
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-tunable silicon nanoparticles synthesized in solution via a redox reaction†
Megan A.
Parker
a,
Maria Letizia
De Marco‡
a,
Alexander
Castro-Grijalba§
a,
Anissa
Ghoridi
 b,
David
Portehault
b,
Stanislav
Pechev
a,
Elizabeth A.
Hillard
a,
Sabrina
Lacomme
c,
Aurélie
Bessière
d,
Frédérique
Cunin
d,
Patrick
Rosa
*a,
Mathieu
Gonidec
*a and
Glenna L.
Drisko
*a
b,
David
Portehault
b,
Stanislav
Pechev
a,
Elizabeth A.
Hillard
a,
Sabrina
Lacomme
c,
Aurélie
Bessière
d,
Frédérique
Cunin
d,
Patrick
Rosa
*a,
Mathieu
Gonidec
*a and
Glenna L.
Drisko
*a
aUniv. Bordeaux, CNRS, Bordeaux-INP, ICMCB, UMR 5026, F-33600, Pessac, France. E-mail: patrick.rosa@icmcb.cnrs.fr; mathieu.gonidec@icmcb.cnrs.fr; glenna.drisko@icmcb.cnrs.fr
bSorbonne Université, CNRS, Laboratoire de Chimie de la Matière Condensée de Paris (LCMCP), F-75005 Paris, France
cInstitut Charles Gerhardt Montpellier, Univ. Montpellier, CNRS, ENSCM, UMR 52531919 route de Mende, 34293 Montpellier Cedex 05, France
dUniv.Montpellier, CNRS, ICGM, ENSCM, UMR-5618, F-34293, Montpellier, France
First published on 2nd April 2024
Abstract
A current challenge in silicon chemistry is to perform liquid-phase synthesis of silicon nanoparticles, which would permit the use of colloidal synthesis techniques to control size and shape. Herein we show how silicon nanoparticles were synthesized at ambient temperature and pressure in organic solvents through a redox reaction. Specifically, a hexacoordinated silicon complex, bis(N,N′-diisopropylbutylamidinato)dichlorosilane, was reduced by a silicon Zintl phase, sodium silicide (Na4Si4). The resulting silicon nanoparticles were crystalline with sizes tuned from a median particle diameter of 15 nm to 45 nm depending on the solvent. Photoluminescence measurements performed on colloidal suspensions of the 45 nm diameter silicon nanoparticles indicated a blue emission signal, attributed to the partial oxidation of the Si nanocrystals or to the presence of nitrogen impurities.
1. Introduction
Photoluminescence has several origins in silicon particles, due to quantum size effects, excitonic radiation recombination pathways, surface traps, charge transfer from silicon to surface-bound ligands, and combinations of these effects.1–4 Efforts to engineer both the silicon nanoparticle and the surface ligands have led to high quantum efficiencies, tunable emission colors, lifetimes and full width at half maximum band emissions.5,6 Partially oxidized amorphous silicon nanostructures have been shown to display photoluminescence via radiative processes from surface or sub-surface defects.7 Thus, surface states offer the possibility to produce photoluminescence in silicon particles across a range of nanoscale dimensions.Colloidal syntheses toward photoluminescent silicon particles are typically low yielding, while high-temperature routes are energy demanding, and require dangerous starting materials or reagents, e.g. gaseous silanes or hydrofluoric acid.8–10 New silicon reactions through colloidal synthesis are needed, ideally yielding silicon nanoparticles under relatively mild conditions. A dual Si precursor system, using electron-poor and electron-rich silicon compounds, provides an interesting and unexplored pathway towards this goal.
A potentially useful class of easily reducible silicon precursors is hypervalent silicon coordination complexes. Hypervalent silicon possesses empty 3d orbitals and forms hexa- or penta- coordinated compounds by accepting the ligands’ electron lone pair.11,12 The electron withdrawing groups in hypervalent silicon complexes shift the electron density away from the central silicon atom and towards the ligands.13 Silicon coordination complexes of this kind merit examination as relatively stable and easy to manipulate precursors, however, they require a reductant to form Si–Si covalent bonds.
Silicon-based Zintl phases are an obvious choice for a reductant because they contain clusters of silicon anions. A variety of Zintl structures provides a range of different reduction potentials. There is vast literature concerning the synthesis of silicon materials by oxidation of Si-based Zintl precursors.14–17 The reactive, covalently bonded anionic subunits lend themselves to the formation of 3D networks and thus can form a variety of nanostructures during solution-based syntheses.18 More specifically, Zintl precursors employed towards silicon nanoparticles have involved the Zintl phases Na4Si4, K4Si4, and heavier phases such as A12Si17 (A = K, Rb, Cs), which require the oxidation of the negatively charged silicon clusters [Si4]4− or [Si9]4− to form Si(0). Alternatively, the use of weak oxidizing agents such as bromine or NH4Br results in the formation of crystalline silicon particles, with particle size limited to average diameters of 4–5 nm.15,17,19 Na4Si4 remains the most practical precursor for the solution synthesis of silicon nanoparticles, thus it was chosen as the reductant in this study.
Standard redox reactions between Zintl phases and silicon halides (SiCl4, SiI4) have been reported,20–22 resulting in silicon nanoparticles below 5 nm in diameter, large clusters of nanostructured silicon or silicon nanodendrimers. In this study, we explore the reaction of the Na4Si4 Zintl phase with a Si coordination complex, silicon bisamidinate (bis(N,N′-dimethylbutylamidinato)dichlorosilane). We synthesize silicon nanoparticles at ambient temperature and pressure (Fig. 1), and evaluate the effect of two different solvents on particle size, crystallinity, and morphology. Finally, we test the fluorescence of these nano-objects.
