
Mechanistic study of the competition between carbon dioxide reduction and hydrogen evolution reaction and selectivity tuning via loading single-atom catalysts on graphitic carbon nitride†
Joel Jie
Foo
ab,
Sue-Faye
Ng
ab,
Mo
Xiong
*c and
Wee-Jun
Ong
 *abdef
*abdef
aSchool of Energy and Chemical Engineering, Xiamen University Malaysia, Sepang, Selangor Darul Ehsan 43900, Malaysia. E-mail: weejun.ong@xmu.edu.my
bCenter of Excellence for NaNo Energy & Catalysis Technology (CONNECT), Xiamen University Malaysia, Sepang, Selangor Darul Ehsan 43900, Malaysia
cMOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, School of Physics, Xi'an Jiaotong University, Xi'an 710049, Shanxi, China. E-mail: xiongmo@xjtu.edu.cn
dState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
eGulei Innovation Institute, Xiamen University, Zhangzhou 363200, China
fShenzhen Research Institute of Xiamen University, Shenzhen, 518057, China
First published on 1st July 2024
Abstract
In the context of catalytic CO2 reduction (CO2RR), the interference of the inherent hydrogen evolution reaction (HER) and the possible selectivity towards CO have posed a significant challenge to the generation of formic acid. To address this hurdle, in this work, we have investigated the impact of different single-atom metal catalysts on tuning selectivity by employing density functional theory (DFT) calculations to scrutinize the reaction pathways. Single-atom catalysts supported on carbon-based systems have proven to be pivotal in altering both the activity and selectivity of the CO2RR. In this study, a series of single-atom-metal-loaded g-C3N4 monolayers (MCN, M = Ni, Cu, Zn, Ga, Cd, In, Sn, Pb, Ag, Au, Bi, Pd and Pt) were systematically examined. Through detailed DFT calculations, we explored their influence on reaction selectivity between the *COOH and *OCHO intermediates. Notably, NiCN favors the reaction via the *OCHO route, with a significantly lower rate-determining potential of 0.36 eV, which is approximately 73.5% lower than that of the CN system (1.36 eV). Most importantly, the Ni single-atom catalyst with lower coordination significantly enhances CO2 adsorption, promoting CO2RR over HER. Overall, this study, guided by DFT calculations, provides a theoretical prediction of how the selection of single-atom metal catalysts can effectively modulate the reaction pathway, thereby offering a potential solution for achieving high product selectivity in CO2RR.
1. Introduction
The development of green and sustainable energy is particularly important in the contemporary era to address devastating environmental issues such as the ever-increasing atmospheric CO2 concentration due to emissions from the combustion of the depleting fossil fuel by the energy sector. In fact, CO2 emission is the main reason that drives global climate change towards an irreparable state, gradually reshaping ecosystems to uninhabitable environments.1–3 Thus, technologies to reduce CO2 emissions and development of alternative green and sustainable fuels are urgently needed to address the deteriorating environmental conditions. Tremendous research efforts have been devoted to capturing and containing greenhouse gases, such as carbon capture and reformation strategies and catalysis technology. However, the conventional hydrogenation of CO2 is highly detrimental from the environmental perspective due to the carbon emission from this thermally activated reaction. Besides, the conventional reaction requires precious metals such as Rh and Ru as the catalyst, thus lowering the profitability of the technology.4,5 Whereas, sustainable catalytic technologies such as electrocatalysis and photocatalysis that convert CO2 into value-added fuels (such as CO,6–9 HCOOH,10–15 CH3OH16–18 and even C2 products19–23) are ideal solutions to reduce the atmospheric CO2 concentration and simultaneously generate sustainable fuels using CO2 as the raw material to replenish the depletion of the fuel, killing two birds with one stone.24–26 Among carbonaceous products, formic acid/formate has received considerable attention due to its potential to serve as a green energy carrier. The high density of formic acid (1.22 g mL−1) endows it with a considerable volumetric H2 capacity (53 g L−1), which boosts its potential as a liquid H2 carrier for hydrogen fuel-cell vehicles.27 In addition, formic acid is also one of the significant intermediates formed during the synthesis of oxygen-containing compounds such as alcohol, ester and acids in the field of syngas production.28,29Yet, the industrial application of catalytic CO2 reduction reactions (CO2RR) is greatly restrained by the inert and linear geometry of CO2, which requires −1.9 V vs. NHE for CO2RR activation.30 The second challenge is the multiple-electron kinetics, which makes it less competitive to the hydrogen evolution reaction (HER), which only requires two electrons.31 Thirdly, the complex reaction pathway and low product selectivity can lead to endless possibilities of carbonaceous products.32,33 Thus, the selectivity of the reaction to specific carbon products is highly dependent on the interactions between the catalyst and the key intermediate, which is governed by the energy barrier of the transition state evolution step in each of the proton-coupled electron transfer processes.
Graphitic carbon nitride (g-C3N4) is an emerging material in the field of catalysis owing to its extraordinary electrical conductivity and low internal resistance, which allows the generated charge carriers to spontaneously transmit to the active sites.34–36 The high thermal and chemical stability demonstrate the robust stability of the catalyst under harsh reaction environments. Its low toxicity ensures safer handling and operation while lowering the destruction risk of the ecosystem. Most importantly, its semiconducting properties, with a band gap of 2.7 eV,37 allow photon harvesting in the visible-light region (which accounts for roughly 43% of the solar spectrum),38 signifying that it can potentially serve as a photocatalyst.
The recent advent of nanomaterials with engineered shapes and dimensions presents promising modification strategies for advancement in the realm of catalysis. Among various classes of nanoarchitectures, 2D g-C3N4 has emerged as one of the prominent materials owing to its intrinsic properties that significantly enhance the catalytic performance in CO2RR. (1) The 2D nanostructure endows ample active sites for the adsorption and activation of the CO2 reactant. It eases the modification strategies, such as defect engineering and heterojunction engineering with another catalyst, by presenting highly exposed surface atoms, which can be removed or substituted.35,39 (2) 2D materials generally have a less light-shielding effect and hence can maximize the light absorption ability.40 (3) The short diffusion distance of the generated charge carrier to travel to the active sites reduces charge recombination before reaching the active sites.40,41 Despite the intrinsic advantages of 2D g-C3N4, carbon nitride is unable to deliver satisfactory catalytic CO2RR performance owing to the poor activation of adsorbed CO2.
Recently, there has been a surge in the investigation of single-atom catalysts (SAC) for maximum atomic utilization to lower the cost and simultaneously overcome the large energy barrier of the first protonation steps of the CO2RR.42–44 The introduction of foreign atoms was found to alter the reaction pathway with a lowered energy barrier and redistribute the electrons on the surface of the catalyst.45,46 He et al. found that the addition of Pd SAC enhances CO2 to CO conversion, with the Pd-N4 moiety sites leading to the *HOCO key intermediate reaction pathway.47 Liu et al. reported that the loading of Co SAC on the graphdiyne system induced charge redistribution; the Co SAC induced a negative net charge of −3.76e, and the neighboring C atoms exhibited positive values of around 0.5–0.7e, indicating that the charge transfer from the substrate contributes to the adsorption and activation of CO2 molecules.48 As such, graphitic carbon nitride with a tri-s-triazine structure has a large interstitial void surrounded by 6 unsaturated nitrogen atoms, which represents a potential anchoring site for SAC.49,50 Although machine learning and computational studies of SAC-loaded g-C3N4 have been done previously,51–58 these studies involve either non-CO2RR applications or the metal selection used for calculations is only limited to one or two SAC(s). Therefore, there is a lack of understanding of the effect of different metal SAC (M-SAC) on the underlying reaction mechanism and selectivity of the complex CO2RR toward the formation of formic acid.
By leveraging the idea of combining SAC and 2D g-C3N4, in this work, we investigated the effect of different M-SACs on the two-proton-coupled CO2RR electron transfer pathway that leads to the formation of formic acid. The formation energy calculation served as the first layer of screening to evaluate the feasibility of the system before the reaction, followed by the evaluation of CO2RR selectivity between HCOOH and CO, HER competition, density of states and Bader charge analysis to unravel the effects of various metals on the product selectivity of the CO2RR via density functional theory (DFT) calculations. Among the studied SACs, Ni SAC was found to be the optimal sample owing to strong CO2 adsorption and a significant reduction in the rate-determining potential (0.36 eV) required for the formation of formic acid via the *OCHO key intermediate. Besides, the findings demonstrate that NiCN is selective towards the CO2RR despite the inevitable competition from the HER of the water splitting process.
