
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Therapeutic applications of responsive organic photocatalytic polymers, enabling in situ drug activation†
Rong
Li
a,
Xueqing
Zhang
a,
Seunghyeon
Kim
 a,
Volker
Mailänder
a,
Katharina
Landfester
*a and
Calum T. J.
Ferguson
*b
a,
Volker
Mailänder
a,
Katharina
Landfester
*a and
Calum T. J.
Ferguson
*b
aMax Planck Institute for Polymer Research, Mainz, Rheinland-Pfalz, Germany. E-mail: Landfester@mpip-mainz.mpg.de
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: c.ferguson.1@bham.ac.uk
First published on 24th July 2024
Abstract
Targeted prodrug activation within the acidic tumour microenvironment is needed to limit off-target effects in chemotherapy. This in combination with photodynamic generation of reactive oxygen species (ROS) can be used for efficient remediation of cancerous tissue. To achieve this, pH-responsive polymers with photocatalytic units that become activated in the acidic pH of the tumour microenvironment have been created. Four model prodrug linkages in small molecule substrates have been investigated along with a model polymer-based prodrug. We have demonstrated the pH-dependent activation of model prodrug molecules, due to conformational changes of the pH-responsive photocatalytic polymers. Additionally, a prodrug of the common skin cancer chemotherapy drug 5-fluorouracil (5FU) could be photocatalytically activated and could induce cell death in cancer cells.
The emergence of photodynamic therapy (PDT) has facilitated the minimally invasive treatment of various diseases (e.g. cancers) with well-understood fundamental mechanisms of operation. Upon light irradiation, a photocatalyst absorbs a photon generating a short-lived excited singlet state (S1) that can undergo intersystem crossing and populate the more stable excited triplet state T1. The energy of this excited triplet state can be further transferred to molecular oxygen (O2), generating reactive singlet oxygen (1O2).1,2 Additionally, other reactive oxygen species (ROS) such as the superoxide radical O2˙− can also be produced via an electron transfer process, which can further interact with water as solvent generating hydroxyl radicals OH˙. These ROS can induce oxidative damage and ultimately kill cancer cells.3–6 However, the performance of many currently developed photocatalyst molecules has significant limitations such as intrinsic hydrophobicity7 and the lack of targeting towards tumour cells.8–11 Therefore, the development of a novel tumour-specific PDT system is highly desired.
To develop a general strategy to selectively target the tumour tissue, ubiquitous features of the tumour microenvironment (for instance a low pH value, high interstitial pressure, or hypoxia conditions) have been frequently selected as alternatives to endogenous biomarkers.12 Typical systems have been designed by incorporating stimuli-responsive moieties to modulate their functionalities in response to either external (e.g. UV light13,14) or endogenous stimuli (e.g. enzymes,15,16 changes in the pH value,17–19 redox,20 and hypoxia conditions21,22). Solid tumours are ubiquitously characterized by the dysregulated pH value, where the pH of the extracellular microenvironment (pHe 6.5–6.9) is slightly lower than that of normal tissues (pH 7.2–7.4). With this consideration, developing a pH-sensitive polymer system may achieve the targeted activation of the photocatalyst in the tumour microenvironment. Diblock polymer chains can cluster to form particles through self-assembly, where the photocatalytic segments are immobilised in the core and remain inactive in the bloodstream. If a pH-responsive group is incorporated into the polymer structure and upon exposing the particle to the acidic extracellular microenvironment, photocatalytic moieties can be revealed and activated due to disassembly, during which the aqueous compatibility of the photocatalyst is also enhanced. Therefore, we propose that a pH-responsive polymer system containing photocatalytic moieties may modulate the tumour-specific production of ROS for cancer therapy.
ROS can act not only as active therapeutic agents to kill cancer cells directly but also as a trigger to control the activation of other treatment processes (e.g., prodrug activation or drug release from nanocarriers), inducing additive or even synergistic efficacies. For example, conjugating ROS-sensitive linkers6,23 such as an aminoacrylate bond,24,25 a thioketal bond,26,27 and phenylboronic ester28–31 with the chemical structures of nanocarriers and/or drugs (generally with –OH, –NH–, or –NH2 functional groups) has been explored for cascade reaction-driven anti-cancer drug release. Therefore, the combination of pH-responsive polymeric photocatalysts, which are capable of ROS generation, and ROS-responsive prodrug molecules could lead to triggered drug release in situ, which may provide a promising strategy to enhance antitumour efficacy through PDT/chemo combination therapy.
Here, we have developed a pH-responsive photocatalytic system that can selectively generate ROS in the tumour tissue and further activate prodrugs through a cascade reaction. To demonstrate the versatility of this photocatalytic system, the activation of four ROS-sensitive linkages, including an aminoacrylate bond, thioketal bond, phenylboronic ester and oxalate, has been examined. As illustrated in Scheme 1, the pH-responsive amphiphilic polymer poly(ethylene glycol)-b-poly(2-azepane ethylmethacrylate)-b-2-(methacryloyloxy)ethyl 2-(2,4,5,7-tetrabromo-3,6-dihydroxy-9H-xanthen-9-yl)benzoate (PEG113-b-PAEMA50-EYHEMA1) was synthesized. The amphiphilic polymer chains self-assemble at pH 7.4 to form polymer particles. Once accumulated in the acidic tumour environment (pH 6.5), the hydrophobic PAEMA block of the polymer becomes protonated, leading to the disassembly of the particles and exposure of the photocatalyst eosin Y. Upon light irradiation, ROS are generated by the active eosin Y and subsequently allow the activation of prodrugs through ROS-induced cleavage of the protecting group.
