
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoswitchable catalysis to inhibit and promote plastic flow in vitrimers†
Filip
Van Lijsebetten
,
Stephan
Maes
 ,
Johan M.
Winne
* and
Filip E.
Du Prez
*
,
Johan M.
Winne
* and
Filip E.
Du Prez
*
Polymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC) and Laboratory of Organic Synthesis, Departement of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281-S4, Ghent, 9000, Belgium. E-mail: Filip.DuPrez@UGent.be; Johan.Winne@UGent.be
First published on 26th March 2024
Abstract
Acid-base catalysis is a common strategy to induce covalent bond exchanges in dynamic polymer networks. Strong acids or strong bases can promote rapid network rearrangements, and are simultaneously preferred catalysts for chemical reactions where maximum efficiency at the lowest possible temperature is aimed for. However, within the context of dynamic polymer networks, the incorporation of highly active catalysts can negatively affect the longer term application potential. Network dynamicity can diminish through catalyst ageing or quenching and highly active catalysts may prematurely activate bond exchanges, leading to dimensional instability and thus low creep resistance of the polymer networks. Herein, we present several examples where we explicitly explored weak acids (carboxylic acids) as catalysts for dynamic bond exchanges, using vinylogous urethanes (VU) as a well-understood protic acid catalysed vitrimer chemistry. Surprisingly, we have found that the sought-after long-term stability offered by a weak acid does not necessarily bring lower activity at high temperature. In fact, the weak acids show a remarkable thermoswitchable catalytic behaviour, going from an inactive hydrogen bonded state to an active state where the polymer matrix is protonated, with a profound impact on the network reactivity and rheology. Carboxylic acids with different electronic or steric environments show clear reactivity trends and their fine-tuning resulted in the most thermally responsive VU vitrimers studied to date. Our findings point out that catalyst choice and design for vitrimers is only poorly informed by catalyst performance in more traditional chemical reactions (in solvent), and that a more tailored catalyst design holds great promise for the field of vitrimers.
Introduction
Hydrogen bonding and proton transfers are vital processes found in chemical and biological systems.1–5 As a result, these related phenomena have received extensive experimental and theoretical recognition. For instance, the acidity of a molecule is a widely used parameter for predicting chemical and physical properties, designing experiments, and understanding reaction mechanisms.6,7 These structure–reactivity relationships have long been recognised, and they frequently form an important part of the undergraduate curriculum, serving as the foundation for modern organic and polymer chemistry. The seminal work of Hammett and Taft, for example, has aided in identifying and predicting the effect of substituents on the acidity of various compounds in different media.4,8–11 Consequently, because many chemical reactions can be explained using simple acid-base chemistry,12 changes in acidity/basicity play a significant role in optimising reactivity and selectivity.13,14More recently, in large part fueled by the impressive progress made in supramolecular and dynamic covalent chemistry,15–22 the scientific community has introduced vitrimers, an intriguing new material platform for studying and directing chemical reactions.23 Specifically, despite remaining a cross-linked polymer network, vitrimers can flow and change structure via thermally activated bond exchange reactions.24–26 During this process, two polymer chains first associate, then form a new network bond and subsequently fragment an existing network bond.15 This allows distinct network segments to diffuse, without losing network connectivity, making vitrimers particularly appealing recyclable alternatives for thermosetting materials. In particular, vitrimers possess the potential to combine dimensional stability and solvent resistance of thermosets with the thermal processing and recycling abilities found in thermoplastics.
Since the introduction of vitrimers, simple acid-base catalysis was shown to be an important design aspect in ensuring that the conditions and timescale of exchange are compatible with the intended use of the polymer material.23,27–35 Therefore, as recently highlighted by Kalow and co-workers,36 a fundamental understanding of acidity/basicity changes in cross-linkers,37–44 additives,45,46 and catalysts47–49 allows one to design vitrimers with optimised material properties.
Brønsted acid catalysis has received particular attention in the field of vitrimers, for example for the design and synthesis of polyester based dynamic networks, with proton transfer reactions between different basic groups being an integral part of network rearrangement pathways.28 Any acid-base equilibrium critically depends on the relative stability of the competing proton acceptors and their protonated forms, as well as on the external environment (solvent or macromolecular backbone). As such, a given proton transfer reaction of a (neutral) acid catalyst HA to a reactive bond can range from full ionisation (almost complete formation of the A(−) conjugate base, or K ≫ 1) to almost no ionisation at all (K ≪ 1) or any intermediate level of ionisation. Weaker acids that mostly remain in their non-ionized state (HA) can still be active as hydrogen bond donating catalysts.50–54 The exact nature of protic catalysis can thus be complicated, and not be fully captured by considering known pKa-values.
In principle, the extent of a given proton transfer (K) can be estimated from the difference in pKa values of the equilibrating protic species in a relevant solvent system.6,7 However, for many situations this approach will only work as a crude approximation, as experimental pKa values – if available at all – are obtained in pure dilute solvents (such as water, DMSO or acetonitrile), in which ion stability is dominated by solvent–solute interactions, while other interactions (e.g. ion–ion) are not considered. Transferring these observations to other more complex systems should only be done with much care. Exact solvent (or matrix) composition, temperature and concentration, can all shift proton exchange equilibria. For instance, it is well appreciated that the formation of ions is significantly less favourable in apolar organic matrices than in aqueous media, and that most ‘strong’ acids do not even fully dissociate in organic solvents.7,55 For example, hydrochloric acid is actually a very weak acid in acetonitrile (pKa = 10.3) and a rather weak acid in DMSO (pKa = 2.0). Also, upon changing solvents, an acid-base equilibrium can undergo a shift. Such context-dependent shifting of acid-base equilibria and pKa-shifts have been extensively studied inside protein binding sites or catalytically active sites.53,56–59 On the other hand, the general impact of such switching proton transfer equilibria has so far not been studied in the context of Brønsted catalysed vitrimers or other covalent adaptable networks (CANs), whereas the impact can be expected to be important in the material design and performance.
The proton transfer between carboxylic acids and basic amines is a good example of solvent- or context-dependent acid-base equilibrium, and has been extensively studied to determine the extent of proton transfer in various media.5,7,55,60 In neutral aqueous media, full proton transfer is observed between most carboxylic acids and most amines (ΔpKa = 5). However, the relative order of pKa values of carboxylic acids and protonated aliphatic amines has been shown to switch order, even in fairly polar solvents such as DMSO, resulting in an equilibrium that favours the non-ionised forms.55,61 As these equilibria usually have a non-zero reaction enthalpy, and ionisation typically carries an entropic cost or penalty, i.e. solvent dipole ordering around ions, the general expectation is that proton transfer equilibria can be shifted to one side as a function of temperature. Yet, an open research question so far is if such thermally responsive proton transfers could be used to control the kinetics of bond exchange in vitrimers (Fig. 1a).
