
Facile synthesis of defective ZnS–ZnO composite nanosheets for efficient piezocatalytic H2 production†
Xiaoxiao
Lu
,
Xiaojing
Zhao
,
Xiangyu
Chen
,
Miaoqiong
Xu
,
Miaoling
Huang
,
Wen-Jie
Chen
,
Yubin
Liu
 * and
Xiaoyang
Pan
*
* and
Xiaoyang
Pan
*
College of Chemical Engineering and Materials, Quanzhou Normal University, Quanzhou 362000, China. E-mail: liu_yubin1989@163.com; xypan@qztc.edu.cn
First published on 7th November 2024
Abstract
A facile approach was developed for the synthesis of ultrathin ZnS–ZnO nanosheets. By simply manipulating the synthesis temperature, ZnS–ZnO composite nanosheets with sulfur vacancies were successfully obtained using ZnS(en)0.5 as the precursor. The formation of the ZnS–ZnO composite leads to the creation of a heterojunction at the interface between the two materials, which enhances the separation of piezogenerated electrons and holes. Additionally, sulfur vacancies are concurrently introduced into the ZnS lattice during the heat treatment process. This defective ZnS with sulfur vacancies exhibits a narrowed bandgap and low excitation energy. Consequently, the defective ZnS–ZnO composite nanosheets demonstrate much higher piezocatalytic activity compared to ZnS and ZnO catalysts, surpassing the performance of most reported piezocatalysts. Furthermore, the ZnS–ZnO composite nanosheets maintain stability over five cycles of catalytic reactions. The study offers a promising approach for enhancing piezocatalytic performance for H2 production.
Introduction
The quest for sustainable and renewable energy sources has been at the forefront of scientific research in recent decades. Hydrogen (H2) production through water splitting has garnered significant attention due to its potential as a clean and abundant energy carrier.1 Piezocatalysis, a process that harnesses mechanical force to drive chemical reactions, has emerged as a promising alternative to conventional photocatalysis and electrocatalysis for H2 production.2–4 This approach offers the advantage of direct conversion of mechanical energy into chemical energy, bypassing the need for external electrical or optical inputs.Recently, a variety of semiconducting materials have been used for piezocatalytic H2 production.5–14 However, their performance is greatly limited by the small surface area, poor charge separation efficiency, and insufficient active sites. To address these challenges, the development of nanosheet-based piezocatalysts has attracted significant attention.15–23 Nanosheets, with their ultrathin structure and high surface-to-volume ratio, offer unique advantages in terms of enhanced charge transport, increased active site availability, and improved catalytic activity.1
ZnS, as an Earth-abundant non-centrosymmetric semiconductor, has been widely investigated in the field of piezocatalysis.24–26 However, due to the non-layered structure of ZnS, the synthesis of ZnS 2D nanosheets is intrinsically difficult.27,28 Recently, Feng et al. revealed an effective method for the synthesis of atomically thin ZnS nanosheets.24 The resulting ZnS catalyst shows efficient piezocatalytic activity for H2 production due to the large mechanical capture area, high piezoelectric coefficient and easy deformation characteristics. However, the ZnS catalyst deactivated after several cycles of reactions. Generally, the performance of ZnS is limited by two main factors. One is that ZnS is a wide-bandgap semiconductor. Therefore, the excitation of charge carriers requires large energy input. The other is that the piezo-generated charge carriers are easily recombined during the catalytic process. Therefore, it is highly desirable to develop efficient ZnS piezocatalysts via bandgap engineering and regulate the behavior of piezo-generated carriers.
Herein, we provide a facile method for the synthesis of ultrathin ZnS nanosheets. By simply adjusting the synthesis temperature, ZnS–ZnO composite nanosheets are obtained. The formation of the ZnS–ZnO composite allows for the creation of a heterojunction at the interface between the two materials. This heterojunction facilitates the separation of piezogenerated electrons and holes. In addition, sulfur vacancies are simultaneously formed in the lattice of ZnS during heat treatment. The defective ZnS with sulfur vacancies possesses a narrowed bandgap with low excitation energy.29 As a result, the defective ZnS–ZnO composite nanosheets show much higher piezocatalytic activity than ZnS and ZnO catalysts, which is also higher than those of most of the reported piezocatalysts. Moreover, the ZnS–ZnO composite nanosheets are also stable during five cycles of catalytic reactions.
Experimental
Materials
Analytical grade zinc acetate (Zn(CH3COO)2), thiourea (SC(NH2)2), ethanol (C2H5OH), methanol (CH3OH), lactic acid (C3H6O3), triethanolamine (C6H15NO3) and ethylenediamine (C2H8N2) were purchased from Sinopharm Chemical Reagent Co., Ltd. Deionized water was supplied using a local device.Synthesis
1 mmol of zinc acetate and 3 mmol of thiourea were dissolved in 30 mL of ethylenediamine, and magnetically stirred at room temperature for 30 minutes. The mixed solution was then transferred to a 50 mL stainless steel autoclave lined with polytetrafluoroethylene. The solution was hydrothermally treated at 373 K for 500 minutes. The product was washed with deionized water and ethanol three times. The product was dried at 333 K. The as-obtained sample was denoted as ZnS(en)0.5.The precursor ZnS(en)0.5 was calcined in a muffle furnace at temperatures of 673, 773, and 873 K respectively for 2 hours. The obtained samples were labeled as ZnS, ZnS–ZnO and ZnO, respectively.
