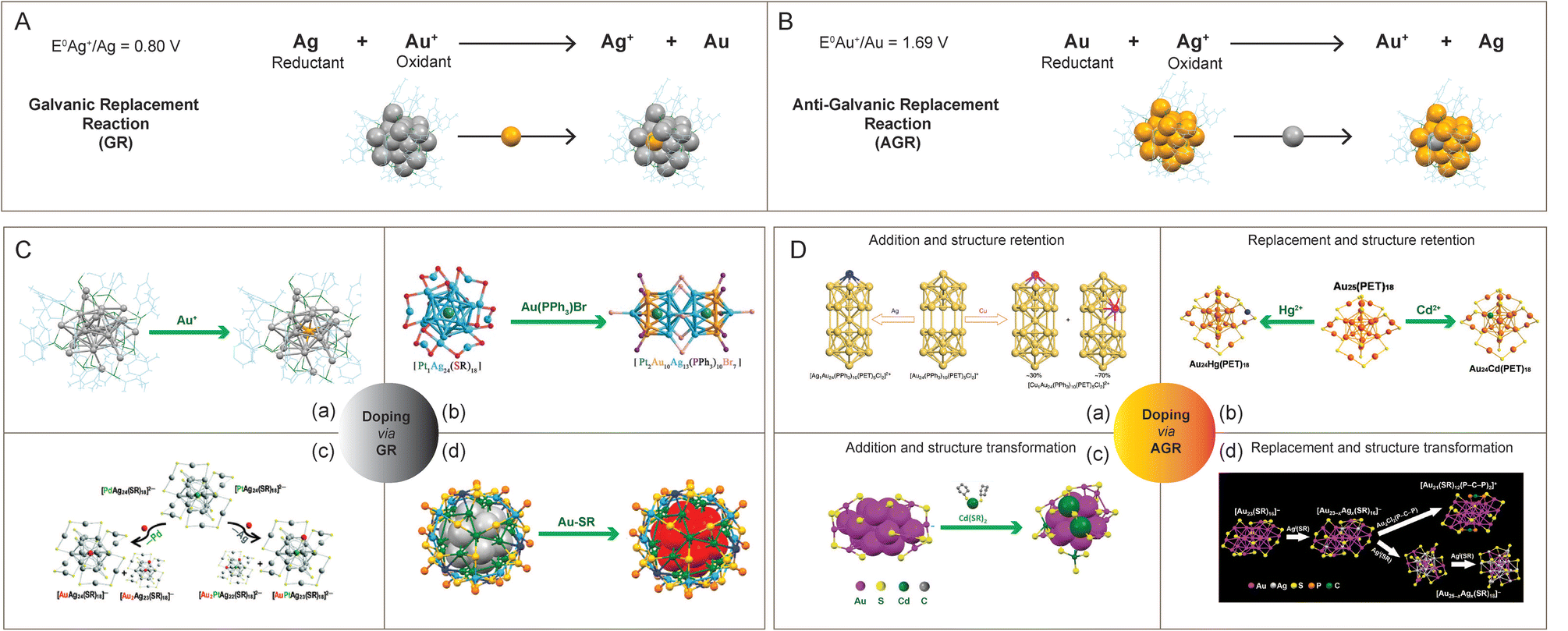 | ||
| Fig. 7 Schematic representation of (A) galvanic replacement and (B) anti-galvanic replacement reaction from the context of doping in Ag25(SR)18 and Au25(SR)18, respectively. Examples showing the preparation of alloy nanoclusters using (C) galvanic and (D) anti-galvanic reaction routes. Reproduced with permission from ref. C-a,234 b,236 c,237 and d,238 and D-a,246 b,248 c,251 and d.249 Copyright 2015 and 2017 American Chemical Society. Copyright 2016 and 2018 John Wiley and Sons. Copyright 2016 Royal Society of Chemistry. Copyright 2017, Springer Nature Group. Copyright 2019 National Academy of Sciences. | ||
An anti-galvanic reaction (AGR) defies the classical galvanic reduction (GR) as the metal ions get reduced by the less reactive (or more noble) metal (Fig. 7B). The driving force for an anti-galvanic reaction can be explained in terms of the difference in the redox potential of the participating entities. In 1985, Plieth proposed a theory on the relationship between the electrode potential of metal nanoparticles and their particle diameter.239 Theoretically, bulk metals (Mbulk) can be transformed into small metal particles (MNP) by dissolving into metal ions (M+) and then redepositing as particles. The reduction of bulk metal (Mbulk) into metal nanoparticles (MNP) can be described by the electrochemical cell reaction,239
| Mbulk → M+(aq) + ze− |
| M+(aq) + ze− → MNP(s) |
Plieth states that the standard reduction potential of a small metal particle undergoes a negative shift, as expressed in the following equation,239
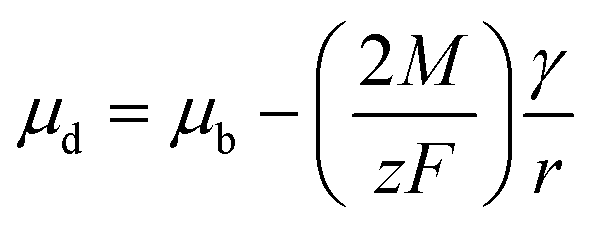
The ultrasmall-sized noble metals nanoclusters have been recently emerging as interesting candidates for anti-galvanic reactions.232,241 The anti-galvanic reaction route provides a more facile and milder method towards alloying with a better control over the composition, structure, and properties of the nanoclusters. Choi et al. reported the first anti-galvanic reaction in nanoclusters with the alloying of [Au25(PET)18] nanocluster.242 The [Au25(PET)18]− nanocluster upon reaction with Ag+ ion resulted in a bimetallic [Au24Ag(PET)18]− nanocluster. From the electrochemical series, it is known that Au is less reactive than Ag, and it was anticipated that the reduction of Au(III) by Ag metal is facile in the ambient conditions but the opposite is not. In 2012, Wu studied the reaction of Au25(PET)18 with Ag+ and Cu2+ ions resulting in bimetallic products as characterized using matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) and XPS.232 Ag+ ions failed to react with 2–3 nm Au NPs, thereby establishing Au25(PET)18 nanoclusters as a unique candidate towards anti-galvanic reaction. The thiolate (–SR) ligand coverage on the nanocluster surface plays a pivotal role in the anti-galvanic reactions. As the ligand attaches to the metal surface, it gains a partial negative charge which further assists in the reduction of less noble ions, like, Ag+ and Cu2+. Here, the anti-galvanic reaction is catalyzed by the highly reactive metal atom on the nanocluster surface. The AGR products are influenced by monolayers on the nanocluster surface. For instance, Wu et al. demonstrated that when exposed to silver ion precursors (such as AgNO3, Ag-PET, Ag-EDTA, and Ag-DTZ; where EDTA stands for ethylenediamine tetraacetic acid disodium salt and DTZ for dithiazone), Au25(PET)18 reacts and forms various Ag–Au alloy nanoclusters.243 However, Au25(SG)18 does not exhibit any reaction with Ag+ ions.244
Using anti-galvanic reaction routes, gold nanoclusters can be alloyed by a heteroatom addition or replacement that also involves a retention or an alteration of the structural framework.241
Here are a few examples of heteroatom addition with retention of the initial nanocluster structural framework. The reaction between Au25(PET)18 and AgNO3 in acetonitrile results in the formation of Au25Ag2(PET)18 nanocluster as a major product.245 The incoming Ag-atoms, instead of replacing Au-atoms, get added to the Au25(PET)18 nanocluster structure. Upon comparison with the Au25(PET)18 nanocluster, the Ag-added species, Au25Ag2(PET)18, has a ∼3.5-fold enhancement in QY while no change was seen in the Ag-replaced species, Au25−xAgx(PET)18 (x = ∼3). Wang et al. observed that a foreign Ag-atom can squeeze into the hollow site of the Au24(PPh3)10(PET)5Cl2 nanocluster without altering its composition or structure (Fig. 7D(a)).246 Other popular examples of metallic replacement with the retention of starting nanocluster structure are Ag, Pt, and Pd-doped Au38(PET)24, Ag-doped Au36(TBBT)24, Cu and Ag-doped Au144(PET)60.247 In 2015, Liao et al. synthesized Au25Hg1(PET)18 and Au25Cd1(PET)18 from Au25(PET)18 using an anti-galvanic reaction (Fig. 7D(b)).248 The single-crystal X-ray diffraction of Au25Hg1(PET)18 revealed the structural similarity with Au25(PET)18 where one of the outer-shell Au-atoms is replaced by a Hg-atom, while for the Au25Cd1(PET)18 structure, the Cd-atom replaces the Au-atom to occupy the central position (Fig. 7B).
Jin et al. pioneered alloying methods where the heteroatomic replacement initiates a structural transformation. Li et al. studied the Ag-doping-induced transformation of the Au23(CHT)16 (CHT = cyclohexanethiolate) nanocluster into Au25−xAgx(CHT)18 (Fig. 7D(d)).249 The alloying reaction between Au23(CHT)16 and Ag(I)-CHT proceeds through a two-step metal-exchange route: (i) Au23(CHT)16 is initially converted to a Au23−xAgx(CHT)16 (x ∼ 1) intermediate, and (ii) then it is allowed to grow into Au25−xAgx(CHT)16 (x ∼ 4), with Ag sitting at the icosahedral inner shell.250 Zhu et al. reported another method of synthesis of the bimetallic nanocluster involving a non-replacement of the heteroatom along with a structural transformation. The Au20Cd4(SH)(CHT)19 nanocluster was prepared from Au23(SR)16 using the anti-galvanic reaction (Fig. 7D(c)).251 The starting Au23(SR)16 nanocluster is known to have an Au15 bi-capped cuboctahedron-based kernel, protected by two Au3(SR)4 trimeric and Au(SR)2 monomeric staples along with four simple bridge –SR– ligands.252 Structural similarity of Au25 and Au20Cd4(SH)(CHT)19 nanocluster system was found to be composed of a centered icosahedral Au13 and Au11Cd2 kernel, respectively. The introduction of two Cd-atoms distorts the Au13 kernel of Au25. The resulting nanocluster structure consists of a distorted central icosahedral Au11Cd2 kernel with the capping of two non-equivalent trimeric staples, one dimeric staple, two monomeric staples, four bridging thiolates (–SR–), and one CdSH unit. Li et al. reported Cd-addition to the Au22(SAdm)16 (SAdm = 1-adamantanethiol) nanocluster (bioctahedral Au10 kernel), which resulted in a structurally transformed Au22Cd1(SAdm)16 nanocluster (cuboctahedral Au12Cd1 kernel).253 Using the reaction between Cl@Ag14 and AgClO, Hau et al. introduced the metal core enlargement in nanoclusters, leading to the formation of bigger Cl6Ag8@Ag30.116 Recent investigations have shown that carboxylate ligands on the surface of Ag nanoclusters provide ligand–shell flexibility, inducing structural modifications in the NCs due to differential coordination of Ag between carboxylate and thiol/alkyl moieties.254,255 For example, monocarboxylate and dicarboxylate ligand triggers structural transformations Mo6O22@Ag44 → Mo8O28@Ag50 (ref. 254) and Ag54 → Ag28, (ref. 255) respectively.