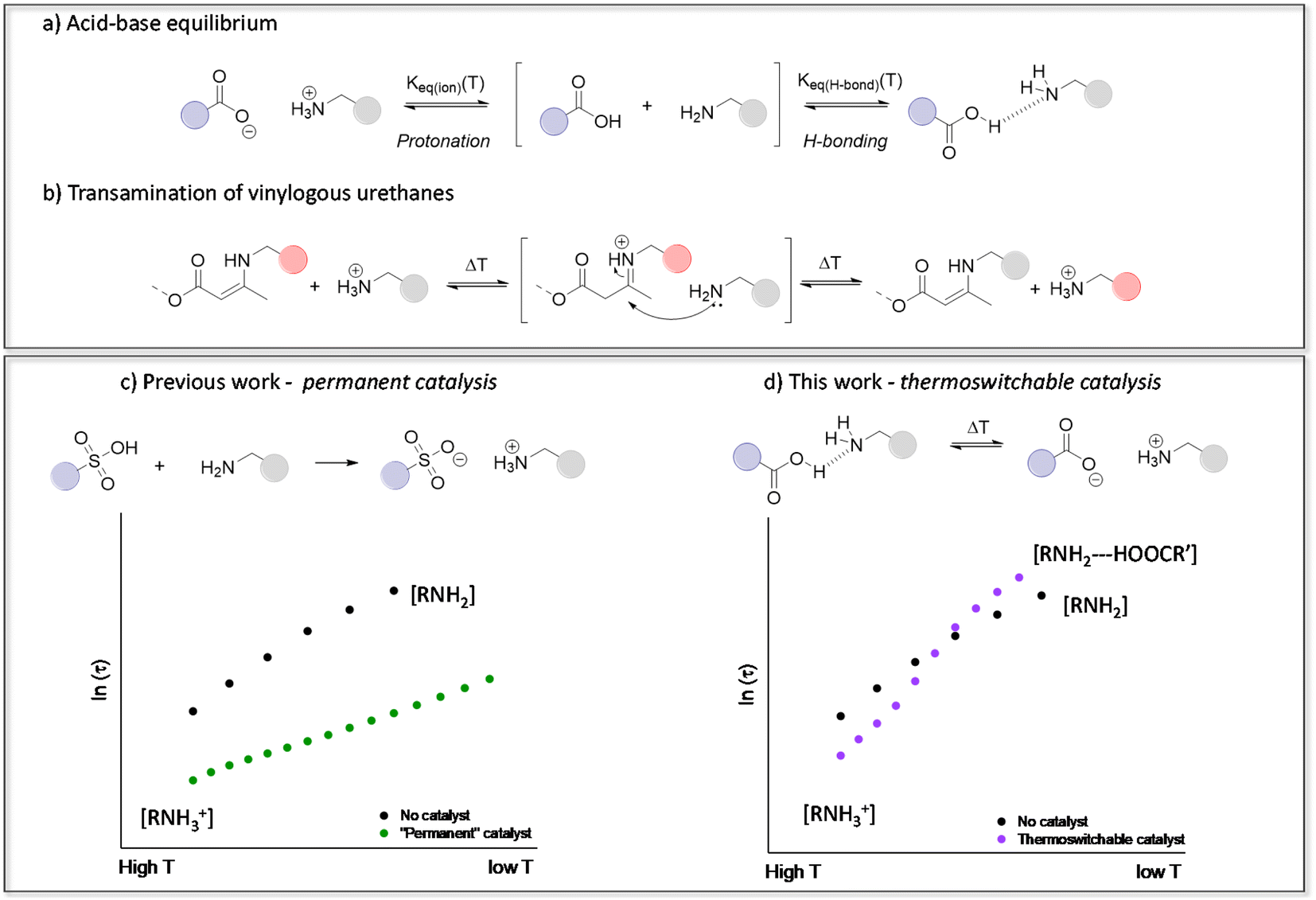 | ||
| Fig. 1 (a) Carboxylic acid–amine equilibrium resulting in either ionisation (protonation) or hydrogen bonding. (b) Transamination of vinylogous urethane vitrimers following a protic iminium-pathway. (c) Our previous work showing that transamination can be significantly accelerated in the presence of strong Brønsted acids, but without any thermal control.46 (d) The thermoswitchable catalysis platform devised in this work to accelerate or inhibit transamination. | ||
Since our introduction of vinylogous urethane chemistry as one of the first associative chemistry platforms for the design of vitrimers, it received a great deal of attention. We have previously demonstrated that the presence of protonated ammonium species actually greatly influences the reaction rate of a benchmark vitrimer system, based on the transamination of vinylogous urethane (VU) cross-links (Fig. 1b and c).46,62–64 Hence, VU-based vitrimers represent an ideal ‘case study’ for assessing the role of proton transfer equilibria in dynamic polymer networks. On the other hand, because proton transfer equilibria are at the root of many vitrimer systems, such detailed studies provide useful insights that can be extrapolated to different types of vitrimeric materials, such as polyimine-based networks.39,65,66 It should be noted that the nitrogen lone pair of the VU moiety is involved in a conjugated system and is thus deactivated as a base. Therefore, the “iminium” intermediate in acid catalysed VUs is obtained after proton transfer onto the alkene moiety of the enamine.
By taking into account the context-dependent shifting of acid-base equilibria, we herein present the first example of a tailor-made vitrimer with a temperature-controlled (or “thermoswitchable”) Brønsted acid catalysis. The materials are conceptually simple, consisting of VU-based vitrimers that incorporate reactive free amines as well as an acid catalyst. Fine-tuning of the catalyst revealed unusual dynamic behaviour, which can be related to subtle pKa changes, and significantly increase the thermal responsivity of the vitrimer network (Fig. 1d), by the combined effect of thermal catalyst activation at high temperatures (>135 °C) and catalyst deactivation at lower temperatures (<135 °C). As demonstrated herein below, this behaviour results in an unusually high apparent activation energy (Ea,flow) for VU-based vitrimers. This previously overlooked design principle offers a fresh perspective on optimising the use of catalysts in vitrimer materials, which allows to obtain reprocessable materials with genuine commercial potential.
In this paper, we describe how proton transfers inside a polymer matrix can be controlled as a function of temperature, by using different readily available substituted carboxylic acids as weakly acidic catalysts. We will identify distinct structural trends, allowing for the development of structure–reactivity relationships based on well-documented Hammett and Taft parameters. The most performant materials were subsequently studied using creep experiments at lower temperatures to identify the impact of the acid catalyst on the structural integrity of the vitrimer material during use.
Results and discussion
Synthesis of vinylogous urethane vitrimers with protic acids and hydrogen bonding additives
The primary goal of this study was to determine if proton transfer/hydrogen bonding can show a thermal responsiveness in VU vitrimers, which would be kinetically relevant for the acid catalysed VU bond exchanges. To that end, a variety of dynamic polymer networks were synthesised as described herein below (Fig. 2). First, a non-catalysed reference material (i.e. a network with ‘no added’ acid catalyst) was prepared by mixing trifunctional 1,1,1-trimethyl-propane trisacetoacetate (1) and bifunctional Priamine 1074 (2) with a theoretical stoichiometric excess of 5 mol% amine groups (VU-ref). This formulation has previously been shown to produce elastomeric VU vitrimer samples with a relatively high cross-linking density and a sufficient amount of pendent primary amines for fast transamination under moderate heating.46,63,64 In a next step, a range of materials with the same formulation was prepared, but with the addition of several different Brønsted acid catalysts, at a 4 mol% doping level, which is enough to theoretically protonate up to 80 mol% of the free pendent amine network defects, i.e. 4 mol% of acid for 5 mol% of reactive amines.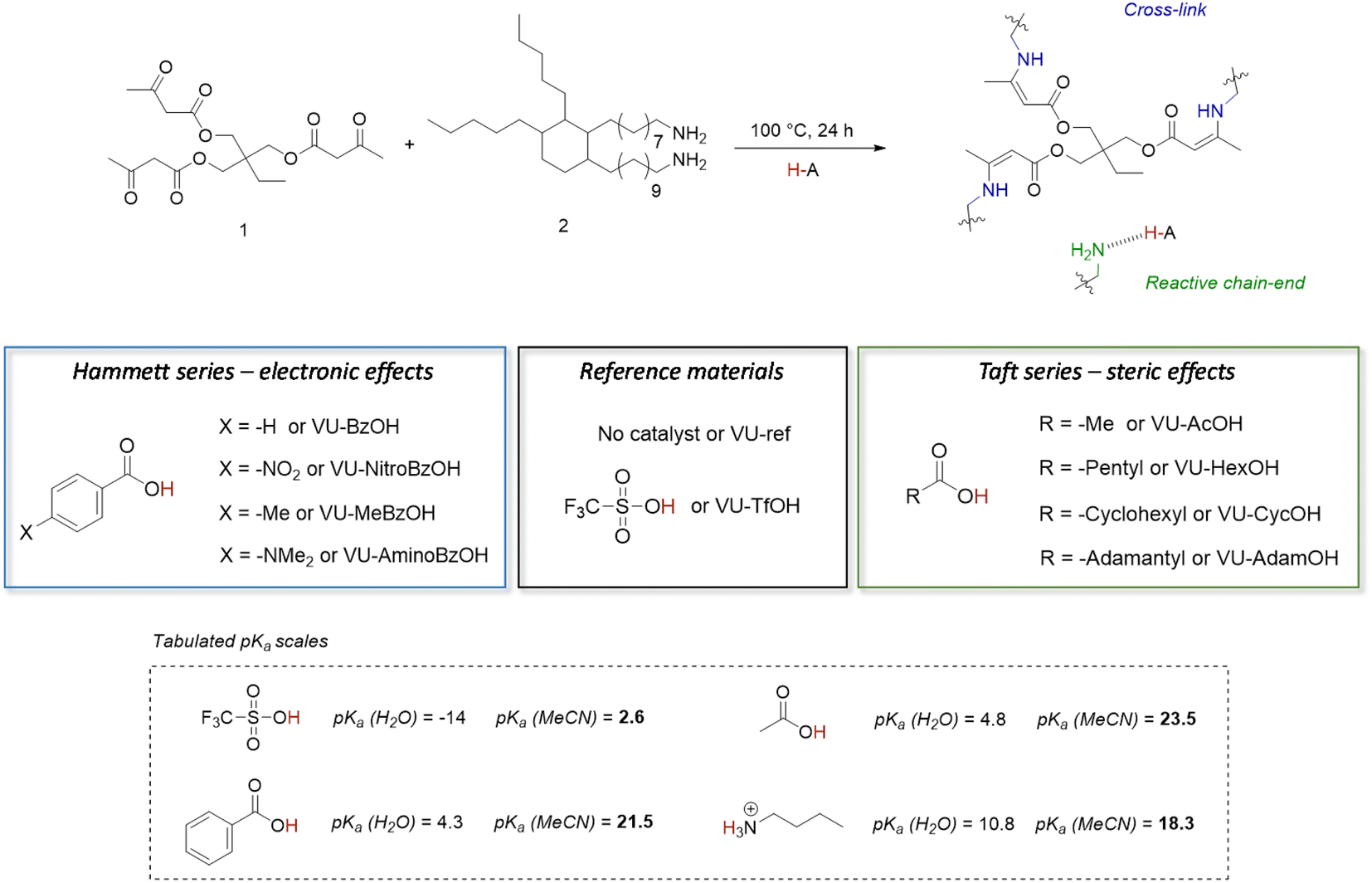 | ||
| Fig. 2 Design of vinylogous urethane based vitrimers with thermoswitchable catalysis of bond exchange. The aim is to control the tendency of protonation or hydrogen bonding by changing the electronic and/or steric environment of the carboxylic acid additive, which results in a change in formal pKa values.67 | ||
In previous studies of protic acid catalysed VU vitrimers, the main focus has typically been on using strong sulfonic acids, which would increase the concentration of available reactive ammonium species (Fig. 1c).46,62,67 This ‘full’ protonation (or ionisation of the sulfonic acid) can indeed be expected from the large difference in pKa between sulfonic acid derivatives and ammonium species, even in non-aqueous solvents. In order to assure full protonation, we added 4 mol% triflic acid (TfOH, VU-TfOH) in the reference material. This serves as an example for “complete proton transfer”, with a catalyst species that should be equally active and available at all temperatures. The VU-ref and VU-TfOH material conveniently serve as a set of reference networks to compare any changes in thermomechanical material properties and viscoelastic behaviour (vide infra).
