DOI:
10.1039/D4NR01196A
(Paper)
Nanoscale, 2024,
16, 14020-14032
ZIF-67-derived Co–N–C supported nickel cobalt sulfide as a bifunctional electrocatalyst for sustainable hydrogen production via alkaline electrolysis†
Received
18th March 2024
, Accepted 3rd July 2024
First published on 4th July 2024
Abstract
As non-renewable resources are finite and cannot be utilized indefinitely, hydrogen (H2) has emerged as a promising alternative for clean and sustainable energy. The cost-effective hydrogen production to meet large-scale commercial demand poses a significant challenge. Water electrolysis, powered by electricity derived from renewable resources, stands out as a viable route towards sustainable hydrogen production, with electrocatalysis playing a pivotal role in this process. Notably, materials derived from metal–organic frameworks (MOFs) exhibit excellent physicochemical properties, making them promising candidates for electrocatalysis. In this study, we synthesized zeolitic imidazolate framework-67 (ZIF-67) and its derived Co–N-doped carbon (Co–N–C) supported NiCo2S4 on nickel foam (NF), namely NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4, using a hydrothermal method. The electrocatalytic activity of these synthesized materials for both oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) was systematically evaluated using various electrochemical techniques. The NF@ZIF-67@NiCo2S4 material demonstrates overpotentials of 248 and 359 mV for OER and HER at the current density of 50 mA cm−2, whereas, NF@Co–N–C@NiCo2S4 exhibits overpotentials of 239 and 351 mV, respectively. Furthermore, the catalysts exhibit excellent stability in both OER and HER even under high applied potentials. Moreover, to assess their catalytic performance in a full-cell configuration, two alkaline electrolyzer cells were assembled: NF@ZIF-67@NiCo2S4(+)∥NF@ZIF-67@NiCo2S4(−) and NF@Co–N–C@NiCo2S4(+)∥NF@Co–N–C@NiCo2S4(−). These two electrolyzers demonstrated cell potentials of 1.62 V and 1.59 V at 10 mA cm−2, respectively, showcasing their efficacy in overall water-splitting.
1. Introduction
For over a century, fossil fuels have fuelled our vehicles, lit our world, and powered industries. Currently, around 80% of our energy needs are fulfilled by using oil, coal, and gas. This reliance on fossil fuels has profoundly affected both life and the environment, causing widespread pollution and contributing to global warming.1 Hence, integrating renewable energy resources like wind and solar into the grid is crucial. However, intermittent power supply and seasonal variations of renewable resources pose challenges for seamless incorporation. Robust energy conversion and storage capabilities are needed to facilitate load-levelling and provide a viable solution. Water electrolysis being an energy conversion technology, breaking water into O2 and H2 with the help of electricity offers a promising approach to address these challenges by producing easy storable green H2 in liquid form.2 There are three primary categories of water electrolysis technologies, distinguished by the type of electrolyte employed: alkaline, proton exchange membrane (PEM), and anion exchange membrane (AEM) electrolyzers. Alkaline and AEM electrolyzers, employing nickel-based catalysts, are cost-effective for H2 production but have lower efficiency. In contrast, PEM electrolyzers are more efficient but come at a higher cost due to the platinum-based metal catalysts.3 In all the cases, the change in free energy for splitting water into H2 and O2 is ΔG = +237.2 kJ mol−1 under standard conditions. Regardless of whether the reaction medium is neutral, acidic, or alkaline, the standard thermodynamic potential (Ecell) for water electrolysis remains constant at 1.23 V at 25 °C.4,5 However, the actual production of H2 and O2 from H2O at the cathode and anode in water electrolysis requires overcoming activation barriers and resistances, including solution and contact resistance. The additional potential required to overcome these barriers is known as overpotential (η). Efficient water splitting depends on minimizing overpotential, especially for the OER at the anode. Using effective OER catalysts with low overpotential enhances the energy efficiency of electrolysis.6 The central challenge lies in determining how OER can be catalyzed efficiently on the electrode surface to minimize overpotential and achieve the highest current density. Benchmark materials, such as noble metal oxides (e.g., RuO2 and IrO2), known for their stability in highly oxidizing environments, have been employed to improve the OER process.6,7 However, their exorbitant cost and limited availability significantly restrict their widespread use. The current research objective is to develop an economical and effective electrocatalyst for OER that not only enhances overall water-splitting performance but also holds promise for commercialization.
The search for efficient electrocatalysts, derived from abundant earth metals, for both the OER and HER is crucial. An extensive literature review reveals numerous electrocatalytic systems for OER and HER, featuring diverse elemental compositions and microstructures synthesized through various methods. Significant research efforts have focused on exploring cost-effective catalysts for OER and HER, spanning a wide range of materials, including transition metal alloys, hydroxides, oxyhydroxides, oxides, sulfides, nitrides, selenides, carbides, phosphides, oxysulphides, borides, hydroxide carbonates, hydroxyl phosphates, and various carbon-based materials.4,6,8–11 Given that an alkaline medium is less corrosive compared to strong acidic conditions, considerable attention has been directed towards developing efficient and durable bifunctional electrocatalysts suitable for both the OER and HER in an alkaline environment. In this context, materials based on transition metals from the 3d series hold special significance owing to their low cost, abundance, and facile synthesis.6,9–11 In recent times, MOFs have emerged as valuable templates for generating a diverse range of materials through additional processing. The utilization of MOFs as a template offers a versatile and tunable platform for creating advanced materials with enhanced properties, making them particularly attractive for various applications in science and technology.12–16 MOFs, with metal centers linked by organic linkers, offer significant advantages. Their ultrahigh porosity (up to 90% free volume) and expansive surface areas (up to 6000 m2 g−1) make them exceptionally attractive for applications like catalysis, gas storage, and sensing.15 Moreover, careful control of morphology and microstructure exposes more active sites, enhancing surface area. This optimization improves ion, or mass transport through the porous architecture, maximizing efficiency and reactivity. This is crucial for applications, especially in electrocatalysis.13,14 Thermal decomposition of MOF in a nitrogen atmosphere produces metal-incorporated N-doped carbon, known as M–N–C, which exhibits higher catalytic activity attributed to enhanced conductivity and surface area.16
Recently, transition metal chalcogenides (TMCs) have become a focal point of interest due to their remarkable catalytic activity, distinctive electronic properties, and versatile structural designs.17,18 Notably, these materials exhibit a wide range of electronic properties, transitioning from insulators such as HfS2, semiconductors like MoS2 and WS2, to semimetals like TiSe2, and metals including NbS2 and VSe2.19 This diversity in electronic characteristics makes TMCs highly promising for a myriad of applications. Recent studies have noted that TMCs demonstrate exceptional catalytic activity compared to other transition metal catalysts. This enhanced performance is attributed to factors such as their thermo-neutral H-binding energy, large surface area, and higher proportion of exposed atoms.20 These traits make TMCs highly efficient in catalysing diverse reactions and hold promise for applications in electrocatalysis and energy conversion.21 Despite notable progress in developing electrocatalytic materials with TMCs, their activities still face significant limitations. Challenges include low electrical conductivity, poor stability, aggregation issues, inert basal plane, and undesirable water adsorption.22 Addressing these factors is paramount for unlocking the full potential of TMCs, particularly in electrocatalysis for sustainable energy conversion. Ongoing research endeavors aim to surmount these limitations and enhance the performance of TMC-based electrocatalytic materials.23–25 Among the transition metal sulfides mentioned earlier, bimetallic sulfides containing Ni and Co have garnered considerable attention in recent reports. For instance, Biswas et al. demonstrated the fabrication of Zn0.76Co0.24S/NiCo2S4 binary composite flowers, showcasing impressive electrocatalytic properties with overpotentials of 248 mV and 141 mV for the OER and HER, respectively, at a current density of 10 mA cm−2.26 Similarly, Li et al. synthesized cerium-doped nickel sulfide nanospheres grown on Ni foam via a two-step hydrothermal approach. This material exhibited exceptional electrocatalytic performance for OER, achieving an overpotential of 283 mV at 10 mA cm−2.27 Considering the aforementioned factors, our study focuses on synthesizing ZIF-67-supported NiCo2S4 and ZIF-67-derived Co–N–C supported NiCo2S4 on NF, denoted as NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4, respectively. We aim to investigate their potential for catalyzing the OER, HER, and overall water-splitting (OWS) processes. In situ growth of ZIF-67@NiCo2S4 and Co–N–C@NiCo2S4 on porous NF eliminates the need for a binder, enhancing contact and electrical conductivity between NF and the catalytic electrode material. This direct integration on the porous substrate optimizes electrocatalyst performance, emphasizing efficient electron transport during electrochemical processes.28,29 The NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials have demonstrated highly effective catalytic activities for OER. Notably, the electrocatalytic activity for HER exhibited a substantial improvement after stability testing. This improvement is attributed to phase changes and morphological alterations in the materials during the hydrogen evolution process. Importantly, the alkaline electrolyzers constructed using these materials exhibited remarkably low cell potentials at a current density of 10 mA cm−2, highlighting their efficiency in facilitating OWS processes.
2. Experimental section
2.1. Materials
Cobalt nitrate hexahydrate (Co(NO3)2·6H2O), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), KOH pellets (85%), HCl (35%), acetone were used of extra pure grade and purchased from FINAR. 2-Methylimidazole, thiourea from SRL, double-deionized (DI) water and absolute ethanol were used for synthesis and cleaning purposes.
2.2. Synthesis
2.2.1. Washing of nickel foam.
A commercially available nickel foam (2 × 4 cm2) was cleaned by sonicating it for 10 minutes in 3 M HCl. The impurities were removed by sonicating the foam in DI water, followed by a mixture of water and acetone, and then pure acetone for 2–3 minutes. The nickel foam was then dried at 40 °C for an hour.