3.3. Reactions with halocarbons
The studies of the reaction of halocarbons with noble metal NPs opened up a new direction in nanocluster chemistry. Nair and Pradeep reported that citrate-capped Ag and Au NPs possess catalytic property for the destruction of halocarbons, resulting in the formation of metal halides and amorphous carbon. This was the first time such properties had been reported.256 The halocarbon, CCl4, upon reaction with an alcoholic solution of Ag and Au NPs resulted in the formation of AgCl and AuCl3, respectively, along with an amorphous carbon residue. This reaction was able to completely mineralize halocarbons like choloro, fluoro, and bromocarbons. Later, Bootharaju and Pradeep identified a pesticide degradation pathway using the citrate-capped Ag and Au NPs.257 Here, chlorpyrifos (CP), an organophosphorothioate pesticide, was used as the model pesticide. After reacting with CP, optical absorption spectroscopy showed a red shift in the surface plasmon of Ag NP, and transmission electron microscopic (TEM) analysis revealed aggregation. Upon reaction with the unsupported and alumina-supported Ag and Au NP, the CP degrades into less toxic by-products like 3,5,6-trichloro-2-pyridinol (TCP) and diethyl thiophosphate (DETP), which was established using mass spectrometric studies. The proposed mechanisms involve steps like: (i) first, the CP binds to the NP surface through an Agn+ ← S bond, (ii) P–O cleavage, (iii) nucleophilic H2O attack at the electrophilic P site, and (iv) finally, electron withdrawal from Agn+ ← N and Agn+ ← S bonds resulting the formation of stable TCP and DETP compounds. The Ag NPs were found to have better catalytic performance over Au NPs. When reacting with CP, the unsupported Ag@citrate NPs tend to aggregate, while the alumina-supported Ag NPs do not. This makes the latter more efficient for water purification, and reusable.In 2013, Bootharaju et al. reported the degradation of halocarbons like CCl4, C6H5CH2Cl, and CHCl3, using atomically precise Ag9MSA7 nanoclusters.258 The reaction products, AgCl, CCl3COOH, amorphous carbon, and acetone, were characterized using XRD, Raman, infrared, optical absorption, X-ray photoelectron spectroscopy, and mass spectrometry. To increase the miscibility of halocarbons like CCl4 in the reaction mixture, isopropyl alcohol (IPA) was used. The precipitate is amorphous carbonaceous material with a graphitic structure, while the supernatant contains acetone from oxidation of IPA. The proposed mechanism (Fig. 8) involves an initial adsorption of IPA and CCl4 on the nanocluster surface, which in turn catalyses the oxidation of IPA into acetone and activation of the C–Cl bond of the CCl4. The surface activities initiate a series of electron transfer reactions like release of H+ and Cl− ions to the medium, making it acidic, Cl− ions replacing the MSA ligands, oxidation of Ag0 to Ag+, and finally, mineralization of CCl4.
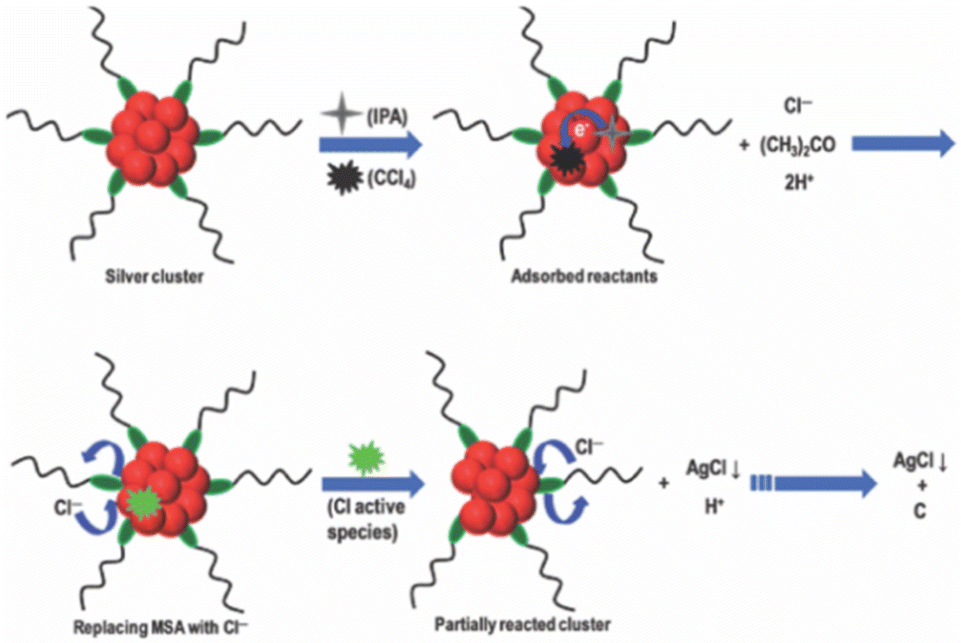 | ||
| Fig. 8 Schematic representation of the proposed mechanism for degradation of halocarbons, like CCl4, by Ag nanocluster. Adapted from ref. 258. Copyright 2013 Royal Society of Chemistry. | ||
3.4 Supramolecular chemistry of metal nanoclusters
The supramolecular chemistry of nanoclusters is an emerging area of research which also highlights the molecular nature of atomically precise nanoclusters. The organic ligands protecting the metal core of the nanocluster can interact with suitable molecules by weak supramolecular interactions. Such interactions include π–π, C–H⋯π, van der Waals and electrostatic interactions. Moreover, metallophilic interactions of the nanocluster core also play an active role in controlling the interactions. Such interactions have been the major driving force in controlling the crystal packing of the nanoclusters. Recently, such interactions have also been explored with other molecules. Mathew et al. reported interaction between Au25SBB18 (SBB = 4-(t-butyl)benzyl mercaptan) and cyclodextrins (CDs), which showed that the SBB ligands were encapsulated in the cavity of CDs, forming inclusion complexes of the nanocluster with CDs.259 Chakraborty et al. reported host–guest complexes of nanoclusters and fullerenes.260 Such interactions were largely dependent on the geometrical compatibility of the two molecules for forming the adducts and further assisted by weak supramolecular interactions. A range of such complexes can be made depending on the structure of the nanoclusters. Ag29(BDT)12 nanocluster can capture C60 molecules on its surfaces, forming adducts such as [Ag29(BDT)12(C60)n]3− (n = 1–9). Structures of [Ag29(BDT)12(C60)4]3− and [Ag29(BDT)12(C60)8]3− are presented in Fig. 9A(a), which reveals that C60 molecules are captured on tetrahedral sites on the nanocluster surface, assisted by interactions with the BDT ligands. Similarly, Ag25(DMBT)18− and Au25(PET)18− nanoclusters also formed adducts with fullerenes.261 Due to a different geometrical structure of M25(SR)18− nanoclusters compared with that of [Ag29(BDT)12]3− nanocluster, the nature of the host–guest adducts with fullerenes were also different in the two cases. Ag25(DMBT)18− and Au25(PET)18− nanoclusters formed aggregates with fullerenes as shown in Fig. 9A(b), and these aggregates were actually dimeric, trimeric, or polymeric adducts of the nanoclusters. Supramolecular interactions of nanoclusters with crown ethers have also been observed, and such complexes were crystallized.262 Crown ethers were captured in the crystal lattice of Ag29 nanoclusters, forming lattice inclusion compounds, as shown in Fig. 9B. Such interactions also resulted in a change in the emission properties compared to the crystals of parent nanocluster. The chemical reactivity of the nanoclusters with other molecules also leads to the emergence of new properties in the host–guest complexes. Nag et al. reported inclusion complexes of Ag29(BDT)123− nanocluster with CDs.263 Such complexes showed isomerism due to the different binding possibilities of CDs on the nanocluster surface, as presented in Fig. 9C. About six CD attachments to the nanocluster were observed and the geometry of the supramolecular adducts resulted in isomerism similar to the octahedral coordination complexes of metals. Water-soluble and red luminescent Ag29(LA)12 (LA is lipoic acid) nanocluster also formed host–guest complexes with cucurbiturils and CDs which resulted in an enhancement in luminescence of the nanoclusters, as shown in Fig. 9D.264 Such luminescent complexes were used for dopamine sensing. Pillar[5]arene-protected nanoclusters, Ag29(LA–P5)12(TPP)2, were reported by Muhammed et al. which formed spherical assemblies with enhanced luminescence.265 The reactivity of the nanoclusters was also reflected in their interaction with nanostructures like Au nanorods and Te nanorods to form a variety of self-assembled hybrid nanostructures (to be discussed later in detail). Recently, Sheng et al. reported the first supramolecular polymorphs of high-nuclearity Ag48 NCs encapsulated in an anionic template via solvent mediation.266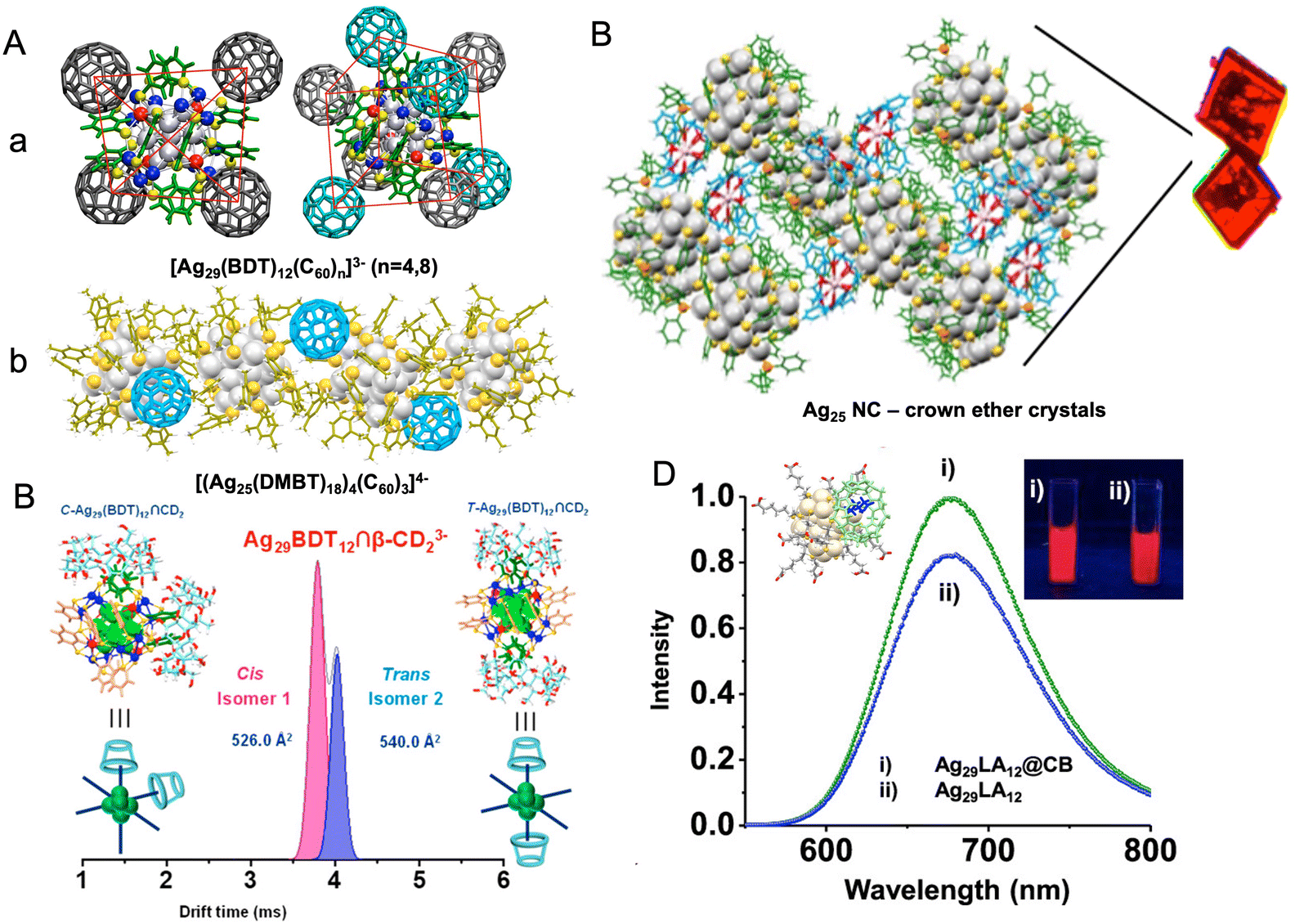 | ||
