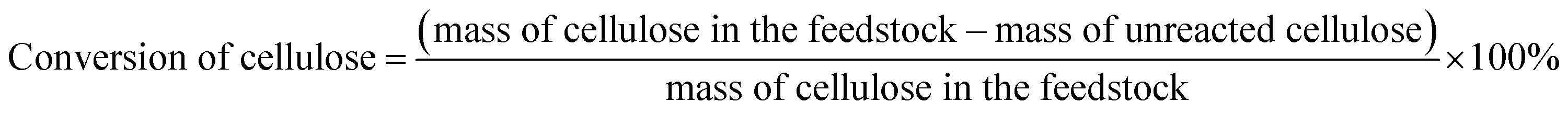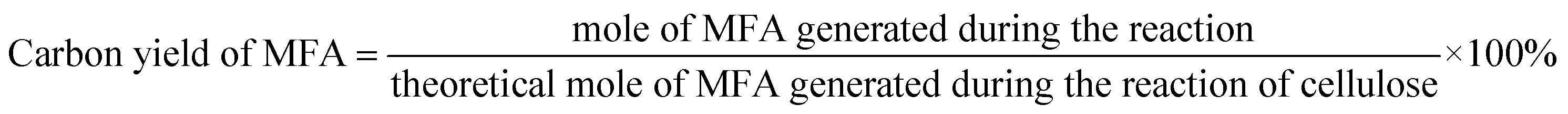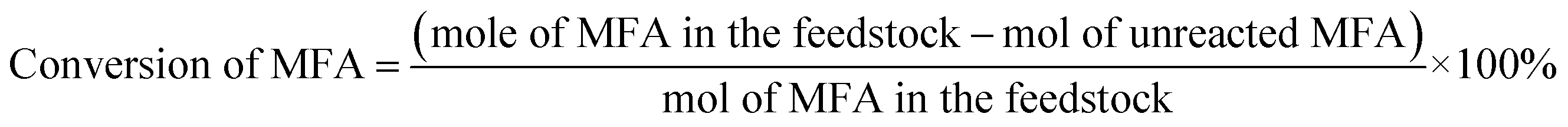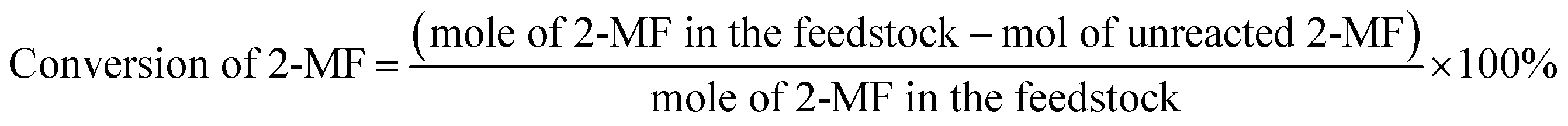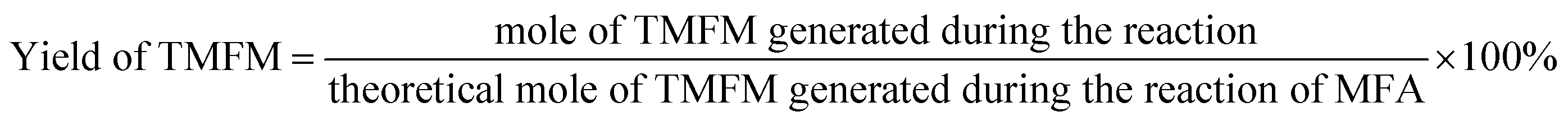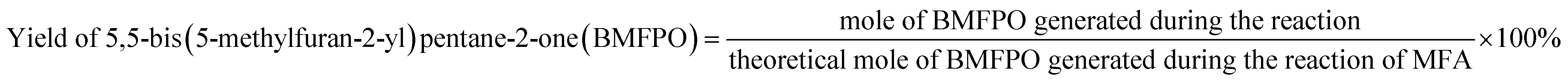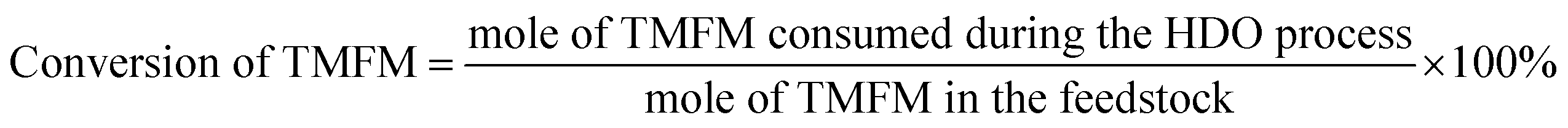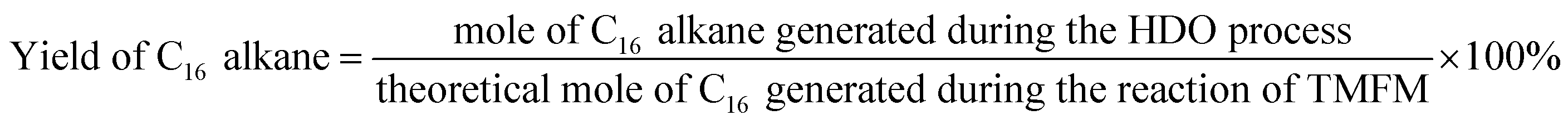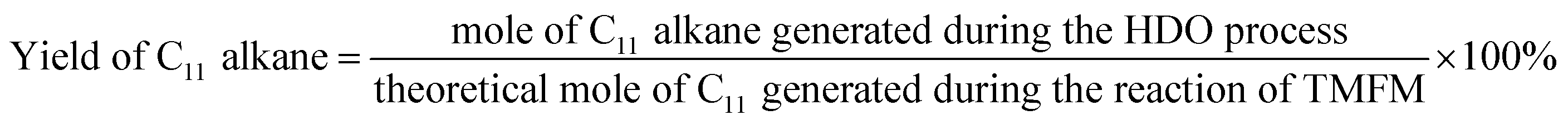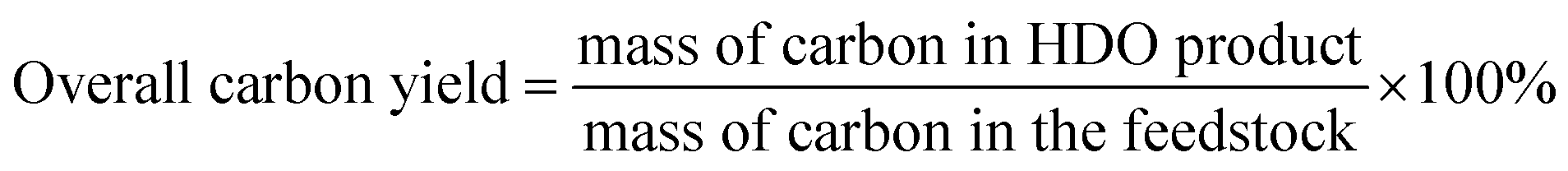Synthesis of jet fuel range paraffins from cellulose†
Received
27th May 2024
, Accepted 30th July 2024
First published on 31st July 2024
Abstract
In this work, a new route was developed for the synthesis of jet fuel range C16 and C11 paraffins with cellulose, the most abundant biomass. In the first step, cellulose was selectively converted to 5-methylfurfural (MFA) by a cascade hydrolysis/isomerization/dehydration/chlorination reaction in a toluene/NaCl aqueous solution biphasic system, followed by the hydrodechlorination over Pd/C catalyst at room temperature. After being decarbonylated, the MFA was converted to 2-methylfuran (2-MF). Among the investigated catalysts, Pd/C exhibited the highest activity for this reaction, which can be rationalized by the higher activity of Pd for the decarbonylation. Subsequently, tris(5-methylfuran-2-yl)methane (TMFM) was obtained by the solvent-free hydroxyalkylation/alkylation (HAA) reaction of MFA and 2-MF over a series of acidic resins. Among them, Nafion resin exhibited the highest activity, which can be rationalized by the high acid strength of this catalyst. Finally, the TMFM as obtained was hydrodeoxygenated to jet fuel range C16 and C11 paraffins under the co-catalysis of Ni/hydroxyapatite (Ni/HAP) and H-ZSM-5.
Introduction
Due to the great social concerns about sustainable development and environment protection, the production of sustainable aviation fuel (SAF) with renewable, CO2 neutral, cheap and abundant biomass has drawn lots of attention.1,2 Cellulose is the most abundant biomass. Paraffins are the most important component of jet fuel. In the pioneering work of Dumesic's group,3 jet fuel range C8–C16 branched alkanes were synthesized by the oligomerization and hydrogenation of butenes obtained by the decarboxylation of γ-valerolactone from hydrogenation of levulinic acid, a platform compound that can be produced by the hydrolysis/dehydration reaction of cellulose. In the latter work of Mascal's group4 and Zhang et al.,5 jet fuel range branched alkanes were produced by the self-condensation of angelica lactone (the further dehydration product of levulinic acid), followed by the hydrodeoxygenation (HDO). As another option, Mascal et al.6 also found jet fuel range branched alkanes and cycloalkanes could be obtained by the HDO and decarboxylation of the angelica lactone self-condensation product over Pd/Al2O3 catalyst. So far, most of the reported routes about cellulose-based SAF were concentrated on the synthesis of jet fuel range branched alkanes with levulinic acid (or its derivates). During the conversion of cellulose to levulinic acid, at least one sixth carbon atom will be lost in the form of formic acid. For the butene oligomerization/hydrogenation route and angelica lactone self-condensation/decarboxylation route, this problem is more serious because additional carbon atoms will be lost in the form of carbon dioxide. From the pointview of atomic economy, the exploration of new routes for the synthesis of jet fuel range paraffins with cellulose derived C6 platform compounds has great significance.