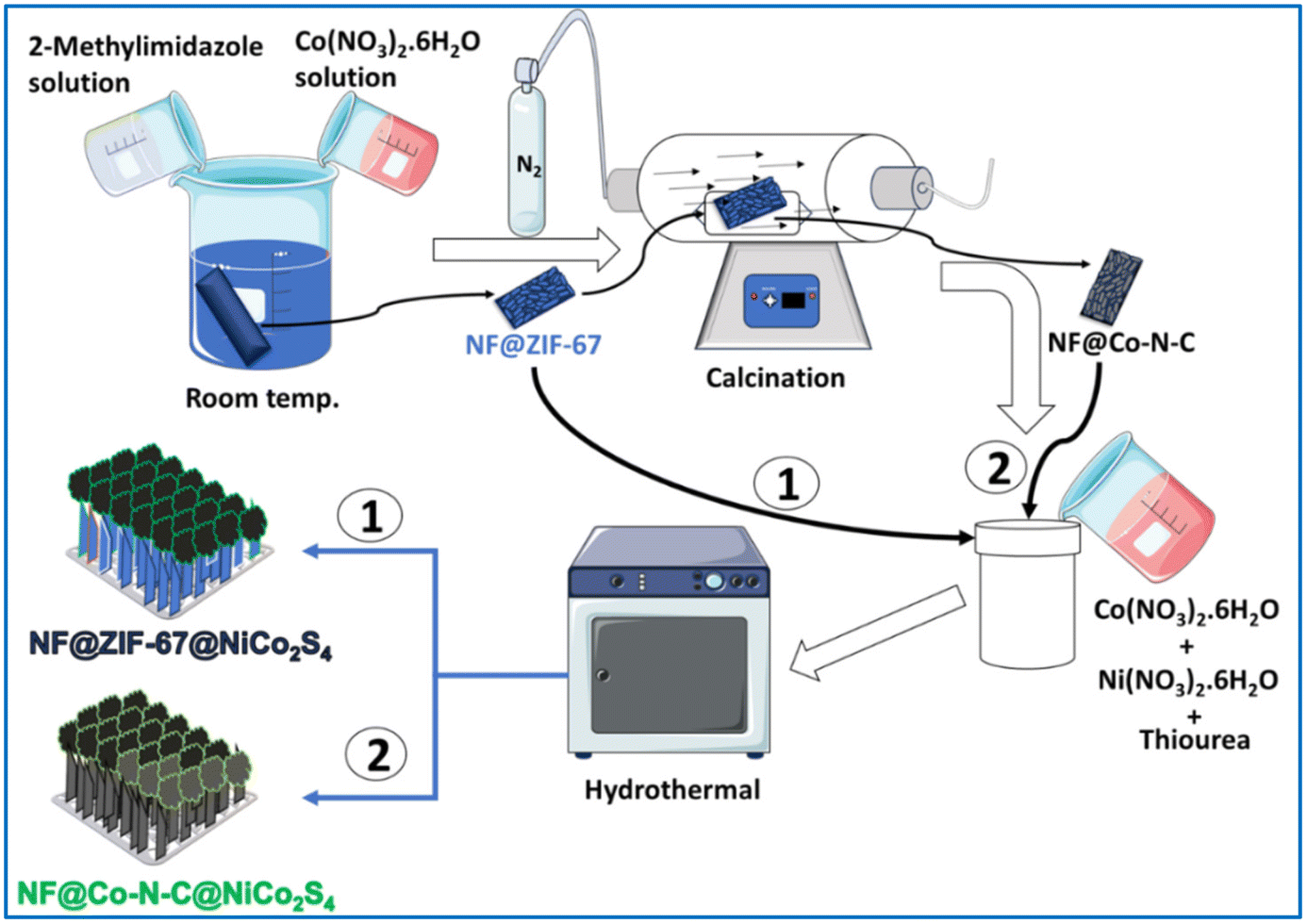
2.2.2. Synthesis of NF@ZIF-67.
In a typical synthesis of ZIF-67, 1 mmol of Co(NO3)2·6H2O and 16 mmol of 2-methylimidazole were dissolved in 40 mL of DI water in separate beakers and stirred for 10 minutes to obtain the homogeneous solution. After that 2-methylimidazole solution was slowly added to the cobalt nitrate solution. After mixing, the color of the solution turned to deep blue. A clean 2 × 4 cm2 NF was kept in the solution for 12 h at room temperature without stirring, and blue-colored material (i.e. ZIF-67) was deposited on the NF named NF@ZIF-67. The deposited ZIF-67 on NF was washed with DI water, a mixture of water and ethanol and finally with ethanol, and then dried at 60 °C for 8 h in a conventional oven. The bare NF and blue-colored ZIF-67 deposited on NF are shown in Fig. S1a and b.†
2.2.3. Synthesis of NF@ZIF-67@NiCo2S4.
The synthesized NF@ZIF-67 was introduced into a Teflon-lined stainless-steel autoclave of 50 mL capacity, replacing the NF used in the previous synthesis, and kept at 120 °C for 12 h in the hot air oven. A dark-colored material was deposited on the NF@ZIF-67 denoted as NF@ZIF-67@NiCo2S4. The final material was washed with DI water, a mixture of water and ethanol, and finally with ethanol. After washing, the material was carefully dried at 60 °C for 8 hours. A visual representation of NF@ZIF-67@NiCo2S4 can be observed in the digital image presented in Fig. S1c.†
2.2.4. Synthesis of NF@Co–N–C@NiCo2S4.
Before the calcination of ZIF-67, its thermal stability was assessed through thermogravimetric analysis (TGA). The TGA results indicated a significant weight loss below 600 °C under a nitrogen atmosphere (Fig. S2†). This observation highlights the necessity of utilizing a calcination temperature when N2 gas is employed as the carrier gas for the synthesis of the desired porous N-doped carbon materials. Prior to initiating the temperature program, N2 at a flow rate of approximately 60 mL min−1 was introduced for 10 minutes to purge the chamber of air. The pre-synthesized NF@ZIF-67 underwent calcination at 500 °C for 2 hours in an N2 atmosphere using a ramp rate of 2 °C min−1 in a tubular furnace. After allowing the material to naturally cool to room temperature, the resulting NF coated with the black-colored sample was designated as NF@Co–N–C. Subsequently, a further coating of NiCo2S4 was applied to NF@Co–N–C following the previously outlined procedure. The resulting material, with NiCo2S4 deposited on NF@Co–N–C, is referred to as NF@Co–N–C@NiCo2S4. A visual depiction of NF@Co–N–C@NiCo2S4 can be found in the digital image presented in Fig. S1d of the ESI.†
2.2.5. Synthesis of NF@NiCo2S4.
For comparison, NF@NiCo2S4 was synthesized as follows: initially, 0.67 mmol of Ni(NO3)2·6H2O, 1.34 mmol of Co(NO3)2·6H2O and 4 mmol of thiourea were dissolved in 30 mL of DI water. The resultant aqueous solution and thoroughly washed NF were transferred into the Teflon-lined stainless-steel autoclave of 50 mL capacity and kept at 120 °C for 12 h in the hot air oven, a dark-colored material was deposited on the NF denoted as NF@NiCo2S4.
2.3. Physicochemical characterizations
The Thermogravimetric Analyzer (TGA) SDT Q600 from TA Instruments is employed for TGA analysis. The powder X-ray diffraction (PXRD) patterns were acquired using Cu Kα radiation with a wavelength (λ) of 1.54 Å, employing the XPERT-PRO diffractometer. For the field emission scanning electron microscopy (FE-SEM) and energy-dispersive X-ray (EDX) spectroscopy analyses, the Thermo Fisher FEI-Quanta 250 FEG (FE-SEM) instrument was utilized. High-resolution transmission electron microscopy (HR-TEM) was conducted with the assistance of the 200 kV Jeol/JEM 2100 instrument. X-ray photoelectron spectroscopy (XPS) analysis was conducted using the ULVAC PHI (VersaProbe III) instrument.
2.4. Electrochemical measurements
All electrochemical experiments were conducted using a BioLogic Science Instrument (SP-150e) workstation. For investigations related to OER and HER, a conventional three-electrode configuration was employed. For overall OWS, a two-electrode configuration was adopted. All electrochemical experiments were carried out at room temperature to maintain consistent conditions across the experiments. In this set up, 1 × 1 cm2 area of NF deposited with NiCo2S4, ZIF-67@NiCo2S4 and Co–N–C@NiCo2S4 materials were directly used as working electrodes while a graphite rod and Hg/HgO (1.0 M KOH) electrodes used as counter and reference electrode, respectively. In the case of OER and HER all the potentials (vs. Hg/HgO) were converted to vs. reversible hydrogen electrode (RHE) using the Nernst equation ERHE = EHg/HgO/1 M KOH + 0.059 pH + 0.098 V. All the LSV curves were iR-corrected using the equation Ecorr = Emea − iRct where Ecorr is iR-corrected potential, Emea is experimentally measured potential, Rct is charge transfer resistance, and i is the corresponding current. The overpotential (η) values for water electrolysis were determined using the following equations: for OER, η = ERHE − 1.23 V and for HER, η = 0 − ERHE. For comparison, commercial Pt/C (20 wt%) and RuO2 (99.9 wt%, Alfa) were coated on a 1 cm2 area of NF. The catalyst inks were prepared by dispersing Pt/C and RuO2 (5 mg each) in separate vials. This dispersion was achieved in a solution mixture consisting of isopropyl alcohol (750 μL), deionized (DI) water (200 μL), and Nafion (50 μL), and the mixture was subjected to ultrasonication for 20 minutes. For practical applications, two alkaline electrolyzers were fabricated. The combinations were NF@ZIF-67@NiCo2S4(+)∥NF@ZIF-67@NiCo2S4(−) and NF@Co–N–C@NiCo2S4(+)∥NF@Co–N–C@NiCo2S4(−). These electrolyzers were then tested for their performance in the OWS. This experimental design allows for a comparative assessment of the catalytic activities of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 in alkaline electrolysis.
