
Achieving a balance of rapid Zn2+ desolvation and hydrogen evolution reaction inertia at the interface of the Zn anode†
Xiaofen
Xiao
 *a,
Deqiang
Wang
a,
Guangyi
Xu
a,
Zhuxiang
Zhang
a,
Jun
Li
ab,
Shun
Wang
a,
Yifei
Yuan
a,
Chuangang
Hu
c and
Huile
Jin
*ab
*a,
Deqiang
Wang
a,
Guangyi
Xu
a,
Zhuxiang
Zhang
a,
Jun
Li
ab,
Shun
Wang
a,
Yifei
Yuan
a,
Chuangang
Hu
c and
Huile
Jin
*ab
aKey Lab of Advanced Energy Storage and Conversion, Zhejiang Province Key Lab of Leather Engineering, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, Zhejiang 325035, China. E-mail: xiaoxf3@mail2.sysu.edu.cn
bZhejiang Engineering Research Center for Electrochemical Energy Materials and Devices, Institute of New Materials and Industrial Technologies, Wenzhou University, Wenzhou, Zhejiang 325035, China
cState Key Laboratory of Organic-Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 19th August 2024
Abstract
It is difficult to achieve fast kinetics of Zn2+(H2O)6 desolvation as well as HER inertia at the same electrolyte/Zn interface during long-term cycling of Zn plating/stripping in aqueous Zn-ion batteries. Herein, an effective interface construction strategy with hydrophilic transition metal oxides was proposed to achieve that balance using a CeO2 layer coating. The hydrophilic CeO2 layer can bring a balance between improving the access to the anode surface for Zn2+(H2O)6 electrolyte ions, providing uniform Zn2+ nucleation sites and HER inertia. What's more, Zn corrosion can be significantly inhibited benefiting from this coating layer. The efficiency of aqueous Zn-ion batteries showed a great enhancement. Ultra-long plating/stripping stability up to 1600 h and excellent recovery (returning to 0.5 from 20 mA cm−2) for the symmetric CeO2@Zn system were observed. A full cell with the MnO2 cathode (CeO2@Zn//MnO2) with good reversibility and stability (∼600 cycles) was fabricated for practical application. Our work provides a fundamental understanding and an essential solution to deal with the balance between rapid desolvation and inhibition of the hydrogen evolution reaction, which is important for promoting the practical application of rechargeable Zn batteries.
1. Introduction
Among various future battery technologies beyond the lithium ion, aqueous zinc-ion batteries (ZIBs) with near-neutral electrolytes have attracted particular attention in recent years due to the following advantages: low cost, good safety, and a high abundance of Zn.1–3 However, the great potential development of Zn metal anodes is severely hampered by the low plating/stripping coulombic efficiency (CE) and poor cycling stability caused by hydrogen evolution, corrosion and dendrite formation in slightly acidic electrolytes.4–6 The side reactions could be expressed as follows:| H2O + e− → ½H2↑ + OH− |
| 4Zn2+ + 6OH− + SO42− + nH2O → Zn4SO4(OH)6·nH2O↓ |
The growth of Zn dendrites originated from the inhomogenous nucleation sites during the long-term cycling process, especially under large current density conditions.7–10 What's more, Zn dendrite growth and the HER would accelerate with each other.11 With the growth of dendrites, more active sites would be exposed and accelerate the HER rate;12 while a rough surface would be formed on the surface of Zn as Zn corrodes in the HER.10 This lowers the energy barrier of the desolvation process of Zn2+(H2O)6 at the interface of the electrolyte/electrode to enhance a fast mass transfer kinetics of Zn plating/stripping, which is important for fostering homogeneous ion distribution and uniform deposition.13 As reported by the research studies, hydrophilic surfaces are beneficial for driving the first trapping process of Zn2+(H2O)6, however, this always occur along with solvent molecule degradation and the HER.7
Though various strategies have been reported on tackling these anode issues, it is still difficult to reach a certain balance between HER inertia and fast kinetics of Zn2+(H2O)6 desolvation4,10,14–17 to achieve continuous regulation of the deposition behavior of Zn during a long-term cycling process. Hence, suppressing the parasitic reaction and inducing homogeneous Zn plating/stripping at the fundamental level are urgently needed.7,11,18
Analyzing the thermodynamic problems, based on the research results of the field of electrocatalysis,19–21 the HER is a typical two-electron transfer reaction with one catalytic intermediate (H*, where * denotes a site on the electrode surface) and occurs via either the Volmer–Tafel or the Volmer–Heyrovsky mechanism:22
Volmer step:
| H+ + e + * → H* | (1) |
Heyrovsky step:
| H* + H+ + e → H2 + * | (2) |
Tafel step:
| 2H* → H2 + 2* | (3) |
The hydrogen adsorption free energy ΔGH above controlled the rate of the overall reaction. Based on the Sabatier's principle that the binding energy between the catalyst and the reactant should be neither too strong nor too weak, if hydrogen binds to the surface too weakly, the adsorption (Volmer) step will limit the overall reaction rate, whereas if the binding is too strong, the rate will be limited by the desorption (Heyrovsky/Tafel) step.23,24 For 2M ZnSO4 aqueous solution, it was weakly acidic because of the reaction Zn2+ + 2H2O = Zn(OH)2 + 2H+.25,26 It is very easy for (1) the Volmer step to occur because that H+ owns an empty 1s orbital which could bind strongly with the surface O.27 However, too much strength of the O–H bond can make the following desorption (2)/(3) (Heyrovsky/Tafel) step very difficult to occur, bringing the high energy barrier of the whole HER process. Therefore, barely transition metal oxides were used for electrocatalytic HER in acidic medium.27,28 Here in the system of aqueous Zn-ion batteries, the HER is taken as an adverse reaction which needs to be inhibited.12
Based on this, CeO2 was deliberately chosen as a coating layer to fabricate an interface bringing a balance between HER inertia and fast kinetics of Zn2+(H2O)6 desolvation. On one hand, CeO2 is a kind of typical indirect transition N-type semiconductor which is unfavorable for HER.17,27,29,30 On the other hand, it has a hydrophilic property.31 This gives the potential of CeO2 that ensures a fast capture of Zn2+(H2O)6 leading to a uniform distribution of Zn2+ nucleation sites as well as inhibiting the HER adverse reaction.4,15,24,32 What's more, the standard electrode potential of Ce4+/Ce3+ in aqueous solutions at 25 °C (vs. NHE) was much higher than φZn2+/Zn as shown in the following:
| Ce4+ + e ⇄ Ce3+ 1.72 v |
| H+ + e ⇄ 1/2 H2 0 v |
| Zn2+ + 2e ⇄ Zn −0.76 v |
It means that during the zinc deposition/stripping process, the CeO2 modified layer is stable and does not allow other side reactions. Furthermore, the semiconductor CeO2 layer can prevent the dissolution of alkaline zinc sulfate by-products into the electrolyte, slowing down the rate of corrosion on the zinc surface, which furthermore prohibits the HER.4,16,24,32 This positive role of CeO2 is shown in Fig. 1.