Characterization
The crystallographic configuration of the freshly prepared samples was probed utilizing a Rigaku Miniflex II X-ray diffractometer, specifically harnessing Cu Kα radiation for analysis. To unravel their morphological features and microscopic architecture, a combination of tools was employed: a field-emission scanning electron microscope (JSM-6700F FESEM) and a transmission electron microscope (JEM-2010, alongside the advanced Tecnai G2 F20 FEG TEM from FEI). Additionally, X-ray photoelectron spectroscopy (XPS) examinations were conducted on a Thermo Scientific ESCA Lab250 spectrometer, equipped with a monochromatic Al Kα X-ray source, a hemispherical analyzer, and a versatile sample stage for multi-axial alignment, allowing for precise compositional analysis of sample surfaces. Calibration of all binding energies was achieved by referencing the C 1s peak of adventitious carbon present on the surface, set at 284.6 eV. The optical characteristics of the samples were then assessed through a Cary 100 UV-visible diffuse reflectance spectrophotometer (DRS), where BaSO4 served as the benchmark for internal reflectance correction.Activity test
To assess the hydrogen production capability, 5 mg of catalyst was meticulously weighed and dispersed within a quartz reactor that contained 10 ml of deionized water. The reactor was then hermetically sealed to ensure airtightness and argon gas was continuously purged through the system under agitation to eliminate any traces of residual air. A sonication process was initiated using an ultrasonic machine (model: KQ-200FHG.DZ from Kunshan). Subsequently, a microinjector precisely extracted 1 μL of gas from the reactor, which was subsequently introduced into a gas chromatograph (GC9790Plus from Fuli) for comprehensive compositional analysis.Results and discussion
Fig. 1a shows the synthetic procedure for the ZnS–ZnO nanocomposite. Briefly, ZnS(en)0.5 was first synthesized by hydrothermal treatment of zinc acetate and thiourea in ethylenediamine solvent. Subsequently, ZnS(en)0.5 was calcined at different temperatures to obtain ZnS, ZnS–ZnO and ZnO samples. Fig. S1† shows the X-ray diffraction (XRD) pattern of ZnS(en)0.5, which is in good agreement with the previously reported data.30 ZnS(en)0.5 shows a sheet-like morphology with ultrathin thickness (Fig. S2†). The SEM images in Fig. 1b–d show the morphological changes of the samples obtained by calcining ZnS(en)0.5 at different temperatures. As shown in Fig. 1b and c, the ZnS and ZnS–ZnO samples obtained through calcination of ZnS(en)0.5 at 673 or 773 K exhibit a flake-like structure similar to that of ZnS(en)0.5, indicating the preservation of the characteristic morphology during the thermal treatment process. When ZnS(en)0.5 is calcined at 873 K, the obtained ZnO presents uniform nanoparticles (Fig. 1d).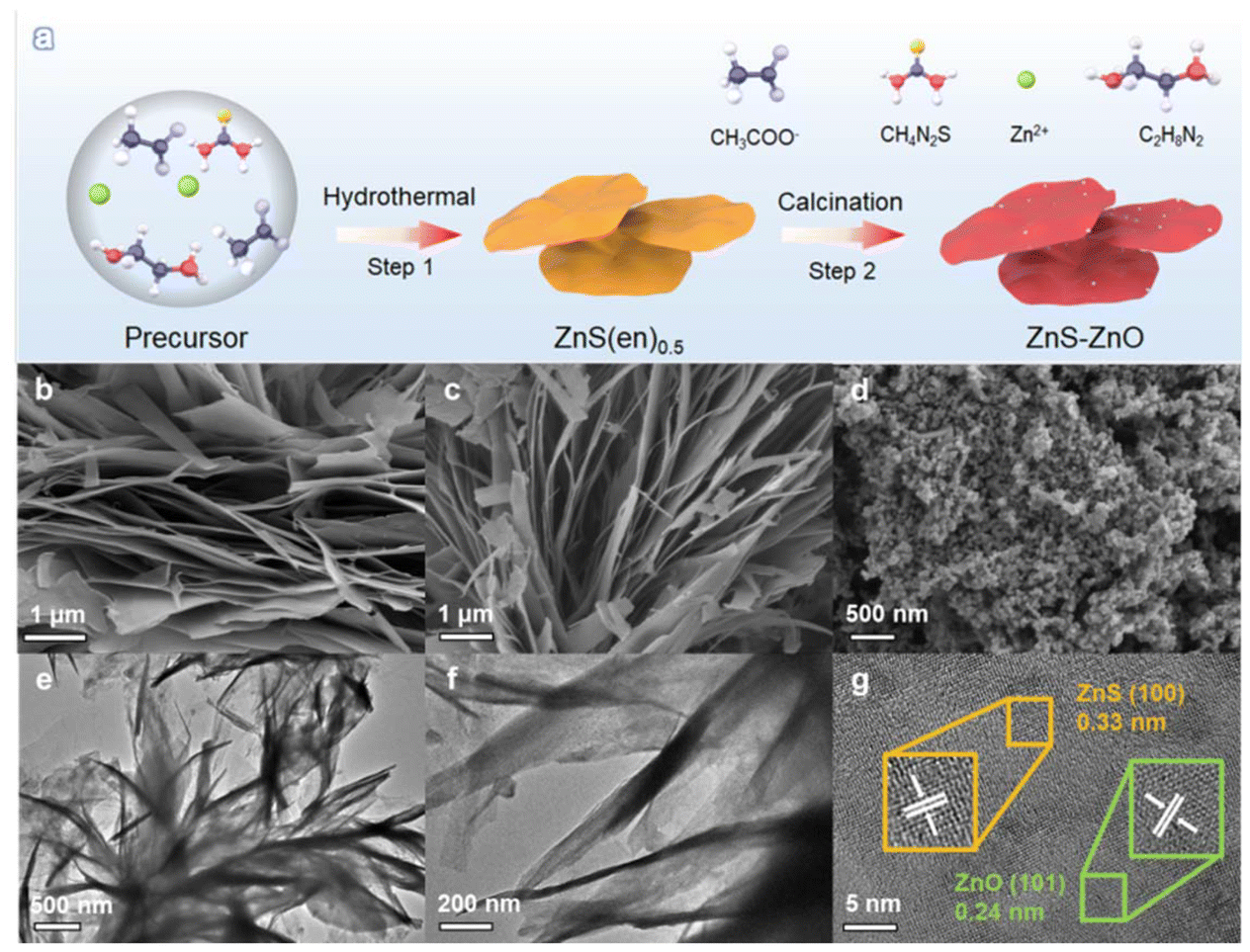 | ||
| Fig. 1 (a) Synthetic procedure of the ZnS–ZnO nanocomposite; SEM images of (b) ZnS, (c) ZnS–ZnO, and (d) ZnO; (e and f) TEM images and (g) HRTEM image of ZnS–ZnO. | ||
A transmission electron microscope (TEM) was used to further investigate the morphology and structure of the ZnS–ZnO sample. TEM images (Fig. 1e and f) show a sheet-like structure. The HRTEM image (Fig. 1g) shows different lattice spacings of 0.33 nm and 0.24 nm, which correspond to the (100) plane of ZnS (JCPDS no. 80-0007) and the (101) plane of ZnO (JCPDS no. 80-0074), respectively. This result indicates the successful formation of a ZnS–ZnO heterojunction. The elemental mapping analysis reveals that the Zn, S, O and N elements are homogeneously distributed over the sample (Fig. S3†). This result indicates that the N element of ethylenediamine in ZnS(en)0.5 was doped into the sample during the heat treatment.