3. Results and discussion
3.1. Structural characterization
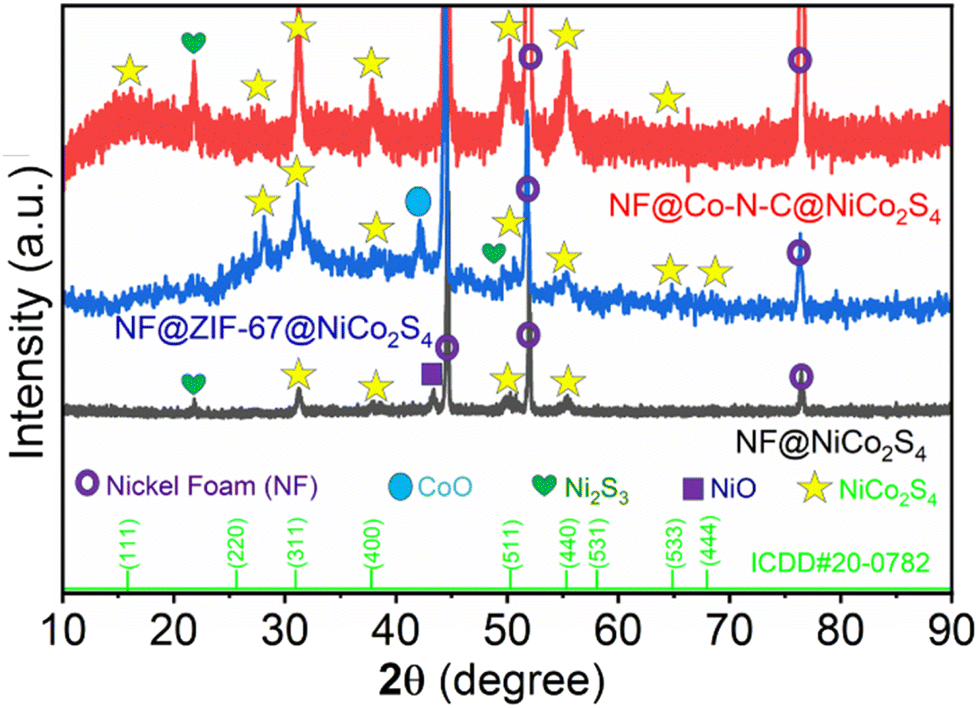
The detailed synthesis procedure is illustrated in Scheme 1. The PXRD pattern of NF@NiCo2S4, NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 are shown in Fig. 1. The diffraction pattern of NF@NiCo2S4 reveals distinct peaks at 2θ values of 31.23°, 38.5°, 50.4°, and 55.4°, corresponding to the crystallographic planes of (311), (400), (511), and (440), respectively. These findings strongly indicate the successful formation of NiCo2S4 (ICDD no. 20-0782) on the NF. Upon closer inspection, in the case of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4, there is a slight shift in these peaks, and some additional peaks are observed at 2θ values of 64.8° (533), 68.5° (444), and 16.4° (111). This shift and the appearance of new peaks suggest potential structural modifications or additional phases introduced by the presence of ZIF-67 and Co–N–C in the composite materials.30 The appearance of additional high intense peaks at 2θ values of 45°, 52°, and 76.9° is indicative of the underlying NF. Moreover, the observed decreasing in the NF diffraction peaks indicates the successful deposition of ZIF-67 and NiCo2S4 on NF. The additional peaks appeared at 21.8°, 42.16° and 43.3° corresponds to Ni2S3, CoO and NiO respectively.9,30 The diffraction analysis gives valuable insights into the structural composition and the interactions between the different components in the synthesized materials.
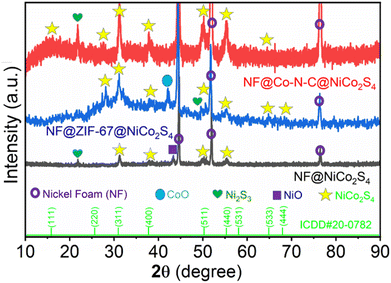 |
| | Fig. 1 PXRD patterns of NF@NiCo2S4, NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4. | |
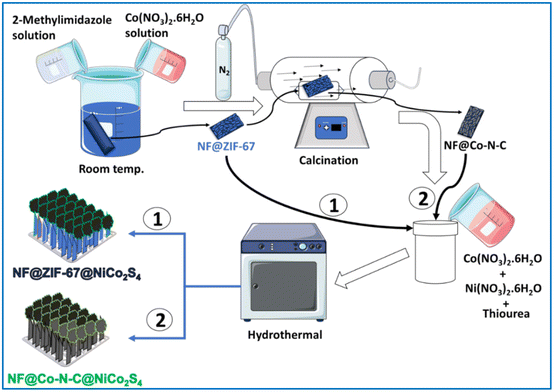 |
| | Scheme 1 Schematic illustration depicting the synthesis process for NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials. | |
Field emission scanning electron microscopy was employed to examine the surface morphological features of all the samples. In the case of the starting material (ZIF-67), both low and high-magnification FE-SEM images depict a microbuilding-like morphology (Fig. S3†). This structure is uniformly grown vertically on the porous NF. Following calcination, the low and high-magnification FE-SEM images of NF@Co–N–C (Fig. S4†) reveal a transformation in the surface characteristics. The initially uniform and smooth surface of Co-MOF microbuildings array underwent a change, resulting in a rough surface with decreased thickness and increased porosity. This transformation is attributed to the carbonization of the organic moiety during the calcination process. The FE-SEM images provide visual insights into the structural changes induced by the calcination treatment in NF@Co–N–C. Following the deposition of NiCo2S4 on ZIF-67, the resulting NF@ZIF-67@NiCo2S4 exhibits a distinctive flake-like morphology, as illustrated in Fig. 2a–c. In Fig. 2c, the circled region specifically highlights the tip of the micro-building blocks, corresponding to the morphology of ZIF-67. This observation strongly indicates that NiCo2S4 is indeed grown on ZIF-67. The FE-SEM images (Fig. 2a–c) further showcase the uniform growth of NiCo2S4 flakes on the micro-buildings of ZIF-67, comprising a cross-linking alignment. The average thickness of these flakes is measured to be ∼23 nm. These FESEM images visuals further confirm the successful growth and uniform distribution of NiCo2S4 on the ZIF-67 microstructures. In Fig. 2d, the X-ray elemental color mapping shows further confirmation of the presence and uniform distribution of Ni, Co, and S elements on the surface. This elemental analysis helps in confirming the homogeneity of the synthesized material and provides valuable insights into its composition. The NF@Co–N–C@NiCo2S4 shows a marigold flower-like morphology, as shown in Fig. 2e–g. Notably, due to the significant porosity of Co–N–C, the petals of NiCo2S4 coalesce and form a distinctive flower-like structure with an average diameter measuring ∼1.1 μm. This unique morphological feature suggests a synergistic interaction between the Co–N–C framework and the deposited NiCo2S4, contributing to the formation of this visually captivating and well-defined structure. In Fig. 2h, the elemental color mapping assists as additional confirmation for the presence and uniform distribution of Ni, Co, and S elements on the surface of the NF@Co–N–C@NiCo2S4 material. The uniform and consistent color distribution across the material in Fig. 2h reinforces the successful incorporation of Ni, Co, and S elements, providing further evidence of their homogeneous presence in the synthesized NF@Co–N–C@NiCo2S4 composite.
 |
| | Fig. 2 Low- and high-magnification FESEM images of NF@ZIF-67@NiCo2S4 (a, b, c) and NF@Co–N–C@NiCo2S4 (e, f, g), (d) elemental color mapping images showing the distribution of Ni, Co, and S in NF@ZIF-67@NiCo2S4, (h) elemental color mapping images showing the distribution of Ni, Co and S in NF@Co–N–C@NiCo2S4. | |
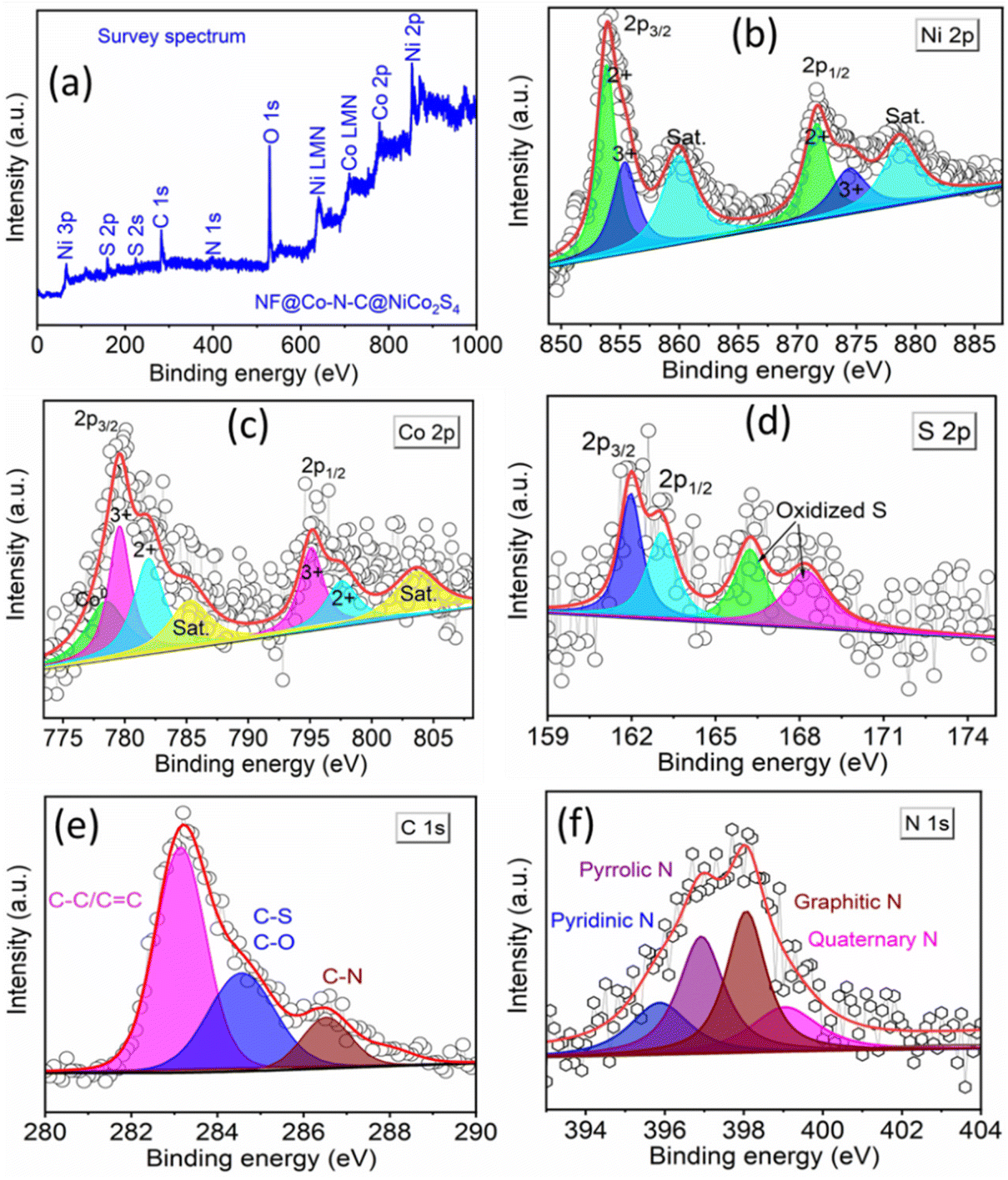
To gain deeper insights into the structural details of NiCo2S4 coated on NF@ZIF-67 and NF@Co–N–C, HR-TEM images were recorded. Fig. 3a and b shows the HR-TEM images of NiCo2S4 coated on NF@ZIF-67, while Fig. 3d and e depicts the corresponding images for NF@Co–N–C@NiCo2S4. These HR-TEM images offer a closer look at the nanostructures, providing valuable information about the size, shape, crystallinity, and arrangement of the materials. This detailed analysis contributes to a comprehensive understanding of the structural-related information for NiCo2S4 deposited on NF@ZIF-67 and NF@Co–N–C. Fig. 3a shows low-magnification TEM images indicating the deposition of NiCo2S4 on ZIF-67. Inset images in Fig. 3b, d and e reveal an interlayer spacing of ∼0.24 nm for both materials, corresponding to the (400) plane of NiCo2S4. Inset of Fig. 3d shows an interlayer spacing of 0.24, 0.217 and 0.204 nm indicates the presence of NiCo2S4, CoO and Co, respectively. This meticulous analysis of high-magnification HRTEM images provides valuable insights into the crystallographic arrangement within in the material. Fig. 3c and f shows the selected area electron diffraction (SAED) pattern of NiCo2S4 coated on NF@ZIF-67 and NF@Co–N–C, respectively. The pattern exhibits bright spots in a ring formation, indicative of the polycrystalline nature of the material. The diffraction rings are attributed to the (511), (440), and (311) planes of NiCo2S4. This SAED pattern aligns with the XRD results, providing further confirmation of the formation of the NiCo2S4 phase in the synthesized material. In Fig. 3g, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image is presented, along with corresponding elemental color mapping indicating the presence of Ni, Co, S, N, and C. This comprehensive analysis aids to confirm both the calcination of ZIF-67 and the successful formation of Co–N–C. The observation of Ni, Co, S, N, and C signals across the material in Fig. 3g, as indicated by the elemental mapping, further supports the successful formation of NF@Co–N–C@NiCo2S4. Additionally, to provide the specific element content ratio, an energy dispersive X-ray (EDX) spectrum has been recorded and is shown in Fig. S5.