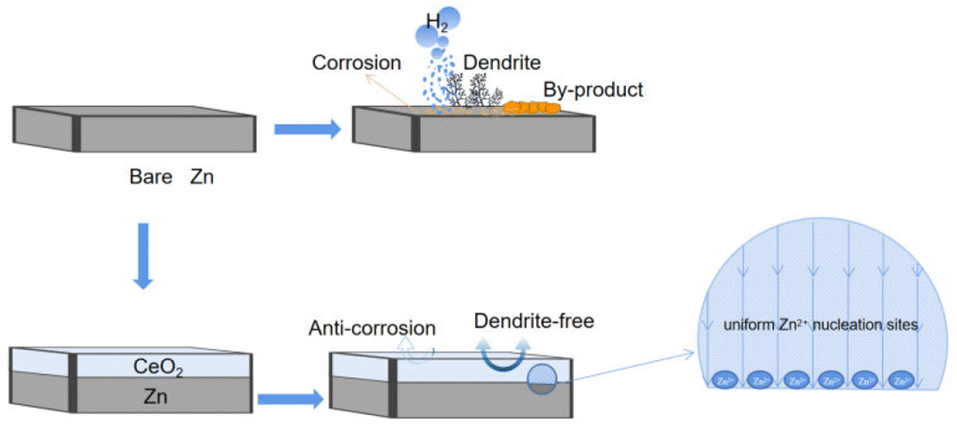 | ||
| Fig. 1 Schematic diagram of the protection mechanism of the CeO2 layer for CeO2@Zn in Zn-ion batteries. | ||
2. Results and discussion
To fabricate the coating layer, CeO2 powder was dispersed in 4% PVDF and then drop cast onto the surface of Zn foil, NMP was removed by drying in the oven as described in the ESI.† The X-ray diffraction pattern in Fig. S1† shows the successful cast of CeO2 without any changes in the crystal structure. Meanwhile, a small amount of PVDF here was used to guarantee the elasticity and flexibility of the coating layer as reported by our group members before.33The hydrophilicity of the interface was confirmed from the contact angle measurements in 2 M ZnSO4 electrolyte. As shown in Fig. 2f and g, the significantly improved hydrophilicity of the CeO2@Zn foil anode was proved by the smaller contact angle of 60.69° compared to that of Zn foil (92.25°), that makes a great contribution to the easier desolvation process for Zn2+(H2O)6 at the interface of the CeO2 layer.
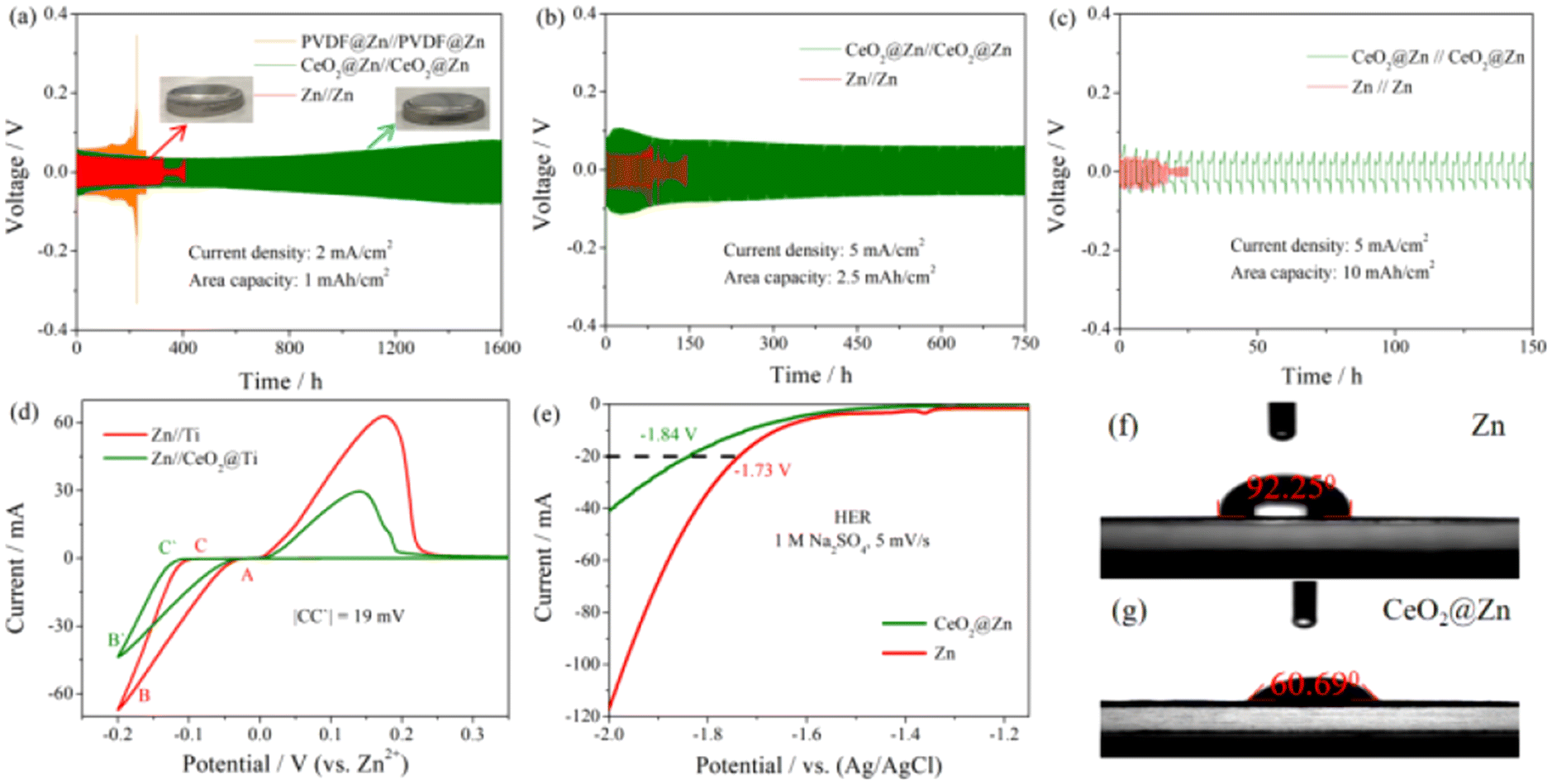 | ||
| Fig. 2 Voltage profiles of symmetric cells based on Zn foil and the CeO2@Zn anode at (a) 2 mA cm−2 with a capacity of 1 mA h cm−2, (b) 5 mA cm−2 with a capacity of 2.5 mA h cm−2 and (c) 5 mA cm−2 with a capacity of 10 mA h cm−2, (d) CV curves of Zn//Ti and CeO2@Zn//Ti (0.2 mA cm−2 for 15 h), (e) LSV curves of bare Zn foil and CeO2@Zn foil under a neutral electrolyte, (f) the contact angle measurement results for (g) bare Zn and (g) the fabricated Zn anodes. | ||
To examine the inhibitory effect on hydrogen evolution of this coating layer, CeO2 modified on Zn foil was examined in a three-electrode system with 1 M Na2SO4 aqueous electrolyte, in which CeO2@Zn foil or Zn foil was used as the working electrode, Ag/AgCl as the reference electrode, and carbon rod as the counter electrode (for more details, see the ESI†). As depicted in Fig. 2e, the LSV curves show that the overpotential for CeO2@Zn foil to reach a current density of 20 mA cm−2 was ∼110 mV, much higher than that of pure Zn foil, which provides direct evidence of the inhibition of the HER by the CeO2 coating layer.