2. Methodology
In this study, all theoretical calculations were performed based on DFT by employing the Vienna Ab initio Simulation Package (VASP) with the post-processing VASPKIT package.59–63 Generalized gradient approximation with the Perdew–Burke–Ernzerhof (GGA-PBE) exchange–correlation functional was adopted in the calculations. The projected augmented wave (PAW) pseudopotentials and the plane-wave basis functions with a plane wave energy cut-off of 500 eV were used.64 A 3 × 3 × 1 Monkhorst–Pack grid k-point mesh was used to sample the surface Brillouin zone.65 For geometric optimization and ionic self-consistent calculation, the energy convergence criterion was set to 1.0 × 10−4 eV per atom, and force convergence at 0.05 eV Å−1 was considered. A 2 × 2 × 1 supercell was constructed with a vacuum space of 15 Å in the z-direction to minimize the interlayer interactions. Furthermore, the thermodynamic stability of the selected SAC was studied by ab initio molecular dynamics (AIMD) simulation with a time step of 1.5 fs. Charge distribution in the form of net Bader net atomic charge was calculated using the code provided by the Henkelman group.66The formation energy of the loading of M-SAC on the g-C3N4 host material is an important indicator of the stability of M-SAC in the system. The following eqn (1) was employed to define the formation energy (Ef) of the SAC-loaded system:
| Ef = ESAC–CN − ESAC − ECN | (1) |
The Gibbs free energy changes in the reaction coordinate are based on the relative energy changes of the transition state. The Gibbs free energy denotes the energy required to overcome the barrier towards the formation of the next transition state, which directly indicates the likelihood of the formation of the transition state from the energy perspective. Eqn (2) was used to define the adsorption energy (Eads) of the reactant and the energy of the intermediate-product-loaded system:
| Eads = Etotal − (ECN or MCN − Eadsorbate) | (2) |
A screening process (Fig. 1) was developed to screen the optimal M-SAC to be loaded on the g-C3N4 system to catalyze the CO2RR via the two-proton coupled electron transfer process. The process consisted of 4 evaluation levels: (1) the thermodynamic and dynamic stability of the M-SAC-loaded system, (2) the CO2RR activity and product selectivity (the competition between the formation of CO and HCOOH), as well as H2 evolution reaction activity, (3) the competition between CO2RR and HER and (4) the optical properties and charge distribution of the M-SAC-loaded system.
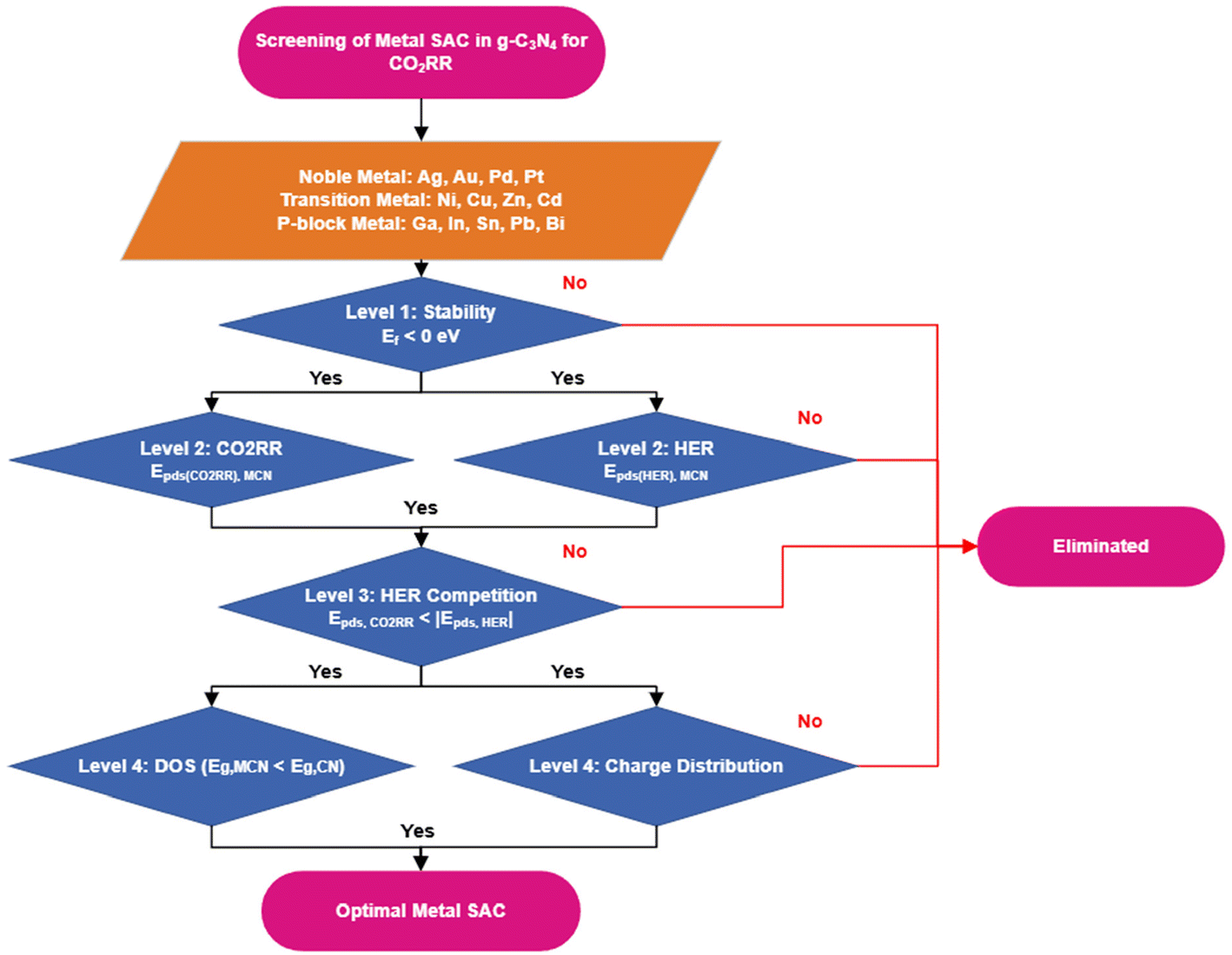 | ||
| Fig. 1 Process flow chart for screening M-SAC to be loaded on g-C3N4 for catalyzing the CO2RR. | ||
3. Results and discussion
3.1 Geometries and the stability of SACs on g-C3N4
It is known from the widely reported nitrogen-coordinated metal-single-atom-loaded graphene-based catalysts67–71 that the unique tri-s-triazine architecture of g-C3N4 exhibits a large internal cavity between the triazine structures and the unsaturated nitrogen atoms, which is particularly suitable for the loading of atoms with large atomic radii. As such, this work is focused on loading various M-SACs at the center of the six-fold cavity of g-C3N4 and evaluating their role in lowering the rate-determining potential of the CO2RR. The well-optimized monolayer structure of g-C3N4 with C–N bond lengths of around 1.33–1.40 Å and 1.47–1.5 Å in the six-membered ring and the heptazine bridging C–N, respectively, and the lattice size of 7.14 Å (a = b) are in good agreement with previous theoretical results.72–74 The formation energies of different M-SACs were calculated to evaluate their stability in the interstitial space of the heptazine structure of g-C3N4, as shown in Fig. 2(a). Apart from the Cd and Zn SACs, the formation energies of all other M-SACs had negative values ranging from −0.39 to −2.54 eV. This implies that the loading of the M-SACs is thermodynamically favorable and spontaneous except for the Cd and Zn SACs, as shown in Fig. 2(b). Therefore, the Cd and Zn SACs were excluded from further investigations.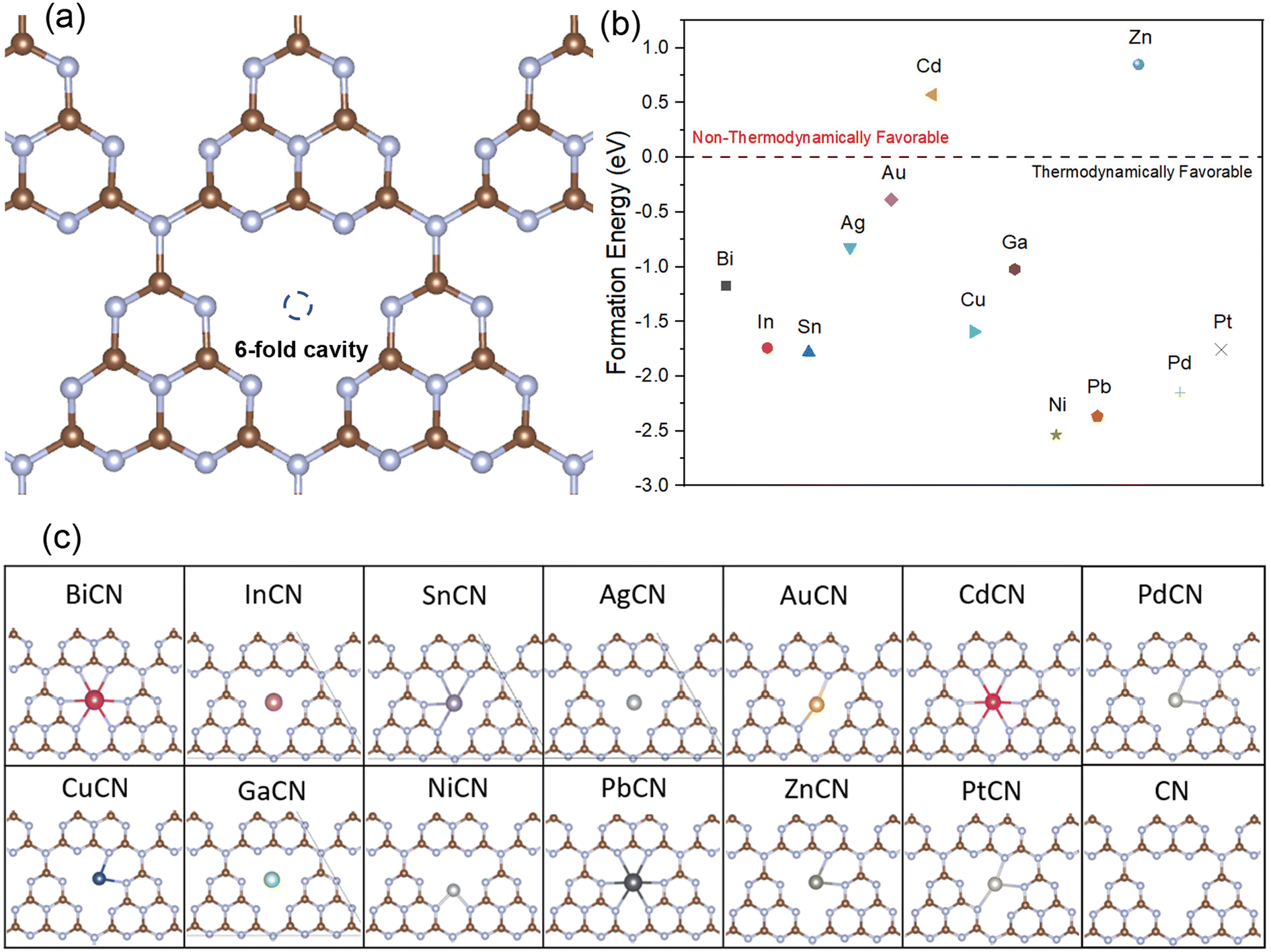 | ||