| Fig. 9 (A) Host–guest complexes of (a) Ag29(BDT)123− and (b) Ag25(DMBT)18− nanocluster with C60, (B) crystal packing of Ag29(BDT)12(TPP)43− nanocluster with 18-crown-6-ether, (C) separation of isomers of the inclusion complex, [Ag29(BDT)12(β-CD)2]3− by ion mobility mass spectrometry, (D) enhancement in emission of Ag29LA12@CB complexes compared with that of Ag29LA12 nanocluster alone. Reproduced with permission from ref. A-a,260 b,261 B,262 C,263 and D.264 Copyright 2018, 2019, and 2020 American Chemical Society. | ||
3.5 Intercluster reactions
Nanoclusters that undergo chemical reactions with one another, also known as intercluster reactions, are a rapidly developing field in nanoscience. Intercluster reactions are now utilized as tools to generate novel hybrid nanoclusters. It is crucial to have an atomic-level understanding of the chemical transformations of nanoclusters in such reactions. In this section, we will be discussing a few such examples of intercluster reactions and their associated mechanisms.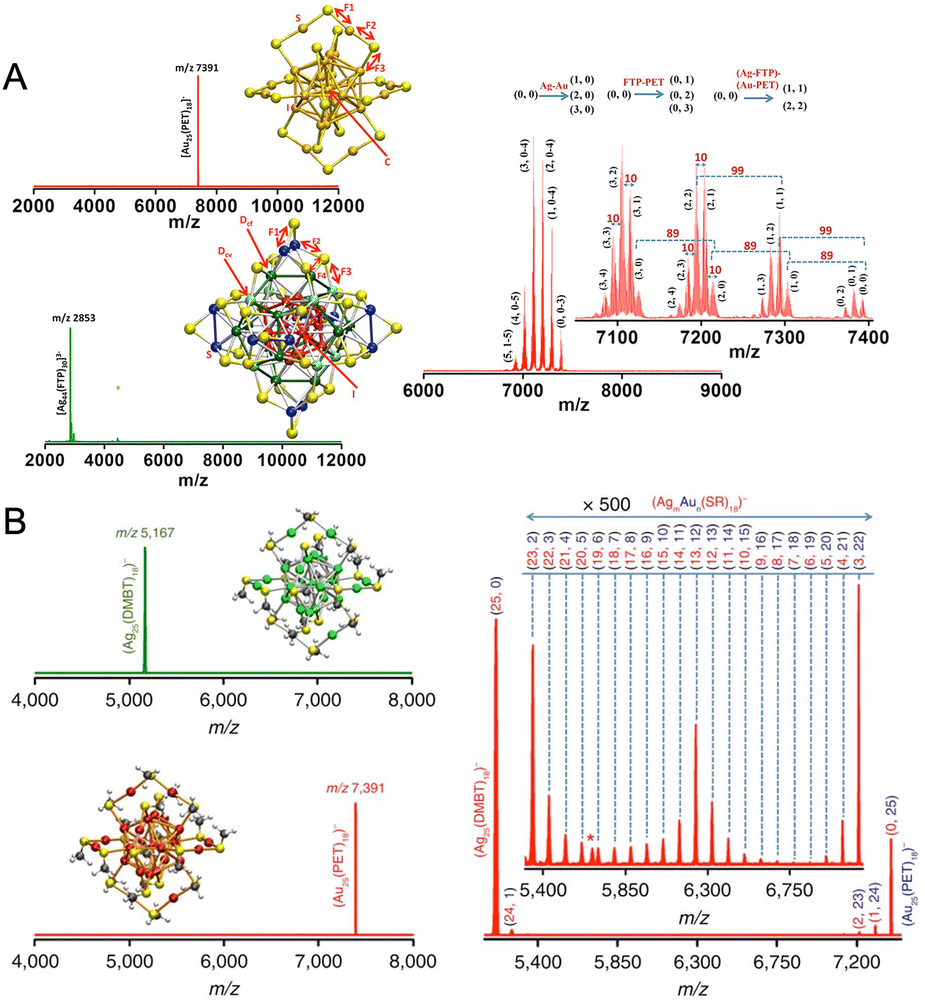 | ||
| Fig. 10 Intercluster reaction of structurally and compositionally (A) different, Au25(PET)18 and Ag44(FTP)30, and (B) analogous Au25(PET)18 and Ag25(DMBT)18 nanoclusters. Adapted from ref. A,54 and B.55 Copyright 2016 American Chemical Society and Springer Nature Group. | ||
Several questions arise, such as (i) how two negatively charged nanoclusters interact despite the electrostatic repulsion and steric hindrance offered by the ligands, (ii) whether the reaction is driven by the entire nanocluster entity or any metal–thiolate fragments, and (iii) whether the reaction involves any intermediate or adduct species. In the next section, we will be discussing answers to a few of the questions, while a few remain unanswered.
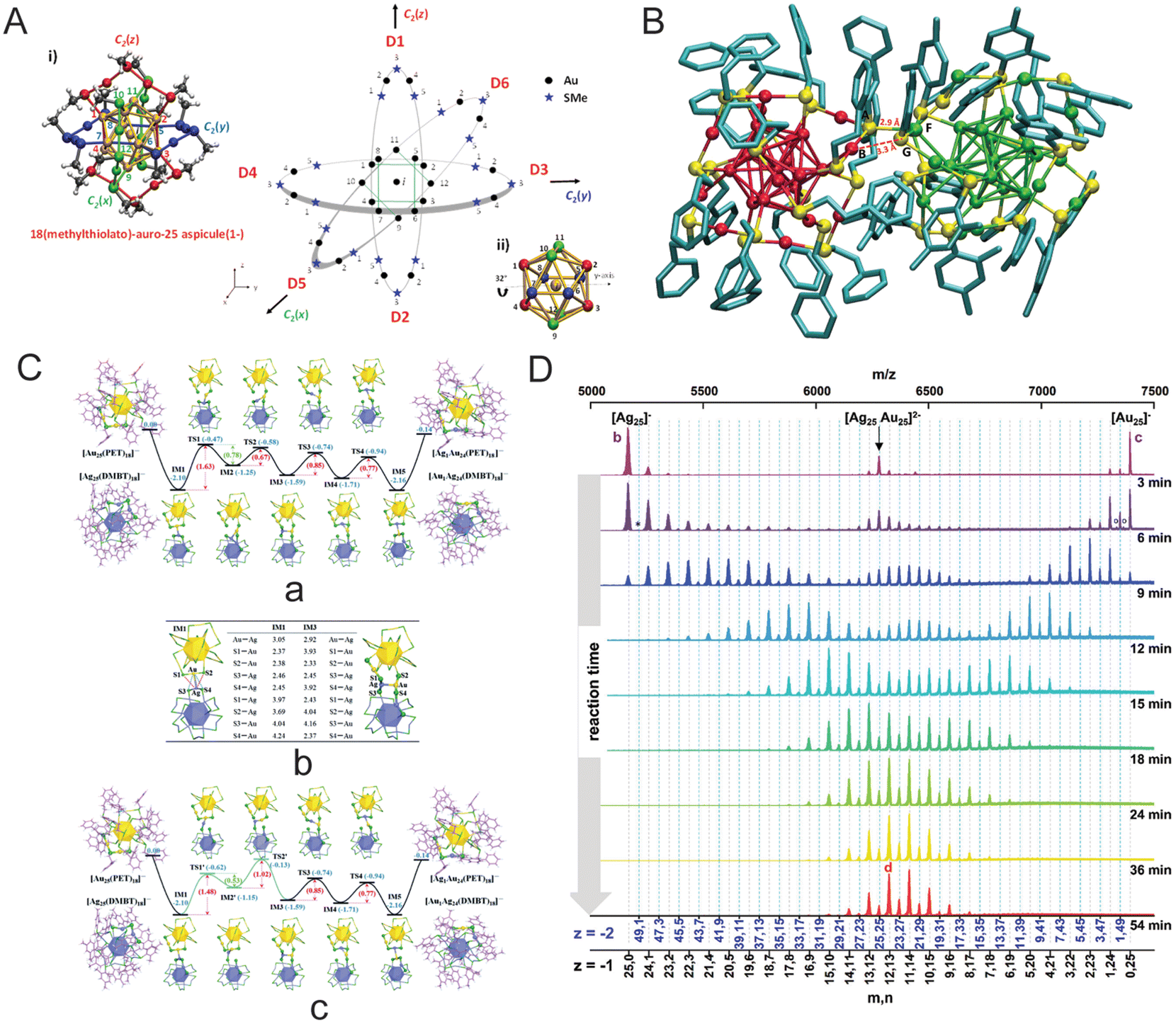 | ||
| Fig. 11 (A) Borromean rings diagram of Au25(SMe)18, (B) DFT-optimization of the structure of [Ag25Au25(DMBT)18(PET)18]2− adduct (with Ag25 on the left and Au25 on the right) as obtained from a force-field-based molecular docking simulation, (C) energy profiles of the metal exchange reaction between the [Au25(PET)18]− and [Ag25(DMBT)18]− nanoclusters; (a) path 1 initiated by Au–SPET bond breaking, (b) DFT optimized metal–metal and metal–S bond lengths (in Angstrom) (c) path 2 initiated by Ag–SDMBT bond breaking, and (D) time-dependent mass spectra of a 1:1 mixture of [Au25(PET)18]− and [Ag25(DMBT)18]−. Reproduced with permissions from ref. A–B,166 C,270 and D.271 Copyright 2015 and 2021 American Chemical Society. Copyright 2016, The Royal Chemical Society. | ||
To study the mechanism of intercluster reactions, one needs to understand the role played by the metal–ligand interface in such reactions. The intercluster reaction is a redox-like reaction triggered by the difference in oxidation states of the metal atoms present in the core and staple. For example, let us consider the intercluster reaction between Ag25(SR)18 and Au25(SR)18.55 Here, an Ag25(SR)18 molecule reacts with the Au2(SR)3 staples of Au25(SR)18, wherein Au in the Au2(SR)3 staples is in the +1 oxidation state. Similarly, an Au25(SR)18 molecule reacts with the Ag2(SR)3 staples of Ag25(SR)18, wherein Ag from the Ag2(SR)3 staple is in the +1 oxidation state. Such redox reactions between M25(SR)18 and M(I) thiolates, where M = Ag/Au, are well studied,267,268 although it is still unclear how this difference in oxidation states contributes to the chemical reaction. Next, the intercluster reaction was studied for two entirely different nanoclusters, Au25(SR)18 and Ag44(SR)30, that resulted in the formation of reactive fragments like Ag(SR)2−, which further reacts with the Au2(SR)3 staples of Au25(SR)18, resulting in an exchange of metal atoms, ligands, and metal–ligand fragments.54 In conclusion, the stability and chemical reactivity of these nanoclusters are characteristics of the nature of the ligand and the bonding present in the metal–ligand oligomeric units.58
New insights into the intercluster reactions of Au25(PET)18 and Ag25(DMBT)18 came up with the detection of [Ag25Au25(DMBT)18(PET)18]2− adduct species.55 Detection of such species indicates a possible pathway involving the formation of an adduct intermediate when two intact nanoclusters participate in these ‘bimolecular’ reactions. Using density functional theory (DFT), the Ag–S bond between the staples of the nanoclusters is observed in an optimized adduct structure (Fig. 11B). Computational studies suggest an interaction at the metal–ligand interface for the reacting nanoclusters at the early stages of the reaction. Furthermore, no metallic exchange was detected when nanoclusters, AgxAu38−x(SR)24 and Au38(SR)24, were separated by a dialysis membrane, and that suggests an intercluster collision as the origin of such reactions.269
In this section, some more mechanistic insights into the intercluster reactions are presented. More studies are needed to understand how two negatively charged nanoclusters collide, overcoming the electrostatic repulsion. One possible explanation for this could be coming from the fact that the anionic nanoclusters are not point charges; the overall negative charge is diffused over the entire nanocluster entity. At the early stages of the reaction, the intercluster interaction could lead to collisions, electron transfer, etc., resulting in nanocluster destabilization. The destabilization of the nanocluster eventually may lead to ring opening (refer to the Borromean ring model) followed by it taking up a flexible elongated conformation, allowing atoms to interact freely with other nanoclusters. At this stage, the nanoclusters with open rings can interact more easily and undergo metal, ligand, and metal–ligand fragment exchanges. The Borromean ring model of Au25(SR)18 suggests the spontaneous inclusion of Ag-atom in the nanocluster core as it is not sterically hindered. Theoretical calculations were performed by Huang et al. to understand the intercluster exchange reaction mechanism between Au25(SR)18 and Ag25(SR)18 (Fig. 11C).270 As per calculations, the intercluster reactions are a two-step mechanism: (i) dianionic adduct [Au25Ag25(PET)18(DMBT)18]2− intermediate formation followed by metal–ligand staple rearranges to facilitate metal exchange, and (ii) then the heterometal atom in the staple swaps with the metal atom present in the icosahedral M13-kernel. Recently, Neumaier et al. reported a detailed experimental and computational study of the intercluster reaction kinetics of Au25(PET)18 and Ag25(DMBT)18 at room temperature.271 During the reaction, the participation of both nanocluster monomer and dimers were observed in mass spectral and collision-induced dissociation (CID) measurements. For an equimolar concentration of nanoclusters, time-dependent mass spectra show a sufficient abundance of both monomers and dimers along with continuous change in overall Ag:Au compositions until a dynamic equilibrium is achieved (Fig. 11D). The kinetic model suggested a three-step reaction route involving dimerization of monomers, metal atom exchange in the transient dimer, and dimer dissociation.