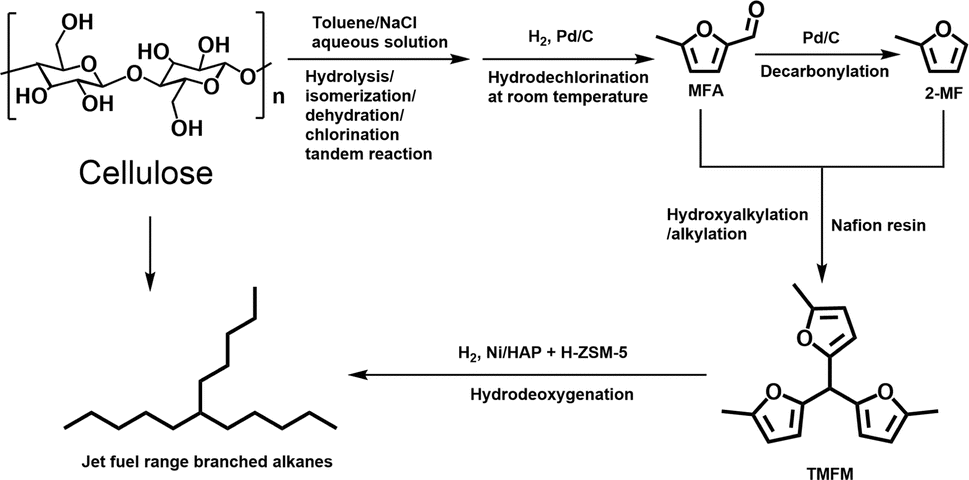
In this work, we developed a new route for the synthesis of jet fuel range paraffins from cellulose with 5-methylfurfural (MFA) as the intermediate. First, MFA was obtained at 77.5% carbon yield by the reaction of cellulose in a toluene/NaCl aqueous solution biphasic system, followed by the hydrodechlorination over Pd/C catalyst at room temperature. Subsequently, the MFA was converted to 2-methylfuran (2-MF) by decarbonylation. To the best of our knowledge, this should be the first report about the synthesis of 2-MF with cellulose. As the final aim of this work, we also explored the synthesis of jet fuel range paraffins by the solid acid catalyzed hydroxyalkylation/alkylation (HAA) reaction of MFA and 2-MF, followed by the hydrodeoxygenation. The strategy of this work was illustrated in Scheme 1.
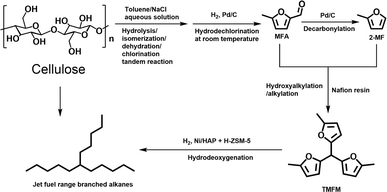 |
| | Scheme 1 Strategy for the synthesis of jet fuel range paraffins from cellulose with MFA as the intermediate. | |
Experimental
Materials
The microcrystalline cellulose used in this work was purchased from ACROS. Before the activity tests, the microcrystalline cellulose was ball-milled by a QM-3SP4 Laboratory Planetary Ball Mill Machine. Typically, 2 g of microcrystalline cellulose was charged in an agate grinding cell and ball-milled at a frequency of 50 Hz with 6 mm agate balls for 24 h. The Pd/C, Ru/C, Rh/C and Pt/C catalysts with a metal loading of 5 wt% were prepared by the incipient wetness impregnation of activated carbon with aqueous solutions of PdCl2, RuCl3, RhCl3 and H2PtCl6. The NaCl, toluene, methyltetrahydrofuran (MTHF), tetrahydrofuran (THF) and γ-valerolactone (GVL), Amberlyst-15 resin used in the work were supplied by Shanghai Aladdin Bio-Chem Technology Co. Nafion and Amberlyst-36 resins were supplied by Nanda Synthetic Co. Ltd. Amberlite IRC-76CRF were supplied by Tianjin Shuanglian Co., Ltd. H-ZSM-5 (SiO2/Al2O3 molar ratio = 25) and HAP were obtained from Nankai University Catalyst Co., Ltd The metal loaded HAP catalysts (denoted as M/HAP, M = Pt, Pd, Ru, Ni) used in this paper were manufactured by the deposition–precipitation method. To facilitate the comparison, the theoretical metal contents in the M/HAP catalysts were controlled as 2 wt%. The detail information for the preparation of the M/HAP catalysts was described in ESI.†
Experimental procedure
Conversion of cellulose to MFA.
The synthesis of MFA with cellulose was carried out by a two-stage process with a stainless-steel batch reactor (50 mL, Anhui Kemi Machinery Technology Co. Ltd). Typically, 0.3 g ball-milled cellulose, 3.0 g NaCl, 10 mL H2O, and 20 mL organic solvent were used for the tests. In the first stage, the reaction was carried out in N2 atmosphere at 473 K under a stirring speed of 400 rpm for 6 h. After cooling down the reactor to room temperature, the product was taken out from reactor and filtrated to remove the unreacted cellulose. The upper organic phase was transferred into another batch reactor with 0.03 g Pd/C catalyst. The reactor was purged with nitrogen for 3 times and charged with hydrogen to 2 MPa H2 at room temperature. The reaction system was stirred at 303 K for 2 h at 400 rpm and cooled down to room temperature. After releasing the unreacted hydrogen, the product was taken out from the reactor, filtrated to remove the catalyst and analyzed by GC and GC-MS using tridecane as an internal standard.
Decarbonylation of MFA to 2-MF.
The decarbonylation of MFA to 2-MF was carried out in a stainless-steel batch reactor (50 mL, Anhui Kemi Machinery Technology Co. Ltd) equipped with a temperature-controlled heating jacket and magnetic stirring. Typically, 1 mmol MFA, 0.05 g catalyst, and 5 mL ethyl acetate were used for the tests. After the reaction, the reactor was quickly cooled to room temperature. The reaction mixture was taken for analysis by GC and GC-MS using tridecane as the internal standard.
Hydroxyalkylation/alkylation (HAA).
The solvent-free HAA reaction of MFA and 2-MF was carried out in a round-bottomed flask equipped with a reflux condenser and a magnetic stirrer. The reaction temperature was controlled with a water bath. Typically, 0.09 g catalyst, 5 mmol MFA, and 10 mmol 2-MF were used for each test. After the reaction, the product was taken out from the flask, diluted with CH2Cl2, and analyzed by GC and GC-MS using tridecane as the internal standard.
Hydrodeoxygenation (HDO).
The HDO process of the HAA product was carried out by a stainless-steel batch reactor. For each reaction, 0.1 g HAA product, 30 mL cyclohexane 0.1 g M/HAP and 0.1 g H-ZSM-5 were used. The reaction was conducted under 4 MPa H2 for 24 h and terminated by quenching the reactor in cool water. After releasing of unreacted H2, the product was taken out from the reactor, filtered and analysed by GC and GC-MS using tridecane as the internal standard.
Analytical procedures.
The products were analyzed by an Agilent 7890A gas chromatograph (GC) equipped with a HP-5 capillary column (30 m, 0.32 mm ID, 0.5 mm film) and a flame ionization detector (FID) using tridecane as the internal standard or an Agilent 1100 HPLC equipped with a ZORBAX SB-C18 column and refractive index detector (RID) using a mixture of methanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the eluent. The densities, freezing point and net heat of combustion (NHOC) of the paraffins obtained from HDO test were measured by a DMA 4500M digital densitometer, a AmericaTA DSC250 and a SDC712 calorimeter, respectively.
:1 v/v) as the eluent. The densities, freezing point and net heat of combustion (NHOC) of the paraffins obtained from HDO test were measured by a DMA 4500M digital densitometer, a AmericaTA DSC250 and a SDC712 calorimeter, respectively.
The conversions of feedstocks and the carbon yields of different products were calculated according to following equations:
Results and discussion
Conversion of cellulose to MFA
MFA is a widely used as flavours and the intermediate in the production of some insecticides. In the previous work of Mascal et al.,7 MFA was obtained at a carbon yield of 62.5% by the reaction of cellulose in a 1,2-dichloroethane/HCl–LiCl aqueous solution biphasic reaction system, followed by the hydrogenolysis under the catalysis of PdCl2. However, the HCl is corrosive and environmental unfriendly. PdCl2 is a homogeneous catalyst that is difficult to be separated from reactant. As a solution to these problems, we developed a more environment friendly two-stage method for the synthesis of MFA with cellulose. In the first stage, cellulose was converted to chloromethylfurfural (CMF) in a toluene/NaCl aqueous solution biphasic system (see Fig. S1 and S2 in ESI†). As what has been reported in our previous work,8 this process is a tandem reaction that involves the hydrolysis of cellulose to glucose, the isomerization of glucose to fructose, the dehydration of fructose to 5-hydroxymethylfurfural (HMF) and the chlorination of HMF to CMF. NaCl is non-toxic, cheap and abundant in seawater. Different from HCl, NaCl is neutral and non-toxic. Therefore, we believe that this method is advantageous in real application. As what has been reported in the previous work of Mascal et al.,7,9 the CMF as obtained is reactive and unstable even at room temperature. To overcome this problem, we converted it to MFA by the hydrodechlorination over a commercial Pd/C catalyst at 303 K (see Fig. S3 and S4 in ESI†). To do this, we immediately transferred the upper organic phase to another batch reactor after the reactor was cooled down to room temperature and conducted hydrodechlorination under the catalysis of Pd/C.