| Fig. 2 (a) The optimized corrugated g-C3N4 structures and the M-SAC binding sites of the six-fold cavity. Brown and blue indicate C and N atoms, respectively. (b) The computed formation energy of different M-SACs supported on the six-fold cavity. (c) Optimized structures of the six-fold cavities in the investigated MCN samples. | ||
Besides evaluating the thermodynamics of the loading of M-SACs, it is generally known that the N atoms at the edge of the triazine ring serve as the active sites of graphitic carbon nitride due to the unsaturated coordination of the N atoms.75 Hence, the interaction between the M atoms and the neighboring unsaturated N atoms plays an important role in retaining the structural integrity and constructing a favorable coordination environment for the activation of CO2.76 Even though all the metals can be spontaneously loaded onto the interstitial space of the tri-s-triazine allotrope except for Cd (0.57 eV) and Zn (0.85 eV) (Fig. 2(c)), it is expected that elements that exhibit poor interaction with the N atoms are unlikely to exhibit good reduction performance owing to the structural deformation during the activation of the CO2 molecules (In, Ag, Cd and Ga SACs). On the other hand, the M-SACs with larger atomic radii, including Bi, Sn, and Pb SACs, were strongly bonded to g-C3N4 with M–N distances ranging from 2.49–2.76 Å while residing in the center of the 6-fold cavity. Therefore, the loaded SACs were less likely to aggregate. Meanwhile, the M-SACs with smaller radii (Cu, Ni, Zn, Pd and Pt) tended to shift from the middle of the interstitial space to one of the corners of the heptazine ring, forming bonds with the two neighboring N atoms (M–N distances of 1.89–2.23 Å) and resulting in a structure with a lower coordination number.
The thermodynamic stability of an M-SAC is generally represented by the formation energy, and it is correlated with the dynamic stability of the MCN system. Thus, to ensure that the M-SAC has been successfully loaded to the six-fold cavity of the CN system, the Au system with the lowest formation energy was tested. In order to verify the stability of the SAC-loaded CN system, we performed an AIMD simulation of the AuCN system at T = 300 K for 4 picoseconds (4 ps). In the simulation, there was no obvious geometry deformation of AuCN (Fig. S1†), and most importantly, the metal atoms did not escape the 6-fold cavity of g-C3N4 throughout the duration of the AIMD simulation, manifesting its stability in the six-fold cavity. This verifies that the interaction between the metal single atom and the CN system can stabilize the metal in its atomic form without aggregation.
3.2 CO2RR pathways
In the first stage of reaction analysis, it is important to identify the relevant reaction pathway of the CO2RR. The first transfer of the proton–electron pair to CO2 will lead to the formation of a carboxyl (*COOH) species or formate species (*OCHO), which can be further reduced to various carbonaceous products.77 In this study, we particularly investigated the role of SAC in tuning the elementary CO2RR steps towards governing the product selectivity of the reaction. Therefore, we limited this study to only the 2-proton/electron CO2RR pathway (i.e., CO2 to HCOOH and then to CO). The scope of the study was narrowed down to the competition between the formation of CO and HCOOH from CO2via the carbonate or the formate reaction route.First, the CO2 molecule is adsorbed to the surface of the system and is reduced to one of the key intermediates, namely *COOH (carbon-bond configuration) or *OCHO (oxygen-bonded configuration), which initiates the reduction reaction. Subsequently, the second transition state is formed with a further reduction of *COOH or *OCHO to *CO or *HCOOH via the 3 reaction pathways given below (eqn (3)–(5)). These are desorbed from the surface, producing CO and HCOOH as the end products of CO2RR, respectively (Fig. 3).