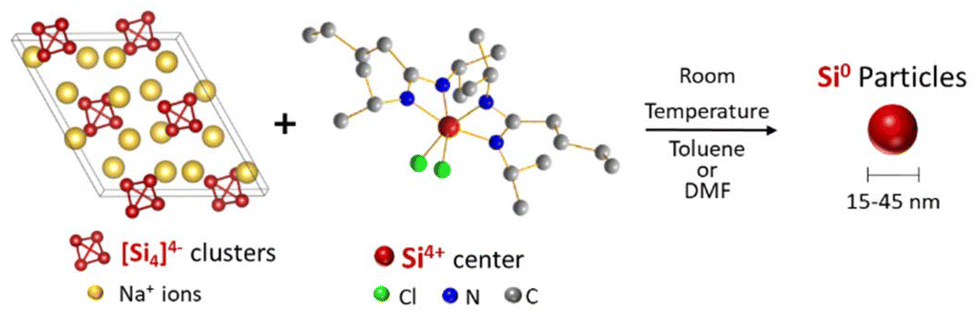 | ||
| Fig. 1 Reaction scheme between sodium silicide and silicon bisamidinate to form silicon nanoparticles. | ||
2. Experimental
2.1 Materials
N,N′-Diisopropylcarbodiimide (99%), silicon tetrachloride (99%), n-butyllithium (2.5 M in hexane), and NaH (90%) were purchased from Sigma-Aldrich and used without further purification. Fomblin Y oil was purchased from Merck (CAS:69991-67-9). NVH-type oil was purchased from Cargille Laboratories, NJ, USA. Silicon particles were purchased from Nanomakers (40 nm). n-Bu4PF6 (electrochemical grade) was purchased from Fluka. Tetrahydrofuran (THF) was obtained from an MBraun MBSPS 5 solvent purification system. Toluene, dimethylformamide (DMF), and formamide were dried using an Innovative Technology solvent purification system, then oxygen was removed by three freeze–pump–thaw cycles. The purified solvents were stored over 3 Å molecular sieves in an argon filled glovebox. Carbon-coated copper grids were purchased from Electron Microscopy Sciences (Hatfield, PA). All the experiments were carried out under inert atmosphere, with Schlenk techniques or in an argon-filled glovebox.2.2 Synthesis of bis-(N,N′-diisopropylbutylamidinate) dichlorosilane ([(i-PrN)2C(n-Bu)]2SiCl2)
The silicon precursor was prepared by a two-step, one-pot synthesis (Fig. 2). First, butyllithium (13 mL, 0.032 mol) was added to a solution of N,N′-diisopropylcarbodiimide (5.0 mL, 0.032 mol) in THF (80 mL) in a cooling bath slurry consisting of a 60/40 v/v% ethanol/water mixture with liquid N2 (−40 °C) under agitation. The yellowish solution was stirred at room temperature overnight. This reaction yields the lithium amidinate; [(i-PrN)2C(n-Bu)]Li·THF (1). 1H NMR (1) (C6D6): δ 0.95 (t, 3H n-Bu, 3JH–H 7.20 Hz, CH3), δ 1.21 (m, 2H n-Bu, CH2), δ 1.33 (d, 12H i-Pr,3JH–H 6.64 Hz, CH3), δ 1.40 (m, 4H THF, CH2), δ 1.62 (m, 2H n-Bu, CH2CN2), δ 2.39 (m, 2H n-Bu, CH2CN2), δ 3.58 (m, 4H THF, CH2O), δ 3.72 (m, 2H i-Pr, Me2CH). In a second step, SiCl4 (1.86 mL, 0.016 mol) was added dropwise in a cooling bath slurry consisting of a 70/30 v/v% ethanol/water mixture (−80 °C) with liquid N2. The solution was allowed to come to room temperature, and stirred overnight. The THF was evaporated under vacuum and substituted with toluene (60 mL). LiCl precipitated and was filtered out of the media via cannula filtration. Most of the toluene (50 mL) was removed via evaporation to concentrate the product. The product was stored at −30 °C until crystallites formed. Then, cold toluene (30 mL) was added and the product was further chilled at −30 °C overnight. The next day, larger, colorless crystals had formed. These were collected and washed with cold toluene. Product 2 was stored at −30 °C under inert atmosphere, to avoid premature degradation, as it is extremely sensitive to moisture and somewhat thermally labile. 1H NMR (2) (C6D6): δ 0.77 (t, 6H n-Bu, 3JH–H 7.44 Hz, CH3), δ 1.10 (d, 6H i-Pr, 3JH–H 6.52 Hz, CH3), δ 1.13 (m, 4H n-Bu, MeCH2), δ 1.39 (d, 6H i-Pr, 3JH–H 6.68 Hz, CH3), δ 1.38 (m, 4H n-Bu, MeCH2CH2), δ 1.60 (d, 6H i-Pr, 3JH–H 7.00 Hz, CH3), δ 1.63 (d, 6H i-Pr, 3JH–H 6.88 Hz, CH3), δ 2.01 (m, 4H n-Bu, CH2CN2), δ 3.50 (sept., 2H i-Pr, 3JH–H 6.76 Hz, Me2CH), δ 4.18 (sept., 2H i-Pr, 3JH–H 8.92, Me2CH).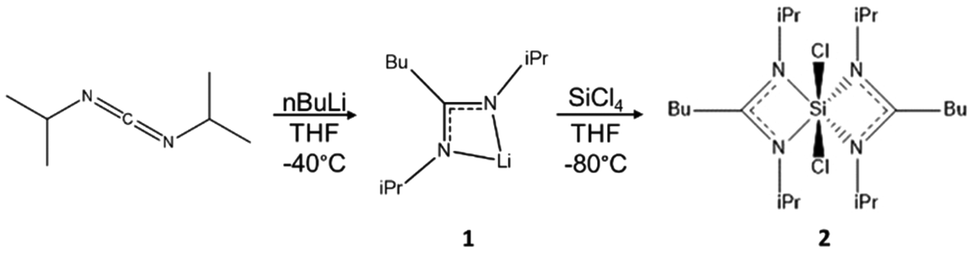 | ||
| Fig. 2 Preparation of intermediate complex lithium (N,N′-diisopropylbutylamidinate) (1). Its reaction with silicon tetrachloride forms the complex bis(N,N’-diisopropylbutylamidinate) dichlorosilane (2). | ||
2.3 Synthesis of sodium silicide, Na4Si4
High purity Na4Si4 was prepared according to a previously published protocol.17 In a typical batch, NaH (19.6 mmol, 470 mg) and silicon nanoparticles (17.9 mmol, 500 mg) were mixed together at 20 Hz for 2 min using a ball mill (Retsch MM400 ball miller, airtight vials of 50 mL, one steel ball of 62.3 g, and a diameter of 23 mm). The powder was recovered in an argon-filled glovebox and put into a h-BN crucible, which was placed in an air-tight quartz tube. The quartz tube was placed inside a vertical oven and connected to an Ar flow of 0.06 L min−1. A heating ramp of 10 °C min−1 was used to 400 °C, followed by a dwell time of 24 h. Finally, the sample was cooled down naturally. The reaction vessel was then transferred back to an argon-filled glovebox and the synthesized powder was recovered and stored in the glovebox.2.4 Reaction of [(i-PrN)2C(n-Bu)]2SiCl2 with Na4Si4
In an argon-filled glove box, 45 mg of 2 (0.01 mmol) was dissolved in 3 mL of solvent in a Schlenk tube and then on a Schlenck line, transferred via cannula to a vessel containing Na4Si4 (20 mg, 0.01 mmol) in 5 mL of solvent under magnetic agitation. The reaction was carried out in DMF and toluene in order to assess the effect of solvent on product formation. After the synthesis, the products were collected under inert atmosphere. The crude product, appearing as a black solid, was washed twice with anhydrous formamide, and twice with anhydrous THF via 10 min centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400g.
400g.
2.5 Proton nuclear magnetic resonance (1H NMR)