In contrast to the situation described for sulfonic acids, proton transfer between carboxylic acids and amines is frequently an unfavourable process in non-aqueous media.7 Indeed, based on the pKa values in acetonitrile shown in Fig. 2,68 the addition of acetic acid (AcOH) or benzoic acid (BzOH) should not increase the concentration of protonated ammonium-type species under standard conditions at room temperature. However, we hypothesised, as explained in the introduction, that small pKa differences (and differences in reaction enthalpy and entropy) between carboxylic acids may translate into quite distinct alteration of a catalyst's behaviour over a wide temperature range. Hence, we theorized that by tuning the ΔpKa, the extent of proton transfer to pendant amines can be shifted from protonated to non-protonated states, as an on/off type of situation.
To study the effect of small pKa differences between carboxylic acids on the concentration of protonated ammonium-type species, we investigated two series of carboxylic acid additives for the VU vitrimers. One series of acids focuses on changes in the electronic environment of the carboxylic acid moiety, while the other series probes the effect of the steric environment of the acidic group. More specifically, a ‘Hammett-based approach’ was used to investigate the effect of a para substituent's electron-donating or electron-withdrawing nature on the acidity of a range of benzoic acids. The pKa-values follow a logically increasing trend in the series –NO2, –H, –Me to –NMe2, as the para-substituent becomes ever more electron donating and anion destabilising. The resulting materials are further referred to as VU-NitroBzOH, VU-BzOH, VU-MeBzOH, and VU-AminoBzOH, respectively (Fig. 2 left). In the second series, a ‘Taft-based approach’ was followed in which acetic acid (AcOH) was compared to more bulky alkanoic acids. More specifically, increasing steric hindrance from –methyl, –pentyl, –cyclohexyl to –adamantyl is expected to result in minor changes in pKa (trending slightly upward with degree of alkyl substitution on the alfa-carbon) but the steric bulk can hinder transfer and diffusivity of the ion pair, as well as impair solvation of the ion. The ‘Taft-series’ of modified VU materials is further referred to as VU-AcOH, VU-HexOH, VU-CycOH, and VU-AdamOH, respectively (Fig. 2 right).
For all prepared VU materials, the same solvent-free curing procedure was followed (100 °C for 24 h). Subsequently, after network formation, full conversion of the acetoacetate monomers was verified via ATR-FTIR analysis (Fig. S1–S3†). Unfortunately, monitoring of any proton transfer (ammonium ions) in the network via IR-spectroscopy was not possible, as any of the expected small changes in carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or ammonium N–H stretch vibrations were obscured by the VU matrix and could thus not be readily identified.
O or ammonium N–H stretch vibrations were obscured by the VU matrix and could thus not be readily identified.
Swelling experiments in THF for 24 h did reveal important trends, as important changes in the networks' swelling capacity and soluble fraction could be seen when compared to VU-ref (Table 1). Interestingly, only the samples containing the relatively strong acids TfOH and NitroBzOH showed a significant increase in swelling degree with values up to ∼350% as well as a larger soluble fraction of 7–8%. In contrast, the swelling degree (∼125–140%) and soluble fraction (∼3–5%) of all other materials were comparable to VU-ref. These results can be rationalised as an indication that most carboxylic acids (except for NitroBzOH) show a similar (low) degree of amine protonation in these VU networks at room temperature.
| Vitrimer | T g (°C) | T d5% (°C) | Swel. rat.c (%) | Sol. frac.c (%) | E a,high (kJ mol−1) | E a,low (kJ mol−1) | τ 160°C (s) | τ 120°C (s) |
|---|---|---|---|---|---|---|---|---|
| a Determined from the second heating run in DSC analysis (10 C° min−1). b TGA onset temperature after 5% weight loss. c Obtained after swelling in THF at room temperature for 24 h. d Obtained by fitting to a stretched single exponential decay. The lower temperature region was analysed separately if a shift in ionisation equilibrium was observed. e Relaxation values obtained by fitting to a stretched single exponential decay. Depending on the completeness of relaxation, a larger standard deviation was observed. f Extrapolated values. | ||||||||
| VU-ref | −5 | 318 | 130 ± 1 | 3.1 ± 0.1 | 150 ± 6 | — | 40 | 2800 |
| VU-TfOH | 0 | 278 | 309 ± 7 | 7.7 ± 0.9 | 75 ± 1 | — | 2 | 20 |
| VU-AcOH | −5 | 315 | 140 ± 4 | 4.8 ± 0.9 | 227 ± 11 | 180 ± 1 | 8 | 7100 |
| VU-HexOH | −4 | 317 | 131 ± 2 | 5.3 ± 0.5 | 307 ± 5 | 164 ± 6 | 17 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
| VU-CycOH | −3 | 316 | 136 ± 1 | 5.0 ± 0.2 | 295 ± 16 | 172 ± 12 | 13 | 15500 |
| VU-AdamOH | 0 | 315 | 132 ± 2 | 4.0 ± 0.1 | 368 ± 17 | 243 ± 1 | 18 | 63000f |
| VU-BzOH | 0 | 316 | 130 ± 1 | 4.8 ± 0.4 | 72 ± 2 | — | 12 | 100 |
| VU-NitroBzOH | 0 | 303 | 350 ± 8 | 7.1 ± 0.7 | 79 ± 1 | — | 1 | 7 |
| VU-MeBzOH | 1 | 317 | 126 ± 1 | 5.3 ± 0.3 | 399 ± 15 | 237 ± 8 | 13 | 60300f |
| VU-AminoBzOH | −1 | 317 | 137 ± 3 | 4.2 ± 0.4 | 410 ± 6 | 242 ± 14 | 20 | 100300f |
Next to the clear trends in swelling degrees, some other changes in material properties could be seen for the networks with more acidic additives. First, the VU-TfOH and VU-NitroBzOH both showed a markedly decreased thermal stability (Table 1 and Fig. S4–S10†). TGA analysis indeed revealed a 15–40 °C decrease in degradation onset temperature (Td5%), which could be attributed to the intrinsically higher reactivity of these networks. Finally, small changes in Tg values could be observed in different networks, with a Tg varying from −5 to 0 °C, as determined via DSC (Fig. S11–S13†). However, these Tg values did not show a clear trend. On the other hand, much more distinct and quite interesting differences could be seen in the next section that is highlighting the rheological behaviour of these networks.