† This spectrum reveals the presence of Ni, Co, N, C, and S. However, the exact metal-to-S ratio cannot be observed due to the deposition of NiCo2S4 on NF@Co–N–C, wherein Co exists in both Co–N–C and NiCo2S4, resulting in a higher atomic percentage than sulfur. Nevertheless, the XRD data clearly shows the formation of the NiCo2S4 phase (Fig. 1). This integrated approach utilizing advanced microscopy techniques and elemental mapping enhances the understanding of the structural and compositional aspects of the synthesized material. Certainly, previous studies have consistently shown that the catalytic activity of materials are significantly influenced by various factors, including the shape, size, conductivity, porosity, and orientation of their micro/nanostructures. In the specific case of NiCo2S4, its hierarchical flower-like architecture holds great promise for electrocatalytic applications, particularly in OWS (OER and HER) reactions. The unique morphology of the NiCo2S4 microflower, grown on the porous micro-buildings of Co–N–C, introduces several advantages. First, the hierarchical structure enhances the contact between the active electrode material and the electrolyte, facilitating efficient charge transfer during electrochemical reactions. Second, the porous micro-buildings of Co–N–C provide a high surface area, creating numerous active sites for electrocatalysis. This increased surface area promotes enhanced interaction between the material and electrolyte, thereby improving electrocatalytic activity. Furthermore, the hierarchical architecture of NiCo2S4 on Co–N–C offers short diffusion pathways for ions/molecules, a crucial factor in improving mass transport during water-splitting reactions. This feature can contribute to the overall efficiency of the electrocatalyst by minimizing diffusion limitations and ensuring rapid access of reactants to the active sites. Additionally, XPS was employed to investigate the surface elemental composition and the chemical states of the electrocatalyst. In Fig. 4a, the XPS survey spectrum shows the presence of C, N, O, Ni, Co, and S as the main elements in NF@Co–N–C@NiCo2S4. The Ni 2p deconvoluted spectrum shown in Fig. 4b reveals peaks at binding energies of 853.9 and 872.7 eV, corresponding to Ni 2p3/2 and Ni 2p1/2, respectively.21,30 The deconvoluted peaks at 853.8 and 872.6 are attributed to Ni2+, while those appeared at 855.4 and 874.3 eV correspond to the characteristics of Ni3+.18,30 The Co 2p XPS spectrum (Fig. 4c) shows the peaks at binding energies of 779.5 and 795.2 corresponds to Co 2p3/2 and Co 2p1/2.31 The deconvoluted peaks at 782 eV and 797.6 eV are assigned to Co2+, while the peaks located at 779.6 eV and 795.2 eV belong to Co3+. The additional peak appeared at 778.4 eV corresponds to Co0.9,31 In Fig. 4d, the S 2p spectrum shows peaks at 161.9 and 163.05 eV correspond to S 2p3/2 and S 2p1/2, respectively. Additional peaks at 166.23 eV and 168.21 eV signify the presence of sulfoxide and sulfate species.21,30 The deconvoluted C 1s XPS spectrum (Fig. 4e) shows three peaks at 283.1 eV, 284.6 eV, and 286.5 eV, assigned to (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C), (C–S/C–O), and (C–N), respectively.14,31 The N 1s XPS spectrum in Fig. 4f, shows four types of N species assigned to pyridinic N (396.9 eV), pyrrolic N (397.8 eV), graphitic N (398.1 eV) and quaternary N (BE = 399.3 eV).9,31 In summary, the designed hierarchical flower-like structure of NiCo2S4 on Co–N–C demonstrates a potential for improved electrocatalytic performance in water splitting applications, highlighting the significance of material morphology and composition in dictating its functional properties.
C/C–C), (C–S/C–O), and (C–N), respectively.14,31 The N 1s XPS spectrum in Fig. 4f, shows four types of N species assigned to pyridinic N (396.9 eV), pyrrolic N (397.8 eV), graphitic N (398.1 eV) and quaternary N (BE = 399.3 eV).9,31 In summary, the designed hierarchical flower-like structure of NiCo2S4 on Co–N–C demonstrates a potential for improved electrocatalytic performance in water splitting applications, highlighting the significance of material morphology and composition in dictating its functional properties.
 |
| | Fig. 3 High-magnification HRTEM images of NF@ZIF-67@NiCo2S4 (a, b) and NF@Co–N–C@NiCo2S4 (d, e), and selected area electron diffraction patterns of (c) NF@ZIF-67@NiCo2S4 and (f) NF@Co–N–C@NiCo2S4, (g) HAA DF-STEM image and corresponding elemental color mapping showing the presence of Ni, Co, S, N and C. | |
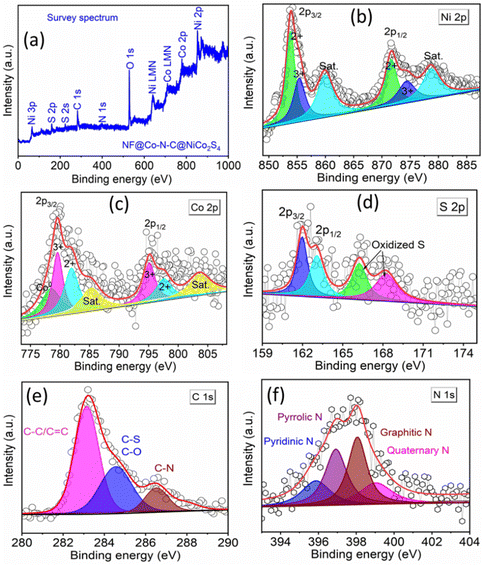 |
| | Fig. 4 X-ray photoelectron spectra of NF@Co–N–C@NiCo2S4: (a) survey spectrum, high-resolution spectra of (b) Ni 2p, (c) Co 2p, (d) S 2p, (e) C 1s, and (f) N 1s. | |
3.2. Electrocatalytic OER study
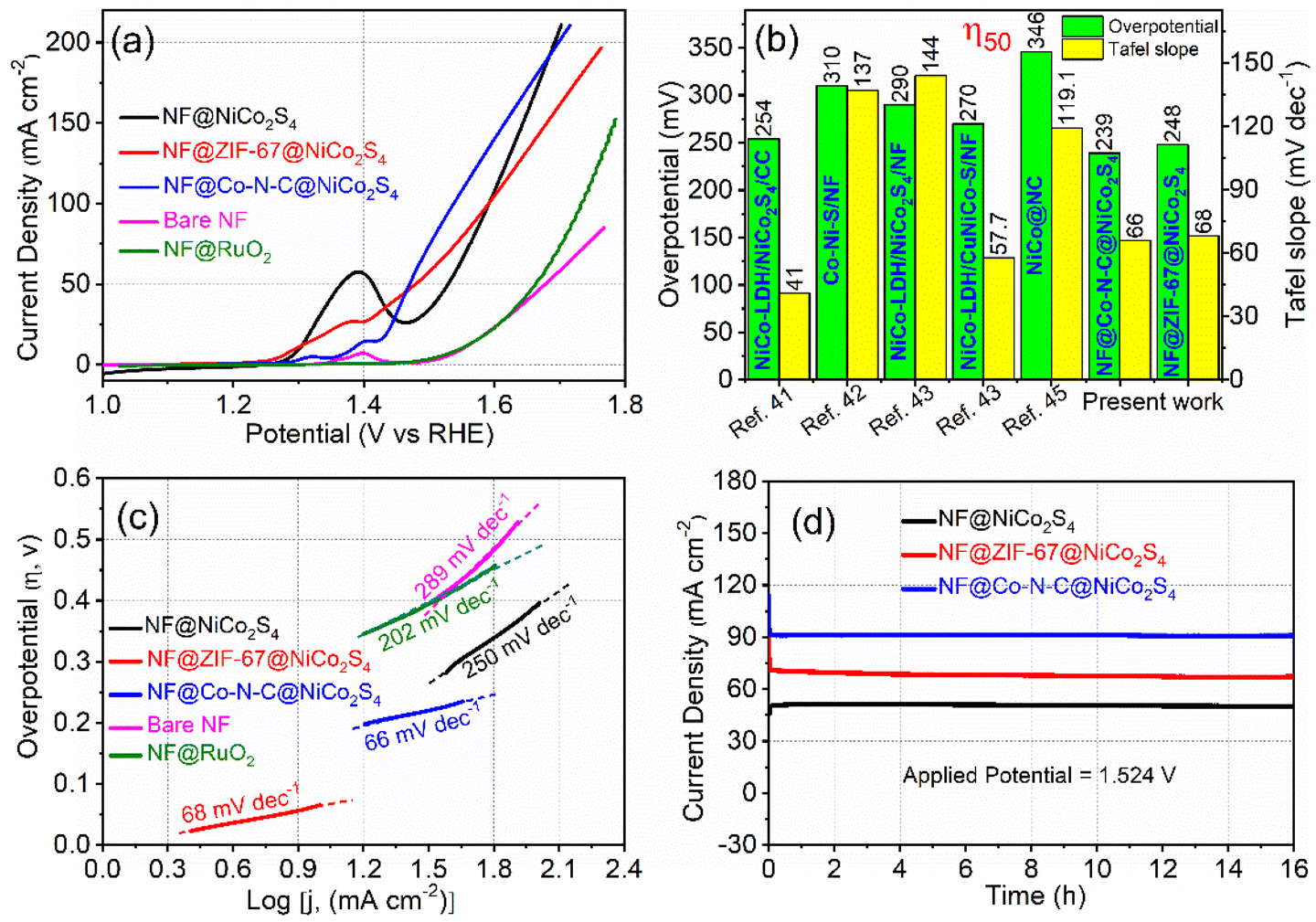
The electrocatalytic performances of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 for the OER were assessed using a standard three-electrode system in a 1 M KOH electrolyte, and all potentials were referenced to the RHE scale. Before conducting the actual OER experiments, each prepared sample underwent activation through 100 consecutive cyclic voltammetry (CV) cycles at a scan rate of 50 mV s−1 and within a potential range of 0.924 to 1.574 V. This activation step is crucial for stabilizing the electrodes and ensuring reproducibility in the electrochemical results. Fig. 5a shows the iR-corrected OER polarization curves of the NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 samples, recorded at a low scan rate of 2 mV s−1. Notably, the NF@ZIF-67@NiCo2S4 material shows an overpotential of 248 mV and 360 mV at current densities of 50 and 100 mA cm−2, respectively. While, the NF@Co–N–C@NiCo2S4 sample displays an overpotential of 239 mV and 307 mV at the corresponding current densities. For comparison, linear sweep voltammetry (LSV) was conducted on bare NF, NF@NiCo2S4 and the benchmark NF@RuO2 catalyst under identical conditions, as depicted in Fig. 5a. The overpotential values for the synthesized materials are depicted in Fig. 5b, and a detailed comparison with relevant literature is given in Table S1.