X-ray diffraction (XRD) was employed to investigate the crystalline structure of the samples. Fig. 2a shows the XRD patterns of ZnS, ZnS–ZnO and ZnO. The XRD peaks of ZnS correspond precisely to the hexagonal wurtzite crystal structure (JCPDS no. 80-0007). The diffraction peaks observed at 27.2°, 28.8°, 30.9°, 40.1°, 48.1°, 52.4°, and 56.2° correspond to reflections from the crystal planes (100), (002), (101), (102), (110), (103), and (200), respectively. For ZnS–ZnO, both the XRD patterns of wurtzite ZnS and hexagonal ZnO (JCPDS no. 80-0074) are observed, indicating the partial transformation of ZnS into ZnO. Notably, after the ZnS(en)0.5 sample was calcined at 873 K, only peaks associated with ZnO were detected, suggesting that ZnS was completely oxidized to ZnO.
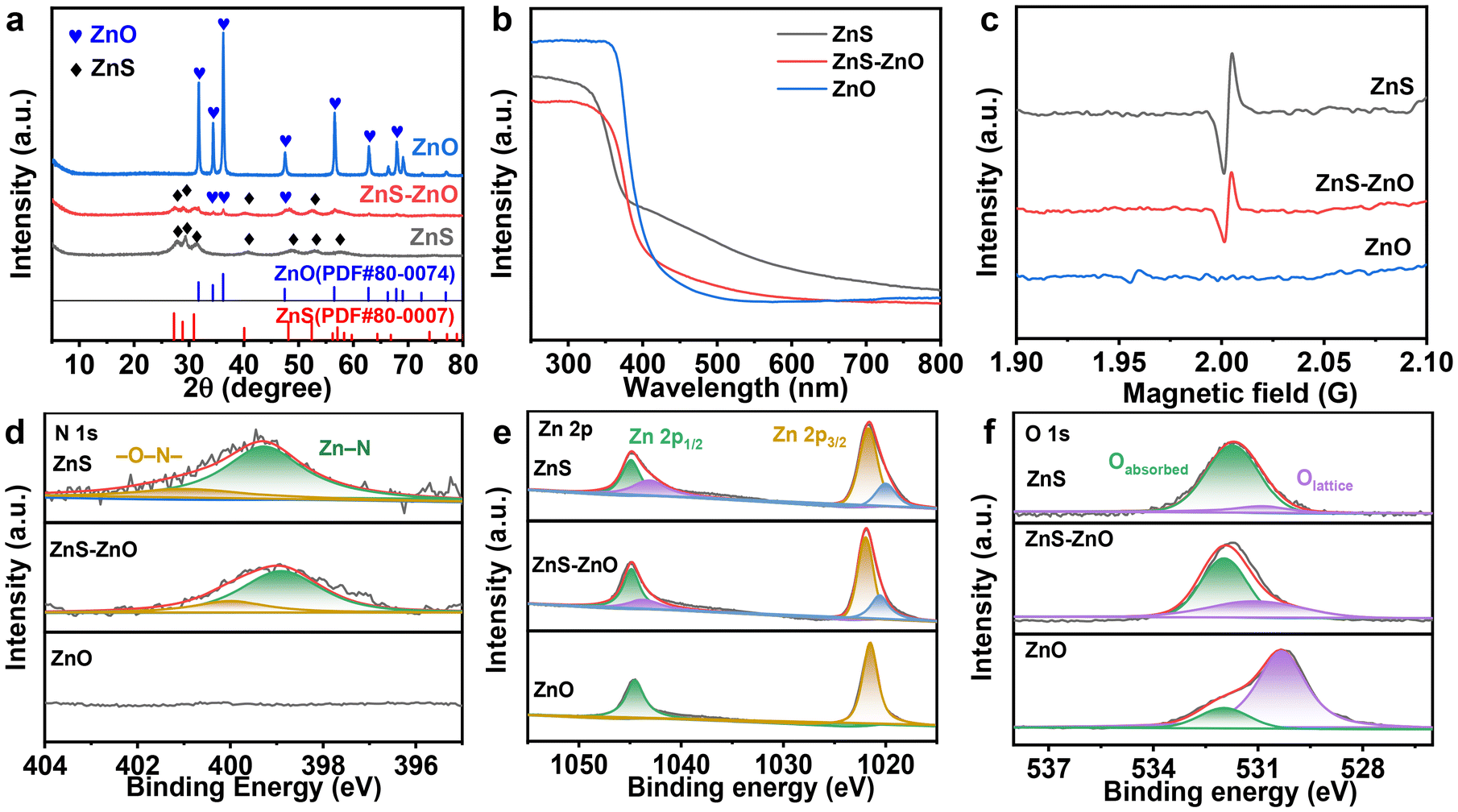 | ||
| Fig. 2 (a) The XRD patterns, (b) UV-vis DRS spectra and (c) EPR spectra of ZnS, ZnS–ZnO and ZnO; (d–f) XPS spectra of N 1s, Zn 2p and O 1s of ZnS–ZnO. | ||
Fig. 2b shows the ultraviolet-visible diffuse reflectance spectra (DRS) of the samples. ZnS and ZnS–ZnO show band-edge absorption around 400 nm and an obvious peak trailing in the visible region. However, the absorption in the visible region decreases with the increase of calcination temperature, presumably because of the decrease of S vacancy content.31 This conjecture was confirmed by electron paramagnetic resonance (EPR) analysis. As shown in Fig. 2c, the EPR spectrum of ZnS has a signal peak at g = 2.0029, which belongs to the S vacancy signal.29 However, this signal peak disappears in ZnO, indicating that there are no S vacancies in ZnO.
