
Lipid nanoparticles for enhancing oral bioavailability
Anushareddy
Gangavarapu
a,
Lillian V.
Tapia-Lopez
 b,
Barnali
Sarkar
b,
Jaqueline
Pena-Zacarias
c,
Abu Zayed Md
Badruddoza
*d and
Md
Nurunnabi
*a
b,
Barnali
Sarkar
b,
Jaqueline
Pena-Zacarias
c,
Abu Zayed Md
Badruddoza
*d and
Md
Nurunnabi
*a
aDepartment of Pharmaceutics and Drug Delivery, School of Pharmacy, University of Mississippi, MS 38677, USA. E-mail: bsnabi@olemiss.edu; Tel: +1-662-915-7341
bDepartment of Pharmaceutical Sciences, School of Pharmacy, University of Texas at El Paso, El Paso, TX 79902, USA
cBiological Sciences Program, College of Science, University of Texas at El Paso, El Paso, TX 79965, USA
dPharmaceutical Sciences Small Molecule, Pfizer Worldwide Research and Development, Groton, CT 06340, USA. E-mail: abuzayedmd.badruddoza@pfizer.com
First published on 29th August 2024
Abstract
In recent studies, lipid nanoparticles have attracted attention as drug delivery systems owing to their preeminent potential in achieving the desired bioavailability of biopharmaceutics (BCS) class II and class IV drugs. The current debate concerns the bioavailability of these poorly absorbed drugs with their simultaneous oral degradation. Lipid nanoparticles, including solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC), are lipid-based carrier systems that can effectively encapsulate both lipophilic and hydrophilic drugs, offering versatile drug delivery systems. The unique properties of lipids (biodegradability and biocompatibility) and their transportation pathways enhance the biological availability of drugs. These particles can increase the gastrointestinal absorption and solubilization of minimally bioavailable drugs via a selective lymphatic pathway. This review mainly focuses on providing a brief update on lipid nanoparticles (LNPs) that synergistically increase the bioavailability of limited permeable drugs and highlight the transversal mechanisms of LNPs across the gastrointestinal hurdles, transmembrane absorption, transport kinetics, and computational tools. Finally, the present hurdles and future perspectives of LNPs for oral drug delivery systems are discussed.
1. Introduction
The use of lipid colloidal particles as drug delivery systems has significantly increased owing to their unique characteristics and ability to overcome biological barriers. Although many pharmaceuticals have the potential to be extremely efficient, they exhibit poor solubility and low absorption levels orally. Considering this, lipid colloidal particles can help overcome these issues and allow pharmaceuticals to perform their function in a much more efficient manner. Lipid colloidal particles or lipid nanoparticles are spherical in shape (Fig. 1) and consist of a lipid monolayer that faces the extracellular environment and surrounds a hydrophobic core that faces the intracellular environment. The interior hydrophobic core allows the integration of pharmaceuticals or materials with limited oral bioavailability, while the exterior lipid monolayer easily integrates into the extracellular environment given the fact that lipids are fundamental building blocks of all cells in the human body. As lipids are a key component of the human body, lipid nanoparticles are recognized by the body, and therefore can be readily absorbed without being rapidly excreted. Currently, the most preferred route of administration for lipid colloidal particles is the oral route given that it is inexpensive and non-invasive, which increases patient compliance and its likelihood of being seen as a prospective mode of treatment. As previously mentioned, the oral route of administration has posed numerous challenges, most of which can be overcome using lipid colloidal particles. Lipid colloidal particles have higher stability in the gastrointestinal environment, exceptional membrane permeability, and low toxicity.1,2 To date, the most attractive lipid-based formulations are microemulsions, nanoemulsions, self-emulsifying materials, liposomes, lipid nanoparticles, and lipid–drug conjugates. While all these formulations have unique advantages and disadvantages, nano-based lipid formulations have gained increasing attention given that they provide the unique benefits of both lipids and nanoparticles. Together, these agents provide formulations that can easily permeate challenging biological barriers, provide targeted delivery, decrease toxicity, and have overall lower production costs. Owing to the size of nanoparticle-based liposomes and their intrinsic properties, lipid nanoparticles undergo endocytosis.3 Lipid nanoparticles exhibit a positive charge at a lower pH (pH < 4.5), which allows RNA complexation, and exhibit a neutral charge at physiological pH, reducing possible toxic effects.3 The positive charge of lipid nanoparticles at a low pH increases the likelihood of endosomal escape, therefore releasing the material inside of the lipid nanoparticle into the cytoplasm shortly after. Usually, lipid-based formulations contain an additional lipid to increase the likelihood of binding to cells, cholesterol to prevent leakage of the therapeutic agent, and polyethylene glycol to increase the circulation half-life.4,5 In this review, the characteristics, components, methodologies, advantages, disadvantages, and future directions of lipid nanoparticles are thoroughly discussed. | ||
| Fig. 1 Classification of lipid nanoparticles in various drug delivery systems. Created with https://www.istockphoto.com/. | ||
2. Classification of lipid-based oral drug delivery systems
Generally, lipid nanoparticles are prepared using four classes of lipids, which differ in structure, properties, and function (Table 1), including phospholipids, sterols, polymer lipids, and ionizable lipids. Phospholipids include phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, and phosphatidylglycerols, which usually contain stabilizers such as cholesterol. Phospholipids contribute to the overall structure of the nanoparticle and endosomal escape. The ability of phospholipids to undergo spontaneous organization in the lipid bilayer enhances the membrane stability in lipid nanoparticles. Phospholipids integrate themselves into the periphery of the nanoparticle and are usually semi-synthetic. Also, sterols such as cholesterol aid in the overall stability of nanoparticles through changes in the membrane integrity and rigidity. The incorporation of cholesterol increases the circulation half-life, reduces the likelihood of surface-bound proteins, and can reduce drug leakage, which were previous issues with early liposome drug delivery.6 Polymer lipid hybrids consist of natural, semi-synthetic or synthetic polymers such as polyethylene glycol, which serve as the solid component in the lipid nanoparticles.7 Usually, there will be a hydrophilic or hydrophobic polymer shell with a lipid surrounding it.8 This hybrid allows favorable drug release, surface functionalization, increased drug loading, and enhanced biocompatibility. Ionizable lipids play a detrimental role in guarding RNA and allowing cytosolic transport and are positively charged in the acidic environment to integrate RNA within the lipid nanoparticles. Currently, there are five types of ionizable lipids including unsaturated, multi-tail, polymeric, biodegradable, and branched tail (Fig. 1).13| Type of lipid nanoparticle | Lipid class | Type of lipids used | Encapsulated molecules | Ref. |
|---|---|---|---|---|
| Nano emulsions | Phospholipids | Phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, and phosphatidylglycerols | Evodiamine | 5 |
| Liposomes | Phospholipids sterols | Distearoylphosphatidylcholine (DSPC), 1,2-distearoyl-sn-glycerol-3-phosphoglycerol (DSPG), cholesterol, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyloxy-3-trimethylammonium propane chloride (DOTAP) | Alendronate | 5 |
| Phospholipid, cationic lipids, sterols | Cholesterol | Plasmid DNA | 9 | |
| SLNPs | Polymer lipids | Synthetic and semi-synthetic polymers | 5 | |
| Triglyceride, nonionic lipids, fatty acids | Dynasan 118, Softisan 154, and Imwitor 900K, Stearic acid, Span 80 | Sulpiride | 10 | |
| Sterol | Compritol ATO, cholesterol | c-Tocotrienol (c-T3) | 139 | |
| NLCs | Triglycerides, fatty acids | Glyceryl tripalmitate, oleic acid | Olanzapine | 11 |
| Glycolipid, ionizable phospholipid | Digalactosyldiacylglycerol (DGDG), phosphatidic acid (PA) | Simvastatin | 12 |
Presently, lipid-based formulations are consistently used as oral drug delivery systems, including liposomes, nanoemulsions, microemulsions, lipid nanoparticles, and lipid–drug conjugates. Generally, the oils and lipids utilized in these drug delivery systems are dietary lipids, making their permeability and excretion much more feasible. Liposomes lack the ability to enhance oral bioavailability. However, through the modulation of their bilayer composition and the addition of polymers or ligands on their surface, this obstacle can be overcome. Although liposomes are composed of cholesterol and phospholipids, they demonstrate limited stability in the gastrointestinal environment when their phase transition temperature is below 37 °C. Therefore, studies have shown that the encapsulation of lipids with transition temperatures of above 37 °C can increase their stability. Specifically, the addition of stearylamine, a steroid lipid with a positive charge, has been demonstrated to survive the harsh acidic conditions of the GI tract. Nanoemulsions are emulsions with sizes in the range of 10–1000 nm. Their most important component is the surfactant, and its selection is the key to their formation and the overall physical stability. Generally, biocompatible oils and surfactants are used given that currently nanoemulsions are utilized in pharmaceuticals, food, and cosmetics. Also, proteins and lipids can be utilized as stabilizers in nanoemulsions.11,14 Microemulsions, similar to nanoemulsions, have a suitable droplet size and require the same three components, i.e., oil, water, and surfactant, and their size is typically less than 100 nm, exhibiting a thermodynamically stable nature. Their incorporation ability helps to improve the research-mediated approaches with hydrophilic and hydrophobic drugs with different preparation approaches. Generally, LNPs are considered to be safe with biodegradable and biocompatible lipids. In the early development stages of solid lipid nanoparticles, they were synthesized using lipids that possessed melting points higher than the regular body temperature.14 The solid components of nanoparticles help protect the content of solid lipid nanoparticles with low chances of their content dispersing elsewhere. Morphologically, solid lipid nanoparticles are sphere-shaped particles with a drug-containing lipid matrix together with a layer of surfactant, which is used as a stabilizer in the aqueous phase. The distribution of the active ingredients in a solid lipid nanoparticle system is the basis for its formulation method. Based on this distribution, there are three models for solid lipid nanoparticles including the solid solution model, drug-enriched shell model, and drug-enriched core model. Solid lipid nanoparticles have various attractive properties such as the ability to increase the stability of the system, protective properties for drugs and therapeutics, controlled release, small size, increased surface area, increased drug loading, and phase interaction. Lastly, lipid drug conjugates consist of drugs with covalently attached lipids. This conjugation increases the attraction of the drug to lipids and modifies other properties associated with the drugs.6,15 Conjugation offers various advantages such as increased oral bioavailability, targeted delivery to the lymphatic system, increased targeting of existing tumors, and a decrease in overall toxicity. The conjugation strategies that are used are dependent on the structure of the drugs, lipids, and their expected interaction. To conjugate drugs and lipids, a few strategies have been developed, including drug conjugation with fatty acids, steroids, glycerides, and phospholipids. These drug conjugates avoid early hydrolysis and increase the lipid–drug interaction with the cell membrane. The lymphatic system is highly targeted given that it is responsible for transporting dietary lipids from the intestine to the lymphatic capillaries.15
3. Composition of lipid nanoparticles
Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) are the most widely used lipid nanoparticles. Solid lipid nanoparticles are attractive due to their advantages of large-scale production, increased bioavailability, low toxicity, and ability to incorporate both water-soluble and non-water-soluble drugs. SLNs are composed of two main materials, lipids and stabilizers. Also, additional materials such as co-surfactants, preservatives, cryoprotectants, and charge modifiers can be incorporated. The most commonly used lipids are monoacid triglycerides, fatty acids, steroids, waxes, fats, and partial glycerides. The commonly used surface stabilizers are phospholipids, bile salts, soybean lecithin, egg lecithin, phosphatidylcholine, poloxamers, and polysorbates. The solid lipid core of solid lipid nanoparticles is responsible for solubilizing the hydrophobic drug components with the aid of the appropriate surface stabilizer. However, despite their advantages, SLNs also have some disadvantages, including the possible leakage of the encapsulated drug, issues with stability due to high their water content, and decreased drug loading efficiency.16 Alternatively, NLCs offer a solution to overcome these disadvantages posed by SLNs. Unlike SLNs, in NLCs, the solid lipid content is replaced with oil, which provides a less structured lipid matrix, therefore allowing an increase in the drug loading efficiency and a decrease in the likelihood of leakage. There are three different types of nanostructured lipid carriers including imperfect type, amorphous type, and multiple type. NLCs are composed of solid/liquid lipids, surfactants, and water.17–19 The lipids and surfactants are essentially similar that of SLNs, which were described above.4. Methods for the preparation of lipid nanoparticles (LNPs)
LNPs made up of a lipid core, also known as lipid core nanoparticles (LCNPs), i.e. microemulsions (MEs) and nanoemulsions (NEs), are extensively utilized. Nanoemulsions (NEs) are characterized as colloidal systems combining two non-miscible liquids, where one liquid is scattered as nanodroplets inside the other liquid and stabilized by an amphiphilic surfactant. Nanoemulsions (NEs) can exist in two forms, oil in water (O/W) or water in oil (W/O).20 NEs can be used directly for delivering drugs and targeting specific areas. They can also serve as a framework for creating polymeric nanoparticles and lipid nanocapsules.21,22 Non-equilibrium systems, such as NEs, necessitate an energy input for their formation. This energy arises from the stored potential energy in the system or from mechanical devices that generate disruptive solid forces. SLNs and NLCs are often synthesized using comparable techniques to that employed for lipid nanocarriers. Therefore, there are two main classifications of approaches to produce micro- and nano-emulsions (Fig. 2), namely, high-energy methods and low-energy methods. High-energy techniques employ powerful disruptive forces to fragment the oil and water phases, forming nanodroplets. Usually, a rough emulsion is initially created by blending both phases. The coarse emulsion is further homogenized using mechanical apparatus such as high-pressure homogenizers, high-shear homogenizers, ultrasonicators, and microfluidizers (Fig. 2).22 Conversely, low-energy methods involve nanoemulsification techniques, which require minimal energy depending on the inherent chemical energy inside the system to create nanodroplets.23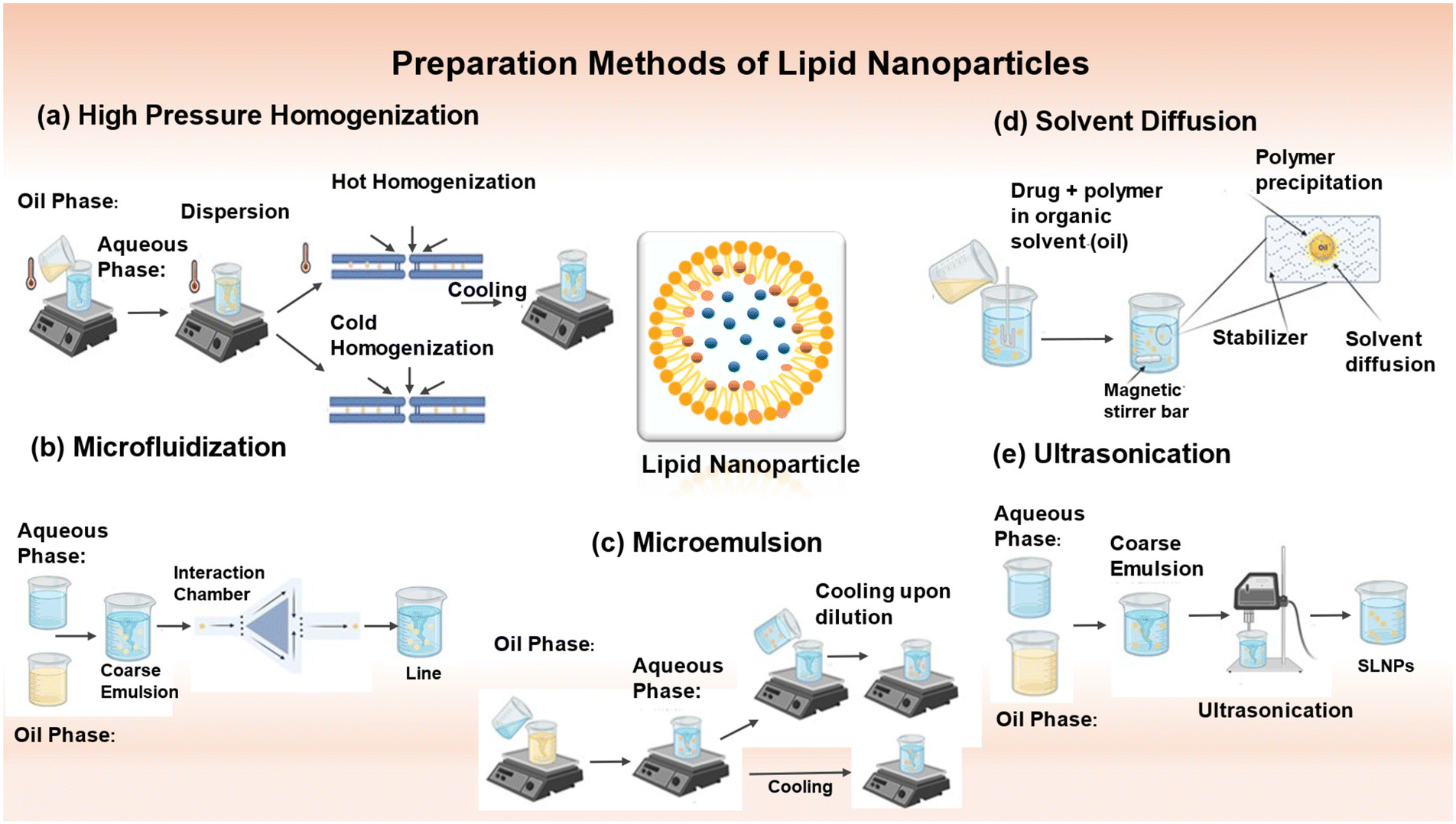 | ||
| Fig. 2 Methods for the preparation of lipid nanoparticles (LNPs). (a) High-pressure homogenization method (hot homogenization and cold homogenization), (b) microfluidization, (c) microemulsion, (d) solvent diffusion, and (e) ultrasonication.28 Adapted with permission. Created with https://www.biorender.com/. | ||
4.1. High-pressure homogenization
The high-pressure homogenization (HPH) process is extensively utilized because it is simple, economical, scalable, and can easily be performed at a high rate. It avoids the use of organic solvents and overuse of surface-active stabilizers. Melt emulsification with high-speed homogenization produces SLN.24 High-pressure homogenizers exert a high pressure (between 100 to 2000 bar) passing through a small opening (a few microns). The fluid accelerates to a remarkably high velocity (over 1000 km h−1) in a noticeably short distance. Particle disruption or breakage occurs at submicron levels due to the extremely high shear stress and cavitation pressures. The droplet size and polydispersity index depend on the number of cycles, pressure, temperature of the system, and the formulation itself.25 Both HPH techniques, hot and cold homogenization, combine drugs with a substantial amount of lipid melt.Substituting media (such as oil and PEG 600) with limited solubility for the drug can also be used to reduce the amount of hydrophilic drug lost to the aqueous phase. The particle size and polydispersity index are both increased during cold homogenization. The first preparation phase involves melting the lipid/drug mixture; therefore, even although cold homogenization reduces the thermal exposure of the drug, it cannot be entirely avoided.
4.2. Microfluidization
A microfluidizer is a patented mixing device that continuously forces a coarse emulsion through a chamber with tiny channels or microchannels using a high-pressure positive displacement pump (5 to 135 MPa) until the desired particle size is achieved.31 Turbulent flow, in conjunction with cavitation, induces droplet fragmentation and the creation of non-equilibrium structures. Subsequently, the bulk emulsion is subjected to filtration to eliminate sizable droplets, leading to the formation of homogeneous non-equilibrium structures. This approach is well-suited for use on an industrial scale.324.3. Microemulsion-based method
The microemulsion-based technique is based on the dilution of microemulsions. Microemulsions consist of two distinct phases, namely, an inner phase and outer phase. A solution composed of a low melting fatty acid (e.g., stearic acid), an emulsifier (e.g., polysorbate 60, polysorbate 20, soy phosphatidylcholine, and taurodeoxycholic acid sodium salt), co-emulsifiers (e.g., sodium monooctyl phosphate and butanol), and water is subjected to stirring at a temperature in the range of 65–70 °C.33 The resulting mixture exhibits optical transparency. Stirring is employed to disperse the heated microemulsion into cold water at a temperature in the range of 2–3 °C. From a technical standpoint, the precipitation of lipid particles in water results in the dilution of the system, reducing the dispersion of solid lipid nanoparticles. An optimal lipid solid concentration of 30% is preferred in specific technological procedures. The use of an SLN dispersion as a granulation fluid for conversion into solid products such as tablets and pellets is a viable approach. However, a drawback is its high-water content, which needs to be eliminated due to the low particle concentration. The microemulsion content and temperature gradients determine the quality of the product. The dilution method reduces the lipid content in formulations compared to formulations based on HPH.344.4. Solvent diffusion method
The initial stage in fabricating lipid nanoparticles via the solvent diffusion method involves the creation of an emulsion consisting of a somewhat water-miscible solvent. This solvent contains the lipid, which is then mixed with water. A study employed low-toxicity, water-miscible solvents, especially benzyl alcohol and butyl lactate. When a transitory oil-in-water emulsion is transferred into water and subjected to continuous stirring, the dispersed phase droplets undergo solidification and transform into lipid nanoparticles because of the diffusion of the organic solvent. Moreover, the suspension undergoes purification through ultrafiltration, removing approximately 99.8% of benzyl alcohol. Trotta et al. employed the above-mentioned process to fabricate solid lipid nanoparticles utilizing glyceryl monostearate in conjunction with various combinations of surfactants. The SLN produced with benzyl alcohol and butyl lactate showed an increase in mean diameter from 205 to 695 nm and from 320 to 368 nm upon increasing the GMS content from 2.5% to 10% when lecithin and taurodeoxycholic acid sodium salt were used, respectively.35 Solid lipid nanoparticles containing clobetasol propionate mixed with monostearin were produced using a unique solvent diffusion process. The drug and lipid compounds were dissolved in a mixture of acetone and ethanol at a temperature of 50 °C. The organic solution was added to an aqueous solution with an acidic pH = 1.1. This aqueous solution contained 1 wt% polyvinyl alcohol and was subjected to mechanical agitation at room temperature. These solid nanoparticles (SLNs) containing the medication were efficiently synthesized and conveniently isolated through the process of centrifugation.364.5. Ultrasonication
The ultrasonication technique employed in this study serves as a dispersing method primarily utilized to manufacture solid lipid nano-dispersions. The underlying mechanism of ultrasonication relies on the phenomenon of cavitation. The experimental procedure involves two main steps. Firstly, the drug is added to a pre-melted solid lipid matrix. As the next step, either probe sonication, a high-speed stirrer, or magnetic stirring is used to bring the heated aqueous phase to the same temperature as the molten lipid, and then the two phases are mixed to form an emulsion.37 The obtained pre-emulsion is ultrasonicated using a probe sonicator in a water bath set at 0 °C. To mitigate recrystallization throughout the production process, the temperature is at least 5 °C higher than the melting point of the lipid.38 The O/W nanoemulsion was obtained after filtration using a 0.45 μm membrane to eliminate any contaminations introduced during ultrasonication. To enhance the longevity of the formulation, it was subjected to lyophilization. Additionally, mannitol (5%) is occasionally included in solid lipid nanoparticles as a cryoprotectant.39This approach is an efficient technique for producing SLNs without the use of organic solvents. However, an additional filtration step is necessary to exclude impurities (such as metals) generated during ultrasonication. Furthermore, the presence of microparticles often poses a challenge to the success of this method.40
4.6. Supercritical fluid technology (SCF)
When the temperature and pressure of a fluid are higher than its critical value, it is referred to as a supercritical fluid, which possesses distinct thermo-physical characteristics. As the pressure increases, the gas becomes denser without a considerable increase in viscosity, but its ability to dissolve compounds also increases.41 The solvation power can be modified by precisely manipulating the temperature and pressure variations. Several gases, including CO2, ammonia, ethene, CHCIF2, and CH2FCF3, were tested. However, CO2 is the most suitable choice for the SCF technique due to its overall safety, convenient critical point (31.5 °C, 75.8 bar), non-oxidizing properties towards drug materials, absence of any residue after the process, low cost, non-flammability, environmental acceptability, and ease of recycling or disposal.42 This approach often employs organic solvents (such as DMSO and DMFA) due to their complete miscibility in SCF-CO2. This technology encompasses various methods for producing nanoparticles, including the rapid expansion of supercritical solution (RESS), particles from gas-saturated solution (PGSS), gas/supercritical antisolvent (ASES), solution-enhanced dispersion by supercritical fluid (SEDS), and supercritical fluid extraction emulsions (SFEE).43In the supercritical antisolvent (SAS) process, the near-critical or supercritical fluid is initially delivered into an organic solvent vessel, where the crystallized solid material is dissolved. This leads to the thorough blending of the fluid and liquid, expanding the liquid and precipitation of particles. Consequently, lysozyme spherical nanoparticles were prepared utilizing a water/ethanol solution.44
During the PGSS procedure, the SCF is mixed with a liquid substrate, which can be a solution of the substrate in a solvent or a suspension of the substrate in a solvent. Subsequently, the combination is rapidly depressurized through a nozzle, creating SLN.45
4.7. Double emulsion method
The double emulsion approach involves dissolving the drug, particularly hydrophilic drugs, in an aqueous solution, which is subsequently emulsified in melting fat. The initial emulsion is stabilized using a stabilizer, such as gelatin and poloxamer-407. Subsequently, the stabilized initial emulsion is disseminated in an aqueous phase that contains a hydrophilic emulsifier, such as PVA. Next, the double emulsion is agitated, and then separated by filtration.46 The double emulsion process eliminates the need to melt the lipid for preparing peptide-loaded lipid nanoparticles. Additionally, the surface of the nanoparticles can be changed to provide steric stabilization by including a lipid/PEG derivative. The addition of steric stabilization greatly enhances the ability of these colloidal systems to withstand the effects of gastrointestinal fluids.47 This approach is mostly employed to encapsulate hydrophilic drugs, specifically peptides. An important limitation of this approach is the production of a significant proportion of microparticles. The preparation of insulin-loaded solid lipid nanoparticles (SLN) was achieved utilizing a unique process called reverse micelle-double emulsion. This technique involved the use of a mixed micelle composed of sodium cholate and phosphatidylcholine.48 Hecq et al. synthesized cationic solid lipid nanoparticles containing insulin for oral administration using this double emulsion method.495. Effect of GI barriers in oral delivery