| CO2 → *CO2 → *COOH → *CO → CO | (3) |
| CO2 → *CO2 → *COOH → *HCOOH → HCOOH | (4) |
| CO2 → *CO2 → *OCHO → *HCOOH → HCOOH | (5) |
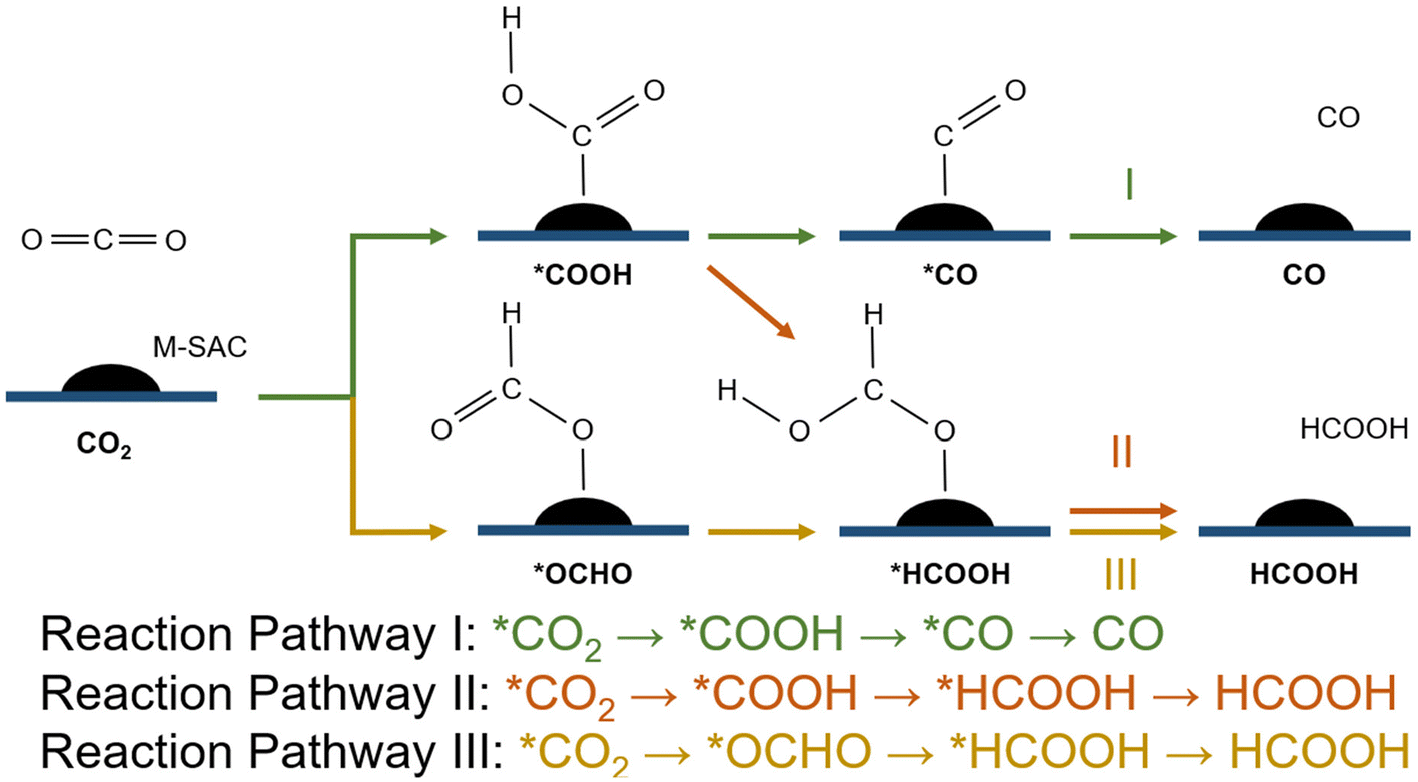 | ||
| Fig. 3 CO2RR mechanism. I: CO2 to CO via the *COOH intermediate; II: CO2 to HCOOH via the *COOH intermediate; III: CO2 to HCOOH via the *OCHO intermediate. | ||
3.3 CO2RR activity and selectivity analysis
As mentioned above, only the two-proton coupled electron transfer process was studied in this investigation. The first protonation of CO2 is generally known as the potential-limiting transition state, which governs the effectiveness of the system in catalyzing the CO2RR. The loading of heteroatoms, particularly SAC, is one of the effective methods to alter the catalytic surface chemistry and enhance the catalytic activity of the system by lowering the energy barrier. Hence, noble metals (Ag, Au, Pt and Pd), post-transition metals (Ni, Cu, Zn and Cd) and p-block metals (Ga, In, Sn, Pb and Bi) SAC were loaded onto the surface of g-C3N4 and investigated for their effect on tuning the key intermediates to yield different carbon products. The findings suggest that the loading of SACs stabilized the *COOH and *OCHO key intermediates, with all samples showing significantly lower relative free energy changes in comparison with the pristine CN system (Fig. S3(a, b) and Table S2†). Owing to the oxophility of p-block metals, all the p-block metals (Ga, In, Sn, Pb and Bi) favored the formation of the O-bonded intermediate *OCHO (over the *COOH intermediate), with the relative Gibbs free energy changes of 0.37, 0.50, −0.82, −0.56 and −0.40 eV, respectively. *OCHO proved to be the key intermediate in the formation of formic acid (Fig. S3(c)†). Unlike transition metals, the post-transition metals possessed p-block-metal-like behavior and mostly showed lower free energy for the formation of the O-bonded intermediate than the C-bonded intermediate.In order to identify the active reaction sites of the MCN, a few potential active sites were investigated to locate the feasibility of the CO2RR. The unsaturated edge nitrogen is not only a potential anchoring spot for the M-SAC; it is also a commonly studied catalytically active site in pristine g-C3N4. Thus, the CO2 molecules were first adsorbed on the unsaturated N atoms on g-C3N4 and the MCN system. In the pristine CN system, the CO2 molecules were adsorbed on the saturated N atoms, and thus the edge N atoms on the heptazine framework were selected as the active sites of the CN system. However, the CO2 reactant is less likely to be adsorbed on the same spots owing to electron redistribution after the loading of the M-SAC. Hence, after the adsorption of CO2 molecules on the M-SAC, it was found that the SAC not only favors the adsorption of CO2 molecules, but it also significantly lowers the energy barrier by destructing the linear and stable structure of CO2.
It has been found previously that the lone electron pair on the oxygen of CO2 tends to interact with the metal single atom, and the molecular behavior is similar to the other M-SAC supported on graphene.78 However, the distance between adsorbed CO2 and the metal single atom (actives) ranged from 1.9 to 3.4 Å due to the weak van der Waals interaction between CO2 and the M-SAC-loaded carbon nitride surface, which is also reflected in the CN structure (Fig. S2(a)†). A similar phenomenon has been observed by Maihom and group who employed empirical correction in Gimme's scheme (DFT-D3).72 Moreover, the Ni SAC, which was shifted to the corner position with lower coordination, exhibited more active CO2 chemisorption because of its strong electron-donating ability. Evidently, the activation of CO2 was revealed by the dimensional changes of the CO2 molecule, with the C–O bond length increasing from 1.17 to 1.21 Å when adsorbed on the surface of the NiCN catalyst.79–82 Another apparent evidence of CO2 activation was observed since the linear angle of CO2 (180°) was significantly reduced to 143.4° after the geometry optimization of CO2 adsorbed on the NiCN catalyst (Fig. S2†).
For the formation of CO, in most cases, the rate-determining step is the first hydrogenation step of CO2, but several exceptions like catalysis on AuCN, CuCN, NiCN, PdCN and PtCN have been reported as they are restricted by the desorption of CO. Hence, it is highly unlikely for the end product of the CO2RR to be CO, which will be further reduced into other products, such as CH4, HCOOH, C2+, and even C3+, which align with the following discussion on HCOOH.14,15,83–86
From the coordination environment perspective, it was observed that the coordination number of the M-SAC plays a significant role in modulating the potential-limiting steps of the reaction. As mentioned above, due to poor interaction between the M-SAC and neighboring N atoms, M-SACs with lower coordination numbers (InCN, AgCN and GaCN) had a huge increment in the first protonation step of the CO2 molecule towards forming both *COOH and *OCHO intermediates. This originates from the deformation of the structure due to weak bonding between the M-SAC and the unsaturated N atoms, causing the structures to heavily deform (i.e. away from the original plane of the monolayer) and form a bond with the intermediates. On the other hand, the highly coordinated M-SACs (BiCN, CdCN and PbCN) generally had difficulty in the adsorption of the CO2 molecule, while the *CO desorption process was easy owing to the weak interaction between the SAC and the intermediate. This phenomenon is caused by the dangling and available bonds on the M-SACs that tend to form bonds with the neighboring N atoms. Interestingly, the 6-coordinated M-SACs aid the activation of CO2 and the formation of *OCHO intermediates. Unlike the M-SACs with high coordination, the low-coordinated SACs (AuCN, CuCN, NiCN and ZnCN) were exquisite in the protonation of CO2 to form the *COOH intermediate while exhibiting difficulty in the CO desorption process as a trade-off.