The presence of sulfur vacancies is attributed to the incorporation of the N element into the ZnS lattice during the calcination of ZnS(en)0.5, where ethylenediamine serves as the dopant. This observation is evidenced by XPS analysis. As depicted in Fig. 2d, the N 1s spectra of the samples indicate that both the ZnS and ZnS–ZnO samples contain the N element, while no N element is detected in ZnO. Further confirmation of N doping is evident in the Zn 2p spectra. As shown in Fig. 2e, the Zn 2p orbitals in ZnS and ZnS–ZnO can be divided into two sets of peaks. Zn 2p3/2 is deconvoluted into two peaks at different binding energies (corresponding to functional groups): 1020.1 eV (Zn–N) and 1021.6 eV (Zn–O/S).29
During heat treatment, the sample undergoes both nitrogen doping and a change in composition. Fig. 2f shows the O 1s spectra, where 531.7 eV and 529.6 eV correspond to adsorbed oxygen and lattice oxygen, respectively. Notably, ZnS also contains a small amount of lattice oxygen. This is attributed to the slight oxidation of ZnS during the heat treatment. When the calcination temperature is increased to 873 K, the lattice oxygen content of the sample obviously increases (Fig. 2f), accompanied by the total disappearance of the S element (Fig. S4†), indicating the complete transformation of the sample into zinc oxide.
The piezocatalytic hydrogen production performance of the samples was studied. Firstly, we investigated the piezocatalytic hydrogen production performance of ZnS–ZnO at different frequencies. As shown in Fig. 3a and b, the hydrogen production rates at ultrasonic frequencies of 40, 45, 80 and 100 kHz are 1.897, 4.645, 0.361 and 0.185 mmol g−1 h−1, respectively. These results indicate that the optimized frequency is 45 kHz.
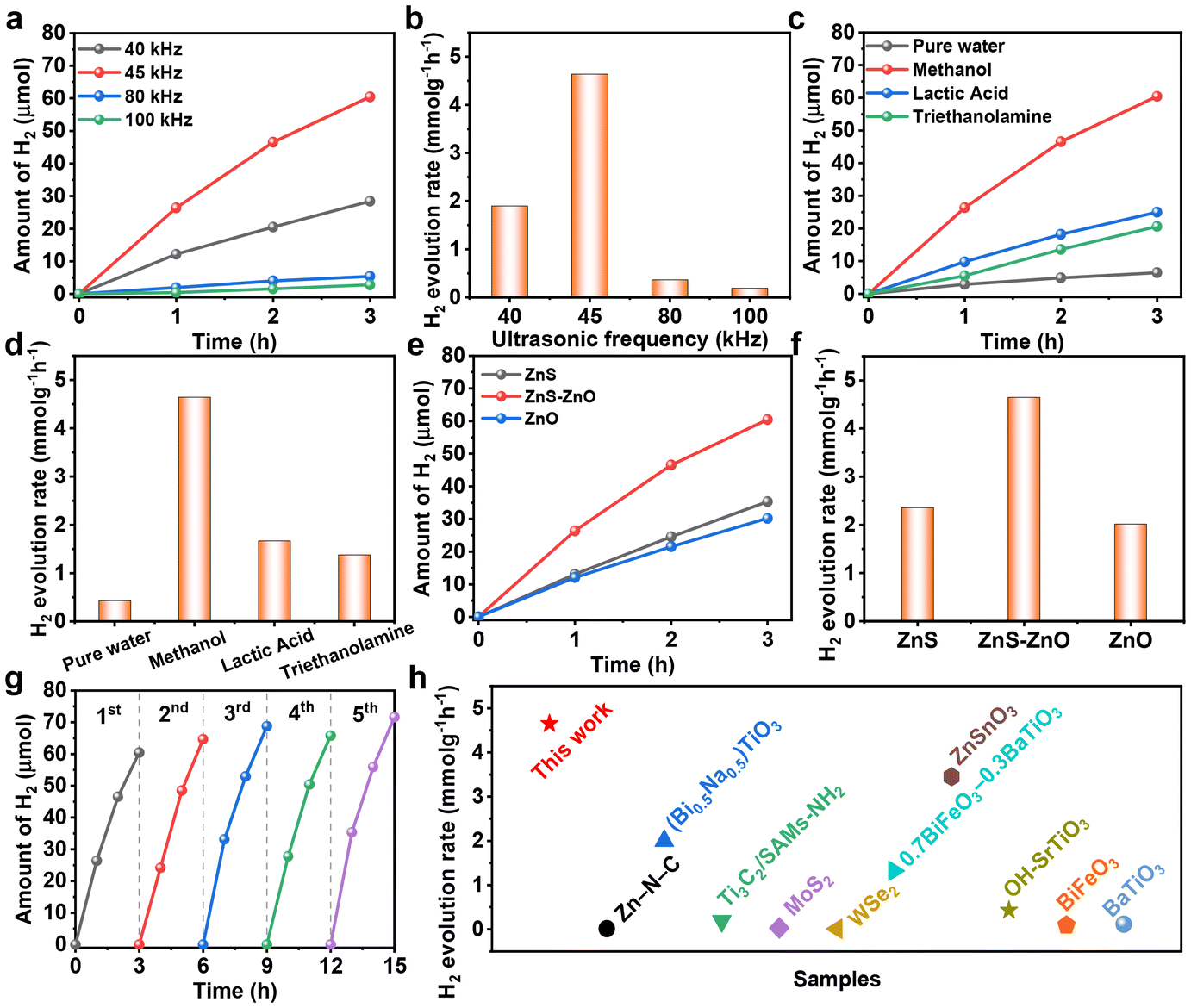 | ||
| Fig. 3 (a and b) Piezocatalytic H2 production over ZnS–ZnO under sonication (300 W) at different frequencies; (c and d) piezocatalytic H2 production over ZnS–ZnO under sonication (300 W, 45 kHz) using different sacrificial agents; (e and f) piezocatalytic H2 production over the samples under sonication (300 W, 45 kHz); (g) stability test of the piezocatalytic H2 production of ZnS–ZnO at 45 kHz and 300 W; (h) H2 evolution performance comparison of ZnS–ZnO with the reported piezocatalysts. | ||
The activity of ZnS–ZnO was also investigated in the presence of different sacrificial agents. As shown in Fig. 3c and d, when methanol, lactic acid and triethanolamine are used as sacrificial agents, the hydrogen production rates are 4.645, 1.666 and 1.376 mmol g−1 h−1, respectively. The results show that methanol is an effective sacrificial agent for hydrogen production. This is due to the fact that CH3OH has the lowest oxidation potential (1.05 eV) and the highest dielectric constant (31.2) among the three sacrificial reagents, so it is easily trapped and oxidized by holes.32 To investigate the contribution of methanol decomposition to H2 production, a blank experiment without the piezocatalyst was also performed. The result shows that CH3OH can undergo ultrasonic decomposition to produce hydrogen in the absence of the piezocatalyst (Fig. S5†). This is also one of the key reasons why methanol exhibits excellent hydrogen production performance when used as a sacrificial agent. Compared to the hydrogen production of ZnS–ZnO, the hydrogen evolution under catalyst-free conditions is obviously less (Fig. S5†). This clearly indicates that the hydrogen production process from methanol as a sacrificial agent is mainly composed of two parts: one is the hydrogen contributed by the piezocatalytic process, and the other is the hydrogen generated from the ultrasonic decomposition of methanol itself.