Chakraborty et al. reported isotopic metal exchanges in nanoclusters, which provided further insights into the mechanism of atom transfer in NPs. Two isotopic Ag25(DMBT)18− nanoclusters, made from 107Ag and 109Ag, reacted spontaneously in solution to form an isotopically mixed nanocluster (Fig. 12A).272 Such isotopic exchanges were similar to isotopic exchanges in H2O/D2O, further supporting the molecular nature of the nanoclusters. The exchange was rapid and occurred within seconds after mixing the solutions at room temperature. The exchange could be controlled by controlling the temperature. The exchange kinetics was better studied using a more rigid nanocluster system, Ag29(BDT)12(TPP)43−, which exhibited slower exchange rates. Similar isotopic 107Ag/109Ag atom exchange was also observed in this case, and a time-resolved study revealed that the process involved an initial fast reaction rate, probably arising from atom exchanges in the staples, followed by a slow reaction rate arising from the diffusion of atoms from staple to core and a final state where the exchanged atoms rearrange until they attain the thermodynamic equilibrium state (Fig. 12B). Such an exchange process was driven by the entropy of mixing. These results suggested the dynamic nature of the metal atom transfer in nanoclusters in solution. The dynamic nature of the ligand monolayers was also reported in a study by Salassa et al.273 The metal–ligand interface also plays an important role in controlling atom transfer and intercluster reactions of nanoclusters.
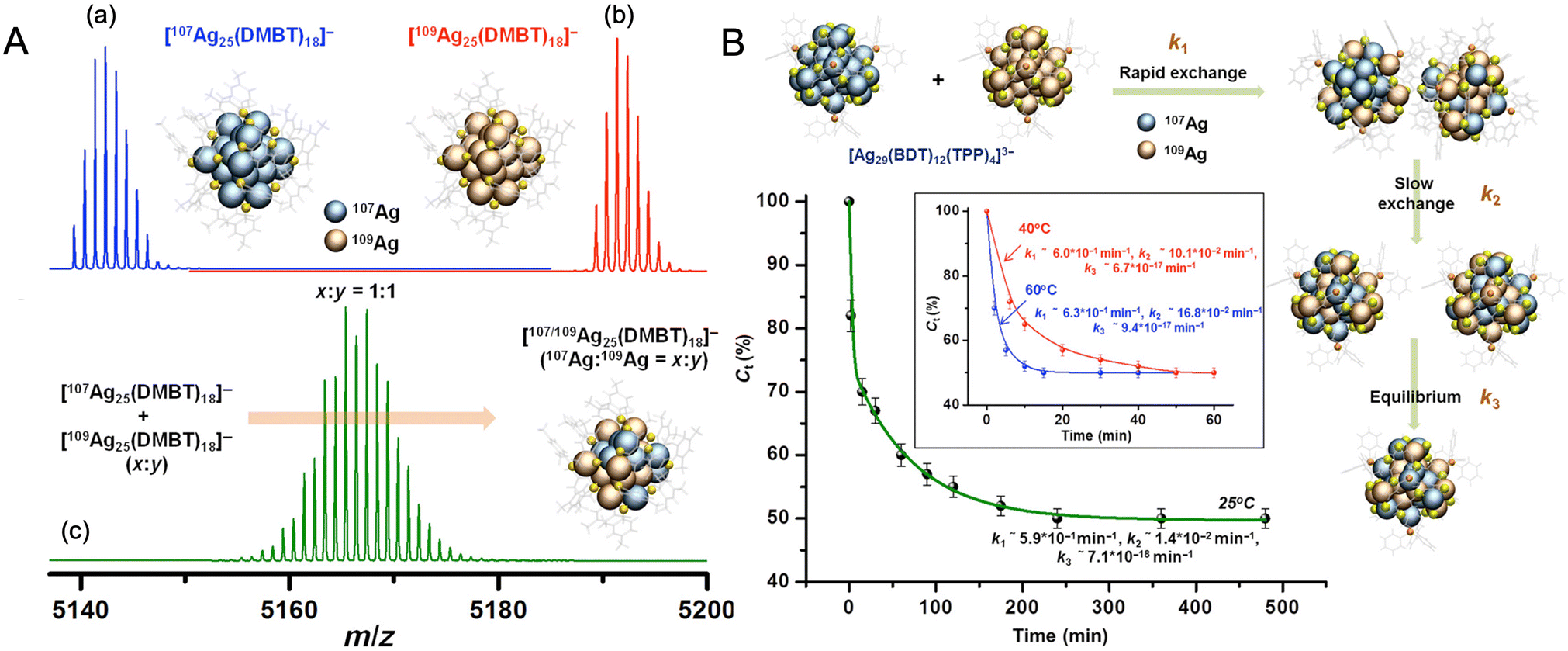 | ||
| Fig. 12 (A) ESI MS of (a) [107Ag25(DMBT)18]−, (b) [109Ag25(DMBT)18]−, and (c) isotopically mixed [Ag25(DMBT)18]− nanocluster with 107Ag:109Ag ratio of 1:1. (B) Kinetics of isotopic Ag atom exchange in [Ag29(BDT)12(TPP)4]3− nanocluster. Adapted from ref. 272. Copyright 2019 American Association for the Advancement of Science. | ||
3.6. Interparticle reactions: reactions with higher dimension materials
In 2014, Ghosh et al. made the first attempt to study the chemical interactions between atomically precise nanoclusters with other nanomaterials.274 In this study, graphene reacted with Au25(PET)18, resulting in the formation of a larger nanocluster, Au135(PET)57. The conversion of Au25(PET)18 nanocluster was monitored using time-dependent matrix-assisted laser desorption ionization mass spectrometry (MALDI MS) (Fig. 13A). As the reaction progressed, the nanocluster peak at m/z 7391 due to Au25(PET)18 gradually disappeared with the simultaneous evolution of the peak at m/z 34.4 kDa corresponding to Au135(PET)57. This conversion process is driven by an overall energy gain of the system due to the entrapment of smaller nanoclusters at the local valleys of the graphenic surface resulting in the reduction in surface curvature and, finally, leading to coalescence. This study gave insights into the utility of surface as a reactive substrate for the chemical transformations of ligand-protected metal nanoclusters.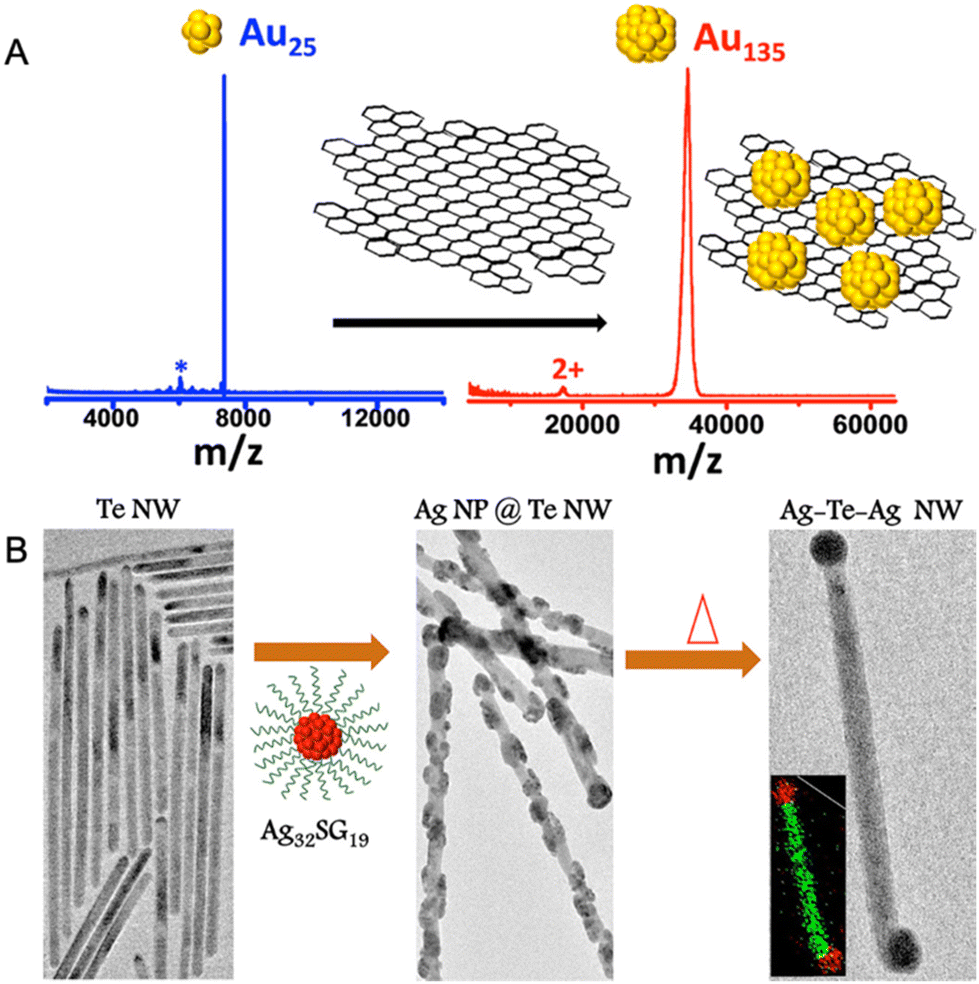 | ||
| Fig. 13 (A) MALDI MS study for the conversion of Au25(PET)18 to Au135(PET)57 entrapped on the on the graphene surface upon reaction with graphene. (B) HRTEM images and corresponding schematic representation of pure Te NW upon reaction with Ag32SG19 and upon further heating yielding Ag nodule-decorated Te NW and dumbbell-shaped Ag–Te–Ag NWs, respectively. Adapted from ref. 274 and 275 for A and B, respectively. Copyright 2014 American Chemical Society. | ||
Unique reactivity of water-soluble Au32SG19 nanocluster to Te nanowires (NWs) was reported by Som et al.275 This reaction results in the formation Ag–Te hybrid NWs with the growth of nodule-shaped Ag NPs on the NW surface (Fig. 13B). Structural analysis of the modified Te NWs using HRTEM-EDS and XRD confirmed the presence of Ag as nodules with Te NW retaining its inherent (001) hexagonal structure. Furthermore, on heating the Ag-decorated Te NWs, the morphology evolves into nano dumbbell-shaped Ag–Te–Ag NWs with Ag NPs specifically located at the tips (Fig. 13B). The ultrasmall size of the Ag32SG19 nanocluster provides an increased surface free energy, thereby inducing a tendency of intercluster coalescence, resulting in bigger particles.