† Remarkably, NF@Co–N–C@NiCo2S4 shows the lowest overpotential among the tested materials. The order of OER performance, based on overpotential, is as follows: NF@Co–N–C@NiCo2S4 > NF@ZIF-67@NiCo2S4 > NF@RuO2 > bare NF. It is noteworthy that among all NF@Co–N–C@NiCo2S4 exhibits a lower overpotential and higher current across all potentials, highlighting its superior catalytic activity in the OER. In NF@Co–N–C@NiCo2S4 material, the Co–N–C porous microstructure, derived from ZIF-67, serves multiple crucial roles in enhancing catalytic activity for the OER. This structure comprises several key features: (i) the derived Co–N–C porous microstructure incorporates N-doped carbon, introducing catalytically active sites. Nitrogen doping enhances the overall conductivity and provides sites for effective electrochemical reactions (ii) the material contains abundant Co species that act as catalytic sites for the OER. The presence of Co species enhances the overall electrocatalytic performance by facilitating the necessary reaction pathways (iii) the Co–N–C structure serves as a framework for efficient electron transfer. The Co–N–C skeletons efficiently transfer electrons, contributing to improved conductivity within the material. This electron transfer capability is crucial for the overall effectiveness of the catalyst in OER. The combination of N-doping, rich Co species, and the conductive Co–N–C skeleton collectively results in a highly efficient and active catalyst for the OER, making it a promising material for electrochemical applications.32 In addition, it is known that metal chalcogenides are active catalysts for HER, as revealed by previous research studies, and also exhibit admirable catalytic activity for the OER. The presence of sulfide in NiCo2S4 is a key factor in enhancing electrocatalytic performance by contributing to the weakening of metal–oxygen–hydrogen (M–OH) bonds, thereby facilitating water dissociation and promoting the formation of reactive oxygen intermediates, which is vital for the OER.33 Also sulfide forms strong bonds with metals like Ni and Co thus creating a stable structure that is robust against corrosion and degradation, particularly in challenging acidic and alkaline environments.34 In the current study, a strategic approach has been employed to further enhance the OER catalytic activity. The coating of metal chalcogenides on Co–N–C serves the purpose of creating a bifunctional catalyst, demonstrating proficiency in both OER and HER. This innovative strategy has potential implications for simplifying the fabrication of alkaline electrolyzers and reducing associated costs. The bifunctional catalyst, represented by the coated NF@Co–N–C@NiCo2S4 material, offers a synergistic effect, making it a promising candidate for dual functionality in both OER and HER. Such advancements contribute to the progress of cost-effective and efficient electrochemical systems for practical applications.35 To gain deeper insights into these electrodes, Tafel plots were derived from the corresponding polarization curves using the Tafel equation η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(j), where η is overpotential, j is the current density, and b is the Tafel slope. These Tafel plots offer a detailed analysis of the electrocatalytic behavior, providing valuable information about the reaction kinetics and the efficiency of the electrodes in the OER.35,36 The Tafel plot (Fig. 5c), illustrating the relationship between the logarithm of current density (j) and overpotential, serves as a crucial tool for determining the Tafel slope and exchange current density. Analyzing the Tafel slope obtained from the LSV curves at the lowest scan rate is particularly significant, as it is assumed to have minimal experimental error.37 The Tafel slopes (Fig. 5c) for NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials are found to be 68 and 66 mV dec−1, respectively. Remarkably, these values are lower than those of the benchmark RuO2 catalyst (108 mV dec−1) and recently reported materials as shown in Table S1.† The lower Tafel slope values signify faster kinetics in the OER, suggesting superior electrocatalytic efficiency compared to the benchmark and other reported materials. This observation reinforces the outstanding performance of these synthesized materials in facilitating the oxygen evolution process. In evaluating the intrinsic catalytic activity of different catalysts, consideration of the electrochemical active surface area (ECSA) is paramount. Understanding ECSA is crucial for precisely assessing the reacting interface region and making effective comparisons of various materials catalytic performance.38 For instance, in the case of platinum catalysts, the active surface area can be reliably measured by determining the charge associated with one monolayer of under-deposited hydrogen desorption using CV.39–44 This becomes especially important for Ni-based electrode materials, where estimation can be challenging due to their complex nature and the presence of side reactions, leading to potential inaccuracies in representing their activity. In this study, the ECSA of the synthesized materials was estimated using the equation ECSA = Cdl/CS, where Cdl represents the electrochemical double-layer capacitance, and CS is the specific capacitance of a flat, smooth surface of the electrode material (assumed to be 40 μF cm−2).28 While methods for determining ECSA can vary, this approach provides a reasonable approximation, enabling a more accurate comparison of the catalytic performances of the different materials under consideration. To estimate the Cdl, cyclic voltammograms were recorded in the non-faradaic region over a potential range of 1.07 to 1.17 V at various scan rates (40, 50, 60, 70, 80, 90, and 100 mV s−1). The difference between the anodic and cathodic current density (Δj = ja − jc) was then plotted against the scan rate, as depicted in Fig. S6.† The Cdl values were determined by calculating half of the slope obtained from the plot of the difference between Δj against scan rate. NF@Co–N–C@NiCo2S4 exhibited a higher Cdl value of 7 mF cm−2 compared to NF@ZIF-67@NiCo2S4 with a Cdl value of 5.3 mF cm−2. This suggests that both NF@Co–N–C@NiCo2S4 and NF@ZIF-67@NiCo2S4 possess a greater number of exposed electrocatalytic active sites. The higher Cdl values in these materials indicate a larger electrochemically active surface area, which is a crucial factor influencing catalytic activity. This increased surface area likely contributes to the higher catalytic activity observed in NF@Co–N–C@NiCo2S4 and NF@ZIF-67@NiCo2S4. The presence of more active sites enhances the materials capability to participate in electrochemical reactions, ultimately leading to improved overall catalytic performance. Evaluating
the stability of materials over an extended period in the OER region is crucial for practical applications in electrolyzer systems. To assess the stability of the materials in alkaline media, chronoamperometry (CA) was employed. In this technique, a constant potential of 1.524 V (vs. RHE) was applied, and the current was continuously measured over time, as shown in Fig. 5d. Both NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 electrode materials demonstrated impressive stability, maintaining nearly the same current even after 16 hours. Initially, a slight decrease in current for NF@ZIF-67@NiCo2S4 was observed due to material peeling from the NF; however, after 12 hours, it became nearly constant. In contrast, NF@Co–N–C@NiCo2S4 maintained a stable current from the start, with no significant decrease observed over time. The cooperative interaction between Co–N–C and NiCo2S4 likely enhances the overall structural integrity and reduces degradation Consequently, the materials exhibit remarkable stability during prolonged exposure to alkaline media, reinforcing their suitability for practical applications in electrolyzer systems.