Following oral delivery, the gastrointestinal epithelium is a physical and biological barrier to permeant absorption. The biochemical barrier is comprised of peptides broken down by enzymes, while the physical barrier is comprised of the impermeable GI epithelium (Fig. 3). Thus, understanding these limitations is critical for obtaining effective oral administration.50 The GI tract absorbs medications differently depending on the nature of the drug and geographical variables such as pH, enzyme activity, mucosal thickness, residence period, and surface area.51 | ||
| Fig. 3 Oral delivery of lipid carriers via different barriers in the GI tract. | ||
5.1. Effect of biochemical barrier in oral delivery
Two features of the biochemical barrier are the complicated intestinal lumen and the pH fluctuations that occur throughout the GI tract. A human stomach has a pH of 1.5–3.5, which is highly acidic. Alternatively, the small intestine has a pH of 6.6–7.5, which is nearly neutral, and the cecum has a pH of 6.4. Thus, the severe pH variations in the GI tract can subject medicine molecules to rigorous testing of their integrity and stability. Strict conditions that also affect the solubility of drug molecules are presented by the complex intestinal lumen components such as pancreatic secretions, bile salt, and proteolytic and digestive enzymes.52 Various locations, including the brush border, intestinal lumen, cytosol, and lysosomes, are susceptible to enzymatic degradation.5.2. Effect of mucosal barrier in oral delivery
Another limiting factor in the intestinal absorption of medication is the mucus layer, which covers the intestinal epithelium. The intestinal epithelium beneath is shielded from damage by the mucus layer, a highly hydrated, viscoelastic fluid. Although it prevents infections and external particles from entering, it permits the free flow of water, tiny molecules, and permeable nutrients. Multiple layers make up the mucus layer. Overlying the intestinal epithelium is a layer called glycocalyx or membrane-attached mucin, which acts as a docking process for the mucus-containing second layer.53 A pH gradient is created throughout the mucus gel layers by the epithelial secretion of bicarbonate ions, resulting in a pH that is almost neutral at the epithelial surface. This mucus layer, rich in bicarbonates, is a barrier to keep luminal acid at bay.Furthermore, by managing the swelling and dispersion characteristics of mucins and mucus, bicarbonate ions are also essential in controlling the viscosity of these substances.54 The mucus layer protects the intestinal epithelium against the GI milieu, infections, and foreign particles. The primary constituents of the mucus layer are mucin fibers, which are released by intestinal epithelial cells known as goblet cells. Glycoproteins called mucin fibers are abundant in hydrophobic domains and negatively charged glycosylated areas. Disulfide bonds and hydrophobic interactions cause these fibers to entangle and crosslink, creating a dense porous structure that can block big molecules and particles sterically.55,56
Mucus secretion is dynamic because it constantly renews itself, recycling, breaking down, or eliminating the old layer. Thus, the mucus layer complexity and removal ability are a significant impediment to achieving ideal drug absorption.53
5.3. Effect of other physical barriers in oral delivery
The intestinal epithelium acts as a physical barrier that prevents the transport of medication molecules. The small intestine has an absorptive solid surface and is the primary location for absorption. The tight junctions (TJs) are obstacles that restrict the pace of paracellular diffusion across the intestinal epithelium by controlling the movement of particles larger than 2 nm. Tight junctions are intricate formations consisting of intracellular plaque proteins (ZO-1, ZO-2, ZO-3, cingulin, and 7H6), transmembrane integral proteins (claudins and occludins) and regulatory proteins.57Efflux transporters serve as additional obstacles that hinder the absorption of oral medicine. Several efflux transporters, including P-glycoprotein (P-gp) and multidrug resistance protein (BCRP), are commonly located in the apical membrane of enterocytes. Efflux transporters and the metabolizing enzyme cytochrome P450 (CYP) have been recognized as significant elements restricting the absorption of substances in the intestines.58
6. Mechanisms to enhance the intestinal permeability and oral bioavailability
6.1. Paracellular transport
Paracellular transport refers to the process by which substances move across the epithelium by traversing the intercellular gaps between epithelial cells, which is a passive process that occurs due to diffusion. Tight junctions facilitate the regulation of this transportation. A tight junction serves as the primary barrier that restricts the movement of ions and giant molecules through the paracellular pathway (Fig. 4).59 The electrical resistance and ionic selectivity of paracellular transport vary significantly among different types of epithelia. Paracellular transport enhances the transcellular process by determining the extent and specificity of backward leakage for ions and solutes, with a significant tissue-specific contribution to total transport.60,61 | ||
| Fig. 4 Mechanisms to enhance the intestinal permeability and oral bioavailability. (a) Carrier-mediated transport, (b) paracellular transport, (c) transcellular transport, and (d) receptor-mediated transport. | ||
Tight junctions have similar biophysical characteristics to conventional ion channels, such as selective permeability based on size and charge, dependence on ion concentration for permeability, sensitivity to pH, competition among permanent molecules, and anomalous mole-fraction effects. The hydrogen bonding capacity and lipophilicity do not significantly regulate the paracellular route.62
6.2. Transcellular transport
Transcytosis is a biological process that allows intestinal epithelial cells to internalize particles (Fig. 4), enabling them to participate in transcellular transport. An illustrative instance is the transfer of glucose from the inner lining of the intestines to the fluid surrounding the cells by specialized cells called epithelial cells. This process commences with an endocytic event occurring at the apical membrane of cells. Afterward, particles are conveyed across the cells and discharged at the basolateral pole.63 Due to its significantly lower protein-to-lipid ratio, the basolateral membrane is both thinner and more permeable than the apical membrane. Several factors determine the success of transcellular transfer in transporting particles, as follows: (a) the physiology of the gastrointestinal system; (b) the animal model chosen to study absorption; and (c) the size, lipophilicity, hydrogen bond potential, charge, surface hydrophobicity and, other physiochemical features of the particles.64,65The essential cells responsible for intestine transport are enterocyte and M cells. Enterocytes constitute most cells that line the gastrointestinal tract. Alternatively, M cells are primarily found in the epithelium of Peyer's patches and make up a tiny portion of the intestinal epithelium (approximately 5% of human follicle-associated epithelium or about 1% of the total intestinal surface66). M cells can take in microbes, particles, and macromolecules by phagocytosis, adsorptive endocytosis through clathrin-coated pits and vesicles, and fluid phase endocytosis.67 However, due to the insufficient endocytic activity of enterocytes, the quantity of the particles propagated via these pathways is typically negligible. Accordingly, Peyer's patches and M cells have developed the ability to absorb various substances efficiently. However, this pathway is restricted to transporting relatively low molecular weight lipophilic medicines. Moreover, research conducted on people has shown that the process of absorption through the transcellular pathway diminishes dramatically in the colon. However, no similar decrease has been observed for the paracellular pathway.68
6.3. Carrier-mediated transport
The carrier-mediated transport process involves the transportation of tiny molecules or macromolecules through membrane proteins known as transporters. This mechanism is alternatively referred to as enhanced diffusion or active transport. Di- and tri-peptides are absorbed in the intestines by carrier-mediated peptide transport mechanisms and have been widely recognized. Initially, Newey and Smyth revealed the existence of a peptide transport mechanism in the mammalian stomach in 1959.69 Oligopeptide transporters facilitate the uptake of peptidomimetics, including animal-lactam antibiotics, renin-inhibitors, and angiotensin-converting enzyme inhibitors, for absorption.70 Comprehensive knowledge of the structural characteristics of peptides is necessary to target these transporters for protein delivery effectively.6.4. Receptor-mediated transport
In case of receptor-mediated transport, protein medicines function either as a receptor for surface-attached ligands or a ligand specific to surface-attached receptors.71 Receptor-mediated transport has been utilized to enhance the oral bioavailability of protein medications through modifications involving receptor-specific ligands with peptide and protein drugs. This process involves the inward folding of the cell membrane (Fig. 4), creating a small sac called a vesicle. The process of transporting substances into a cell is called endocytosis, which involves many mechanisms such as pinocytosis, phagocytosis, receptor-mediated endocytosis (clathrin-mediated), and potocytosis (non-clathrin-mediated).72 Once they reach the gastronomical tract, there are two absorption mechanisms for protein medicines, portal blood and intestinal lymphatics.7. Facilitated oral absorption by lipid particulates (enhanced permeation)
The trend of using oral lipid nanoparticles is to enhance the bioavailability of ineffectively absorbed drugs. These systems encapsulate hydrophilic and lipophilic drugs and preserve their therapeutic efficacy in the GI tract.2 Researchers have considered the physicochemical properties of lipids and how they are metabolized in organisms73–75 to create nanoparticles to increase the absorption of drugs orally.75,76 LNPs facilitate the solubility of drugs in their core structure, enhance membrane permeability, overcome significant barriers of absorption, and inhibit efflux transporters, avoiding a reduction in the drug concentration in cells.77 The size of nanoparticles increases their surface area, improving the drug uptake and delivery. Smaller nanoparticles have a higher surface area, where the optimal nanoparticle size depends on the application.76,78 Additionally, the control of drug release confers drug stability, reducing plasma accumulation and decreasing tissue toxicity.18 Finally, some lipid-based nanoparticle formulations can induce drug delivery via lymphatic transport, evading the initial drug metabolism in the liver.76,112 Overall, the characteristics of LNPs contribute to the optimization of drug absorption and bioavailability.H. Hassan et al. optimized a lipid formulation for encapsulating the antiviral drug acyclovir in SLNs and estimated its pharmacokinetic profile in vivo. These researchers intended to improve the loading capacity and controlled release of acyclovir by improving the composition between the solid lipid and surfactant formulations, focusing on size, polydispersity, and zeta potential. According to the results, the encapsulated drug showed higher oral bioavailability than the regular suspension.79
8. Factors affecting the permeability-limited oral bioavailability
Lipid-based formulations are directly related to the behavior and performance of nanoparticles, including stability, drug loading capacity, release profile, and absorption pathway.80 Additionally, the physicochemical characteristics of nanoparticles such as their size, surface modification, and superficial charge influence the permeability and oral bioavailability (Fig. 6).2,81 | ||
| Fig. 5 (A) Schematic of how surface charge affects the intestinal transportation and plasma concentration of LNPs. (B) CLSM observation of jejunum cryosections following in situ perfusion of unmodified, anionic, cationic, and net neutral SLNs. Red: SLNs; blue: cell nuclei. This figure has been adapted/reproduced from ref. 94 with permission from the American Chemical Society, copyright 2019. | ||
 | ||
| Fig. 6 Factors affecting the permeability-limited oral bioavailability. (a) Solubilization of drug into mixed micelles and the formation of chylomicrons, (b) LNP surface charge, (c) LNP size, (d) LNP with mucoadhesive improvement, and (e) LNP functionalized with different ligands. | ||
8.1. Lipid types