The samples were dissolved in deuterated benzene in the glovebox, and then placed within a screw-cap NMR tube. The spectra were collected using a Bruker Avance III-HD 400 MHz SB spectrometer (Wissembourg, France) equipped with a 5 mm broadband SmartProbe at 25 °C. 1H NMR spectra were acquired at 400.13 MHz using a single pulse sequence (π/2 pulse width of 10 μs, recycling delay of 2 s, acquisition time of 1.6 s, spectral window of 25 ppm and 24 scans).2.6 Single crystal X-ray diffraction (XRD)
A small amount of air sensitive silicon bisamidinate 2 was shielded by Fomblin Y oil and taken out of its storage under inert atmosphere. A diffraction-suitable single crystal was fixed on a micromount using NVH-type oil, then transferred into the nitrogen-gas stream of an Oxford Cryosystems Cryostream 700 Plus. Single crystal diffraction data were collected at 120 K, using a Bruker Kappa APEX II diffractometer with a sealed tube Mo radiation source (λ = 0.071073 nm). Indexing and data integration were performed using APEX3 (Bruker 2016. Bruker AXS Inc., Madison, Wisconsin, USA.). The final data set was corrected for absorption using the multi-scan method and SADABS.23 The single-crystal structure of 2 was resolved with the intrinsic phasing method and SHELXT structure solution program.24 The resulting model was refined with SHELXL and least-squares minimization. Olex225 was used as an interface to edit the results. Crystallographic parameters for the structural analysis reported in this work (Table S1†) are also deposited in the Cambridge Crystallographic Data Centre and can be freely accessed at https://www.ccdc.cam.ac.uk (CCDC no. 2157265).2.7 Powder X-ray diffraction (XRD)
Powder samples were prepared under argon in 0.5 mm quartz capillaries. The capillaries were filled with 2 cm of powder and were then sealed with epoxy resin. XRD measurements were realized with a PANalytical X'Pert 3 diffractometer equipped with a capillary spinner, using Cu Kα wavelength (λ = 0.15418 nm) and operating at 40 mA per 45 kV. Measurements were made within an angular range of 8–80° over 15 h.2.8 Electrochemistry
Electrochemical experiments were performed with an Autolab PGSTAT101 potentiostat. Experiments were performed in an argon-filled glove box using a three-electrode configuration in a single compartment cell. The electrodes consisted of a 1 mm diameter platinum disc as the working electrode, a Ag/AgCl pseudo-reference electrode, and a platinum wire as the counter electrode. A 0.1 M electrolyte solution consisting of n-Bu4PF6 in anhydrous DMF was prepared. Standard cyclic voltammetry experiments and square wave voltammetry experiments were performed at 100 mV s−1. Chemical reversibility was checked by increasing rates up to 500 mV s−1. A weighed amount of ferrocene was added subsequently to provide an internal reference26 and to estimate the amount of electrons implicated in the observed redox processes (Fig. S1†).2.9 DFT calculations
The Raman spectrum of 2 was simulated by density functional theory calculations with the Gaussian09 program.27 The geometry of the complexes were first pre-optimized by a semi-empirical calculation with PM628 followed by a geometry optimization and a frequency calculation with the B3LYP functional29 the 6-31G(d,p) basis set.30,31 Finally, the simulated spectra were generated with the GaussSum program32 using the NIST reference vibrational scaling factor for B3LYP/6-31G(d,p) of 0.961.2.10 Raman spectroscopy
Raman spectra were recorded on an Xplora spectrometer by Horiba, equipped with a confocal microscope. An objective lens with a magnification of 10× was used to observe the sample inside the capillary and choose the measurement area. An Olympus LM Plan FLN 100× objective lens with 0.80 numerical aperture and 3.4 mm working distance was employed to focus the laser beam on the sample and collect the scattered light. A longpass edge filter was used to remove the fundamental line from the collected scattered light. The spectra were run using a 632.81 nm HeNe gas laser or a 785 nm laser diode excitation, using a filter to reduce the laser power to 50% of the total power. A 600 lines per mm grating was used to separate the scattered light into its components that were collected onto a Syncerity TE-cooled FI-UV-VIS detector. Baseline subtraction was performed on each spectrum, using a linear baseline. Five measurements in five different zones were collected for each sample, to assess the reproducibility of the measurement and the uniformity of the sample.2.11 Transmission electron microscopy (TEM)
A colloidal dispersion of nanoparticles in THF was sonicated for 3 min, and then one drop was cast onto a copper grid covered with a carbon film (previously activated by UV light, carbon film, face up) for analysis by TEM. Grids were subjected to plasma cleaning (30% power, 30 sccm 100% argon gas flow for 10 s) prior to being analysed. Bright field and dark field TEM images were taken with a Talos F200S G2 TEM (ThermoFisher, Eindhoven), using an accelerating voltage of 2000 V and current of 0.06 nA. Dark field images were taken in scanning (STEM) mode with a current of 0.255 nA.2.12 Photoluminescence measurements