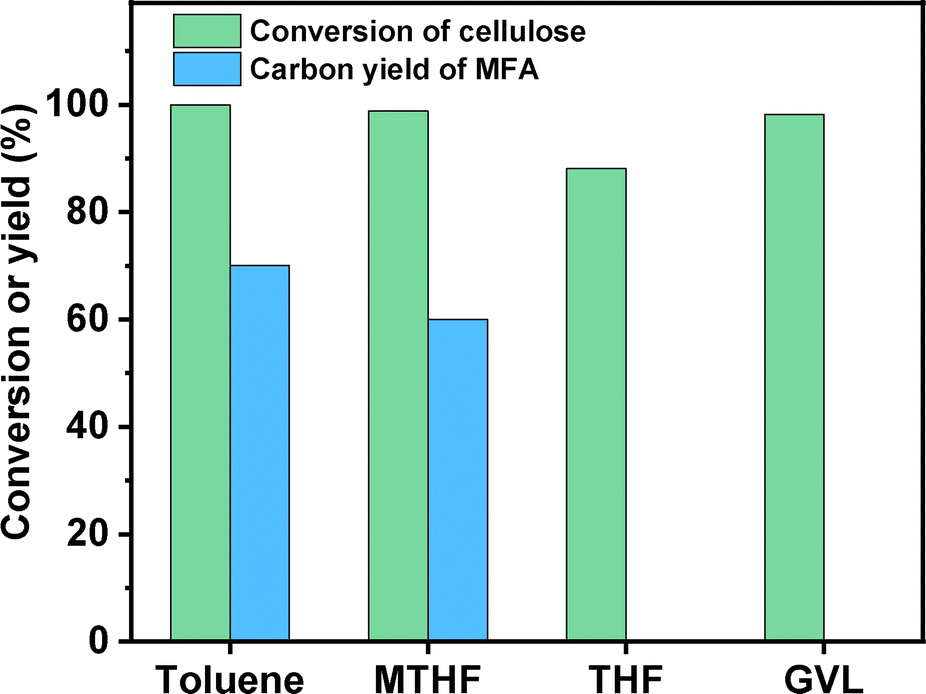
Based on Fig. 1, an evident solvent effect was observed in the conversion of cellulose to MFA. Among the investigated organic solvents, toluene and methyltetrahydrofuran (MTHF) demonstrated evident promotion effects on the conversion of cellulose to MFA. Compared with MTHF, the promotion effect of toluene is more evident. In contrast, the promotion effects of THF and GVL are not so evident under the investigated conditions. After being heated under a N2 atmosphere for 6 h and hydrodechlorinated for 2 h at 303 K, cellulose was completely converted, high carbon yield (70.1%) of MFA was obtained (see Fig. 1). In order to figure out the reaction pathway of cellulose to MFA, we did some additional experiments. Firstly, we investigated the effect of reaction temperature on the product distribution of the first stage. As we can see from the Fig. S5,† HMF was obtained as the predominate product at a relatively lower reaction temperature (e.g. 403 K). This is consistent with the previous work of Wang et al.10 and Hu's group.11 With the increment of reaction temperature from 403 K to 453 K, the major product switched from HMF to CMF. Based on this phenomenon, we can see that CMF was generated from the further chlorination of HMF (promoted by the proton in the high temperature water that can act as Brønsted acid catalyst for many reactions12). In the second stage, the CMF was converted to MFA and HCl by hydrodechlorination. To verify this speculation, we passed the gas phase product towards a moist litmus paper and found it turned red (indicating HCl was generated during the reaction).
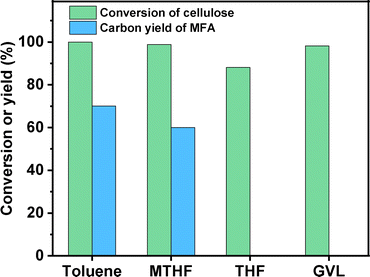 |
| | Fig. 1 Cellulose conversion and the carbon yield of MFA as the function of organic solvent. Reaction conditions: first stage: 473 K, 1 MPa N2, 400 rpm, 6 h; 0.3 g cellulose, 3 g NaCl, 10 mL H2O, 20 mL organic solvent were used in each test. Second stage: 303 K, 2 MPa H2, 400 rpm, 2 h; the organic phase product from the first stage and 0.03 g Pd/C was used in each test. | |
Based on above results, a reaction pathway was proposed for the conversion of cellulose to MFA (see Fig. S6†). Firstly, cellulose is hydrolyzed and isomerized to fructose under the promotion of NaCl.10 Fructose is dehydrated and chlorinated to CMF under the catalysis of high temperatures. Due to the salting out effect of NaCl, the CMF generated in the first stage was extracted from the aqueous phase to the organic phase and converted to MFA and HCl through hydrodechlorination over the Pd/C catalyst. As we know, HCl has high solubility in water. In real application, the HCl generated during the second stage can be easily adsorbed by NaCl aqueous solution in the first stage and used again for the production of CMF.
Subsequently, we also investigated the effect of heating time in the first stage on the synthesis of MFA from cellulose (see Fig. 2). When the heating time was extended from 4 h to 8 h, the carbon yield of MFA was increased from 46.4% to 77.5%. Further increment of heating time led to the decrease of MFA carbon yield. This can be rationalized by the low stability of CMF. Under the investigated conditions, it may be polymerized when the reaction time is too long. To verify this speculation, we compared the organic phase products that were generated from the reaction of cellulose at different reaction time. From Fig. S7,† we can see that with the extension of reaction time, the color of organic phase become darker and darker. This can be considered as evidence for the generation of polymer (or coke) at long reaction time.
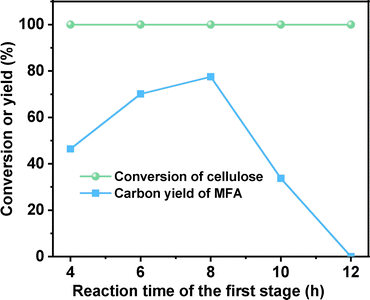 |
| | Fig. 2 Cellulose conversion and the carbon yield of MFA as the function of reaction time in the first stage. Reaction conditions: first stage: 473 K, 1 MPa N2, 400 rpm, 0.3 g cellulose, 3 g NaCl, 10 mL H2O, 20 mL toluene were used in each test; second stage: 303 K, 2 MPa H2, 400 rpm, 2 h; the organic phase product from the first stage and 0.03 g Pd/C was used in each test. | |
Decarbonylation of MFA to 2-MF
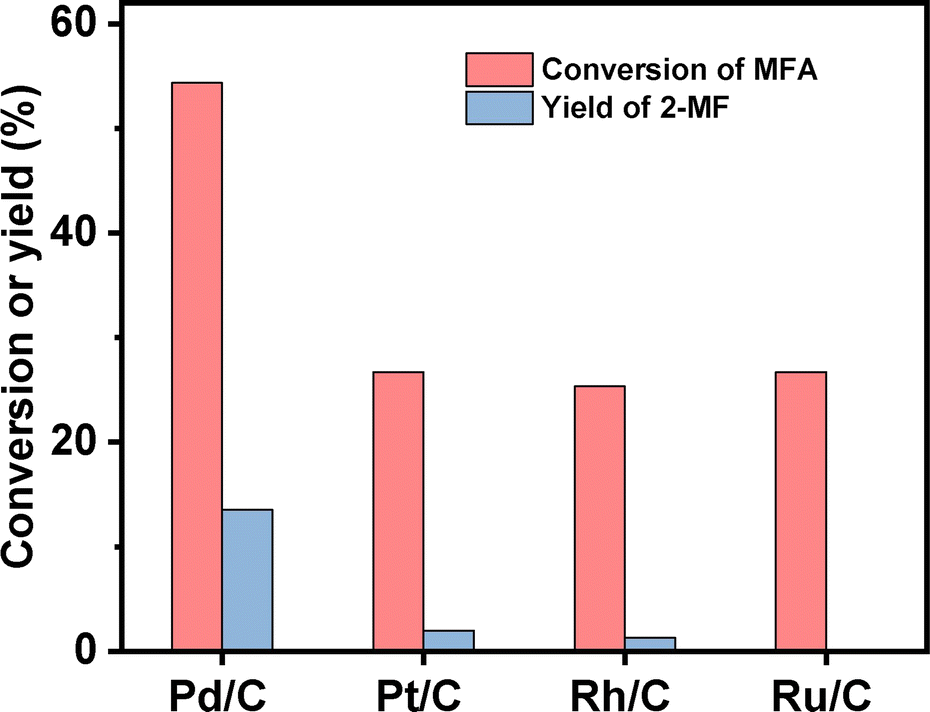
2-MF is a widely used intermediate in the production of drugs (such as vitamin B1, chloroquine phosphate and primaquine phosphate), pyrethroid pesticides and essence. In the previous work of Corma et al.2,13,14 and our group,15 a series of jet fuel range paraffins were synthesized by the HAA (or alkylation) of 2-MF followed by hydrodeoxygenation (HDO). Currently, 2-MF is produced in industrial scale by the hydrogenolysis of furfural from the hydrolysis/dehydration of hemicellulose that only account for small part of agriculture waste and forest residues. As a solution to this problem, it is still necessary to develop some new route for the synthesis of 2-MF with cellulose (the most abundant biomass). In the second part of this work, we studied the decarbonylation of MFA to 2-MF over a series of activated carbon loaded noble metal catalysts. Among them, the Pd/C exhibited the highest activity (see Fig. 3, S8 and S9 in ESI†). Over it, MFA was completely converted to 2-MF after reaction was carried out at 443 K for 4 h. According to literature,16 this result can be rationalized by the higher activity of Pd for the decarbonylation reaction.