In this analysis, a comparison of the rate-determining potential between the CO2RR and HER served as an indicator of reaction selectivity; this was evaluated based on the thermodynamic reaction free energy of the CO2RR step involving the formation of either CO or HCOOH and the reduction of water to hydrogen. Among noble metals, Au SAC showed a significantly low thermodynamic reaction free energy of 0.63 eV, and therefore, <0.63 eV was chosen as the passing criterion for the M-SAC that can substitute the cost-inefficient noble metals. In this stage, there were only 7 metals SACs that met this criterion, namely Bi, In, Cu, Ga, Ni, Pb and Pd SACs. Among them, only 4 SACs exhibited energy barriers less than 0.45 eV, including Ga, Pb and Ni SACs with energy barriers of 0.21, 0.36 and 0.44 eV, respectively. Yet, Ga SAC had poor CO2 adsorption on the surface, which is higher than any of the transition states in the evolution of CO2. Thus, it is excluded from property evaluation in the later discussion. Thus, NiCN showed optimal performance among the 13 metal SACs studied. This showcases the potential of Ni SACs supported on carbon nitride as an efficient catalyst for reducing CO2 to formic acid (Table 1).87
| Systems | ΔG [*COOH] (eV) | ΔG [*OCHO] (eV) |
|---|---|---|
| CN | 1.36 | 2.44 |
| BiCN | 0.32 | −0.40 |
| InCN | 1.24 | 0.50 |
| SnCN | 0.77 | −0.82 |
| AgCN | 0.73 | 0.63 |
| AuCN | −0.82 | −0.93 |
| CuCN | 0.23 | −0.13 |
| GaCN | 0.87 | 0.37 |
| NiCN | 0.45 | 0.22 |
| PbCN | 0.51 | −0.56 |
| PdCN | 0.64 | 0.53 |
| PtCN | −0.45 | −0.95 |
3.4 HER competition
The CO2RR is usually conducted in the liquid–gas phase or the solid–gas (moist CO2) phase, where the interaction between the catalyst and water is inevitable. Hence, the competition between the HER and CO2RR is detrimental to the catalytic progression of the CO2RR. Both HER and CO2RR will consume the generated h+ + e− pairs during the protonation process. In addition to that, the H atoms will occupy and cause an active site poisoning effect, demoting the CO2RR efficiency. Hence, a comparison between the free energy barriers of HER and CO2RR serves as a great selectivity descriptor in understanding which reaction is more likely to occur on the surface of the MCN system. The reaction with a lower free energy barrier is more favorable than the reaction with a higher free energy barrier owing to the large overpotential requirement in driving the latter reaction (Table 2).| Systems | Reaction path | PDS | Potential-limiting energy barrier (eV) |
|---|---|---|---|
| *PDS = potential-determining steps (the transition state that requires the highest potential in the entire reaction path). | |||
| CN | I or II | *CO2 → *COOH | 1.36 |
| BiCN | I or II | *CO2 → *COOH | 0.32 |
| InCN | III | *CO2 → *OCHO | 0.50 |
| SnCN | I or II | *CO2 → *COOH | 0.77 |
| AgCN | III | *CO2 → *OCHO | 0.63 |
| AuCN | II | *COOH → HCOOH | 1.11 |
| CuCN | II or III | *HCOOH → HCOOH | 0.52 |
| GaCN | III | *CO2 → *OCHO | 0.37 |
| NiCN | III | *OCHO → *HCOOH | 0.36 |
| PbCN | III | *OCHO → *HCOOH | 0.44 |
| PdCN | III | *CO2 → *OCHO | 0.53 |
| PtCN | II | *COOH → *HCOOH | 0.75 |
By comparing the HER and the two CO2RR (two-proton/electron pairs) reaction pathways, it was found that there is a strong correlation between the *H and *COOH intermediates, which is similar to the previously reported DFT work.88 A similar trend was observed for the formation of CO through the carboxyl (*COOH) intermediate, with the inevitable and strong HER competition causing the formation of CO to be less competitive to the formation of H2. A majority of CO-selective MCN systems (BiCN, SnCN, CdCN, ZnCN) had strong HER competition, as exemplified by the much lower free energy barrier of the HER reaction. A similar phenomenon was found in the noble metal systems AuCN, PdCN and PtCN, which had rate-determining potentials of 0.87, 0.39 and 0.28 eV, respectively. On the other hand, the CO2RR pathway with the formate intermediate (*OCHO) was found to retain selectivity towards the CO2RR over the HER (Table 3).
| Systems | ΔGpds HER (eV) | ΔGpds CO2RR (eV) | Selectivity |
|---|---|---|---|
| *HER = Hydrogen evolution reaction, CO2RR = Carbon dioxide reduction to carbon monoxide via *COOH as the key intermediate or to formic acid via *OCHO as the key intermediate (depending on which has a lower free energy barrier). | |||
| CN | 1.80 | 1.36 | CO2RR |
| BiCN | 0.18 | 0.31 | HER |
| InCN | 1.02 | 0.50 | CO2RR |
| SnCN | 0.51 | 0.77 | HER |
| AgCN | 0.67 | 0.63 | CO2RR |
| AuCN | 0.87 | 1.11 | HER |
| CdCN | 0.16 | 0.32 | HER |
| CuCN | 0.42 | 0.52 | HER |