The different types of lipids include digestible lipids such as fatty acids, triglycerides, phospholipids, and cholesterol, which are mostly broken down through hydrolysis into digestion products the body assimilates. These lipids enhance the oral bioavailability of drugs due to the improvement in their solubilization and absorption. Also, there are indigestible lipids, including mineral, essential, and flavor oils, which do not tend to undergo lipase hydrolysis, and the solubility of the drug is compromised.80,82 Consequently, digestible lipids enhance the oral bioavailability of hydrophobic drugs, and combining both can be beneficial because indigestible lipids can improve the stability of the nanoparticle structure.83 The length of the fatty acid chains must be considered for drug oral bioavailability. Long-chain fatty acids, with 14 or more carbon atoms, participate more in forming mixed micelles for the solubilization of drugs and their delivery using lymphatic transport, enhancing the drug bioavailability.2 Studies indicate that a small amount of long-chain lipids with 18 carbon atoms triggers gall bladder contraction, expelling a mixture of bile components,83 which are required to form mixed micelles. Conversely, short- and medium-chain fatty acids tend to be more miscible in water and can diffuse directly to the enterocyte membranes and enter the bloodstream.2,74,83 Moreover, long-chain fatty acids degrade more slowly,5 slowing the drug release.83Porter et al. investigated lipid formulations and their potential effects on the dispersion and digestion of danazol, as well as its bioavailability when administered orally. This study compared three formulations, including a triglyceride with long-chain (LCT-solution) and two self-micro emulsifying systems (SMEDDS) derived from lipids with long-chain (LC-SMEDDS) and medium-chain lipids (MC-SMEDDS). The LCT-solution and LC-SMEDDS formulation improved the drug oral bioavailability related to the micronized formulation. On the contrary, MC-SMEDDS led to a slight improvement in drug bioavailability. Moreover, this medium-chain lipid formulation showed more drug precipitation in the in vitro studies than the long-chain lipid formulations.84
Besides, fatty acids are more easily absorbed than triglycerides because they do not require digestive hydrolysis. They can be integrated into micelles and transported through the lymphatic system more efficiently than triglycerides. This increases the drug bioavailability and reduces the time to its peak plasma concentration.74,85
Furthermore, the saturation degree is also related to the drug oral bioavailability. Unsaturated fatty acids form larger lipoproteins than their saturated counterparts, contributing to enhanced drug delivery through the lymphatic pathway.74,85 Likewise, increasing the unsaturation degree of lipid chains lowers the lipid melting point, enhances the drug solubility, and improves the drug release.74 Furthermore, phospholipids can contribute to lymphatic transport as an excipient in the lipid nanoparticle formulation.5 Patel et al. investigated the use of SLNs prepared with the phospholipid TPGS for the encapsulation of asenapine maleate, a drug employed in the treatment of schizophrenia. The results indicated that the drug bioavailability increased in vivo 50-fold compared to the free drug. Furthermore, the researchers administered cycloheximide as a pre-treatment, reducing the drug plasma concentration and corroborating the use of the lymphatic transport pathway.85
Recently, researchers investigated LNP formulations for the effective encapsulation and delivery of genetic drugs, including ionizable lipids. This type of lipid enhances the genetic drug loading capacity and delivery into the cytosol due to the lipid-positive charge within an acidic environment and a nearly neutral charge at physiological pH.82,86
Therefore, the appropriate lipid selection is intricately tied to the application and the characteristics of the drug. A combination of lipid types, including chain length and degree of saturation, is recommended.7,10,62
8.2. Effect of size and shape in intestinal transportation and pharmacokinetics
The size of lipid nanoparticles is essential because it is directly related to how they behave and perform in the body. The size and distribution of nanoparticles are associated with their adhesive properties, sustained drug release, targeted distribution,18 and cellular uptake.8,72 The size distribution of nanoparticles is determined through polydisperse index (PI) measurements with values ranging from 0 to 1.18 A monodisperse distribution has values smaller than 0.05,88 where a value below 0.3 can be considered the optimum value.86 Nanoparticles of specific sizes that are both stable and effective have a homogeneous size distribution.18 Moreover, the size distribution of nanoparticles is indispensable to consider their stability in the gastrointestinal fluid. As the size of nanoparticles decreases, their vulnerability to enzymatic degradation increases due to their larger exposed surface area, leading to earlier drug release.18Furthermore, the size of nanoparticles is a variable parameter that changes according to the drug dosage and the route of administration.88 The size of solid lipid nanoparticles has been reported to be 50–1000 nm.79 Nanoparticles with a size below 300 nm are adequate for gastrointestinal transit.86 Alternatively, nanoparticles between 200 and 500 nm diffuse through the mucus lining,2 SLNs around 120–200 nm avoid filtration by the liver and spleen,18 and below 200 nm can be transported through endocytosis.90 Huipeng Li et al. studied the impact of NLCs with different sizes of 100, 200, and 300 nm on oral drug delivery. The 100 nm nanoparticles showed increased cellular uptake and permeability against the intestinal barrier according to the in vitro studies. The in vivo studies indicated that this size nanoparticles had greater bioavailability in imaging experiments, while the pharmacokinetic studies showed their better oral absorption and bioavailability.91 Waheed M. Ibrahim et al. optimized the preparation of SLN to encapsulate sulpiride for oral delivery administration. Sulpiride is a psychiatric drug with low bioavailability, while treatments require large doses. They obtained SLNs having a narrow size distribution 147.8–298.8 nm, spherical shape, and adequate drug release profile. Overall, they observed an improvement in the intestinal permeability of sulpiride according to the everted intestine sac model.92 The shape of nanoparticles can also influence their cell uptake.87 Different forms interact differently biologically. M cells have shown differences in the internalization and transport of particles according to their shape as determined in the CaCo-2/Raji-B model, where particles with rod and disc shapes had a slightly higher percentage of transport compared to spherical ones.2
Banerjee et al. studied how the size, shape, and use of a biotin-targeting agent on the surfaces of polystyrene nanoparticles affected their cellular uptake and transport within intestinal cells in vitro. The study exposed that the shape of nanoparticles played a significant role. Specifically, rod-shaped nanoparticles were more efficiently taken up than spherical and disc nanoparticles, even without the active targeting moieties, as demonstrated by the co-cultured Caco-2 and HT-29 cell lines. After the biotin was coated, the rod-shaped nanoparticles also presented higher cellular uptake. Regarding transport, rods and discs showed better results compared to spheres. This suggests that using non-spherical geometries, which offer a higher surface area per unit volume, can be an effective strategy for drug delivery systems. Additionally, smaller nanoparticles (50–200 nm) are more efficient in cellular uptake and transport than larger nanoparticles. The authors further suggested that in vivo studies be performed and the use of biodegradable materials to validate their findings.81 The discussion above demonstrates that the size and shape of nanoparticles are critical factors in enhancing their cellular uptake. However, the type of materials used, with their unique properties, will produce different behaviors in organisms. Plaza-Oliver et al. underlined that the precise effect of the shape and even size on diffusion through the gastrointestinal mucus requires further evaluation given that other factors affect this phenomenon.5
8.3. Effect of surface charge/zeta potential in intestinal transportation and pharmacokinetics
The zeta potential (ZP) is a parameter related to the superficial charge of particles in contact with a liquid. It predicts the physical stability of colloidal suspensions and the electrostatic attractions or repulsions between particles.93A stable dispersion has a ZP higher than 30 mV in absolute value. A ZP higher than −60 mV for SLNs and −30 mV for NLCs represent stability.18 Other authors stated that a value of −25 mV signifies stability, while −15 mV suggests the commencement of gelation.92 In the case of nanoparticle formulations, it is essential to consider that their surface coating lowers the ZP because it diminishes the electrophoretic mobility of particles.89 Recent studies on SLNs revealed that surface charge modifications enhance the oral absorption of intact SLNs, presumably through improved mucus penetration (Fig. 5).94
The surface charge of nanoparticles plays a pivotal role in their behaviour in biological systems. The absolute value of ZP influences nanoparticle aggregation. An increase in ZP, indicating high surface charge, is a desirable characteristic, which prevents the aggregation and enhances the interaction of particles. Conversely, a low ZP, reducing the repulsions between particles, can lead to their aggregation. The ZP value itself is also significant. A positive ZP triggers the intriguing process of proteins in the bloodstream adhering to the particle surface, developing a protective layer. This process minimizes organism clearance, thereby prolonging the circulation time of nanoparticles.95 However, a negative ZP shows lower toxicity.96
8.4. Surface modification
Different nanoparticle surface modifications with polymeric and non-polymeric materials enhance their permeability.2 These modifications contribute to the stability, integrity, and specificity of nanoparticles.84 Enzymes found in the digestive tract and bile salts can affect the stability of the nanoparticles and induce the early release of the drug, compromising its oral bioavailability. Furthermore, the electrostatic balance between suspended nanoparticles must be considered to avoid their agglomeration.5 The use of hydrophilic polymeric coatings has been studied to surpass some GI barriers.97 The low molecular weight polymer PEG creates a barrier that prevents aggregation, which contributes to balancing the electrostatic instability of nanoparticles.5,79 In addition, PEG < 10 kDa diminishes mucoadhesion by reducing the influence of electrostatic or hydrophobic forces.5,79 Moreover, PEGylation is also used to prevent or reduce the absorption of enzymes on the surface of nanoparticles.Likewise, the positively charged polymer chitosan can create electrostatic attraction with the negatively charged mucin, increasing the mucoadhesion. M. Mendes et al. designed NLCs coated with specific polymers and encapsulating two complementary drugs, olanzapine (OL) and simvastatin, for the treatment of schizophrenia and bipolar disorder, respectively. The results indicated their greater intestinal permeability compared to commercial tablets.11
Yuan et al. analyzed the mucus penetrating ability of PEGylated solid lipid nanoparticles (PEG2000 – stearic acid) (pSLN) versus SLN. The PEGylation reduced the particle size from 230 to 153 nm and the absolute zeta potential from 20 to 15 mV, thereby enhancing the mucus penetration by the nanoparticles. Furthermore, the stability of the nanoparticles was thoroughly tested in simulated intestinal fluid, and the result was improved by increasing the percentage of PEGylation. The pSLN had a less negative charge and smaller particle size, improving the mucus penetration by the nanoparticles. Also, the stability of the nanoparticles in simulated intestinal fluid improved by increasing PEGylation. In terms of pharmacokinetic studies, pSLN demonstrated superior absorption and prolonged duration in the bloodstream with its relative bioavailability being 1.99-fold higher than that of SLN.89
X. Cheng and R. J. Lee proposed the use of helper lipids and cationic lipids to deliver oligonucleotides. Among the helpers, the authors mentioned the PEG polymer for nanoparticle stability and prolonged circulation time. However, they noted the importance of destabilizing the nanoparticle once in contact with cells; in this case, reversible PEGylation, to increase cell uptake and promote endosomal release, favoring the delivery of the oligonucleotide into the cytoplasm. For this, it is crucial to consider which PEGylating agent is appropriate and in what percentage it will be used including methoxy polyethylene glycol (M.W. 2000)-distearoylphosphatidylethanolamine (mPEG-DSPE), N-[(methoxy poly(ethylene glycol)2000)carbamoyl]-1,2-dimyristyloxlpropyl-3-amine (PEG-C-DMA), D-alpha tocopheryl polyethylene glycol 10000 succinate (TPGS), polysorbate 80 (Tween-80), and PEG-CHOL. The choice of the PEGylating agent is a critical decision that can significantly impact the effectiveness of the delivery system.96
Besides, adding targeting ligands improves the specificity and uptake by target cells or tissues.81 Short peptides can be attached to the surface of nanoparticles due to their ability to transport small and large molecules throughout cell membranes.2 Therefore, lipid-based formulations and surface modification of nanoparticles are directly related to their permeability and oral bioavailability.