Long-term cycling stability of the symmetric batteries under different current densities and area capacities were tested as shown in Fig. 2a, b and Fig. S9–S12.† The symmetric CeO2@Zn cells exhibited a prolonged lifespan over 1600 h (2 mA cm−2, 1 mA h cm−2), 750 h (5 mA cm−2, 2.5 mA h cm−2), 150 h (5 mA cm−2, 10 mA h cm−2) and 75 h (20 mA cm−2, 20 mA h cm−2), respectively, compared to that of the bare Zn anode at about ∼360 h, ∼80 h, ∼20 h and ∼40 h. According to the above experimental results, this might be ascribed to the stable hydrophilicity and the formation of an HER inert solid electrolyte interface layer; all the symmetric CeO2@Zn cells lasted longer than the symmetric bare Zn cells. The corresponding CE curves are also presented in Fig. S11–S13.†
The nucleation stage is a pivotal step for understanding the transformation course of Zn dendrite formation. The positive role played by the CeO2 coating layer on this aspect was proven by the overpotential evolution of symmetric Zn//Zn and CeO2@Zn//CeO2@Zn cells at a current density of 2.0 mA cm−2 with a total capacity of 1.0 mA h cm−2 at the initial 3 cycles.11 As can be seen from Fig. S14,† each Zn2+ deposition process has a typical nucleation overpotential in the Zn//Zn cell. However, there is no obvious nucleation overpotential for the CeO2@Zn//CeO2@Zn cell except the initial nucleation overpotential, which shows that the CeO2 coating layer reduces the energy barrier of Zn2+ nucleation and changes the deposition process of Zn2+ compared with that of bare Zn. The positive role in inhibiting the HER played by the CeO2 coating layer can also be proven by the phenomenon that the symmetric Zn//Zn batteries split much quickly than CeO2@Zn//CeO2@Zn cells because of the gas inside during long-term cycling.