| GaCN | 0.88 | 0.37 | CO2RR |
| NiCN | 0.75 | 0.36 | CO2RR |
| PbCN | 0.59 | 0.44 | CO2RR |
| ZnCN | 0.43 | 0.45 | HER |
| PdCN | 0.39 | 0.53 | HER |
| PtCN | 0.28 | 0.75 | HER |
3.5 Property investigation and charge distribution
For an emerging photocatalytic material, the bandgap indicates its capability to harvest light and the absorption range of the light to induce the formation of electron–hole pairs for the photocatalytic redox reaction. The bandgap of g-C3N4 was calculated to be 1.85 eV, which is way below the experimental value, when the PBE functional was formulated to obtain the ground-state properties of atoms, molecules and condensed matter. However, it could not predict the exact bandgaps of semiconductors. The fundamental bandgap of a system with N electrons is given by the differences in the total ground-state energies of systems with different numbers of electrons. The ground-state total energy can be estimated by DFT but this does not imply that the bandgap of the Kohn–Sham system is the fundamental gap of the interacting electron system. Correction using HSE03 can be implemented to rectify the underestimation of the bandgap, but it requires a lot of computational resources and is less suitable for calculating a 2 × 2 × 1 supercell to compare the pristine and the metal-loaded systems. Yet, the PBE generated can serve well to compare the relative bandgap changes between pristine CN and the metal-loaded counterpart. In our analysis, the N 2p orbital dominated the valence band maximum (VBM), while the conduction band minimum (CBM) was mainly composed of a combination of the 2p orbitals of C and N atoms, similar to that reported in the literature.72 Two M-SACs, namely Pb and Ni SACs, had narrower bandgaps, indicating that the metal-loaded systems have better light adsorption capability than pristine CN (Fig. 5). Furthermore, it was observed that the mid-gap state, which is also known as the impurities state of the metal-loaded SAC, was located closer to the CB. As such, the impurity states will serve as the electron capture sites and thus active sites for the reduction process, once again verifying the better adsorption of the reactant and other molecules to the M-SAC. Besides, the loading of M-SAC was found to have a band-shifting effect that governs the reducibility of the catalyst system. All three M-SAC-loaded CN samples had upshifted peaks, indicating the stronger reduction potential of the systems. The two M-SAC-loaded systems PbCN and NiCN appeared to have strong light harvesting ability with lowered bandgaps of 1.51 and 1.71 eV, respectively.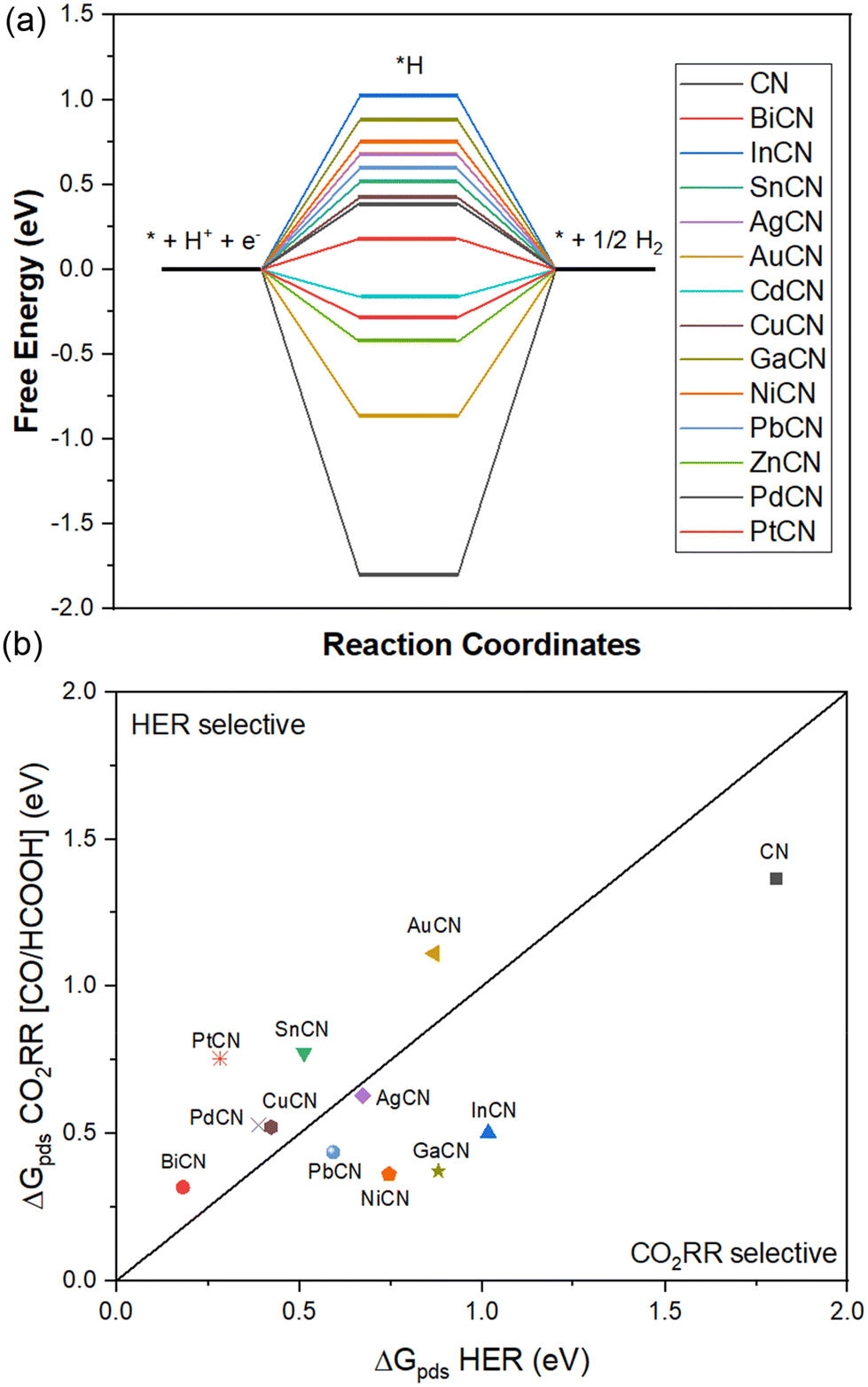 | ||
| Fig. 4 (a) Free energy diagram of HER on the SAC-loaded CN systems (M = Bi, In, Sn, Ag, Au, Cd, Cu, Ga, Ni, Pb and Zn). (b) Competition between CO2RR and HER based on the rate-determining potentials of the CN and MCN systems. The area below the linear lines indicates CO2RR-selective catalysts, whereas the area above the linear line indicates HER-selective catalysts. | ||
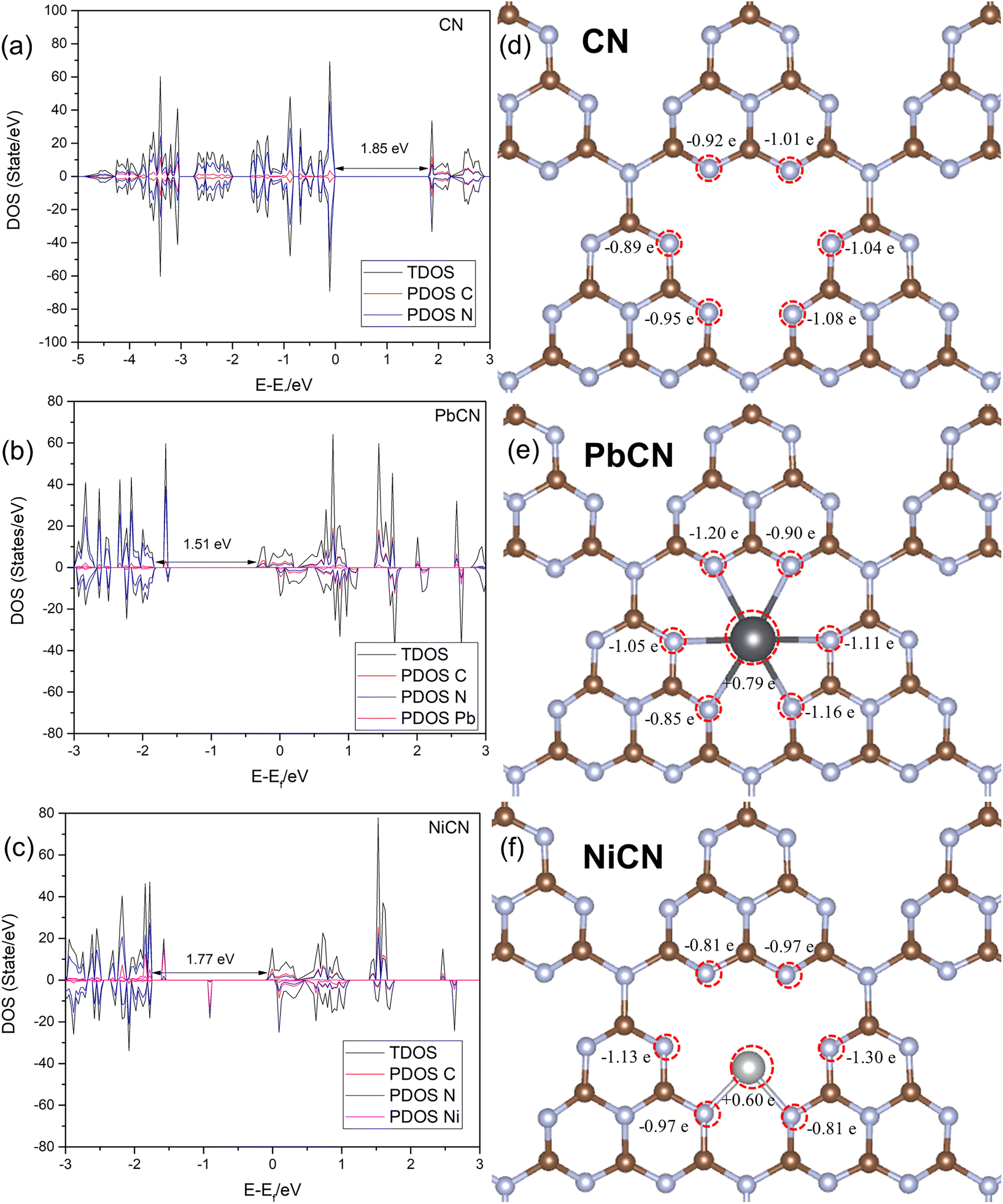 | ||
| Fig. 5 The density of states of the metal-loaded CN systems: (a) pristine CN, (b) PbCN and (c) NiCN. Bader net atomic charges on the catalytic systems: (d) pristine CN, (e) PbCN and (f) NiCN. The brown balls represent carbon atoms; the light blue balls represent nitrogen atoms; the black ball represents the Pb atom and the silvery grey ball represents the Ni atom. | ||
Bader charge analysis was conducted to investigate the charge distribution on the catalyst surface to elucidate the charge transfer process after the loading of the M-SACs. It was observed that the loading of different metal-SACs leads to different charge redistribution on the catalyst and that the neighboring N atoms contribute electrons to the M-SACs, which is in coherence with the discussion on the density of states of the system that the M-SAC serves as the electron-trapping zone. Both the systems showed charge migration effect on the Pb and Ni SAC sites with positive charges at +0.79 |e| and +0.60 |e|, respectively, while the total charge changes in the surrounding N atoms were ±0.38 |e| and ±0.10 |e| in comparison with the pristine CN system (Fig. 5). This reflects that charges are contributed by the neighboring N atoms to the loaded M-SAC, easing the adsorption of the CO2 molecule, as reflected by the lowered energy barrier in the previous discussion. Electron transfer from the metal atoms to g-C3N4 leads to a more positive charge value on the metal single atoms, once again confirming the strong covalent interactions between the metal single atoms and g-C3N4.