log(j), where η is overpotential, j is the current density, and b is the Tafel slope. These Tafel plots offer a detailed analysis of the electrocatalytic behavior, providing valuable information about the reaction kinetics and the efficiency of the electrodes in the OER.35,36 The Tafel plot (Fig. 5c), illustrating the relationship between the logarithm of current density (j) and overpotential, serves as a crucial tool for determining the Tafel slope and exchange current density. Analyzing the Tafel slope obtained from the LSV curves at the lowest scan rate is particularly significant, as it is assumed to have minimal experimental error.37 The Tafel slopes (Fig. 5c) for NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials are found to be 68 and 66 mV dec−1, respectively. Remarkably, these values are lower than those of the benchmark RuO2 catalyst (108 mV dec−1) and recently reported materials as shown in Table S1.† The lower Tafel slope values signify faster kinetics in the OER, suggesting superior electrocatalytic efficiency compared to the benchmark and other reported materials. This observation reinforces the outstanding performance of these synthesized materials in facilitating the oxygen evolution process. In evaluating the intrinsic catalytic activity of different catalysts, consideration of the electrochemical active surface area (ECSA) is paramount. Understanding ECSA is crucial for precisely assessing the reacting interface region and making effective comparisons of various materials catalytic performance.38 For instance, in the case of platinum catalysts, the active surface area can be reliably measured by determining the charge associated with one monolayer of under-deposited hydrogen desorption using CV.39–44 This becomes especially important for Ni-based electrode materials, where estimation can be challenging due to their complex nature and the presence of side reactions, leading to potential inaccuracies in representing their activity. In this study, the ECSA of the synthesized materials was estimated using the equation ECSA = Cdl/CS, where Cdl represents the electrochemical double-layer capacitance, and CS is the specific capacitance of a flat, smooth surface of the electrode material (assumed to be 40 μF cm−2).28 While methods for determining ECSA can vary, this approach provides a reasonable approximation, enabling a more accurate comparison of the catalytic performances of the different materials under consideration. To estimate the Cdl, cyclic voltammograms were recorded in the non-faradaic region over a potential range of 1.07 to 1.17 V at various scan rates (40, 50, 60, 70, 80, 90, and 100 mV s−1). The difference between the anodic and cathodic current density (Δj = ja − jc) was then plotted against the scan rate, as depicted in Fig. S6.† The Cdl values were determined by calculating half of the slope obtained from the plot of the difference between Δj against scan rate. NF@Co–N–C@NiCo2S4 exhibited a higher Cdl value of 7 mF cm−2 compared to NF@ZIF-67@NiCo2S4 with a Cdl value of 5.3 mF cm−2. This suggests that both NF@Co–N–C@NiCo2S4 and NF@ZIF-67@NiCo2S4 possess a greater number of exposed electrocatalytic active sites. The higher Cdl values in these materials indicate a larger electrochemically active surface area, which is a crucial factor influencing catalytic activity. This increased surface area likely contributes to the higher catalytic activity observed in NF@Co–N–C@NiCo2S4 and NF@ZIF-67@NiCo2S4. The presence of more active sites enhances the materials capability to participate in electrochemical reactions, ultimately leading to improved overall catalytic performance. Evaluating
the stability of materials over an extended period in the OER region is crucial for practical applications in electrolyzer systems. To assess the stability of the materials in alkaline media, chronoamperometry (CA) was employed. In this technique, a constant potential of 1.524 V (vs. RHE) was applied, and the current was continuously measured over time, as shown in Fig. 5d. Both NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 electrode materials demonstrated impressive stability, maintaining nearly the same current even after 16 hours. Initially, a slight decrease in current for NF@ZIF-67@NiCo2S4 was observed due to material peeling from the NF; however, after 12 hours, it became nearly constant. In contrast, NF@Co–N–C@NiCo2S4 maintained a stable current from the start, with no significant decrease observed over time. The cooperative interaction between Co–N–C and NiCo2S4 likely enhances the overall structural integrity and reduces degradation Consequently, the materials exhibit remarkable stability during prolonged exposure to alkaline media, reinforcing their suitability for practical applications in electrolyzer systems.
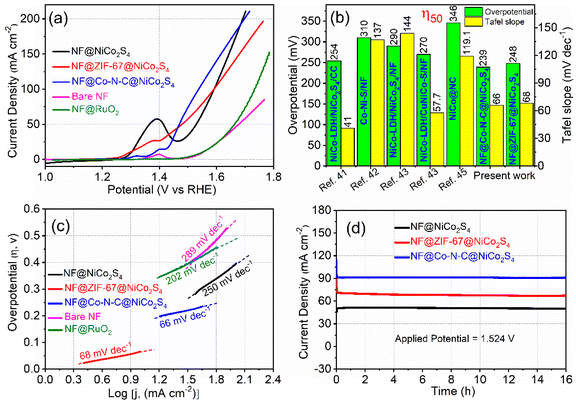 |
| | Fig. 5 OER analysis: (a) LSV curves recorded at a scan rate of 2 mV s−1 (iR-corrected), (b) overpotential and Tafel slopes comparison with the recent studies41–45 at 50 mA cm−2 (c) Tafel plots and (d) CA curves. | |
3.3. Electrocatalytic HER study
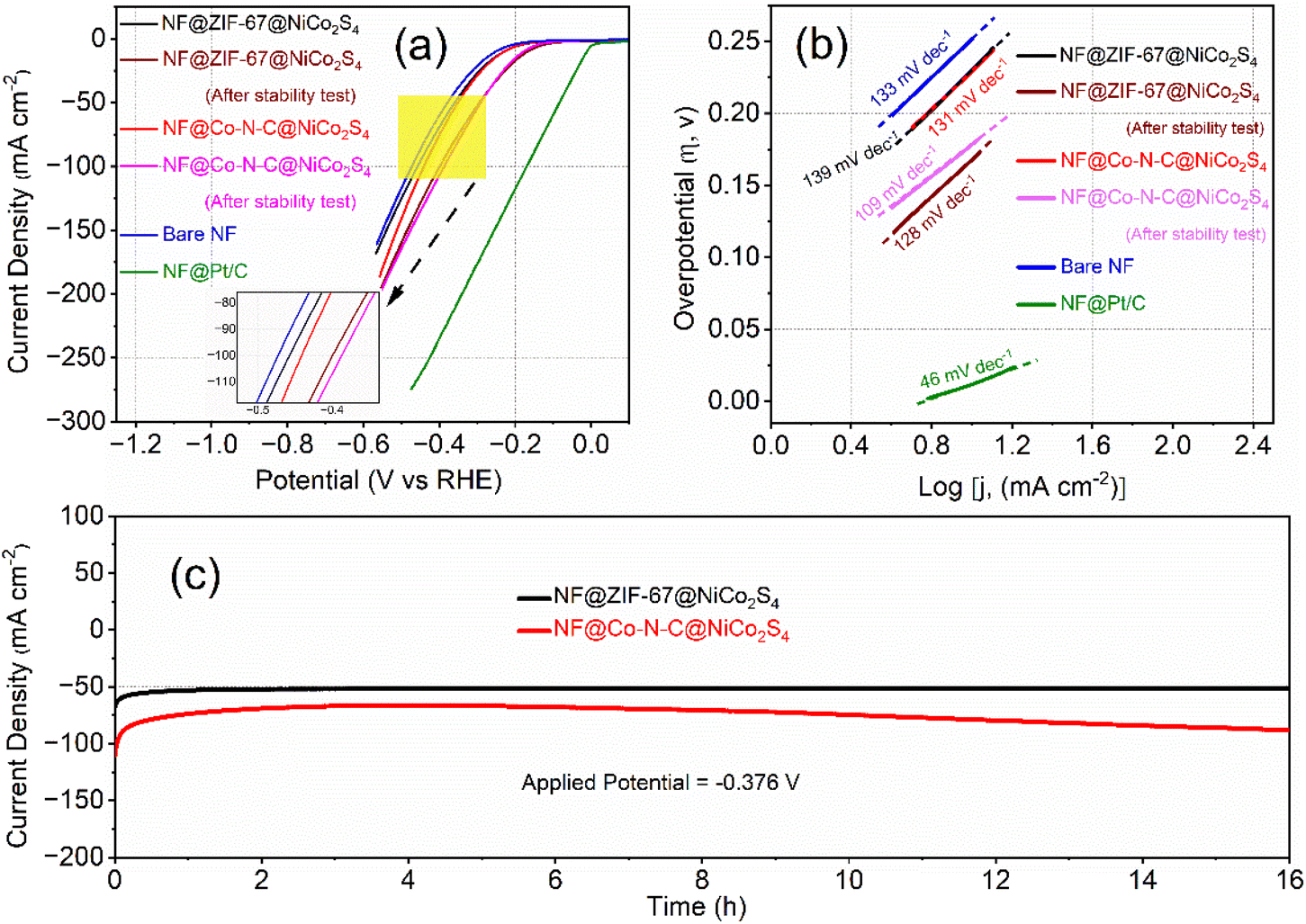
Recognizing the superior OER catalytic activity, the bifunctional properties of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials were further studied by assessing their catalytic activity for the HER. The HER performance of the synthesized materials was evaluated using a three-electrode system, and all potentials were normalized to the RHE scale. In Fig. 6a, the iR-corrected HER polarization curves of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials are presented, recorded at a scan rate of 2 mV s−1. Similar to OER activity, the NF@Co–N–C@NiCo2S4 sample exhibits superior HER activity compared to NF@ZIF-67@NiCo2S4. Specifically, NF@Co–N–C@NiCo2S4 exhibits lower overpotentials of 272 mV and 351 mV at current densities of 20 and 50 mA cm−2, respectively. In contrast, NF@ZIF-67@NiCo2S4 shows slightly higher overpotentials of 276 mV and 359 mV at the same current densities. Initially, there is a slight difference in catalytic activity among NF, NF@ZIF-67@NiCo2S4, and NF@Co–N–C@NiCo2S4. However, following the CA stability tests, a significant enhancement in catalytic activity is observed for the NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials. This improvement is shown through the comparison of LSV curves before and after the CA stability test, as shown in Fig. 6a. Notably, both materials exhibit relatively lower overpotentials compared to some recently reported materials (Table S2†), indicating their admirable electrocatalytic performance for HER. For comparison, bare NF and NF@Pt/C were included in the study under the same alkaline conditions, as shown in Fig. 6a. NF@Pt/C exhibited the lowest onset potential, while NF displayed a higher overpotential in comparison to the synthesized materials. The HER electrocatalytic activity of the nonprecious NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials is slightly lower to that of Pt/C. However, it is noteworthy that these materials still outperform some recently reported nonprecious materials, as indicated in Table S2.† Despite not matching the performance of NF@Pt/C, NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 exhibit competitive HER activity among nonprecious catalysts, emphasizing their potential as effective and economical alternatives for hydrogen evolution in alkaline media. In Fig. 6b, the Tafel plots of NF@ZIF-67@NiCo2S4, NF@Co–N–C@NiCo2S4, bare NF, and Pt/C are presented, and derived from the corresponding polarization curves. The Tafel slopes for NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 are measured at 139 and 131 mV dec−1, respectively. This indicates that the NF@Co–N–C@NiCo2S4 material exhibits better electrode kinetics than NF@ZIF-67@NiCo2S4, pointing to its enhanced HER catalytic activity. The lower Tafel slope for NF@Co–N–C@NiCo2S4 suggests more efficient reaction kinetics, reflecting its superior performance in promoting the hydrogen evolution process compared to NF@ZIF-67@NiCo2S4. To evaluate the durability of the materials, CA was conducted on both electrodes at a high potential of −0.376 V vs. RHE, as shown in Fig. 6c. Surprisingly, the HER catalytic activity of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 improved after 16 hours under rapid hydrogen evolution. This highlights the potential for improved stability and performance over prolonged periods for these materials. The ability of NF@Co–N–C@NiCo2S4 to exhibit a more substantial increase in electrocatalytic activity highlights its promising characteristics for long-term applications, making it an exciting and durable candidate for practical use in alkaline electrolyzer systems.