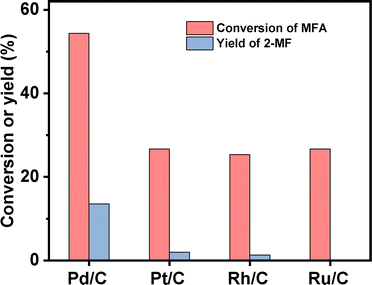 |
| | Fig. 3 Conversions of MFA and the yield of 2-MF over different activated carbon loaded noble metal catalysts. Reaction conditions: 403 K, 12 h, 400 rpm; 1 mmol MFA, 0.03 g catalyst, 5 mL ethyl acetate were used for each test. | |
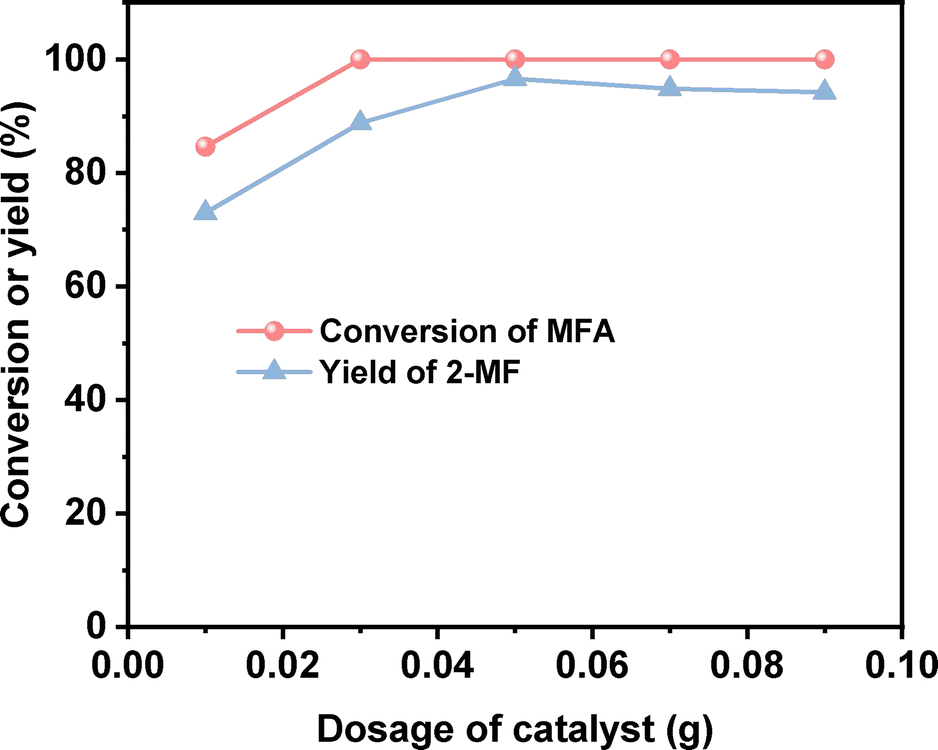
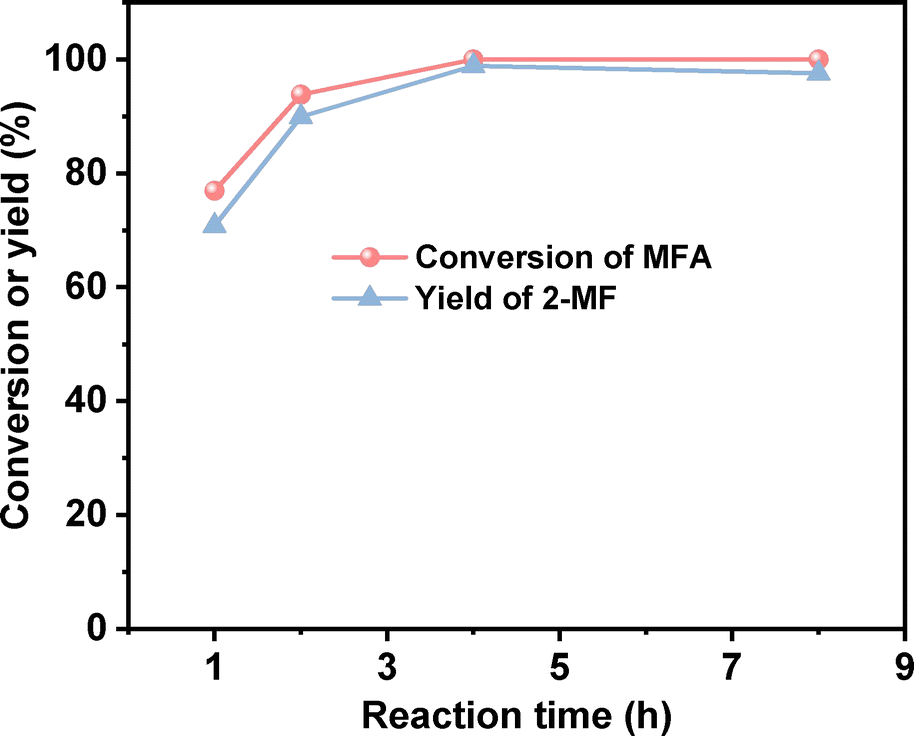
After the further optimization of reaction conditions (such as reaction temperature, catalyst dosage and reaction time), high yield (98.9%) of 2-MF was achieved when the reaction was carried out at 443 K for 4 h using 0.05 g Pd/C catalyst (see Fig. 4–6).
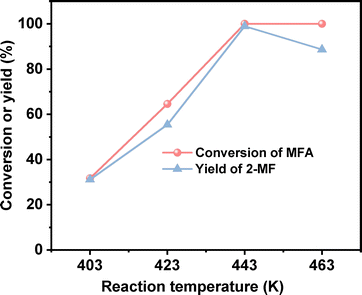 |
| | Fig. 4 Conversion of MFA and the yield of 2-MF over the Pd/C catalyst as the function of reaction temperature. Reaction conditions: 4 h, 400 rpm; 1 mmol MFA, 0.05 g Pd/C, 5 mL ethyl acetate were used for each test. | |
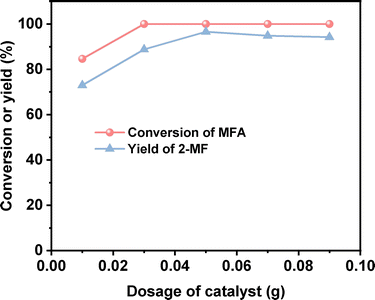 |
| | Fig. 5 Conversion of MFA and the yield of 2-MF over the Pd/C catalyst as the function of reaction temperature. Reaction conditions: 443 K, 4 h, 400 rpm; 1 mmol MFA and 5 mL ethyl acetate were used for each test. | |
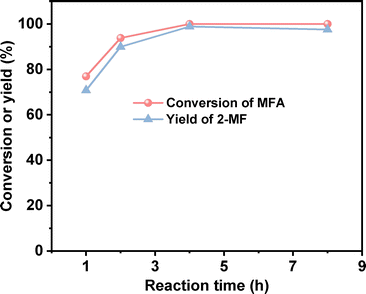 |
| | Fig. 6 Conversion of MFA and the yield of 2-MF over the Pd/C catalyst as the function of reaction time. Reaction conditions: 443 K, 400 rpm; 1 mmol MFA, 5 mL ethyl acetate and 0.05 g Pd/C were used for each test. | |
Hydroxyalkylation/alkylation (HAA) of MFA and 2-MF
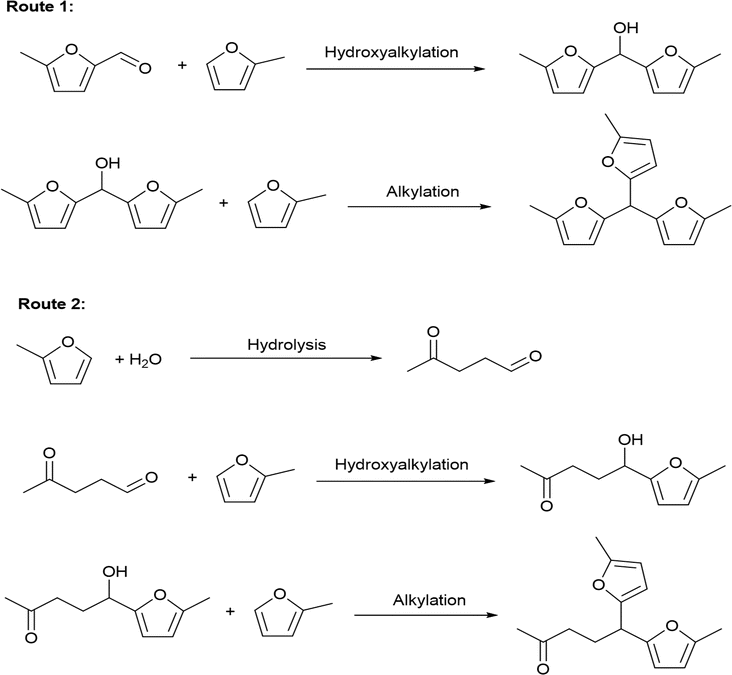
The solvent-free HAA reaction of MFA and 2-MF was carried out using acidic resins as the catalysts. Based on the analysis results from GC-MS and NMR (see the Fig. S10–S12 in ESI†), tri-(5-methylfuran-2-yl)-methane (TMFM) was identified as the main product. Besides TMFM, small amount of 5,5-bis(5-methylfuran-2-yl)pentane-2-one (BMFPO) was also identified as a by-product (see the Fig. S10, S13 and S14 in ESI†). According to the previous work of Corma et al.,13 BMFPO was generated from the HAA of 2-MF and its hydrolysis product 4-pentanone (see Route 2 of Scheme 2). Based on their carbon chain length and structure. Both TMFM and BMFPO can be used as the precursors for the synthesis of jet-fuel range branched alkanes.