4. Conclusion
In summary, we applied periodic DFT calculations to investigate how CO2 is adsorbed and reduced to formic acid in the MCN systems. The metal active sites attract CO2 through the electron withdrawal from the neighbouring N atoms and accumulation on the metal active sites. From the reaction kinetics perspective, NiCN showed outstanding CO2 activation ability, as observed from the bent structure of CO2 from 180° to 143.3° and the lengthened C–O bond from 1.17 to 1.21 Å after adsorption on the catalyst surface. This is mainly because of the electrons that migrate from the neighboring N atoms to the Ni atoms (0.60 |e|), which drastically improve the adsorption and activation of the CO2 molecule on the surface. By comparing the potential-determining steps across various pathways leading to the formation of carbon monoxide and formic acid, it was found that NiCN shows optimal performance with the lowest Gibbs Free reaction energy change of 0.36 eV, which is approximately a 73.5% improvement compared with pristine CN. Moreover, NiCN is also selective towards the formation of formic acid, with a significantly lowered rate-determining potential of <0.5 eV; It selectively converts CO2 to formic acid in two steps (Pathway 3): (i) CO2 is first hydrogenated to the formate intermediate state; (ii) the formate intermediate is then further reduced to formic acid. The hydrogenation of the formate intermediate to formic acid is the potential-determining step of the NiCN system. The lower activation energy of the hydrogenation steps in NiCN is due to the greater electron delocalization between the catalyst active site and the C–O bond of CO2, which leads to a more stable transition state. Apart from that, the free energy change of the CO2RR potential-determining step to generate formic acid (0.36 eV) is lower than that of the highly competitive HER (0.75 eV), thus inferring that the catalytic system favors CO2RR over the HER. In conclusion, our results have established an inspiring framework based on CO2 adsorption and hydrogenation to produce formic acid on graphitic carbon nitride with metal-single-atom modifications for advancing the design of future catalysts.Data availability
ESI† associated with this article can be found, in the online version, at doi: 10.1016/Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to acknowledge the financial support provided by the Ministry of Higher Education (MOHE) Malaysia under the Fundamental Research Grant Scheme (FRGS) (Ref no: FRGS/1/2020/TK0/XMU/02/1). The authors would like to thank the Ministry of Science, Technology and Innovation (MOSTI) Malaysia under Strategic Research Fund (SRF) (S.22015). The authors gratefully acknowledge Agilent Technologies Malaysia Sdn Bhd for their contribution through chromatography. The authors would also like to acknowledge the financial support provided by the National Natural Science Foundation of China (Ref no: 22202168) and Guangdong Basic and Applied Basic Research Foundation (Ref no: 2021A1515111019). This work is supported by the Embassy of the People's Republic of China in Malaysia (EENG/0045). We would like to acknowledge the financial support from the State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University (Ref. no: 2023X11). This work is also funded by Xiamen University Malaysia Investigatorship Grant (Grant no: IENG/0038), Xiamen University Malaysia Research Fund (ICOE/0001 and XMUMRF/2021/C8/IENG/0041), and Hengyuan International Sdn. Bhd. (Grant no: EENG/0003).References
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CAS.
- Z.-X. Bi, R.-T. Guo, X. Hu, J. Wang, X. Chen and W.-G. Pan, Nanoscale, 2022, 14, 3367–3386 RSC.
- E. Pugliese, P. Gotico, I. Wehrung, B. Boitrel, A. Quaranta, M.-H. Ha-Thi, T. Pino, M. Sircoglou, W. Leibl, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2022, 61, e202117530 CrossRef CAS PubMed.
- A. Jaleel, S.-H. Kim, P. Natarajan, G. H. Gunasekar, K. Park, S. Yoon and K.-D. Jung, J. CO2 Util., 2020, 35, 245–255 CrossRef CAS.
- A. Erdőhelyi, Catalysts, 2020, 10, 155 CrossRef.
- S. Lu, S. Zhang, L. Li, C. Liu, Z. Li and D. Luo, Chem. Eng. J., 2024, 483, 149058 CrossRef CAS.
- J. Liang, H. Zhang, Q. Song, Z. Liu, J. Xia, B. Yan, X. Meng, Z. Jiang, X. W. Lou and C.-S. Lee, Adv. Mater., 2024, 36, 2303287 CrossRef CAS PubMed.
- P. Zhu, H. Cao, H. Yang, M. Geng, S. Qin, L. Tan, X. Gao and C. Wang, Appl. Surf. Sci., 2024, 652, 159348 CrossRef CAS.
- Z. Zhou, W. Guo, T. Yang, D. Zheng, Y. Fang, X. Lin, Y. Hou, G. Zhang and S. Wang, Chin. J. Struct. Chem., 2024, 43, 100245 CrossRef CAS.
- L.-Y. Peng, G.-N. Pan, W.-K. Chen, X.-Y. Liu, W.-H. Fang and G. Cui, Angew. Chem., Int. Ed., 2024, 63, e202315300 CrossRef CAS.
- Y. Cao, C. Zhang, W. Wang, Y. Liu, Y. Tao, J. Fan, M. Chen, D. Zhang and G. Li, Appl. Catal., B, 2024, 349, 123867 CrossRef CAS.
- J. Zhou, X. Tian, S. Yu, Z. Zhao, Y. Ji, U. Schwanebrg, B. Chen, T. Tan, Z. Cui and M. Wang, Chem. Eng. Sci., 2024, 285, 119613 CrossRef CAS.
- Y. Sun, L. Tao, M. Wu, D. Dastan, J. Rehman, L. Li and B. An, Nanoscale, 2024, 16, 9791–9801 RSC.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- J. Ma, H. Gong, T. Zhang, H. Yu, R. Zhang, Z. Liu, G. Yang, H. Sun, S. Tang and Y. Qiu, Appl. Surf. Sci., 2019, 488, 1–9 CrossRef CAS.
- H. Jian, K. Deng, T. Wang, C. Huang, F. Wu, H. Huo, B. Ouyang, X. Liu, J. Ma, E. Kan and A. Li, Chin. Chem. Lett., 2024, 35, 108651 CrossRef CAS.
- S. M. Albukhari, L. A. Al-Hajji and A. A. Ismail, Environ. Res., 2024, 241, 117573 CrossRef CAS.
- X. Guo, C. Qiu, Z. Zhang, J. Zhang, L. Wang, J. Ding, J. Zhang, H. Wan and G. Guan, J. Environ. Chem. Eng., 2024, 12, 111990 CrossRef CAS.
- S.-F. Ng, J. J. Foo and W.-J. Ong, InfoMat, 2022, 4, e12279 CrossRef CAS.
- P. Li, L. Liu, W. An, H. Wang, H. Guo, Y. Liang and W. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS.
- H. Li, M. Xia, X. Wang, B. Chong, H. Ou, B. Lin and G. Yang, Appl. Catal., B, 2024, 342, 123423 CrossRef CAS.
- J. Yin, X. Song, C. Sun, Y. Jiang, Y. He and H. Fei, Angew. Chem., Int. Ed., 2024, 63, e202316080 CrossRef CAS PubMed.
- X. Cao, C. Liu, Y. Dong, T. Yang, X. Chen and Y. Zhu, Catal. Sci. Technol., 2024, 14, 2003–2011 RSC.
- A. Bagger, O. Christensen, V. Ivaništšev and J. Rossmeisl, ACS Catal., 2022, 12, 2561–2568 CrossRef CAS.
- Z. Wang, Z. Yang, R. Fang, Y. Yan, J. Ran and L. Zhang, Chem. Eng. J., 2022, 429, 132322 CrossRef CAS.
- X. Xuan, M. Wang, M. Zhang, Y. V. Kaneti, X. Xu, X. Sun and Y. Yamauchi, J. CO2 Util., 2022, 57, 101883 CrossRef CAS.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC.