The nanoparticles were dispersed in ethanol (96%) and placed in ultrasonic bath for 30 s. The dispersion was then placed in an air-tight 10 mm quartz cuvette (Hellma, GL14 screw cap with silicone seal). The emission and excitation photoluminescence spectra were recorded at 25 °C on a fluorescence spectrophotometer (FS920, Edinburgh Instruments), equipped with an air cooled photomultiplier operating at −20 °C (R928P, Hamamatsu) and a 450 W continuous xenon arc lamp as the excitation source. The spectra were corrected for detection and the optical spectral response of the spectrophotometer. The photoluminescence quantum yield was measured using a Quanta-phi integrating sphere mounted on the FS920, Edinburgh Instruments spectrophotometer.2.13 Absorbance measurements
The UV-vis absorbance spectra were recorded using a SPECORD 210 PLUS spectrophotometer.2.14 Fourier-transform infrared spectroscopy (FTIR)
Infrared spectra were recorded on a Nicolet IS5 spectrometer with the ATR ID5 module.3. Results and discussion
3.1 Synthesis and crystal structure of the silicon coordination complex
The synthesis of 2 proceeds through a one-pot, two-step reaction, as shown in Fig. 2. The synthesis is straightforward, reproducible, and yields about 3.0 g (6.5 mmol, 41% molar yield) of crystalline product, enough for all the analyses presented in this article. The resulting single crystals allowed us to determine the X-ray structure of 2 (Fig. S2 and Table S1†). We can see that the Si atom is hexacoordinated, with amidinate and chlorine ligands positioned at the vertices of a distorted octahedron. The distortion parameters are calculated using the OCTADIST software and the Continuous Shape Measures tool (Tables S2 and S3†).33,34 The calculated values show that complex 2 shows C2 symmetry, with the coordination sphere varying with respect to a perfect octahedron, in terms of both the angular distortions and Si-ligand bond lengths. The two amidinate ligands chelate the silicon atom through both of their nitrogen atoms. N(1) atoms are in the apical positions of the distorted octahedron with a bond length of 1.845 Å, while N(2) and Cl occupy its equatorial plane with bond lengths of 1.902 Å and 2.209 Å respectively. By applying the valence bond concept, the formal hybridization of silicon centers goes from sp3 for tetravalent centers to sp3d for pentavalent centers to sp3d2 for hexavalent centers. With this trend, the electron density at the silicon center decreases due to reduced s-character, while the electropositive character increases.15 The reactivity of the complex can be assessed by determining its reduction potential.3.2 Electrochemistry
The cyclic voltammogram of 2 was recorded in DMF (Fig. S1†). It indicates that the complex undergoes a chemically irreversible, single electron reduction. Electrochemistry experiments were performed in only DMF and not compared to that in toluene, due to the insolubility of the electrolyte in toluene. We hypothesize that the reduction of 2 results in a chemical decomposition releasing a highly reactive Si species. Since Na4Si4 is expected to have a much lower reduction potential, when 2 comes into contact with Na4Si4, 2 entirely decomposes. The reduction potential of silicon bisamidinate 2 is −2.1 V vs. Fc/Fc+, whereas for SiCl4 it is −2.60 V and for SiBr4 −2.43 V.35 This upward shift of the redox potential for the bisamidinate evidences that the bisamidinate compound 2 is easier to reduce in comparison to silicon halides. This could explain why the reaction between silicon bisamidinate and the reductant Na4Si4 forming silicon nanoparticles is spontaneous at room temperature and even occurs at −30 °C (Fig. S3†).3.3 Formation and characterization of silicon nanoparticles
The high charge:size ratio of the [Si4]4− clusters results in low solubility in most solvents,36 while the silicon bisamidinate complex 2 was found to be soluble in a wide range of organic solvents (benzene, hexane, toluene, THF, glyme, DMF, and NMP). Hence, a redox reaction occurs in a biphasic mixture between the [Si4]4− clusters in sodium silicide and the Si4+ center of the bisamidinate coordination complex, to produce Si(0) nanoparticles. The synthesis was performed in DMF and toluene at room temperature for 16 h under argon.The observed powder X-ray diffraction peaks (Fig. 3a) are consistent with the diamond cubic structure of crystalline Si. Calculated patterns for the precursors (Fig. S4†) were compared to those of the products. The absence of peaks attributed to Na4Si4 and silicon bisamidinate in the product diffraction patterns confirms precursor consumption during the reaction. The silica capillary yields a large, broad peak centered at 2θ = 21°. This peak was baseline removed, but uncorrected patterns can be seen in Fig. S5.† However, this broad contribution could also indicate the presence of amorphous silicon or partially oxidized nanoparticles. HRTEM confirmed the presence of crystalline silicon (Fig. 3b). Dark field TEM images can be found in Fig. S6.† The Scherrer equation was used to calculate the average coherent domain length based on peak broadening in powder XRD patterns (eqn (S1) and Table S4†). Coherent domain lengths of 15.6 nm were evaluated for the products from DMF and 21.9 nm for the products from toluene.