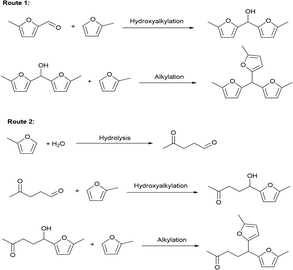 |
| | Scheme 2 Reaction pathways for the generation of TMFM and BMFPO during the reaction of MFA and 2-MF. | |
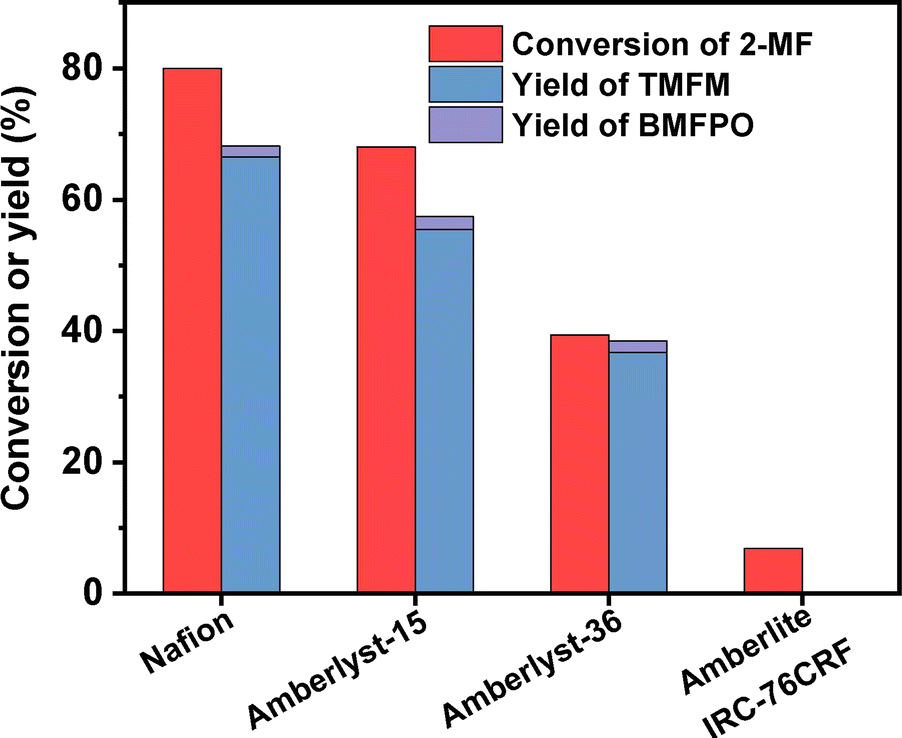
According to Fig. 7, Nafion resin exhibited the highest activity for the HAA of MFA and 2-MF among the investigated catalysts. Over it, ∼60% yield of TMFM was achieved after the reaction was carried out at 323 K for 2 h. Besides Nafion resin, Amberlyst-15 and Amberlyst-36 resins also exhibited good performances for the HAA of MFA and 2-MF. Although their activities are evidently lower than that of Nafion resin. In contrast, Amberlite IRC-76CRF resin is inactive for the HAA of MFA and 2-MF under the investigated conditions.
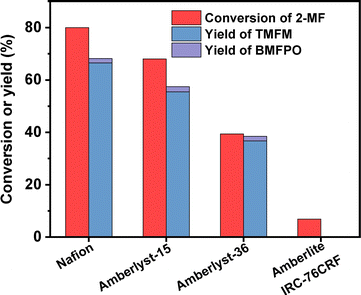 |
| | Fig. 7 Conversion of 2-MF and the yield of TMFM and BMFPO over different acidic resins. Reaction conditions: 323 K, 400 rpm, 2 h; 5 mmol MFA, 10 mmol MF, 0.05 g catalyst were used for each test. | |
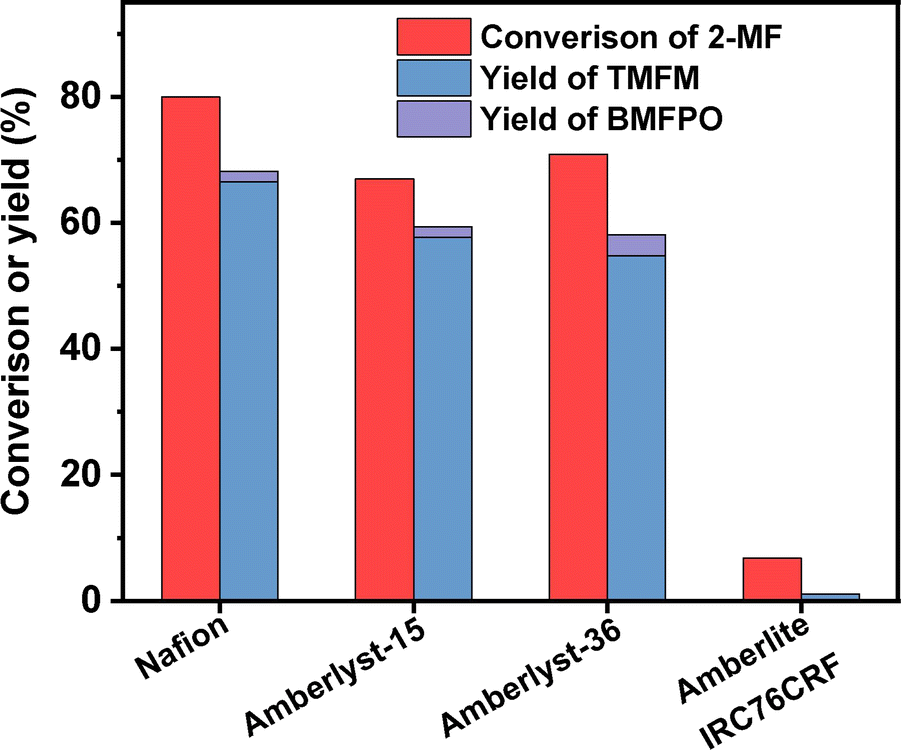
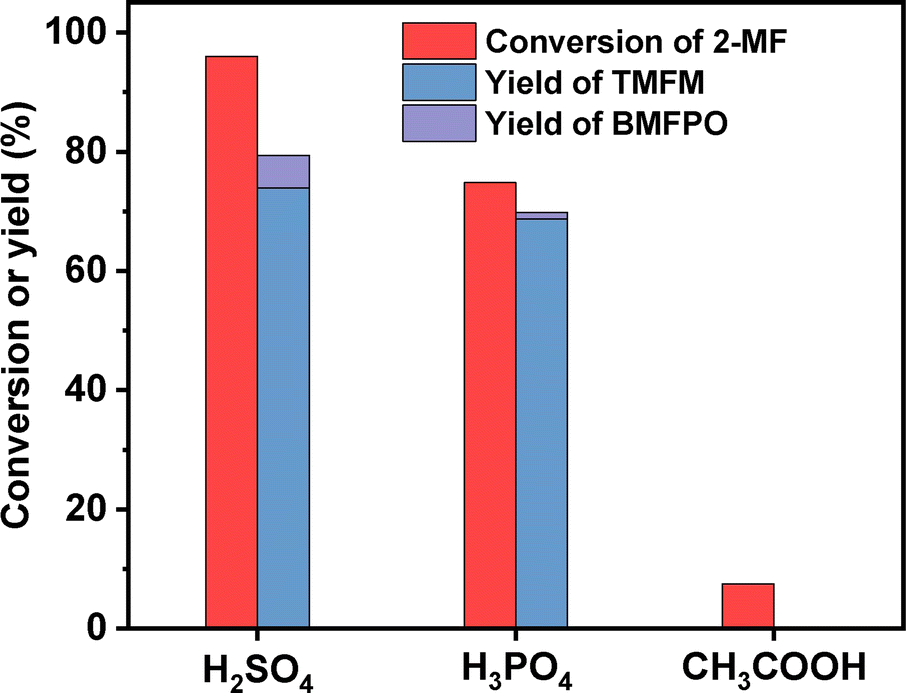
To figure out the reason for excellent activity of Nafion resin, we characterized the investigated acid resins by different technologies. Based on the results of NH3-chemisorption and N2-physisorption (see Table 1), no clear relationship was observed between the amounts of acid sites (or specific surface areas) of acid resins and their catalytic performances in HAA reaction. According to our previous work,17 the acid strengths of investigated catalysts in the order of Nafion > Amberlyst-15 > Amberlyst-36 > Amberlite IRC-76CRF. This is consistent with their activity sequence in the HAA of MFA and 2-MF. Amberlite IRC-76CRF is a weak acidic resin, specifically a carboxylic macroporous polypropylene resin.18 Amberlyst-15 and Amberlyst-36 are sulfonated cross-linked polystyrene ion exchange resins.19 Based on the chemical structures of acidic functional groups (–COOH and –SO3H), we can understand why the acid strength of Amberlite IRC-76CRF resin is lower than those of Amberlyst-15 and Amberlyst-36 resins. Nafion is a perfluorinated sulfonic acid resin that is also known as a super acid.20 The electron absorption effect of fluorine enhances the acid strength of –SO3H group. Consequently, the acid strength of Nafion is higher than that of Amberlyst-15 and Amberlyst-36. Therefore, we believe that the outstanding performance of Nafion resin should be comprehended by its higher acid strength that is resulted from chemical structure. To verify this hypothesis, we did some additional experiments. First, we compared the activities of H2SO4, H3PO4 and acetic acid (three representatives of strong acid, medium strong acid and weak acid). As we expected, the activities of these acids decreased in the order of H2SO4 > H3PO4 > CH3COOH (see Fig. 9). This is consistent with the sequence of their acid strength. Furthermore, we also compared the catalytic performances of Nafion resin, Amberlyst-15, Amberlyst-36 and Amberlite IRC-76CRF at the fixed amount of acid sites. From the result of Fig. 8, the activity of investigated catalysts decreased in the order of Nafion > Amberlyst-15, Amberlyst-36 > Amberlite IRC-76CRF. This is consistent with the sequence of their acid strengths.