8.5. Cytotoxicity
The cytotoxicity of LNPs mainly depends on the particle parameters such as hydrophobicity, surface area, and surface charge, which influence the dispersion and aggregation of the particles. Therefore, it is essential to determine the ratio between the components. Additionally, it is vital to consider the number of nanoparticles that accumulate in the cell membrane.98One of the most considerable parameters of cytotoxicity in recent research studies is particle surface charge. The delivery of positively charged LNPs together with nucleic acids has been widely used as potential therapeutics for delivery to the cytoplasm of the cell. However, most of the reports are contradictory, and necessary steps should be taken to determine their toxic effects.99 The resistance to nuclease degradation increases with the delivery of lipids with positive charge together with encapsulated nucleic acids at the desired target cells. Nucleic acids enter the cells by the adsorption of LNPs on the cell surface, followed by endocytosis, and thereby the release of the nucleic acids. This entire process is electrostatically promoted between the cell membranes and LNPs, driving the process, followed by membrane fusion and endocytosis.98,100,101
Despite this process, the cationic surface charge causes cytotoxicity and cell damage by the release of mediators. For instance, N-decyl-N,N-dimethyldecan-1-aminium bromide is a quaternary ammonium lipid that has been added to mRNA vaccines as an adjuvant to induce the intended immune response. However, research demonstrated that the mediators secreted by these nanoparticle forms exhibit cytotoxicity. Theoretically, interactions between cationic nanoparticles and the membranes of cells lead to membrane rupture and subsequent Ca2+ influx. Classified lipids are useful in stimulating gene delivery and cellular absorption, but they can also set off an immunological reaction that results in inflammation and possible tissue damage. Consequently, safe and protective lipids with a neutral charge have been employed as vaccine therapeutic delivery systems by replacing cationic lipids.98
9. Lipid nanoparticles for the oral delivery of various drugs
The different physicochemical properties of drugs require developing a specific delivery system to adequately encapsulate the drug molecules, considering their size, shape, charge, hydrophobicity, and hydrophilicity, ensuring their optimal delivery and therapeutic efficacy. Li et al. recommend the use of the combinatorial library synthesis approach to rapidly screen a wide range of lipid materials to identify the optimal composition for the specific drug, including small molecules, nucleic acids, proteins, and ribonucleoprotein complexes.1029.1. Small drug molecules
Small-molecule drugs are organic compounds with a simple chemical structure. Their low molecular weight below 500 Da facilitates their easy penetration into the cell. In addition, they may interact with multiple body targets, and through this mechanism, activate or inhibit the target protein function, modulate signaling pathways, or interfere with enzymatic activity.103 Besides, the drug molecule with effective therapeutic activity must have adequate solubility and bioavailability. Some strategies are used to overcome the low solubility and GI permeability limitations related to some drugs, including solid lipid nanoparticles.104Tsai et al. conducted research to determine the oral bioavailability of apomorphine and how this drug was distributed in the brain when it was encapsulated in SLNs. Apomorphine is used for treating Parkinson's disease. The researchers utilized glyceryl monostearate (GMS) and polyethylene glycol monostearate (PMS) emulsifiers, which impacted the physicochemical properties of the lipid nanoparticles. The results indicated a nanoparticle mean diameter of 155 nm (GMS incorporation) and 63 nm (PMS incorporation). Besides, the PMS system showed increased stability in simulated intestinal medium considering the particle size and drug loading compared with the GMS system. Both systems showed improved oral bioavailability; in vivo, experimentation utilizing rats showed that the oral bioavailability was higher compared to the drug solution, and the drug was well distributed at the site of therapeutic interest.105
Fang et al. prepared NLCs to encapsulate the anticancer drug docetaxel (DNLCs) and improve its oral bioavailability. They evaluated different properties such as stability, drug release profile, absorption in the GI tract, and drug transport pathway. The results indicated an increment in drug stability and prolonged drug release profile for DNLCs than the docetaxel solution. After intraduodenal administration, in situ, the drug concentrated in the plasma was higher for DNLCs than that from the docetaxel solution. Besides, the drug absorption mostly occurred by endocytosis. Finally, the researchers found that the drug was transported mainly using the lymphatic pathway. According to the results, NLCs are an effective drug delivery system to improve the oral bioavailability of docetaxel.106
9.2. Proteins and peptides
Proteins and peptides are molecules susceptible to enzyme degradation, have a low plasma half-life, and induce cell-mediated immune responses. These molecules can adhere to cell surfaces and other biological structures in the body due to their specific binding affinities to receptors. Also, their size, shape, charge, overall hydrophilicity, and hydrophobicity induce their accumulation in tissue.107However, peptides are quickly eliminated from the body, which limits their therapeutic effectiveness. Because of the hydrophilic nature of most peptides, the use of a hydrophobic ion pair (HIP) can improve their encapsulation. Camille Dumont et al. analyzed the encapsulation of a water-soluble peptide using SLN and NLCs as a drug delivery system. They used the HIP and evaluated the encapsulation efficiency. Also, they evaluated the size, morphology, and release rates using FaSSiF-V2, a relevant medium, and the protection against proteases. The results showed that the HIP technique increased the encapsulation of the peptide, but the liberation was fast when the nanoparticles were dispersed in the medium. Regarding peptide protection, NLC was effective against trypsin degradation but not against α-chymotrypsin. SLN failed to offer any protection. They concluded the effectiveness of NLC and the HIP technique for peptide encapsulation. However, it is a challenge to control the release and protection.108
Protein therapy has been investigated to treat different diseases with the advantage that proteins can interact with biomolecules that are complicated to target with conventional small drug molecules. The effective delivery of therapeutic proteins into specific cell regions can expand their applications.109 Hirai et al. investigated the intracellular delivery of proteins and their biodistribution using pH-responsive, charge-reversible LNPs through in vitro studies. The size of the nanoparticle was less than 200 nm and its polydispersity index was less than 0.2. Consequently, efficient cellular uptake of the protein was observed.109
9.3. Nucleic acids
Gene therapy research continues to progress, expanding its applications and improving its security and effectiveness in treating different diseases and disorders109 by gene silencing and protein expression.110 The use of nucleic acids is a medical approach that aims to modulate gene expression to achieve the desired therapeutic effects.82 Research is being conducted to develop oral LNPs for mRNA treatments, including cationic lipids, ionizable lipids, phospholipids, cholesterol, and PEG-functionalized lipids.111 These materials offer unique properties and functions for the delivery of mRNA and other nucleic acid-based therapeutics, such as aptamers, antisense oligonucleotides (AOSs), microRNA (miRNA), plasmid DNA (pDNA), and interfering RNA (siRNA).89,111 One of the challenges is delivering these materials into the cytosol to harness the potential of RNA-based molecules.82 Schlich et al. reviewed the use of ionizable lipids to encapsulate RNA. Their aim was to investigate the use of ionizable nanoparticle formulations for effective RNA delivery and stimulate endosomal destabilization to improve the delivery of genetic materials into the cellular cytosol.82 Rebecca L. Ball et al. investigated the delivery of siRNA encapsulated in lipid-based nanoparticles into epithelial cells. The interest in this research is due to the potential of siRNA in treating intestinal diseases. The studies were carried out in vitro, where they determined how stable the nanoparticles were in the acidic gastric environment, whose effect was diminished with pepsin, bile salts, and Caco-2 cell mucin. According to the in vivo results, the nanoparticles remained in the GI for around 8 h, and then moved into epithelial cells from the small intestine and colon. They recommended LNP protection to prevent the inactivation of pepsin using a pH-responsive polymer.113 Moreover, the U.S. Food and Drug Administration (FDA) approved the LNP siRNA for gene silencing in 2018.112 The lipid-based nanoparticle was designed to treat the condition transthyretin-induce amyloidosis, which is characterized by the buildup of abnormal protein deposits called amyloids in tissues.112Sung et al. encapsulated IL-22-mRNA, assisted by a cationic polymer, to treat ulcerative colitis. IL-22-mRNA is the messenger RNA molecule that encodes interleukin-22 (IL-22) protein production. This cytokine is crucial in regulating epithelial homeostasis and maintaining intestinal health. The LNPS obtained were around 200 nm in size, had a zeta potential of −18 mV, and provided efficacy for treating ulcerative colitis, accelerating the healing process. Nevertheless, the authors suggested short-term administration to prevent the risk of developing cancer associated with colitis.12
In the study by Zhiyu et al., they proposed a one-step method based on turbulent mixing conditions for the encapsulation of pDNA with better control of the particle size and distribution, encapsulation efficiency, and stability compared to traditional methods. The authors used two different lipid nanoparticle compositions to encapsulate the pDNA. In the first, the pDNA was condensed by cationic lipids and this composition was named lipo-complex. The other was the encapsulation of pDNA and a polycation with neutral lipids, which the authors named lipo-polyplexes.