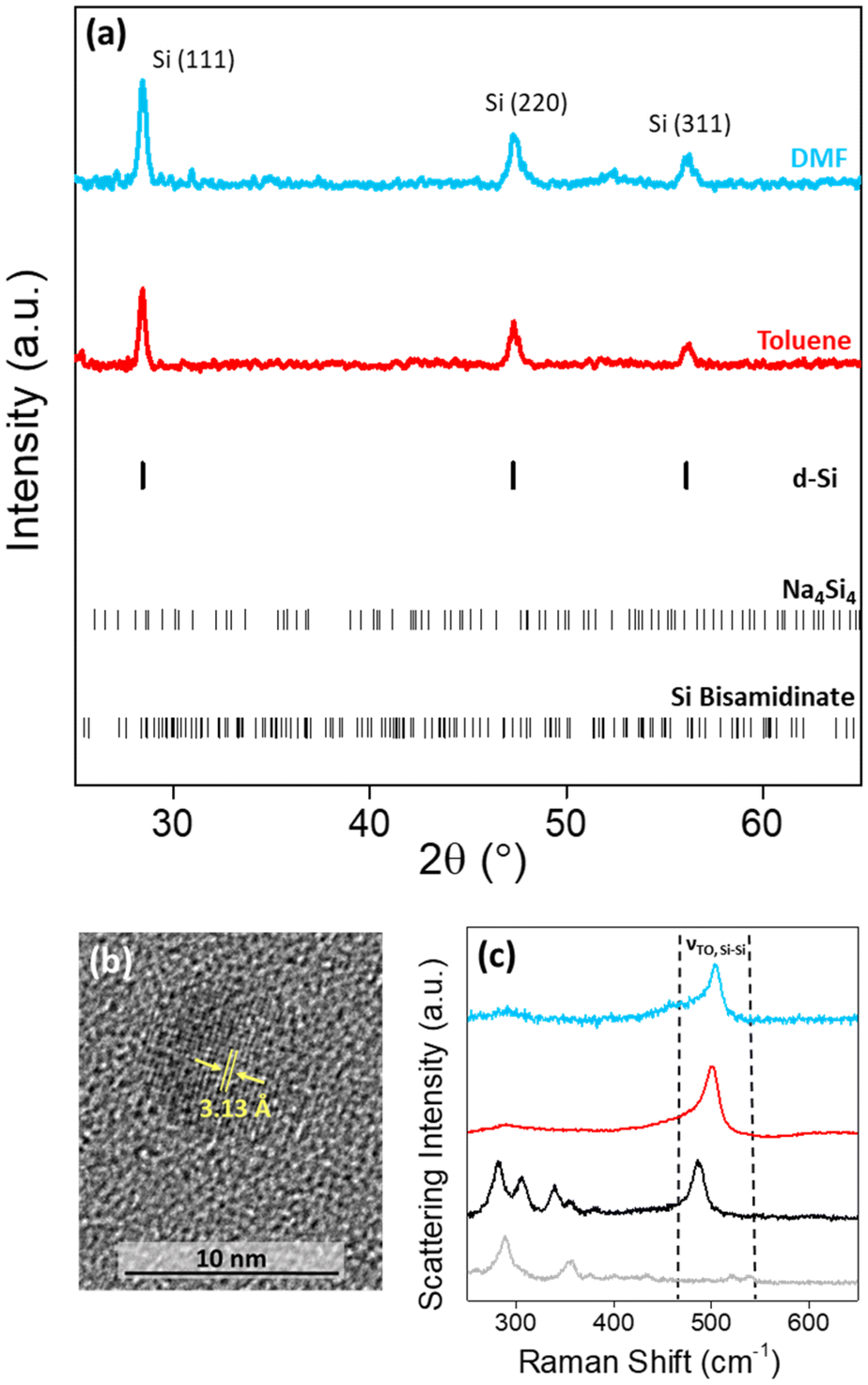 | ||
| Fig. 3 (a) Powder XRD patterns of product produced in (blue) DMF and (red) toluene with Bragg peak positions plotted for diamond cubic Si, Na4Si4 and silicon bisamidinate precursors. (b) HRTEM image of an individual particle produced in DMF showing lattice fringes corresponding to d-spacing of (111) cubic Si plane. (c) Raman spectra of (black) silicon bisamidinate, (grey) sodium silicide, and particles produced in (blue) DMF and (red) toluene. | ||
Raman spectroscopy was performed for a more complete understanding. The Raman spectra of the two precursors and the nanoparticles formed in these two different solvents were recorded (Fig. 3c; Fig. S7†). For the sodium silicide Zintl phase, three signals in the region of interest for silicon bonding were present at 284, 307, and 489 cm−1 corresponding to the [Si4]4− cluster vibrations, which have previously been reported.37 For silicon bisamidinate, the experimental spectrum was compared to a DFT simulation which reproduces well the experimental results (Fig. S8†). The Si–Cl asymmetrical stretching mode is observed at 358 cm−1 (experimental) vs. 342 cm−1 (calculated), which is comparable to other hypervalent silicon complexes.38 The bands associated with the precursors are not found in the Raman spectra of the silicon nanoparticles. However, in all three cases, a prominent band is observed at ∼500 cm−1, which is associated with the transverse optical (TO) mode for silicon, typically between 480 cm−1 and 520 cm−1.