Table 1 Specific BET surface areas (SBET) and the amounts of acid sites of the acid resins used in this work
| Catalyst |
S
BET
(m2 g−1) |
Acid site amountb (mmol g−1) |
|
Measured by N2-physisorption results.
Measured by NH3 chemisorption.
|
| Nafion |
5.0 |
3.60 |
| Amberlyst-15 |
31.4 |
5.74 |
| Amberlyst-36 |
1.6 |
1.11 |
| Amberlite IRC76CRF |
<1 |
0.21 |
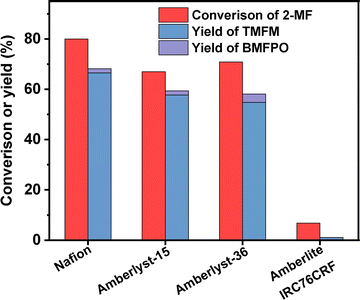 |
| | Fig. 8 Conversions of 2-MF and the yields of TMFM and BMFPO over different acidic resins with the fixed amount of acid sites. Reaction conditions: 323 K, 2 h, 400 rpm; 5 mmol MFA, 10 mmol 2-MF and acidic resin (the mass of acidic resin was calculated based on NH3-chemisorption result to keep the amount of acid site at 0.18 mmol) were used for each test. | |
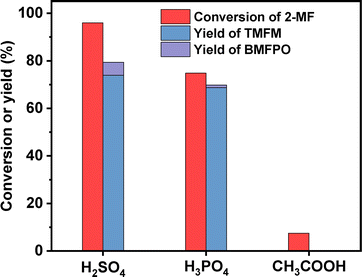 |
| | Fig. 9 Conversion of 2-MF and the yields of TMFM and BMFPO over different proton acids. Reaction conditions: 323 K, 2 h, 400 rpm; 5 mmol MFA, 10 mmol 2-MF, 0.001 equivalent of acid were used for each test. | |
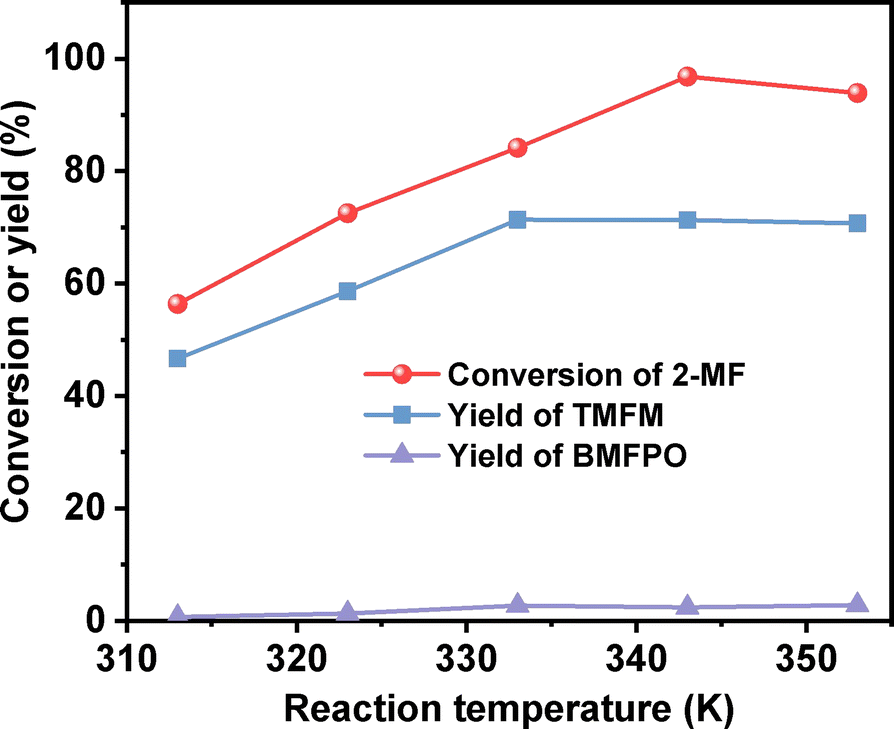
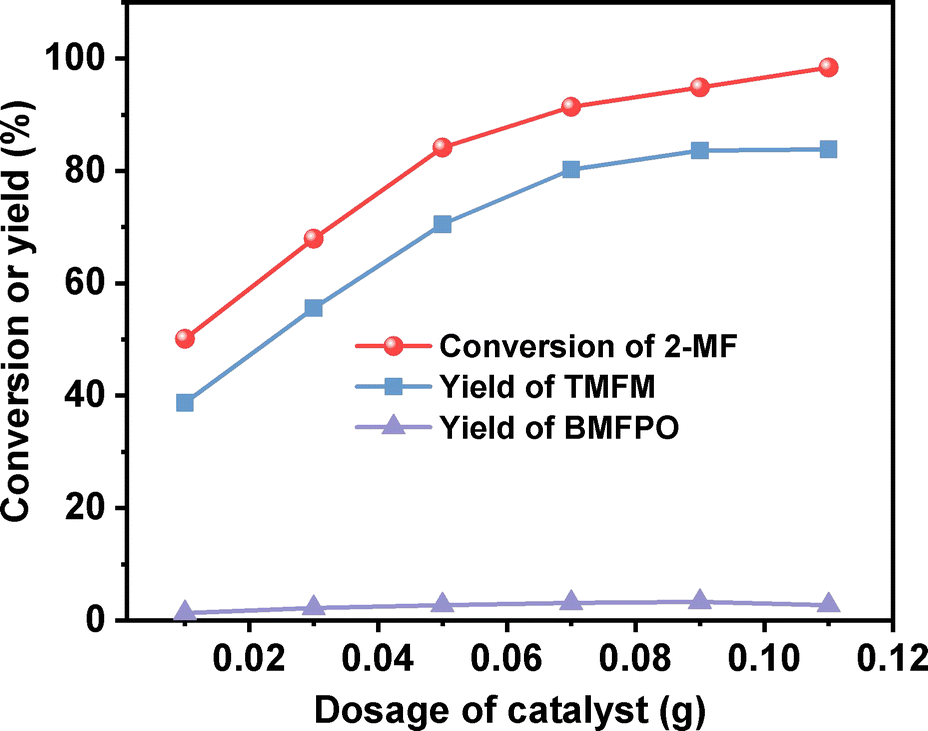
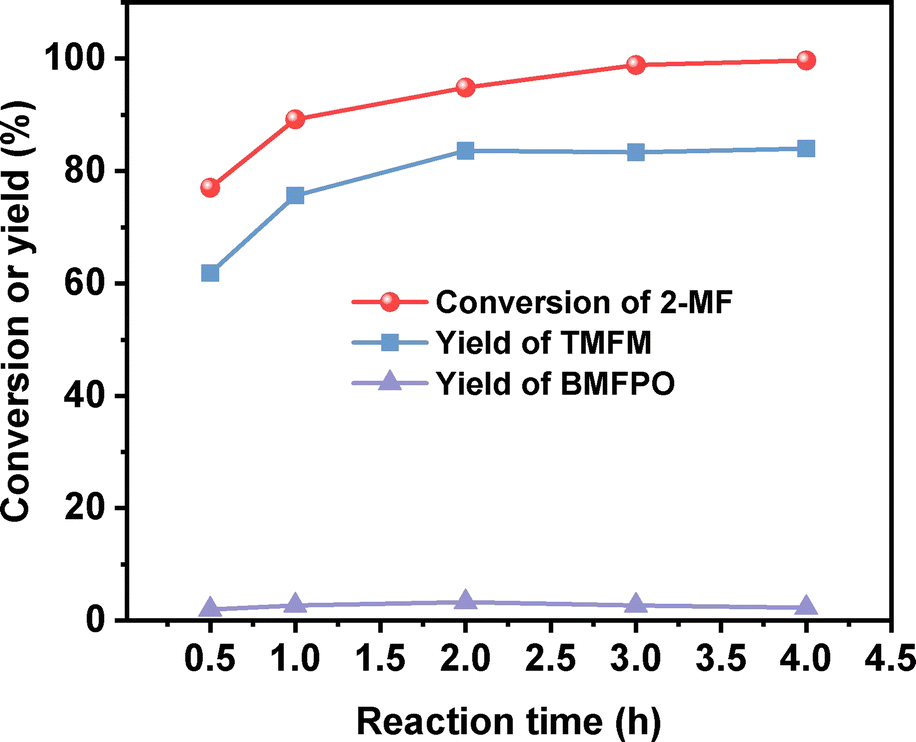
After further optimization of the reaction temperature and the dosage of Nafion resin (see Fig. 10–12), a good yield of 84% for TMFM was achieved after reaction was carried out at 333 K for 4 h at a Nafion resin dosage of 0.09 g.