Influence of catalysis on stress-relaxation of vinylogous urethane vitrimers
Polymer networks are a challenging medium in terms of reactivity monitoring. Indeed, a direct follow-up of the reactivity or the protonation state of reactive intermediates at different temperatures is not straightforward. In fact, the presented concept exists of two equilibria, a first acid-base equilibrium controlling the amount of reactive ammonium species and a second equilibrium that determines the rearrangements in the VU networks. Nonetheless, since it has been demonstrated that the presence of protonated ammonium species has a strong influence on the rate of bond exchange through transamination in VU vitrimers, rheology experiments can be used as an indirect measure of the ionisation equilibrium of each of the protic acid additives. In other words, a network that behaves as the reference material VU-ref, will have most of the protons residing on the carboxylic acid moiety, with only a limited amount of catalytically active ammonium species. On the other hand, a network that behaves rheologically more like the VU-TfOH network, should also have a pronounced proton transfer from the acid to the amine functions. Hence, the extent of proton transfer between an acid and amine, traditionally assessed by a ΔpKa, determines how fast stresses can be relaxed by the rearranging network. In this way, and quite interestingly, stress-relaxation experiments can be used to monitor an acid-base equilibrium, as it is expected to alter the molecular-level movement or diffusion of polymer chains within the polymer network.20 It should be noted that other factors can obviously also influence the rheological behaviour.Before measuring stress-relaxation, each (modified) VU vitrimer was compression moulded into the required dimensions, which simultaneously demonstrated the possibility to reprocess these cross-linked polymer networks. All samples were shredded into smaller pieces of ∼1 mm and subsequently pressed at 150 °C for 5–10 min, depending on the expected rate of material flow. Next, the obtained samples were subjected to stress-relaxation experiments using a 0.5% deformation and measuring the stress decrease over time (Fig. S14–S16†). The probed temperature ranges spanned between 160 and 90 °C, as appropriate for the observed rates of stress decay. More specifically, rheological measurements were stopped at the temperature at which, after 4 hours, less than 30–40% of the stress could be decayed. Slower processes can be detected with other rheology tests, such as creep experiments (vide infra).32
The stress-relaxation curves shown in Fig. 3 immediately reveal the significant and expected difference in dynamic behaviour between the vitrimers including the sulfonic acid (VU-TfOH, Fig. 3a) and carboxylic acid (VU-AcOH, Fig. 3b and VU-BzOH, Fig. 3c) additives. In line with the work of Bates and co-workers on Brønsted acid catalysis of polyester vitrimers,28 a stronger acid resulted in faster relaxation and up to a 20-fold increase in reaction rate (TfOH > BzOH > AcOH). This is in line with the basic expectation that a weaker acid is a less strong proton donor, and that less reactive ammonium species are formed subsequently. In contrast, when compared to VU-ref (dashed lines), the different spacing between the relaxation curves of each acid and the pronounced shifts in timescale of exchange (see arrows) highlight a remarkable and unexpected difference in the response of each material to temperature.
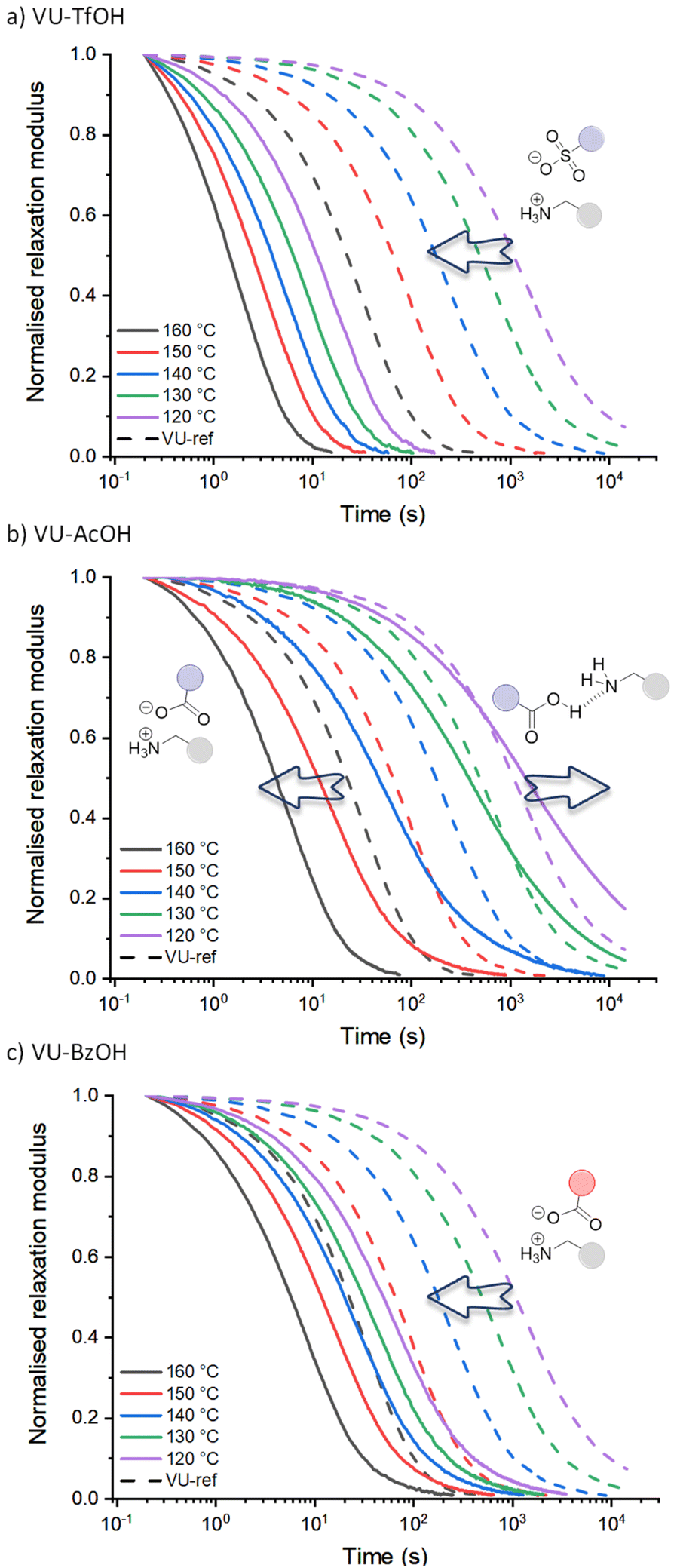 | ||
| Fig. 3 Normalised stress-relaxation curves of (a) VU-TfOH, (b) VU-AcOH and (c) VU-BzOH measured from 160 to 120 °C and compared to VU-ref (dashed lines). Different viscoelastic behaviour could be observed depending on the acid strength of the additive. | ||
Given what is known about the molecular basis of vitrimer rheology,15,19,36,69 we propose that the observed peculiar dynamic behaviour can be explained by a change in the availability of protonated ammonium species [RNH3+]. More particularly, it can be rationalised for stronger (sulfonic) acids that the amount of available acidic protons remains the same at each investigated temperature and that the acid-base equilibrium is shifted to the catalytic active protonated side and subsequently leads to an Arrhenian behaviour. However, effective proton transfer is hampered with weaker (carboxylic) acids, and the amount of protonated species only increases at higher temperatures, as the proton transfer equilibrium can shift to the endothermic side, resulting in an increasing availability of reactive ammonium species. In the case of AcOH (Fig. 3b), this would imply that at 120 °C, the thermoreversible but unfavourable equilibrium yields no significant additional ammonium groups, and that the relaxation curve would be similar to that of VU-ref (i.e., the relaxation curves should coincide). Yet, strikingly, while the relaxation curves do cross (e.g. at a normalised relaxation modulus of 0.35 for 130 °C), exchange for VU-AcOH actually becomes three times slower at lower temperatures, hinting at a pertinent inhibitory effect. Drawing from the well-examined propensity of carboxylic acids and aliphatic amines to shift towards a non-ionised, hydrogen-bonded state in small molecules (see introduction), we attribute the observed inhibitory effect to a comparable tendency. Specifically, we propose that (supramolecular) hydrogen bonding factors become significant at lower temperatures. In this scenario, the acid additive, in its neutral form, restricts the mobility and reactivity of a participating amine chain-end.
For BzOH (Fig. 3c), the catalytic effect is obvious at all temperatures, and here a ‘thermoswitchable catalyst’ behaviour cannot be observed in the range of 120 to 160 °C. Nonetheless, also in this system a pronounced decrease in reactivity could be detected in lower temperature measurements from 110 to 90 °C (Fig. S16†). These results align with the anticipated trends derived from the pKa values of BzOH and AcOH. Benzoic acid is a slightly stronger acid compared to acetic acid, resulting in a less endergonic equilibration to its deprotonated state and therefore occurs at lower temperatures.