In addition, it is noteworthy that during the piezocatalytic process, nanocatalysts absorb ultrasonic energy to disperse aggregated nanoparticles, while solid particles also absorb and scatter ultrasonic waves.33 These two factors jointly contribute to the obvious attenuation of ultrasound in the reaction solution. To ensure consistency in experimental conditions, we replaced the piezocatalyst with non-piezoelectric titanium dioxide. Our experiments revealed that the hydrogen production of this catalyst was obviously lower than that of blank methanol (Fig. S5†). This result indicates that the hydrogen production from blank methanol does not fully equate to the hydrogen production from ultrasonic decomposition of methanol under piezocatalytic conditions.
The performance of ZnS–ZnO was compared with that of ZnS and ZnO. As shown in Fig. 3e and f, ZnS–ZnO exhibits the highest hydrogen production rate among the three samples, which may be attributed to the synergistic effect of the heterojunction structure and sulfur vacancy creation.34–36 The stability experiment of ZnS–ZnO demonstrates that the hydrogen production performance remains stable after five cycles, as shown in Fig. 3g. Furthermore, the ZnS–ZnO sample demonstrates a much higher piezocatalytic hydrogen production activity than most previously reported piezocatalysts (Fig. 3h and Table S1†).
Piezoresponse force microscopy (PFM) analysis was conducted to investigate the possible influence of piezoelectric properties on the performance of the samples. As shown in Fig. S6,† the typical amplitude-voltage butterfly loops with a 180 phase switching signal confirm the piezoelectricity of the samples. The piezoelectric coefficient values of ZnS, ZnS–ZnO and ZnO are determined to be approximately 188, 201 and 158 pm V−1, respectively. These results demonstrate that ZnS–ZnO has relatively higher piezoelectricity than ZnO and ZnS, which is beneficial for charge separation.37
To reveal the charge separation process, the piezoelectric current response measurements were conducted on the ZnS, ZnS–ZnO and ZnO electrodes. As shown in Fig. 4a, each electrode exhibited rapid and consistent piezoelectric current responses upon switch-on and switch-off events. Specifically, when the sonication was halted, the current promptly returned to zero. ZnS–ZnO shows an obvious enhancement in the current response compared to ZnS and ZnS. This enhancement in photocurrent generation suggests a favorable electronic interaction between ZnS and ZnO, which facilitates charge transfer and separation, ultimately leading to improved piezo-electrochemical performance. The electrochemical impedance spectrum (EIS) depicted in Fig. 4b provides insights into the charge transfer resistance of the samples. Obviously, ZnS–ZnO shows a smaller arc radius than ZnS and ZnO. This result indicates the lower charge transfer resistance in the heterostructure. Our results indicate that the ZnS–ZnO heterojunction with S vacancies could promote more efficient carrier separation and transfer, thus leading to improved piezocatalytic performance.