Zn deposition on a foreign substrate such as bare Ti foil was investigated as shown in Fig. 2d. A crossover characteristic of nucleation processes was observed while sweeping the potential toward the positive direction and labelled as “A” in Fig. 2d, and the potential value of which is also known as the crossover potential (Eoc). The potential difference between the crossover point (A) and the point (B/B′) where Zn2+ starts to be reduced on the substrate is regarded as the polarization voltage difference. No excess decomposition current on the CeO2 interphase was observed, indicating that chemically stable films did not bring about other side reactions during cycling.11,34
To better understand the behavior of the CeO2 coating layer during the Zn electrodeposition process, an optical microscope was used to monitor in situ the surface morphology of the dendritic growth for Zn electrodes (CeO2@Zn and Zn). Homemade optical cells were fabricated with a transparent glass plate and 10 mA cm−2 was applied to the cells as shown in Fig. 3 and the ESI.† It can be seen from Fig. 3a that Zn dendrites were distributed randomly on the bare Zn surface after 10 min, and formed tree-like Zn dendrites after 30 min deposition. These Zn dendrites cannot be reused and might puncture the diaphragm. The formation of these fragile dendrites (dead Zn) resulted in short circuits during the cycling stability test for Zn//Zn symmetry cells (Fig. 2a–c and Fig. S5, S9, and S10†). In contrast, it could be observed through the in situ deposition video (Fig. 3b and ESI video 1 and 2†) that CeO2 uniformly covered the surface of Zn foil at the beginning; Zn2+ tended to electrodeposit above the surface of Zn but under the CeO2 layer when the voltage was applied, which prevented Zn4SO4(OH)6·nH2O from dissolving into the electrolyte. This could also be proved from the XRD patterns of bare Zn and CeO2@Zn after 250 cycling tests as seen from Fig. 5. The CeO2 layer pushed slowly and evenly with no observed dendrites on the surface of CeO2@Zn even after 30 min. Its uniform deposition could be attributed to the homogenization of the nucleation site by the CeO2 layer alongside the inhibition of the HER as discussed earlier in Fig. 1.
 | ||
| Fig. 3 Optical microscope images of (a) bare Zn and (b) CeO2@Zn in the aqueous ZnSO4 electrolyte to characterize the dendrite growth during plating at a current density of 10 mA cm−2. | ||
The feasibility of CeO2@Zn anodes in the practical application was further verified by full cells using MnO2 as the cathode. MnO2 was synthesized by a hydrothermal method according to a previous reports,35 with its purity and phase state characterized by XRD as shown in Fig. S1.† The featured peaks of the sample corresponded to the standard α-MnO2 (JCPDS: 44-0141) spectrogram, which indicated that the synthesized MnO2 was in the α crystal phase. It indicated a nanorod morphology for the α-MnO2 used in this experiment (Fig. S2†). For the full cell test, 0.1 M MnSO4 was added to 2 M ZnSO4 electrolyte to suppress the disproportion reaction of MnO2.35 As seen in Fig. 4a, both Zn//MnO2 and CeO2@Zn//MnO2 showed the same two pairs of anodic/cathodic peaks in the CV curves derived from the two-step reverse oxidation/reduction between MOOH and MnO2, in agreement with those reported in other literature studies.4
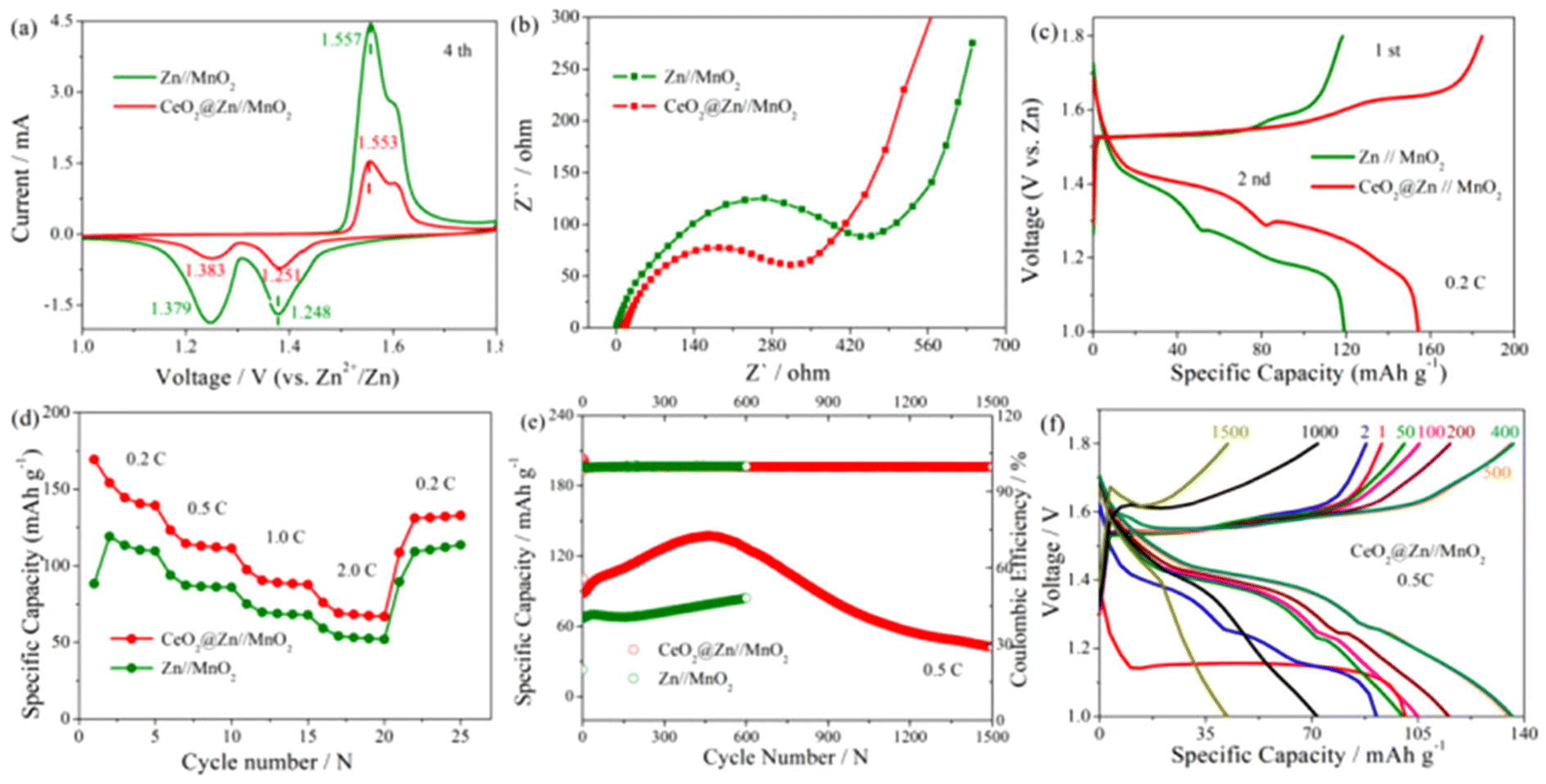 | ||
| Fig. 4 Performance of Zn/MnO2 and CeO2@Zn//MnO2 batteries: (a) CV curves at a scanning rate of 0.1 mV s−1, (b) Nyquist plots, (c) DC curves at 0.2 C derived from (d) rate performance at various current densities, (e) cycling performance at 0.5 C and (f) DC curves derived from (e). | ||