The investigation of the Taft-based series (i.e. including sterically hindered alkanoic acids) also revealed a clear trend (Fig. 4). The increase in bulkiness of the counterion from VU-HexOH (Fig. 4a) to VU-AdamOH (Fig. 4b) further slows down stress-relaxation at lower temperatures, and prevents full stress-relaxation within the normal temperature window (all stress-relaxation curves are shown in the ESI†). One explanation that can be put forward here is that the actual availability and mobility of free amines is further hampered by hydrogen bonding to the bulky acid group (Scheme S1†). As this effect is much more pronounced at lower temperatures than at higher temperatures, a much stronger temperature response (and apparent activation energy) is observed in these systems (Table 1).
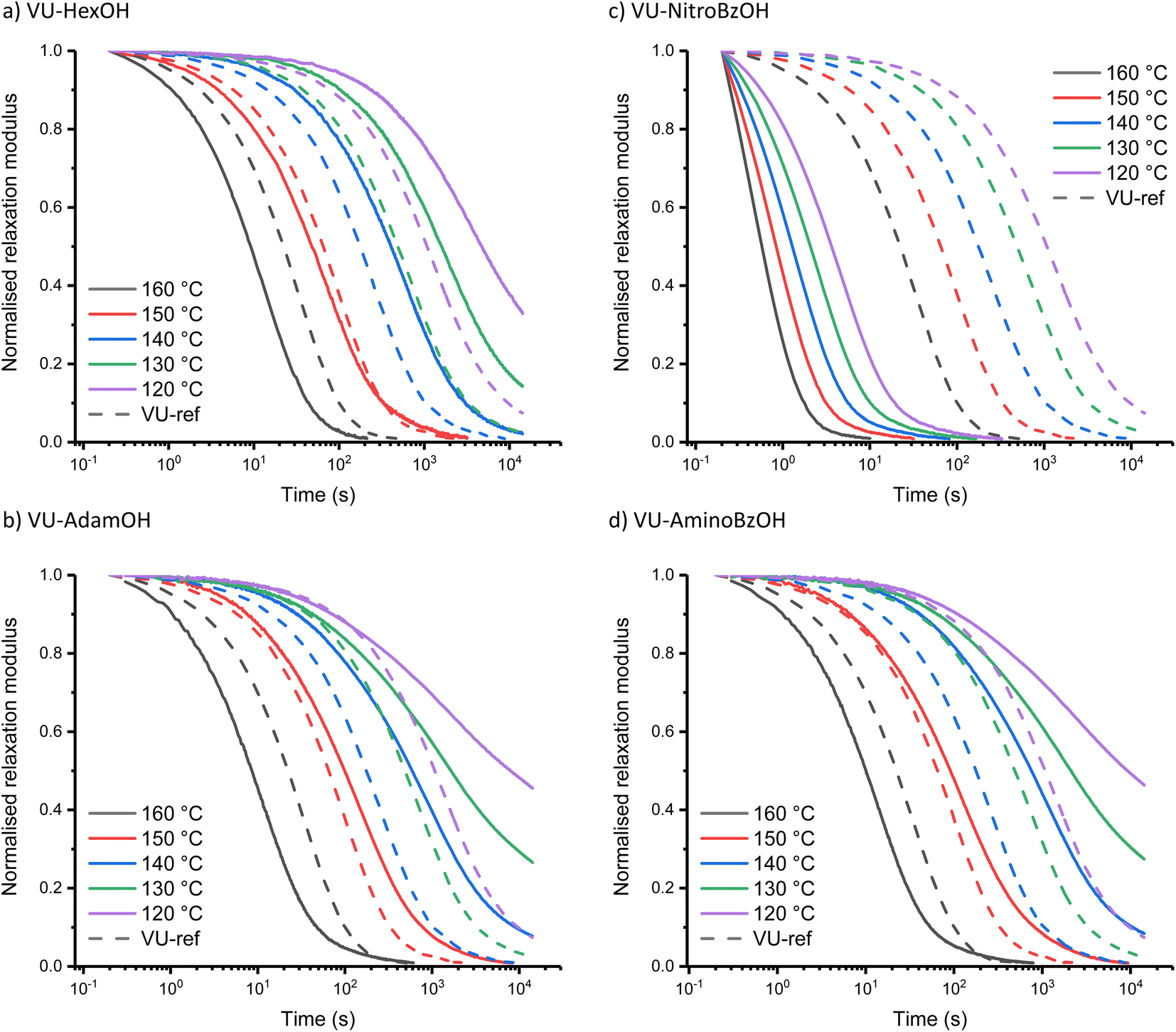 | ||
| Fig. 4 Normalised stress-relaxation curves of (a) VU-HexOH, (b) VU-AdamOH, (c) VU-NitroBzOH and (d) VU-AminoBzOH measured from 160 to 120 °C and compared to VU-ref (dashed lines), highlighting the effect of the steric and electronic environment of the additive on the accelerating and inhibiting behaviour. | ||
In the Hammett-series of benzoic acids with different electron donating and withdrawing groups, again a clear trend could be observed. As expected, for the electron-withdrawing –NO2 substituent at the para position of the aromatic ring (Fig. 4c), the rheological curves further shift towards those observed for VU-TfOH, even in the 90 to 110 °C temperature range. Actually, and somewhat surprisingly, stress-relaxation induced in VU-NitroBzOH becomes even faster than that in VU-TfOH, which cannot be explained by a higher concentration of ammonium species. This could be ascribed to the improved diffusivity of the organic para-NO2-benzoate counter ion, compared to the mostly inorganic triflate counter ion. Indeed, nitro-benzoate anions can be expected to show better ‘solubility’ in organic media. Moving to the less acidic benzoic acid derivatives in the Hammett series, we observe an expected drop in catalytic activity at lower temperatures, while the stress-relaxation remains fast at higher temperatures. For the para-NMe2-BzOH network, the rheology profile starts to coincide with that of the less acidic aliphatic carboxylic acids in the Taft-series, as expected based on trends in the pKa values. In fact, VU-AminoBzOH most closely resembles the VU-AdamOH network for its rheological profile (Fig. 4d). This can be rationalised by the fact that VU-AminoBzOH and VU-AdamOH have a comparable acidity and steric bulkiness or mobility of the hydrogen bonded amine–acid pair.
Investigation of the rheology curves revealed that the relaxation data did not follow a simple single exponential stress decay, especially at lower temperatures (Fig. S17 and S18†). This could be explained by the fact that the modified VU vitrimer's composing network segments will be made up of faster relaxing acid rich and slower relaxing acid poor domains. A quantitative analysis of this “chemical heterogeneity” was expected to yield further mechanistic insights into the network rheology. In a first assumption, a stretched exponential fit was used to determine the rate of bond exchange via a characteristic relaxation time ( , Table 1), which covered the distribution or dispersity in relaxation behaviour (eqn (1)).36,70
, Table 1), which covered the distribution or dispersity in relaxation behaviour (eqn (1)).36,70
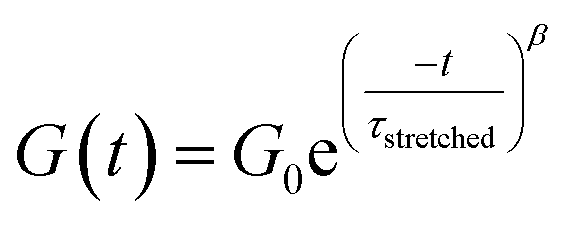 | (1) |
To model the temperature variation of  , Arrhenius curves were constructed by plotting
, Arrhenius curves were constructed by plotting 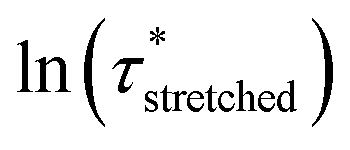 against 1000/T (Fig. 5 and S19†). A first observation was that each curve for which a shift in the ionisation equilibrium is expected in the investigated temperature window, showed a clear deviation from linearity (Fig. 5b). Based on the involvement of an ammonium species in the rate determining step for the VU exchange, such a sudden decrease in viscosity is expected as soon as sufficient protonated ammonium species are generated at elevated temperatures (Fig. 5a). This explains why two viscoelastic regimes appear with different slopes and a transition point between faster and slower relaxation than the one of VU-ref. Based on the Arrhenius plots in Fig. 5b and c, this transition point can be seen to shift based on the acidity and steric hindrance of the protic acid.
against 1000/T (Fig. 5 and S19†). A first observation was that each curve for which a shift in the ionisation equilibrium is expected in the investigated temperature window, showed a clear deviation from linearity (Fig. 5b). Based on the involvement of an ammonium species in the rate determining step for the VU exchange, such a sudden decrease in viscosity is expected as soon as sufficient protonated ammonium species are generated at elevated temperatures (Fig. 5a). This explains why two viscoelastic regimes appear with different slopes and a transition point between faster and slower relaxation than the one of VU-ref. Based on the Arrhenius plots in Fig. 5b and c, this transition point can be seen to shift based on the acidity and steric hindrance of the protic acid.