The in vivo measurements, confirming the efficacy of the proposed method, revealed significantly enhanced transgene expression levels in the lungs and liver after oral administration with no signs of toxicity detectable. These promising results were particularly notable for the lipo-complexes composition, underscoring the potential of this approach.9
Moreover, in recent times, innovative methods have facilitated the in vivo screening of LNP formulations, improving traditional in vitro assay studies and obtaining more accurate results. El-Mayta et al. created a library of 96 LNP formulations with DNA barcodes (b-DNAs). They screened them in vivo using molecular barcoding, polymerase chain reaction (PCR), and genetic sequencing. This screening form enabled the evaluation of LNP behavior in the GI tract. It allowed the researchers to select better formulations or improve them to overcome oral delivery barriers and deliver nucleic acids effectively.112 Another retrospective study of LNP demonstrated the promising delivery of siRNA to intestinal epithelial cells by maintaining greater stability in the GI tract (Fig. 7).114
 | ||
| Fig. 7 LNPs visualized in the small intestine and colon epithelial cells of mice. Mice received an oral gavage of either naked or LNP-encapsulated Cy5.5-labeled siRNA (0.5 mg kg−1). Mice were sacrificed after 6 h and tissue sections were fixed and stained for DNA (blue) and actin (green). siRNA appears in red. Scale bars are 20 μm (magnification = 63×). This figure has been adapted/reproduced from ref. 114 with permission from Nature Springer, copyright 2018. | ||
10. Improving encapsulation efficiency and drug loading capacity
The amount of drug that is encapsulated in the final LNP product is called the encapsulation efficiency, where all the lipid components play a major role.115 The ionizable lipids (under acidic pH) promote electrostatic interaction and facilitate the encapsulation of the drug within the dense LNP core.The recent studies on LNP formulations for the efficient encapsulation of RNA cargos revealed encapsulation efficiency (EE%) values greater than 90%.116 This efficiency of LNPs while loading RNA is a contradictory result with the established calculation of encapsulation efficiency (EE%). EEinput% was shown to be <50% for all the formulations, whereas the traditional calculation EE% was consistently >85%. This can be minimized by increasing the lipid concentration but not up to the coverage of EE%. This major difference appeared because of the loss of RNA, which was not calculated in the case of the traditional method.117 Research also compared the LNP size (Z-average) and polydispersity index (PDI) in formulations, with results showing that the LNP size is not affected by the particle cargo size, which is a contradictory report. Thus, studies to gain insight into LNPs and their behavior in vitro and in vivo should be necessary for the advancement and development of future RNA therapeutics.
The drug loading (DL) capacity of lipid nanoparticles (LNPs) is defined as the required amount of drug that can be loaded into a delivery system, which is evaluated by the ratio of the total amount of entrapped drug and the total weight of the nanoparticle. This is expressed as a percentage.118 The DL capacity of LNPs varies with the type and the model compound. For example, trimyristin-based dispersions generally have a high loading capacity compared with other dispersions, and solid lipid nanoparticles also have the highest drug loading capacity for betamethasone-17-valerate.
Most LNPs used in clinical settings are limited in terms of their DL capacity, which is less than 10 wt%. In this case, the emulsion/solvent evaporation method can produce NPs with a drug loading of up to 14%, but other methods, such as traditional nanoprecipitation, can only achieve less than 5%. The DL capacity of NLCs can be increased by combining solid and liquid lipids with different properties. Imperfect crystal types, which are created by mixing different lipids, can also have a high drug loading capacity.119,120
11. Computational tools to predict oral absorption
Given that orally administered drugs should pass across the gastrointestinal barrier for the body to absorb them, the absorption of drugs mostly depends on their solubility and intestinal permeability, often known as intestinal absorption.121 The Biopharmaceutical Drug Disposition and Classification System (BDDCS) has incorporated solubility and permeability, indicating a strong and close relation between the intestinal permeability and metabolic rate.122 However, the intestinal permeability is very complex for drugs to pass the gut epithelium and reach the blood vessels.The most used in vitro platforms for determining gastrointestinal permeability are human colorectal adenocarcinoma cells (Caco-2), Madin–Darby canine kidney cells (MDCK), and parallel artificial membrane permeability assay (PAMPA).121 These systems are conceivably influenced by factors resembling various cell lines and culture conditions. The most widely used assay to measure potent permeability (Peff) within different regions of the GI tract (duodenum, jejunum, ileum, and colon) in humans, rats, and mice is the in situ single-pass intestinal perfusion (SPIP) model.123 The Peff, measured as cm s−1, is calculated using the following equation:
Peff = −Q![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(C′out/C′in)/A ln(C′out/C′in)/A |
| A = 2πRL |
To facilitate drug development, the pharmaceutical industry and regulatory bodies typically employ in silico models to compute the intestinal parameters. In general, these models are classified as quantitative structure–property relationships (QSPR), where the association between molecular parameters and permeability is calculated, and simple models depend on general trends by associating permeability properties.125 The former encompasses classification schemes such as Lipinski Ro5 and is mainly based on essential physicochemical characteristics. Using more intricate mathematical chemometric models, the QSPR techniques made a connection within the molecular descriptors and desired absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties. With varying degrees of effectiveness, this method has been frequently utilized to develop models that predict intestinal permeability or absorption using either observed or derived chemical descriptors of pharmacological molecules.126
Regression analysis of many descriptors and their significance is the major background for statistical models that have grown in popularity, in part because of how quickly and easily they can make predictions. These multivariable models are created based on quantitative structure–activity relationships (QSAR), which are used to evaluate the characteristics of ligands and novel analogs.127 The partial least squares projection to latent structures (PLS), support vector regression/machines (SVR/SVM), random forest (RF), and artificial neural network (ANN) are potential methods used for predicting absorption-related concerns such as solubility, and permeability. Occasionally, these models are used together with consensus models to obtain more reliable forecasts.128 The most popular linear correlation technique is multiple linear regression (MLR), which explains the relationship between two or more explanatory variables (X) and a response variable (Y). Linear discriminant analysis (LDA) divides the space of chemical descriptors into discriminant functions or hyperplanes to provide the best possible separation between various classes. Using a kernel function K(xi, xj), the support vector machine (SVM) translates the input variable into a high-dimensional feature space. SVM uses a sparse subset of training examples to construct a result.
12. Stability, safety, and regulatory concerns
Lipid-based drug delivery systems (LBDDS) enhance oral drug delivery by regulating various metabolic pathways, from enhancing the residence time in the gastric environment to lymphatic transport by extending the permeability in the intestine.129 It is widely accepted that lipid-based excipients improve the absorption of medication through their physiological effects, which include the transportation of drugs via lymphatic vessels, increasing membrane fluidity, opening tight junctions, prolonging gastric emptying,130 and pancreatic juice secretion by stimulating bile.131Triglycerides, cholesterol, and phospholipids are some examples of lipid constituents that are present in lipid-based nanocarriers. Because of the recognized safety of their lipid components and the expanding significance of nanotechnology, lipid-based nanocarriers have attracted increasing attention in pharmaceutical research. These nanocarriers have the potential to significantly overcome drug-related toxicities by reducing non-specific biodistribution with surface charge modification.132 In addition, they offer safe and effective vehicles to boost the solubility and stability of drugs. Lipid-based nanocarriers shield the therapeutic molecules from the gastric environment by encapsulation. This controls the drug precipitation in vivo and increases the stability of hydrophobic drugs in particular aqueous conditions. Furthermore, the intrinsic pharmacokinetic characteristics of pharmacological molecules are usually altered when encapsulated or associated with lipid nanocarriers.133 The extended drug release profile is primarily determined by the rate of lipid degradation and/or drug diffusion through the lipid structures.
The usage of solubilization techniques for FDA-approved medications has expanded in tandem with the extending hydrophobic nature of therapeutic candidates. A recent review stated that solubilization technologies accounted for about 6% of all recognized novel molecular entities used between 1975 and 2013, and among the solubilization techniques, LBDDS was the most popular.134 Another review performed by Strickley stated that oral LBDDS medication items make up 2–4% of all commercially sold pharmaceuticals.135 The LBDDS, known as an adaptable platform, can aid a broad range of therapeutic substances. The variety of excipients utilized in LBDDS contributes to its adaptability. The major categories included in the LBDDS lipids are hydrophilic cosolvents and water-insoluble (HLB < 12) and soluble surfactants (HLB > 1). The drug solubility, LBDDS dispersibility, and formulation characteristics are influenced by the selection and proportion of excipients in LBDDS. This helps explain its widespread use in formulating drugs with low solubility. According to Savla,136 LBDSS was deliberated for the solubilization of drug molecules in a formulation before being administrated, thereby transitioning the drug to a mixed micellar phase.
A new product cannot be released on the commercial market unless all regulatory requirements have been met. These regulatory requirements cover equipment and procedures utilized in the validation to get better potential results in addition to the components employed in the formulation. These systems are intended to be used in the market commercially and are governed by three separate United States Food and Drug Administration (USFDA) units including the Center for Devices and Radiological Health (CDRH), the Center for Drug Evaluation and Research (CDER), and the Center for Biologics Evaluation and Research (CBER). The regulatory authorities accept goods or substances designated as GRAS (generally recognized as safe) if the certification is validated for usage within certain acceptable limitations in concentrations. To determine the safety of formulations, a separate toxicity study should be conducted if the drug dosage form calls for the integration of ingredients with greater concentration than allowed limitations. Various publications and patents have addressed the manufacturing techniques used for production that are acceptable to regulatory authorities, as well as their concerns regarding the market.137 Particularly for lipid nano-formulations (LNFs), presently, there are insufficiently clear regulatory processes or parameters in place to determine their risk to performance, health, safety, and the environment. It seems that there is no consensus on generally accepted data on these formulations in defining toxicity-associated metrics. Alternatively, as innovative products, nanoformulations in general will be controlled by previous proof of safety and effectiveness as well as the components of their lipid matrix. Given this, it becomes feasible to assume that medications that have already received approval for use in traditional oral formulations or lipids categorized as GRAS materials will likely be easier to gain approval through the regulatory process to demonstrate that their efficacy and safety are comparable to that of the earlier formulations.138
13. Conclusions and outlook
The design and delivery of biologicals and treatments through oral administration have long presented hurdles. The persistent difficulties include permeability, low solubility, drug degradation, intra-enterocyte metabolism, first-pass metabolism, and enzymatic degradation. Given that LNP formulations can potentially improve the oral bioavailability and absorption of drugs, they are valuable for treating water-insoluble medications. The process and parameters of these particles such as their digestion and solubilization alter the chemical nature and association between lipid digestion products and water contents of the GI tract. Literature research has made it clear that lipid nanoparticle formulations with exceptional therapeutic applicability, bio-acceptability, and biodegradability have been developing widely. These formulations are extremely suitable and intended to be used for the delivery of insoluble drugs orally. Furthermore, they exhibit reduced toxicity, and are regarded as the most effective for encapsulating lipophilic medications in the pharmaceutical industry; however, this is a challenging task.Physiological lipids make up lipid nanoparticle-based therapeutic systems. Their fate under in vivo conditions mainly resembles the biological fate of lipids, which means they decrease toxicity, while improving the stability in the harsh GI tract environment. In addition, their nano-scaled size will help to improve the bioavailability and help enhance the drug concentration in the systemic circulation via the transport system. As a result, they are regarded as a secure and effective delivery method for poorly soluble drugs. These systems can recapitulate the intricate gastrointestinal environment, including mucus, peristaltic motions, and gut bacteria. LNP-derived mRNA therapeutics are considered one of the most appropriate therapeutic platforms to achieve advancement in LNP-based mRNA vaccines. Endosomal escape plays a pivotal role in nucleic acid-based therapeutics, but studies are lacking in terms of cytotoxicity and in vivo imaging of endosomal escape. A concern has also been expressed in the encapsulation efficiency, EEinput%, while loading RNA, which has been demonstrated to be significantly lower than the traditionally expressed EE%. Accordingly, it is necessary to evaluate the endosomal escape, cytotoxicity, and encapsulation efficiency for the development of better LNP-based mRNA formulations. The clinical translation of these technologies will be significantly impacted by the application of these methodologies, which will also contribute to the eventual progressive closing of the existing and present space by connecting academic research with the industrial development of lipid-based nanocarriers. Thus, to better comprehend the way lipid-based formulations interact with their biological environments, additional research is required in terms of agglomeration, enzymatic degradation, adsorption/desorption processes, and interaction with endogens. Lipid-based formulations as drug nanocarriers exhibit the capability to accomplish wide-ranging goals in the treatment of many diseases. Future lipid-based formulations can potentially showcase a broader range of lipid materials.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
Md Nurunnabi serves as Scientific Advisory Board member for KB BioMed Inc., and an inventor on patents related to the tools and methods discussed in this manuscript (owned and managed by University of Texas at El Paso). Other authors declare no conflict of interest.Acknowledgements
We acknowledge the funding received from Lizanell Colbert Coldwell Foundation.References
- Editorial, Nat. Rev. Mater., 2021, 6, 99 CrossRef.