The similar CV curves of CeO2@Zn//MnO2 compared to that of the Zn//MnO2 full cell revealed similar redox behavior and indicated that the CeO2 coating layer did not affect the redox reactions in MnO2. Furthermore, as shown in Fig. 4b the charge transfer resistance Rct value of CeO2@Zn//MnO2 was much lower than that of Zn//MnO2, which indicates a more profound Zn2+ ion transfer at the electrolyte/anode interface of CeO2@Zn//MnO2, in consistence with the results of the previous contact angle test (Fig. 2f and g).
Of interest, while Zn//MnO2 (∼165 mA h g−1) and CeO2@Zn//MnO2 (∼169 mA h g−1) have almost the same specific capacity for the initial discharge profile at 0.2 C (Fig. 4c and Fig. S6†), however, there is an obvious faster fading of the capacity for the uncoated Zn//MnO2 (discharge capacity ∼120 mA h g−1) at the second discharge step compared to CeO2@Zn//MnO2 (discharge capacity ∼159 mA h g−1). The possible mechanism was controversial and has been deeply discussed by researchers in recent years.4
H+ plays a vital role for the charge storage mechanism in our rechargeable Zn//MnO2 system, and our experimental results can be explained according to the H+ conversion reaction mechanism reported recently.4 For the first discharge step, MnO2 reacts with a proton from water to form MnOOH (MnO2 + H+ + e− + MnOOH) in both cells, since the electrolyte was the same for both Zn//MnO2 and CeO2@Zn//MnO2 at the beginning. Counter to the H+ ions reacting with MnO2, the resulting OH− ions reacted with ZnSO4 and H2O in the aqueous electrolyte to form large flake-like ZnSO4[Zn(OH)2]3·xH2O and reached a neutral charge in the system. As shown in Fig. 2e, the coating layer of the anticatalytic HER CeO2, thereby alleviating H+ to form H2 and promoting the formation of MnOOH. This supports the longer cycling stability and shorter decreasing of specific capacity for CeO2@Zn//MnO2 compared to that of Zn//MnO2 observed in Fig. 4f.
The rate capability of full cells was evaluated at current densities from 0.2 to 2.0 C (Fig. 4d and Fig. S5†). It was observed that the modified CeO2@Zn//MnO2 showed better capacity than that of bare Zn//MnO2 at various current densities, both of which exhibited excellent reversibility (returning to 2.0 from 0.2 C). This illustrates the high feasibility of this energy storage system. The long-term cycling performance of full cells (CeO2@Zn//MnO2 and Zn//MnO2) was tested at 0.5 C between 1.0 and 1.8 V (Fig. 4e). For CeO2@Zn//MnO2, the capacity gradually increased with every activation before 500 cycles, but then gradually decreased since ∼500 laps and led to 30% capacity retention rate after 1500 cycles. However, the capacity of Zn//MnO2 steadily lasted around 600 cycles and then cut off.
To further validate our analysis on the performances above, we disassembled the tested batteries and analyzed their structures and morphologies. Scanning electron microscopy (SEM) images revealed the surface morphology of the bare Zn and CeO2@Zn anodes in the symmetric cells before and after cycling (Fig. 5a–d). Initially, the bare Zn anode exhibit a smooth and shiny metallic sheen surface (Fig. 5a), CeO2@Zn was also smooth but with a thin layer of the CeO2 solid covering the surface (Fig. 5c). After 250 cycles, a layer of coarse nanoflakes covered the surface of the bare Zn layer (Fig. 5b and Fig. S4†) which was randomly distributed. However, the SEM images show that CeO2@Zn still maintained a relatively smooth and strong solid surface with a homogeneous Zn deposition, with no tree-like nanoflakes being observed (Fig. 5d). XRD patterns have proven that the sediment on CeO2@Zn was Zn4SO4(OH)6·4H2O. In contrast, much less Zn4SO4(OH)6·xH2O (x = 4,5) was observed for bare Zn since it was dissolved in the electrolyte and this has been verified previously in the literature.36 This intuitively proves the positive role of the CeO2 layer in preventing zinc surface corrosion and formation of dendrites.