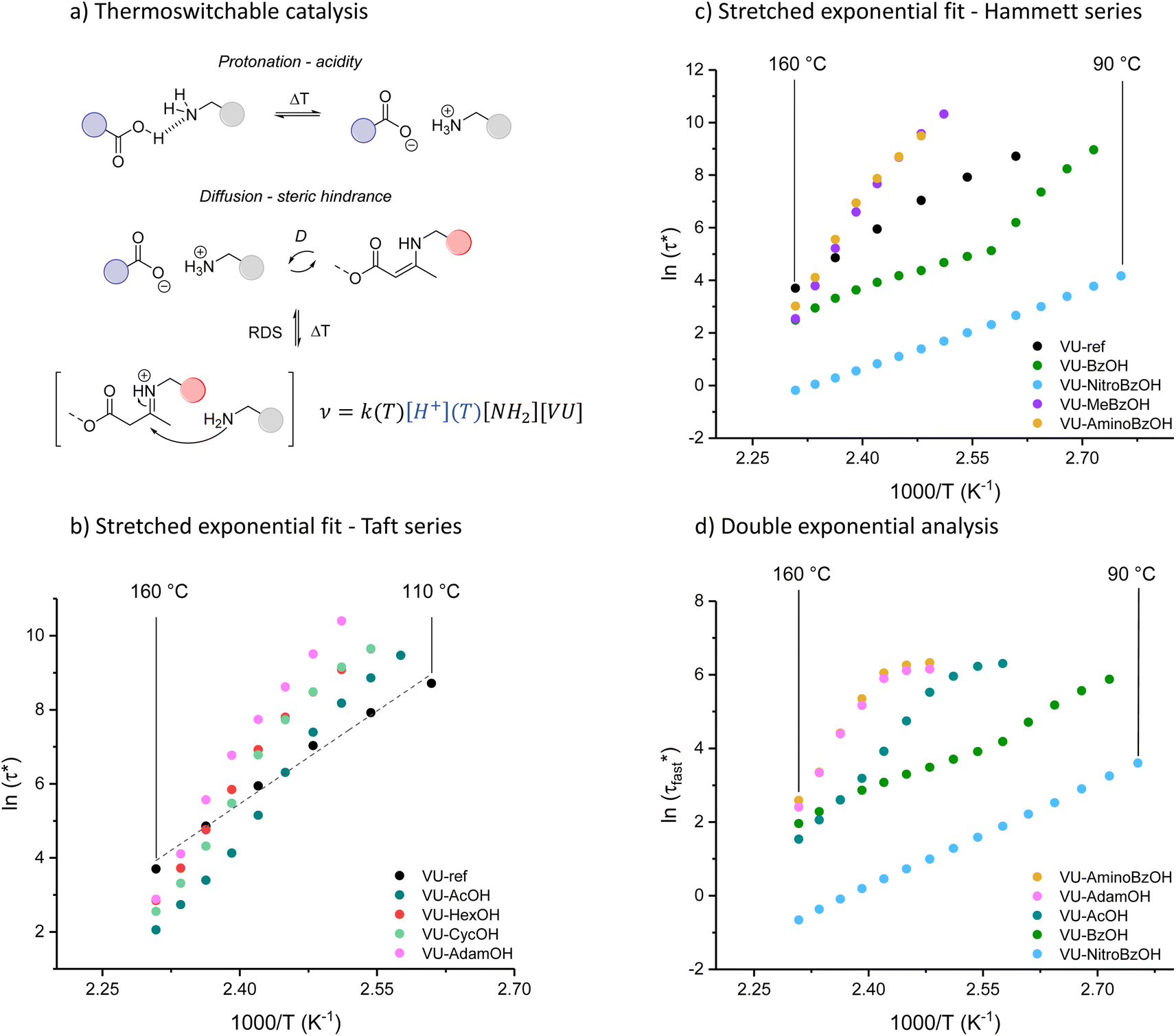 | ||
| Fig. 5 (a) General scheme illustrating the proposed effect of the thermoswitchable carboxylic acid catalyst on the rate of VU transamination. Arrhenius plot of (b) the Taft series (c) the Hammett series after fitting the relaxation data to a stretched exponential decay. (d) Arrhenius plot after fitting the relaxation data to a double exponential decay (fast and slow). The depicted data originates from the fastest relaxing element, which can be attributed to acid-rich polymer segments. | ||
As a result of the combined effects of shifting acid/base equilibria and steric factors, some of the newly developed VU vitrimers showed unprecedented apparent activation energies (Ea,flow) from 150 up to 410 kJ mol−1 in a relatively narrow temperature range, amounting to the most thermally responsive VU vitrimers studied to our knowledge.
In order to verify the applicability of this strategy for recyclable thermosets, the VU-AcOH sample was shredded and remoulded three times and stress-relaxation experiments were repeated (Fig. S20†). The Arrhenius plots of these experiments clearly show that the enhanced thermal responsiveness and dual stress-relaxation behaviour persists and that similar  values can be extracted within error (Fig. S21†). Conveniently, these reprocessing and an additional heat treatment (3 hours at 150 °C) trials provided additional evidence that no other factors, such as acid evaporation or amidation side reactions (as evidenced by FT-IR, Fig. S22†), could be at the root of the observed viscoelastic behaviour, but that the observed effects are intrinsically related to the specific material design. On the other hand, a small deviation can be observed in the lower temperature region (140–120 °C), that can be attributed to a certain extent of oxidation of the free amine chain-ends due to longer exposure to elevated temperatures.71 Importantly, these longer exposure times are needed to have full relaxation of a slower relaxing material, which becomes increasingly worse for more creep-resistant materials. While less reactive chains are available for transamination/network rearrangements, both the pristine and reprocessed network showed similar relaxation behaviour between 160 and 140 °C, which proves that both materials could relax fast at elevated temperatures because of the acid catalyst.
values can be extracted within error (Fig. S21†). Conveniently, these reprocessing and an additional heat treatment (3 hours at 150 °C) trials provided additional evidence that no other factors, such as acid evaporation or amidation side reactions (as evidenced by FT-IR, Fig. S22†), could be at the root of the observed viscoelastic behaviour, but that the observed effects are intrinsically related to the specific material design. On the other hand, a small deviation can be observed in the lower temperature region (140–120 °C), that can be attributed to a certain extent of oxidation of the free amine chain-ends due to longer exposure to elevated temperatures.71 Importantly, these longer exposure times are needed to have full relaxation of a slower relaxing material, which becomes increasingly worse for more creep-resistant materials. While less reactive chains are available for transamination/network rearrangements, both the pristine and reprocessed network showed similar relaxation behaviour between 160 and 140 °C, which proves that both materials could relax fast at elevated temperatures because of the acid catalyst.
The concept of a thermoswitchable catalyst is new to the field of vitrimers and therefore merits further investigation. Thus, we wanted to verify which “components” of the polymer network actively contribute to the observed stress-relaxation behaviour. In previous studies, we have found that analysing the same relaxation data with a double exponential fit allows to associate faster and slower exchanging chain segments (e.g. acid rich or poor domains) to a distinct relaxation time (eqn (2) and Fig. S23–S25†).70 As a result, when the amount of acid rich domains start to deplete as a function of temperature, it should significantly decrease the contribution of the fast segments to the overall relaxation process. Indeed, as indicated in the Arrhenius plot for the fast Maxwell element in Fig. 5d, at temperatures beyond the transition point (as observed in Fig. 5b and c), the fast exchanging segments became significantly less important, further supporting our findings (Table S1†).