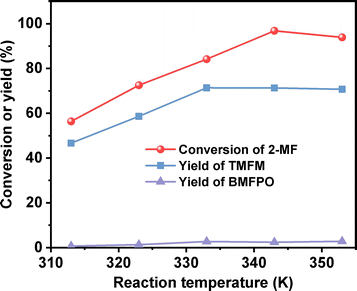 |
| | Fig. 10 Conversion of 2-MF and the yields of TMFM and BMFPO over the Nafion resin as the function of reaction temperature. Reaction conditions: 2 h, 400 rpm; 5 mmol MFA, 10 mmol 2-MF, 0.05 g Nafion resin were used for each test. | |
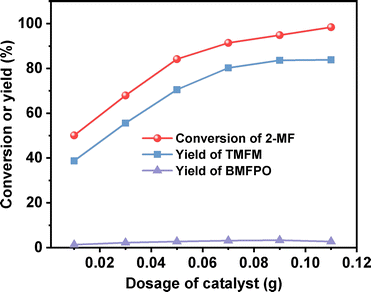 |
| | Fig. 11 Conversion of 2-MF and the yields of TMFM and BMFPO over the Nafion resin as the function of catalyst dosage. Reaction conditions: 333 K, 2 h, 400 rpm; 5 mmol MFA and 10 mmol 2-MF were used for each test. | |
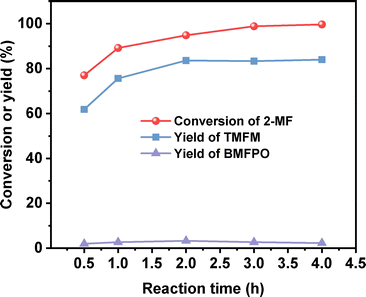 |
| | Fig. 12 Conversion of 2-MF and the yields of TMFM and BMFPO over the Nafion resin as the function of reaction time. Reaction conditions: 333 K, 400 rpm; 5 mmol MFA, 10 mmol 2-MF and 0.09 g Nafion resin were used for each test. | |
To fulfil the need of real applications, we also checked the reusability of Nafion, Amberlyst-15 and Amberlyst-36 resins. As shown in Fig. S15,† the Nafion resin is more stable than Amberlyst-15 and Amberlyst-36 resins. After being repeatedly used for 5 times, no evident deactivation was observed over it. This can be considered as another advantage of Nafion resin.
HDO of TMFM
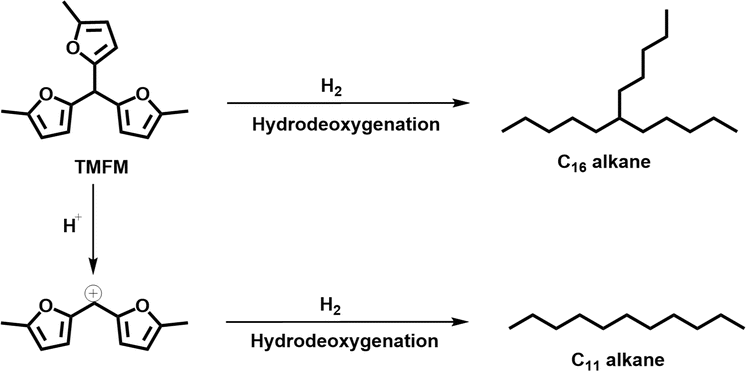
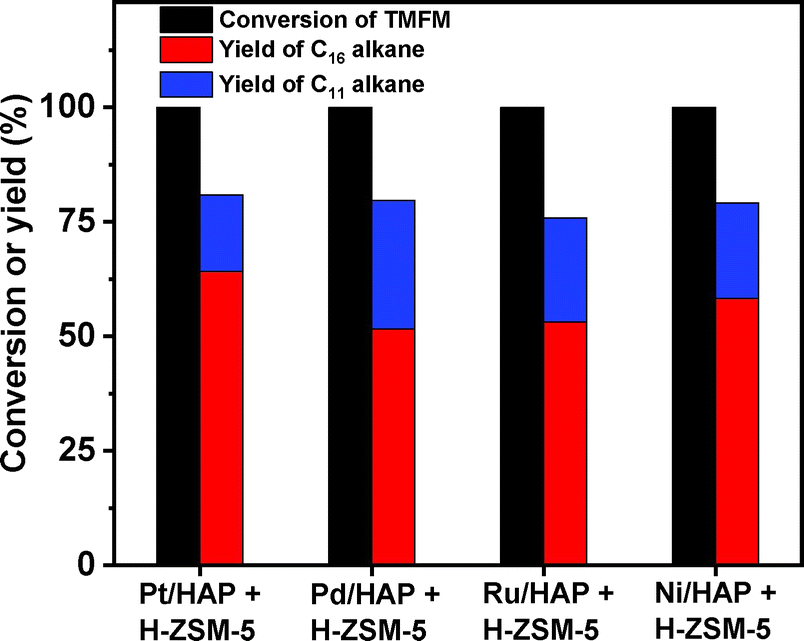
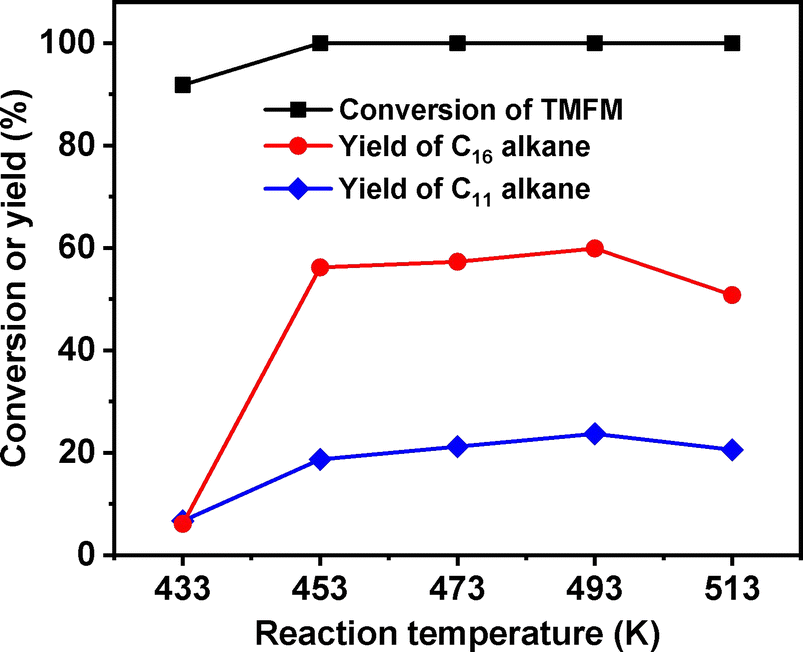
As the aim of this work, we investigated the HDO of TMFM under the co-catalysis of M/HAP (M = Pt, Pd, Ru, Ni) and H-ZSM-5 using cyclohexane as the solvent. To exclude the interference effect of solvent, we did a blank experiment in the absence of substrate. As we can see from Fig. S16,† no evident change of cyclohexane was observed after the blank experiment was carried out for 24 h under the investigated conditions. Therefore, we believe that the interference effect of cyclohexane can be exclude. Based on our analysis (see Fig. S17 and S18 in ESI†), jet fuel range C16 alkane was obtained as the main product. Besides C16 alkane, C11 alkane was also identified as a by-product from the HDO of TMFM (see Fig. S17 and S19 in ESI†). According to the literature2 and our previous work,21 C16 alkane was produced from the HDO of TMFM, while C11 alkane was generated from the hydrocracking reactions during the hydrodeoxygenation process (see Scheme 3). Due to the conjugation effect of the two furan rings in TMFM molecule, the carbocation generated from acid catalyzed cracking reaction at the branched site is more stable, which may be the reason why C11 alkane was obtained as the major by-product during the HDO of TMFM. Among the investigated catalysts, the combinations of Pt/HAP + H-ZSM-5 and Ni/HAP + H-ZSM-5 exhibited the best performances for the HDO of TMFM (see Fig. 13). Over them, high total carbon yields (∼80%) of C16 and C11 alkanes was achieved after the reaction was carried out at 453 K for 24 h. Taking into consideration of the lower cost and higher reserves of Ni than those of Pt, we think that the Ni/HAP + H-ZSM-5 catalyst is more promising in future application.