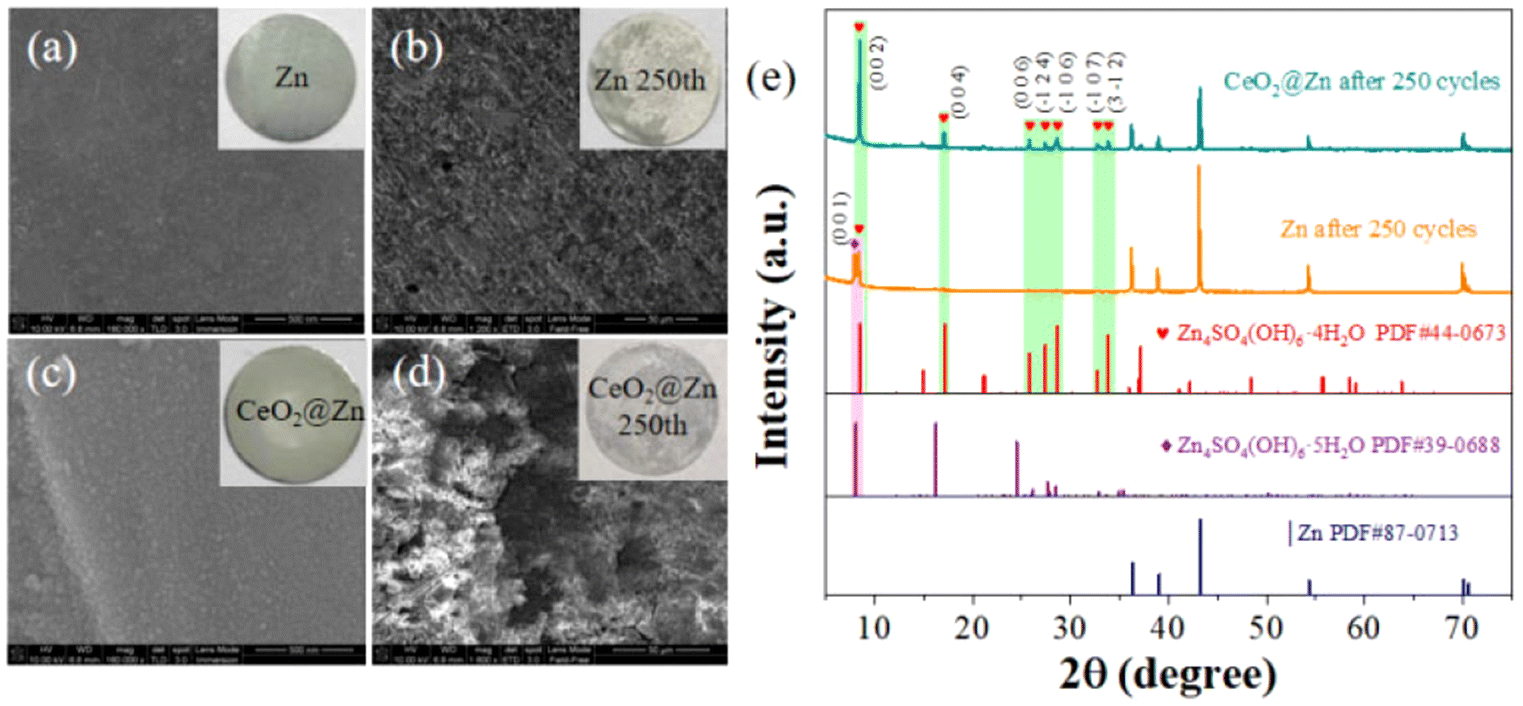 | ||
| Fig. 5 SEM photos of : (a) Zn and (b) Zn after 250th cycles, (c) CeO2@Zn and (d) CeO2@Zn electrode after 250th cycles, respectively; (e) the XRD patterns of the corresponding electrodes in (a). | ||
3. Conclusions
In summary, an efficient hydrophilic interface strategy that can bring a balance between the rapid desolvation kinetics of Zn2+(H2O)6 and inhibition of the HER by CeO2 layer coating was developed. It greatly improved the easy access of electrolyte ions to the anode surface, provided uniform Zn2+ nucleation sites, inhibited the HER and Zn corrosion desolvation, leading to a significant enhancement of the efficiency of aqueous Zn-ion batteries (Table S1 in the ESI†). The long-term stability of symmetrical CeO2@Zn//CeO2@Zn batteries was observed as they lasted for ∼1600 h without obvious dendrite formation, which is almost 4.4 times than that of bare Zn//Zn. The modified CeO2@Zn coupled with α-MnO2 cathode presented excellent reversibility and greatly improved long cycling stability with almost no decay after 600 cycles compared with bare Zn. Our work provides a fundamental understanding and an essential solution to deal with the balance between rapid desolvation and inhibition of the hydrogen evolution reaction, which is important for promoting the practical application of rechargeable Zn batteries.4. Experimental and methods
4.1. Chemicals
Deionized water (18.2 MΩ) was used for all experiments. Zn foil (20 μm, 80 μm, 99.9%) was bought from Guangsheng New Materials Co. Ltd. ZnSO4·7H2SO4 (99.0%), MnSO4 and Ti foil (10 μm, 99.99%) were bought from Aladdin Reagent Co. LTD. Hydrophobic carbon paper was bought from Shanghai Hesen Electric Co., Ltd. Glass fiber membrane (Whatman GF/D, glass microfiber filters, diameter 47 mm) was brought from Shanghai Yuling Filtering Equipment Ltd. N-Methyl-2-pyrrolidone (NMP, >99.5%) and PVDF (Mw = 8900–9800) were bought from Shanghai Aladdin Bio-tech Co. Ltd.4.2. Characterization of the morphology and structure
XRD patterns were recorded using a Bruker-AXS micro-diffractometer (D8 ADVANCE) with Cu-Kα radiation (λ = 1.5406 Å) from 10° to 90°. The contact angle between the electrolyte and the Zn anode was measured using a contact angle measuring instrument (JC2000D1, Shanghai Powereach Digital Technology Equipment Co., Ltd.) An optical microscope (6XB-PC, Optical Instrument Factory) was used to observe in situ the deposition and exfoliation of Zn. A homemade optical cell was fabricated using Zn plates (100 mm × 100 mm × 50 mm) fixed on a transparent glass plate for the in situ optical analysis.4.3. The synthesis of electrode materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVDF (9:1) and then 100 μl of the mixture was drop cast onto Zn or Ti foil, and the resultant electrodes were dried in an oven at 80 °C, overnight to remove the NMP completely.