Ligand-protected metal nanoclusters make excellent building blocks to create self-assembled hierarchical frameworks.192,276,277 An interesting phenomenon of self-assembly arises in Te NWs when the surface is modified with Ag44(p-MBA)30 nanocluster. The p-MBA ligand shells initiate a H-bonding interaction among themselves, leading to the formation of a bilayer assembly of NWs oriented at an angle of 81° w.r.t. each other (Fig. 14A).278 Nonappa et al. reported the unique ability of p-MBA-protected gold nanoclusters to undergo intercluster H-bonding resulting in monolayer-thick 2D nanosheets and spherical capsids.276,279 Som et al. showed that the Na4Ag44-pMBA30 nanoclusters can be self-assembled into a large-area freestanding elastic membrane via entrapping them in a transient solvent layer at the air–water interface.280 The patchy distribution of ligands around the metal core facilitates symmetry breaking and eventually directs a preferential interlayer H-bonding between the carboxylic acid groups of the p-MBA ligands.276,277,279 Chakraborty et al. showed hydrogen bonding-induced chemical interaction between the Ag44(p-MBA)30 nanocluster and a plasmonic gold nanorod (GNR), leading to the encapsulation of the latter (Fig. 14B), here denoted as GNR@Ag44.281 The nanocage-like hybrid material was found to have an octahedral morphology as studied using a series of highly sophisticated microscopes, like transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and 3D reconstruction using electron tomography. The anisotropic growth was credited to the preferential anchoring of the nanoclusters to Au 〈110〉 over Au 〈100〉 facet of GNR@p-MBA. Ag25(DMBT)18 nanocluster-mediated site-selective etching of anisotropic planar gold nanotriangles (NTs) was reported.282 Due to differential surface energies, the Au NTs interacting with the nanocluster underwent metallic etching at the edges and doping at the tips while the core remained unaltered. Roy et al. reported a polydispersed CuO NP and Au25(PET)18 reaction-mediated formation of spherical-shaped nanoaggregates of Cu-doped-Au-nanoclusters (Fig. 14C).283 The reaction involved an NP–NC atom transfer reaction route. Rival et al. recently demonstrated that the thiolated azobenzene-protected Au25 nanoclusters form reversible assemblies triggered by light.284 With the right choice of nanocluster system, such hybrid nanomaterials having extraordinary stability at room temperature can significantly improve the limit of detection of nanocluster-based sensors.
 | ||
| Fig. 14 (A) Composite Ag44@Te NW bilayer oriented at an 81° angle w.r.t to each other and (B) its corresponding packing driven by the intercluster H-bonding. (C) Schematic representation and HRTEM micrographs illustrating the assembly of Ag44 on the GNR. Adapted from ref. A,278 B,281 and C.283 Copyright 2014 and 2018, John Wiley and Sons. Copyright 2023, The Royal Society of Chemistry. | ||
Bose et al. extended the interparticle chemistry to the reactions involving isotropic Ag NPs.285 A spontaneous reaction between Au25(PET)18 nanocluster and polydispersed Ag NPs with a core diameter of 4.4 ± 2.3 nm, protected with 2-phenylethanethiol (PET), resulted in monodispersed alloy NPs with a core diameter of 3.4 ± 1.2 nm under ambient conditions. The resulting NPs also underwent a spontaneous self-assembly to form a 2D superlattice which was analysed using HRTEM. Using STEM-EDS analysis, the reacted NPs were found to be Ag–Au alloy NPs. A 3D reconstruction of the 2D assembly using electron tomography further revealed that the assembly was composed of reacted alloy NPs arranged in a hexagonal close-packed (hcp) lattice with an interparticle distance of ∼4.5 nm (Fig. 15A). The mechanism involved an interparticle atomic exchange (metal–ligand species), and the metal–ligand interface was found to be crucial in controlling the reaction. Systematic analysis using time-dependent ESI MS showed that the reaction proceeds through a transient Au25−xAgx(PET)18 (x = 1, 2, 3,…,) species along with other alloy nanocluster intermediates. Similar reactions were performed for Au25(PET)18 nanocluster with differently sized Ag@PET NPs, in which the interparticle reactivity was enhanced upon decreasing the size of NPs. The nanocluster–NP reactions can thereby open up an entirely new way of generating alloy NPs in the solution phase with better control over the NP size distribution.
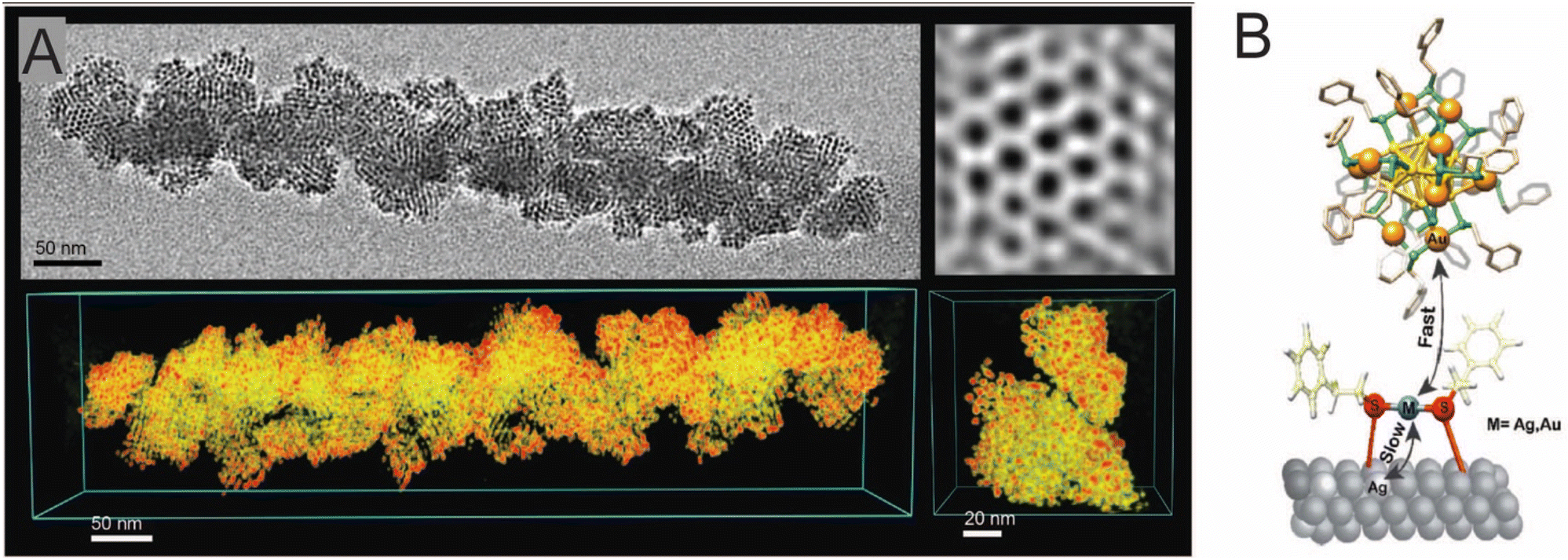 | ||
| Fig. 15 (A) TEM micrograph, its corresponding 3D reconstruction, and inverse fast Fourier transform image of 2D superlattice assembly (hcp) of the Au–Ag alloy NP resulting from the NP–nanocluster reaction. (B)The number of Ag-doped Au nanocluster versus time plot for the nanocluster–Ag surface reaction and the schematic representation metallic exchange at the metal–ligand interface on the bulk surface. Reproduced with permission A285 and B.288 Copyright 2020 Royal Society of Chemistry. Copyright 2008 American Chemical Society. | ||
As we move from noble metal nanosystems to their bulk counterparts, chemical properties are altered due to changes in energy levels. Baksi et al. explored the glucose-mediated extraction of Ag from bulk silver surfaces otherwise considered inert.286 Later, Nag et al. reported the solution phase synthesis of Ag nanoclusters from bulk metallic silver in the presence of carbohydrate and glutathione followed by chemical reduction.287 Kazan et al. utilized the bulk thiolated silver surface to understand intercluster reaction as an interfacial phenomenon of metal–ligand exchange.288 The study involved Au25(PET)18 and Au38(PET)24 nanoclusters, and pure silver foil as model systems. MALDI MS and XPS characterization techniques were used to study the effect of reaction on the nanocluster and foil, respectively. Upon time-dependent monitoring of the Au25(PET)18 and PET-monolayered Ag foil, Au25−xAgx(PET)18 (x = 1–4) appeared just after 2 min and a higher Ag-doping was detected for a longer reaction time. With the neat Ag foil, the reaction was found to be slow, as Au24Ag(PET)18 appeared only after 3 h. Hence, the kinetics of pre-adsorbed and neat silver foils are very different. The average number of doped Ag plotted as a function of time shows that the substitution follows a 2-phase kinetics (initially fast followed by slower exchange) and a sigmoidal trend (initially delayed followed by faster exchange) in preadsorbed and neat foils, respectively. This sigmoidal kinetics can be related to the autocatalytic reactions where the starting 3 h is an induction period for the thiolate, here acting as a catalyst, to deposit on the surface. As the time progressed, thiolate deposition happened, and this is reflected with an increased reaction rate. The mechanism behind the 2-phase kinetics in an atomic exchange reaction was explained using the scheme in which thiolated-Ag on the surface exchanges faster in the early stages of the reaction (Fig. 15B). At the later stages of the reaction, kinetics become slower due to less availability of exchangeable sites on the Ag foil surface. XPS measurements of the reacted Ag foil surface showed the presence of the metallic Au. Similar trends were observed when the Au38(PET)18 nanocluster reacts with both the pre-adsorbed and neat Ag surfaces. XPS measurements of the reacted pre-adsorbed thiolated and neat foils also confirmed the presence of metallic Au. The Au25(PET)18 and Au38(PET)18 reaction was also explored with pre-adsorbed and free surface of other metals like Cd and Cu. This study concluded that for a feasible Au doping in the Ag nanocluster, thiol plays a key role. Recently, Chakraborty et al. reported the dynamics of isotopic exchange reactions of isotopically pure Ag nanocluster with different dimensions of metallic Ag, like nanoclusters, plasmonic NPs, and bulk.289 Isotopically pure 107Ag25(DMBT)18 and 108Ag25(DMBT)18 was reacted with different sizes of Ag@DMBT NPs (naturally abundant Ag). With the increase in the NP size, the rate of atomic exchange was reduced. The exchange rate further decreased when the nanocluster was reacted with bulk Ag samples, such as foil and micron-sized powder. The kinetics of isotopic exchange, i.e., reaction timescales, was analyzed by fitting the reactant concentration as a function of time to a reaction model. Under similar reaction conditions, the reaction timescale was longer for the nanocluster-NP reactions compared with intercluster reactions. This suggests that suchreactions can be controlled by careful engineering the reacting nanostructures.
4. Insights from nanocluster chemistry
From the results presented, we list below the factors determining the feasibility of reactions of nanoclusters.(1) The metal–ligand interface primarily controls the atomic exchange reactions and their feasibility.
(2) The geometry and stereochemistry of the protecting groups play crucial roles in the intercluster and interparticle reactions.
(3) Thermodynamics drives the interparticle and isotopic exchange reactions.
(4) Entropy of mixing drives the isotopic metal exchange in intercluster reactions.
(5) Nanoclusters exhibit redox-like reactions triggered by the difference in oxidation states of the metal atoms present in the core and staple.
(6) The core–shell geometry of a nanocluster influences the nanocluster–analyte reaction primarily via weak interactions, such as metallophilic and supramolecular interactions.
(7) Supramolecular interactions like C–H⋯π, π⋯π, van der Waals, hydrogen bonding, and electrostatic interactions guide the formation of the nanocluster assemblies.
(8) Reactive intermediates, such as adducts and fragments, vary depending on the reacting species.
(9) LEIST, a method to prepare new nanoclusters, is a ligand-controlled phenomenon. The rate of ligand exchange depends also on the electron-donating or withdrawing nature of the ligand.
(10) Chemical reactions occur between atomically precise nanoclusters and a range of systems such as ions, clusters, NPs, and bulk metals, and therefore we may suggest that nanoclusters are reactive to the whole range of chemical systems.
(11) The chemistry is highly sensitive to reaction conditions such as concentration, time, and temperature, and is expected to be influenced by other factors such as the medium, ionic strength, etc.
5. Conclusions and future perspectives
Nanoclusters, with their inherent molecule-like properties, show a wide range of chemical reactions with counterparts such as metal ions, clusters, NPs and bulk metals. These chemical reactions yield well-defined alloys which may be nanoclusters, NPs or bulk materials, depending on the systems involved. Such reactions also lead to ligand exchange. The processes yield supramolecular interactions forming assemblies and superstructures. Thermodynamics and kinetics govern the chemistry, and the underlying processes can be modelled with greater accuracy. The chemistry is sensitive to various process conditions such as concentration, temperature, solvent, etc., as typical of molecular events. The products formed and their kinetics and dynamics confirm their molecular nature. In the coming years, such cluster chemistry of atomically precise metal nanoclusters will be explored with oxides, sulfides, and others in the form of clusters, NPs, and bulk materials, yielding new materials. Similar science will be possible between materials of different size regimes of oxides and sulfides themselves, expanding the diversity of the area. The science presented will be greatly influenced by the experimental and computational methodologies used, which can reveal the intricate details of the processes involved. The science at this stage has provided us with many insights into the phenomena at the nanoscale as revealed by the fast isotopic exchange between NPs. Applications of such science are still at infancy.Author contributions
All authors contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Department of Science and Technology (DST), Government of India, and Centre of Excellence program of the Indian Institute of Technology Madras, on the theme of Molecular Materials and Functions, under the Institutions of Eminence of Ministry of Education, India, for supporting our research.References
- R. Feynman, Feynman And Computation, CRC Press, 2002 Search PubMed.