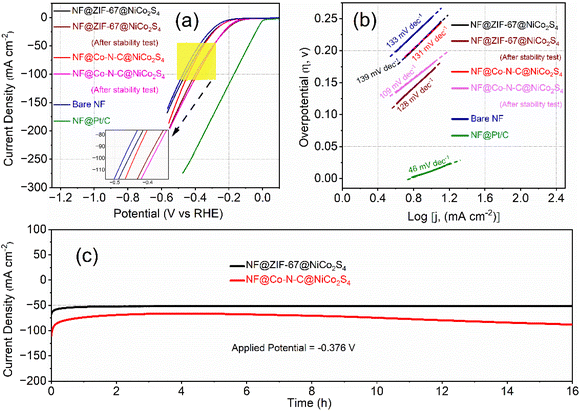 |
| | Fig. 6 HER analysis: (a) LSV curves recorded at a scan rate of 2 mV s−1 (iR corrected), (b) Tafel curves and (c) CA curves. | |
3.4. Electrochemical overall water-splitting study
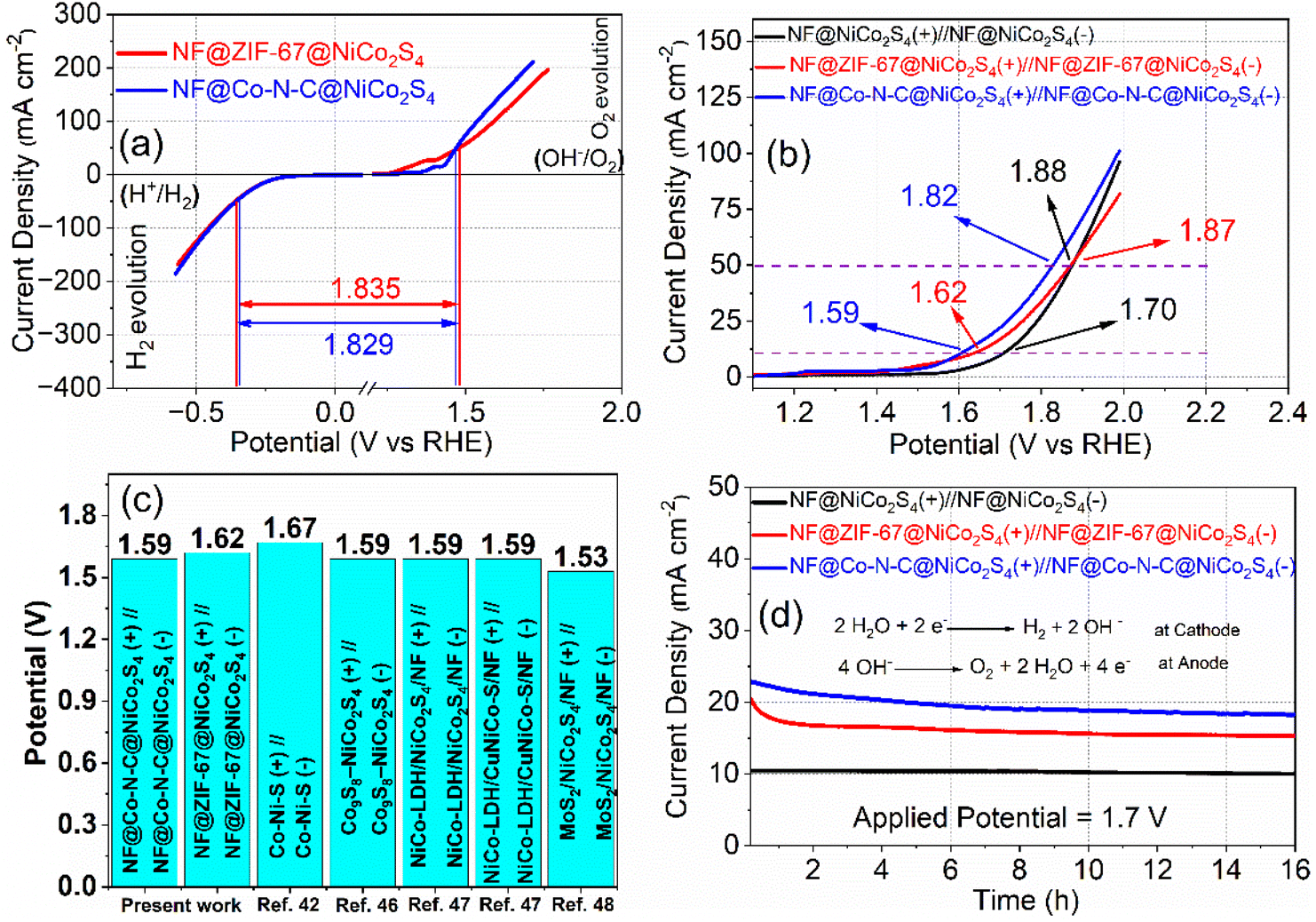
The half-cell studies reveal that both NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 exhibit bifunctional properties, demonstrating catalytic activity for both the OER and the HER. This dual functionality suggests that these materials have the potential to serve as both cathode and anode in electrolyzer systems. This bifunctional nature enhances their versatility and positions them as valuable components for advancing the efficiency and performance of electrolyzer systems. In practical applications, achieving a very small voltage difference between the OER and HER is crucial for an efficient water-splitting process. Fig. 7a illustrates the voltage difference (i.e. cell potential, Ecell) between OER and HER for NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4. At a current density of 50 mA cm−2, the cell potential for NF@ZIF-67@NiCo2S4∥NF@ZIF-67@NiCo2S4 is 1.835 V, and for NF@Co–N–C@NiCo2S4∥NF@Co–N–C@NiCo2S4 is 1.829 V. Indeed, the half-cell electrode measurements demonstrate the efficient bifunctional properties of both NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 for the OER and HER. While these findings are promising, it is crucial to move beyond half-cell studies and evaluate their real performance in an alkaline electrolyzer for practical applications. Full-cell evaluations provide a more comprehensive understanding of the materials behaviour in realistic electrochemical systems, accounting for potential interactions and synergies between the cathode and anode sides during overall water-splitting. This approach allows for a more accurate assessment of their performance and durability under conditions that mimic real-world applications. Two types of electrolyzers have been constructed using NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 electrodes. These electrolyzers are configured with NF@ZIF-67@NiCo2S4 as both the anode and cathode in one setup (NF@ZIF-67@NiCo2S4(+)∥NF@ZIF-67@NiCo2S4(−)), and NF@Co–N–C@NiCo2S4 as both the anode and cathode in the other setup (NF@Co–N–C@NiCo2S4(+)∥NF@Co–N–C@NiCo2S4(−)). The corresponding LSV curves for the electrolyzers are shown in Fig. 7b. At a current density of 10 mA cm−2, the cell potential for the NF@ZIF-67@NiCo2S4(+)∥NF@ZIF-67@NiCo2S4(−) and NF@Co–N–C@NiCo2S4(+)∥NF@Co–N–C@NiCo2S4(−) electrolyzers is measured to be 1.62 V and 1.59 V, respectively. In the two electrolyzers, the cell potentials obtained at 50 mA cm−2 are 1.87 and 1.82 V, respectively. The similarity between the cell potential values obtained in the two-electrode system and those in the three-electrode system enhances the credibility and reliability of the results. The obtained cell potentials are relatively higher to a simple NF@NiCo2S4(+)∥NF@NiCo2S4(−) electrolyzer (Fig. 7b) and to those recently reported for nonprecious metals based on other electrolyzers, as shown in Table S3† and Fig. 7c. The results from CA test reveal that the current density of the electrolyzers, with NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 electrodes, remains almost constant with minimal loss of current over 16 hours (Fig. 7d). These findings highlight the potential of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 as efficient electrocatalysts for water electrolysis. The observed stability and sustained current density over an extended duration position these materials as promising candidates for practical applications in renewable energy technologies and H2 production systems. The consistent performance demonstrated in CA studies further supports their feasibility for long-term and continuous use in alkaline electrolyzer systems.