Bright field TEM images show that the silicon particles vary in size and shape (Fig. 4a and c and Fig. S5†). In comparison to particles produced in toluene (median particle size = 43.8 nm), the particles produced in DMF were much smaller (median particle size = 15.8 nm). The change in particle size is possibly due to a difference in reduction potential as a function of solvent.39 Even so, the silicon nanoparticles produced were crystalline, with a Raman shift of ∼500 cm−1, indicating that the degree of crystallinity is not very sensitive to the solvent composition (Fig. 3c).
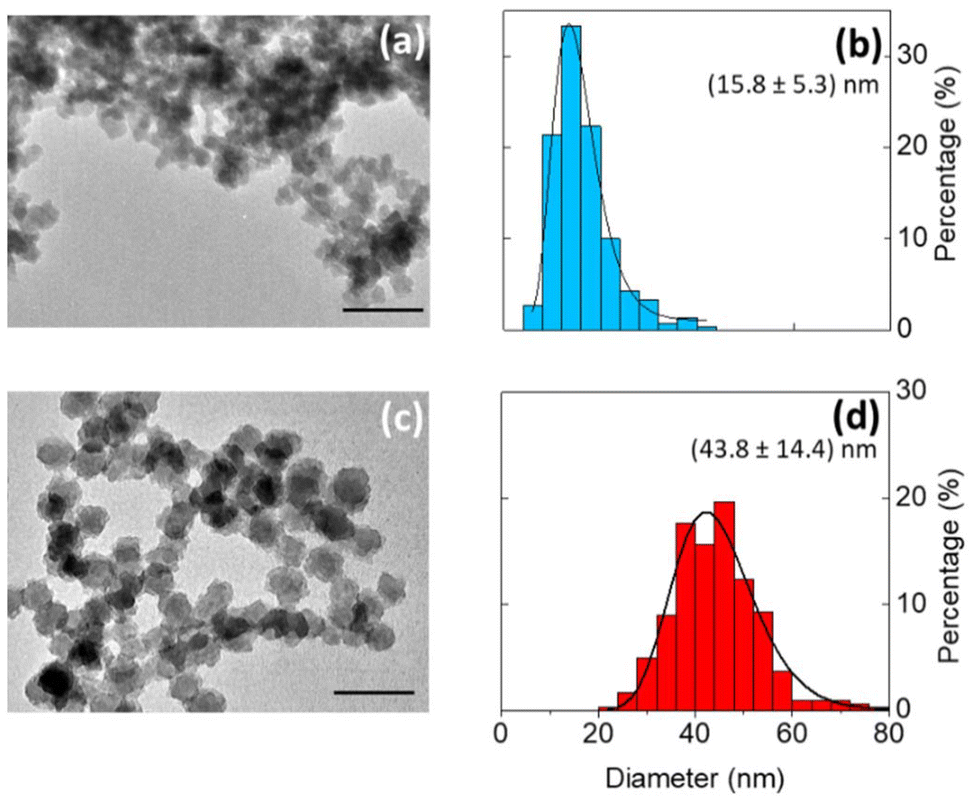 | ||
Fig. 4 (a and c) Bright field TEM images, and (b and d) particle size distribution of silicon particles produced in (a and b) DMF and (c and d) toluene. Scale bars represent 100 nm. All particle sizes were determined by analysis of TEM images (300 particles counted), sizes are given as (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ± σx) nm and fitted using a log–normal distribution. ± σx) nm and fitted using a log–normal distribution. | ||
The silicon particles produced in both DMF and toluene were dispersed in ethanol for photoluminescence measurements, but only the nanoparticles prepared in toluene exhibited a detectable photoluminescence emission signal. The recorded photoluminescence spectra displayed an excitation wavelength-dependent emission band in the UV-blue spectral region (Fig. 5). The ethanol solvent alone was also measured and peaked at a significantly lower intensity at 340 nm. (Fig. 5a and Fig. S9†). The excitation and absorption spectra for the silicon nanoparticles produced in toluene and for ethanol are provided in Fig. S10 and S11.† Here, the blue photoluminescence emission from the silicon nanoparticles, commonly named F-band because of its fast decay, is not attributed to the direct bandgap transition as observed in ultrasmall silicon nanocrystals 1–2 nm in diameter. It is rather ascribed to recombinations through silicon particle defects due to surface oxidation and nitrogen impurities, that, in the present study, could arise from the bisamidinate precursor and/or the formamide solvent during nanoparticle washing.