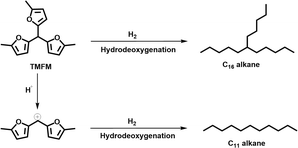 |
| | Scheme 3 Reaction pathways for the generation of C16 branched alkanes and C11 straight alkane from the HDO of TMFM. | |
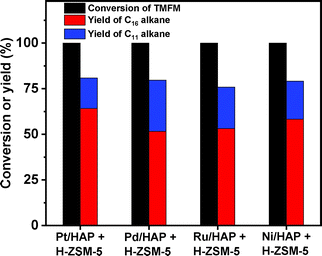 |
| | Fig. 13 Conversions of TMFM and the carbon yields of different alkanes from the HDO of TMFM over different catalysts. Reaction conditions: 453 K, 4 MPa H2, 500 rpm, 24 h; 0.1 g TMFM, 30 mL cyclohexane, 0.1 g M/HAP (M = Pt, Pd, Ru and Ni) and 0.1 g H-ZSM-5 were used for each test. | |
After further optimization of reaction temperature, higher total carbon yield (83.5%) of C16 and C11 alkanes was achieved after the HDO reaction was carried out at 493 K for 24 h (see Fig. 14). On the basis of our measurements, the density, freezing point and mass heat value of the C16 and C11 alkanes mixture as obtained were determined as 0.78 g mL−1, 207.8 K and 47.8 kJ g−1, respectively.
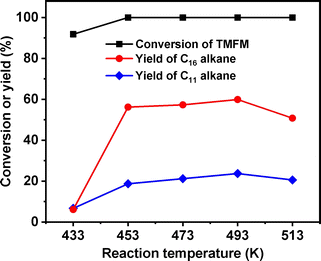 |
| | Fig. 14 Conversion of TMFM and the carbon yields of C16 and C11 alkanes over the Ni/HAP + H-ZSM-5 catalyst as the function of reaction temperature. Reaction conditions: 4 MPa H2, 24 h, 500 rpm; 0.1 g TMFM, 30 mL cyclohexane, 0.1 g Ni/HAP and 0.1 g H-ZSM-5 were used for each test. | |
Finally, we also compared this route with those reported in literature about the synthesis of jet fuel range alkanes with cellulose. As shown in Table S1,† our new route has an evidently higher overall carbon yield (54% vs. 25–47%) of jet fuel range alkanes from cellulose than the routes that used the levulinic acid (or its derivates) as intermediate. At the same time, it is also higher than the one (48%) reported in our recent work about the synthesis of jet fuel range polycycloalkanes with cellulose (using 2,5-hexanedione as the intermediate).22 In the very excellent work of Samec's group,23 jet fuel range moncycloalkane with strained four-membered ring was obtained by the self [2 + 2] cycloaddition of 6-hydroxy-2H-pyran-3(6H)-one derived from the hemicellulose in prehydrolysis liquors. In the future application, we can combine this route and Samec's route to improve the atomic economy for the production of jet fuel range alkanes with lignocellulose.
Conclusions
A new method was reported for the synthesis of jet fuel range C16 and C11 paraffins with cellulose. Firstly, MFA was obtained at a 77.5% carbon yield by the reaction of cellulose in toluene/NaCl aqueous solution at 473 K for 8 h, followed by the hydrodechlorination over the Pd/C catalyst at 303 K for 2 h. After the decarbonylation over the Pd/C catalyst, MFA was converted to 2-MF. A high 2-MF carbon yield (98.9%) was obtained after the reaction was carried out at 443 K for 4 h. Under the catalysis of acidic resins, the MFA and MF were converted to TMFM by HAA reaction. Among the investigated catalysts, Nafion resin exhibited the best performance for the HAA reaction, which can be explained by its higher acid strength. After being hydrodeoxygenated under the co-catalysis of Ni/HAP and H-ZSM-5, TMFM was converted to a mixture of C16 and C11 paraffins. As a potential application, the C16 and C11 paraffins obtained in this work can be used as sustainable aviation fuel.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
F. Cong and Z. Yu performed the catalyst preparation and activity tests. A. Wang and Y. Cong analyzed the data. N. Li and T. Zhang conceived the overall direction of the project. F. Cong., N. Li and T. Zhang co-wrote the paper. All the authors discussed the results and provided input for the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the supports from the National Key R&D Program of China (No. 2022YFA1504900), National Natural Science Foundation of China (No. 22078318).
Notes and references
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed; N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef PubMed; B. G. Harvey and R. L. Quintana, Energy Environ. Sci., 2010, 3, 352–357 RSC; C. Zhao, D. M. Camaioni and J. A. Lercher, J. Catal., 2012, 288, 92–103 CrossRef; E. R. Sacia, M. Balakrishnan, M. H. Deaner, K. A. Goulas, F. D. Toste and A. T. Bell, ChemSusChem, 2015, 8, 1726–1736 CrossRef PubMed; Z. Zhao, H. Shi, C. Wan, M. Y. Hu, Y. Liu, D. Mei, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 9178–9185 CrossRef PubMed; G. Li, R. Wang, J. Pang, A. Wang, N. Li and T. Zhang, Chem. Rev., 2024, 124, 2889–2954 CrossRef PubMed; S. Wang, A. Cheng, F. Liu, J. Zhang, T. Xia, X. Zeng, W. Fan and Y. Zhang, Industrial Chemistry & Materials, 2023, 1, 188–206 Search PubMed.
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef CAS PubMed.
- J. Xin, S. Zhang, D. Yan, O. Ayodele, X. Lu and J. Wang, Green Chem., 2014, 16, 3589–3595 RSC.
- F. Chang, S. Dutta and M. Mascal, ChemCatChem, 2017, 9, 2622–2626 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS PubMed.
- Z. Yu, G. Li, F. Lu, X. Si, A. Wang, X. Wang, Y. Cong, T. Zhang and N. Li, Chem Catal., 2022, 2, 595–609 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370–373 RSC.
- X. Li, Y. Zhang, Q. Xia, X. Liu, K. Peng, S. Yang and Y. Wang, Ind. Eng. Chem. Res., 2018, 57, 3545–3553 CrossRef CAS.
- Z. Jiang, J. Yi, J. Li, T. He and C. Hu, ChemSusChem, 2015, 8, 1901–1907 CrossRef CAS PubMed.
- N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725–2750 CrossRef CAS PubMed.
- A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chem., Int. Ed., 2011, 50, 2375–2378 CrossRef CAS PubMed.
- A. Corma, O. de la Torre and M. Renz, ChemSusChem, 2011, 4, 1574–1577 CrossRef CAS PubMed.
- G. Li, N. Li, Z. Wang, C. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, ChemSusChem, 2012, 5, 1958–1966 CrossRef CAS PubMed; G. Li, N. Li, S. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Chem. Commun., 2013, 49, 5727–5729 RSC; W. Wang, N. Li, S. Li, G. Li, F. Chen, X. Sheng, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2016, 18, 1218–1223 RSC; G. Ren, G. Li, Y. Zhang, A. Wang, X. Wang, Y. Cong, T. Zhang and N. Li, Sustain. Energy Fuels, 2022, 6, 1156–1163 RSC.
- Y. Hu, N. Li, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, ChemSusChem, 2017, 10, 2880–2885 CrossRef CAS PubMed.
- S. Li, N. Li, G. Li, L. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Green Chem., 2015, 17, 3644–3652 RSC; H. Tang, N. Li, G. Li, W. Wang, A. Wang, Y. Cong and X. Wang, ACS Sustainable Chem. Eng., 2018, 6, 5645–5652 CrossRef CAS.
- P. A. Riveros, Hydrometallurgy, 2004, 72, 279–290 CrossRef CAS.
- B. G. Harvey, M. E. Wright and R. L. Quintana, Energy Fuels, 2010, 24, 267–273 CrossRef CAS; R. Weingarten, G. A. Tompsett, W. C. Conner and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef.
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef CAS PubMed; K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef PubMed.
- G. Li, N. Li, J. Yang, L. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2014, 16, 594–599 RSC.
- Y. Liu, G. Li, Y. Hu, A. Wang, F. Lu, J.-J. Zou, Y. Cong, N. Li and T. Zhang, Joule, 2019, 3, 1028–1036 CrossRef CAS.
- D. Lebedeva, L. W. Schick, D. Cracco, W. Sangsuwan, G. Castiella-Ona, D. O. Silva, A. Marson, E. Svensson Grape, A. K. Inge, L. M. Rossi, E. Subbotina, A. Manzardo and J. S. M. Samec, Green Chem., 2024, 26, 7258–7267 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ad,
Yu
Cong
a,
Tao
Zhang
ad and
Ning
Li
ad,
Yu
Cong
a,
Tao
Zhang
ad and
Ning
Li