:PVDF (9:1) and then 100 μl of the mixture was drop cast onto Zn or Ti foil, and the resultant electrodes were dried in an oven at 80 °C, overnight to remove the NMP completely.
4.4. The assembly of batteries and electrochemical measurements
All cell measurements (galvanostatic charge–discharge, linear polarization and chronoamperograms) were performed on a Neware Battery Tester (Shenzhen, China). Corrosion analysis and cyclic voltammetry of full cells were tested at a scan rate of 0.1 mV s−1 and the EIS spectra were recorded on an electrochemical workstation (VMP-300, Bio-Logic Science Instruments Co.) with a frequency range from 100 kHz to 0.01 Hz using a BioLogic electrochemistry workstation.HER measurements were conducted in a three electrode configuration, in which bare Zn and coated Zn plate were used as the working electrode, the Zn plate as the counter electrode, and saturated Ag/AgCl as the reference electrode. The corrosion potential and corrosion current were calculated from the Tafel fit system in the electrochemical workstation.
:2:1 in NMP solvent, then coating the slurry onto hydrophobic carbon paper using a razor blade. The α-MnO2 mass loading was about 1–3 mg cm−2. Zn/MnO2 (or CeO2@Zn//MnO2) batteries were performed as CR2032 type coin cells with the α-MnO2 electrodes as the cathode, glass fiber as the separator and Zn foil as the anode. The electrolyte was 2 M ZnSO4 with 0.1 M MnSO4 aqueous solution (200 μl) at 0.2 C with a voltage range of 1.0–1.8 V.
where x (μm) is the thickness of the Zn foil and y (mA h cm−2) represents the Zn areal capacity used in the electrochemical testing. Here the DoD was 1% as shown in Fig. 4e.
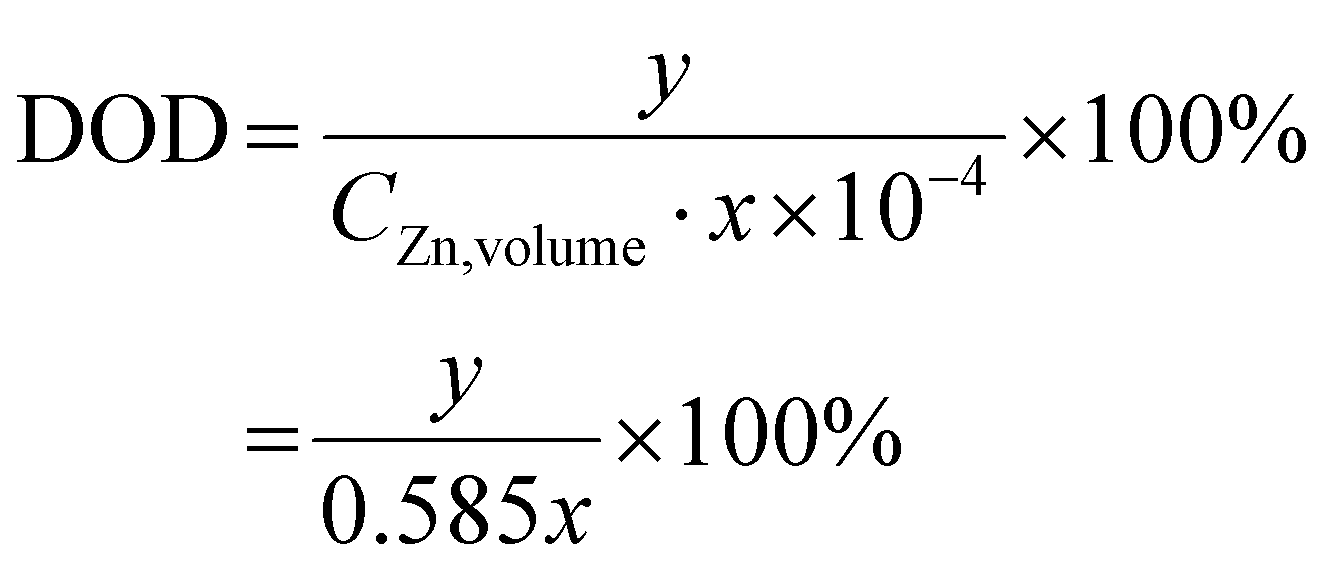
Author contributions
J. Li. and C. H. conceptualised the study. X. X. and H. J. obtained the funding. S. W. and Y. Y. provided resources. X. X., Z. Z., G. X. and D. W. conducted the experiments.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52272088) and the China Postdoctoral Science Foundation (220708). We further thank Tingting He for the help with the schematic part in Fig. 1.References
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- J. S. Kim, S.-W. Heo, S. Y. Lee, J. M. Lim, S. Choi, S.-W. Kim, V. J. Mane, C. Kim, H. Park, Y. T. Noh, S. Choi, T. van der Laan, K. Ostrikov, S.-J. Park, S. G. Doo and D. H. Seo, Nanoscale, 2023, 15, 17270–17312 RSC.
- Y. Du, R. Kang, B. Zhang, H. Wang, G. Chen and J. Zhang, ACS Energy Lett., 2024, 9, 967–975 CrossRef CAS.
- C. Xu, B. Li, H. Du and A. F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS.