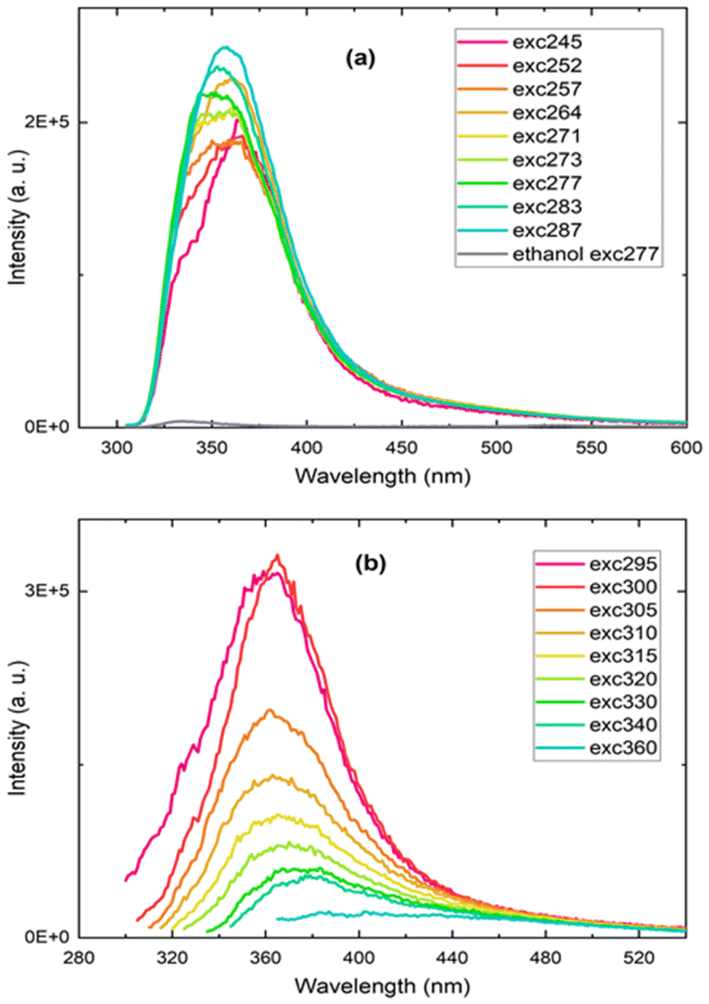 | ||
| Fig. 5 Photoluminescence emission spectra of silicon nanoparticles prepared in toluene and further dispersed in ethanol. The photoluminescence spectra were obtained with different excitation wavelengths : (a) excitation from 245 nm to 287 nm (b) excitation from 295 nm to 360 nm. The PL emission spectrum of ethanol alone is shown in (a) for comparison. | ||
The origin of the blue photoluminescence from surface defect states was previously established by several groups, in particular for silicon nanocrystals obtained by liquid-phase synthesis methods involving relatively mild temperature and pressure conditions, consistent with the conditions of the particles synthesized in this work (room temperature and pressure).2,40–43 In order to confirm the presence of silicon oxide on the silicon nanoparticles, Fourier Transform Infrared (FTIR) spectra of the silicon nanoparticles were recorded (Fig. S12†). We observe a band centered at 1100 cm−1 assigned to the stretching vibration mode of Si–O–Si bonds, confirming partial surface oxidation.
The evaluation of the photoluminescence quantum yield was performed on silicon nanoparticles suspended in ethanol, indicating a low quantum yield below 1%. The photoluminescence quantum efficiency of silicon nanostructures relies on their surface state and reactivity and thus can be potentially boosted by means of more controlled surface functionality.
4. Conclusions
This one-step solution synthesis offers a facile and bottom-up route toward the production of silicon nanoparticles. We have synthesized crystalline silicon nanoparticles up to 44 nm in diameter via an original redox reaction between Na4Si4, and a hypervalent silicon precursor under ambient conditions. Because this reaction is performed at room temperature, it is possible to use common organic solvents as the dispersive media and particle size can be adapted by changing the solvent, while allowing crystallinity to be preserved in all studied systems. We are currently investigating surfactants and reaction conditions to gain further control over nanoparticle morphology. We foresee ample opportunities for control over particle size, shape, and surface modification by slight modification of the synthesis procedure.Photoluminescence tests show that it is possible for relatively large particles (44 nm) with large crystalline domains (22 nm) to display photoluminescence signal in the blue region of the visible range, associated with oxide- and possibly nitrogen-related defects. Further surface chemical treatments would be necessary to improve the photoluminescence properties of the nanoparticles and enhance their potential in light emitting applications.
Author contributions
MLDM and GLD conceptualized the project. MAP, GLD, MG, PR, and DP developed the methodology. MAP, MLDM, AG, ACG, SP, EAH, SL, AB, FC, MG, and PR participated in investigations. PR, MG, and GLD supervised the project. MAP, MLDM, FC, and GLD wrote the original draft. All authors contributed to the reviewing and editing of the submitted version. GLD and DP acquired funding for the research. GLD was responsible for project administration. All authors have given approval to the final version of the manuscript.Conflicts of interest
MAP, MLDM, DP, PR, MG and GLD have submitted a patent, entitled “Process for Preparing Crystalline Silicon Nanoparticles”, number EP23306951.Acknowledgements
MAP, ACG and GLD were supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement No. 948319). MAP also acknowledges support from the LIGHT S&T Graduate Program (PIA3 Investment for the Future Program, ANR-17-EURE-0027). MLDM and GLD recognize the LabEx AMADEus (ANR-10-LABX-42) in the framework of IdEx Bordeaux (ANR-10-IDEX-03-02), i.e. the Investissements d'Avenir program of the French government managed by the Agence Nationale de la Recherche. A. G. and D. P. received funding from the European Research Council (ERC) Consolidator Grant GENESIS under the European Union's Horizon 2020 research and innovation program (grant agreement No 864850). Computer time for this study was provided by the computing facilities MCIA (Mésocentre de Calcul Intensif Aquitain) of the Université de Bordeaux. 1H NMR measurements were obtained at the Institut Europeén de Chimie et Biologie (IECB) NMR platform with the help of Estelle Morvan and Axelle Grélard. Eric Lebraud assisted in collecting powder XRD data through the XRD service of the ICMCB. Photoluminescent quantum yield evaluation was realized with the help of Sebastien Clement, and Emilie Lambert assisted in performing UV-vis absorption spectra at the Institut Charles Gerhardt Montpellier (ICGM).References
- A. N. Thiessen, L. Zhang, A. O. Oliynyk, H. Yu, K. M. O'Connor, A. Meldrum and J. G. C. Veinot, Chem. Mater., 2020, 32, 6838 CrossRef CAS.
- R. Sinelnikov, M. Dasog, J. Beamish, A. Meldum and J. G. C. Veinot, ACS Photonics, 2017, 4, 1920 CrossRef CAS.
- G. M. Carroll, R. Limpens and N. R. Neale, Nano Lett., 2018, 18, 3118 CrossRef CAS PubMed.