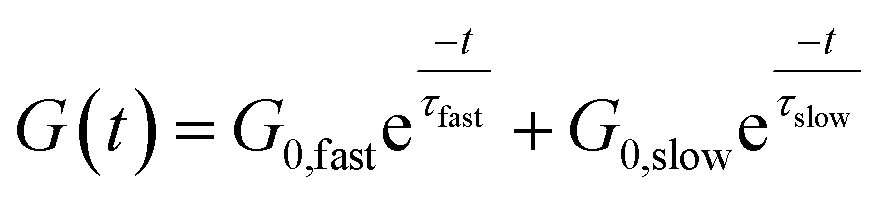 | (2) |
According to our analysis and rationalisation of the data, the carboxylic acid additives work as rheological inhibitors at lower temperatures (ascribed to hydrogen bonding) and as rheological promotors (transamination catalysts) at high temperatures. Therefore, one might expect to see more pronounced effects in formulations that have more of this additive, as it will both inhibit and catalyse more. Thus, a new set of materials was prepared using the same VU curing formulation (with 5 mol% excess pendent primary amines) but this time with 6 mol% of AcOH, BzOH or AdamOH (Fig. S26–S29†). According to Le Chatelier's principle, protonation can be shifted more to the endothermic side with an excess of acid, and mobility or availability of amines should also be maximally quenched by an excess of acid. The stress-relaxation measurements are gathered in the ESI (Fig. S30†). Arrhenius plots of the corresponding networks (VU-AcOHexcess, VU-BzOHexcess and VU-AdamOHexcess) revealed that the excess acid did not generally result in a significant improvement of high temperature network exchange reactions (in the case of AdamOH, even some inhibition could be seen), but rather shifted the transition point between the two viscoelastic regimes (Fig. 6a and S31†).
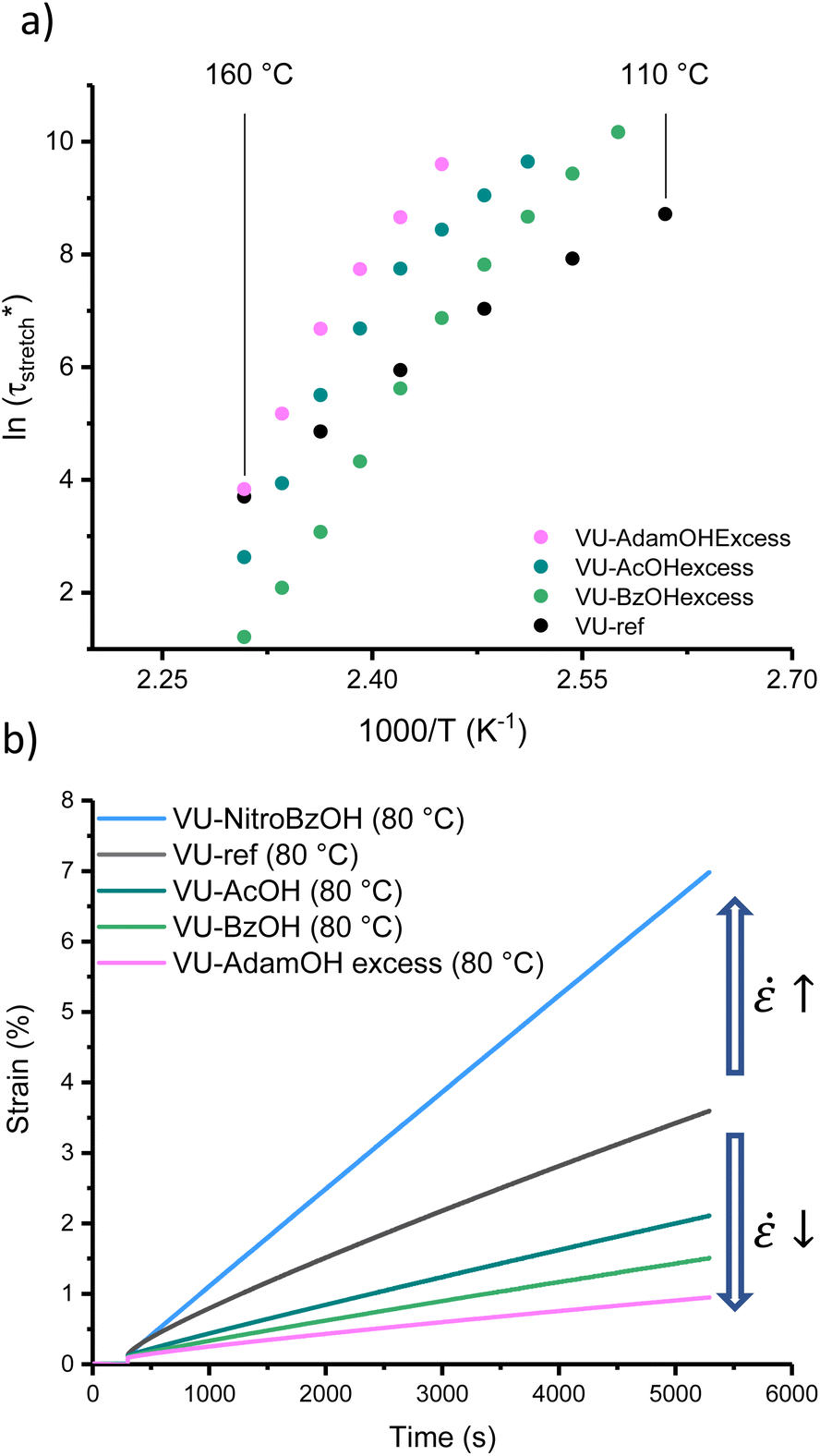 | ||
| Fig. 6 (a) Arrhenius plot obtained after fitting the relaxation data of the VU materials with an excess of acid additives to a stretched exponential decay. (b) Creep curves at 80 °C, highlighting creep reduction for weaker acids (VU-AcOH, VU-BzOH and VU-AdamOHexcess) and creep increase for more potent acids (VU-NitroBzOH). | ||
Both the VU-AcOHexcess and VU-BzOHexcess networks showed a significant slowing down of stress-relaxation at low temperatures. The most remarkable effect was observed for BzOH. Here, a much sharper temperature response could be observed with a strongly inhibited stress-relaxation below 140 °C, and even a 10-fold faster relaxation at 160 °C, with a shift in the transition point from ∼90 to ∼135 °C. Furthermore, the transition point between each regime differed only by 5 °C between VU-AcOHexcess and VU-AdamOHexcess but by around 20 °C between VU-AcOHexcess and VU-BzOHexcess (Table S2†), which was consistent with the expected difference in pKa between each acid.
An important aspect of vitrimer design is finding approaches to balance fast bond exchange at (re)processing temperatures with a high throughput while avoiding degradation, and at the same time very slow exchanges at service temperatures to guarantee creep resistance.32,45 Thus, the thermoswitchable catalysis approach presented here provides a new tool for controlling the cold flow of vitrimers. To demonstrate the extent of creep resistance of these modified VU vitrimers, viscosity changes can be conveniently evaluated below 100 °C via creep experiments. A selection of materials (see Fig. S32 and S33†) was investigated using a constant shear stress of 2 kPa for 5000 s in the range of 30 to 80 °C while monitoring the shear strain (ε) (Fig. 6b). Next, from the slope of the strain curve in the steady-state regime (above 4500 s), creep rate (ε![[small epsi, Greek, dot above]](https://www.rsc.org/images/entities/char_e0a1.gif) ) values could be calculated and compared to VU-ref (Fig. S34†). Except for VU-NitroBzOH, in which protonated ammonium species were readily available at all investigated temperatures, all modified materials demonstrated a relative decrease in creep rate in comparison to the reference sample (between 10% and 65% at 80 °C). It should be noted that based on the measurements and the observed differences, the main reason for the observed cold flow could be ascribed to the presence of non-elastic (reactive) chain-ends rather than bond exchange.63,72
) values could be calculated and compared to VU-ref (Fig. S34†). Except for VU-NitroBzOH, in which protonated ammonium species were readily available at all investigated temperatures, all modified materials demonstrated a relative decrease in creep rate in comparison to the reference sample (between 10% and 65% at 80 °C). It should be noted that based on the measurements and the observed differences, the main reason for the observed cold flow could be ascribed to the presence of non-elastic (reactive) chain-ends rather than bond exchange.63,72
Conclusion
In summary, we have reported a straightforward thermoswitchable Brønsted acid catalyst system, exemplified for VU-based vitrimers and widely applicable to other dynamic polymer network types. The acid additives can be thermally switched between activation and inhibition of bond exchange in the presented vitrimers, which is related to an endothermic proton transfer reaction that generates a catalytic species. Here, the transamination of vinylogous urethanes was chosen as the dynamic covalent chemistry because of the ease of synthesis of the polymer networks and the well-known influence of protonated ammonium species on the rate of dynamicity. Specifically, the generally endothermic carboxylic acid–amine ionization equilibrium was thermally forced to generate protonated intermediates and activate bond exchange. Furthermore, the tendency of the carboxylic acid to transfer a proton to an amine chain-end could be modulated by changing the electronic (Hammett parameters) or steric (Taft parameters) environment, which allows for a rational design of rheological parameters.Rheology experiments revealed that the strong thermal control of the amount of protonated species resulted in a double relaxation behaviour with extremely high apparent activation energies of up to 400 kJ mol−1. This approach is attractive in vitrimer design as the implementation does not present the classical trade-off between enhanced network dynamics and reduced network stability. In other words, the dynamic behaviour at higher temperatures can be significantly accelerated, while being inhibited by the same species at lower temperatures. In addition, when changing the concentration of the acid additive with respect to the calculated number of dangling primary amines, this effect becomes even more pronounced.