- F. A. Cotton and J. T. Mague, Inorg. Chem., 1964, 3, 1094–1098 CrossRef CAS.
- F. A. Cotton, Inorg. Chem., 1964, 3, 1217–1220 CrossRef CAS.
- R. W. Murray, Chem. Rev., 2008, 108, 2688–2720 CrossRef CAS PubMed.
- G. Chen, J. Seo, C. Yang and P. N. Prasad, Chem. Soc. Rev., 2013, 42, 8304–8338 RSC.
- H. Zhu, Y. Yang and T. Lian, Acc. Chem. Res., 2013, 46, 1270–1279 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- T. Tsukuda and H. Hakkinen, Protected Metal Clusters: From Fundamentals to Applications, 2013, vol. 53 Search PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- V. G. Albano, P. L. Bellon, M. Manassero and M. Sansoni, J. Chem. Soc. D, 1970, 1210–1211 RSC.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- Atomically precise metal clusters, ed. T. Pradeep, Elsevier, 1st edn, 2023 Search PubMed.
- S. S. Narayanan and S. K. Pal, J. Phys. Chem. C, 2008, 112, 4874–4879 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- J. Yu, S. Choi and R. M. Dickson, Angew. Chem., Int. Ed., 2009, 48, 318–320 CrossRef CAS.
- P. L. Xavier, K. Chaudhari, A. Baksi and T. Pradeep, Nano Rev., 2012, 3, 14767 CrossRef CAS.
- X. Wen, P. Yu, Y. R. Toh, A. C. Hsu, Y. C. Lee and J. Tang, J. Phys. Chem. C, 2012, 116, 19032–19038 CrossRef CAS.
- D. M. Chevrier, V. D. Thanthirige, Z. Luo, S. Driscoll, P. Cho, M. A. MacDonald, Q. Yao, R. Guda, J. Xie, E. R. Johnson, A. Chatt, N. Zheng and P. Zhang, Chem. Sci., 2018, 9, 2782–2790 RSC.
- T. Vosch, Y. Antoku, J. C. Hsiang, C. I. Richards, J. I. Gonzalez and R. M. Dickson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616–12621 CrossRef CAS PubMed.
- H. C. Yeh, J. Sharma, J. J. Han, J. S. Martinez and J. H. Werner, Nano Lett., 2010, 10, 3106–3110 CrossRef CAS PubMed.
- J. T. Petty, B. Sengupta, S. P. Story and N. N. Degtyareva, Anal. Chem., 2011, 83, 5957–5964 CrossRef CAS PubMed.
- J. Sharma, H. C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2011, 47, 2294–2296 RSC.
- E. Gwinn, D. Schultz, S. M. Copp and S. Swasey, Nanomaterials, 2015, 5, 180–207 CrossRef PubMed.
- D. J. E. Huard, A. Demissie, D. Kim, D. Lewis, R. M. Dickson, J. T. Petty and R. L. Lieberman, J. Am. Chem. Soc., 2019, 141, 11465–11470 CrossRef CAS.
- L. A. Peyser, A. E. Vinson, A. P. Bartko and R. M. Dickson, Science, 2001, 291, 103–106 CrossRef CAS PubMed.
- L. A. Peyser, T. H. Lee and R. M. Dickson, J. Phys. Chem. B, 2002, 106, 7725–7728 CrossRef CAS.
- J. Zheng and R. M. Dickson, J. Am. Chem. Soc., 2002, 124, 13982–13983 CrossRef CAS PubMed.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed , 8AD.
- M. C. Paau, C. K. Lo, X. Yang and M. M. F. Choi, J. Phys. Chem. C, 2010, 114, 15995–16003 CrossRef CAS.
- E. S. Shibu and T. Pradeep, Chem. Mater., 2011, 23, 989–999 CrossRef CAS.
- A. George, E. S. Shibu, S. M. Maliyekkal, M. S. Bootharaju and T. Pradeep, ACS Appl. Mater. Interfaces, 2012, 4, 639–644 CrossRef CAS.
- M. Mayer, M. Rohdenburg, V. van Lessen, M. C. Nierstenhöfer, E. Aprà, S. Grabowsky, K. R. Asmis, C. Jenne and J. Warneke, Chem. Commun., 2020, 56, 4591–4594 RSC.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- H. W. Kroto, A. W. Allaf and S. P. Balm, Chem. Rev., 1991, 91, 1213–1235 CrossRef CAS.
- J. Almutlaq, J. Yin, O. F. Mohammed and O. M. Bakr, J. Phys. Chem. Lett., 2018, 9, 4131–4138 CrossRef CAS PubMed.
- Y. Yan, J. Gong, J. Chen, Z. Zeng, W. Huang, K. Pu, J. Liu and P. Chen, Adv. Mater., 2019, 31, 1808283 CrossRef.
- R. B. Wyrwas, M. M. Alvarez, J. T. Khoury, R. C. Price, T. G. Schaaff and R. L. Whetten, Eur. Phys. J. D, 2007, 43, 91–95 CrossRef CAS.
- H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847–1859 RSC.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 99–104 CrossRef CAS.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- Q. Tang, G. Hu, V. Fung and D. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS.
- R. S. Ingram, M. J. Hostetler, R. W. Murray, T. G. Schaaff, J. T. Khoury, R. L. Whetten, T. P. Bigioni, D. K. Guthrie and P. N. First, J. Am. Chem. Soc., 1997, 119, 9279–9280 CrossRef CAS.
- D. Lee, R. L. Donkers, J. M. DeSimone and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 1182–1183 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- D. Jiang, M. L. Tiago, W. Luo and S. Dai, J. Am. Chem. Soc., 2008, 130, 2777–2779 CrossRef CAS.
- P. Yu, X. Wen, Y.-R. Toh, X. Ma and J. Tang, Part. Part. Syst. Charact., 2015, 32, 142–163 CrossRef CAS.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS PubMed.
- K. R. Krishnadas, A. Ghosh, A. Baksi, I. Chakraborty, G. Natarajan and T. Pradeep, J. Am. Chem. Soc., 2016, 138, 140–148 CrossRef CAS PubMed.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS PubMed.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- K. R. Krishnadas, G. Natarajan, A. Baksi, A. Ghosh, E. Khatun and T. Pradeep, Langmuir, 2019, 35, 11243–11254 CrossRef CAS PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- S. Biswas, A. K. Das and S. Mandal, Acc. Chem. Res., 2023, 56, 1838–1849 CrossRef CAS PubMed.
- Y. Xiao, Z. Wu, Q. Yao and J. Xie, Aggregate, 2021, 2, 114–132 CrossRef CAS.
- Y. Negishi, H. Tsunoyama, M. Suzuki, N. Kawamura, M. M. Matsushita, K. Maruyama, T. Sugawara, T. Yokoyama and T. Tsukuda, J. Am. Chem. Soc., 2006, 128, 12034–12035 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 CrossRef CAS PubMed.
- S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, J. Am. Chem. Soc., 2013, 135, 15585–15594 CrossRef CAS PubMed.
- M. Agrachev, S. Antonello, T. Dainese, J. A. Gascón, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, Chem. Sci., 2016, 7, 6910–6918 RSC.
- M. Agrachev, S. Antonello, T. Dainese, M. Ruzzi, A. Zoleo, E. Aprà, N. Govind, A. Fortunelli, L. Sementa and F. Maran, ACS Omega, 2017, 2, 2607–2617 CrossRef CAS.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed.
- M. Galchenko, A. Black, L. Heymann and C. Klinke, Adv. Mater., 2019, 31, 1–6 CrossRef PubMed.
- P. Yuan, R. Zhang, E. Selenius, P. Ruan, Y. Yao, Y. Zhou, S. Malola, H. Häkkinen, B. K. Teo, Y. Cao and N. Zheng, Nat. Commun., 2020, 11, 2229 CrossRef CAS.
- G. Zhang, Y. Li, J. Xu, C. Zhang, S. Shuang, C. Dong and M. M. F. Choi, Sens. Actuators, B, 2013, 183, 583–588 CrossRef CAS.
- X. Yuan, Y. Tay, X. Dou, Z. Luo, D. T. Leong and J. Xie, Anal. Chem., 2013, 85, 1913–1919 CrossRef CAS PubMed.
- X. Chen, J. B. Essner and G. A. Baker, Nanoscale, 2014, 6, 9594–9598 RSC.
- A. Mathew and T. Pradeep, Part. Part. Syst. Charact., 2014, 31, 1017–1053 CrossRef CAS.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909–1930 RSC.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- S. Choi, R. M. Dickson and J. Yu, Chem. Soc. Rev., 2012, 41, 1867–1891 RSC.
- X. R. Song, N. Goswami, H. H. Yang and J. Xie, Analyst, 2016, 141, 3126–3140 RSC.
- L. Zhang and E. Wang, Nano Today, 2014, 9, 132–157 CrossRef CAS.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS.
- P. Liu, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2017, 139, 2122–2131 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Chem. Rev., 2023, 123, 5948–6002 CrossRef CAS PubMed.
- A. Henglein, J. Phys. Chem., 1979, 83, 2209–2216 CrossRef CAS.
- A. Henglein and R. Tausch-Treml, J. Colloid Interface Sci., 1981, 80, 84–93 CrossRef CAS.
- A. Henglein, J. Phys. Chem., 1993, 97, 5457–5471 CrossRef CAS.
- A. Henglein, Chem. Rev., 1989, 89, 1861–1873 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- P. Chakraborty and T. Pradeep, NPG Asia Mater., 2019, 11, 48 CrossRef.
- W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Y. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141–2143 CrossRef CAS.
- R. E. Leuchtner, A. C. Harms and A. W. Castleman Jr., J. Chem. Phys., 1989, 91, 2753–2754 CrossRef CAS.
- D. E. Bergeron, A. W. Castleman, T. Morisato and S. N. Khanna, Science, 2004, 304, 84–87 CrossRef CAS.
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS PubMed.
- T. P. Martin, Phys. Rep., 1996, 273, 199–241 CrossRef CAS.
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2002, 116, 4094–4101 CrossRef CAS.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982–6990 CrossRef CAS.
- A. Lechtken, C. Neiss, M. M. Kappes and D. Schooss, Phys. Chem. Chem. Phys., 2009, 11, 4344–4350 RSC.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403 CrossRef CAS PubMed.
- Z. Li, H. Y. T. Chen, K. Schouteden, T. Picot, T. W. Liao, A. Seliverstov, C. Van Haesendonck, G. Pacchioni, E. Janssens and P. Lievens, Sci. Adv., 2020, 6, eaay4289 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. McPartlin, R. Mason and L. Malatesta, J. Chem. Soc. D, 1969, 334–334 RSC.
- B. K. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2745 CrossRef CAS.
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis and J. W. A. van der Velden, Chem. Ber., 1981, 114, 3634–3642 CrossRef CAS.
- N. Jian, C. Stapelfeldt, K. J. Hu, M. Fröba and R. E. Palmer, Nanoscale, 2015, 7, 885–888 RSC.
- T. G. M. M. Kappen, P. P. J. Schlebos, J. J. Bour, W. P. Bosman, J. M. M. Smits, P. T. Beurskens and J. J. Steggerda, Inorg. Chem., 1994, 33, 754–758 CrossRef CAS.
- M. Bodiuzzaman and T. Pradeep, in Atomically Precise Metal Nanoclusters, ed. T. Pradeep, Elsevier, 2023, pp. 271–298 Search PubMed.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed.
- T. Carducci and R. Murray, in Nanoelectrochemistry, ed. S. A. Michael and V. Mirkin, CRC Press, Boca Raton, 2015, pp. 73–124 Search PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem. B, 2006, 110, 9927–9931 CrossRef PubMed.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- S. C. K. Hau, P.-S. Cheng and T. C. W. Mak, J. Am. Chem. Soc., 2012, 134, 2922–2925 CrossRef CAS PubMed.
- Z. Wang, H. F. Su, X. P. Wang, Q. Q. Zhao, C. H. Tung, D. Sun and L. S. Zheng, Chem. – Eur. J., 2018, 24, 1640–1650 CrossRef CAS PubMed.