- J. J. Romero, M. L. Dell'Arciprete, H. B. Rodríguez, E. Gonik, D. Cacciari, A. L. Moore and M. C. Gonzalez, ACS Appl. Nano Mater., 2022, 5, 8105 CrossRef CAS.
- J. L. Heinrich, C. L. Curtis, G. M. Homsy, K. L. Kavanagh and M. J. Sailor, Science, 1992, 255, 66 CrossRef CAS PubMed.
- Q. Li and R. Jin, Nanotechnol. Rev., 2017, 6, 601 CAS.
- A. Guleria, S. Neogy, D. K. Maurya and S. Adhikari, J. Phys. Chem. C, 2017, 121, 24302 CrossRef CAS.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, Chem. Mater., 2006, 18, 6139 CrossRef CAS.
- W. Sun, C. Qian, X. S. Cui, L. Wang, M. Wei, G. Casillas, A. S. Helmy and G. A. Ozin, Nanoscale, 2016, 8, 3678 RSC.
- T. Popelář, P. Galář, F. Matějka, G. Morselli, P. Ceroni and K. Kůsová, J. Phys. Chem. C, 2023, 127, 20426 CrossRef.
- R. Tacke and T. Ribbeck, Dalton Trans., 2017, 46, 13628 RSC.
- H. H. Karsch, P. A. Schlüter and M. Reisky, Eur. J. Inorg. Chem., 1998, 4, 433 CrossRef.
- S. Rendler and M. Oestreich, Synthesis, 2005, 1727 CAS.
- D. Neiner, H. W. Chiu and S. M. Kauzlarich, J. Am. Chem. Soc., 2006, 128, 11016 CrossRef CAS PubMed.
- K. A. Pettigrew, Q. Liu, P. P. Power and S. M. Kauzlarich, Chem. Mater., 2003, 15, 4005 CrossRef CAS.
- B. M. Nolan, T. Henneberger, M. Waibel, T. F. Fässler and S. M. Kauzlarich, Inorg. Chem., 2015, 54, 396 CrossRef CAS PubMed.
- Y. Song, I. Gómez-Recio, R. Kumar, C. C. Diogo, S. Casale, I. Génois and D. Portehault, Dalton Trans., 2021, 50, 16703 RSC.
- M. Beekman, S. M. Kauzlarich, L. Doherty and G. S. Nolas, Materials, 2019, 12, 1139 CrossRef CAS PubMed.
- X. Zhang, D. Neiner, S. Wang, A. Y. Louie and S. M. Kauzlarich, Nanotechnology, 2007, 18, 095601 CrossRef PubMed.
- D. Mayeri, B. L. Phillips, M. P. Augustine and S. M. Kauzlarich, Chem. Mater., 2001, 13, 765 CrossRef CAS.
- C. S. Yang, R. A. Bley, S. M. Kauzlarich, H. W. H. Lee and G. R. Delgado, J. Am. Chem. Soc., 1999, 121, 5191 CrossRef CAS.
- S. K. Bux, M. Rodriguez, M. T. Yeung, C. Yang, A. Makhluf, R. G. Blair, J. P. Fleurial and R. B. Kaner, Chem. Mater., 2010, 22, 2534 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. Gritzner and J. Kuta, J. Pure Appl. Chem., 1984, 56, 461 CrossRef.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada and D. Fox, Gaussian 16, Revision A02, 2009 Search PubMed.
- J. J. P. Stewart, J. Mol. Model., 2013, 19, 1 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- N. M. O′boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef PubMed.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086 RSC.
- S. Alvarez, Chem. Rev., 2015, 115, 13447 CrossRef CAS PubMed.
- A. D. Beck, L. Schäffer, S. Haufe and S. R. Waldvogel, Eur. J. Org. Chem., 2022, e202201253 CrossRef CAS.
- Y. Wang, J. E. McGrady and Z. M. Sun, Acc. Chem. Res., 2021, 54, 1506 CrossRef CAS PubMed.
- K. Annou, M. Pelosi, G. Gershinsky, G. Favier, Y. Cuminal, M. Tillard and D. Zitoun, Mater. Renew. Sustain. Energy, 2014, 3, 32 CrossRef.
- D. Boal and G. A. Ozin, Can. J. Chem., 1973, 51, 609 CrossRef CAS.
- S. Semlali, B. Cormary, M. L. D. Marco, J. Majimel, A. Saquet, Y. Coppel, M. Gonidec, P. Rosa and G. L. Drisko, Nanoscale, 2019, 11, 4696 RSC.
- J. Fuzell, A. Thibert, T. M. Atkins, M. Dasog, E. Busby, J. G. C. Veinot, S. M. Kauzlarich and D. S. Larsen, J. Phys. Chem. Lett., 2013, 4, 3806 CrossRef CAS.
- M. Dasog, Z. Yang, S. Regli, T. M. Atkins, A. Faramus, M. P. Singh, E. Muthuswamy, S. M. Kauzlarich, R. D. Tilley and J. G. C. Veinot, ACS Nano, 2013, 7, 2676 CrossRef CAS PubMed.
- L. Canham, Faraday Discuss., 2020, 222, 10–81 RSC.
- J. Valenta, A. Fucikova, I. Pelant, K. Kusova, K. Dohnalova, A. Aleknavicius, O. Cibulka, A. Fojtik and G. Kada, New J. Phys., 2008, 10, 073022 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD, Raman spectra, TEM of samples at varying temperatures, DFT simulations. CCDC 2157265. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nr05793c |
| ‡ Currently: Institut de Physique et de Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504, Université de Strasbourg-CNRS, 23, Rue du Loess, 67034 Strasbourg, France. E-mail: maria-letizia.demarco@ipcms.unistra.fr |
| § Present address: Orleans. E-mail: alexander.castro-grijalba@univ-orleans.fr |
| This journal is © The Royal Society of Chemistry 2024 |