- T. Lin, X. Qi, X. Wang, L. Xia, C. Wang, F. Yu, H. Wang, S. Li, L. Zhong and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 4627–4631 CrossRef CAS.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2021, 7, 849–881 CAS.
- Y. Wang, E. Chen and J. Tang, ACS Catal., 2022, 12, 7300–7316 CrossRef CAS PubMed.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- W. Ju, A. Bagger, X. Wang, Y. Tsai, F. Luo, T. Möller, H. Wang, J. Rossmeisl, A. S. Varela and P. Strasser, ACS Energy Lett., 2019, 4, 1663–1671 CrossRef CAS.
- E. C. Ra, K. Y. Kim, E. H. Kim, H. Lee, K. An and J. S. Lee, ACS Catal., 2020, 10, 11318–11345 CrossRef CAS.
- F. M. Yap, G. Z. S. Ling, B. J. Su, J. Y. Loh and W.-J. Ong, Nano Res. Energy, 2024, 3, e9120091 CrossRef.
- K. J. Wong, J. J. Foo, T. J. Siang and W.-J. Ong, Adv. Funct. Mater., 2023, 33, 2306014 CrossRef CAS.
- Z. Gu, Z. Shan, Y. Wang, J. Wang, T. Liu, X. Li, Z. Yu, J. Su and G. Zhang, Chin. Chem. Lett., 2024, 35, 108356 CrossRef CAS.
- Y. Liu, L. Chen, X. Liu, T. Qian, M. Yao, W. Liu and H. Ji, Chin. Chem. Lett., 2022, 33, 1385–1389 CrossRef CAS.
- W. Zhang, H. He, H. Li, L. Duan, L. Zu, Y. Zhai, W. Li, L. Wang, H. Fu and D. Zhao, Adv. Energy Mater., 2021, 11, 2003303 CrossRef CAS.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, J. Environ. Sci., 2024, 140, 103–112 CrossRef CAS.
- W. Mo, J. J. Foo and W.-J. Ong, Electron, 2024, 2, e20 CrossRef.
- S.-F. Ng, X. Chen, J. J. Foo, M. Xiong and W.-J. Ong, Chin. J. Catal., 2023, 47, 150–160 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS.
- Y. Ren, J. Wang, M. Zhang, Y. Wang, Y. Cao, D. H. Kim and Z. Lin, Angew. Chem., Int. Ed., 2024, 63, e202315003 CrossRef CAS.
- M.-K. Wong, J. J. Foo, J. Y. Loh and W.-J. Ong, Adv. Energy Mater., 2024, 14, 2303281 CrossRef CAS.
- X. Ma, Q. Chen, C. Han, S. Zhou, Z. Li, J. Liu, F. Hu, J. Wang, N. Wang, Y. Zhu and J. Zhu, Adv. Funct. Mater., 2024, 34, 2307733 CrossRef CAS.
- Q. He, J. H. Lee, D. Liu, Y. Liu, Z. Lin, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Adv. Funct. Mater., 2020, 30, 2000407 CrossRef CAS.
- T. Liu, G. Wang and X. Bao, J. Phys. Chem. C, 2021, 125, 26013–26020 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- C.-L. Chen, R. Chen, L.-S. Long, L.-S. Zheng and X.-J. Kong, Chin. Chem. Lett., 2024, 35, 108795 CrossRef CAS.
- C. Zhao, M. Xi, J. Huo, C. He and L. Fu, Chin. Chem. Lett., 2023, 34, 107213 CrossRef CAS.
- Y. Qin, Y. Li, W. Zhao, S. Chen, T. Wu and Y. Su, Nano Res., 2023, 16, 325–333 CrossRef CAS.
- Y. Huang, C. Tang, Q. Li and J. Gong, Appl. Surf. Sci., 2023, 616, 156440 CrossRef CAS.
- A. Banerjee, Catal. Today, 2023, 418, 114079 CrossRef CAS.
- A. Hassan and M. A. Dar, New J. Chem., 2023, 47, 7225–7231 RSC.
- S. Liu, G. Xing and J.-Y. Liu, Appl. Surf. Sci., 2023, 611, 155764 CrossRef CAS.
- F. Chen, Y. Yang and X. Chen, ACS Appl. Nano Mater., 2024, 7, 11862–11870 CrossRef CAS.
- R. Shakir, H.-P. Komsa, K. K. Paswan, A. S. K. Sinha and J. Karthikeyan, J. Phys. Chem. C, 2024, 128, 5505–5514 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- S. Ali, R. Iqbal, A. Khan, S. U. Rehman, M. Haneef and L. Yin, ACS Appl. Nano Mater., 2021, 4, 6893–6902 CrossRef CAS.
- M. D. Esrafili and B. Nejadebrahimi, Appl. Surf. Sci., 2019, 475, 363–371 CrossRef CAS.
- C. Jia, S. Li, Y. Zhao, R. K. Hocking, W. Ren, X. Chen, Z. Su, W. Yang, Y. Wang, S. Zheng, F. Pan and C. Zhao, Adv. Funct. Mater., 2021, 31, 2107072 CrossRef CAS.
- F. Pan, B. Li, E. Sarnello, Y. Fei, X. Feng, Y. Gang, X. Xiang, L. Fang, T. Li, Y. H. Hu, G. Wang and Y. Li, ACS Catal., 2020, 10, 10803–10811 CrossRef CAS.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- K. Homlamai, T. Maihom, S. Choomwattana, M. Sawangphruk and J. Limtrakul, Appl. Surf. Sci., 2020, 499, 143928 CrossRef CAS.
- J. Wirth, R. Neumann, M. Antonietti and P. Saalfrank, Phys. Chem. Chem. Phys., 2014, 16, 15917–15926 RSC.
- S. Lu, Z. W. Chen, C. Li, H. H. Li, Y. F. Zhao, Y. Y. Gong, L. Y. Niu, X. J. Liu, T. Wang and C. Q. Sun, J. Mater. Chem. A, 2016, 4, 14827–14838 RSC.
- G. Z. S. Ling, V. B.-Y. Oh, C. Y. Haw, L.-L. Tan and W.-J. Ong, Energy Mater. Adv., 2023, 4, 0038 CrossRef CAS.
- Y. Ren, J. Wang, M. Zhang, Y. Wang, Y. Cao, D. H. Kim, Y. Liu and Z. Lin, Small, 2024, 20, 2308213 CrossRef CAS.
- R. Nankya, A. Elgazzar, P. Zhu, F.-Y. Chen and H. Wang, Mater. Today, 2024, 76, 94–109 CrossRef CAS.
- J. Sirijaraensre and J. Limtrakul, Appl. Surf. Sci., 2016, 364, 241–248 CrossRef CAS.
- G. Peng, L. Xu, V.-A. Glezakou and M. Mavrikakis, Catal. Sci. Technol., 2021, 11, 3279–3294 RSC.
- P. Lozano-Reis, H. Prats, P. Gamallo, F. Illas and R. Sayós, ACS Catal., 2020, 10, 8077–8089 CrossRef CAS.
- B. Kreitz, G. D. Wehinger, C. F. Goldsmith and T. Turek, J. Phys. Chem. C, 2021, 125, 2984–3000 CrossRef CAS.
- H. Chen, M. Yang, J. Liu, G. Lu and X. Feng, Catal. Sci. Technol., 2020, 10, 5641–5647 RSC.
- Q. Wang, X. Zheng, J. Wu, Y. Wang, D. Wang and Y. Li, Small Struct., 2022, 3, 2200059 CrossRef CAS.
- D. Chen, Z. Chen, Z. Lu, J. Tang, X. Zhang and C. V. Singh, J. Mater. Chem. A, 2020, 8, 21241–21254 RSC.
- T. Tang, Z. Wang and J. Guan, Exploration, 2023, 3, 20230011 CrossRef CAS PubMed.
- H. Ou, G. Li, W. Ren, B. Pan, G. Luo, Z. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 22075–22082 CrossRef CAS PubMed.
- Z. Zhang, C. Shen, K. Sun, X. Jia, J. Ye and C.-J. Liu, J. Mater. Chem. A, 2022, 10, 5792–5812 RSC.
- J. S. Yoo, R. Christensen, T. Vegge, J. K. Nørskov and F. Studt, ChemSusChem, 2016, 9, 358–363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01932f |
| This journal is © The Royal Society of Chemistry 2024 |