Based on additional rheology experiments and previous literature on carboxylic acid–amine equilibria in various solid, liquid, or gaseous media,5,11,59,73 it was hypothesised that this unusual behaviour resulted from the transition from a protonated (charged) to a hydrogen-bonded (neutral) state of the acid–amine complex, which significantly influenced both chemical kinetics and physical chain diffusion.
In short, the carboxylic acid–amine thermoswitchable catalyst system described herein presents a promising example of achieving precise control of dynamic behaviour in vitrimers. The strategy may be applicable more generally in vitrimer design, and once again points out the need to fundamentally understand the chemical mechanisms that are operating in a reactive polymer network, which are by no means captured by small molecule kinetic model studies. One remarkable conclusion from this study is that ‘weaker’ catalysts may actually be much better suited for the activation of polymer network rearrangements than ‘stronger’ catalysts. In fact, classical catalysts from organic chemistry are generally not optimized for poor performance at lower temperatures. The design of catalyst systems for vitrimers can thus be hampered by an overreliance on classical catalyst species.
Data availability
Experimental details and material characterisation including FT-IR spectra, DSC and TGA thermograms, stress-relaxation and creep data and NMR spectra are available in the ESI† of this article. The raw data is available upon request from the corresponding author.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. D. P. thanks the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (CiMaC project – grant agreement no 101021081). F. D. P. and J. W. thank BOF-UGent for GOA-funding. F. V. L. acknowledges the Research Foundation-Flanders (FWO) for his PhD (application 1S49122N) fellowship. S. M. acknowledges Ghent University for funding of his doctoral research (DOCT/009009). We would like to thank Bernhard De Meyer for technical support and Dr Nezha Badi for fruitful discussions.Notes and references
- P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef CAS.
- F. M. Menger, Acc. Chem. Res., 1993, 26, 206–212 CrossRef CAS.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
- D. F. Detar and R. W. Novak, J. Am. Chem. Soc., 1970, 92, 1361–1365 CrossRef CAS.
- Z. Wang, H. Deng, X. Li, P. Ji and J. P. Cheng, J. Org. Chem., 2013, 78, 12487–12493 CrossRef CAS PubMed.
- S. T. Heller and T. P. Silverstein, ChemTexts, 2020, 6, 1–17 CrossRef.
- N. F. Hall, Chem. Rev., 1931, 8, 191–212 CrossRef CAS.
- J. Mitsky, L. Joris and R. W. Taft, J. Am. Chem. Soc., 1972, 94, 3442–3445 CrossRef CAS.
- T. B. McMahon and P. Kebarle, J. Am. Chem. Soc., 1977, 99, 2222–2230 CrossRef CAS.
- I. Alkorta, I. Rozas, O. Mó, M. Yáñez and J. Elguero, J. Phys. Chem. A, 2001, 105, 7481–7485 CrossRef CAS.
- J. N. Brönsted, Chem. Rev., 1928, 5, 231–338 CrossRef.
- K. Kaupmees, N. Tolstoluzhsky, S. Raja, M. Rueping and I. Leito, Angew. Chem., Int. Ed., 2013, 52, 11569–11572 CrossRef CAS PubMed.
- M. L. Bender, J. Am. Chem. Soc., 1957, 79, 1258–1259 CrossRef CAS.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- B. Marco-Dufort, R. Iten and M. W. Tibbitt, J. Am. Chem. Soc., 2020, 142, 15371–15385 CrossRef CAS PubMed.
- S. P. O. Danielsen, H. K. Beech, S. Wang, B. M. El-Zaatari, X. Wang, L. Sapir, T. Ouchi, Z. Wang, P. N. Johnson, Y. Hu, D. J. Lundberg, G. Stoychev, S. L. Craig, J. A. Johnson, J. A. Kalow, B. D. Olsen and M. Rubinstein, Chem. Rev., 2021, 121, 5042–5092 CrossRef CAS PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS PubMed.
- D. N. Barsoum, V. C. Kirinda, B. Kang and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 10168–10173 CrossRef CAS PubMed.
- M. O. Saed and E. M. Terentjev, ACS Macro Lett., 2020, 9, 749–755 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- F. Van Lijsebetten, T. Debsharma, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202210405 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 22358–22377 CrossRef CAS PubMed.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Polym. Chem., 2020, 11, 5339–5345 RSC.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef CAS PubMed.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Chem. Sci., 2021, 12, 293–302 RSC.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- L. Zhang and S. J. Rowan, Macromolecules, 2017, 50, 5051–5060 CrossRef CAS.
- N. J. Bongiardina, K. F. Long, M. Podgórski and C. N. Bowman, Macromolecules, 2021, 54, 8341–8351 CrossRef CAS.
- K. M. Herbert, P. T. Getty, N. D. Dolinski, J. E. Hertzog, D. de Jong, J. H. Lettow, J. Romulus, J. W. Onorato, E. M. Foster and S. J. Rowan, Chem. Sci., 2020, 11, 5028–5036 RSC.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- M. Podgórski, N. Spurgin, S. Mavila and C. N. Bowman, Polym. Chem., 2020, 11, 5365–5376 RSC.
- C. Dertnig, G. Guedes de la Cruz, D. Neshchadin, S. Schlögl and T. Griesser, Angew. Chem., Int. Ed., 2023, 62, e202215525 CrossRef CAS PubMed.
- D. Reisinger, S. Kaiser, E. Rossegger, W. Alabiso, B. Rieger and S. Schlögl, Angew. Chem., Int. Ed., 2021, 60, 14302–14306 CrossRef CAS PubMed.
- J. S. Stevens, L. K. Newton, C. Jaye, C. A. Muryn, D. A. Fischer and S. L. M. Schroeder, Cryst. Growth Des., 2015, 15, 1776–1783 CrossRef CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef CAS.
- H. B. Bürgi and J. D. Dunitz, Acc. Chem. Res., 1983, 16, 153–161 CrossRef.
- G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1–15 CrossRef CAS.
- H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC.
- H. Ishikita and K. Saito, J. R. Soc., Interface, 2014, 11, 20130518 CrossRef PubMed.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2013, 16, 1150–1160 RSC.
- J. P. Layfield and S. Hammes-Schiffer, Chem. Rev., 2014, 114, 3466–3494 CrossRef CAS PubMed.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharm., 2007, 4, 317–322 CrossRef PubMed.
- J. Zielińska, M. Makowski, K. Maj, A. Liwo and L. Chmurzyński, Anal. Chim. Acta, 1999, 401, 317–321 CrossRef.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharm., 2007, 4, 323–338 CrossRef CAS PubMed.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- F. Van Lijsebetten, K. De Bruycker, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202113872 CrossRef CAS PubMed.
- F. Van Lijsebetten, K. De Bruycker, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2022, 11, 919–924 CrossRef CAS PubMed.
- S. K. Schoustra, T. Groeneveld and M. M. J. Smulders, Polym. Chem., 2021, 12, 1635–1642 RSC.
- S. K. Schoustra, V. Asadi, H. Zuilhof and M. M. J. Smulders, Eur. Polym. J., 2023, 195, 112209 CrossRef CAS.
- P. Haida and V. Abetz, Macromol. Rapid Commun., 2020, 41, 2000273 CrossRef CAS PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- F. Van Lijsebetten, K. De Bruycker, E. Van Ruymbeke, J. M. Winne and F. E. Du Prez, Chem. Sci., 2022, 13, 12865–12875 RSC.
- Y. Spiesschaert, M. Guerre, I. De Baere, W. Van Paepegem, J. M. Winne and F. E. Du Prez, Macromolecules, 2020, 53, 2485–2495 CrossRef CAS.
- H. Zhou, E. M. Schön, M. Wang, M. J. Glassman, J. Liu, M. Zhong, D. Díaz Díaz, B. D. Olsen and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 9464–9470 CrossRef CAS PubMed.
- H. G. Brittain, Cryst. Growth Des., 2009, 9, 3497–3503 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00417e |
| This journal is © The Royal Society of Chemistry 2024 |