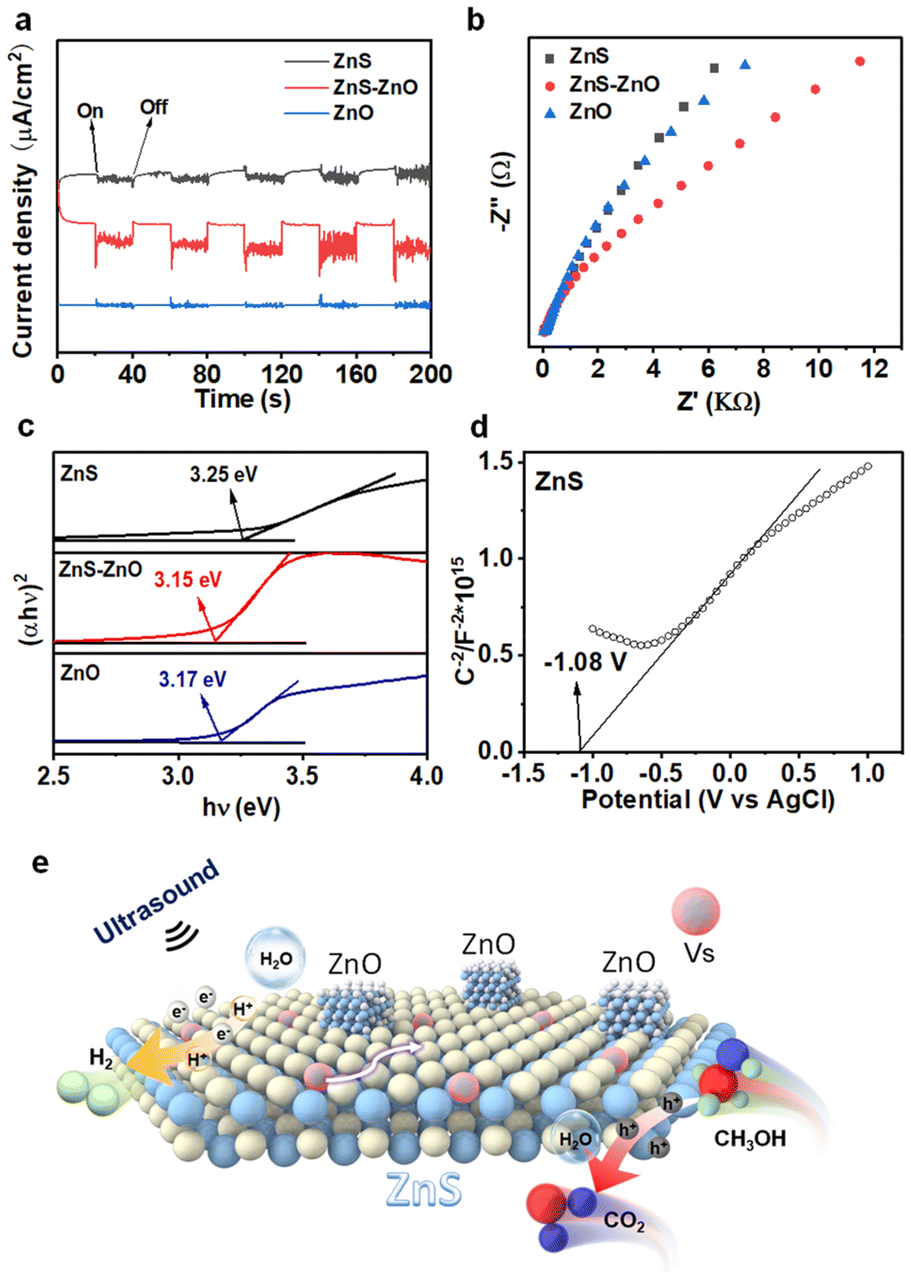 | ||
| Fig. 4 (a) Piezo-current response of different catalysts; (b) EIS Nyquist plots of the samples; (c) Tauc plots of the samples; (d) Mott–Schottky plot of ZnS; (e) proposed reaction mechanism for piezocatalytic H2 production over ZnS–ZnO composite nanosheets. | ||
A variety of experiments were performed to investigate the role of sulfur vacancies in the performance of the sample. To eliminate the influence of other factors, a hydrothermal method was used to synthesize ZnS spheres (ZnS-S) and sulfur vacancies were subsequently introduced by calcining ZnS-S under a nitrogen atmosphere (Fig. S7 and S8a†). The results revealed that the introduction of S vacancies obviously enhanced the activity of ZnS-S (Fig. S8b†). When comparing the samples before and after the introduction of sulfur vacancies, no significant change was observed in the stability of the samples (Fig. S8c and S8d†). This indicates that S vacancies primarily improve the activity of the sample without significantly affecting its stability. Based on piezo-current and impedance analyses, it was found that the introduction of S vacancies in ZnS-S obviously improved charge carrier separation efficiency (Fig. S9a†). Simultaneously, the introduction of S vacancies reduced the material's impedance (Fig. S9b†), facilitating charge carrier transport. This result is in accordance with the previous report, in which S vacancies act as electron traps to promote the separation of carriers like a metal cocatalyst.38
DFT calculations were performed to investigate the H2O adsorption on the sample (Fig. S10†). They revealed that the adsorption energy of H2O on ZnS with sulfur (S) vacancies is −3.05 eV, which is more negative than that of H2O on ZnS without S vacancies (−2.14 eV), as shown in Table S2.† Therefore, the result shows that the S vacancy promotes the adsorption of H2O. In a previous report, Huo et al. also revealed that S vacancies could promote the dissociation of water through theoretical calculations.39
The bandgaps of ZnS, ZnS–ZnO, and ZnO were determined to be 3.25 eV, 3.15 eV, and 3.17 eV, respectively (Fig. 4c). The formation of S vacancies reduces the band gap width of ZnS (3.25 eV), as compared to ZnS in a previous report (3.6 eV).40 As a result, relatively low excitation energy is required for the catalytic process. Fig. 4d and Fig. S11† show the Mott–Schottky curves of the samples. The positive slopes indicate that all the samples are n-type semiconductors. The estimation of the flat-band potential (Vfb) from the Mott–Schottky plots reveals values of approximately −1.08 V, −0.70 V, and −0.80 V (vs. Ag/AgCl) for ZnS, ZnS–ZnO, and ZnO, respectively. Applying the conversion factor E(NHE) = E(Ag/AgCl) + 0.197 V, the conduction band potentials are approximately −0.88 V, −0.50 V, and −0.60 V (vs. NHE). Finally, based on the equation EVB = ECB + Eg, the valence band potentials were calculated to be 2.25 V, 2.45 V, and 2.37 V (vs. NHE), respectively.41
Based on these results, the reaction mechanism underlying the piezocatalysis is illustrated in Fig. 4e. Upon sonication, electrons are excited from the valence band to the conduction band, and the sulfur vacancies act as electron traps, facilitating their migration to the nanosheet surface. In addition, the ZnO–ZnS heterojunction further improves the charge separation efficiency. Here, the excited electrons participate in the reduction of water molecules or protons, leading to the formation of hydrogen gas. The presence of sulfur vacancies not only modifies the electronic band structure, lowering the excitation energy, but also accelerates the charge transfer processes, inhibiting electron–hole recombination and enhancing the overall catalytic activity. In addition, the S vacancy facilitates the adsorption of the H2O molecule, which could capture the electron trapped in the S vacancy for H2 production. In this process, S vacancies serve as active sites for H2 evolution like metal co-catalysts.42
Conclusions
In conclusion, we provide a facile method to synthesize ZnS–ZnO composite nanosheets with defects. The heterojunction is beneficial for the separation of electrons and holes, and the defective structure with sulfur vacancies lowers the excitation energy, which enhances the piezocatalytic activity of the catalyst. The ZnS–ZnO composite nanosheets show much higher piezocatalytic activity than ZnS and ZnO catalysts and exhibit excellent stability. This study offers a promising method for hydrogen production through piezoelectric catalysis.Author contributions
X. Zhao and X. Pan designed and supervised the whole experiments. X. Lu, X. Chen, M. Xu and M. Huang performed the experiments. X. Lu, Y. Liu and X. Pan wrote the original draft. X. Zhao, Y. Liu, W. Chen and X. Pan contributed to data analysis and improved the paper quality by discussions.Data availability
Data available on request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (21802085), the Undergraduate Training Program for Innovation and Entrepreneurship (S202410399031), the Innovation and Entrepreneurship Projects for High-Level Talents of Quanzhou (no. 2024QZC003R) and the Natural Science Foundation of Fujian Province of China (2019J01730, 2023J01889).References
- X. Yang, X. Yang, M. Yu, X. Lu, H. Kang, M. Yang, Q. Qian, X. Zhao, S. Liang and Z. Bian, Nat. Commun., 2023, 14, 4183 CrossRef.
- Z. Zhao, L. Wei, S. Li, L. Zhu, Y. Su, Y. Liu, Y. Bu, Y. Lin, W. Liu and Z. Zhang, J. Mater. Chem. A, 2020, 8, 16238–16245 RSC.
- S. Li, Z. Zhao, J. Zhao, Z. Zhang, X. Li and J. Zhang, ACS Appl. Nano Mater., 2020, 3, 1063–1079 CrossRef CAS.
- F. Yang, P. Wang, J. Hao, J. Qu, Y. Cai, X. Yang, C. Li and J. Hu, Nano Energy, 2023, 118, 108993 CrossRef CAS.
- Y. Wang and J. M. Wu, Adv. Funct. Mater., 2020, 30, 1907619 CrossRef CAS.
- K. Zhang, X. Sun, H. Wang, Y. Ma, H. Huang and T. Ma, Nano Energy, 2024, 121, 109206 CrossRef CAS.
- S. Park, C. W. Lee, M. Kang, S. Kim, H. J. Kim, J. E. Kwon, S. Y. Park, C. Kang, K. S. Hong and K. T. Nam, Phys. Chem. Chem. Phys., 2014, 16, 10408–10413 RSC.
- X. Qiu, J. Xie, X. Ning, Y. Cao, Z. Lu, J. Hu and A. Hao, Appl. Surf. Sci., 2024, 670, 160694 CrossRef CAS.
- X. Zhao, X. Lu, W. Chen, M. Yang, X. Pan and Z. Bian, J. Colloid Interface Sci., 2024, 659, 11–20 CrossRef CAS.
- Y. Zhao, Z.-B. Fang, W. Feng, K. Wang, X. Huang and P. Liu, ChemCatChem, 2018, 10, 397–3401 Search PubMed.
- Y. C. Wang and J. M. Wu, Adv. Funct. Mater., 2019, 30, 1907619–1907620 CrossRef.
- K. Hong, H. Xu, H. Konishi and X. Li, J. Phys. Chem. Lett., 2010, 1(6), 997–1002 CrossRef CAS.
- W. Yang, Y. Yu, M. B. Starr, X. Yin, Z. Li, A. Kvit, S. Wang, P. Zhao and X. Wang, Nano Lett., 2015, 15, 7574–7580 CrossRef CAS PubMed.
- K. Wang, D. Shao, L. Zhang, Y. Zhou, H. Wang and W. Wang, J. Mater. Chem. A, 2019, 7, 20383–20389 RSC.
- H. You, Z. Wu, L. Zhang, Y. Ying, Y. Liu, L. Fei, X. Chen, Y. Jia, Y. Wang, F. Wang, S. Ju, J. Qiao, C. Lam and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 11779–11784 CrossRef CAS.
- Q. Tang, J. Wu, D. Kim, C. Franco, A. Terzopoulou, A. Veciana, J. Puigmartí-Luis, X. Chen, B. J. Nelson and S. Pané, Adv. Funct. Mater., 2022, 30, 2202180 CrossRef.
- S. Li, Z. Zhao, J. Li, H. Liu, M. Liu, Y. Zhang, L. Su, A. I. Pérez-Jiménez, Y. Guo, F. Yang, Y. Liu, J. Zhao and J. Zhang, Small, 2022, 18, 220507 Search PubMed.
- K. Ke, J. Wu, Z. Kang, E. Lin, N. Qin and D. Bao, Nanoscale, 2024, 16, 15652–15662 RSC.
- Q. T. Hoang, K. A. Huynh, T. G. Cao, J. H. Kang, X. N. Dang, V. Ravichandran, H. C. Kang, M. Lee, J. Kim, Y. T. Ko and T. Lee, Adv. Mater., 2023, 35, 2300437 CrossRef.
- C. Wang, F. Chen, C. Hu, T. Ma, Y. Zhang and H. Huang, Chem. Eng. J., 2022, 431, 133930 CrossRef CAS.
- J. M. Wu, W. E. Chang, Y. T. Chang and C. Chang, Adv. Mater., 2016, 28, 3718–3725 CrossRef CAS PubMed.
- M. Wu, J. Lee, Y. J. Chung, M. Srinivaas and J. Wu, Nano Energy, 2017, 40, 369–375 CrossRef CAS.
- Y. Su, L. Zhang, W. Wang, X. Li, Y. Zhang and D. Shao, J. Mater. Chem. A, 2018, 6, 11909–11915 RSC.
- W. Feng, J. Yuan, L. Zhang, W. Hu, Z. Wu, X. Wang, X. Huang, P. Liu and S. Zhang, Appl. Catal., B, 2020, 277, 119250 CrossRef CAS.
- X. Zhou, B. Shen, J. Zhai and J. C. Conesa, Small, 2021, 5, 2100269 CrossRef CAS.
- Z. Ren, X. Li, L. Guo, J. Wu, Y. Li, W. Liu, P. Li, Y. Fu and J. Ma, Mater. Lett., 2021, 292, 129635 CrossRef CAS.
- X. Chen, H. Xu, N. Xu, F. Zhao, W. Lin, G. Lin, Y. Fu, Z. Huang, H. Wang and M. Wu, Inorg. Chem., 2003, 42, 3100–3106 CrossRef CAS PubMed.
- S. H. Yu and M. Yoshimura, Adv. Mater., 2002, 14, 296–300 CrossRef CAS.
- J. Xiong, Y. Li, S. Lu, W. Guo, J. Zou and Z. Fang, Chem. Commun., 2021, 57, 8186–8189 RSC.
- X. Ouyang, T. Tsai, D. Chen, Q. Huang, W. Cheng and A. Clearfiled, Chem. Commun., 2003, 886–887 RSC.
- J. Xiong, X. Wang, J. Wu, J. Han, Z. Lan and J. Fan, Molecules, 2022, 27, 8544 CrossRef CAS.
- M. Wang, S. Shen, L. Li, Z. Tang and J. Yang, J. Mater. Sci., 2017, 52, 5155–5164 CrossRef CAS.
- B. A. McCandless, K. Raum and M. Muller, Phys. Med. Biol., 2024, 69, 115018 CrossRef CAS.
- X. Hao, J. Zhou, Z. Cui, Y. Wang, Y. Wang and Z. Zou, Appl. Catal., B, 2018, 229, 41–51 CrossRef CAS.
- C. Liu, P. Wu, J. Wu, J. Hou, H. Bai and Z. Liu, Chem. Eng. J., 2019, 359, 58–68 CrossRef CAS.
- X. H. Chen, X. L. Li, L. L. Wu, H. C. Fu, J. Luo, L. Shen, Q. Zhang, J. L. Lei, H. Q. Luo and N. B. Li, J. Mater. Chem. A, 2021, 9, 11563–11570 RSC.
- C. Wang, F. Chen, C. Hu, T. Ma, Y. Zhang and H. Huang, Chem. Eng. J., 2022, 431, 133930 CrossRef CAS.
- T. Su, C. Men, L. Chen, B. Chu, X. Luo, H. Ji, J. Chen and Z. Qin, Adv. Sci., 2021, 9, 2103715 CrossRef.
- S. Yin, X. Zhao, E. Jiang, Y. Yan, P. Zhou and P. Huo, Energy Environ. Sci., 2022, 15, 1556–1562 RSC.
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, Prog. Mater. Sci., 2011, 56, 175–287 CrossRef CAS.
- Z. Chen, D. Li and C. Chen, ACS Appl. Nano Mater., 2023, 6, 21897–21908 CrossRef CAS.
- W. Tang, L. Cheng, L. Zhang, X. Xue, D. Zhou, B. Li, G. Wang, Y. Zeng, X. Xing, X. Zhang, W. Dong and C. Hou, J. Colloid Interface Sci., 2023, 629, 1015–1026 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03733b |
| This journal is © The Royal Society of Chemistry 2024 |