- S. Talegaonkar and A. Bhattacharyya, AAPS PharmSciTech, 2019, 20, 1–15 CrossRef PubMed.
- I. V. Zhigaltsev and P. R. Cullis, Langmuir, 2023, 39, 3185–3193 CrossRef CAS PubMed.
- K. Suri, J. Wolfram, H. Shen and M. Ferrari, Novel Approaches and Strategies for Biologics, Vaccines and Cancer Therapies, Elsevier, 2015, pp. 41–58 Search PubMed.
- M. Plaza-Oliver, M. J. Santander-Ortega and M. V. Lozano, Drug Delivery Transl. Res., 2021, 11, 471–497 CrossRef CAS PubMed.
- C. H. Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, Adv. Drug Delivery Rev., 2022, 188, 114416 CrossRef PubMed.
- S. Jain, M. Kumar, P. Kumar, J. Verma, J. M. Rosenholm, K. K. Bansal and A. Vaidya, J. Funct. Biomater., 2023, 14, 437 CrossRef CAS PubMed.
- Y. Zhang, C. Sun, C. Wang, K. E. Jankovic and Y. Dong, Chem. Rev., 2021, 121, 12181–12277 CrossRef CAS PubMed.
- Z. He, Y. Hu, T. Nie, H. Tang, J. Zhu, K. Chen, L. Liu, K. W. Leong, Y. Chen and H.-Q. Mao, Acta Biomater., 2018, 81, 195–207 CrossRef CAS PubMed.
- W. M. Ibrahim, A. H. AlOmrani and A. E. B. Yassin, Int. J. Nanomed., 2014, 129–144 Search PubMed.
- M. Mendes, H. T. Soares, L. G. Arnaut, J. J. Sousa, A. Pais and C. Vitorino, Int. J. Pharm., 2016, 515, 69–83 CrossRef CAS PubMed.
- J. Sung, Z. Alghoul, D. Long, C. Yang and D. Merlin, Biomaterials, 2022, 288, 121707 CrossRef CAS PubMed.
- X. Han, H. Zhang, K. Butowska, K. L. Swingle, M.-G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2021, 12, 7233 CrossRef CAS PubMed.
- K. S. Yadav, G. Soni, D. Choudhary, A. Khanduri, A. Bhandari and G. Joshi, Med. Drug Discovery, 2023, 100162 CrossRef CAS.
- L. Xu, X. Wang, Y. Liu, G. Yang, R. J. Falconer and C.-X. Zhao, Adv. NanoBiomed Res., 2022, 2, 2100109 CrossRef CAS.
- P. Ghasemiyeh and S. Mohammadi-Samani, Res. Pharm. Sci., 2018, 13, 288–303 CrossRef PubMed.
- D. Irby, C. Du and F. Li, Mol. Pharm., 2017, 14, 1325–1338 CrossRef CAS PubMed.
- N. Dhiman, R. Awasthi, B. Sharma, H. Kharkwal and G. T. Kulkarni, Front. Chem., 2021, 9, 580118 CrossRef CAS PubMed.
- V. N. Thuy, T. V. Van, A. H. Dao and B.-J. Lee, OpenNano, 2022, 8, 100064 CrossRef.
- Y. Singh, J. G. Meher, K. Raval, F. A. Khan, M. Chaurasia, N. K. Jain and M. K. Chourasia, J. Controlled Release, 2017, 252, 28–49 CrossRef CAS PubMed.
- C. Vauthier and K. Bouchemal, Pharm. Res., 2009, 26, 1025–1058 CrossRef CAS PubMed.
- N. Anton, J.-P. Benoit and P. Saulnier, J. Controlled Release, 2008, 128, 185–199 CrossRef CAS PubMed.
- C. Lovelyn and A. A. Attama, J. Biomater. Nanobiotechnol., 2011, 2, 626 CrossRef CAS.
- S. Mukherjee, S. Ray and R. S. Thakur, Indian J. Pharm. Sci., 2009, 71, 349 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, T. A. Ataei, P. S. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- C. Grumbach, V. Krüger and P. Czermak, Pharmaceutics, 2022, 14, 1603 CrossRef CAS PubMed.
- R. Lander, W. Manger, M. Scouloudis, A. Ku, C. Davis and A. Lee, Biotechnol. Prog., 2000, 16, 80–85 CrossRef CAS PubMed.
- P. Graván, A. Aguilera-Garrido, J. A. Marchal, S. A. Navarro-Marchal and F. Galisteo-González, Adv. Colloid Interface Sci., 2023, 102871 CrossRef PubMed.
- I. Chauhan, M. Yasir, M. Verma and A. P. Singh, Adv. Pharm. Bull., 2020, 10, 150 CrossRef CAS PubMed.
- S. V. Khairnar, P. Pagare, A. Thakre, A. R. Nambiar, V. Junnuthula, M. C. Abraham, P. Kolimi, D. Nyavanandi and S. Dyawanapelly, Pharmaceutics, 2022, 14, 1886 CrossRef CAS PubMed.
- S. J. Shepherd, D. Issadore and M. J. Mitchell, Biomaterials, 2021, 274, 120826 CrossRef CAS PubMed.
- S. Uluata, E. A. Decker and D. J. McClements, Food Biophys., 2016, 11, 52–59 CrossRef.
- M. R. Gasco, US Pat., USS 188837, 1993 Search PubMed.
- S. Bala and A. Nair, Int. J. Pharm. Sci. Rev. Res., 2010, 5, 78–90 Search PubMed.
- M. Trotta, F. Debernardi and O. Caputo, Int. J. Pharm., 2003, 257, 153–160 CrossRef CAS PubMed.
- K. Manjunath, J. S. Reddy and V. Venkateswarlu, Methods Find. Exp. Clin. Pharmacol., 2005, 27, 127–144 CrossRef CAS PubMed.
- Z. Mei, X. Li, Q. Wu, S. Hu and X. Yang, Pharmacol. Res., 2005, 51, 345–351 CrossRef CAS PubMed.
- C. Puglia, P. Blasi, L. Rizza, A. Schoubben, F. Bonina, C. Rossi and M. Ricci, Int. J. Pharm., 2008, 357, 295–304 CrossRef CAS PubMed.
- D. Zhang, T. Tan and L. Gao, Nanotechnology, 2006, 17, 5821 CrossRef CAS.
- D. Hou, C. Xie, K. Huang and C. Zhu, Biomaterials, 2003, 24, 1781–1785 CrossRef CAS PubMed.
- I. R. Dos Santos, J. Richard, B. Pech, C. Thies and J. P. Benoit, Int. J. Pharm., 2002, 242, 69–78 CrossRef PubMed.
- M. Banchero, Pharmaceuticals, 2021, 14, 562 CrossRef CAS PubMed.
- R. Parhi and P. Suresh, J. Adv. Pharm. Sci. Technol., 2013, 1, 13–36 CrossRef.
- A. Bouchard, N. Jovanović, G. W. Hofland, W. Jiskoot, E. Mendes, D. J. A. Crommelin and G.-J. Witkamp, Eur. J. Pharm. Biopharm., 2008, 68, 781–794 CrossRef CAS PubMed.
- A. J. Almeida and E. Souto, Adv. Drug Delivery Rev., 2007, 59, 478–490 CrossRef CAS PubMed.
- G. B. Singhal, R. P. Patel, B. G. Prajapati and N. A. Patel, Int. Res. J. Pharm., 2011, 2, 20–52 Search PubMed.
- M. Garcıa-Fuentes, D. Torres and M. J. Alonso, Colloids Surf., B, 2003, 27, 159–168 CrossRef.
- J. Liu, T. Gong, C. Wang, Z. Zhong and Z. Zhang, Int. J. Pharm., 2007, 340, 153–162 CrossRef CAS PubMed.
- J. Hecq, K. Amighi and J. Goole, J. Drug Delivery Sci. Technol., 2016, 36, 192–200 CrossRef CAS.
- S. M. Catnach, P. D. Fairclough and S. M. Hammond, Gut, 1994, 35, 441 CrossRef CAS PubMed.
- U. B. Kompella and V. H. L. Lee, Adv. Drug Delivery Rev., 2001, 46, 211–245 CrossRef CAS PubMed.
- P. Langguth, V. Bohner, J. Heizmann, H. P. Merkle, S. Wolffram, G. L. Amidon and S. Yamashita, J. Controlled Release, 1997, 46, 39–57 CrossRef CAS.
- R. A. Cone, Adv. Drug Delivery Rev., 2009, 61, 75–85 CrossRef CAS PubMed.
- E. Y. T. Chen, N. Yang, P. M. Quinton and W.-C. Chin, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2010, 299, L542–L549 CrossRef CAS PubMed.
- S. K. Lai, Y.-Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158–171 CrossRef CAS PubMed.
- J. K. Sheehan, K. Oates and I. Carlstedt, Biochem. J., 1986, 239, 147–153 CrossRef CAS PubMed.
- N. N. Salama, N. D. Eddington and A. Fasano, Adv. Drug Delivery Rev., 2006, 58, 15–28 CrossRef CAS PubMed.
- A. Nusrat, J. R. Turner and J. L. Madara, Am. J. Physiol., 2000, 279, G851–G857 CAS.
- J. L. Madara, Annu. Rev. Physiol., 1998, 60, 143–159 CrossRef CAS PubMed.
- D. W. Powell, Am. J. Physiol.: Gastrointest. Liver Physiol., 1981, 241, G275–G288 CrossRef CAS PubMed.
- P. H. Barry, Methods Enzymol., 1989, 171, 678–715 CAS.
- V. W. Tang and D. A. Goodenough, Biophys. J., 2003, 84, 1660–1673 CrossRef CAS PubMed.
- E. Sanders and C. T. Ashworth, Exp. Cell Res., 1961, 22, 137–145 CrossRef CAS PubMed.
- P. S. Burton, R. A. Conradi and A. R. Hilgers, Adv. Drug Delivery Rev., 1991, 7, 365–385 CrossRef CAS.
- A. T. Florence, J. Drug Targeting, 2004, 12, 65–70 CrossRef CAS PubMed.
- P. J. Giannasca, K. T. Giannasca, A. M. Leichtner and M. R. Neutra, Infect. Immun., 1999, 67, 946–953 CrossRef CAS PubMed.
- A. Buda, C. Sands and M. A. Jepson, Adv. Drug Delivery Rev., 2005, 57, 123–134 CrossRef CAS PubMed.