- Z. Wang, J. W. Liu, H. F. Su, Q. Q. Zhao, M. Kurmoo, X. P. Wang, C. H. Tung, D. Sun and L. S. Zheng, J. Am. Chem. Soc., 2019, 141, 17884–17890 CrossRef CAS PubMed.
- Z. Wang, Y. J. Zhu, Y. Z. Li, G. L. Zhuang, K. P. Song, Z. Y. Gao, J. M. Dou, M. Kurmoo, C. H. Tung and D. Sun, Nat. Commun., 2022, 13, 1802 CrossRef CAS.
- Z. G. Jiang, K. Shi, Y. M. Lin and Q. M. Wang, Chem. Commun., 2014, 50, 2353–2355 RSC.
- S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784 CrossRef CAS PubMed.
- K. M. Harkness, D. E. Cliffel and J. A. McLean, Analyst, 2010, 135, 868–874 RSC.
- T. Chen, Q. Yao, R. R. Nasaruddin and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 11967–11977 CrossRef CAS PubMed.
- B. L. Nannenga and T. Gonen, Emerging Top. Life Sci., 2018, 2, 1–8 CrossRef CAS PubMed.
- S. Vergara, D. A. Lukes, M. W. Martynowycz, U. Santiago, G. Plascencia-Villa, S. C. Weiss, M. J. de la Cruz, D. M. Black, M. M. Alvarez, X. López-Lozano, C. O. Barnes, G. Lin, H. C. Weissker, R. L. Whetten, T. Gonen, M. J. Yacaman and G. Calero, J. Phys. Chem. Lett., 2017, 8, 5523–5530 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. C, 2008, 112, 19797–19800 CrossRef CAS.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- K. L. D. M. Weerawardene and C. M. Aikens, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef CAS PubMed.
- P. Bose, G. Natarajan and T. Pradeep, Atomically Precise Metal Nanoclusters, Elsevier, 2023, pp. 313–343 Search PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- N. A. Sakthivel, S. Theivendran, V. Ganeshraj, A. G. Oliver and A. Dass, J. Am. Chem. Soc., 2017, 139, 15450–15459 CrossRef CAS PubMed.
- N. A. Sakthivel, M. Stener, L. Sementa, A. Fortunelli, G. Ramakrishna and A. Dass, J. Phys. Chem. Lett., 2018, 9, 1295–1300 CrossRef CAS.
- C. Kumara, X. Zuo, J. Ilavsky, D. Cullen and A. Dass, J. Phys. Chem. C, 2015, 119, 11260–11266 CrossRef CAS.
- S. K. Eswaramoorthy, N. A. Sakthivel and A. Dass, J. Phys. Chem. C, 2019, 123, 9634–9639 CrossRef CAS.
- H. Yang, Y. Wang, X. Chen, X. Zhao, L. Gu, H. Huang, J. Yan, C. Xu, G. Li and J. Wu, Nat. Commun., 2016, 7, 12809 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696 CrossRef CAS.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580 CrossRef CAS PubMed.
- S. Vergara, U. Santiago, C. Kumara, D. Alducin, R. L. Whetten, M. J. Yacaman, A. Dass and A. Ponce, J. Phys. Chem. C, 2018, 122, 26733–26738 CrossRef CAS.
- C. Kumara, M. M. Hoque, X. Zuo, D. A. Cullen, R. L. Whetten and A. Dass, J. Phys. Chem. Lett., 2018, 9, 6825–6832 CrossRef CAS PubMed.
- J. H. Beynon, Microchim. Acta, 1956, 44, 437–453 CrossRef.
- E. McDaniel, D. Martin and W. Barnes, Rev. Sci. Instrum., 1962, 33, 2–7 CrossRef CAS.
- F. W. McLafferty and T. A. Bryce, Chem. Commun., 1967, 1215–1217 RSC.
- A. Baksi, P. Chakraborty, A. Nag, D. Ghosh, S. Bhat and T. Pradeep, Anal. Chem., 2018, 90, 11351–11357 CrossRef CAS PubMed.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D. E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS PubMed.
- A. Baksi, P. Chakraborty, S. Bhat, G. Natarajan and T. Pradeep, Chem. Commun., 2016, 52, 8397–8400 RSC.
- A. Baksi, A. Ghosh, S. K. Mudedla, P. Chakraborty, S. Bhat, B. Mondal, K. Krishnadas, V. Subramanian and T. Pradeep, J. Phys. Chem. C, 2017, 121, 13421–13427 CrossRef CAS.
- P. Chakraborty, A. Baksi, S. K. Mudedla, A. Nag, G. Paramasivam, V. Subramanian and T. Pradeep, Phys. Chem. Chem. Phys., 2018, 20, 7593–7603 RSC.
- P. Chakraborty, S. Malola, M. Neumaier, P. Weis, H. Häkkinen and M. M. Kappes, Angew. Chem., 2023, e202305836 CAS.
- P. Chakraborty, M. Neumaier, P. Weis and M. M. Kappes, J. Am. Soc. Mass Spectrom., 2023, 34, 676–684 CrossRef CAS PubMed.
- R. Yost and C. Enke, J. Am. Chem. Soc., 1978, 100, 2274–2275 CrossRef CAS.
- F. W. McLafferty, Acc. Chem. Res., 1980, 13, 33–39 CrossRef CAS.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- S. Malola, L. Lehtovaara, S. Knoppe, K.-J. Hu, R. E. Palmer, T. Bürgi and H. Häkkinen, J. Am. Chem. Soc., 2012, 134, 19560–19563 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, C. Liu, K. Nobusada, N. L. Rosi and R. Jin, Sci. Adv., 2015, 1, e1500425 CrossRef PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 8710–8713 CrossRef CAS PubMed.
- A. Dass, S. Theivendran, P. R. Nimmala, C. Kumara, V. R. Jupally, A. Fortunelli, L. Sementa, G. Barcaro, X. Zuo and B. C. Noll, J. Am. Chem. Soc., 2015, 137, 4610–4613 CrossRef CAS PubMed.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten and U. Landman, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. D. Gobbo, R. G. AbdulHalim, M. Eddaoudi, D. E. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS PubMed.
- L. Cheng, Y. Yuan, X. Zhang and J. Yang, Angew. Chem., Int. Ed., 2013, 52, 9035–9039 CrossRef CAS PubMed.
- G. Natarajan, A. Mathew, Y. Negishi, R. L. Whetten and T. Pradeep, J. Phys. Chem. C, 2015, 119, 27768–27785 CrossRef CAS.
- W. W. Xu, B. Zhu, X. C. Zeng and Y. Gao, Nat. Commun., 2016, 7, 13574 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- Y. Negishi, K. Munakata, W. Ohgake and K. Nobusada, J. Phys. Chem. Lett., 2012, 3, 2209–2214 CrossRef CAS.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Phys. Chem. C, 2013, 117, 7914–7923 CrossRef CAS.
- K. Kwak, Q. Tang, M. Kim, D. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- K. Kwak, V. D. Thanthirige, K. Pyo, D. Lee and G. Ramakrishna, J. Phys. Chem. Lett., 2017, 8, 4898 CrossRef CAS PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- J. P. Wilcoxon, J. E. Martin, F. Parsapour, B. Wiedenman and D. F. Kelley, J. Chem. Phys., 1998, 108, 9137–9143 CrossRef CAS.
- Z. Wu and R. Jin, ACS Nano, 2009, 3, 2036–2042 CrossRef CAS PubMed.
- H. Qian, M. Zhu, C. Gayathri, R. R. Gil and R. Jin, ACS Nano, 2011, 5, 8935–8942 CrossRef CAS PubMed.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104, 2630–2641 CrossRef CAS.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- S. Tian, Y. Z. Li, M. B. Li, J. Yuan, J. Yang, Z. Wu and R. Jin, Nat. Commun., 2015, 6, 8667 CrossRef CAS PubMed.
- I. Dolamic, B. Varnholt and T. Bürgi, Phys. Chem. Chem. Phys., 2013, 15, 19561–19565 RSC.
- B. Varnholt, P. Oulevey, S. Luber, C. Kumara, A. Dass and T. Bürgi, J. Phys. Chem. C, 2014, 118, 9604–9611 CrossRef CAS.
- E. Khatun, P. Chakraborty, B. R. Jacob, G. Paramasivam, M. Bodiuzzaman, W. A. Dar and T. Pradeep, Chem. Mater., 2020, 32, 611–619 CrossRef CAS.
- Y. Negishi, W. Kurashige and U. Kamimura, Langmuir, 2011, 27, 12289–12292 CrossRef CAS PubMed.
- W. Kurashige, S. Yamazoe, K. Kanehira, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2013, 4, 3181–3185 CrossRef CAS.
- Q. Xu, S. Wang, Z. Liu, G. Xu, X. Meng and M. Zhu, Nanoscale, 2013, 5, 1176–1182 RSC.
- I. Chakraborty, W. Kurashige, K. Kanehira, L. Gell, H. Häkkinen, Y. Negishi and T. Pradeep, J. Phys. Chem. Lett., 2013, 4, 3351–3355 CrossRef CAS PubMed.
- D. M. Chevrier, X. Meng, Q. Tang, D. Jiang, M. Zhu, A. Chatt and P. Zhang, J. Phys. Chem. C, 2014, 118, 21730–21737 CrossRef CAS.
- P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi and T. Tsukuda, J. Am. Chem. Soc., 2013, 135, 9450–9457 CrossRef CAS PubMed.
- Z. Lei, X. K. Wan, S. F. Yuan, J. Q. Wang and Q. M. Wang, Dalton Trans., 2017, 46, 3427–3434 RSC.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- V. Linko, H. Zhang, Nonappa, M. A. Kostiainen and O. Ikkala, Acc. Chem. Res., 2022, 55, 1785–1795 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CrossRef CAS.
- M. J. Hostetler, S. J. Green, J. J. Stokes and R. W. Murray, J. Am. Chem. Soc., 1996, 118, 4212 CrossRef CAS.
- Y. Song, T. Huang and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 11694–11701 CrossRef CAS PubMed.
- R. Guo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143 CrossRef CAS PubMed.
- J. F. Parker, J. E. F. Weaver, F. McCallum, C. A. Fields-Zinna and R. W. Murray, Langmuir, 2010, 26, 13650–13654 CrossRef CAS PubMed.
- R. Guo, Y. Song, G. Wang and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 2752–2757 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS PubMed.
- A. Dass, A. Stevenson, G. R. Dubay, J. B. Tracy and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 5940–5946 CrossRef CAS PubMed.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS PubMed.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS PubMed.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS PubMed.
- L. G. Abdulhalim, N. Kothalawala, L. Sinatra, A. Dass and O. M. Bakr, J. Am. Chem. Soc., 2014, 136, 15865–15868 CrossRef CAS PubMed.
- Y. Chen, M. Zhou, Q. Li, H. Gronlund and R. Jin, Chem. Sci., 2020, 11, 8176–8183 RSC.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS PubMed.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- S. Yang, S. Chen, L. Xiong, C. Liu, H. Yu, S. Wang, N. L. Rosi, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2018, 140, 10988–10994 CrossRef CAS PubMed.
- A. Das, T. Li, G. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, Nanoscale, 2014, 6, 6458–6462 RSC.
- M. S. Bootharaju, C. P. Joshi, M. J. Alhilaly and O. M. Bakr, Chem. Mater., 2016, 28, 3292–3297 CrossRef CAS.
- M. S. Bootharaju, V. M. Burlakov, T. M. D. Besong, C. P. Joshi, L. G. AbdulHalim, D. M. Black, R. L. Whetten, A. Goriely and O. M. Bakr, Chem. Mater., 2015, 27, 4289–4297 CrossRef CAS.
- E. Khatun, A. Ghosh, D. Ghosh, P. Chakraborty, A. Nag, B. Mondal, S. Chennu and T. Pradeep, Nanoscale, 2017, 9, 8240–8248 RSC.
- M. S. Bootharaju, R. Dey, L. E. Gevers, M. N. Hedhili, J.-M. Basset and O. M. Bakr, J. Am. Chem. Soc., 2016, 138, 13770–13773 CrossRef CAS PubMed.
- M. Bodiuzzaman, A. Ghosh, K. S. Sugi, A. Nag, E. Khatun, B. Varghese, G. Paramasivam, S. Antharjanam, G. Natarajan and T. Pradeep, Angew. Chem., Int. Ed., 2019, 58, 189–194 CrossRef CAS PubMed.