- J. Yin, Y. Wang, Y. Zhu, J. Jin, C. Chen, Y. Yuan, Z. Bayhan, N. Salah, N. A. Alhebshi, W. Zhang, U. Schwingenschlögl and H. N. Alshareef, Nano Energy, 2022, 99, 107331 CrossRef CAS.
- L. Wang, B. Zhang, W. Zhou, Z. Zhao, X. Liu, R. Zhao, Z. Sun, H. Li, X. Wang, T. Zhang, H. Jin, W. Li, A. Elzatahry, Y. Hassan, H. J. Fan, D. Zhao and D. Chao, J. Am. Chem. Soc., 2024, 146, 6199–6208 CrossRef CAS PubMed.
- F. Bu, Z. Sun, W. Zhou, Y. Zhang, Y. Chen, B. Ma, X. Liu, P. Liang, C. Zhong, R. Zhao, H. Li, L. Wang, T. Zhang, B. Wang, Z. Zhao, J. Zhang, W. Li, Y. S. Ibrahim, Y. Hassan, A. Elzatahry, D. Chao and D. Zhao, J. Am. Chem. Soc., 2023, 145, 24284–24293 CrossRef CAS PubMed.
- Y. Xu, X. Zheng, J. Sun, W. Wang, M. Wang, Y. Yuan, M. Chuai, N. Chen, H. Hu and W. Chen, Nano Lett., 2022, 22, 3298–3306 CrossRef CAS PubMed.
- K. Liang, S. Huang, H. Zhao, W. Liu, X. Huang, W. Chen, Y. Ren and J. Ma, Adv. Mater. Interfaces, 2022, 9, 2200564 CrossRef CAS.
- R. Zhao, X. Dong, P. Liang, H. Li, T. Zhang, W. Zhou, B. Wang, Z. Yang, X. Wang, L. Wang, Z. Sun, F. Bu, Z. Zhao, W. Li, D. Zhao and D. Chao, Adv. Mater., 2023, 35, 2209288 CrossRef CAS.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- H. Li, R. Zhao, W. Zhou, L. Wang, W. Li, D. Zhao and D. Chao, JACS Au, 2023, 3, 2107–2116 CrossRef CAS PubMed.
- C. Xie, Y. Li, Q. Wang, D. Sun, Y. Tang and H. Wang, Carbon Energy, 2020, 2, 540–560 CrossRef CAS.
- S. So, Y. N. Ahn, J. Ko, I. T. Kim and J. Hur, Energy Storage Mater., 2022, 52, 40–51 CrossRef.
- P. Li, J. Ren, C. Li, J. Li, K. Zhang, T. Wu, B. Li and L. Wang, Chem. Eng. J., 2023, 451, 138769 CrossRef CAS.
- F. Li, D. Ma, K. Ouyang, M. Yang, J. Qiu, J. Feng, Y. Wang, H. Mi, S. Sun, L. Sun, C. He and P. Zhang, Adv. Energy Mater., 2023, 13, 2204365 CrossRef CAS.
- Q. Ren, X. Tang, K. He, C. Zhang, W. Wang, Y. Guo, Z. Zhu, X. Xiao, S. Wang, J. Lu and Y. Yuan, Adv. Funct. Mater., 2024, 34, 2312220 CrossRef CAS.
- X. Xiao, C. Hu, Q. Dai, C. Xiong, D. Liu and H. Jin, J. Electroanal. Chem., 2022, 925, 116862 CrossRef CAS.
- X. Xiao, C.-T. He, S. Zhao, J. Li, W. Lin, Z. Yuan, Q. Zhang, S. Wang, L. Dai and D. Yu, Energy Environ. Sci., 2017, 10, 893–899 RSC.
- X. Xiao, Z. Fang and D. Yu, Front. Mater., 2019, 6, 219 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. T. Xu, X. Xiao, Z. M. Ye, S. Zhao, R. Shen, C. T. He, J. P. Zhang, Y. Li and X. M. Chen, J. Am. Chem. Soc., 2017, 139, 5285–5288 CrossRef CAS PubMed.
- H. Liu, J. G. Wang, W. Hua, H. Sun, Y. Huyan, S. Tian, Z. Hou, J. Yang, C. Wei and F. Kang, Adv. Sci., 2021, 8, 2102612 CrossRef CAS PubMed.
- T. Shoji, M. Hishinuma and T. Yamamoto, J. Appl. Electrochem., 1988, 18, 521–526 CrossRef CAS.
- P. K. Bowen, J. Drelich and J. Goldman, Adv. Mater., 2013, 25, 2577–2582 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- A. Vojvodic and J. K. Nørskov, Natl. Sci. Rev., 2015, 2, 140–143 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- X. Huang, K. Zhang, B. Peng, G. Wang, M. Muhler and F. Wang, ACS Catal., 2021, 11, 9618–9678 CrossRef CAS.
- K. Wang, M. Qin, C. Wang, T. Yan, Y. Zhen, X. Sun, J. Wang and F. Fu, J. Colloid Interface Sci., 2023, 629, 733–743 CrossRef CAS PubMed.
- R. Zhao, Y. Yang, G. Liu, R. Zhu, J. Huang, Z. Chen, Z. Gao, X. Chen and L. Qie, Adv. Funct. Mater., 2020, 31, 2100445 Search PubMed.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- R. Zhao, Y. Yang, G. Liu, R. Zhu, J. Huang, Z. Chen, Z. Gao, X. Chen and L. Qie, Adv. Funct. Mater., 2020, 31, 2001867 CrossRef.
- X. Zhang, L. Zhang, X. Jia, W. Song and Y. Liu, Nano-Micro Lett., 2024, 16(1), 75 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02385d |
| This journal is © The Royal Society of Chemistry 2024 |