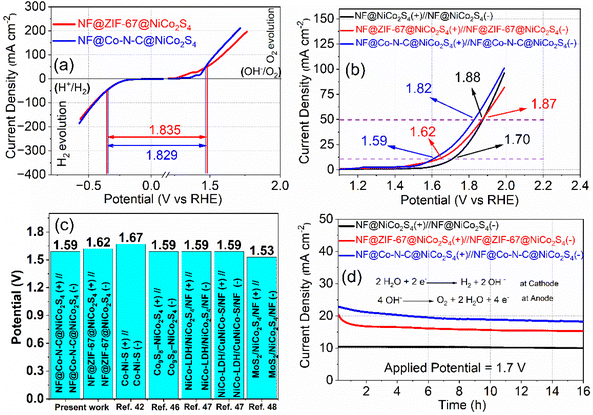 |
| | Fig. 7 Overall water-splitting analysis: (a) half-cell OER and HER LSV curves at a scan rate 2 mV s−1, (b) LSV curves of NF@ZIF-67@NiCo2S4(+)∥NF@ZIF-67@NiCo2S4(−), NF@Co–N–C@NiCo2S4(+)∥NF@Co–N–C@NiCo2S4(−) and NF@NiCo2S4(+)∥NF@NiCo2S4(−) electrolyzers recorded at a scan rate of 2 mV s−1, (c) cell potential comparison with the recent studies42,46–48 and (d) CA curves. | |
After completing all the electrochemical tests XPS was conducted to analyze the changes in the surface elemental composition. Fig. S7a and S7b† show the post-OER and post-HER XPS survey spectrum of NF@Co–N–C@NiCo2S4, respectively. Both the survey spectrums show the presence of Ni, Co, S, N, C and O with a noticeable decrease in the intensity of S. It is anticipated that after prolonged testing and extended exposure to the atmosphere, partial surface oxidation will occur in metal sulfides.21,49 This may result in diminished S peak intensity in XPS characterization due to the highly surface-sensitive nature of the technique.21 Fig. S8a and S9a† show the high-resolution XPS Ni 2p spectrum recorded after OER and HER testing, respectively. The spectra display two main peaks at ∼854 and ∼873 eV, along with two shake-up satellites, confirming the coexistence of Ni2+ and Ni3+.46 The Co 2p spectrum in Fig. S8b and S9b† show two main peaks at ∼781 and ∼796 eV and two shake-up satellites, indicating the coexistence of Co2+ and Co3+. After HER (Fig. S9b, ESI†), an additional peak at ∼778.85 eV corresponds to the Coδ+. The Co0 peak BE shifts from 778.4 to 778.85 eV, indicating partial oxidation of Co0 (Coδ+, 0 < δ < 2).50 After OER test, the electrode material expected to undergo oxidation, resulting in the absence of Co0 peak (Fig. S8b, ESI†). The C 1s XPS spectrum (Fig. S8c and S9c, ESI†) shows peaks at approximately 283.1, 284.6, and 286.5 eV, assigned to (CC/C–C), (C–S/C–O), and (C–N), respectively.14,31 The O 1s XPS spectrum (Fig. S8d and S9d, ESI†) shows peaks at 528.9, 530.2 and 530.6 eV corresponds to metal oxide, -hydroxide and adsorbed water, respectively.9,31
4. Conclusions
In summary, NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials were successfully synthesized using a straightforward room temperature, and one-step hydrothermal method. The OER and HER catalytic activities of the synthesized materials were separately tested in an alkaline medium. Due to numerous beneficial features, the NF@Co–N–C@NiCo2S4 electrode has demonstrated superior catalytic activity for both the OER and HER. Both NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 materials exhibited stable OER catalytic activities and demonstrated improved catalytic activity toward the HER during stability tests. These positive outcomes can be attributed to favorable structural and electronic changes that occurred during the stability tests. It is noteworthy that following the half-cell studies, two types of alkaline electrolysis cells were constructed: NF@ZIF-67@NiCo2S4(+)∥NF@ZIF-67@NiCo2S4(−) and NF@Co–N–C@NiCo2S4(+)∥NF@Co–N–C@NiCo2S4(−). These two electrolyzers require only 1.62 and 1.59 V, respectively, to drive a 10 mA cm−2 current density for the production of H2 and O2. This low voltage requirement highlights the efficiency and practical applicability of NF@ZIF-67@NiCo2S4 and NF@Co–N–C@NiCo2S4 in alkaline electrolysis. The development of these electrodes and electrolyzers is characterized by their cost-effectiveness, environmental friendliness, and simplicity in synthesis, design, and fabrication. These attributes are pivotal in enabling the realization of clean renewable energy infrastructure.
Data availability
Due to privacy restrictions, the raw data underlying this study are not publicly available. However, the data will be available from the corresponding author, Dr Gaddam Rajeshkhanna, upon reasonable request.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors gratefully acknowledge the support of the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India, under sanction order no. SRG/2021/001028, for funding this research. Additionally, the authors express their appreciation to the National Institute of Technology Warangal (NITW) for providing research seed money (P1144 Plan-Gen.-RSM).
References
- F. Martins, C. Felgueiras, M. Smitkova and N. Caetano, Energies, 2019, 12, 964 CrossRef CAS.
- W. Li, H. Tian, L. Ma, Y. Wang, X. Liu and X. Gao, Mater. Adv., 2022, 3, 5598 RSC.
- S. P. S. Badwal, S. S. Giddey, C. Munnings, A. I. Bhatt and A. F. Hollenkamp, Front. Chem., 2014, 2, 1–28 CrossRef CAS PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587 RSC.
- K. Zhang, M. Ma, P. Li, D. H. Wang and J. H. Park, Adv. Energy Mater., 2016, 6, 1600602 CrossRef.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- Y. Shia and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529 RSC.
- X. Duan, S. Ren, F. Ge, X. Zhu, M. Zhang and H. Zhen, Nanoscale, 2021, 13, 17655 RSC.
- M. S. Kim, B. Lamichhane, J.-H. Lee, J.-G. Bae, J. Y. Heo, H. J. Lee, S. Kattel and J. H. Lee, J. Energy Chem., 2023, 87, 89–97 CrossRef CAS.
- A. Dymerska, B. Środa, K. Sielicki, G. Leniec, B. Zielińska, R. Zairov, R. Nazmutdinov and E. Mijowska, J. Energy Chem., 2023, 86, 263–276 CrossRef CAS.
- W. Niu and Y. Yang, ACS Appl. Energy Mater., 2018, 1, 2440–2445 CrossRef CAS.
- C. Zhu, D. Du, A. Eychmüller and Y. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS.
- F. Yang, P. Zhao, X. Hua, W. Luo, G. Cheng, W. Xing and S. Chen, J. Mater. Chem., 2016, 4, 16057 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang and M. Bosch, Adv. Mater., 2018, 30, 1704303 CrossRef.
- S. G. Peera, R. Koutavarapu, C. Liu, G. Rajeshkhanna, A. Asokan and Ch. V. Reddy, Energies, 2021, 14, 1320 CrossRef CAS.
- C. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713 RSC.
- C. Yin, F. Yang, S. Wang and L. Feng, Chin. J. Catal., 2023, 51, 225–236 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- J. Jin, T. Xiao, Y.-f. Zhang, H. Zheng, H. Wang, R. Wang, Y. Gong, B. He, X. Liu and K. Zhou, Nanoscale, 2021, 13, 19740 RSC.
- R. He, C. Wang and L. Feng, Chin. Chem. Lett., 2023, 34, 107241 CrossRef CAS.
- A. Arulraj, P. K. Murugesan, C. Rajkumar, A. T. Zamorano and R. V. Mangalaraja, Energies, 2023, 16, 1669 CrossRef CAS.
- B. Qu, C. Li, C. Zhu, S. Wang, X. Zhang and Y. Chen, Nanoscale, 2016, 8, 16886 RSC.
- S. Mao, Z. Wen, S. Ci, X. Guo, K. K. Ostrikov and J. Chen, Small, 2015, 11, 414–419 CrossRef CAS PubMed.
- Y. Pei, J. Cheng, H. Zhong, Z. Pi, Y. Zhao and F. Jin, Green Chem., 2021, 23, 6975 RSC.
- R. Biswas, P. Thakur, I. Ahmed, T. Rom, M. S. Ali, R. A. Patil, B. Kumar, S. Som, D. Chopra, A. K. Paul, Y.-R. Ma and K. K. Haldar, Energy Fuels, 2023, 37, 604–613 CrossRef CAS.
- D. Li, H. Guo, H. Wang, L. Pan and J. Lin, ChemSusChem, 2024, 202400751 CrossRef PubMed.
- G. Rajeshkhanna, E. Umeshbabu, P. Justin and G. Ranga Rao, Int. J. Hydrogen Energy, 2015, 40, 12303–12314 CrossRef CAS.
- A. Borah, Sumit, S. Palaniyappan and G. Rajeshkhanna, Sustainable Energy Fuels, 2024, 8, 2265–2279 RSC.
- X. Zhao, H. Liu, Y. Rao, X. Li, J. Wang, G. Xia and M. Wu, ACS Sustainable Chem. Eng., 2019, 7, 2610–2618 CrossRef CAS.
- T. I. Singh, G. Rajeshkhanna, U. N. Pan, T. Kshetri, H. Lin, N. H. Kim and J. H. Lee, Small, 2021, 17, 2101312 CrossRef CAS.
- B. Wu, H. Meng, D. M. Morales, F. Zeng, J. Zhu, B. Wang, M. Risch, Z. J. Xu and T. Petit, Adv. Funct. Mater., 2022, 32, 2204137 CrossRef CAS.
- M. Wang, L. Zhang, Y. Hec and H. Zhu, J. Mater. Chem. A, 2021, 9, 5320 RSC.
- U. Aftab, A. Tahira, R. Mazzaro, V. Morandi, M. I. Abro, M. M. Baloch, C. Yu and Z. H. Ibupoto, RSC Adv., 2020, 10, 22196–22203 RSC.
- L. Wei, Q. Wu and J. Li, J. Mater. Sci.: Mater. Electron., 2021, 32, 12966–12990 CrossRef CAS.
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843 RSC.
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
- S. Ntais, A. Serov, N. I. Andersen, A. J. Roy, E. Cossar, A. Allagui, Z. Lu, X. Cui, E. A. Baranova and P. Atanassov, Electrochim. Acta, 2016, 222, 1455–1463 CrossRef CAS.
- G. Jerkiewicz, Electrocatalysis, 2010, 1, 179–199 CrossRef CAS.
- Y. Liu, Y. Bai, W. Yang, J. Ma and K. Sun, Electrochim. Acta, 2021, 367, 137534 CrossRef CAS.
- S. Gopalakrishnan, V. Saranya, G. Anandha Babu, S. Harish, E. Senthil Kumar and M. Navaneethan, J. Alloys Compd., 2023, 965, 171124 CrossRef CAS.
- E. Cossar, M. S. E. Houache, Z. Zhang and E. A. Baranova, J. Electroanal. Chem., 2020, 870, 114246 CrossRef CAS.
- M. Shao, J. H. Odell, S. Il Choi and Y. Xia, Electrochem. Commun., 2013, 31, 46–48 CrossRef CAS.
- Y. Zhang, Y. Jia, M. Song, N. Xiao, C. Dai, Y. Sun, L. Wang, Y. Zhao, J. Yu and Y. Qu, Colloids Surf., A, 2023, 658, 130665 CrossRef CAS.
- M. Ali, M. Wahid and M. Kowsar, New J. Chem., 2023, 47, 13888 RSC.
- X. Yu, J. Shen, Q. Chen and Q. Zhong, Dalton Trans., 2022, 51, 17743 RSC.
- X. Xu, W. Zhong, L. Zhang, G. Liu, W. Xu, Y. Zhan and Y. Du, Surf. Coat. Technol., 2020, 397, 126 Search PubMed.
- P. Ganesan, A. Sivanantham and S. Shanmugam, J. Mater. Chem. A, 2016, 4, 16394 RSC.
- D. Yin, J. Tang, R. Bai, S. Yin, M. Jiang, Z. Kan, H. Li, F. Wang and C. Li, Nanoscale Res. Lett., 2021, 16, 11 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a