- C. K. Manju, D. Ghosh, M. Bodiuzzaman and T. Pradeep, Dalton Trans., 2019, 48, 8664–8670 RSC.
- A. Jana, P. Chakraborty, W. A. Dar, S. Chandra, E. Khatun, M. P. Kannan, R. H. A. Ras and T. Pradeep, Chem. Commun., 2020, 56, 12550–12553 RSC.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- S. Wang, X. Zhu, T. Cao and M. Zhu, Nanoscale, 2014, 6, 5777–5781 RSC.
- A. Kim, C. Zeng, M. Zhou and R. Jin, Part. Part. Syst. Charact., 2017, 34, 1600388 CrossRef.
- E. Khatun, A. Ghosh, P. Chakraborty, P. Singh, M. Bodiuzzaman, P. Ganesan, G. Nataranjan, J. Ghosh, S. K. Pal and T. Pradeep, Nanoscale, 2018, 10, 20033–20042 RSC.
- X. Kang, M. Zhou, S. Wang, S. Jin, G. Sun, M. Zhu and R. Jin, Chem. Sci., 2017, 8, 2581–2587 RSC.
- E. Reyes, R. Madueño, M. Blázquez and T. Pineda, J. Phys. Chem. C, 2010, 114, 15955–15962 CrossRef CAS.
- S. Knoppe, A. C. Dharmaratne, E. Schreiner, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2010, 132, 16783–16789 CrossRef CAS PubMed.
- S. Knoppe, R. Azoulay, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 20302–20305 CrossRef CAS PubMed.
- M. A. Habeeb Muhammed and T. Pradeep, Chem. Phys. Lett., 2007, 449, 186–190 CrossRef CAS.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2011, 27, 8134–8143 CrossRef CAS PubMed.
- W. Chen, X. Tu and X. Guo, Chem. Commun., 2009, 1736–1738 RSC.
- Z. Wu, Angew. Chem., Int. Ed., 2012, 51, 2934–2938 CrossRef CAS PubMed.
- J. Sun, H. Wu and Y. Jin, Nanoscale, 2014, 6, 5449–5457 RSC.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS PubMed.
- X. Kang, L. Xiong, S. Wang, H. Yu, S. Jin, Y. Song, T. Chen, L. Zheng, C. Pan, Y. Pei and M. Zhu, Chem. – Eur. J., 2016, 22, 17145–17150 CrossRef CAS PubMed.
- X. Kang, X. Wei, S. Jin, Q. Yuan, X. Luan, Y. Pei, S. Wang, M. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18834–18840 CrossRef CAS PubMed.
- M. S. Bootharaju, L. Sinatra and O. M. Bakr, Nanoscale, 2016, 8, 17333–17339 RSC.
- W. Du, S. Jin, L. Xiong, M. Chen, J. Zhang, X. Zou, Y. Pei, S. Wang and M. Zhu, J. Am. Chem. Soc., 2017, 139, 1618–1624 CrossRef CAS PubMed.
- W. J. Plieth, Surf. Sci., 1985, 156, 530–535 CrossRef CAS.
- R. A. Masitas and F. P. Zamborini, J. Am. Chem. Soc., 2012, 134, 5014–5017 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- J. P. Choi, C. A. Fields-Zinna, R. L. Stiles, R. Balasubramanian, A. D. Douglas, M. C. Crowe and R. W. Murray, J. Phys. Chem. C, 2010, 114, 15890–15896 CrossRef CAS.
- S. Tian, C. Yao, L. Liao, N. Xia and Z. Wu, Chem. Commun., 2015, 51, 11773–11776 RSC.
- Z. Wu, M. Wang, J. Yang, X. Zheng, W. Cai, G. Meng, H. Qian, H. Wang and R. Jin, Small, 2012, 8, 2028–2035 CrossRef CAS PubMed.
- C. Yao, J. Chen, M.-B. Li, L. Liu, J. Yang and Z. Wu, Nano Lett., 2015, 15, 1281–1287 CrossRef CAS PubMed.
- S. Wang, H. Abroshan, C. Liu, T.-Y. Luo, M. Zhu, H. J. Kim, N. L. Rosi and R. Jin, Nat. Commun., 2017, 8, 848 CrossRef PubMed.
- X. Yuan, X. Dou, K. Zheng and J. Xie, Part. Part. Syst. Charact., 2015, 32, 613–629 CrossRef.
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 9511–9514 CrossRef CAS PubMed.
- Q. Li, S. Wang, K. Kirschbaum, K. J. Lambright, A. Das and R. Jin, Chem. Commun., 2016, 52, 5194–5197 RSC.
- Q. Li, T. Y. Luo, M. G. Taylor, S. Wang, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- M. Zhu, P. Wang, N. Yan, X. Chai, L. He, Y. Zhao, N. Xia, C. Yao, J. Li, H. Deng, Y. Zhu, Y. Pei and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 4500–4504 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS PubMed.
- Y. Li, M. J. Cowan, M. Zhou, T.-Y. Luo, Y. Song, H. Wang, N. L. Rosi, G. Mpourmpakis and R. Jin, J. Am. Chem. Soc., 2020, 142, 20426–20433 CrossRef CAS PubMed.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2016, 120, 14948 CrossRef CAS.
- Z. Wang, R. K. Gupta, F. Alkan, B.-L. Han, L. Feng, X.-Q. Huang, Z.-Y. Gao, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2023, 145, 19523–19532 CrossRef CAS PubMed.
- A. Sreekumaran Nair and T. Pradeep, Curr. Sci., 2003, 84, 1560–1564 Search PubMed.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2012, 28, 2671–2679 CrossRef CAS PubMed.
- M. S. Bootharaju, G. K. Deepesh, T. Udayabhaskararao and T. Pradeep, J. Mater. Chem. A, 2013, 1, 611–620 RSC.
- A. Mathew, G. Natarajan, L. Lehtovaara, H. Häkkinen, R. M. Kumar, V. Subramanian, A. Jaleel and T. Pradeep, ACS Nano, 2014, 8, 139–152 CrossRef CAS PubMed.
- P. Chakraborty, A. Nag, G. Paramasivam, G. Natarajan and T. Pradeep, ACS Nano, 2018, 12, 2415–2425 CrossRef CAS PubMed.
- P. Chakraborty, A. Nag, B. Mondal, E. Khatun, G. Paramasivam and T. Pradeep, J. Phys. Chem. C, 2020, 124, 14891–14900 CrossRef CAS.
- P. Chakraborty, A. Nag, K. S. Sugi, T. Ahuja, B. Varghese and T. Pradeep, ACS Mater. Lett., 2019, 1, 534–540 CrossRef CAS.
- A. Nag, P. Chakraborty, G. Paramasivam, M. Bodiuzzaman, G. Natarajan and T. Pradeep, J. Am. Chem. Soc., 2018, 140, 13590–13593 CrossRef CAS PubMed.
- A. Nag, P. Chakraborty, A. Thacharon, G. Paramasivam, B. Mondal, M. Bodiuzzaman and T. Pradeep, J. Phys. Chem. C, 2020, 124, 22298–22303 CrossRef CAS.
- M. A. H. Muhammed, L. K. Cruz, A. H. Emwas, A. M. El-Zohry, B. Moosa, O. F. Mohammed and N. M. Khashab, Angew. Chem., Int. Ed., 2019, 58, 15665–15670 CrossRef CAS PubMed.
- K. Sheng, Z. Wang, L. Li, Z. Y. Gao, C. H. Tung and D. Sun, J. Am. Chem. Soc., 2023, 145, 10595–10603 CrossRef CAS PubMed.
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS PubMed.
- K. R. Krishnadas, T. Udayabhaskararao, S. Choudhury, N. Goswami, S. K. Pal and T. Pradeep, Eur. J. Inorg. Chem., 2014, 2014, 908–916 CrossRef CAS.
- B. Zhang, G. Salassa and T. Bürgi, Chem. Commun., 2016, 52, 9205–9207 RSC.
- B. Huang and Y. Pei, J. Mater. Chem. A, 2020, 8, 10242–10251 RSC.
- M. Neumaier, A. Baksi, P. Weis, E. K. Schneider, P. Chakraborty, H. Hahn, T. Pradeep and M. M. Kappes, J. Am. Chem. Soc., 2021, 143, 6969–6980 CrossRef CAS PubMed.
- P. Chakraborty, A. Nag, G. Natarajan, N. Bandyopadhyay, G. Paramasivam, M. K. Panwar, J. Chakrabarti and T. Pradeep, Sci. Adv., 2019, 5, eaau7555 CrossRef PubMed.
- G. Salassa, A. Sels, F. Mancin and T. Bürgi, ACS Nano, 2017, 11, 12609–12614 CrossRef CAS PubMed.
- A. Ghosh, T. Pradeep and J. Chakrabarti, J. Phys. Chem. C, 2014, 118, 13959–13964 CrossRef CAS.
- A. Som, A. K. Samal, T. Udayabhaskararao, M. S. Bootharaju and T. Pradeep, Chem. Mater., 2014, 26, 3049–3056 CrossRef CAS.
- Nonappa and O. Ikkala, Adv. Funct. Mater., 2018, 28, 1704328 CrossRef.
- Nonappa, Chem. Commun., 2023, 59, 13800–13819 RSC.
- A. Som, I. Chakraborty, T. A. Maark, S. Bhat and T. Pradeep, Adv. Mater., 2016, 28, 2827–2833 CrossRef CAS PubMed.
- Nonappa, T. Lahtinen, J. S. Haataja, T. R. Tero, H. Häkkinen and O. Ikkala, Angew. Chem., Int. Ed., 2016, 55, 16035–16038 CrossRef CAS PubMed.
- A. Som, A. Griffo, I. Chakraborty, H. Hähl, B. Mondal, A. Chakraborty, K. Jacobs, P. Laaksonen, O. Ikkala, T. Pradeep and Nonappa, Small, 2022, 18, 2201707 CrossRef CAS PubMed.
- A. Chakraborty, A. C. Fernandez, A. Som, B. Mondal, G. Natarajan, G. Paramasivam, T. Lahtinen, H. Häkkinen, Nonappa and T. Pradeep, Angew. Chem., Int. Ed., 2018, 57, 6522–6526 CrossRef CAS PubMed.
- A. Chakraborty, M. M. Stanley, B. Mondal, Nonappa, M. Bodiuzzaman, P. Chakraborty, M. P. Kannan and T. Pradeep, Nanoscale, 2023, 15, 2690–2699 RSC.
- J. Roy, B. Mondal, G. Vishwakarma, Nonappa, N. V. Sridharan, P. Krishnamurthi and T. Pradeep, Nanoscale, 2023, 15, 8225–8234 RSC.
- J. V. Rival, Nonappa and E. S. Shibu, ACS Appl. Mater. Interfaces, 2020, 12, 14569–14577 CrossRef CAS PubMed.
- P. Bose, P. Chakraborty, J. S. Mohanty, Nonappa, A. Ray Chowdhuri, E. Khatun, T. Ahuja, A. Mahendranath and T. Pradeep, Nanoscale, 2020, 12, 22116–22128 RSC.
- A. Baksi, M. Gandi, S. Chaudhari, S. Bag, S. S. Gupta and T. Pradeep, Angew. Chem., Int. Ed., 2016, 55, 7777–7781 CrossRef CAS PubMed.
- A. Nag, A. Baksi, K. C. Krishnapriya, S. S. Gupta, B. Mondal, P. Chakraborty and T. Pradeep, Eur. J. Inorg. Chem., 2017, 2017, 3072–3079 CrossRef CAS.
- R. Kazan, U. Muller and T. Burgi, Nanoscale, 2019, 11, 2938–2945 RSC.
- P. Chakraborty, P. Bose, J. Roy, A. Nag, B. Mondal, A. Chakraborty and T. Pradeep, J. Phys. Chem. C, 2021, 125, 16110–16117 CrossRef CAS.
- I. Chakraborty, T. Udayabhaskararao and T. Pradeep, J. Hazard. Mater., 2012, 211–212, 396–403 CrossRef CAS PubMed.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
Footnotes |
| † Present address: School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Vithura, Thiruvananthapuram-695551, Kerala, India. |
| ‡ Present address: Institute of Nanotechnology, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, and Institute of Physical Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |