DOI:
10.1039/D4QI01563K
(Research Article)
Inorg. Chem. Front., 2024,
11, 7040-7048
A redox-active ligand combines a PCP pincer site with a bidentate N–N donor in opposition†
Received
21st June 2024
, Accepted 22nd August 2024
First published on 28th August 2024
Abstract
A binucleating ligand (2) combining a monoanionic PCP pincer cleft with a monoanionic N–N cleft has been prepared on the basis of bis(imidazolyl)methane (1). Installation of divalent Pd or Pt into the PCP cleft proceeded smoothly, with the formation of square planar (PCP)MCl complexes (M = Pd or Pt). The “C” in the PCP is the central carbon of the β-diketiminate-like N–N cleft. The N–N cleft in these complexes was obtained in three levels of protonation. In 4-Pd and 4-Pt, the N–N cleft carries one proton and is neutral; in 3-Pd or 3-Pt, it is additionally protonated by HCl, and in 6-Pd or 6-Pt, the proton has been removed by the (Me3Si)2NLi base and replaced by Li. The chloride in 4-Pd/Pt was replaced with an iodide to make 6-Pd/Pt in a metathesis with Me3SiI. Compounds 4-Pd/Pt and 6-Pd/Pt exhibited tautomerism reminiscent of acetylacetone and β-diketimines, possessing either an NH or a CH bond. Compounds 6-Pd/Pt possess a lithiated N–N cleft and serve as convenient transmetallation agents. Their reactions with BF3(OEt2), ½ZnCl2, or TaCl5 produced new compounds 7-Pd/Pt, 8-Pd/Pt, and 9-Pd/Pt that carry a BF2, ½Zn, or TaCl4 units in the N–N cleft. Compounds 8-Pd/Pt are trimetallic with a central Zn coordinated by two N–N clefts each of which carries a (PCP)MCl on the opposite side. Structures of 3-Pd, 7-Pt, 8-Pd, and 9-Pd in the solid state were established by X-ray diffractometry. They demonstrate the remarkable planarity of the extended conjugated organic π-system into which the N–N and the PCP binding sites are incorporated. Electrochemical studies on 7–9 established that each of these extended π-systems can be oxidized twice quasi-reversibly. Compounds 9-Pd/Pt could be additionally reduced quasi-reversibly by a single electron, which was ascribed to the reduction of the TaV center to TaIV. They also differed from 7 and 8 in possessing apparent lower-energy LMCT bands responsible for the blue or purple colors.
10th anniversary statement
I (Oleg Ozerov) was among the first crop of associate editors for ICF, serving during 2013–2022. I recall that at one of our first Board meetings taking place in Beijing, there was a spirited discussion regarding the place and the future for ICF in the chemistry publishing world. We optimistically resolved to make ICF the best inorganic chemistry journal because any other outlook seemed lacking. Now, ten years later, that resolve seems well justified and I am glad I got to play a modest role in the journal's rise. Being an ICF editor allowed me to expand my scientific horizons, and to learn to face the challenges of this different facet of academic life. But it also allowed me to make a number of new friends, and to experience new places in China and Asia in general. For that I am grateful and wish ICF a continued path of success.
|
Introduction
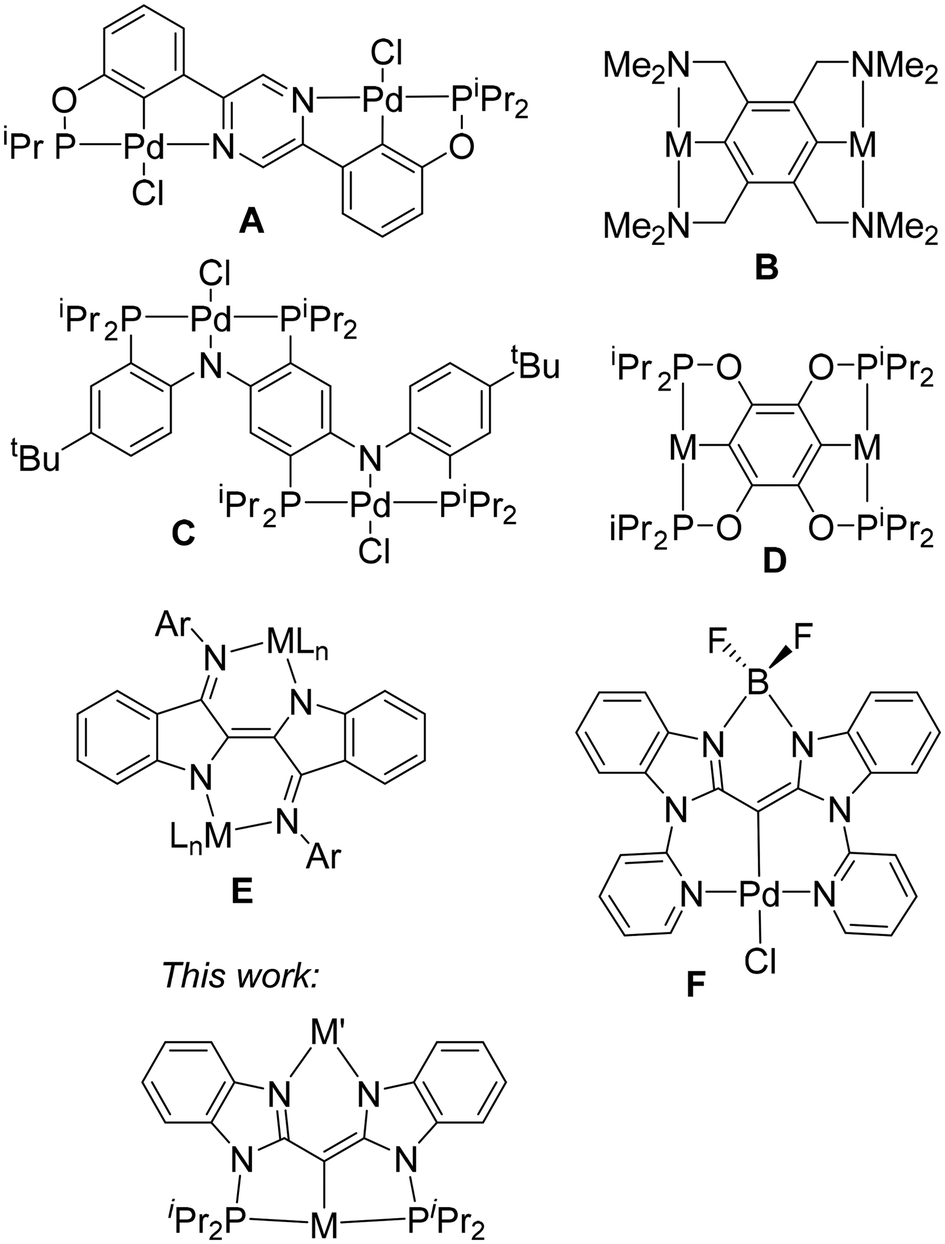
Pincer ligands have become ubiquitous in the studies of organometallic structure and reactivity.1–3 Pincer-supported transition metal complexes have been studied as catalysts for a large variety of applications,1–3 including alkane dehydrogenation,4,5 transfer (de)hydrogenation of compounds with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/C–N and CO/C–O bonds,6–8 C–C coupling,9,10 C–H borylation,11,12 and nucleophilic addition to ketones and aldehydes.13 The tridentate pincer cleft provides a robust environment for the transition metal, in some cases enabling catalytic performance at a temperature of 200 °C or higher,14 which is unusual for organometallic transformations. The most obvious way to influence the transition metal center in a pincer environment is through the modification of the pincer donor sites. However, substantial attention has been paid to the more remote influences, including the use of redox non-innocent ligands15–20 and the provision of distal binding sites for the coordination of additional metals.21,22 The so-called Janus bis-pincer systems (A–D, Fig. 1) can be included within this context, with one pincer-metal unit providing a remote influence on the other.23–30 The Janus bis-pincers provide two opposing binding cleft sites of the same kind, naturally leading to symmetric bimetallic complexes. There also exist examples of non-Janus bis(pincer) ligands with a shared atom or unit linking the two pincer domains.31–34 However, it is synthetically difficult to prepare heterobimetallic complexes with symmetric bis(pincer) ligands. Considering ways to introduce two very different metals into the same molecule, we became interested in preparing a “dissymmetric Janus” type of ligand, with one phosphine-anchored pincer site suitable for a late transition metal, and another N–N bidentate site amenable to binding a broad variety of metals and metalloids. This arrangement provides a way to influence the structure and reactivity of the pincer ligated transition metal center through variation of the other element in the N–N cleft, but without changing the immediate coordination environment about the transition metal. We envisaged an N–N site similar to β-diketiminates (also known as “nacnac” ligands)35 that is integrated into a redox-active pincer framework. In considering this design, we were influenced by the binucleating “nindigo” ligands (E, Fig. 1) pioneered by the Hicks group,36 and also explored by Caulton and Mindiola,37 and by Kaim and Lahiri with co-workers.38 The “nindigo” ligands are essentially a redox non-innocent Janus bis-“nacnac” system. Herein we report on the chemistry of a binucleating ligand fusing a PCP pincer with a “nacnac”-like N–N site. A similar binucleating ligand combining an NCN pincer with a bidentate N–N site (F, Fig. 1) was recently explored by Ong, Frenking, Zhao, and coworkers.39 In this work, we report our exploration of how the nature of the element in the N–N cleft influences the coordination environment about the transition metal in the pincer cleft, along with the electrochemical and spectroscopic properties of the molecule as a whole. This synthetic solution for the remote modulation of the properties of a transition metal pincer complex may provide new way for exploring and tuning catalytic activity in the future.
N/C–N and CO/C–O bonds,6–8 C–C coupling,9,10 C–H borylation,11,12 and nucleophilic addition to ketones and aldehydes.13 The tridentate pincer cleft provides a robust environment for the transition metal, in some cases enabling catalytic performance at a temperature of 200 °C or higher,14 which is unusual for organometallic transformations. The most obvious way to influence the transition metal center in a pincer environment is through the modification of the pincer donor sites. However, substantial attention has been paid to the more remote influences, including the use of redox non-innocent ligands15–20 and the provision of distal binding sites for the coordination of additional metals.21,22 The so-called Janus bis-pincer systems (A–D, Fig. 1) can be included within this context, with one pincer-metal unit providing a remote influence on the other.23–30 The Janus bis-pincers provide two opposing binding cleft sites of the same kind, naturally leading to symmetric bimetallic complexes. There also exist examples of non-Janus bis(pincer) ligands with a shared atom or unit linking the two pincer domains.31–34 However, it is synthetically difficult to prepare heterobimetallic complexes with symmetric bis(pincer) ligands. Considering ways to introduce two very different metals into the same molecule, we became interested in preparing a “dissymmetric Janus” type of ligand, with one phosphine-anchored pincer site suitable for a late transition metal, and another N–N bidentate site amenable to binding a broad variety of metals and metalloids. This arrangement provides a way to influence the structure and reactivity of the pincer ligated transition metal center through variation of the other element in the N–N cleft, but without changing the immediate coordination environment about the transition metal. We envisaged an N–N site similar to β-diketiminates (also known as “nacnac” ligands)35 that is integrated into a redox-active pincer framework. In considering this design, we were influenced by the binucleating “nindigo” ligands (E, Fig. 1) pioneered by the Hicks group,36 and also explored by Caulton and Mindiola,37 and by Kaim and Lahiri with co-workers.38 The “nindigo” ligands are essentially a redox non-innocent Janus bis-“nacnac” system. Herein we report on the chemistry of a binucleating ligand fusing a PCP pincer with a “nacnac”-like N–N site. A similar binucleating ligand combining an NCN pincer with a bidentate N–N site (F, Fig. 1) was recently explored by Ong, Frenking, Zhao, and coworkers.39 In this work, we report our exploration of how the nature of the element in the N–N cleft influences the coordination environment about the transition metal in the pincer cleft, along with the electrochemical and spectroscopic properties of the molecule as a whole. This synthetic solution for the remote modulation of the properties of a transition metal pincer complex may provide new way for exploring and tuning catalytic activity in the future.
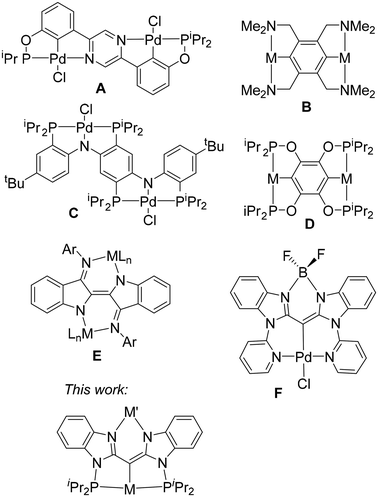 |
| | Fig. 1 Literature examples of Janus binucleating pincer (A–D) and “nacnac” (E) ligands, Ong's compound F, and the target of investigation in this work. | |
Results and discussion
Synthesis of the Janus ligand
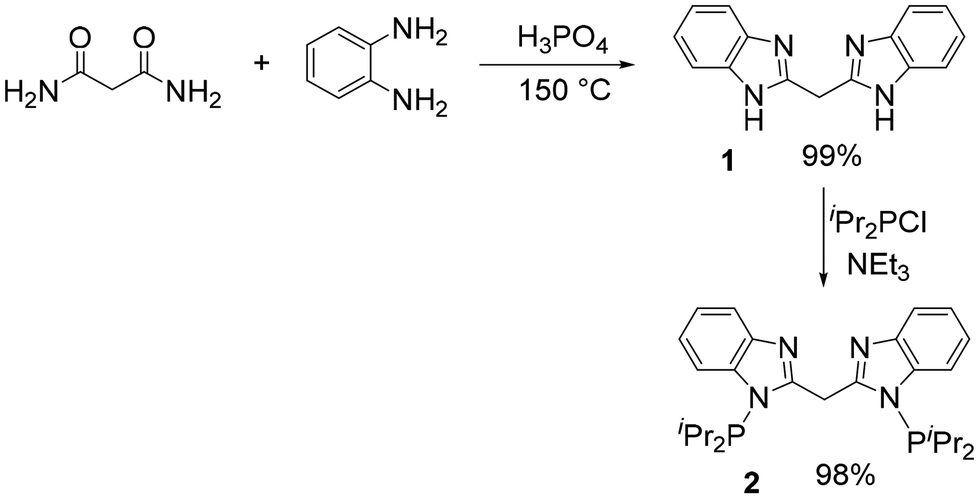
Kamble et al. recently reported a convenient synthesis of benzimidazoles by a thermal or microwave reaction of acid amides with diaminobenzenes in aqueous hydrochloric acid.40 Substituting hydrochloric acid with 85% H3PO4 allowed us to carry out an analogous synthesis of bis(3-benzamidazolyl)methane (1) at a higher temperature without a need for a microwave reactor. We were able to isolate 1 in near-quantitative yield. Treatment of 1 with 2.4 equiv. of ClPPri2 and 2.4 equiv. of Et3N in THF overnight at ambient temperature resulted in the formation of the proto-ligand 2, (Scheme 1) which was isolated in excellent yield upon workup. Compound 2 was characterized by 1H, 13C, and 31P NMR spectroscopy, with a singlet resonance at 68.2 ppm in the 31P{1H} NMR spectrum. The protons of the methylene linkage give rise to a singlet at δ 5.21 ppm in the 1H NMR spectrum and a triplet (JC–P = 17 Hz) at δ 31.8 ppm in the 13C{1H} spectrum. The PCP cleft of compound 2 can be seen as a member of a class of proto-pincers with a central CH2 moiety that connects (hetero)arene rings bearing the two side phosphine arms.41–47 On the other hand, the N–N cleft of 2 is related to a number of monoanionic N–N bidentate ligands.48
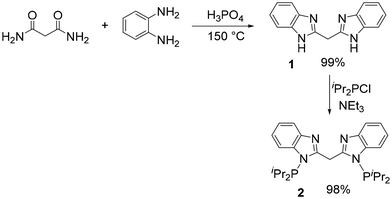 |
| | Scheme 1 Synthesis of binucleating ligand 2. | |
Synthesis and spectroscopic characterization of bi- and trimetallic compounds
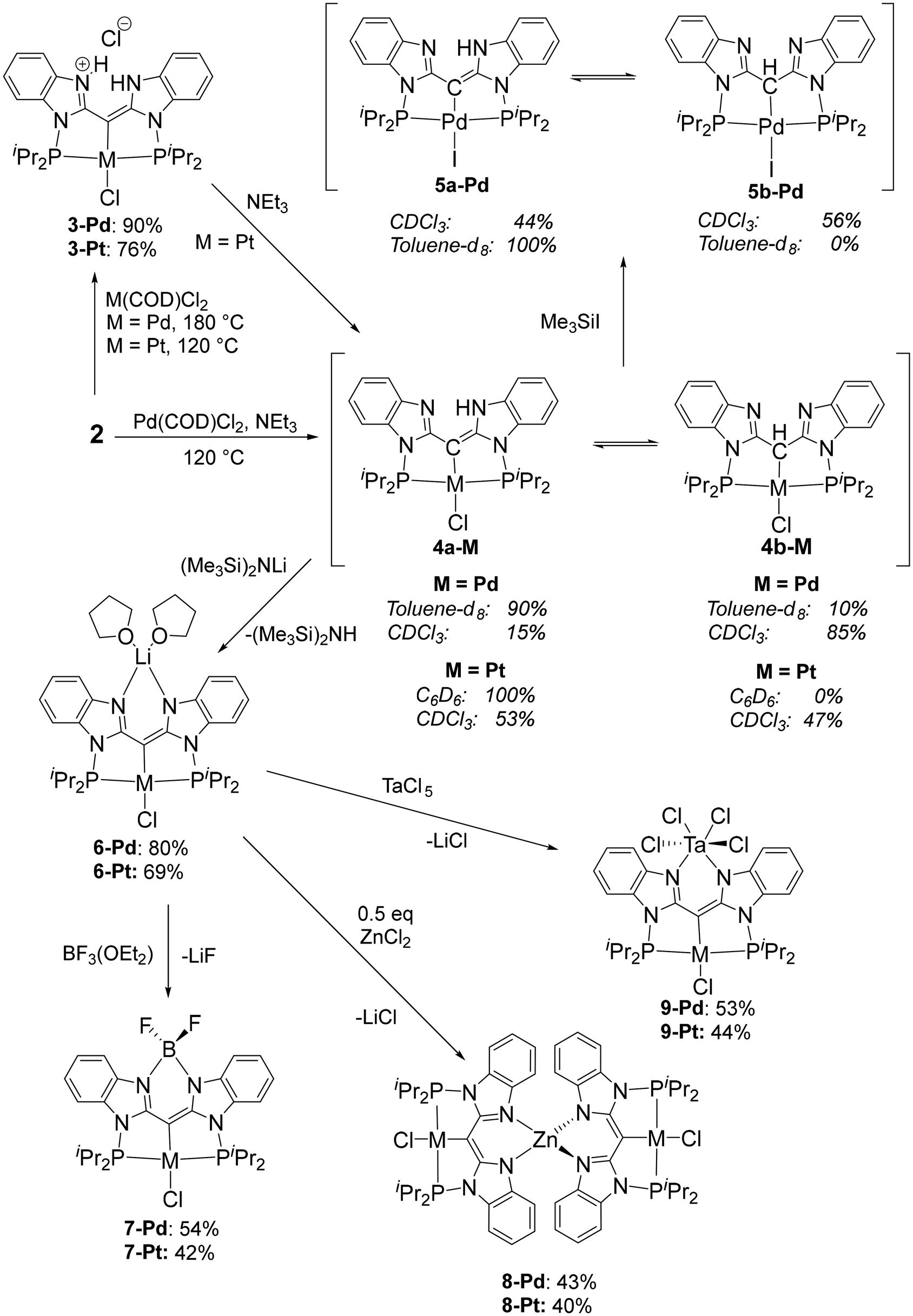
The metalation of the PCP pincer can be carried out with or without the addition of external base, such as Et3N. In the absence of added Et3N, compounds 3-Pd and 3-Pt were isolated from reactions with (COD)MCl2 (M = Pd, Pt). The synthesis of 3-Pd required a rather high temperature (180 °C in o-C6H4Cl2) to ensure high conversion, but the Pt reaction proceeded well at 120 °C. The use of Et3N during the metalation with (COD)PdCl2 resulted in the formation of 4-Pd after 12 h at 120 °C in toluene; however, in the case of Pt, use of NEt3 resulted in decomposition to multiple unidentified products. Compound 4-Pt was accessed by deprotonation of isolated 3-Pt with NEt3 (Scheme 2). Treatment of 4-Pd with Me3SiI in toluene resulted in halogen exchange, allowing for the isolation of 5-Pd in 82% yield after workup.
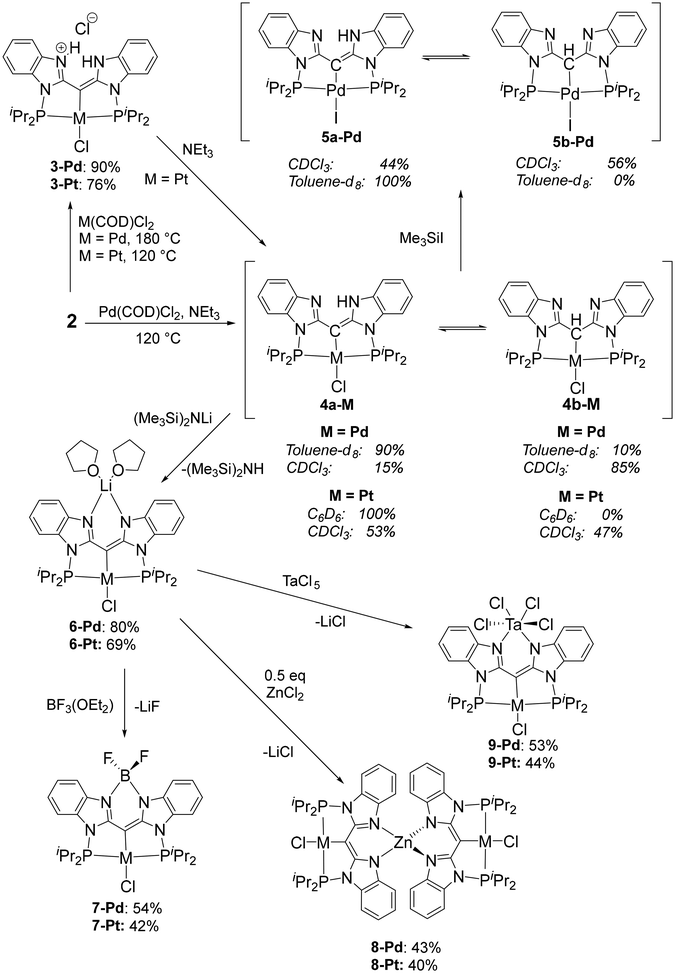 |
| | Scheme 2 Synthesis of mono-metallic complexes at the PCP-pincer cleft with solvent-dependent tautomer ratios and the synthesis of bi- and tri-metallic complexes. | |
Complexes 4-Pd/4-Pt/5-Pd exist as an equilibrium mixture of two tautomers, in which the proton is located either on the nitrogens of the N–N cleft (4a-Pd/4a-Pt/5a-Pd), or on the central carbon (4b-Pd/4b-Pt/5b-Pd). The different locations of H are evident from the 1H and 13C NMR spectra. For example, in 4a-Pd, this N–H proton resonates at δ 10.23 ppm (C6D6 solvent) as a broad singlet, while the C–H of 4b-Pd resonates at δ 5.74 ppm as a sharper singlet. The Pd-bound carbons of the two isomers also have distinct chemical shifts (δ 68.0 ppm, 4a-Pd; δ 48.9 ppm, 4b-Pd, CDCl3 solvent). In 4b-Pt, the C–H resonance (δ 5.51 in CDCl3) displays telltale 195Pt satellites (2JH–Pt = 155 Hz). The two tautomers also possess 31P NMR chemical shifts that are different by ca. 15–20 ppm in each pair, with the more upfield resonance belonging to the C–H tautomers (4b-Pd/4b-Pt/5b-Pd). The observed equilibrium ratios are solvent-dependent, with chloroform favoring the C–H tautomer to a greater degree than benzene or toluene. Compounds 4-Pt and 5-Pd displayed a slightly greater predilection towards the NH tautomer (4a or 5a) than did 4-Pd, in both arene solvents and CDCl3. It should be noted that we saw no evidence for tautomerism in compounds 2 or 3.
Deprotonation of 4a/b-Pd or 4a/b-Pt with (Me3Si)2NLi led to the formation of the Li–Pd/Pt compounds 6-Pd/6-Pt. Both 6-Pd/Pt were isolated in high yields as bis-THF adducts, with presumed THF binding to Li. The N–N cleft of the Janus ligand in 3 can be viewed as accepting both protons of the CH2 group in 2 upon its metalation (and one of the chlorides from PdCl2 or PtCl2). Compounds 4 and 6 can be viewed as products of successive dehydrochlorination and then deprotonation of 3. In other words, 3, 4, and 6 represent three different protonation states with 2, 1, or 0 protons from the original CH2 group retained. The presence of chloride in the isolated 6-Pd and 6-Pt was confirmed by observing formation of one equiv. of Me3SiCl when each was treated with two equiv. of Me3SiI.
The Li compounds 6 served as convenient precursors for the synthesis of bi- and trimetallic compounds 7, 8, and 9 in decent yields via reactions with BF3·OEt2, ZnCl2 (0.5 equiv.), or TaCl5, respectively. Compounds 7–9 show the maximum possible symmetry (C2v for 7 and 9, and for each pincer fragment in 8 locally; D2d for 8 as a whole) in their NMR spectra at ambient temperature. The 1H and 13C NMR resonances for the PCP/NN ligand backbone in these compounds are quite similar (Tables S1 and S2†). Interestingly, the 31P NMR chemical shifts and the 31P–195Pt coupling constant vary considerably in the 3–9 series, Table 1. These differences may be related to the perturbation of the P–Pd–P/P–Pt–P angles and the Pd/Pt–P distances as a function of the size of the moiety in the N–N cleft, as well as the different electronic effects of those moieties. The 11B NMR chemical shifts in 7-Pd and 7-Pt of ca. 3 ppm, the 11B–19F coupling constants of 29–30 Hz, and the 19F NMR chemical shift of ca. −138 ppm are similar to those observed in BODIPY and the related compounds with a BF2 unit in an amido/enamine cleft.39,49–51
Table 1 Selected NMR data for palladium and platinum compounds
| |
δ
13C for C–M, ppm |
δ
31P, ppm |
1
J
Pt–P, Hz |
| Spectra were collected in C6D6 unless otherwise noted. Spectra collected in CDCl3. Spectra collected in C7D8. Spectra collected in CD2Cl2. The Pd/Pt–C resonances were not observed owing to the poor solubility of 9-Pd/Pt and the low achievable concentration of 4b-Pt and 5b-Pd. |
|
3-Pd
|
70.2 |
110.7 |
— |
|
3-Pt
|
59.4 |
102.6 |
3077 |
|
4a-Pd
|
75.2 |
108.6 |
— |
|
4b-Pd
|
45.9 |
93.1 |
|
|
4a-Pt
|
64.0 |
98.9 |
3182 |
|
4b-Pt
|
—d |
90.3 |
3048 |
|
5a-Pd
|
68.0 |
114.6 |
|
|
5b-Pd
|
—d |
97.8 |
|
|
6-Pd
|
76.6 |
99.1 |
— |
|
6-Pt
|
64.9 |
92.9 |
3214 |
|
7-Pd
|
64.7 |
124.4 |
— |
|
7-Pt
|
55.3 |
115.3 |
3268 |
|
8-Pd
|
74.3 |
106.2 |
— |
|
8-Pt
|
62.4 |
99.0 |
3182 |
|
9-Pd
|
—d |
114.4 |
— |
|
9-Pt
|
—d |
106.8 |
3074 |
Compounds 3–8 are yellow, orange, or red, while 9-Pd is purple, and 9-Pt is blue. The properties of 4 and of 7–9 were further studied by UV-Vis spectroscopy. All of these compounds possess strong absorption features in the 300–450 nm range. We surmise that they correspond to the π–π* transitions in the extended organic π-system of the ligand. The UV-Vis spectra of 4 were collected in toluene where 4a-Pd and 4a-Pt are the dominant species in solution that contain an extended organic π-system that is not present in 4b-Pd and 4b-Pt. It appears that a switch from Pd to Pt incurs a bathochromic shift for analogous compounds by about 15–20 nm. For the same metal in the PCP cleft, the order of increasing absorption energy for these features is 8 < 4 < 7 < 9, which can be correlated with the increasing electron-withdrawing power in the series of ½Zn2+ < H+ < BF2+ < TaCl4+. In addition to these π–π* transition features, distinct lower energy broad bands were also observed for 9-Pd (λ = 581 nm) and 9-Pt (λ = 602 nm). These are presumably responsible for the blue or purple colors of these compounds. We tentatively ascribe LMCT character to them, for the transition from the filled π-orbital of the ligand to an empty d-orbital at the d0 Ta center.
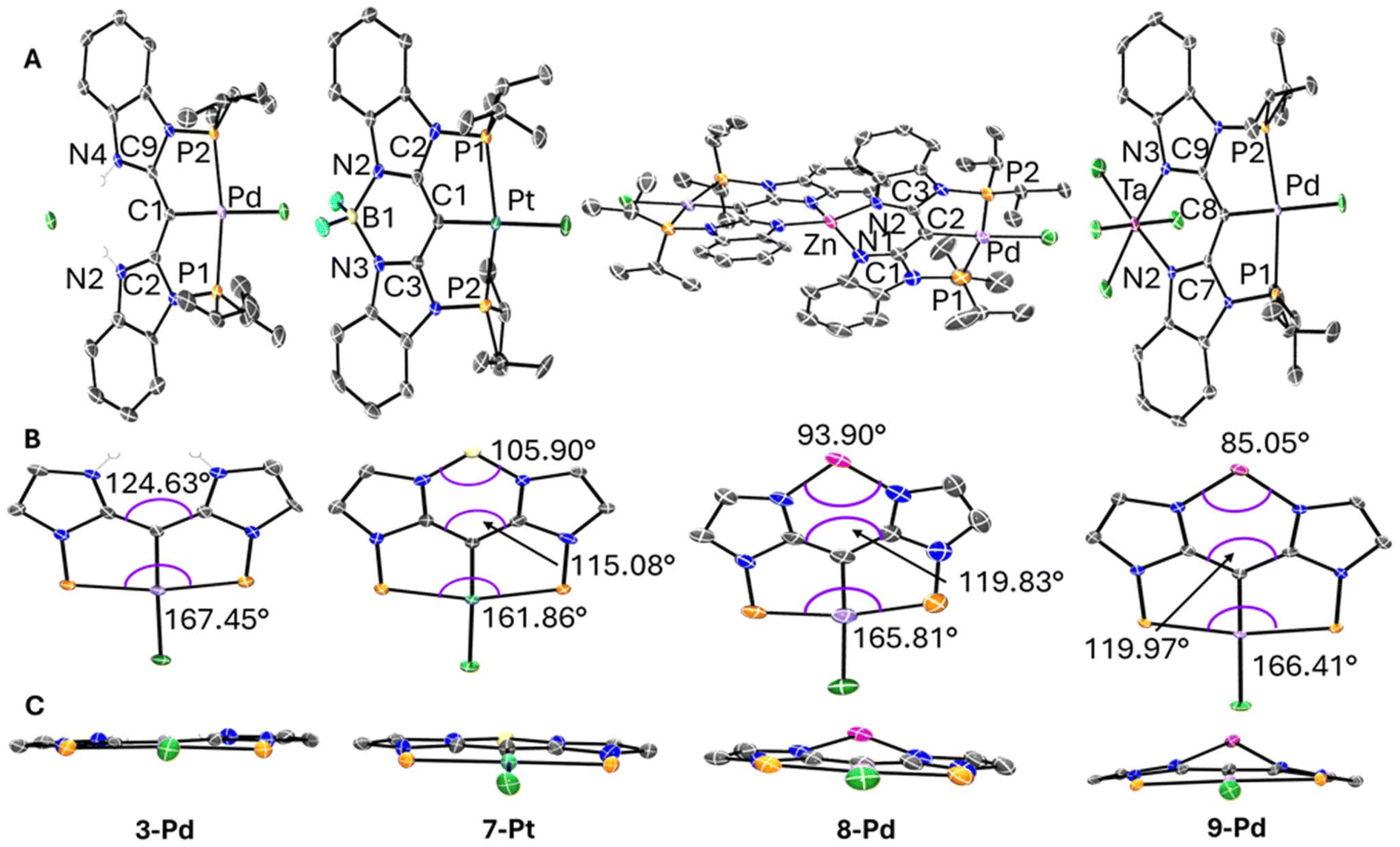
Structural characterization
The structures of 3-Pd, 7-Pt, 8-Pd, and 9-Pd were determined by single-crystal X-ray diffractometry (Fig. 2 and Table 2). In all compounds, the geometry about Pd or Pt is approximately square-planar, and that plane is also approximately the plane of the flat, conjugated organic framework. The H2Cl unit in 3-Pd and the boron atom in 7-Pt are also positioned in that plane. The structure of 3-Pd in the N–N cleft is that of two N–H bonds, with these protons hydrogen-bonded to the chloride. The coordination geometry about boron in 7-Pt closely mimics BODIPY and other related compounds where a monoanionic, bidentate N–N ligand is bound to the BF2 unit.49,51 The Zn atom in 8-Pd is approximately in the plane with one “half” of the ligand framework, but is displaced by ca. 0.65 Å from the other half (shown in Fig. 2C). It is likely a consequence of the need to minimize the considerable crowding brought upon by the two metallaligands binding to the same Zn atom. This also manifests itself in the twisting distortion of one the conjugated “halves”, in that the two “halves” are not strictly perpendicular to each other, and ultimately in the dissymmetry of the distorted tetrahedral environment about Zn. The coordination environment about Ta in 9-Pd is quite close to octahedral, but Ta is displaced from the ligand plane by ca. 0.62 Å. We believe this can be partly attributed to that the hypothetical positioning of Ta “in-plane” would result in the steric clash between the pair of “in-plane” chlorides and the hydrogens ortho to the nitrogens. In a way, this demonstrates that although the N–N cleft is not “bulky” in the usual three-dimensional sense, it can be thought of as “2D-bulky” in its own plane. Thus, there is little steric congestion with a tetrahedral moiety bound in the N–N cleft, but an octahedral moiety must necessarily contend with this “2D-bulkiness”.
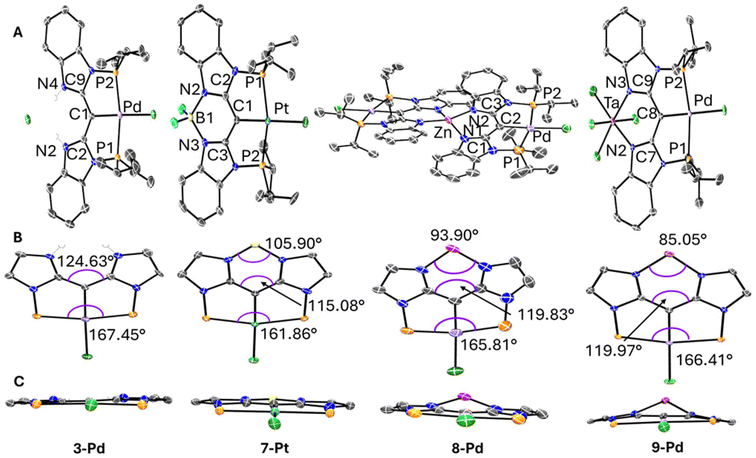 |
| | Fig. 2 (A) POV-Ray renditions of ORTEP plots (50% probability ellipsoids) of 3-Pd, 7-Pt, 8-Pd, and 9-Pd showing selected atom labeling. Hydrogen atoms, solvent molecules, disorder in isopropyl arms are omitted for clarity. An additional molecule of 7-Pt in the asymmetric unit is also omitted for clarity in 7-Pt structure. (B) Highlights of selected approximate bond angles for 3-Pd, 7-Pt, 8-Pd, and 9-Pd. Arenes, isopropyl arms, hydrogen atoms, and disorder are omitted for clarity. (C) Alternate view of each structure in (B) down the M–Cl axis (M = Pd or Pt) to highlight the planarity or lack thereof. | |
Table 2 Selected XRD-derived bond distances in compounds 3-Pd, 7-Pt, 8-Pd, and 9-Pd (M = Pd or Pt, M′ = B, Zn, or Ta) that were studied in solid state by X-ray crystallography
| |
3-Pd
|
7-Pt
|
7-Pt
|
8-Pd
|
8-Pd
|
9-Pd
|
|
Data for both independent molecules of 7-Pt in the unit cell are given.
Data for the two crystallographically different Pd-bound ligand frameworks in 8-Pd are given.
|
| M–P |
2.280(2) |
2.2897(12) |
2.2974(13) |
2.297(3) |
2.296(3) |
2.2862(10) |
| M–P |
2.271(2) |
2.2962(12) |
2.3098(13) |
2.272(3) |
2.299(3) |
2.2917(10) |
| M–C |
2.028(2) |
1.971(4) |
1.964(5) |
2.008(10) |
2.018(10) |
1.991(4) |
| N–M′ |
N/A |
1.561(6) |
1.562(7) |
1.996(10) |
1.963(9) |
2.107(3) |
| N–M′ |
N/A |
1.572(6) |
1.569(7) |
1.984(8) |
2.008(8) |
2.104(3) |
Nonetheless, the series of prepared complexes demonstrate well the ability of the ligand to accommodate a variety of sizes in the N–N cleft of the ligand. The C–C–C bond angle that is highlighted in Fig. 2B serves as a good metric to showcase the flexibility. The angle varies by almost 10° across the series of four structures with H2Cl in 3-Pd giving rise to the largest angle, followed by the similar values engendered by Zn in 8-Pd and TaCl4 in 9-Pd, and finally the BF2 moiety in 7-Pt giving the smallest C–C–C bond angle. The flexing of the N–N cleft portion of the ligand can be seen to affect the geometry on the opposing side of the ligand, in the PCP portion, as illustrated by the changes in the P–M–P angle (Fig. 2B). When the N–N cleft of the ligand has to contract when binding the smaller boron atom in 7-Pt, this causes the phosphines used in the PCP cleft to be pulled back which contracts the P–M–P angle. The binding of the larger Zn (8-Pd) or Ta (9-Pd), or especially the more open N–N cleft in 3-Pd, corresponds to larger P–M–P angles. The changes in the N–N cleft can clearly influence the PCP side without changes in the groups immediately connected to the PCP-bound transition metal. However, the spread of the P–M–P angles is only ca. 5°, and they are all well within the common range52,53 for the [5,5-PXP]54 pincer complexes. The modest degree of PCP perturbation illustrates that the NN/PCP ligand system is flexible enough to adapt to the significant variation in the N–N cleft without making the binding of the transition metal on the PCP side impossible.
Electrochemistry
The electrochemical properties of compounds 4, and 7–9 were studied by means of cyclic voltammetry (CV) experiments (Table 3). A similar picture was observed within each pair of Pd vs. Pt analogs, with two quasi-reversible oxidations per ligand. The Pt compounds generally appeared easier to oxidize by ca. 0.1 V. Previously studied oxidations of conventional (POCOP)MCl or (PCP)MCl complexes were found to be irreversible.55,56 These considerations suggest that the oxidation events in 7–9 are primarily ligand- and not Pd- or Pt-based. DFT calculations performed on the closely related F (Fig. 1) indicated that its HOMO is primarily ligand-based.39 We previously studied the reversible oxidation of Pd and Pt complexes supported by diarylamido-based pincer ligands, which was also ligand-based, and where the Pt analogs were also easier to oxidize by several hundredths of a volt.57,58 The Pd–Pt difference likely reflects the greater π-interaction of the filled 5d-orbitals of Pt with the π-system of the ligand vs. the 4d-orbitals of Pd, driving the energy of the ligand HOMO slightly higher. This notion is also consistent with the slightly lower energy of the LMCT transition in 9-Pt compared to 9-Pd (vide supra). Compounds 4-Pd and 4-Pt appeared to give rise to two quasi-reversible oxidations; however, the recorded CVs had additional unidentified features (see ESI†). They may be owing to the presence of both the 4a and 4b tautomers, or to the facile loss of H+ after oxidation. With the BF2 unit in the N–N cleft, the CV waves were well-defined and quasi-reversible for both 7-Pd and 7-Pt. The trimetallic species 8-Pd and 8-Pt each presented three quasi-reversible waves, one of which was a two-electron wave. We assign the two one-electron waves closer to 0 V as corresponding to the 8+/8 and 82+/8+ events, with the third wave corresponding to the 84+/82+ event. Ostensibly, initial one-electron oxidation of one of the PCP/NN ligands influences the subsequent oxidation of the other PCP/NN ligand. It is interesting that the second oxidation events are not likewise differentiated. It is possible that the second oxidation affects the electron density farther away from the N–N cleft.
Table 3 Reduction potentials (V) of the events observed by cyclic voltammetry for compounds 7–9
| Complex N |
N2+/N+ |
N+/N0 |
N0/N− |
| Cyclic voltammograms were recorded in CH2Cl2 at 1 mM of analyte and referenced to the Fc+/Fc redox couple. Unless otherwise noted CVs were run with 0.1 M of [Bu4N][PF6] as the supporting electrolyte. Corresponds to a two-electron event N4+/N2+. Two events corresponding to N2+/N+ and N+/N0 because of the presence of two ligands in the molecules. CVs run with 0.1 M of [Bu4N][BArF24] as the supporting electrolyte. |
|
7-Pd
|
1.18 |
0.25 |
— |
|
7-Pt
|
1.03 |
0.11 |
— |
|
8-Pd
|
0.92a |
0.02b |
— |
| −0.22b |
|
8-Pt
|
0.82a |
−0.09b |
— |
| −0.32b |
|
9-Pd
|
1.18 |
0.57 |
−0.87 |
|
9-Pt
|
1.07 |
0.47 |
−0.88 |
In the case of the Ta complexes 9-Pd and 9-Pt, while the first oxidation is well-behaved, the second oxidation appeared only partially reversible. It is possible that the higher potential needed for the second oxidation of 9 leads to a less stable dicationic species than in the case of 7 (or the tetracationic in the case of 8). In addition, both 9-Pd and 9-Pt also displayed a quasi-reversible wave corresponding to a 9/9− event at −0.87 and −0.88 V. We tentatively ascribe it to the reduction of the TaV center to TaIV. These values for 9 fall in between the reduction potentials for the [TaCl6]−/[TaCl6]2− (−1.11 V) and the [TaCl5(NCMe)]0/[TaCl5(NCMe)]− (−0.65 V) couples, as reported by Bursten et al.59
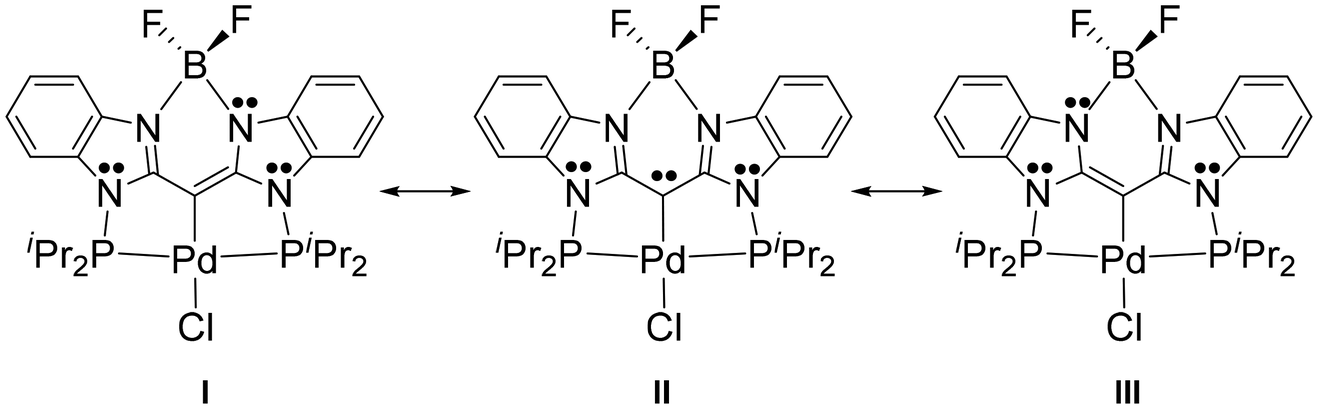
Two oxidations per ligand in 7–9 can be understood by using simple resonance structures, a few of which are shown in Fig. 3 for 7-Pd. The lone pair localized on the central carbon in resonance structure II is delocalized, primarily in the “nacnac” fashion (I and III), but also beyond, into the six-membered aromatic rings (not shown). The two successive oxidations can be thought to convert this central lone pair first into a radical, and then into an empty orbital site, with extensive delocalization. The importance of the resonances structure similar to II in compound F (Fig. 1) led Ong, Frenking, Zhao, and coworkers to point out the similarity of the central carbon-metal interaction in F to the transition metal complexes of “carbodicarbenes”.39 That similarity also exists for 7–9. The importance of the resonance structure II is also related to the PCP pincer complexes featuring a central diarylcarbene, explored by the Piers43–45,60 and Iluc46 groups.
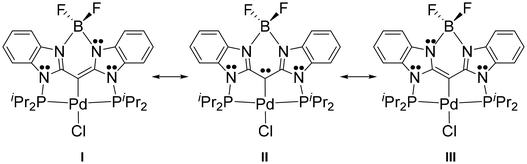 |
| | Fig. 3 Main resonance structures for 7-Pd. | |
Conclusion
In summary, a new binucleating ligand system has been presented. It combines a monoanionic PCP pincer on one side with a monoanionic N–N bidentate cleft on the opposite side. The PCP pincer cleft was accommodated with square-planar, d8 Pd or Pt centers, while the N–N cleft has proven adaptable to binding such divergent set of elements as boron, zinc, or tantalum. Two N–N clefts can bind a single Zn center, resulting in trimetallic (ZnPd2 or ZnPt2) complexes. Structural studies showed that the ligand stays nearly flat in the coordination plane of Pd or Pt, indicating a high degree of conjugation throughout. When bound to metals (or metalloids) via both the N–N and the PCP clefts, the ligand system can undergo two quasi-reversible oxidations, ascribed primarily to the oxidation of the extended organic π-system.
Data availability
Details of experimental procedures, graphical NMR spectra, and details of X-ray structural determination, CV experiments, and graphic UV-Vis spectra have been included as ESI.† CCDC 2351193–2351196† contain crystallographic data for 3-Pd, 7-Pt, 8-Pd, and 9-Pd.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are thankful for the support of this work by the Welch Foundation (grant A-1717 to O. V. O.). We thank Ms Ruth Ann Gholson for assistance with manuscript preparation. We are also grateful to Prof. Michael Nippe and Zhen Ni for helpful discussions.
References
-
Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Heidelberg, 2013 Search PubMed.
- E. Peris and R. H. Crabtree, Key factors in pincer ligand design, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- M. Vogt and R. Langer, The Pincer Platform Beyond Classical Coordination Patterns, Eur. J. Inorg. Chem., 2020, 3885–3898 CrossRef CAS.
- A. Kumar, T. Bhatti and A. S. Goldman, Dehydrogenation of Alkanes and Aliphatic Groups by Pincer-Ligated Metal Complexes, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Catalytic Alkane Metathesis by Tandem Alkane Dehydrogenation-Olefin Metathesis, Science, 2006, 312, 257–261 CrossRef CAS PubMed.
- K. K. Manar, J. Chemg, Y. Yang, X. Yang and P. Ren, Promising Catalytic Application by Pincer Metal Complexes: Recent Advances in Hydrogenation of Carbon-Based Molecules, ChemCatChem, 2023, 15, e202300004 CrossRef CAS.
- A. Kumar, P. Daw and D. Milstein, Homogeneous Catalysis for Sustainable Energy: Hydrogen and Methanol Economies, Fuels from Biomass, and Related Topics, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Bond Activation and Catalysis by Ruthenium Pincer Complexes, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- V. Arora, H. Narjinari, P. G. Nandi and A. Kumar, Recent advances in pincer–nickel catalyzed reactions, Dalton Trans., 2021, 50, 3394–3428 RSC.
- S. D. Timpa, C. J. Pell and O. V. Ozerov, A Well-Defined (POCOP)Rh Catalyst for the Coupling of Aryl Halides with Thiols, J. Am. Chem. Soc., 2014, 136, 14772–14779 CrossRef CAS PubMed.
- L. P. Press, A. J. Kosanovich, B. J. McCulloch and O. V. Ozerov, High-Turnover Aromatic C-H Borylation Catalyzed by POCOP-type Pincer Complexes of Iridium, J. Am. Chem. Soc., 2016, 138, 9487–9497 CrossRef CAS PubMed.
- B. J. Foley, N. S. Bhuvanesh, J. Zhou and O. V. Ozerov, Combined Experimental and Computational Studies of the Mechanism of Dehydrogenative Borylation of Terminal Alkynes (DHBTA) Catalyzed by PNP Complexes of Iridium, ACS Catal., 2020, 10, 9824–9836 CrossRef CAS.
- N. Selander and K. J. Szabó, Catalysis by Palladium Pincer Complexes, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- D. Bézier and M. Brookhart, Applications of PC(sp3)P Iridium Complexes in Transfer Dehydrogenation of Alkanes, ACS Catal., 2014, 4, 3411–3420 CrossRef.
- V. Lyaskovskyy and B. de Bruin, Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- P. J. Chirik, Preface: Forum on Redox-Active Ligands, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed.
-
B. de Bruin, P. Gualco and N. D. Paul, Ligand Design in Metal Chemistry, John Wiley & Sons, Ltd, Hoboken, NJ, 2016, pp. 176–204 Search PubMed.
- A. I. Nguyen, R. A. Zarkesh, D. C. Lacy, M. K. Thorson and A. F. Heyduk, Catalytic nitrene transfer by a zirconium(IV) redox-active ligand complex, Chem. Sci., 2011, 2, 166–169 RSC.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Comparison of the Electronic Properties of Diarylamido-Based PNZ Pincer Ligands: Redox Activity at the Ligand and Donor Ability Toward the Metal, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS PubMed.
- D. M. Beagan, N. A. Maciulis, M. Pink, V. Carta, I. J. Huerfano, C.-H. Chen and K. G. Caulton, A Redox-Active Tetrazine-Based Pincer Ligand for the Reduction of N-Oxyanions Using a Redox-Inert Metal, Chem. – Eur. J., 2021, 27, 11676–11681 CrossRef CAS PubMed.
- S. Acosta-Calle and A. J. M. Miller, Tunable and Switchable Catalysis Enabled by Cation-Controlled Gating with Crown Ether Ligands, Acc. Chem. Res., 2023, 56(8), 971–998 CrossRef CAS PubMed.
- H. Valdés, J. M. Germán-Acacio, G. van Koten and D. Morales-Morales, Bimetallic complexes that merge metallocene and pincer-metal building blocks: synthesis, stereochemistry and catalytic reactivity, Dalton Trans., 2022, 51, 1724–1744 RSC.
- C.-H. Yu, C. Zhu, X. Ji, W. Hu, H. Xie, N. Bhuvanesh, L. Fang and O. V. Ozerov, Palladium bis-pincer complexes with controlled rigidity and inter-metal distance, Inorg. Chem. Front., 2020, 7, 4357–4366 RSC.
- D. W. Leong, Y. Shao, N. Zhen, N. Bhuvanesh and O. V. Ozerov, A bis(PCN) palladium pincer complex with a remarkably planar 2,5-diarylpyrazine core, Dalton Trans., 2024, 53, 6520–6523 RSC.
- P. Steenwinkel, H. Kooijman, W. J. J. Smeets, A. L. Spek, D. M. Grove and G. van Koten, Intramolecularly Stabilized 1,4-Phenylene-Bridged Homo- and Heterodinuclear Palladium and Platinum Organometallic Complexes Containing N,C,N-Coordination Motifs; η1-SO2 Coordination and Formation of an Organometallic Arenium Ion Complex with Two Pt-C σ-Bonds, Organometallics, 1998, 17, 5411–5426 CrossRef CAS.
- S. L. Jeon, D. M. Loveless, W. C. Yount and S. L. Craig, Thermodynamics of Pyridine Coordination in 1,4-Phenylene Bridged Bimetallic (Pd, Pt) Complexes Containing Two N,C,N′ Motifs, 1,4-M2-[C6(CH2NR2)4-2,3,5,6], Inorg. Chem., 2006, 45, 11060–11068 CrossRef CAS PubMed.
- S. J. Loeb and G. K. H. Shimizu, Dimetallated thioether complexes as building blocks for organometallic coordination polymers and aggregates, J. Chem. Soc., Chem. Commun., 1993, 1395–1397 RSC.
- S. J. Loeb, G. K. H. Shimizu and J. A. Wisner, Mono- versus Dipalladation of the Durene-Based Tetrathioether Ligand 1,2,4,5-(tBuSCH2)4C6H2. Structures of [PdCl((tBuSCH2)4C6H)] and [Pd2((tBuSCH2)4C6)(MeCN)2][BF4]2, Organometallics, 1998, 17, 2324–2327 CrossRef CAS.
- D. Das, P. Singh, M. Singh and A. K. Singh, Tetradentate selenium ligand as a building block for homodinuclear complexes of Pd(II) and Ru(II) having seven membered rings or bis-pincer coordination mode: high catalytic activity of Pd-complexes for Heck reaction, Dalton Trans., 2010, 39, 10876–10882 RSC.
- N. P. N. Wellala, H. T. Dong, J. A. Krause and H. Guan, Janus POCOP Pincer Complexes of Nickel, Organometallics, 2018, 37, 4031–4039 CrossRef CAS.
- D. Wang, S. V. Lindeman and A. T. Fiedler, Bimetallic Complexes Supported by a Redox-Active Ligand with Fused Pincer-Type Coordination Sites, Inorg. Chem., 2015, 54, 8744–8754 CrossRef CAS PubMed.
- J. I. van der Vlugt, S. Demeshko, S. Dechert and F. Meyer, Tetranuclear CoII, MnII, and CuII Complexes of a Novel Binucleating Pyrazolate Ligand Preorganized for the Self-Assembly of Compact [2 × 2]-Grid Structures, Inorg. Chem., 2008, 47, 1576–1585 CrossRef CAS PubMed.
- M. Li, S. K. Gupta, S. Dechert, S. Demeshko and F. Meyer, Merging Pincer Motifs and Potential Metal–Metal Cooperativity in Cobalt Dinitrogen Chemistry: Efficient Catalytic Silylation of N2 to N(SiMe3)3, Angew. Chem., Int. Ed., 2021, 60, 14480–14487 CrossRef CAS PubMed.
- C.-W. Chan, D. M. P. Mingos, A. J. P. White and D. J. Williams, Multi-domain hydrogen-bond forming metal chelates: X-ray crystal structures of dicyclopalladated 2,3-bis[6-(2-amino-4-phenylamino-1,3,5-triazinyl)]pyrazine (H2L)[Pd2Br2L] and 2,6-bis[6-(2-amino-4-phenylamino-1,3,5-triazinylium)] pyridine dichloride, Chem. Commun., 1996, 81–86 RSC.
-
(a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, The Chemistry of β-Diketiminatometal Complexes, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed;
(b) P. L. Holland, Electronic Structure and Reactivity of Three-Coordinate Iron Complexes, Acc. Chem. Res., 2008, 41, 905–914 CrossRef CAS PubMed;
(c) D. J. Mindiola, Oxidatively Induced Abstraction Reactions. A Synthetic Approach to Low-Coordinate and Reactive Early Transition Metal Complexes Containing Metal−Ligand Multiple Bonds, Acc. Chem. Res., 2006, 39, 813–821 CrossRef CAS PubMed;
(d) D. J. Mindiola, Nacnac … Are You Still There? The Evolution of β-Diketiminate Complexes of Nickel, Angew. Chem., Int. Ed., 2009, 48, 6198–6200 CrossRef CAS PubMed;
(e) Y.-C. Tsai, The chemistry of univalent metal β-diketiminates, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS;
(f) R. L. Webster, β-Diketiminate complexes of the first row transition metals: applications in catalysis, Dalton Trans., 2017, 4483–4498 RSC.
- S. R. Oakley, G. Nawn, K. M. Waldie, T. D. MacInnis, B. O. Patrick and R. G. Hicks, “Nindigo”: synthesis, coordination chemistry, and properties of indigo diimines as a new class of functional bridging ligands, Chem. Commun., 2010, 46, 6753–6755 RSC.
- S. Fortier, O. González-del Moral, C. H. Chen, M. Pink, J. J. LeRoy, M. Murugesu, D. J. Mindiola and K. G. Caulton, Probing the redox non-innocence of dinuclear, three-coordinate Co(II) nindigo complexes: not simply β-diketiminate variants, Chem. Commun., 2012, 48, 11082–11084 RSC.
- P. Mondal, F. Ehret, M. Bubrin, A. Das, S. M. Mobin, W. Kaim and G. K. Lahiri, A Diruthenium Complex of a “Nindigo” Ligand, Inorg. Chem., 2013, 52, 8467–8475 CrossRef CAS PubMed.
- C.-H. Yu, K. C. Au-Yeung, R. Liu, C.-H. Lee, D. Jiang, B. S. Aweke, C.-H. Wu, Y.-J. Wang, T.-H. Wang, K. V. Kong, G. P. A. Tap, W.-C. Chen, G. Frenking, L. Zhao and T.-G. Ong, Diversification of the Carbodicarbene Class by Embedding an Anionic Component in its Scaffold, Chem. – Eur. J., 2023, 29, e202302886 CrossRef CAS PubMed.
- P. P. Kattimani, R. R. Kamble and G. Y. Meti, Expedient synthesis of benzimidazoles using amides, RSC Adv., 2015, 5, 29447–29455 RSC.
- W. Weng, S. Parkin and O. V. Ozerov, Double C–H Activation Results in Ruthenium Complexes of a Neutral PCP Ligand with a Central Carbene Moiety, Organometallics, 2006, 25, 5345–5354 CrossRef CAS.
- W. Weng, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Palladium Complexes of a P2C= Ligand Containing a Central Carbene Moiety, Organometallics, 2007, 26, 3315–3320 CrossRef CAS.
- M. L. Clapson, J. K. Kirkland, W. E. Piers, D. H. Ess, B. Gelfand and J.-B. Lin, Carbene Character in a Series of Neutral PCcarbeneP Cobalt(I) Complexes: Radical Carbenes versus Nucleophilic Carbenes, Organometallics, 2022, 41, 235–245 CrossRef CAS.
- J. D. Smith, G. Durrant, D. H. Ess, B. S. Gelfand and W. E. Piers, Carbene Character in a Series of Neutral PCcarbeneP Cobalt(I) Complexes: Radical Carbenes versus Nucleophilic Carbenes, Chem. Sci., 2020, 11, 10705–10717 RSC.
- E. A. LaPierre, W. E. Piers and C. Gendy, Redox-state dependent activation of silanes and ammonia with reverse polarity (PCcarbeneP)Ni complexes: electrophilic vs. nucleophilic carbenes, Dalton Trans., 2018, 47, 16789–16797 RSC.
- P. Cui and V. M. Iluc, Redox-induced umpolung of transition metal carbenes, Chem. Sci., 2015, 6, 7343–7354 RSC.
- M. R. Hoffbauer, C. C. Comanescu, B. J. Dymm and V. M. Iluc, C–H Activation Reactions of a Nucleophilic Palladium Carbene, Organometallics, 2018, 37, 2086–2094 CrossRef CAS.
- J. Kretsch, I. Koehne, M. Lõkov, I. Leito and D. Stalke, Bis(benzoxazol-2-yl)methanes Hounding NacNac: Varieties and Applications in Main Group Metal Coordination, Eur. J. Inorg. Chem., 2019, 3256–3351 Search PubMed.
- K. Perumal, J. A. Garg, O. Blacque, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, β-Iminoenamine-BF2 Complexes: Aggregation-Induced
Emission and Pronounced Effects of Aliphatic Rings on Radiationless Deactivation, Chem. – Asian J., 2012, 7, 2670–2677 CrossRef CAS PubMed.
- B. R. Grove, S. M. Crawford, T. Lundrigan, C. F. Matta, S. Sowlati-Hashijin and A. Thompson, Synthesis and characterisation of the unsubstituted dipyrrin and 4,4-dichloro-4-bora-3a,4a-diaza-s-indacene: improved synthesis and functionalisation of the simplest BODIPY framework, Chem. Commun., 2013, 49, 816–818 RSC.
- S. Madhu, M. R. Rao, M. S. Shaikh and M. Ravikanth, 3,5-Diformylboron Dipyrromethenes as Fluorescent pH Sensors, Inorg. Chem., 2011, 50, 4392–4400 CrossRef CAS PubMed.
- J.-F. Gong, Y.-H. Zhang, M.-P. Song and C. Xu, New PCN and PCP Pincer Palladium(II) Complexes: Convenient Synthesis via Facile One-Pot Phosphorylation/Palladation Reaction and Structural Characterization, Organometallics, 2007, 26, 6487–6492 CrossRef CAS.
- L. González-Sebastián and D. Morales-Morales, Cross-coupling reactions catalysed by palladium pincer complexes. A review of recent advances, J. Organomet. Chem., 2019, 893, 39–51 CrossRef.
- M. D. Fryzuk and P. A. MacNeil, Hybrid multidentate ligands. Tridentate amidophosphine complexes of nickel(II) and palladium(II), J. Am. Chem. Soc., 1981, 103, 3592–3593 CrossRef CAS.
- N. Á. Espinosa-Jalapa, S. Hernández-Ortega, D. Morales-Morals and R. Le Lagadec, Facile synthesis of heterobimetallic compounds from the cyclopentadienyl-ruthenium moiety and group 10 POCOP pincer complexes, J. Organomet. Chem., 2012, 716, 103–109 CrossRef CAS.
- S. Bonnet, M. Lutzs, A. L. Spek, G. van Koten and R. J. M. Klien Gebbink, Bimetallic η6,η1 SCS- and PCP-Pincer Ruthenium Palladium Complexes: Synthesis, Structure, and Catalytic Activity, Organometallics, 2010, 29, 1157–1167 CrossRef CAS.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Comparison of the Electronic Properties of Diarylamido-Based PNZ Pincer Ligands: Redox Activity at the Ligand and Donor Ability Towards the Metal, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS PubMed.
- C.-H. Yu, X. Yang, X. Ji, C.-H. Wang, Q. Lai, N. Bhuvanesh and O. V. Ozerov, Redox Communication between Two Diarylamido/Bis(phosphine) (PNP)M Moieties Bridged by Ynediyl Linkers (M = Ni, Pd, Pt), Inorg. Chem., 2020, 59, 10153–10162 CrossRef CAS PubMed.
- B. E. Bursten, M. R. Green, V. Katovic, J. R. Kirk and D. Lightner, Jr., Electrochemistry of niobium(IV) and tantalum(IV) complexes: ligand additivity in d1 octahedral complexes, Inorg. Chem., 1986, 25, 831–834 CrossRef CAS.
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, Activation of Water, Ammonia, and Other Small Molecules by PCcarbeneP Nickel Pincer Complexes, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*