 | ||
| Scheme 1 Illustration of the pH-responsive polymer photocatalyst that can respond to a subtle pH difference between normal tissue and the slightly acidic tumour microenvironment, which allows the controlled activation of prodrug molecules. | ||
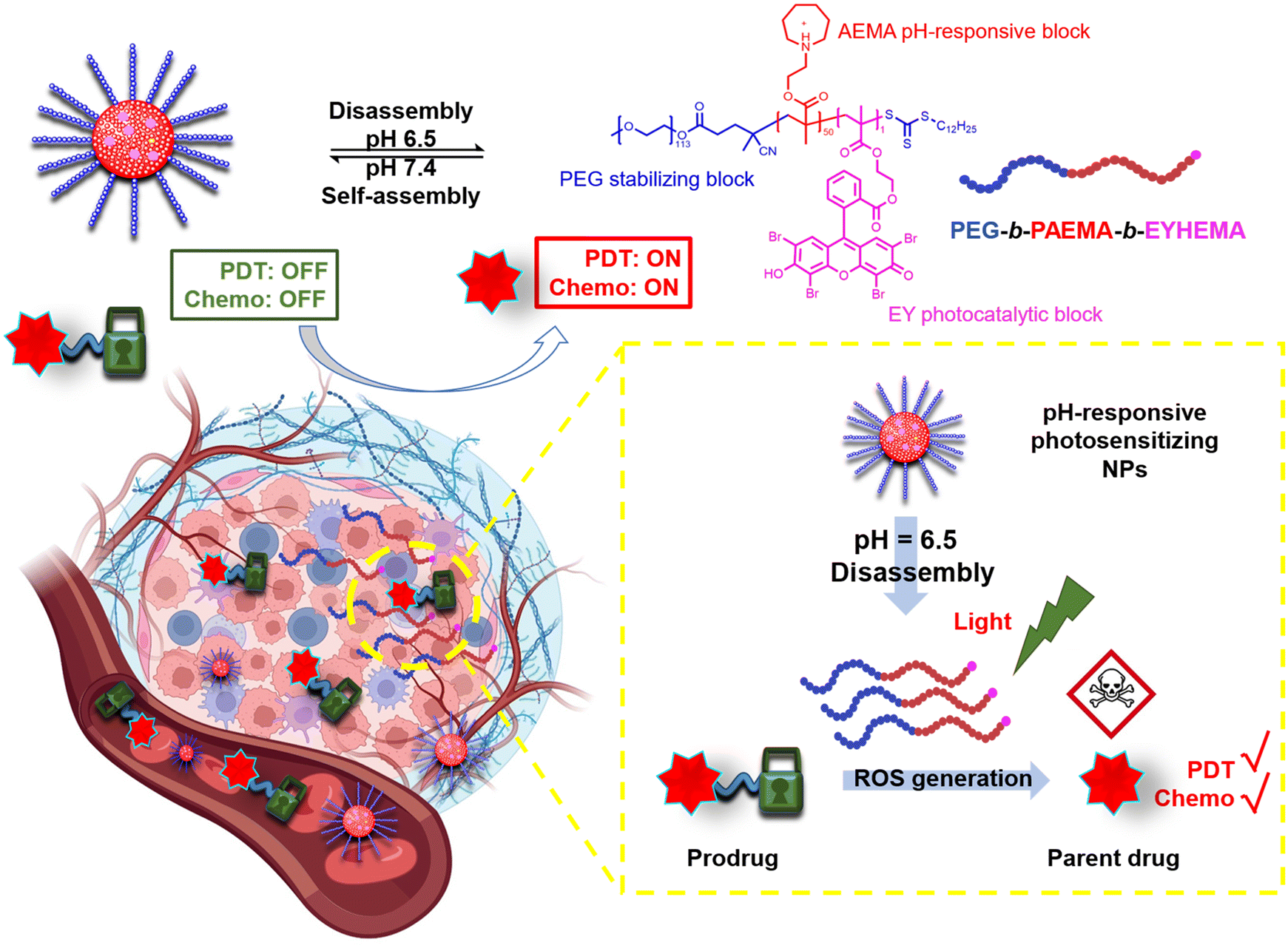
Dual-responsive copolymers PEG113-b-PAEMA50-EYHEMA1 were synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization using poly(ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (mPEG113-CPDTC) as the macro-chain transfer agent (macro-CTA) and 2-azepane ethylmethacrylate (AEMA) and 2-(2-(2,4,5,7-tetrabromo-6-hydroxy-3-oxo-3H-xanthen-9-yl) benzamido) ethyl methacrylate (EYHEMA) as the pH-responsive and light-responsive monomers, respectively (Fig. 1a). The resulting polymer was confirmed by 1H NMR spectroscopy and gel permeation chromatography (GPC) suggesting a number averaged molar mass of 13.9 kDa (ESI, Fig. S1 and S2†). A bimodal distribution was observed, which is believed to be due to unfunctionalized PEG in macroCTA. FTIR (Fig. S3†) was used to examine the chemical compositions of PEG113-b-PAEMA50 and PEG113-b-PAEMA50-EYHEMA1 polymer chains. The fingerprint peaks of these polymer chains, including –CH2–, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, and C–N functional groups, are clearly visible at 2750–3100 cm−1, 1725 cm−1, 1470 cm−1, and 1150 cm−1, respectively. However, the effect of the photocatalytic moiety was not observed in the FTIR spectrum due to the low loading of the photocatalytic monomer (0.49 mol%). The optical properties of the dual-responsive polymers were visualized from the UV/Vis spectrum, displaying absorption in the green light region (Fig. 1b) in agreement with the literature.32
O, C–O, and C–N functional groups, are clearly visible at 2750–3100 cm−1, 1725 cm−1, 1470 cm−1, and 1150 cm−1, respectively. However, the effect of the photocatalytic moiety was not observed in the FTIR spectrum due to the low loading of the photocatalytic monomer (0.49 mol%). The optical properties of the dual-responsive polymers were visualized from the UV/Vis spectrum, displaying absorption in the green light region (Fig. 1b) in agreement with the literature.32
 | ||
| Fig. 1 Characterization of pH-responsive polymers. (a) Molecular structure of the designed pH-responsive photocatalytic copolymer PEG113-b-PAEMA50-EYHEMA1. (b) UV/Vis absorbance and emission spectra of the PEG113-b-PAEMAH+50-EYHEMA1 polymer chains in phosphate buffer at pH 6.5 (1 mg mL−1). (c) The hydrodynamic diameter (PEG113-b-PAEMA50) changes as a function of pH measured by DLS (pH 6.0, 6.5, 6.8, 7.2, 7.4, and 7.8, 0.1 mM) (laser: 632.8 nm). (d) TEM image of the NP-AEMA-EY nano-assembly in PBS at pH 7.4. | ||
PAEMA has been specifically developed as an ultra-pH-sensitive polymer that responds to a pH increment of ∼0.3,17,19,33 aligning perfectly with the pH difference of the extracellular microenvironment (pHe 6.5–6.9) compared to normal tissues (pH 7.2–7.4).34 Taking advantage of this ultrasensitive pH-responsiveness of PAEMA, PEG113-b-PAEMA50 can be protonated at pH 6.5, leading to the solvation of the polymer chains, while being deprotonated at pH 7.4, resulting in an amphiphilic diblock copolymer and self-assembly. Therefore, pH-sensitive nano-assemblies were prepared by self-assembly of photocatalytically active PEG113-b-PAEMA50-EYHEMA1 (NP-AEMA-EY) or photocatalytically inactive PEG113-b-PAEMA50 (NP-PAEMA) copolymers, respectively. The photocatalytic unit was selectively polymerised at the end of the hydrophobic chain to minimise its activity until activated by the change in pH.35,36 The RAFT end group of the polymer in this test was not removed as it has previously been shown to be non-toxic.37 The hydrodynamic diameter of the inactive nano-assemblies was determined by dynamic light scatting (DLS). As shown in Fig. 1c, the diameter of the nano-assemblies peaked at approximately 164 nm at pH 7.4, which decreased to 13 nm at pH 6.5. This sharp size change indicates the disassembly of the nano-assemblies under slightly acidic conditions, which agrees well with the pKa value (∼7.2) of the block copolymer (Fig. S4†). Additionally, UV-Vis transmittance (Fig. S5†) measurement and the digital image of PEG113-b-PAEMA50 nano-assembly dispersions showed that turbidity increased when increasing the pH value of the buffer solution. These findings further demonstrated the pH responsiveness of these nano-assemblies. Furthermore, the transmission electron microscopy (TEM) image has confirmed that at pH 7.4 the polymer chains are assembled into spherical gel-like nano-assemblies (Fig. 1d). Additionally, TEM images also revealed the size change of pH-responsive photocatalytic nano-assemblies PEG113-b-PAEMA50-EYHEMA1 from pH 7.4 to pH 6.5 (Fig. S6†).
The photocatalyst eosin Y is an inexpensive and biocompatible material that has been used extensively in biological applications.38,39 Eosin Y is capable of efficiently generating 1O2 (Fig. 2), which has been widely used for PDT.40 In our preliminary study, with blue light irradiation, molecular eosin Y (eosin Y disodium salt) can efficiently activate prodrug model compounds containing various ROS-sensitive linkers/caps, including thiol ketal, aminoacrylate, boronic acid pinacol ester, and oxalate (Table S1, Fig. S8†). After 2 to 4 h of light irradiation, the prodrug model compounds were activated with over 70% yield, suggesting that the eosin Y photocatalyst is effective in activating a broad range of ROS-sensitive linkages.
 | ||
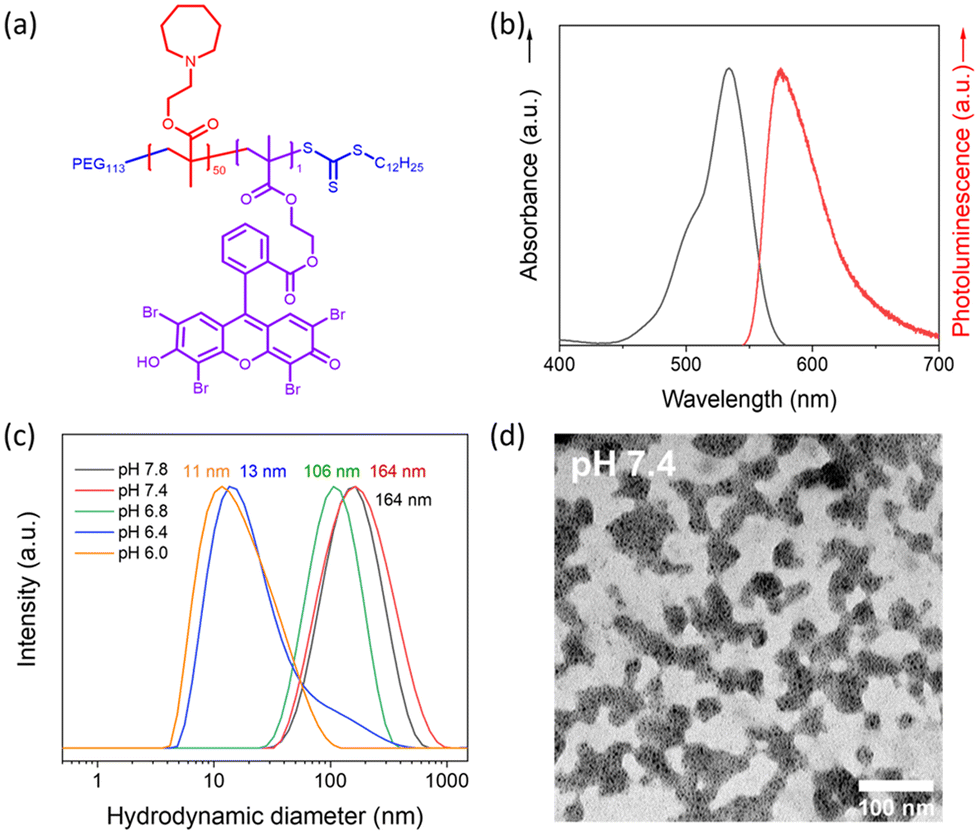
| Fig. 2 Electron paramagnetic resonance (EPR) spin trapping spectra of TEMP 1O2 generated under different conditions. (a) Eosin Y disodium salt (1 mg mL−1), tetramethylpiperidine (TEMP, 0.1 M), O2. (b) Eosin Y disodium salt (1 mg ml−1), TEMP (0.1 M), air. (c) TEMP (0.1 M), O2. (d) Eosin Y disodium salt (1 mg mL−1), TEMP (0.1 M), O2, darkness. All the samples were irradiated with a blue LED for 30 min before measurement. | ||
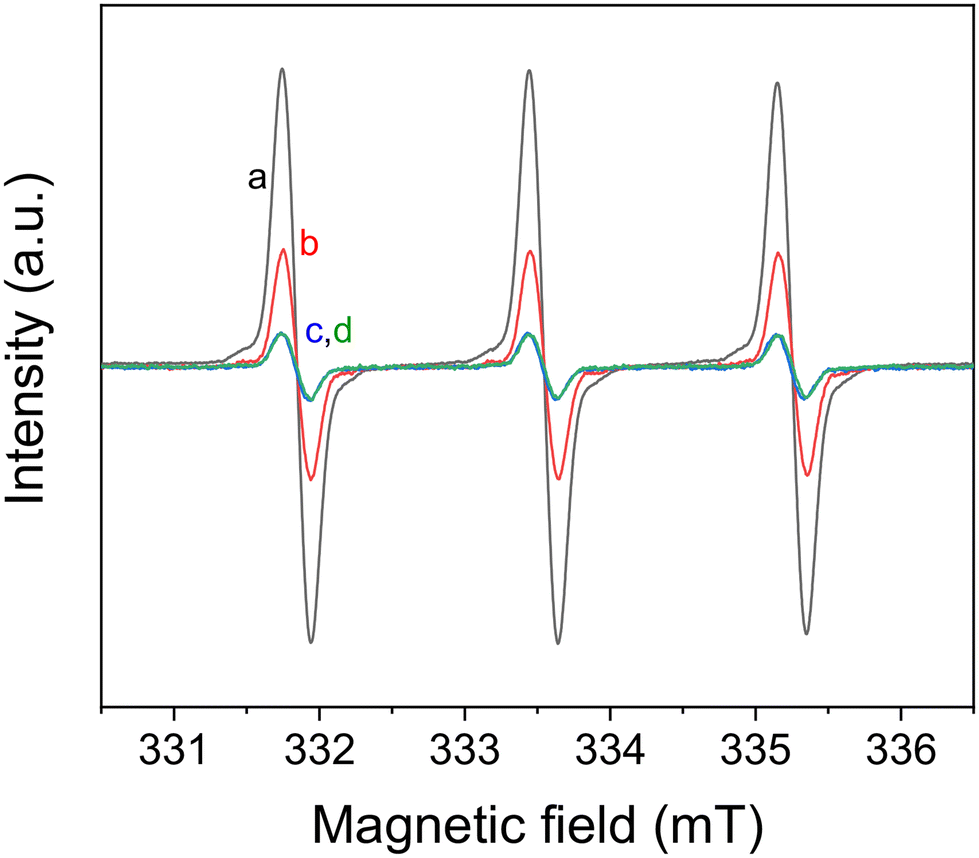
The promising performance of the molecular eosin Y photocatalyst in activating the prodrug model molecules has encouraged us to further examine eosin Y-based pH-responsive photocatalytic nanoparticles (NP-AEMA-EY) for the activation of prodrug model compounds. Initially, the photocatalytic activation of a prodrug model compound containing boronic acid pinacol ester was carried out using the PEG113-b-PAEMA50-EYHEMA1 polymer as a polymeric photocatalyst. In a typical experimental set-up, PEG113-b-PAEMAH+50-EYHEMA1 was dissolved in phosphate buffer at pH 6.5 and combined with the prodrug model compound in a glass vial prior to light irradiation. The prodrug activation kinetic profiles were monitored by GCMS in triplicate. As illustrated in Fig. 3, over 90% yield of activation was obtained using the PEG113-b-PAEMAH+50-EYHEMA1 polymer as a polymeric photocatalyst (2.5 mol% eosin Y moiety) after 1 h of light irradiation. When the oxygen atmosphere was replaced with air, the hydroxylation reaction progressed with a slower reaction rate than the reaction in the presence of oxygen. Only 16% yield was delivered after 1 h under ambient conditions, suggesting that the presence of oxygen is crucial for photocatalytic hydroxylation to occur efficiently. No conversion was detected for controlled reactions without a photocatalyst under light and with a photocatalyst in the dark, respectively.
 | ||
| Fig. 3 Prodrug model compound, phenylboronic acid pinacol ester, activation kinetic profiles obtained using PEG113-b-PAEMA+50-EYHEMA1 polymeric photocatalyst (PC, 2.5 mol%) solution in phosphate buffer solution at pH 6.5 (black). Control reactions: without a photocatalyst under light (green) and with a photocatalyst in the dark (blue). | ||
Furthermore, we have also investigated the controlled activation of a prodrug model compound that contains a 1O2-sensitive aminoacrylate linker by applying pH-responsive photocatalytic nanoparticle NP-AEMA-EY dispersions at pH 7.4 and PEG113-b-PAEMAH+50-EYHEMA1 dissolved in phosphate buffer at pH 6.5, respectively. In a typical experimental set-up, the NP-AEMA-EY dispersion or PEG113-b-PAEMAH+50-EYHEMA1 solution and prodrug were combined in a glass vial prior to irradiation with blue LED light, respectively. The prodrug activation kinetic profiles were monitored by GCMS in triplicate. As we can see from Fig. 4, 68% of the prodrug model compound was activated by the solvated PEG113-b-PAEMAH+50-EYHEMA1 polymeric photocatalyst at pH 6.5 after 1.5 hours of light irradiation, whereas only 2.7% yield of activation was obtained by using NP-AEMA-EY at pH 7.4. This result strongly suggests that the photocatalyst eosin Y moieties were more accessible when the polymer chains were fully solvated under mildly acidic conditions, thereby leading to targeted activation of the prodrug model compound in the mildly acidic tumour microenvironment.
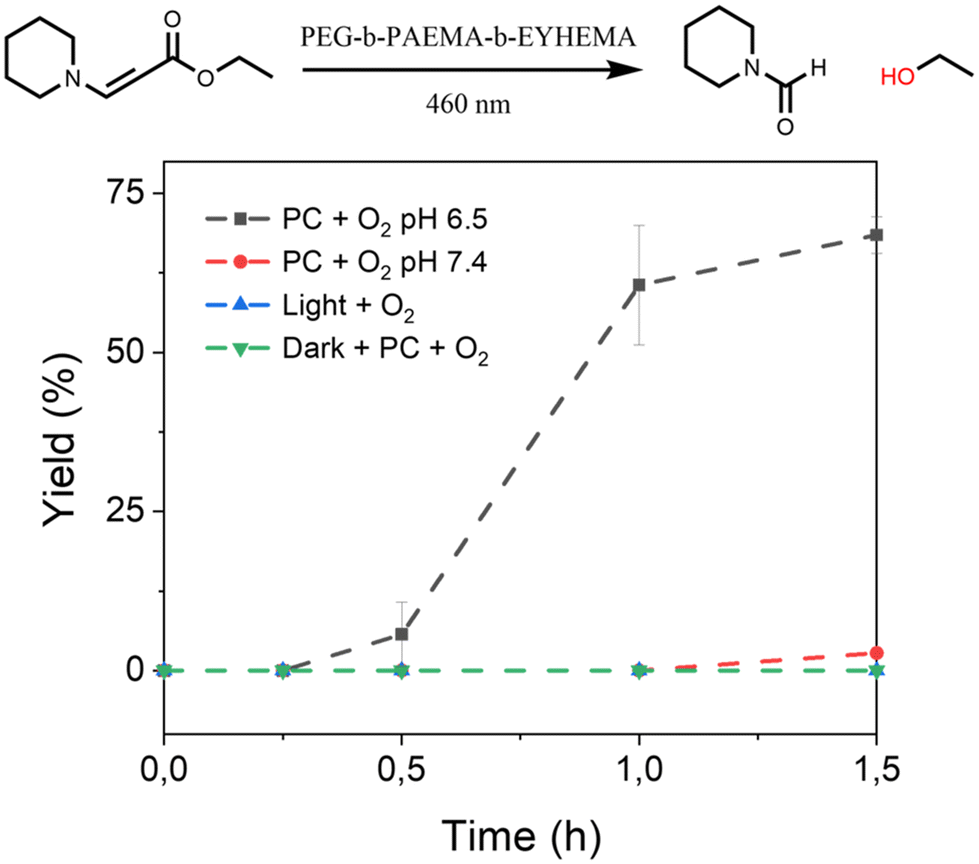 | ||
| Fig. 4 The controlled activation kinetic profile of the prodrug model compound ethyl (E)-3-(piperidin-1-yl)acrylate obtained using PEG113-b-PAEMA50-EYHEMA1 polymeric photocatalyst (PC, 2.5 mol%) solution in phosphate buffer solution at pH 6.5 (black) and NP-AEMA-EY in PBS buffer at pH 7.4 (red). Control reactions: without a photocatalyst in light (green) and with a photocatalyst in the dark (blue). | ||
The excellent performance of the polymeric photocatalyst in activating ROS-sensitive capping groups in a controlled manner has boosted our interest in investigating the activation of an anticancer drug molecule. 5-Fluorouracil (5FU) is an FDA-approved chemotherapeutic drug that has been widely prescribed alone or in combination with other chemotherapeutics for the treatment of various solid tumours (e.g. breast cancer, pancreatic cancer, colorectal cancer, stomach cancer, cervical cancer, and skin cancer). Over the past decades, mechanisms of action of 5FU in the human body have been intensively studied and clearly demonstrated.41–43 Briefly, 5FU molecules can inhibit the activity of the nucleotide synthesis enzyme thymidylate synthase (TS), which is crucial for catalyzing the reductive methylation of deoxyuridine monophosphate to deoxythymidine monophosphate. By blocking the function of TS, DNA replication and repair are interrupted.41,43 Despite the excellent anticancer activity, major side effects of 5FU, including central neurotoxicity, gastrointestinal toxicity, and myelosuppression, as well as being metabolically unstable, still significantly limit its clinical use.44 Therefore, prodrug strategies have been actively investigated to overcome these limitations, where several 5FU-prodrugs among many analogues have been successfully applied in clinical use.45
Here, 5FU was selected as an example anticancer drug and we are interested in the creation of a ROS-sensitive 5FU-prodrug that can be selectively activated by the pH-responsive photocatalyst at the tumour site. As we have demonstrated the remarkable performance of the pH-responsive photocatalyst in activating the boronic acid pinacol ester group, an arylboronate-based prodrug of 5FU (5-fluoro-1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)pyrimidine-2,4(1H,3H)-dione) has been synthesized by introducing a ROS-sensitive p-boronate-benzyl group into the N1 position of 5FU according to the literature.46 Similar to the aforementioned prodrug model compound activation procedures, the 5FU prodrug was mixed with PEG113-b-PAEMAH+50-EYHEMA1 solution in phosphate buffer at pH 6.5 before being subjected to light irradiation. The conversion of the 5FU prodrug was monitored by 19F-NMR, where >99% conversion of the 5FU prodrug into an intermediate was obtained after half an hour of light irradiation (Fig. S8†). As reported in the literature,46 this intermediate is subsequently activated spontaneously under cell culture conditions, leading to the release of active 5FU and the death of cancerous tissue.
A cell viability study was undertaken to verify that the photocatalytically activated 5FU prodrug intermediate could subsequently be converted to 5FU and induce cell death. Here, the prodrug and photocatalyst were either irradiated or kept in the dark prior to incubation with cancer cells. Additionally, control experiments of the photocatalyst and the prodrug were performed separately (Fig. 5). The cell viability results showed that only the combination of the prodrug, photocatalyst, and light-induced cell death. This suggests that the photoactivated prodrug intermediate undergoes further activation and forms 5FU as expected, leading to cell death. As the concentration of the activated prodrug loading increased, cell viability of the group treated with the combination of the prodrug and the photocatalytic polymer PEG113-b-PAEMAH+50-EYHEMA1 after blue light LED irradiation significantly decreased, indicating prominent antitumour efficacy through 5FU chemotherapy. However, the effect of photocleaved 5FU was not as strong as free 5FU, which suggests that not all the prodrug is activated. Meanwhile, the individual components of the prodrug, the photocatalytic polymer PEG113-b-PAEMAH+50-EYHEMA1, as well as the prodrug and photocatalytic polymer PEG113-b-PAEMAH+50-EYHEMA1 mixture in the absence of light irradiation (dark) showed negligible toxicity until high concentrations (100 μM) were applied.
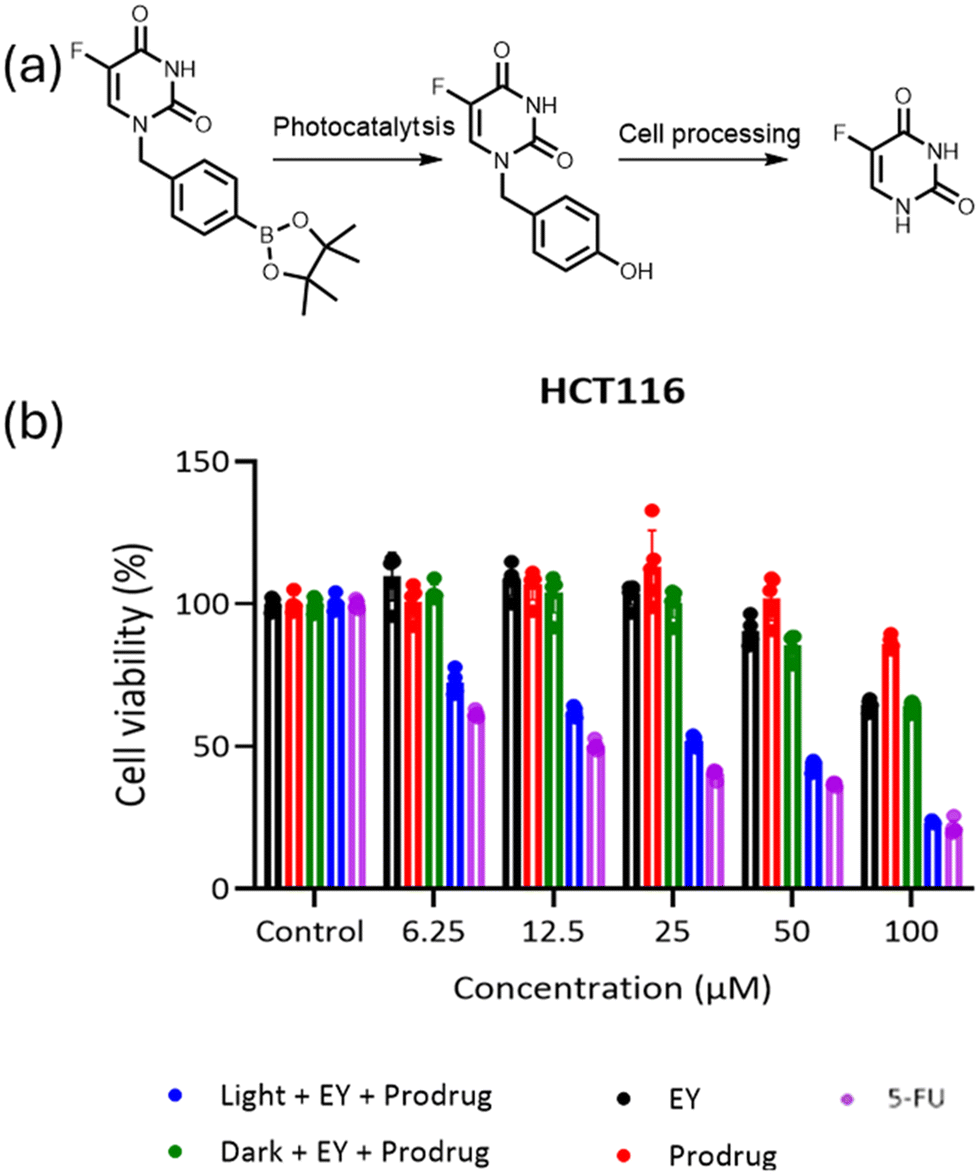 | ||
| Fig. 5 The viability of HCT116 cancer cells treated with varying concentrations of the prodrug, PEG113-b-PAEMAH+50-EYHEMA1 (shortened as EY), the combination of the prodrug and PEG113-b-PAEMA50-EYHEMA1 before and after blue light LED irradiation, and a control of just 5-FU. The cells were treated for 72 h with the indicated concentrations of the compounds. Data are presented as mean ± S.D, n = 5. | ||
In conclusion, we have designed and synthesized a novel pH-responsive polymeric photocatalyst consisting of azepane moieties as pH-responsive functional groups and a small loading of eosin Y as a photocatalyst. This polymeric photocatalytic material exhibited excellent reactivity in the activation of prodrug model compounds with different ROS-sensitive protecting groups under mildly acidic conditions (pH 6.5). Moreover, the controlled activation of the prodrug model compound has been achieved, taking advantage of the ultra pH-sensitive nature (0.3 pH increment) of the polymeric photocatalyst, where a 25 times high yield of release has been obtained at pH 6.5 compared to the reaction at pH 7.4. Furthermore, this polymeric photocatalyst has efficiently activated the 5FU prodrug into an intermediate, which can be spontaneously activated into the active parent 5FU in cell cultivation. These findings demonstrate that the pH-responsive polymeric photocatalyst provides an effective approach to selectively activate prodrugs with various ROS-sensitive linkers/caps, which can potentially enhance the antitumour efficacy through PDT/chemo combination therapy.
The proof-of-principle work demonstrated here has shown the potential of using pH-responsive polymer photocatalysts to activate prodrug molecules. However, several critical factors still need to be overcome for the medical application of these systems, namely the reaction time in low-oxygen environments, the wavelength of light used, and the codelivery of the photocatalytic system and the prodrug. The polymer system demonstrated here is modular, and each component can be replaced. Therefore, future work will investigate different photocatalytic species in order to increase their applicability. This is an exciting emerging area with the potential to create new therapeutic strategies.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely thank Christoph Sieber and Katrin Kirchhoff for taking TEM images. We are grateful for the funding granted by the Max Planck Graduate Centre (MPGC).References
- C. T. J. Ferguson and K. A. I. Zhang, ACS Catal., 2021, 11, 9547–9560 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- S. Wang, G. Yu, Z. Wang, O. Jacobson, L.-S. Lin, W. Yang, H. Deng, Z. He, Y. Liu, Z.-Y. Chen and X. Chen, Angew. Chem., Int. Ed., 2019, 58, 14758–14763 CrossRef CAS PubMed.
- X. Zhang, S. Wang, G. Cheng, P. Yu, J. Chang and X. Chen, Matter, 2021, 4, 26–53 CrossRef CAS PubMed.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- H. O. Alsaab, M. S. Alghamdi, A. S. Alotaibi, R. Alzhrani, F. Alwuthaynani, Y. S. Althobaiti, A. H. Almalki, S. Sau and A. K. Iyer, Cancers, 2020, 12, 2793 CrossRef CAS PubMed.
- D. Bechet, P. Couleaud, C. Frochot, M.-L. Viriot, F. Guillemin and M. Barberi-Heyob, Trends Biotechnol., 2008, 26, 612–621 CrossRef CAS PubMed.
- H. Sun and Z. Zhong, ACS Macro Lett., 2020, 9, 1292–1302 CrossRef CAS PubMed.
- R. Wei, S. Liu, S. Zhang, L. Min and S. Zhu, Anal. Cell. Pathol., 2020, 2020, 6283796 Search PubMed.
- R. Tong, H. H. Chiang and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19048–19053 CrossRef CAS PubMed.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS PubMed.
- S. Ruan, X. Cao, X. Cun, G. Hu, Y. Zhou, Y. Zhang, L. Lu, Q. He and H. Gao, Biomaterials, 2015, 60, 100–110 CrossRef CAS PubMed.
- Q. Zhou, Nat. Nanotechnol., 2019, 14, 16 Search PubMed.
- Y. Dong, Y. Tu, K. Wang, C. Xu, Y. Yuan and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 7168–7172 CrossRef CAS PubMed.
- J. Li, Y. Li, Y. Wang, W. Ke, W. Chen, W. Wang and Z. Ge, Nano Lett., 2017, 17, 6983–6990 CrossRef CAS PubMed.
- H.-J. Li, J.-Z. Du, J. Liu, X.-J. Du, S. Shen, Y.-H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS PubMed.
- S. Zhou, X. Hu, R. Xia, S. Liu, Q. Pei, G. Chen, Z. Xie and X. Jing, Angew. Chem., Int. Ed., 2020, 59, 23198–23205 CrossRef CAS PubMed.
- P. Kulkarni, M. K. Haldar, P. Katti, C. Dawes, S. You, Y. Choi and S. Mallik, Bioconjugate Chem., 2016, 27, 1830–1838 CrossRef CAS PubMed.
- P. Wang, Q. Gong, J. Hu, X. Li and X. Zhang, J. Med. Chem., 2021, 64, 298–325 CrossRef CAS PubMed.
- M. Bio, G. Nkepang and Y. You, Chem. Commun., 2012, 48, 6517–6519 RSC.
- Y. Yuan, C.-J. Zhang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 11419–11423 CrossRef CAS PubMed.
- Z. Cao, Y. Ma, C. Sun, Z. Lu, Z. Yao, J. Wang, D. Li, Y. Yuan and X. Yang, Chem. Mater., 2018, 30, 517–525 CrossRef CAS.
- S. Z. F. Phua, C. Xue, W. Q. Lim, G. Yang, H. Chen, Y. Zhang, C. F. Wijaya, Z. Luo and Y. Zhao, Chem. Mater., 2019, 31, 3349–3358 CrossRef CAS.
- Y. Kuang, K. Balakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- W. Chen, Y. Han and X. Peng, Chem. – Eur. J., 2014, 20, 7410–7418 CrossRef CAS PubMed.
- W. Chen, K. Balakrishnan, Y. Kuang, Y. Han, M. Fu, V. Gandhi and X. Peng, J. Med. Chem., 2014, 57, 4498–4510 CrossRef CAS PubMed.
- W. Zhang, X. Hu, Q. Shen and D. Xing, Nat. Commun., 2019, 10, 1704 CrossRef PubMed.
- S. Jradi, L. Balan, X. H. Zeng, J. Plain, D. J. Lougnot, P. Royer, R. Bachelot, S. Akil, O. Soppera and L. Vidal, Nanotechnology, 2010, 21, 095605 CrossRef CAS PubMed.
- K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
- R. Li, J. Heuer, T. Kuckhoff, K. Landfester and C. T. J. Ferguson, Angew. Chem., Int. Ed., 2023, 62, e202217652 CrossRef CAS PubMed.
- J. Heuer, T. Kuckhoff, R. Li, K. Landfester and C. T. J. Ferguson, ACS Appl. Mater. Interfaces, 2023, 15, 2891–2900 CrossRef CAS PubMed.
- D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef CAS PubMed.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- C. Wu, N. Corrigan, C.-H. Lim, K. Jung, J. Zhu, G. Miyake, J. Xu and C. Boyer, Macromolecules, 2019, 52, 236–248 CrossRef CAS PubMed.
- B. Fan, W. Peng, Y. Zhang, P. Liu and J. Shen, Biomater. Sci., 2023, 11, 4930–4937 RSC.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- G. D. Heggie, J. P. Sommadossi, D. S. Cross, W. J. Huster and R. B. Diasio, Cancer Res., 1987, 47, 2203–2206 CAS.
- M. Scartozzi, E. Maccaroni, R. Giampieri, M. Pistelli, A. Bittoni, M. Del Prete, R. Berardi and S. Cascinu, Pharmacogenomics, 2011, 12, 251–265 CrossRef CAS PubMed.
- J. S. Macdonald, Oncology, 1999, 13, 33–34 CAS.
- J. Shelton, X. Lu, J. A. Hollenbaugh, J. H. Cho, F. Amblard and R. F. Schinazi, Chem. Rev., 2016, 116, 14379–14455 CrossRef CAS PubMed.
- Y. Ai, O. N. Obianom, M. Kuser, Y. Li, Y. Shu and F. Xue, ACS Med. Chem. Lett., 2019, 10, 127–131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00493k |
| This journal is © The Royal Society of Chemistry 2024 |