- J. M. Hebden, C. G. Wilson, R. C. Spiller, P. J. Gilchrist, E. Blackshaw, M. E. Frier and A. C. Perkins, Pharm. Res., 1999, 16, 1087–1092 CrossRef CAS PubMed.
- H. Newey and D. H. Smyth, J. Physiol., 1959, 145, 48 CrossRef CAS PubMed.
- J. P. F. Bai and G. L. Amidon, Pharm. Res., 1992, 9, 969–978 CrossRef CAS PubMed.
- G. J. Russell-Jones, Adv. Drug Delivery Rev., 1996, 20, 83–97 CrossRef CAS.
- P. W. Swaan, Pharm. Res., 1998, 15, 826–834 CrossRef CAS PubMed.
- G. H. Hansen, L.-L. Niels-Christiansen, L. Immerdal, B. T. Nystrøm and E. M. Danielsen, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 293, G1325–G1332 CrossRef CAS PubMed.
- J. A. Yáñez, S. W. J. Wang, I. W. Knemeyer, M. A. Wirth and K. B. Alton, Adv. Drug Delivery Rev., 2011, 63, 923–942 CrossRef PubMed.
- J. Iqbal and M. M. Hussain, Am. J. Physiol.: Endocrinol. Metab., 2009, 296, E1183–E1194 CrossRef CAS PubMed.
- H. Yuan, C.-Y. Chen, G. Chai, Y.-Z. Du and F.-Q. Hu, Mol. Pharm., 2013, 10, 1865–1873 CrossRef CAS PubMed.
- Y. Xu, C. B. Michalowski and A. Beloqui, Curr. Opin. Colloid Interface Sci., 2021, 52, 101414 CrossRef CAS.
- E. Salah, M. M. Abouelfetouh, Y. Pan, D. Chen and S. Xie, Colloids Surf., B, 2020, 196, 111305 CrossRef CAS PubMed.
- H. Hassan, R. O. Bello, S. K. Adam, E. Alias, M. M. R. Meor Mohd Affandi, A. F. Shamsuddin and R. Basir, Nanomaterials, 2020, 10, 1785 CrossRef CAS PubMed.
- B. K. Nanjwade, D. J. Patel, R. A. Udhani and F. V. Manvi, Sci. Pharm., 2011, 79, 705–728 CrossRef CAS PubMed.
- A. Banerjee, J. Qi, R. Gogoi, J. Wong and S. Mitragotri, J. Controlled Release, 2016, 238, 176–185 CrossRef CAS PubMed.
- M. Schlich, R. Palomba, G. Costabile, S. Mizrahy, M. Pannuzzo, D. Peer and P. Decuzzi, Bioeng. Transl. Med., 2021, 6, e10213 CrossRef CAS PubMed.
- A. Ashkar, A. Sosnik and M. Davidovich-Pinhas, Biotechnol. Adv., 2022, 54, 107789 CrossRef CAS PubMed.
- C. J. H. Porter, A. M. Kaukonen, B. J. Boyd, G. A. Edwards and W. N. Charman, Pharm. Res., 2004, 21, 1405–1412 CrossRef CAS PubMed.
- M. Patel, V. Mundada and K. Sawant, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 144–153 CrossRef CAS PubMed.
- S. Das and A. Chaudhury, AAPS PharmSciTech, 2011, 12, 62–76 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- H. Yuan, C.-Y. Chen, G. Chai, Y.-Z. Du and F.-Q. Hu, Mol. Pharm., 2013, 10, 1865–1873 CrossRef CAS PubMed.
- S. K. Basha, R. Dhandayuthabani, M. S. Muzammil and V. S. Kumari, Mater. Today: Proc., 2021, 36, 313–324 CAS.
- H. Li, M. Chen, Z. Su, M. Sun and Q. Ping, Int. J. Pharm., 2016, 511, 524–537 CrossRef CAS PubMed.
- W. M. Ibrahim, A. H. AlOmrani and A. E. B. Yassin, Int. J. Nanomed., 2014, 129–144 Search PubMed.
- W. L. Masiiwa and L. L. Gadaga, J. Drug Delivery, 2018, 2018, 3021738 Search PubMed.
- Z. Yu, W. Fan, L. Wang, J. Qi, Y. Lu and W. Wu, Mol. Pharm., 2019, 16, 5013–5024 CrossRef CAS PubMed.
- S. Samimi, N. Maghsoudnia, R. B. Eftekhari and F. Dorkoosh, Characterization and Biology of Nanomaterials for Drug Delivery: Nanoscience and Nanotechnology in Drug Delivery, 2019, pp. 47–76 Search PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- M. Plaza-Oliver, M. J. Santander-Ortega and M. V. Lozano, Drug Delivery Transl. Res., 2021, 11, 471–497 CrossRef CAS PubMed.
- J. B. Strachan, B. P. Dyett, Z. Nasa, C. Valery and C. E. Conn, J. Colloid Interface Sci., 2020, 576, 241–251 CrossRef CAS PubMed.
- R. Kedmi, N. Ben-Arie and D. Peer, Biomaterials, 2010, 31, 6867–6875 CrossRef CAS PubMed.
- C. J. H. Porter, A. M. Kaukonen, B. J. Boyd, G. A. Edwards and W. N. Charman, Pharm. Res., 2004, 21, 1405–1412 CrossRef CAS PubMed.
- Y. Lee, M. Jeong, J. Park, H. Jung and H. Lee, Exp. Mol. Med., 2023, 55, 2085–2096 CrossRef CAS PubMed.
- Y. Li, Z. Ye, H. Yang and Q. Xu, Acta Pharm. Sin. B, 2022, 12, 2624–2639 CrossRef CAS PubMed.
- Q. Li and C. Kang, Int. J. Mol. Sci., 2020, 21(15), 5262 CrossRef CAS PubMed.
- D. V. Bhalani, B. Nutan, A. Kumar and A. K. Singh Chandel, Biomedicines, 2022, 10, 2055 CrossRef CAS PubMed.
- M.-J. Tsai, Y.-B. Huang, P.-C. Wu, Y.-S. Fu, Y.-R. Kao, J.-Y. Fang and Y.-H. Tsai, J. Pharm. Sci., 2011, 100, 547–557 CrossRef CAS PubMed.
- G. Fang, B. Tang, Y. Chao, Y. Zhang, H. Xu and X. Tang, RSC Adv., 2015, 5, 96437–96447 RSC.
- S. Srivastava, V. Sharma, B. Bhushan, R. Malviya, R. Awasthi and G. T. Kulkarni, J. Res. Pharm., 2021, 25, 99–116 CAS.
- C. Dumont, S. Bourgeois, H. Fessi, P.-Y. Dugas and V. Jannin, Int. J. Pharm., 2019, 565, 409–418 CrossRef CAS PubMed.
- Y. Hirai, H. Hirose, M. Imanishi, T. Asai and S. Futaki, Sci. Rep., 2021, 11, 19896 CrossRef CAS PubMed.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed.
- E. Fattal and F. Fay, Adv. Drug Delivery Rev., 2021, 175, 113809 CrossRef CAS PubMed.
- R. El-Mayta, R. Zhang, S. J. Shepherd, F. Wang, M. M. Billingsley, V. Dudkin, D. Klein, H. D. Lu and M. J. Mitchell, Adv. Ther., 2021, 4, 2000111 CrossRef CAS.
- R. L. Ball, K. A. Hajj, J. Vizelman, P. Bajaj and K. A. Whitehead, Nano Lett., 2018, 18, 3814–3822 CrossRef CAS PubMed.
- R. L. Ball, P. Bajaj and K. A. Whitehead, Sci. Rep., 2018, 8, 2178 CrossRef PubMed.
- M. Mehta, T. A. Bui, X. Yang, Y. Aksoy, E. M. Goldys and W. Deng, ACS Mater. Au, 2023, 3, 600–619 CrossRef CAS PubMed.
- M. Maeki, S. Uno, A. Niwa, Y. Okada and M. Tokeshi, J. Controlled Release, 2022, 344, 80–96 CrossRef CAS PubMed.
- G. B. Schober, S. Story and D. P. Arya, Sci. Rep., 2024, 14, 2403 CrossRef CAS PubMed.
- M. Mehta, T. A. Bui, X. Yang, Y. Aksoy, E. M. Goldys and W. Deng, ACS Mater. Au, 2023, 3, 600–619 CrossRef CAS PubMed.
- K. M. Rosenblatt and H. Bunjes, Eur. J. Pharm. Biopharm., 2017, 117, 49–59 CrossRef CAS PubMed.
- Y. Seo, H. Lim, H. Park, J. Yu, J. An, H. Y. Yoo and T. Lee, Pharmaceutics, 2023, 15, 772 CrossRef CAS PubMed.
- P. V. Balimane, S. Chong and R. A. Morrison, J. Pharmacol. Toxicol. Methods, 2000, 44, 301–312 CrossRef CAS PubMed.
- C.-Y. Wu and L. Z. Benet, Pharm. Res., 2005, 22, 11–23 CrossRef CAS PubMed.
- P. Zakeri-Milani, H. Valizadeh, H. Tajerzadeh, Y. Azarmi, Z. Islambolchilar, S. Barzegar and M. Barzegar-Jalali, J. Pharm. Pharm. Sci., 2007, 10, 368–379 CAS.
- I. Komiya, J. Y. Park, A. Kamani, N. F. H. Ho and W. I. Higuchi, Int. J. Pharm., 1980, 4, 249–262 CrossRef CAS.
- M. Á. Cabrera-Pérez and H. Pham-The, Expert Opin. Drug Discovery, 2018, 13, 509–521 CrossRef PubMed.
- C. A. S. Bergström, W. N. Charman and C. J. H. Porter, Adv. Drug Delivery Rev., 2016, 101, 6–21 CrossRef PubMed.
- J. M. Mayer and H. van de Waterbeemd, Environ. Health Perspect., 1985, 61, 295–306 CrossRef CAS PubMed.
- J. Glassey, Measurement, monitoring, modelling and control of bioprocesses, 2013, pp. 167–191 Search PubMed.
- M. Schwab, G. Sax, S. Schulze and G. Winter, J. Controlled Release, 2009, 140, 27–33 CrossRef CAS PubMed.
- P. G. Welling, J. Pharmacokinet. Biopharm., 1977, 5, 291–334 CrossRef CAS PubMed.
- B. N. Singh, Encyclopedia of Pharmaceutical Science and Technology, Six Volume Set (Print), CRC Press, 2013, pp. 1122–1149 Search PubMed.
- P. P. Constantinides, M. V. Chaubal and R. Shorr, Adv. Drug Delivery Rev., 2008, 60, 757–767 CrossRef CAS PubMed.
- S. Martins, B. Sarmento, D. C. Ferreira and E. B. Souto, Int. J. Nanomed., 2007, 2, 595–607 CAS.
- M. Crew, Drug Dev. Delivery, 2014, 14, 22 Search PubMed.
- R. G. Strickley, Drugs Pharm. Sci., 2007, 170, 1 CAS.
- R. Savla, J. Browne, V. Plassat, K. M. Wasan and E. K. Wasan, Drug Dev. Ind. Pharm., 2017, 43, 1743–1758 CrossRef CAS PubMed.
- L. Battaglia and E. Ugazio, J. Nanomater., 2019, 2019, 2834941 Search PubMed.
- S. Banerjee and J. Pillai, Expert Opin. Drug Metab. Toxicol., 2019, 15, 499–515 CrossRef PubMed.
- B. S. Abuasal, C. Lucas, B. Peyton, A. Alayoubi, S. Nazzal, P. W. Sylvester and A. Kaddoumi, Lipids, 2012, 47, 461–469 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |