
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddressing the persistence of per- and poly-fluoroalkyl substances (PFAS): current challenges and potential solutions
Emeka J.
Itumoh
 ab,
Shailja
Data
ab,
Jack L.-Y.
Chen
bcd,
Melanie
Kah
*e,
Lokesh P.
Padhye
*fg and
Erin M.
Leitao
*ab
ab,
Shailja
Data
ab,
Jack L.-Y.
Chen
bcd,
Melanie
Kah
*e,
Lokesh P.
Padhye
*fg and
Erin M.
Leitao
*ab
aSchool of Chemical Sciences, The University of Auckland, New Zealand
bThe MacDiarmid Institute for Advanced Materials and Nanotechnology, Victoria University of Wellington, New Zealand. E-mail: erin.leitao@auckland.ac.nz
cSchool of Science, Auckland University of Technology, New Zealand
dDepartment of Biotechnology, Chemistry and Pharmaceutical Sciences, Università degli Studi di Siena, Siena, Italy
eSchool of Environment, The University of Auckland, New Zealand. E-mail: melanie.kah@auckland.ac.nz
fDepartment of Civil and Environmental Engineering, The University of Auckland, New Zealand. E-mail: l.padhye@auckland.ac.nz
gThe New York State Center for Clean Water Technology, Stony Brook University, USA
First published on 14th August 2024
Abstract
The combined stability, mobility, and bioaccumulation of per- and poly-fluoroalkyl substances (PFAS) has prompted a global environmental crisis. PFAS have unique properties owing to their strong, hydrophobic C–F bonds, which result in their resistance to water, oil, chemicals, and heat. Applications of PFAS include their use as water-, grease-, and fire-proof coatings, emulsifiers, and surfactants, spanning most manufacturing sectors. The continued regulation of specific PFAS provides significant research opportunities for chemists and their collaborators across environmental, social, engineering, and materials sciences. Solutions in the areas of detection and analysis, immobilisation and destruction, and the creation of viable and safe alternatives are urgently needed. In this tutorial review, PFAS and their associated challenges are described, followed by a summary of existing solutions and future research opportunities.
Sustainability spotlightThe persistence of PFAS in the environment and their bioaccumulation in humans has prompted their regulation, improved detection and analysis, remediation and destruction, as well as their replacement by safer alternatives. The tutorial review has highlighted areas where chemists and collaborators can help to manage problems associated with PFAS. Specifically, in alignment with the UN sustainability goals, remediation of PFAS from water (Goal 6, clean water and sanitation), finding safe alternatives to replace PFAS in consumer products (Goal 3, good health and well-being) and regulators limiting the manufacture and use of PFAS which in turn, lowers the concentrations we are exposed to (Goal 12, responsible consumption and production). |
1 Overview of PFAS
1.1. Background and synthesis
The specific definition of per- and polyfluoroalkyl substances (PFAS) has evolved over time from “highly fluorinated substances” to a more concise definition that reflects the chemical structure of the compound.1 Accordingly, PFAS are organofluorine compounds that contain at least one CF3 or CF2 moiety in their structure (red, Fig. 1).2,3 Many of these substances are amphiphilic in nature as they also contain a hydrophilic head (blue, Fig. 1a). They have important applications in everyday products such as non-stick protective coatings, carpets, clothing, lubricants and as fire suppressors in aqueous film-forming foams (AFFFs).4 PFAS are characterised by strong C–F bonds and thermal and chemical stability, making PFAS resistant to environmental degradation.5 Their inertness can be explained by the electronegative fluorine atoms clustering around the carbon atom, shielding the atoms from chemical reactions. As a result of their widespread use, environmental persistence, high mobility, bioaccumulation, and toxicity, PFAS are now regarded as high-risk environmental chemicals,6,7 and are widely referred to as “forever chemicals”.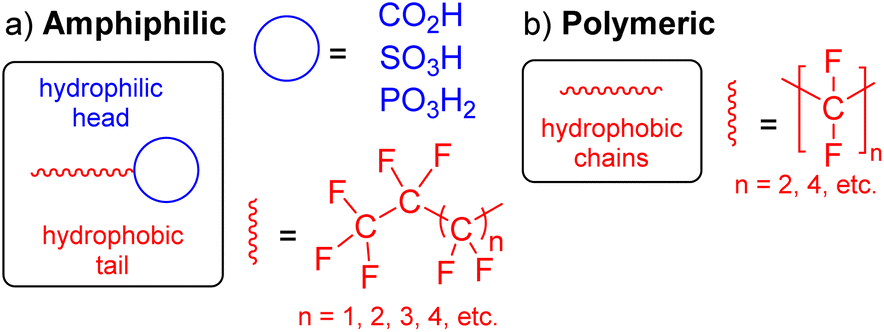 | ||
| Fig. 1 Generic structure of (a) amphiphilic (non-polymeric), and (b) polymeric PFAS. | ||
The accidental discovery of poly(tetrafluoroethylene) (PTFE) ([CF2]n, n = length of polymer chain; Fig. 1b) on April 6, 1938, at DuPont by Roy J. Plunkett was the first ever PFAS produced.8 At DuPont, tetrafluoroethylene (C2F4), made by zinc dichlorination of ClF2CCF2Cl, was stored in cylinders where it polymerised slowly over time to form a white solid, PTFE. After controlled polymerisation and polymer fabrication techniques, DuPont commercialised PTFE under the trade name Teflon® in 1948.8
PFAS are synthesised through two main processes: free radical electrochemical fluorination (ECF, also known as Simons Electrochemical Fluorination)9 and fluorotelomerisation (Scheme 1).3 In the ECF process, an organic compound (e.g., sulfonyl halide, CxHySO2X, X = F or Cl,10Scheme 1a) is dissolved in anhydrous HF for fluorination at the anode, usually nickel. As all of the C–H bonds are replaced with C–F bonds and the C–C multiple bonds are saturated with F atoms, the resulting acid, H+, is concomitantly reduced to hydrogen gas at the cathode.3,11 ECF was a dominant synthetic process for PFAS until the 1990s and usually results in a mixture of linear (70%) and branched (30%) PFAS.3,12 The yields of the products decrease with an increase in chain-length.9 Using ECF, 3M (formerly known as Minnesota Mining and Manufacturing Company) commercially synthesised perfluorooctanesulfonic acid (PFOS)13 from the perfluorooctanesulfonyl fluoride (POSF) precursor, as well as perfluorooctanoic acid (PFOA),14 to be widely applied in aqueous film forming foams (AFFF), treated textiles, and cookware coatings.
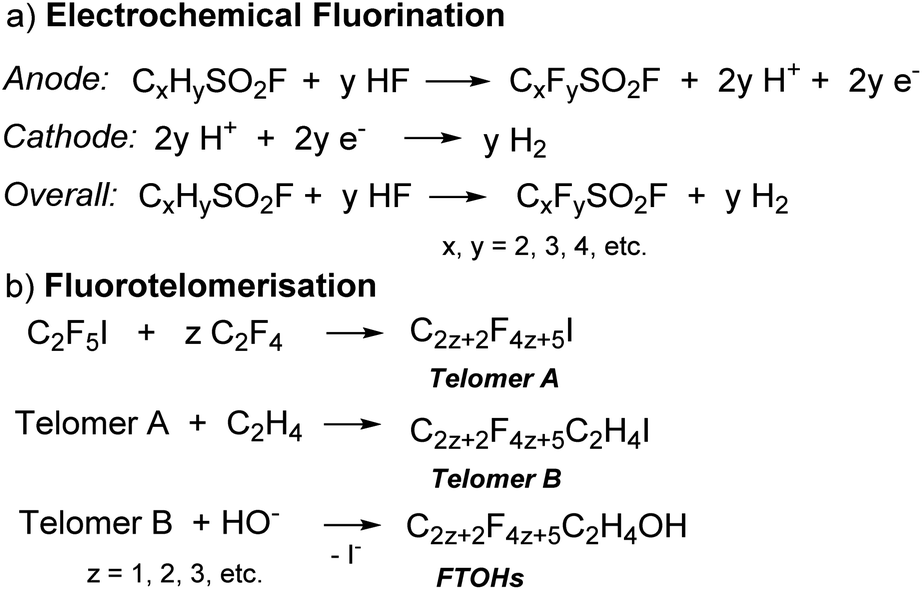 | ||
| Scheme 1 Synthetic routes to PFAS. | ||
A typical fluorotelomerisation process involves the reaction of perfluoroethyl iodide (C2F5I), the starting transfer agent or talogen,9,15 with the unsaturated tetrafluoroethylene (C2F4) (taxogen) to yield a mixture of even-numbered carbon linear perfluoroalkyl iodides (Scheme 1b, Telomer A). Telomer A is then reacted with ethylene (C2H4) to form fluorotelomer iodide (Scheme 1b, Telomer B). Telomer B further reacts with water or oleum to make fluorotelomer alcohols (FTOHs). Telomer A, Telomer B and FTOHs are the basic raw materials used to manufacture fluorotelomer-based surfactants (non-polymer) and polymer products.3
1.2. Classes
The functional groups and the length of the fluorocarbon chain dictate the class and subclass of a specific PFAS (Fig. 2).3 Polymeric PFAS have a longer chain backbone consisting of several repeat units (i.e., >10). There are three subclasses of polymeric PFAS: fluoropolymers, side-chain fluorinated polymers, and perfluoropolyethers (Fig. 2). Non-polymeric PFAS typically contain a backbone with 2–10 fluorocarbon repeat units and fall into either perfluoroalkyl (fully fluorinated carbon chain) or polyfluoroalkyl (partially fluorinated carbon chain) subclasses. Further classification is based on the different hydrophilic head functional groups attached to the fluorocarbon backbones, and most commonly are carboxylic (CA), sulfonic (SA), and phosphonic acids (PA). Ultrashort chain PFAS are classified based on the number of units (i.e., 1–3).17,18 Typical examples are perfluoroethane sulfonate (PFEtS, Et = ethyl = 2 units, CF3CF2SO3−), and perfluoropropanoic acid (PFPrA, Pr = propyl = 3 units, note that in the case of CAs the C in the COOH head group counts as 1 unit, CF3CF2CO2H). By contrast, short-chain PFAS typically contain 4–6 perfluorocarbon units,19 and long-chain PFAS contain ≥6 or 7 perfluorocarbon units.20 While many PFAS are open-chain molecules (linear, straight or branched), cyclic PFAS also exist. Examples include perfluoromethylcyclohexane sulfonate (PFMeCHS, CF3C([CF]2)5SO3H) and perfluoroethylcyclohexane sulfonate (PFEtCHS, CF3CF2C([CF]2)5SO3H).21,22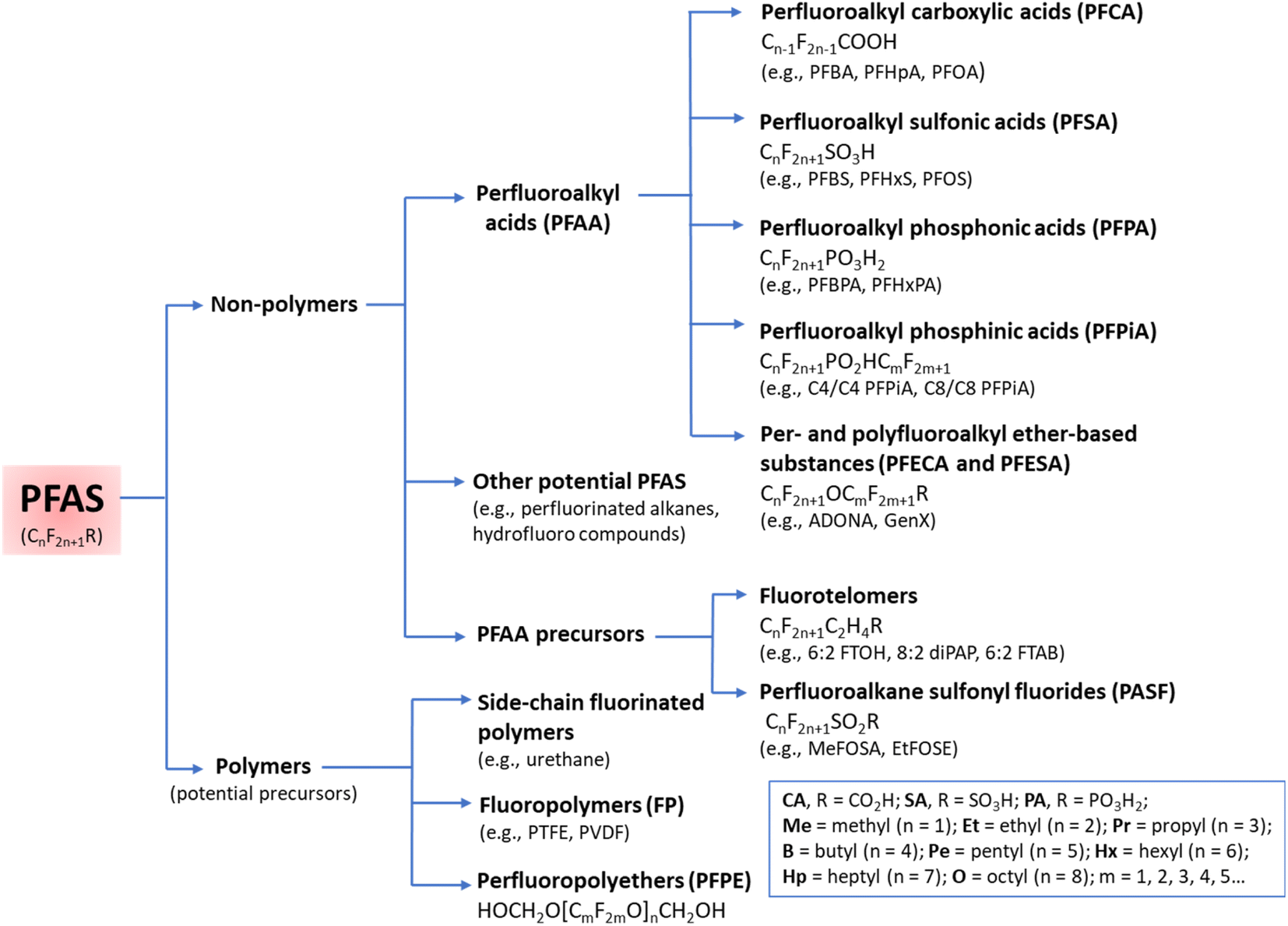 | ||
| Fig. 2 A summary of PFAS classes and subclasses. Adapted with permission from Z. Wang, J. C. DeWitt, C. P. Higgins and I. T. Cousins, Environ. Sci. Technol., 2017, 51, 2508–2518. Copyright 2017 American Chemical Society.16 | ||
1.3. Uses
PFAS possess important physical and chemical properties desired by chemical manufacturing industries, such as high thermal and chemical stability. The strong C–F bonds in PFAS (e.g., 485 kJ mol−1c. f. 346 kJ mol−1 for C–C) requires temperatures >1000 °C to degrade and help to impart these features.8,23 Their complementary hydrophobic and hydrophilic structure (red tail and blue head, Fig. 1) is ideal for generating surfactants, and their hydrophobicity and lipophobicity are useful in water-proof24 and grease-proof25 coatings. Hence, PFAS are used in numerous sectors for the manufacture of everyday items (Table 1). Recent regulations (see Section 2), have prompted the development of safer alternatives for both essential (e.g., selected medical applications, occupational clothing, Nafion™ membranes) and non-essential (e.g., food packaging, personal care products) uses of PFAS (Table 1 and ensuing discussion in Section 5).31,34| Sector | Desirable properties | Uses | Potential alternatives |
|---|---|---|---|
| Food26,27 | • Grease/oil-repellent | • Fast-food, food, and microwavable wrappers, containers, trays, and bowls | • Physical: cellulose based, clay, bamboo, wheat straw, aluminium |
| • Thermally stable | • Coatings for non-stick cookware | • Chemical: silicones, synthetic biopolymers, bio-waxes | |
| • Supercritical fluids used in ceramic powders | |||
| Textiles28 | • Water/oil/stain- repellent | • Occupational protective and durable outdoor clothing | • Silicones, hydrocarbon-based, dendrimer chemistry, inorganic nanoparticles |
| • Thermally and chemically stable | • Coatings for carpets and furniture | ||
| Personal care products29,30 | • Film forming | • Sunscreen | • Silicones, synthetic waxes, bio-based oils, fats |
| • Stabilising | • Hair, face, and body products | ||
| • Surfactant | |||
| • Emulsifier | |||
| Firefighting31 | • Surfactant | • Aqueous film-forming foams (AFFFs) | • Class B F3 foams: Hydrocarbons, detergents, siloxanes, proteins |
| • Fire-suppressant | |||
| Oil and gas mining26,32 | • Surfactant | • Enhance oil recovery | • Silicone/siloxane-based antifoaming agents |
| • mine floating | • Halogenated and radioactive tracers | ||
| • Well simulation additives | • Fluoropolymer material alternatives: metal alloys, ceramic-based, epoxy-based, nylon | ||
| • Solution for hydrostatic blockage | |||
| • Tracers in geological communication | |||
| Electronics26 | • Wetting agents | • Low-foaming noncorrosive components in solders | • Silicone based materials, bio-based polymers, fluorine-free coatings, glass-reinforced composites |
| • Water/oil-repellent | • Coating surfaces or casing | ||
| Agriculture26 | • Surfactant | • Pesticide products | • Non-fluorinated surfactants |
| • Coatings in fertilisers | |||
| Metal plating and finishing processes33 | • Surfactant | • Electro plating and electroless plating | • Non-fluorinated surfactants and fume suppressants |
| • Fume suppressant | • Metal treatment (cleaning, etching, etc.) | • Mechanical controls | |
| • Corrosion inhibitor | |||
| • Wetting agents | |||
| • Improved bath stability |
1.4. Environmental concerns
One of the most significant contributors to PFAS in the environment is from fire-suppressants in the form of aqueous film-forming foams (AFFFs).35,36 It is estimated that the US military was the source of more than 70% of AFFFs used in the US. This resulted in the release of elevated concentrations of PFAS into the environment through AFFFs, mainly on military bases, at training sites, or at municipal airports.36 PFAS are mobile in the environment and are transported away from these sites contaminating soil, groundwater and surface water resources.
Wastewater treatment plant (WWTP) effluent is another significant contributor of PFAS in the environment due to PFAS-containing product usage, industrial discharge, and their incomplete removal during water treatment. In fact, recent studies have revealed an increase in PFAS concentrations in the liquid phase after secondary treatment, as PFAS precursors can biotransform to form PFAS. Additionally, due to their hydrophobicity, a significant portion of PFAS partitions to the solid phase in WWTPs.37 Indeed, reports have indicated that PFAS are present at detectable levels in biosolids used in agricultural soils in Australia.38 Plants and crops grown on biosolid-applied soils uptake PFAS and could serve as a significant exposure route of PFAS to humans and animals.39 Apart from the use of biosolids in agricultural soils, it used to be common practice to bury PFAS-contaminated products in landfills, which directly results in contamination of groundwater sources through leachate,40–42 and the pollution of air around landfills through landfill off-gassing.41
In addition to AFFFs and WWTP effluent, incineration of materials containing PFAS is another major contributor of PFAS in the environment. As of one of the main destruction methods of PFAS, incineration requires high temperatures (>1000 °C) to achieve chemical breakdown.35,43 Specifically, incineration temperatures of up to 1200 °C are required to destroy waste containing PFOS, while over 1000 °C is required to breakdown PFAS adsorbed on spent activated carbon, and ca. 1100 °C is required to destroy PFAS in the gas-phase.44 According to the US-EPA, CF4, which is the most difficult fluorinated organic compound to decompose, requires temperatures over 1400 °C.45 Despite this energy intensive process, incomplete destruction can occur, leaving small molecule PFAS and fluorine-containing by-products in the vicinity of the incineration facilities. A study of the concentration and distribution of PFAS in surface water and soil samples around a PFAS incinerator facility in the US found significant measurable amounts of PFOS (up to 8.3 μg kg−1), PFOA (up to 1.3 μg kg−1) and hexafluoropropylene oxide (GenX) (up to 1.5 μg kg−1) in soil samples and up to 19 ng L−1 and 11 ng L−1 of PFOS and PFOA, respectively, in surface water.46 Moreover, in southern China, a study of three municipal solid waste incineration plants found a high concentration of PFAS (up to 0.7 μg mL−1) in leachate, with variability attributed to the type of waste.47
PFAS in the atmosphere originate from the industrial stack and fugitive emissions from manufacturers and allow distribution far away from their direct sources.52 For example, a study on the non-targeted screening of PFAS in China found at least 34 emerging PFAS in airborne particulate matter.53 Since precipitation (rainfall or snowfall) is an effective scavenger of gas-phase and particle-bound pollutants from the atmosphere, atmospheric PFAS are deposited on soil, surface water, and vegetation.52,54 Moreover, melting glaciers serve as secondary sources of PFAS for the receiving lakes where PFAS are accumulated in lake sediments.54 More importantly, emerging evidence suggests PFAS accumulate at the air–water interface (AWI), through an interaction of the hydrophilic head with the surface of the water and the hydrophobic tail with the air. As sea spray aerosols (SSA) form from wind and waves, the aerosols become enriched with a surface microlayer containing PFAS.55,56 This is problematic as PFAS concentrations have been reported to be >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times higher in SSA when compared with seawater57 and are stable, mobile, long-term sources of PFAS.58
000 times higher in SSA when compared with seawater57 and are stable, mobile, long-term sources of PFAS.58
PFAS concentration, the length of the C–F chain, the presence of specific functional groups, the biotransformation of PFAS precursors, and the presence of organic matter are some of the factors that can affect the accumulation of PFAS in an organism.60 Analysis of the bioaccumulation factors (BAFs) for 27 genera of agricultural crops from 24 studies revealed that 45 PFAS accumulated less in reproductive and storage organs than in vegetative organs (e.g., leaves, shoot buds, root, and stem).61 This is likely because the vegetative organs are mainly responsible for the transport of plant nutrients, thereby accumulating PFAS in the process. A study of bioaccumulation of PFAS by benthic macroinvertebrates (worms, snails, and mussels) found that BAFs in worms (412.84 L kg−1) were higher than in snails (8.08 L kg−1) and mussels (27.12 L kg−1).62 In a human study of autopsy tissues, perfluorobutanoic acid was found to have accumulated in lung tissues (median value: 807 ng g−1) and in kidneys (median value: 263 ng g−1). Similarly, perfluorohexanoic acid was found to have accumulated in the liver tissues (median value: 68.3 ng g−1) and in the brain tissues (median value: 141 ng g−1). In addition, PFOA was found to have accumulated in the bone (median value: 20.9 ng g−1).63 Other reports have suggested that protein–PFAS interactions in humans could be the major molecular mechanisms responsible for the bioaccumulation of PFAS in specific human tissues. For example, the liver-type-fatty acid binding proteins in the liver and serum albumin have high binding affinity to PFAS, hence their accumulation in these tissues.64
Bioaccumulation of PFAS in human body tissues has been linked to some adverse health conditions.63,65 Toxicological studies in animals (mice) have correlated high concentrations of PFAS to endocrine disruption, delay in physical development, cancer and neonatal mortality.65–67 A study in zebrafish (Danio rerio) exposed to PFAS found abnormal ventroflexion of the tail and failed swim bladder inflation.68 In humans, PFAS have been reported to activate receptors associated with carcinogenesis, e.g. peroxisome proliferator-activated receptors (PPARs), due to their structural resemblance to fatty acids and can disrupt lipid metabolism, resulting in dyslipidemia.65,69 The binding of PFAS to PPAR has been linked to poor fetal growth and immune function.65
2 Policies, management, and regulation
The persistence of PFAS in the environment has prompted government agencies to adopt international agreements that aim to restrict the use of these chemicals (Stockholm Convention on Persistent Organic Pollutants) and to issue regulatory frameworks on the use of PFAS (Fig. 3). For example, in 2006 the US-EPA issued a PFOA stewardship program aimed at a 95% reduction in emission of PFOA and related chemicals by 2015, including a health advisory level of 70 ng L−1 for both PFOA and PFOS in 2016.71 In 2018, the US Agency for Toxic Substances and Disease Registry issued provisional daily oral minimal intake risk levels for PFAS as a guideline for the public. Similar regulatory frameworks have been issued in other countries.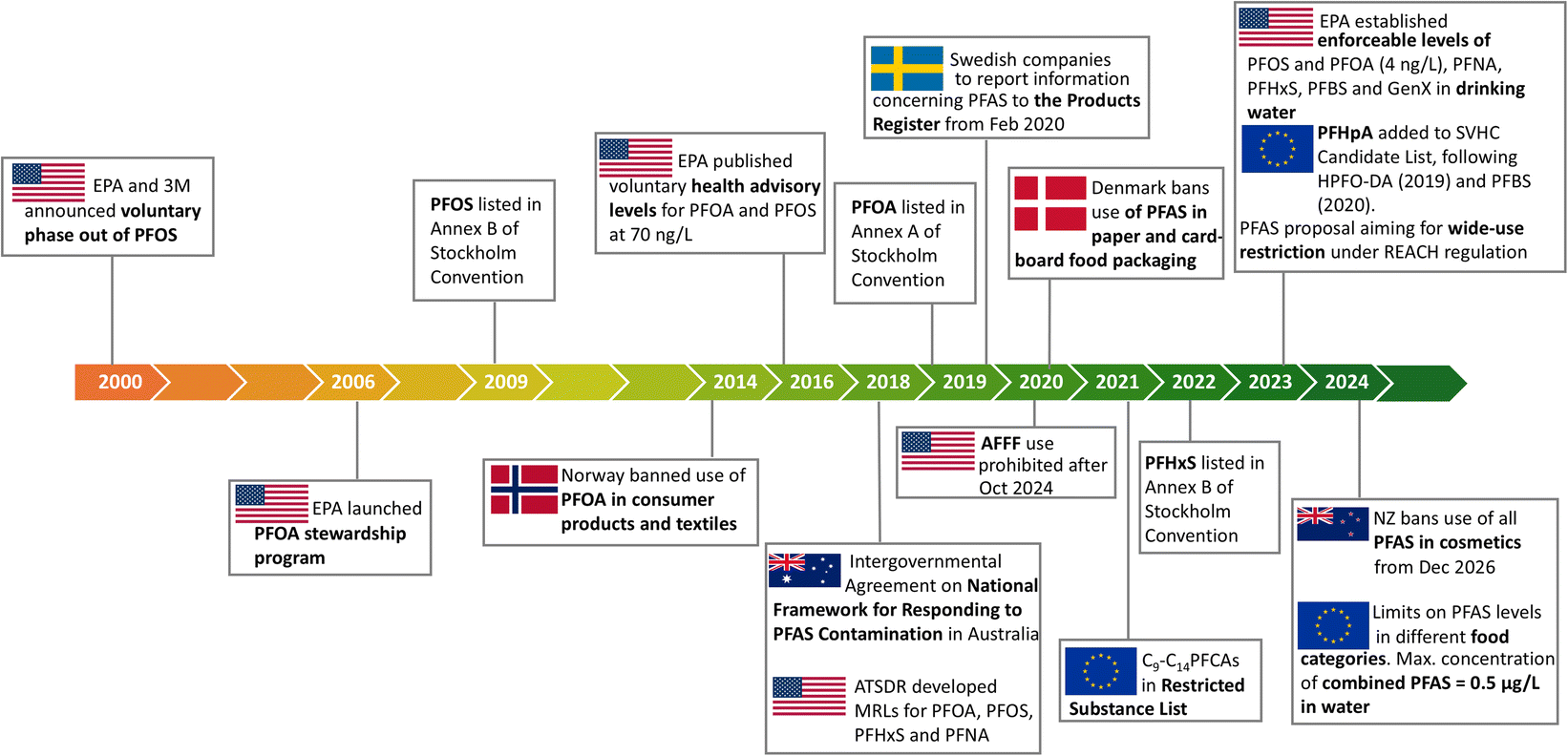 | ||
| Fig. 3 Examples of the evolving PFAS regulatory landscape. Adapted from C. Schiavone and C. Portesi, Applied Sciences, 2023, 13, 6696. Copyright 2023 MDPI.70 | ||
The US-EPA has also provided guidelines for the remediation and destruction of PFAS contamination and has recently dramatically reduced the drinking water health advisory levels.72 An enforceable drinking water maximum contaminant level (MCL) of 4 ng L−1 for PFOS and PFOA was proposed by the US-EPA in March 2023. Similarly, the US-EPA set a chronic oral reference dose of 3 ng kg−1 daily limit for the PFOA replacement, (GenX).35 In July 2022, the US House of Representatives passed a bill to limit PFAS in wastewater discharge from organic chemical manufacturers and mandated the manufacturers to provide supply production, use, and other data to the EPA. The law is expected to come into effect in 2024.73 The European Union (EU) stated that it was necessary to introduce a “strategy that addressed all PFAS through regulatory and non-regulatory interventions”, at the latest by 2025 and to be in effect by 2030, with the ultimate aim of phasing out PFAS at the EU-level.74 Under the EU's Persistent Organic Pollutants Regulation, PFOS was restricted for use in the EU, according to European Chemicals Agency (ECHA). In 2023, the ECHA also introduced a proposal to restrict the use of around 10000 PFAS in materials with the aim of reducing PFAS emissions into the environment, thereby making products and processes safer for people.75 In addition, to ensure global elimination from products, PFOS, and its derivatives were included in the international Stockholm Convention in 2009, while PFOA and its derivatives were banned in 2020. Similarly, perfluorohexane sulfonate (PFHxS), which was a replacement for PFOS, has been included in the Stockholm Convention, with a global ban expected to follow.76
Regulations on PFAS are country and region dependent (Fig. 3). For example, in 2014, Norway prohibited PFOA in solid and liquid consumer products, including carpets and textiles and set regulatory limits of 10 mg kg−1 PFOA in liquid mixtures, 1000 mg kg−1 PFOA in solid products and 1 μg m−2 PFOA in textile materials. Food contact materials including paper and cardboard with any level of fluorine content were banned by Denmark in 2020.77 While in the USA, the use of AFFF will be prohibited by Oct 202478 and NZ is one of the first to ban the use of PFAS in all cosmetics, from Dec 2026.79 These initiatives, while location specific, are crucial to generate international momentum towards further regulating PFAS in specific applications as well as across sectors.70,80
3 Detection and analysis
Detecting and analysing PFAS presents formidable challenges. Current analytical techniques have mainly centred on a limited subset of PFAS, perfluoroalkyl acids (PFAAs).81 The complex landscape of proprietary or mixed PFAS complicates the situation, as does the potential for a single PFAS parent to generate a mixture of intermediate transformation products. Targeted liquid chromatography coupled with high-resolution mass spectrometry (LC-HRMS) or tandem mass spectrometry (LC-MS/MS) captures many known PFAS, while non-targeted HRMS identifies additional suspected or previously uncharacterised PFAS (Fig. 4).82 Surrogate techniques through bulk organofluorine measurements, such as total fluorine and total organic fluorine (TOF) analysis, provide complementary information about the unidentified fraction of PFAS in environmental samples.83,84 This multifaceted approach,83 while not without challenges, is essential for unravelling the complex landscape of PFAS contamination, identifying precursors, and ultimately contributing to informed environmental management and health risk assessments.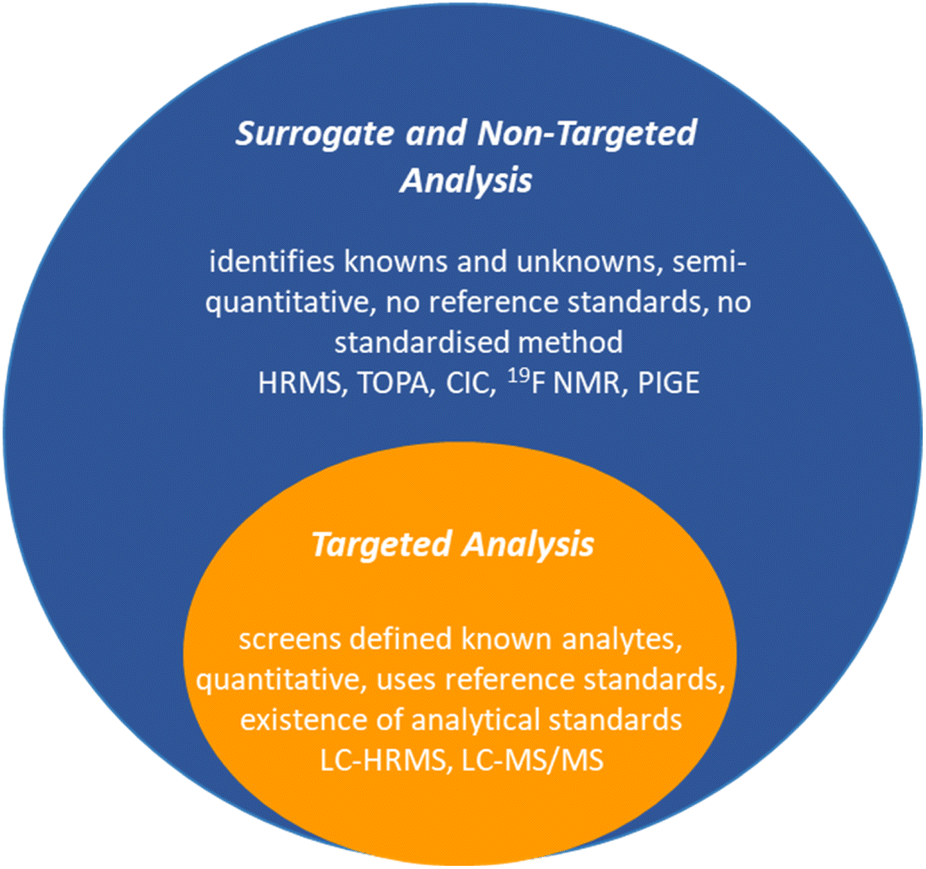 | ||
| Fig. 4 A generic summary of the analytical methods for PFAS detection and analysis. | ||
3.1. Targeted methods
Targeted analysis of PFAS screens analytes against a library of known analytical standards, typically <50 PFAS. LC-HRMS or LC-MS/MS are the most conventional, sensitive, and selective techniques for analysing PFAS from environmental matrices due to the presence of ionisable groups (e.g., COO− or SO3−). Mass spectrometry has an extremely low detection limit (in the ng L−1); however, it is cumbersome (usually requiring solid-phase extraction processes), expensive (ca. $200–300 USD per sample), time-consuming, and requires highly trained personnel to operate, which prohibits using this method for routine measurements.85As a result, complementary surrogate analytical methods aimed at screening and identifying unknown PFAS through their signature molecular masses (i.e., HRMS), or characteristic C–F bonds (i.e., TOF), are developing.86 With all of these analyses in hand, the list of PFAS and their transformation products continues to increase. A database of PFAS according to US-EPA has a list of over 12034 PFAS compounds (as of August 2021).87
3.2. Surrogate and non-targeted methods
In the realm of PFAS analysis, both surrogate techniques and advanced analytical methods play pivotal roles. The Total Oxidizable Precursor Assay (TOPA) selectively targets compounds that can be oxidised to form specific PFAAs, offering insights into PFAS precursor content by comparing PFAA concentrations before and after oxidation.88 Total fluorine analysis, achieved through techniques like combustion ion chromatography (CIC), quantifies the overall fluorine content in a sample, including both organic and inorganic fluorine species.89 Additionally, Fluorine-19 Nuclear Magnetic Resonance Spectroscopy (19F NMR) is a versatile tool for characterising organofluorine compounds and quantifying TOF,90 with a more recent adaptation focusing on the selective measurement of PFAS-related compounds through chemical shift monitoring.91 Meanwhile, Particle-Induced Gamma Ray Emission (PIGE) spectroscopy provides a unique surface analysis technique for elemental fluorine quantification through proton bombardment and gamma-ray emission.81,92 Other methods include, inductively coupled plasma/mass spectrometry (ICP-MS/MS), used to detect fluorine in unknown organofluorine compounds,93 and surface-enhanced Raman spectroscopy (SERS), a sensitive technique able to identify the unique molecular signatures in PFAS after interacting with a substrate (e.g., Au nanoparticles).94It is no surprise that the number of analytical methods to detect and analyse PFAS in environmental samples, such as drinking water, soils, sediments, biota, and biosolids is expanding. Importantly, this includes adapting existing technologies or developing new technologies towards field deployable sensing to allow for real-time on-site monitoring of PFAS.
3.3. Real-time on-site monitoring
A cheap, compact, and effective way to detect low levels of PFAS in the field is desirable. Despite its cost, mass spectrometry is an attractive method of on-site PFAS detection. Iodide time-of-flight chemical ionization mass spectrometry (iodide-ToF-CIMS), in particular, has shown promise for the more challenging to detect gas and aerosol phase PFAS in indoor air,95 or atmosphere in a semi-continuous online fashion.96,97 A significant benefit of this technique is that it avoids the need to perform the aforementioned cumbersome solid-phase extraction processes.In addition to investigating portable solutions for instrumentation,98 sensors are being developed for rapid PFAS identification, typically using optical or electrochemical detectors despite the fact that PFAS are not optically or electrochemically active.85,99
Colourimetric strategies use nanoparticles, which show a change in surface properties100–102 or redox dyes, which change intensity103,104 upon interacting with PFAS. Sensors based on fluorescence or luminescence use fluorescent dyes with molecularly imprinted polymers (MIPs) containing pockets designed to capture specific PFAS, appended to nanoparticles105–108 or luminescent metal–organic frameworks (MOFs) containing a chromophore ligand.109 When PFAS, in concentrations as low as μg L−1, enter the cavities, the fluorescence or luminescence is quenched or modified.
Electrochemical methods use redox probes, such as ferrocene carboxylic acids, which see a change in electron transfer resistance when PFAS molecules enter the cavities of MIPs, such as poly(o-phenylenediamine).110–114 A recent report of a sensor operating via PFAS interrupted energy transfer between fluorinated poly(p-phenylene ethynylene) and a fluorinated squaraine dye demonstrated PFAS detection in water with concentrations in the μg L−1 range.115 Detection by electrochemiluminescence, achieved through electrochemically imprinting ultra-thin graphitic carbon nitride nanosheets, generated a sensitive sensor selective for PFOA.116 It has been shown that switching to an impedimetric detection method by employing nanoporous MOF geometry with interdigitated microelectrodes dramatically increased sensitivity with detection limits in the ng L−1 range.117 Photochemical sensors, comprised of BiOI nanoflake arrays on screen-printed electrodes containing grafted MIP in the form of a disposable sensing strip, have been developed to analyse μg L−1 concentrations of PFOSF in real water samples.118
In another study, researchers have leveraged the amphiphilic properties of PFAS to develop a water-based sensor capable of measuring surface and interfacial tensions. The tensions exhibit an increase in emission intensity, based on the differential behaviour at the interfaces of complex droplets, when PFAS surfactants in mg L−1 concentrations are present.119
Despite being very promising, these techniques for detecting and analysing PFAS require more validation to demonstrate their applicability to real samples with low concentrations and matrix effects.
3.4. Opportunities for contributions from chemists
In order to address the current limitations in effectively detecting and analysing PFAS, and to remain current with the decreasing allowable concentrations of PFAS, particularly in drinking water, analytical instrumentation will continue to become smaller, cheaper, and more effective over time. As this occurs, there are significant opportunities for chemists to contribute to the improved detection (lower limits) and analysis of PFAS (managing the increasing library of PFAS and their derivatives). Examples of these include:(1) Increased understanding of PFAS exposure sources, including consideration of potential contamination from sampling, use of laboratory consumables, and instrumentation parts.120
(2) Development of the lab-scale PFAS detection methods into practical commercial-scale real-time on-site PFAS monitoring in all media with very low detection limits (from mg L−1 to μg L−1 to ng L−1).
(3) Improved analysis of environmentally induced PFAS by-products alongside an increased understanding of their thermal, oxidative, mechanical, and biological degradation mechanisms.
4 Separation and destruction technologies
The major sites for PFAS point source contamination are PFAS manufacturing plants, consumer product manufacturing plants, firefighting foam discharge locations (e.g., military training areas), large-scale cleaning facilities, wastewater treatment facilities, waste disposal facilities, and landfills.121 All of these result in the widespread deposition of PFAS in our soils and in our waterways, enabling them to enter the food chain.Current methods used commercially to remediate PFAS include sorptive removal from aqueous solution, burying waste in lined landfills (with or without stabilisation), or incineration for contaminated spent sorbents, soils, and products.122 Unfortunately, all these techniques have important limitations and they rarely apply across all PFAS. For instance, incineration suffers from high energy consumption, products of incomplete combustion, and cannot be used for liquid samples.123–126 The destruction of PFAS is complicated by the strength of the C–F bond (which increases with an increase in number of fluorine bonds on C), the different types (i.e., variety of tail lengths, head groups) and concentrations, as well as where they exist in the environment (i.e., waters, solids, air).127,128
This presents an opportunity to develop more cost-effective, selective, and efficient technologies. Ideally, a treatment train or tandem approach to managing PFAS in the environment can be employed to achieve both their separation from complex media and their complete destruction. The desired characteristics of an effective technology for managing PFAS include its ability to remove different types of PFAS, including long-chain, (ultra) short-chain, and more complex derivatives, and stage of development (maturity) as well as practicality across different media (e.g. water, solids) (Table 2). From this summary, it is clear that there is significant room for improvement (yellow and red) across all technologies.

|
4.1. Separation technologies
Separation of PFAS, from aqueous solution by immobilisation, or soil and sediments by mobilisation, involves the use of natural (e.g., clay, plants),172,173 semi-natural (e.g., activated carbon)134,174,175 or synthetic (e.g., imprinted polymer)167,168 materials as sorbents or resins. These technologies rely on either adsorption, or absorption.Adsorption is a reversible process whereby adsorbate molecules are transferred from a fluid bulk phase and stick to a solid surface phase.43,185 Adsorption of PFAS onto solid surfaces has been explored in literature as one of the effective methods of PFAS removal from contaminated media because of their easy design, cost-effectiveness and simple operation.135 Popular adsorbent materials include granular activated carbon, biochar, aminated rice husk, clay, molecular imprinted polymer, and zeolite. Granular activated carbon, clay, and biochar materials were used in situ to immobilise short and long-chain PFAS in contaminated artificial soil samples.134 One study reported that the addition of granular activated carbon to contaminated soils reduced leachability through chemical stabilisation between 55.8–99.9%. In addition, the leachability was further reduced to 87.1–99.9% by binding with cement. Foam fractionation is an adsorptive separation technique that functions without the use of solid adsorbents.186 The use of the foam fractionation method in PFAS remediation is based on the surface-active properties of PFAS which facilitates the separation of PFAS from contaminated liquid samples by extraction using rising foam. The foam is collected and collapsed (known as foamate) to give a low-volume residual liquid waste stream.187 In a typical setup for foam fractionation, gas bubbles are introduced into a liquid containing surface-active substances, where the surface-active substances attach to the gas–liquid interface of the bubbles to form foam. The foam is stabilised by decreasing the surface tension of the gas–liquid interface, thereby enabling the formed bubbles to create an emerging foam column above the liquid level. The foam column is collected and then collapsed mechanically or by reduced pressure.188 Foam fractionation can be operated by batch or continuous modes.188 Buckley et al.140 explored foam fractionation to remove long-chain PFAS from contaminated water. The study found 10–90% removal efficiency for the PFHxS, PFOA, and PFOS studied. However, the method was unable to remove short-chain PFAS.
In a study using three native Australian plant species, the study suggested that the plants have potential to reduce PFAS concentration in stormwater, with mean overall percentage removal efficiency of about 53%.194 In general, the report found that less PFOA than PFOS accumulated in the roots, while more PFOA than PFOS accumulated in the shoots.
4.2. Destruction technologies
While incineration is industrially viable and widely used to degrade PFAS on solids, it results in the formation of toxic HF gas and other gaseous fluorocarbons.41,43,155 In addition, incineration requires high temperatures (>1000 °C), enough to break the hydrophobic C–F bonds in PFAS, and is an energy intensive process.35,43 To this end, the US-EPA challenged researchers to discover new technologies that can remove >99% PFAS in unused AFFF with less harmful by-products than incineration.195 Several PFAS destruction technologies have since emerged (Table 2).In an electrochemical oxidation process, a potential difference or an electric current is applied between an anode and a cathode to generate reactive oxidising radicals at the electrode surface, which are used to degrade pollutants.146 Electrochemical oxidation processes can occur through direct and indirect oxidation mechanisms. In the indirect oxidation mechanism, reactive oxidising radicals are electrochemically generated in situ at the electrode, which are used to degrade PFAS. However, in the direct oxidation mechanism, PFAS are degraded at the anode through a direct electron transfer reaction between the PFAS and the anode surface.145–147
During photochemical oxidation reactions, PFAS are degraded as a result of their interactions with excited state oxidising species (e.g., HO˙, 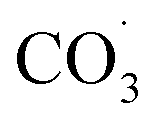 , H˙) in the presence of light.196,197 Efficient photodegradation of PFAS can occur at UV light wavelengths <220 nm (in the vacuum UV range). However, at longer wavelengths (>220 nm), PFAS does not absorb UV light,197 thereby direct photodegradation of PFAS becomes ineffective.198 Liu et al. reported 93–100% total defluorination of perfluorinated and fluorotelomer carboxylates and sulfonates through integrated redox photochemical processes148 as well as near complete defluorination and mineralisation of most PFAS in AFFF.199 The oxidation process was through hydroxyl radical treatment, while the reduction process was through UV-sulfite treatment.
, H˙) in the presence of light.196,197 Efficient photodegradation of PFAS can occur at UV light wavelengths <220 nm (in the vacuum UV range). However, at longer wavelengths (>220 nm), PFAS does not absorb UV light,197 thereby direct photodegradation of PFAS becomes ineffective.198 Liu et al. reported 93–100% total defluorination of perfluorinated and fluorotelomer carboxylates and sulfonates through integrated redox photochemical processes148 as well as near complete defluorination and mineralisation of most PFAS in AFFF.199 The oxidation process was through hydroxyl radical treatment, while the reduction process was through UV-sulfite treatment.
In sonochemistry, ultrasound radiation in the range of 20–1000 kHz is applied to molecules to achieve chemical reactions through a physical phenomenon called acoustic cavitation. This process creates localised spots for chemical reactions, which can reach pressures and temperatures of 500 atm and 5000 K, respectively.200 Reactive radical species such as HO˙ and Cl˙ can be generated in the hot spots (e.g., from the decomposition of H2O to yield HO˙ and H˙)197,200 and are able to degrade pollutants, including PFAS. Sonochemical reactions can occur as quickly as a fraction of a second.201 A study of low frequency (20 kHz) sonochemical degradation of PFOS and PFOA in water showed >90% degradation efficiency at 20 °C under acidic conditions.149
Supercritical water oxidation is regarded as an advancement in wet air oxidation,202 and is postulated to mineralise organic waste samples.203 This method, based on chemical oxidation in supercritical H2O (>374 °C and 22.11 MPa) uses H2O2, air, or O2 as the oxidising agent.204 In a typical supercritical water oxidation process, H2O and CO2 are formed from the oxidation of organic compounds, while heteroatoms such as F, S, P and Cl react to form HF, H2SO4, H3PO4 and HCl, respectively.202 The supercritical water oxidation technology was applied to successfully achieve 99.99% destruction of PFAS in aqueous matrices, and its efficiency was not hampered by co-contaminants such as volatile organic compounds and petroleum hydrocarbons.150
The degradation of PFOA using photoionization in a UV/sulfite system (254 nm) was found to be dependent on the pH of the initial solution in generating eaq− species. In acidic media, eaq− are quenched by reacting with H+ thereby suppressing the decomposition of PFOA in water while under alkaline conditions the H˙ and HO− react to regenerate the eaq− which enhances the defluorination of PFOA (Scheme 2).208 Additionally, the distribution of sulfite species (e.g., SO32− and HSO3−) varies with pH. Hydrogen sulfite, HSO3−, dominates at pH values lower than its pKa (ca. 7) and at pH 9.2 the concentration of sulfite, SO32−, is at its maximum (99%) efficiently able to reduce PFOA. Sulfite is more photoactive, hence able to generate more eaq−, as compared to hydrogen sulfite due to its more significant spectral overlap at wavelengths typical of UV irradiation sources.209 Overall, basic conditions are favored in UV/sulfite system.
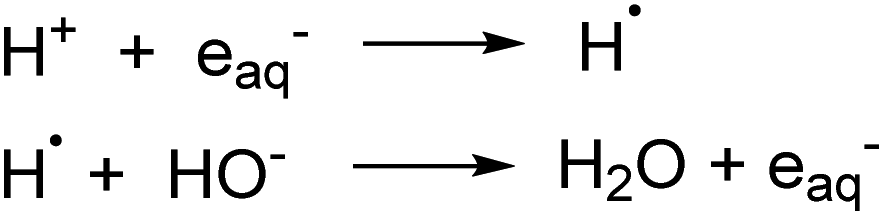 | ||
| Scheme 2 pH dependence on the regeneration of eaq− in UV/sulfite system. | ||
Typical degradation mechanisms include shortening of the chain and H/F exchange. Also, degradation efficiency is affected in the presence of anions which quench eaq−. Production of eaq− can be improved by coupling the ARP process with chemical agents. For example, in high energy vacuum ultraviolet light systems (VUV) at 185 nm, coupling with iron(III) in acid aqueous solution increased the defluorination rate for PFOA by the formation of a complex between ferric ions and PFOA.210 It is important to note that eaq− can also be generated by other sources to decompose PFAS, for example iodide,211 ferrocyanide,212 dithionite and indole derivatives213,214 and the effect of pH depends on the specific system and reductive species. Recently, electrochemical reductive treatment215 has also appeared as a feasible alternative due to ease of operation and milder reaction conditions.
Though ARP has emerged as a promising option as oxidation processes; more understanding of their degradation pathways and mechanisms, types of materials/catalysts, effectiveness of degradation, pH adjustment and implication of solution chemistry is needed. For realistic applications, the influence of effluent total dissolved solids (TDS) needs to be considered because the formation of inert salt residuals, during ARP would increase TDS levels which makes it impractical for drinking water treatment or municipal wastewater reuse (but okay for other PFAS contaminated water applications).151 The concentration of dissolved oxygen in water also needs to be controlled, which is challenging for real-world scenarios.206
Hydrothermal alkaline treatment thermochemically degrades pollutants by leveraging the alkaline properties of H2O (in the presence of NaOH) along with high pressures (2–22 MPa) and temperatures (170–350 °C).217,218 Originally used in the degradation of halogenated waste, HALT has been recently applied to degrade PFAS, with more than 90% reported efficiency.153,219 Proposed mechanisms for hydrothermal alkaline treatment include the nucleophilic substitution of the charged head (e.g., SO3−),154 or of F− from the C–F tail of PFAS with HO− from the alkaline solution to form a less thermodynamically stable C–OH bond.153 These unstable intermediates undergo hydrolysis, decarboxylation or cleavage to release F− as HF or fluoride salts until the PFAS is completely degraded.152–154
Mechanochemical degradation involving high-energy ball milling has been shown to produce sufficient energy to achieve the degradation of PFAS.186 Although partial degradation of halogenated compounds occurred in previous studies using this technique,220 the introduction of tribomaterials or co-milling agents (e.g., CaO, NaOH, SiO2, KOH, La2O3) has improved the degradation efficiency significantly. The co-milling agents generate triboplasma that emit high energy particles at the sites where intramolecular bonds are broken,186,220 thereby facilitating the degradation of PFAS into safe inorganic salts (e.g., KF, K2CO3 and K2SO4, R3SiF).155 Mechanochemical degradation is able to mineralise halogenated pollutants, such as PFAS, in short reaction times.169 For example, MCD of 6:2 fluorotelomer sulfonate (6:2 FTS) with KOH reported nearly 100% mineralisation in less than 1 h.170 Furthermore, it is possible for real-world samples of contaminated soil, to directly undergo destruction using this method.171
Bioremediation is another potential way to degrade PFAS, particularly with partially fluorinated substances.221 However, as with other reductive dehalogenation routes, bacterial defluorination depends heavily on the bacterial strain and, generally does not completely degrade PFAS.156,157 The transformation products are typically shorter-chain derivatives. The process is quite slow compared to other routes (e.g. requires days).158,159 Recent developments in this area have shown significant improvements, for example, using a biomimetic multifunctional lignocellulosic nano-framework to concentrate the PFAS prior to fungal bioremediation yielding shorter chain derivatives;160 and the use of a dual biocatalysed microbial electrosynthesis system resulting in 91% biodegradation of PFOA.161
Plasma is a state of matter with charged gaseous molecules able to degrade PFAS. The ionization can be induced by adding energy which leads to the formation of highly reactive species. Depending on the energy of the electrons compared to the temperature of the background gas, plasma can be categorized as either thermal (same temperature) or non-thermal (NTP, cold plasma, lower gas temperature).122 NTP is preferable due to its lower energy. The effectiveness of the degradation of PFAS by NTP can be correlated with the length of the perfluorinated carbon backbone. Kavanagh et al. investigated the NTP degradation of various PFAS in aqueous solution and observed a more effective degradation of long chain PFAS compared to shorter PFBA and PFBS.162 This is due to lower accumulation of hydrophilic SC-PFAS at the liquid–plasma interface thereby limiting their degradation. To increase the surface activity of SC-PFAS, Thagard et al. used a cationic surfactant, hexadecyltrimethylammonium bromide (CTAB) to destroy the recalcitrant PFBS in contaminated water.163 The PFBS–CTAB complex increased the PFBS mass transport to the interface via argon bubbles thereby increasing its degradation. Overall, plasma treatment is an effective technology with short treatment times and has minimal impact from the presence of organic or inorganic contaminants. Further research is required with regards to PFAS mineralisation pathways, observed formation of SC-PFAS, and acidification of treated water.
Smouldering is a thermal degradation technique that is flameless and occurs on the surface of condensed fuel. It is self-sustained once ignited and is more energy and cost efficient compared to incineration, which requires continuous energy input.164 Conventionally, contaminants are the fuel for smouldering combustion of hydrocarbon contaminated soils, but as PFAS cannot support smouldering on its own, some surrogate fuels (like carbon particles) are needed in small concentrations. Gerhard et al. mixed GAC (fresh or PFAS-loaded) with PFAS contaminated soil to support sufficient smouldering temperatures that could destroy PFAS.165 This method could be promising to capture emitted or transformed PFAS on the sorbent. The same researchers also used the smouldering technique to treat PFAS in sewage sludge by adding CaO (5–10 mg kg−1 of dried sludge), which served the dual purpose of mineralising the fluorine, as well as minimising hazardous PFAS and HF emissions.166 Though smouldering combustion is a promising and energy efficient thermal decomposition technique; the amount of surrogate fuel needed, completeness of PFAS combustion, removal of decomposed PFAS and the careful management of the amount of HF emissions need to be further evaluated. In addition, the solid spent sorbent is destroyed in the process of smouldering combustion, preventing it from reuse.
4.3. Opportunities for contributions from chemists
The complete destruction of PFAS is incredibly challenging, due to a wide variety of samples containing different concentrations, types of PFAS, and contaminated media to manage. Some techniques are practical, yet not well developed, while others look promising for long chain PFAS but have not shown efficacy for short chain PFAS. It is too early to tell with some of the emerging techniques whether or not they will compete with the current state of the art (i.e., adsorption plus incineration or landfilling). There is not yet one technique that checks all of the boxes. Ways that chemists can support the existing destruction and removal of PFAS in products and the environment are:(1) Develop robust protocols to investigate and measure adsorption capacity of both control samples (e.g., deionized water) and realistic samples (e.g., drinking water, waste water) using realistic concentrations of PFAS.
(2) Develop selective, efficient, and reusable separation strategies, particularly for hydrophilic short- and ultra-short chain PFAS from liquid media that can be integrated into existing processes.
(3) Find or develop new solutions for the complete destruction of multiple C–F bonds across all media (e.g., soil, sediment, leachate, water, air) in the environment as well as spent products.
(4) Improved analysis of thermal- or chemical-induced PFAS destruction by-products alongside an increased understanding of the mechanisms involved.
(5) Develop on-site solutions to manage existing and future waste, including contaminated equipment, soil, waters, and PFAS-containing products.
5 The need for alternatives to PFAS
PFAS have been detected in environmental media with confirmed pernicious effects on human health.5,66 The risks associated with PFAS, alongside government regulations, have stimulated the development of alternative substances with comparable chemical and physical properties. Initially, the historic long-chain PFAS (e.g., PFOA, PFOS) were replaced by shorter chain PFAS (e.g., GenX, PFHxS). This strategy is problematic as the shorter-chain alternatives are highly mobile, persistent, difficult to remediate222 and there is no evidence that they are less toxic.223 Substituting PFAS with alternatives without a proper assessment is deleterious for health, ecology and the economy and results in the alternatives being regrettable.224–228 A different strategy is a benign-by-design approach, where alternatives that are safer, sustainable and which can perform the necessary functions to create lasting solutions are sought.165.1. PFAS-free certification
To promote the use of sustainable alternatives, GreenScreen has certified PFAS-free products based on hazard endpoints to help consumers in minimising their PFAS footprint by making healthier and informed choices. Another approach in the global phaseout of PFAS is grouping them into non-essential, substitutable, and essential types based on societal needs and the availability of alternatives.31,34 In contrast to non-essential (e.g., ski wax) and substitutable categories (e.g., aqueous fire-fighting foams), certain applications of PFAS are specialised and essential (e.g., occupational protective clothing) and will require resources for future innovation and product development as no established alternatives currently exist.31To this end, some manufacturers of consumer products are aiming to avoid intentionally adding PFAS to their products, where possible. Progress has been made in developing non-fluorinated alternatives for PFAS in various categories. These alternatives typically target specific properties which are desired for an application, such as water-repellency, surfactant structure, or thermal stability (Fig. 5). For example, melamine and its derivatives are nonfluorinated chemicals used as textile additives to achieve water-repellency. In addition, commercial PFAS-free AFFFs have been developed, such as ENVIRO 2–3% FFF and NFD 20-391.227 Biobased materials are also attractive choices as alternatives, such as starch and zein, which have been employed in food packaging as degradable water- and oil-repellent paper packaging products.230
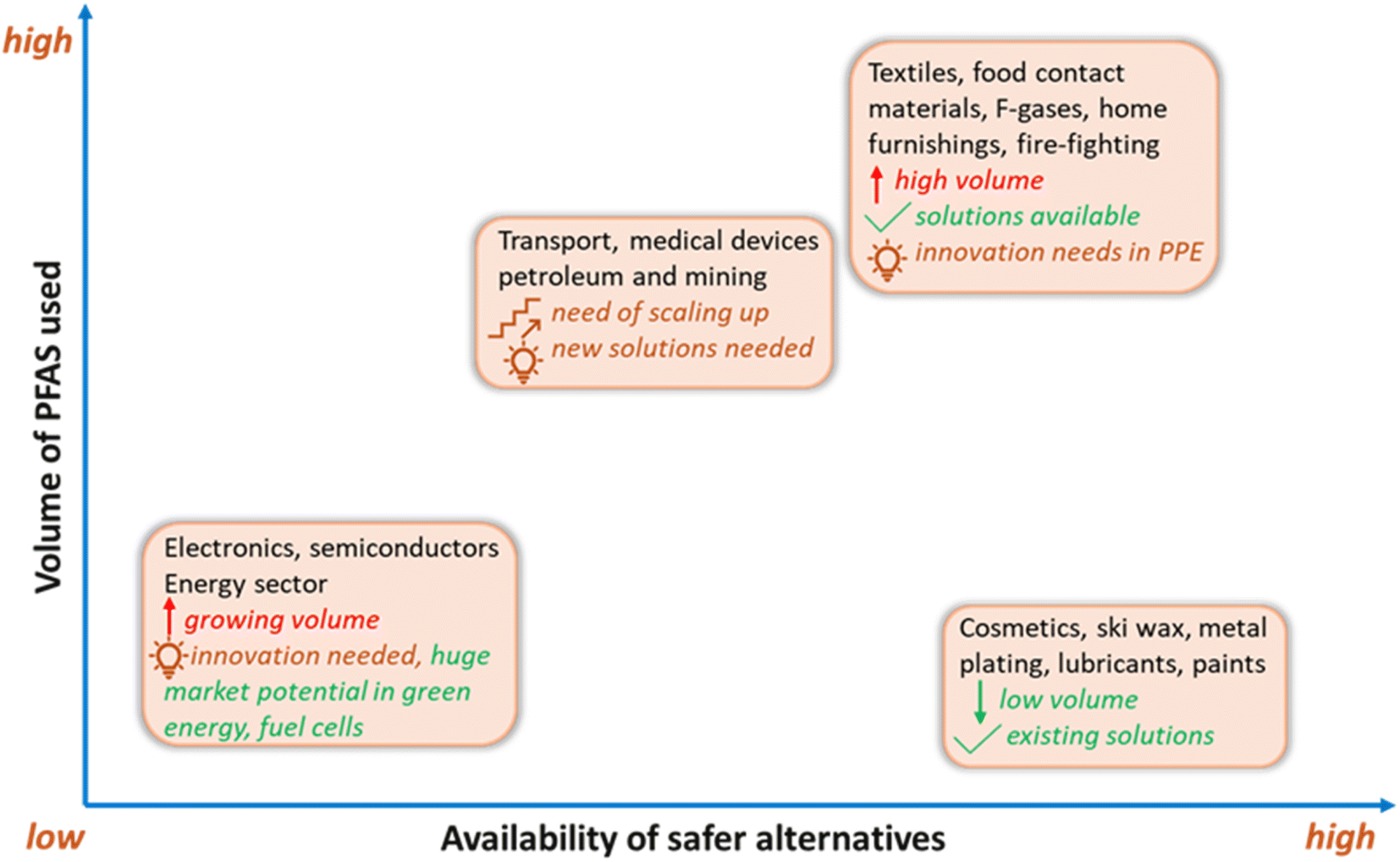 | ||
| Fig. 5 The volume of PFAS used across sectors and the availability of suitable alternatives. Original source credit: ChemSec, 2024.229Note: there are uncertainties in volume use and available alternatives in the fluoropolymers, pharmaceuticals, and medical sectors. PPE = personal protective equipment. | ||
5.2. Safer alternatives across sectors
For cookware, several non-stick alternatives are available, including ceramic, cast iron, and stainless-steel options that does not contain PFAS-based coatings which have been linked to harmful health effects.
There are some alternatives available for PFAS use in the electronics sector;239 however, PFAS still have several specialised uses in the production of semiconductors, fuel cells, lithium-ion batteries, high-speed telecommunication, ICT and acoustic equipment, high voltage cable insulation and wiring. Further research and development are needed to innovate viable alternatives for these essential uses.240
Due to their effective hydrophobic properties, PFAS are used in drilling and fracking liquids to enhance the flow of oil and gas from underground geological formations.241 A number of alternatives are available, including non-fluorinated silicone/siloxane-based anti-foaming agents.
5.3. Opportunities for contributions from chemists
Currently, there is a growing awareness of the environmental and health risks associated with PFAS, which has led to the ongoing regulations to limit their non-essential use. Concurrently, collaborative research efforts are being made to develop methods for destruction and remediation as well as identifying safer PFAS alternatives. A combination of (i) changes in consumer behaviours, (ii) action by manufacturers, (iii) legislation, and (iv) litigation will see an increase in uptake of alternatives to PFAS. However, substituting PFAS needs to be done responsibly by switching to safer alternatives after a thorough evaluation of their hazard profiles, sustainability, performance, and practicality. Specifically, chemists are challenged to work with collaborators to:(1) Perform life-cycle analyses on PFAS, products containing intentionally added PFAS, and alternatives to PFAS.
(2) Develop new molecules and materials to be used in as alternatives to PFAS that can be transitioned in an efficient and cost-effective manner.
(3) Perform assessment of the key material properties (e.g., fire-, water-, oil-, grease-, chemical- and thermal- resistance) for new and existing alternatives to ensure they meet performance standards set by PFAS-containing products.
(4) Determine the environmental fate and effect of new and existing alternatives to support robust risk assessments prior to placement on the market.
6 Summary and future outlook
The exceptional properties of PFAS, combined with the challenges of finding viable alternatives, mean they will continue to be used in numerous essential applications. The unique properties of PFAS also prevent their degradation, which, in light of their potential for bioaccumulation and toxicity, has resulted in a dramatic shift in policy around PFAS production and usage. Importantly, the US EPA has recently finalised a rule (effective since November 2023) requiring manufacturers to report and keep records of PFAS under the Toxic Substances Control Act.242 This mandates includes disclosing usage, production volumes, disposal, exposure, and hazards for PFAS manufacturing as well as the manufacture of PFAS-containing articles. Thus, there is an urgent need to develop novel methods for the effective management of PFAS, using best practice strategies,120 and to design safe and suitable alternatives. This multifaceted problem requires contributions from multiple areas of chemistry and provides an opportunity for chemists to work alongside engineers, epidemiologists, social scientists, and policymakers to develop sustainable solutions to this global issue.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
E. J. I. – conceptualization, investigation, writing – original draft. S. D. – investigation, visualization, writing – original draft. J. L.-Y. C. – conceptualization, validation, writing – review & editing, supervision, funding acquisition. M. K. – conceptualization, validation, writing – review & editing, supervision, funding acquisition. L. P. P. – conceptualization, validation, writing – review & editing, supervision, funding acquisition. E. M. L. – conceptualization, visualization, validation, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Royal Society Te Apārangi Marsden Grant (MFP-UOA2328) and the Warwick & Judy Smith Engineering Fund from the Faculty of Engineering of the University of Auckland. The authors also acknowledge the Faculty of Science of the University of Auckland and the MacDiarmid Institute for funding support.References
- Z. Wang, A. M. Buser, I. T. Cousins, S. Demattio, W. Drost, O. Johansson, K. Ohno, G. Patlewicz, A. M. Richard, G. W. Walker, G. S. White and E. Leinala, Environ. Sci. Technol., 2021, 55, 15575–15578 CrossRef CAS PubMed.
- B. Langenbach and M. Wilson, Int. J. Environ. Res. Public Health, 2021, 18, 11142 CrossRef CAS PubMed.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS.
- G. T. Ankley, P. Cureton, R. A. Hoke, M. Houde, A. Kumar, J. Kurias, R. Lanno, C. McCarthy, J. Newsted, C. J. Salice, B. E. Sample, M. S. Sepúlveda, J. Steevens and S. Valsecchi, Environ. Toxicol. Chem., 2021, 40, 564–605 CrossRef CAS.
- E. Panieri, K. Baralic, D. Djukic-Cosic, A. Buha Djordjevic and L. Saso, Toxics, 2022, 10, 44 CrossRef CAS.
- A. F. Ojo, C. Peng and J. C. Ng, J. Hazard. Mater., 2021, 407, 124863 CrossRef CAS PubMed.
- R. A. Dickman and D. S. Aga, J. Hazard. Mater., 2022, 436, 129120 CrossRef CAS.
- R. E. Banks and J. C. Tatlow, J. Fluorine Chem., 1986, 33, 71–108 CrossRef CAS.
- H.-J. Lehmler, Chemosphere, 2005, 58, 1471–1496 CrossRef CAS.
- N. V. Ignatev, A. Kucherina and P. Sartori, Acta Chem. Scand., 1999, 53, 1110–1116 CrossRef CAS.
- Y. W. Alsmeyer, W. V. Childs, R. M. Flynn, G. G. I. Moore and J. C. Smeltzer, in Organofluorine Chemistry: Principles and Commercial Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Springer, US, Boston, MA, 1994, pp. 121–143, DOI:10.1007/978-1-4899-1202-2_5.
- E. Karbassiyazdi, M. Kasula, S. Modak, J. Pala, M. Kalantari, A. Altaee, M. R. Esfahani and A. Razmjou, Chemosphere, 2023, 311, 136933 CrossRef CAS.
- A. G. Paul, K. C. Jones and A. J. Sweetman, Environ. Sci. Technol., 2009, 43, 386–392 CrossRef CAS PubMed.
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Environ. Sci. Technol., 2006, 40, 32–44 CrossRef CAS.
- G. Kostov and B. Ameduri, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds, ed. H. Groult, F. R. Leroux and A. Tressaud, Elsevier, 2017, pp. 533–559, DOI:10.1016/B978-0-12-803740-9.00018-4.
- Z. Wang, J. C. DeWitt, C. P. Higgins and I. T. Cousins, Environ. Sci. Technol., 2017, 51, 2508–2518 CrossRef CAS PubMed.
- I. J. Neuwald, D. Hübner, H. L. Wiegand, V. Valkov, U. Borchers, K. Nödler, M. Scheurer, S. E. Hale, H. P. H. Arp and D. Zahn, Environ. Sci. Technol., 2022, 56, 6380–6390 CrossRef CAS.
- M. K. Björnsdotter, L. W. Y. Yeung, A. Kärrman and I. E. Jogsten, Environ. Sci. Technol., 2019, 53, 11093–11101 CrossRef.
- M. Ateia, M. Arifuzzaman, S. Pellizzeri, M. F. Attia, N. Tharayil, J. N. Anker and T. Karanfil, Water Res., 2019, 163, 114874 CrossRef CAS.
- F. Li, J. Duan, S. Tian, H. Ji, Y. Zhu, Z. Wei and D. Zhao, Chem. Eng. J., 2020, 380, 122506 CrossRef CAS.
- A. O. De Silva, C. Spencer, B. F. Scott, S. Backus and D. C. G. Muir, Environ. Sci. Technol., 2011, 45, 8060–8066 CrossRef CAS.
- D. Szabo, D. Moodie, M. P. Green, R. A. Mulder and B. O. Clarke, Environ. Sci. Technol., 2022, 56, 8231–8244 CrossRef CAS.
- L. P. Wackett, mSphere, 2021, 6, e00721 CrossRef CAS.
- M. Trojanowicz, A. Bojanowska-Czajka, I. Bartosiewicz and K. Kulisa, Chem. Eng. J., 2018, 336, 170–199 CrossRef CAS.
- R. W. Helmer, D. M. Reeves and D. P. Cassidy, Water Res., 2022, 210, 117983 CrossRef CAS PubMed.
- L. G. T. Gaines, Am. J. Ind. Med., 2023, 66, 353–378 CrossRef CAS PubMed.
- M. Sajid and M. Ilyas, Environ. Sci. Pollut. Res., 2017, 24, 23436–23440 CrossRef CAS.
- H. Holmquist, S. Schellenberger, I. van der Veen, G. M. Peters, P. E. G. Leonards and I. T. Cousins, Environ. Int., 2016, 91, 251–264 CrossRef CAS.
- Y. Fujii, K. H. Harada and A. Koizumi, Chemosphere, 2013, 93, 538–544 CrossRef CAS.
- L. Schultes, R. Vestergren, K. Volkova, E. Westberg, T. Jacobson and J. P. Benskin, Environ. Sci.: Processes Impacts, 2018, 20, 1680–1690 RSC.
- I. T. Cousins, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, C. A. Ng, S. Patton, M. Scheringer, X. Trier, L. Vierke, Z. Wang and J. C. DeWitt, Environ. Sci.: Processes Impacts, 2019, 21, 1803–1815 RSC.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- M. Gilchrist, Minnesoa Pollution Control Agency, PFAS in the Metal Plating and Finishing Industry, 2022, https://www.pca.state.mn.us/sites/default/files/gp3-05.pdf Search PubMed.
- I. T. Cousins, J. C. De Witt, J. Glüge, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, C. A. Ng, S. Patton, M. Scheringer, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2021, 23, 1079–1087 RSC.
- J. Wang, Z. Lin, X. He, M. Song, P. Westerhoff, K. Doudrick and D. Hanigan, Environ. Sci. Technol., 2022, 56, 5355–5370 CrossRef CAS.
- W. J. Backe, T. C. Day and J. A. Field, Environ. Sci. Technol., 2013, 47, 5226–5234 CrossRef CAS PubMed.
- H. T. Nguyen, M. S. McLachlan, B. Tscharke, P. Thai, J. Braeunig, S. Kaserzon, J. W. O'Brien and J. F. Mueller, Chemosphere, 2022, 293, 133657 CrossRef CAS PubMed.
- P. Hopewell and K. Darvodelsky, Assessment of Emergent Contaminants in Biosolids, 2017, https://www.biosolids.com.au/wp-content/uploads/Emerging-Contaminants-in-Biosolids-Research-report.pdf Search PubMed.
- N. Bolan, B. Sarkar, M. Vithanage, G. Singh, D. C. W. Tsang, R. Mukhopadhyay, K. Ramadass, A. Vinu, Y. Sun, S. Ramanayaka, S. A. Hoang, Y. Yan, Y. Li, J. Rinklebe, H. Li and M. B. Kirkham, Environ. Int., 2021, 155, 106600 CrossRef CAS PubMed.
- J. R. Lang, B. M. Allred, J. A. Field, J. W. Levis and M. A. Barlaz, Environ. Sci. Technol., 2017, 51, 2197–2205 CrossRef CAS.
- E. W. Tow, M. S. Ersan, S. Kum, T. Lee, T. F. Speth, C. Owen, C. Bellona, M. N. Nadagouda, A. M. Mikelonis, P. Westerhoff, C. Mysore, V. S. Frenkel, V. deSilva, W. S. Walker, A. K. Safulko and D. A. Ladner, AWWA Water Sci., 2021, 3, e1233 CrossRef CAS PubMed.
- E. Hepburn, C. Madden, D. Szabo, T. L. Coggan, B. Clarke and M. Currell, Environ. Pollut., 2019, 248, 101–113 CrossRef CAS.
- M. A. Uriakhil, T. Sidnell, A. De Castro Fernández, J. Lee, I. Ross and M. Bussemaker, Chemosphere, 2021, 282, 131025 CrossRef CAS.
- L. J. Winchell, J. J. Ross, M. J. M. Wells, X. Fonoll, J. W. Norton Jr and K. Y. Bell, Water Environ. Res., 2021, 93, 826–843 CrossRef CAS PubMed.
- Per- and Polyfluoroalkyl Substances (PFAS): Incineration to Manage PFAS Waste Streams, 2019, https://www.epa.gov/sites/default/files/2019-09/documents/technical_brief_pfas_incineration_ioaa_approved_final_july_2019.pdf.
- K. V. Martin, T. J. Hilbert, M. Reilly, W. J. Christian, A. Hoover, K. G. Pennell, Q. Ding and E. N. Haynes, Environ. Sci. Pollut. Res., 2023, 30, 80643–80654 CrossRef CAS.
- S. Liu, S. Zhao, Z. Liang, F. Wang, F. Sun and D. Chen, Sci. Total Environ., 2021, 795, 148468 CrossRef CAS.
- S. Tartu, J. Aars, M. Andersen, A. Polder, S. Bourgeon, B. Merkel, A. D. Lowther, J. Bytingsvik, J. M. Welker, A. E. Derocher, B. M. Jenssen and H. Routti, Environ. Sci. Technol., 2018, 52, 3211–3221 CrossRef CAS.
- K. R. Miner, H. Clifford, T. Taruscio, M. Potocki, G. Solomon, M. Ritari, I. E. Napper, A. P. Gajurel and P. A. Mayewski, Sci. Total Environ., 2021, 759, 144421 CrossRef CAS.
- W. F. Hartz, M. K. Björnsdotter, L. W. Y. Yeung, A. Hodson, E. R. Thomas, J. D. Humby, C. Day, I. E. Jogsten, A. Kärrman and R. Kallenborn, Sci. Total Environ., 2023, 871, 161830 CrossRef CAS PubMed.
- D. Q. Andrews, T. Stoiber, A. M. Temkin and O. V. Naidenko, Sci. Total Environ., 2023, 901, 165939 CrossRef CAS PubMed.
- M. S. Shimizu, R. Mott, A. Potter, J. Zhou, K. Baumann, J. D. Surratt, B. Turpin, G. B. Avery, J. Harfmann, R. J. Kieber, R. N. Mead, S. A. Skrabal and J. D. Willey, Environ. Sci. Technol. Lett., 2021, 8, 366–372 CrossRef CAS.
- N. Yu, H. Guo, J. Yang, L. Jin, X. Wang, W. Shi, X. Zhang, H. Yu and S. Wei, Environ. Sci. Technol., 2018, 52, 8205–8214 CrossRef CAS PubMed.
- M. Chen, C. Wang, X. Wang, J. Fu, P. Gong, J. Yan, Z. Yu, F. Yan and J. Nawab, J. Geophys. Res.: Atmos., 2019, 124, 7442–7456 CrossRef.
- G. Casas, A. Martínez-Varela, J. L. Roscales, M. Vila-Costa, J. Dachs and B. Jiménez, Environ. Pollut., 2020, 267, 115512 CrossRef CAS.
- B. Sha, J. H. Johansson, P. Tunved, P. Bohlin-Nizzetto, I. T. Cousins and M. E. Salter, Environ. Sci. Technol., 2022, 56, 228–238 CrossRef CAS PubMed.
- B. Sha, J. H. Johansson, M. E. Salter, S. M. Blichner and I. T. Cousins, Sci. Adv., 2024, 10, eadl1026 CrossRef CAS PubMed.
- R. McGregor, Rem. J., 2023, 33, 265–278 CrossRef.
- I. T. Cousins, J. C. DeWitt, J. Glüge, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, M. Scheringer and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2307–2312 RSC.
- A. J. Lewis, X. Yun, D. E. Spooner, M. J. Kurz, E. R. McKenzie and C. M. Sales, Sci. Total Environ., 2022, 822, 153561 CrossRef CAS.
- L. Lesmeister, F. T. Lange, J. Breuer, A. Biegel-Engler, E. Giese and M. Scheurer, Sci. Total Environ., 2021, 766, 142640 CrossRef CAS PubMed.
- X. Yun, A. J. Lewis, G. Stevens-King, C. M. Sales, D. E. Spooner, M. J. Kurz, R. Suri and E. R. McKenzie, Sci. Total Environ., 2023, 866, 161208 CrossRef CAS.
- F. Pérez, M. Nadal, A. Navarro-Ortega, F. Fàbrega, J. L. Domingo, D. Barceló and M. Farré, Environ. Int., 2013, 59, 354–362 CrossRef PubMed.
- W. Cheng, J. A. Doering, C. LaLone and C. Ng, Toxicol. Sci., 2021, 180, 212–223 CrossRef CAS.
- K. S. Betts, Environ. Health Perspect., 2007, 115, A250–A256 CAS.
- S. E. Fenton, A. Ducatman, A. Boobis, J. C. DeWitt, C. Lau, C. Ng, J. S. Smith and S. M. Roberts, Environ. Toxicol. Chem., 2021, 40, 606–630 CrossRef CAS.
- Per- and Polyfluoroalkyl Substances (PFAS) Report, National Science and Technology Council, 2023, https://www.whitehouse.gov/wp-content/uploads/2023/03/OSTP-March-2023-PFAS-Report.pdf.
- S. Gaballah, A. Swank, J. R. Sobus, X. M. Howey, J. Schmid, T. Catron, J. McCord, E. Hines, M. Strynar and T. Tal, Environ. Health Perspect., 2020, 128, 047005 CrossRef CAS.
- K. Roth, Z. Imran, W. Liu and M. C. Petriello, Front. Toxicol., 2020, 2, 601149 CrossRef PubMed.
- C. Schiavone and C. Portesi, Appl. Sci., 2023, 13, 6696 CrossRef CAS.
- US-EPA, 2016, 81, Lifetime Health Advisories and Health Effects Support Documents for Perfluorooctanoic Acid and Perfluorooctane Sulfonate, https://www.gpo.gov/fdsys/pkg/FR-2016-05-25/pdf/2016-12361.pdf.
- US-EPA, 2022, https://www.epa.gov/newsreleases/epa-announces-new-drinking-water-health-advisories-pfas-chemicals-1-billion-bipartisan.
- C. Hogue, Chem. Eng. News, 2022, https://cen.acs.org/policy/legislation/US-legislation-limit-PFAS-industrial/100/web/2022/07 Search PubMed.
- Environmental Officials from Sweden, the Netherlands, Germany, and Denmark, Elements for an EU-strategy for PFAS, 2019, https://www.documentcloud.org/documents/6586418-EUStrategy-for-PFASs-FINAL-VERSION-December-19.html.
- ECHA publishes PFAS restriction proposal, 2023, https://echa.europa.eu/-/echa-publishes-pfas-restriction-proposal.
- https://echa.europa.eu/hot-topics/perfluoroalkyl-chemicals-pfas .
- https://www.pfasfree.org.uk/regulations#1527174044222-65be56c6-8e65 .
- USA Department of Defense, 2023, https://media.defense.gov/2023/Jan/12/2003144157/-1/-1/1/MILITARY-SPECIFICATION-FOR-FIRE-EXTINGUISHING-AGENT-FLUORINE-FREE-FOAM-F3-LIQUID-CONCENTRATE-FOR-LAND-BASED-FRESH-WATER-APPLICATIONS.PDF.
- Cosmetics Products Group Standard 2020, Report HSR002552, New Zealand Environmental Protection Authority, 2024, https://www.epa.govt.nz/assets/RecordsAPI/Cosmetic-Products-Group-Standard-2020-Amended-January-2024.pdf.
- M. Wahlström, E. Pohjalainen, E. Yli-Rantala, D. Behringer, D. Herzke, S. Mudge, M. Beekman, A. Blaeij, J. Devilee, S. Gabbert, M. Kuppevelt, M. Zare Jeddi, P. Gabrielsen and X. Trier, Fluorinated Polymers in a Low Carbon, Circular and Toxic-free Economy, 2021 Search PubMed.
- C. A. McDonough, J. L. Guelfo and C. P. Higgins, Curr. Opin. Environ. Sci. Health., 2019, 7, 13–18 CrossRef PubMed.
- R. A. Brase, E. J. Mullin and D. C. Spink, Int. J. Mol. Sci., 2021, 22, 995 CrossRef CAS PubMed.
- L. J. Winchell, M. J. M. Wells, J. J. Ross, X. Fonoll, J. W. Norton, S. Kuplicki, M. Khan and K. Y. Bell, Sci. Total Environ., 2021, 774, 145257 CrossRef CAS.
- Best Practices for Non-Targeted Analysis BP4NTA, https://nontargetedanalysis.org/reference-content/methods/study-design/.
- R. F. Menger, E. Funk, C. S. Henry and T. Borch, Chem. Eng. J., 2021, 417, 129133 CrossRef CAS.
- R. Gonzalez de Vega, A. Cameron, D. Clases, T. M. Dodgen, P. A. Doble and D. P. Bishop, J. Chromatogr. A, 2021, 1653, 462423 CrossRef CAS PubMed.
- PFAS Master list of PFAS Substances, https://comptox.epa.gov/dashboard/chemical-lists/pfasmaster.
- E. F. Houtz and D. L. Sedlak, Environ. Sci. Technol., 2012, 46, 9342–9349 CrossRef CAS.
- R. Aro, U. Eriksson, A. Kärrman, I. Reber and L. W. Y. Yeung, iScience, 2021, 24, 102968 CrossRef CAS.
- C. A. Moody, W. C. Kwan, J. W. Martin, D. C. G. Muir and S. A. Mabury, Anal. Chem., 2001, 73, 2200–2206 CrossRef CAS PubMed.
- D. Camdzic, R. A. Dickman and D. S. Aga, J. Hazard. Mater. Lett., 2021, 2, 100023 CrossRef CAS.
- E. E. Ritter, M. E. Dickinson, J. P. Harron, D. M. Lunderberg, P. A. DeYoung, A. E. Robel, J. A. Field and G. F. Peaslee, Nucl. Instrum. Methods Phys. Res., Sect. B, 2017, 407, 47–54 CrossRef CAS.
- S. Heuckeroth, T. N. Nxumalo, A. Raab and J. Feldmann, Anal. Chem., 2021, 93, 6335–6341 CrossRef CAS.
- M. B. Bhavya, N. Rhakho, S. R. Jena, S. Yadav, A. Altaee, M. Saxena and A. K. Samal, Sustain. Chem. Environ., 2023, 3, 100031 CrossRef.
- M. J. Davern, G. V. West, C. M. A. Eichler, B. J. Turpin, Y. Zhang and J. D. Surratt, Analyst, 2024, 149, 3405–3415 RSC.
- B. B. Bowers, J. A. Thornton and R. C. Sullivan, Environ. Sci.: Processes Impacts, 2023, 25, 277–287 RSC.
- J. M. Mattila, J. D. Krug, W. R. Roberson, R. P. Burnette, S. McDonald, L. Virtaranta, J. H. Offenberg and W. P. Linak, Environ. Sci. Technol., 2024, 58, 3942–3952 CrossRef CAS PubMed.
- M. Hemida, A. Ghiasvand, V. Gupta, L. J. Coates, A. A. Gooley, H.-J. Wirth, P. R. Haddad and B. Paull, Anal. Chem., 2021, 93, 12032–12040 CrossRef CAS.
- S. Garg, P. Kumar, G. W. Greene, V. Mishra, D. Avisar, R. S. Sharma and L. F. Dumée, J. Environ. Manage., 2022, 308, 114655 CrossRef CAS.
- R. Khan, D. Andreescu, M. H. Hassan, J. Ye and S. Andreescu, Angew. Chem., Int. Ed., 2022, 61, e202209164 CrossRef CAS.
- H. Niu, S. Wang, Z. Zhou, Y. Ma, X. Ma and Y. Cai, Anal. Chem., 2014, 86, 4170–4177 CrossRef CAS PubMed.
- M. Takayose, K. Akamatsu, H. Nawafune, T. Murashima and J. Matsui, Anal. Lett., 2012, 45, 2856–2864 CrossRef CAS.
- C. Fang, X. Zhang, Z. Dong, L. Wang, M. Megharaj and R. Naidu, Chemosphere, 2018, 191, 381–388 CrossRef CAS PubMed.
- R. F. Menger, J. J. Beck, T. Borch and C. S. Henry, ACS Environ. Sci. Technol., 2022, 2, 565–572 CAS.
- H. Feng, N. Wang, T. Tran.T, L. Yuan, J. Li and Q. Cai, Sens. Actuators, B, 2014, 195, 266–273 CrossRef CAS.
- Z. Zheng, H. Yu, W.-C. Geng, X.-Y. Hu, Y.-Y. Wang, Z. Li, Y. Wang and D.-S. Guo, Nat. Commun., 2019, 10, 5762 CrossRef CAS.
- Q. Liu, A. Huang, N. Wang, G. Zheng and L. Zhu, J. Lumin., 2015, 161, 374–381 CrossRef CAS.
- M. Takayose, K. Nishimoto and J. Matsui, Analyst, 2012, 137, 2762–2765 RSC.
- H.-Q. Yin, K. Tan, S. Jensen, S. J. Teat, S. Ullah, X. Hei, E. Velasco, K. Oyekan, N. Meyer, X.-Y. Wang, T. Thonhauser, X.-B. Yin and J. Li, Chem. Sci., 2021, 12, 14189–14197 RSC.
- V. Pichon and F. Chapuis-Hugon, Anal. Chim. Acta, 2008, 622, 48–61 CrossRef CAS.
- R. B. Clark and J. E. Dick, Chem. Commun., 2021, 57, 8121–8130 RSC.
- M. H. Hassan, R. Khan and S. Andreescu, Electrochem. Sci. Adv., 2022, 2, e2100184 CrossRef CAS.
- N. Karimian, A. M. Stortini, L. M. Moretto, C. Costantino, S. Bogialli and P. Ugo, ACS Sens., 2018, 3, 1291–1298 CrossRef CAS.
- R. Kazemi, E. I. Potts and J. E. Dick, Anal. Chem., 2020, 92, 10597–10605 CrossRef CAS PubMed.
- A. Concellón and T. M. Swager, Angew. Chem., Int. Ed., 2023, 62, e202309928 CrossRef PubMed.
- S. Chen, A. Li, L. Zhang and J. Gong, Anal. Chim. Acta, 2015, 896, 68–77 CrossRef CAS PubMed.
- Y. H. Cheng, D. Barpaga, J. A. Soltis, V. Shutthanandan, R. Kargupta, K. S. Han, B. P. McGrail, R. K. Motkuri, S. Basuray and S. Chatterjee, ACS Appl. Mater. Interfaces, 2020, 12, 10503–10514 CrossRef CAS.
- X. Li, X. Wang, T. Fang, L. Zhang and J. Gong, Talanta, 2018, 181, 147–153 CrossRef CAS PubMed.
- V. Trinh, C. S. Malloy, T. J. Durkin, A. Gadh and S. Savagatrup, ACS Sens., 2022, 7, 1514–1523 CrossRef CAS PubMed.
- C. M. Taylor, M. C. Breadmore and N. L. Kilah, Chem.: Methods, 2024, 4, e202300017 CAS.
- M. Zhang, X. Zhao, D. Zhao, T.-Y. Soong and S. Tian, Waste Manage., 2023, 155, 162–178 CrossRef CAS PubMed.
- J. N. Meegoda, B. Bezerra de Souza, M. M. Casarini and J. A. Kewalramani, Int. J. Environ. Res. Public Health, 2022, 19, 16397 CrossRef.
- D. Lu, S. Sha, J. Luo, Z. Huang and X. Zhang Jackie, J. Hazard. Mater., 2020, 386, 121963 CrossRef CAS PubMed.
- T. Yamada, P. H. Taylor, R. C. Buck, M. A. Kaiser and R. J. Giraud, Chemosphere, 2005, 61, 974–984 CrossRef CAS.
- W. Tsang, D. R. Burgess and V. Babushok, Combust. Sci. Technol., 1998, 139, 385–402 CrossRef CAS.
- P. H. Taylor, T. Yamada, R. C. Striebich, J. L. Graham and R. J. Giraud, Chemosphere, 2014, 110, 17–22 CrossRef CAS PubMed.
- N. Bolan, B. Sarkar, Y. Yan, Q. Li, H. Wijesekara, K. Kannan, D. C. W. Tsang, M. Schauerte, J. Bosch, H. Noll, Y. S. Ok, K. Scheckel, J. Kumpiene, K. Gobindlal, M. Kah, J. Sperry, M. B. Kirkham, H. Wang, Y. F. Tsang, D. Hou and J. Rinklebe, J. Hazard. Mater., 2021, 401, 123892 CrossRef CAS PubMed.
- S. Takagi, F. Adachi, K. Miyano, Y. Koizumi, H. Tanaka, I. Watanabe, S. Tanabe and K. Kannan, Water Res., 2011, 45, 3925–3932 CrossRef CAS.
- Y. Dahman, in Nanotechnology and Functional Materials for Engineers, ed. Y. Dahman, Elsevier, 2017, pp. 121–144, DOI:10.1016/B978-0-323-51256-5.00006-X.
- S. J. Zaidi and H. Saleem, in Reverse Osmosis Systems, ed. S. J. Zaidi and H. Saleem, Elsevier, 2022, pp. 141–177, DOI:10.1016/B978-0-12-823965-0.00006-7.
- A. L. Kohl and R. B. Nielsen, in Gas Purification, 5th edn, ed. A. L. Kohl and R. B. Nielsen, Gulf Professional Publishing, Houston, 1997, pp. 187–277, DOI:10.1016/B978-088415220-0/50003-3.
- S. Woodard, J. Berry and B. Newman, Rem. J., 2017, 27, 19–27 CrossRef.
- F. Dixit, R. Dutta, B. Barbeau, P. Berube and M. Mohseni, Chemosphere, 2021, 272, 129777 CrossRef CAS.
- E. Barth, J. McKernan, D. Bless and K. Dasu, J. Environ. Manage., 2021, 296, 113069 CrossRef CAS.
- J. O'Connor, N. S. Bolan, M. Kumar, A. S. Nitai, M. B. Ahmed, S. S. Bolan, M. Vithanage, J. Rinklebe, R. Mukhopadhyay, P. Srivastava, B. Sarkar, A. Bhatnagar, H. Wang, K. H. M. Siddique and M. B. Kirkham, Process Saf. Environ. Prot., 2022, 164, 91–108 CrossRef.
- V. Franke, P. McCleaf, K. Lindegren and L. Ahrens, Environ. Sci.: Water Res. Technol., 2019, 5, 1836–1843 RSC.
- C. C. Murray, H. Vatankhah, C. A. McDonough, A. Nickerson, T. T. Hedtke, T. Y. Cath, C. P. Higgins and C. L. Bellona, J. Hazard. Mater., 2019, 366, 160–168 CrossRef CAS PubMed.
- S. Lath, E. R. Knight, D. A. Navarro, R. S. Kookana and M. J. McLaughlin, Chemosphere, 2019, 222, 671–678 CrossRef CAS PubMed.
- S. Garg, J. Wang, P. Kumar, V. Mishra, H. Arafat, R. S. Sharma and L. F. Dumée, J. Environ. Chem. Eng., 2021, 9, 105784 CrossRef CAS.
- T. Buckley, K. Karanam, X. Xu, P. Shukla, M. Firouzi and V. Rudolph, Sep. Purif. Technol., 2022, 286, 120508 CrossRef CAS.
- D. Q. Zhang, M. Wang, Q. He, X. Niu and Y. Liang, Environ. Pollut., 2020, 257, 113575 CrossRef CAS.
- A. Gredelj, C. Nicoletto, S. Valsecchi, C. Ferrario, S. Polesello, R. Lava, F. Zanon, A. Barausse, L. Palmeri, L. Guidolin and M. Bonato, Sci. Total Environ., 2020, 708, 134766 CrossRef CAS PubMed.
- E. Yamazaki, S. Taniyasu, K. Noborio, H. Eun, P. Thaker, N. J. I. Kumar, X. Wang and N. Yamashita, Chemosphere, 2019, 231, 502–509 CrossRef CAS.
- B. Wen, L. Li, H. Zhang, Y. Ma, X.-Q. Shan and S. Zhang, Environ. Pollut., 2014, 184, 547–554 CrossRef CAS.
- A. Chavoshani, M. Hashemi, M. Mehdi Amin and S. C. Ameta, in Micropollutants and Challenges, ed. A. Chavoshani, M. Hashemi, M. Mehdi Amin and S. C. Ameta, Elsevier, 2020, pp. 35–90, DOI:10.1016/B978-0-12-818612-1.00002-7.
- L. Martín-Pozo, M. d. C. Gómez-Regalado, M. T. García-Córcoles and A. Zafra-Gómez, in Emerging Contaminants in the Environment, ed. H. Sarma, D. C. Dominguez and W.-Y. Lee, Elsevier, 2022, pp. 381–406, DOI:10.1016/B978-0-323-85160-2.00015-9.
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Crit. Rev. Environ. Sci. Technol., 2019, 49, 866–915 CrossRef.
- Z. Liu, M. J. Bentel, Y. Yu, C. Ren, J. Gao, V. F. Pulikkal, M. Sun, Y. Men and J. Liu, Environ. Sci. Technol., 2021, 55, 7052–7062 CrossRef CAS.
- D. Panda, V. Sethu and S. Manickam, Chem. Eng. Process., 2019, 142, 107542 CrossRef.
- C. G. Scheitlin, K. Dasu, S. Rosansky, L. E. Dejarme, D. Siriwardena, J. Thorn, L. Mullins, I. Haggerty, K. Shqau and J. Stowe, ACS Environ. Sci. Technol., 2023, 3, 2053–2062 CAS.
- E. Houtz, D. Kempisty and Y. Lester, Curr. Opin. Chem. Eng., 2024, 44, 101022 CrossRef.
- J. Li, B. R. Pinkard, S. Wang and I. V. Novosselov, Chemosphere, 2022, 307, 135888 CrossRef CAS PubMed.
- S. Hao, Y.-J. Choi, B. Wu, C. P. Higgins, R. Deeb and T. J. Strathmann, Environ. Sci. Technol., 2021, 55, 3283–3295 CrossRef CAS PubMed.
- B. Wu, S. Hao, Y. Choi, C. P. Higgins, R. Deeb and T. J. Strathmann, Environ. Sci. Technol. Lett., 2019, 6, 630–636 CrossRef CAS.
- M. Ateia, L. P. Skala, A. Yang and W. R. Dichtel, J. Hazard. Mater. Adv., 2021, 3, 100014 CrossRef CAS.
- B. G. Kwon, H.-J. Lim, S.-H. Na, B.-I. Choi, D.-S. Shin and S.-Y. Chung, Chemosphere, 2014, 109, 221–225 CrossRef CAS PubMed.
- S. P. Chetverikov, D. A. Sharipov, T. Y. Korshunova and O. N. Loginov, Appl. Biochem. Microbiol., 2017, 53, 533–538 CrossRef CAS.
- S. Huang and P. R. Jaffé, Environ. Sci. Technol., 2019, 53, 11410–11419 CrossRef CAS PubMed.
- S. P. Chetverikov and O. N. Loginov, Microbiology, 2019, 88, 115–117 CAS.
- J. Li, X. Li, Y. Da, J. Yu, B. Long, P. Zhang, C. Bakker, B. A. McCarl, J. S. Yuan and S. Y. Dai, Nat. Commun., 2022, 13, 4368 CrossRef CAS PubMed.
- K. Tahir, A. S. Ali, J. Kim, J. Park, S. Lee, B. Kim, Y. Lim, G. Kim and D. S. Lee, Chemosphere, 2023, 328, 138584 CrossRef CAS PubMed.
- D. Alam, S. Lee, J. Hong, D. F. Fletcher, D. McClure, D. Cook, P. J. Cullen and J. M. Kavanagh, J. Environ. Chem. Eng., 2023, 11, 111588 CrossRef CAS.
- O. Isowamwen, R. Li, T. Holsen and S. M. Thagard, J. Hazard. Mater., 2023, 456, 131691 CrossRef CAS PubMed.
- B. Améduri and H. Hori, Chem. Soc. Rev., 2023, 52, 4208–4247 RSC.
- A. L. Duchesne, J. K. Brown, D. J. Patch, D. Major, K. P. Weber and J. I. Gerhard, Environ. Sci. Technol., 2020, 54, 12631–12640 CrossRef CAS.
- T. Fournie, T. L. Rashwan, C. Switzer and J. I. Gerhard, Waste Manage., 2023, 164, 219–227 CrossRef CAS.
- F. Cao, L. Wang, Y. Tian, F. Wu, C. Deng, Q. Guo, H. Sun and S. Lu, J. Chromatogr. A, 2017, 1516, 42–53 CrossRef CAS.
- Y. Wu, Y. Li, A. Tian, K. Mao and J. Liu, Int. J. Photoenergy, 2016, 2016, 7368795 CrossRef.
- G. Cagnetta, H. Liu, K. Zhang, J. Huang, B. Wang, S. Deng, Y. Wang and G. Yu, Sci. Rep., 2016, 6, 28394 CrossRef CAS PubMed.
- M. Lu, G. Cagnetta, K. Zhang, J. Huang and G. Yu, Sci. Rep., 2017, 7, 17180 CrossRef.
- K. Gobindlal, E. Shields, A. Whitehill, C. C. Weber and J. Sperry, Environ. Sci.: Adv., 2023, 2, 982–989 CAS.
- R. Mukhopadhyay, B. Sarkar, K. N. Palansooriya, J. Y. Dar, N. S. Bolan, S. J. Parikh, C. Sonne and Y. S. Ok, Adv. Colloid Interface Sci., 2021, 297, 102537 CrossRef CAS.
- L. Zhao, J. Bian, Y. Zhang, L. Zhu and Z. Liu, Chemosphere, 2014, 114, 51–58 CrossRef CAS.
- Q. Yu, R. Zhang, S. Deng, J. Huang and G. Yu, Water Res., 2009, 43, 1150–1158 CrossRef CAS.
- D. Zhang, Q. Luo, B. Gao, S.-Y. D. Chiang, D. Woodward and Q. Huang, Chemosphere, 2016, 144, 2336–2342 CrossRef CAS.
- F. Dixit, B. Barbeau, S. G. Mostafavi and M. Mohseni, Environ. Sci.: Water Res. Technol., 2019, 5, 1782–1795 RSC.
- M. F. Rahman, W. B. Anderson, S. Peldszus and P. M. Huck, ACS Environ. Sci. Technol., 2022, 2, 1195–1205 CAS.
- Y.-L. Liu and M. Sun, Water Res., 2021, 207, 117781 CrossRef CAS PubMed.
- Y. Gao, S. Deng, Z. Du, K. Liu and G. Yu, J. Hazard. Mater., 2017, 323, 550–557 CrossRef CAS.
- A. Maimaiti, S. Deng, P. Meng, W. Wang, B. Wang, J. Huang, Y. Wang and G. Yu, Chem. Eng. J., 2018, 348, 494–502 CrossRef CAS.
- F. Dixit, B. Barbeau, S. G. Mostafavi and M. Mohseni, J. Hazard. Mater., 2020, 384, 121261 CrossRef CAS PubMed.
- A. Nickerson, A. E. Rodowa, D. T. Adamson, J. A. Field, P. R. Kulkarni, J. J. Kornuc and C. P. Higgins, Environ. Sci. Technol., 2021, 55, 313–323 CrossRef CAS PubMed.
- F. Dixit, G. Munoz, M. Mirzaei, B. Barbeau, J. Liu, S. V. Duy, S. Sauvé, B. Kandasubramanian and M. Mohseni, Environ. Sci. Technol., 2022, 56, 6212–6222 CrossRef CAS PubMed.
- C. Ching, M. J. Klemes, B. Trang, W. R. Dichtel and D. E. Helbling, Environ. Sci. Technol., 2020, 54, 12693–12702 CrossRef CAS.
- Y. Artioli, in Encyclopedia of Ecology, ed. S. E. Jørgensen and B. D. Fath, Academic Press, Oxford, 2008, pp. 60–65, DOI:10.1016/B978-008045405-4.00252-4.
- S. C. E. Leung, P. Shukla, D. Chen, E. Eftekhari, H. An, F. Zare, N. Ghasemi, D. Zhang, N.-T. Nguyen and Q. Li, Sci. Total Environ., 2022, 827, 153669 CrossRef CAS PubMed.
- T. Buckley, X. Xu, V. Rudolph, M. Firouzi and P. Shukla, Sep. Sci. Technol., 2022, 57, 929–958 CrossRef CAS.
- B. Burghoff, J. Biotechnol., 2012, 161, 126–137 CrossRef CAS.
- T. Le, E. Jamshidi, M. Beidaghi and M. R. Esfahani, ACS Appl. Mater. Interfaces, 2022, 14, 25397–25408 CrossRef CAS PubMed.
- Y. Gogotsi and B. Anasori, ACS Nano, 2019, 13, 8491–8494 CrossRef CAS PubMed.
- G. S. Park, D. H. Ho, B. Lyu, S. Jeon, D. Y. Ryu, D. W. Kim, N. Lee, S. Kim, Y. J. Song, S. B. Jo and J. H. Cho, Sci. Adv., 2022, 8, eabl5299 CrossRef CAS PubMed.
- Á. Soriano, D. Gorri and A. Urtiaga, Ind. Eng. Chem. Res., 2019, 58, 3329–3338 CrossRef.
- T. G. Reichenauer and J. J. Germida, ChemSusChem, 2008, 1, 708–717 CrossRef CAS PubMed.
- J. Awad, G. Brunetti, A. Juhasz, M. Williams, D. Navarro, B. Drigo, J. Bougoure, J. Vanderzalm and S. Beecham, J. Hazard. Mater., 2022, 429, 128326 CrossRef CAS.
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Crit. Rev. Environ. Sci. Technol., 2019, 49, 866–915 CrossRef.
- O. C. Olatunde, A. T. Kuvarega and D. C. Onwudiwe, Heliyon, 2020, 6, e05614 CrossRef CAS PubMed.
- M. B. Ahmed, M. M. Alam, J. L. Zhou, B. Xu, M. A. H. Johir, A. K. Karmakar, M. S. Rahman, J. Hossen, A. T. M. K. Hasan and M. A. Moni, Process Saf. Environ. Prot., 2020, 136, 1–14 CrossRef CAS.
- M. Umar, Water, 2021, 13, 3185 CrossRef CAS.
- Y. Guan, Z. Liu, N. Yang, S. Yang, L. E. Quispe-Cardenas, J. Liu and Y. Yang, Nat. Water., 2024, 2, 443–452 CrossRef.
- N. Tran, P. Drogui and S. K. Brar, Environ. Chem. Lett., 2015, 13, 251–268 CrossRef CAS.
- C. G. Joseph, G. Li Puma, A. Bono and D. Krishnaiah, Ultrason. Sonochem., 2009, 16, 583–589 CrossRef CAS.
- P. Kritzer and E. Dinjus, Chem. Eng. J., 2001, 83, 207–214 CrossRef CAS.
- Z. Chen, K. Tong, F. Xu, M. Xue, H. Chen, Q. Chen, D. Wang and Y. Xu, J. Environ. Chem. Eng., 2021, 9, 105296 CrossRef CAS.
- C. Berg, B. Crone, B. Gullett, M. Higuchi, M. J. Krause, P. M. Lemieux, T. Martin, E. P. Shields, E. Struble, E. Thoma and A. Whitehill, J. Air Waste Manage. Assoc., 2022, 72, 540–555 CrossRef PubMed.
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Environ. Sci. Technol., 2019, 53, 3718–3728 CrossRef CAS PubMed.
- J. Cui, P. Gao and Y. Deng, Environ. Sci. Technol., 2020, 54, 3752–3766 CrossRef CAS.
- M. J. Bentel, Y. Yu, L. Xu, H. Kwon, Z. Li, B. M. Wong, Y. Men and J. Liu, Environ. Sci. Technol., 2020, 54, 2489–2499 CrossRef CAS PubMed.
- Z. Ren, U. Bergmann and T. Leiviskä, Water Res., 2021, 205, 117676 CrossRef CAS PubMed.
- M. Fischer and P. Warneck, J. Phys. Chem., 1996, 100, 15111–15117 CrossRef CAS.
- I. M. F. Cardoso, L. Pinto da Silva and J. C. G. Esteves da Silva, Nanomaterials, 2023, 13, 1668 CrossRef CAS PubMed.
- N. O'Connor, D. Patch, D. Noble, J. Scott, I. Koch, K. G. Mumford and K. Weber, Sci. Total Environ., 2023, 888, 164137 CrossRef PubMed.
- W. A. Maza, J. A. Ridenour, B. L. Chaloux, A. Epshteyn and J. C. Owrutsky, Environ. Sci.: Adv., 2023, 2, 1641–1650 CAS.
- X. Jin, Z. Wang, R. Hong, Z. Chen, B. Wu, S. Ding, W. Zhu, Y. Lin and C. Gu, Water Res., 2022, 225, 119147 CrossRef CAS PubMed.
- H. Tian, J. Gao, H. Li, S. A. Boyd and C. Gu, Sci. Rep., 2016, 6, 32949 CrossRef CAS.
- J. F. King and B. P. Chaplin, Curr. Opin. Chem. Eng., 2024, 44, 101014 CrossRef.
- B. Trang, Y. Li, X.-S. Xue, M. Ateia, K. N. Houk and W. R. Dichtel, Science, 2022, 377, 839–845 CrossRef CAS.
- S. Hao, Y. J. Choi, R. A. Deeb, T. J. Strathmann and C. P. Higgins, Environ. Sci. Technol., 2022, 56, 6647–6657 CrossRef CAS PubMed.
- B. R. Pinkard, C. Austin, A. L. Purohit, J. Li and I. V. Novosselov, Chemosphere, 2023, 314, 137681 CrossRef CAS.
- B. R. Pinkard, J. Environ. Eng., 2022, 148, 05021007 CrossRef CAS.
- L. P. Turner, B. H. Kueper, K. M. Jaansalu, D. J. Patch, N. Battye, O. El-Sharnouby, K. G. Mumford and K. P. Weber, Sci. Total Environ., 2021, 765, 142722 CrossRef CAS PubMed.
- O. Tiedt, M. Mergelsberg, K. Boll, M. Müller, L. Adrian, N. Jehmlich, M. v. Bergen and M. Boll, mBio, 2016, 7, e00990 CrossRef CAS PubMed.
- M. Ateia, A. Maroli, N. Tharayil and T. Karanfil, Chemosphere, 2019, 220, 866–882 CrossRef CAS PubMed.
- C. Hogue, Chem. Eng. News, 2021, vol. 99, p. 40, https://cen.acs.org/environment/persistent-pollutants/US-EPA-deems-two-GenX-PFAS-chemicals-more-toxic-than-PFOA/99/i40 Search PubMed.
- C. Ng, I. T. Cousins, J. C. DeWitt, J. Glüge, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, S. Patton, M. Scheringer, X. Trier and Z. Wang, Environ. Sci. Technol., 2021, 55, 12755–12765 CAS.
- S. V. Kabadi, J. Fisher, J. Aungst and P. Rice, Food Chem. Toxicol., 2018, 112, 375–382 CrossRef CAS.
- D. K. Jones, K. A. Quinlin, M. A. Wigren, Y. J. Choi, M. S. Sepúlveda, L. S. Lee, D. L. Haskins, G. R. Lotufo, A. Kennedy, L. May, A. Harmon, T. Biber, N. Melby, M. K. Chanov, M. L. Hudson, P. B. Key, K. W. Chung, D. W. Moore, J. G. Suski, E. F. Wirth and J. T. Hoverman, Environ. Sci. Technol., 2022, 56, 6078–6090 CrossRef CAS.
- F. Hossain, N. M. Dennis, S. Subbiah, A. Karnjanapiboonwong, J. L. Guelfo, J. Suski and T. A. Anderson, Environ. Toxicol. Chem., 2022, 41, 2003–2007 CrossRef CAS PubMed.
- X. Wu, H. Nguyen, D. Kim and H. Peng, Sci. Total Environ., 2022, 850, 158100 CrossRef CAS PubMed.
- https://www.slideshare.net/slideshows/oecd-global-forum-on-the-environment-dedicated-to-per-and-polyfluoroalkyl-substances-available-pfas-alternatives-and-innovation-needs-anna-lennquist/266308319, accessed Feb 16, 2024.
- G. Glenn, R. Shogren, X. Jin, W. Orts, W. Hart-Cooper and L. Olson, Compr. Rev. Food Sci. Food Saf., 2021, 20, 2596–2625 CrossRef CAS PubMed.
- Firefighting Foams, https://pfas-1.itrcweb.org/3-firefighting-foams/.
- F. F. F. Coalition, https://www.fffc.org/.
- J. Horst, J. Quinnan, J. McDonough, J. Lang, P. Storch, J. Burdick and C. Theriault, Groundwater Monit. Rem., 2021, 41, 19–26 CrossRef.
- S. R. Fernández, C. Kwiatkowski and T. Bruton, 2021. https://greensciencepolicy.org/docs/pfas-building-materials-2021.pdf.
- S. Schellenberger, P. J. Hill, O. Levenstam, P. Gillgard, I. T. Cousins, M. Taylor and R. S. Blackburn, J. Cleaner Prod., 2019, 217, 134–143 CrossRef CAS.
- H. D. Whitehead, M. Venier, Y. Wu, E. Eastman, S. Urbanik, M. L. Diamond, A. Shalin, H. Schwartz-Narbonne, T. A. Bruton, A. Blum, Z. Wang, M. Green, M. Tighe, J. T. Wilkinson, S. McGuinness and G. F. Peaslee, Environ. Sci. Technol. Lett., 2021, 8, 538–544 CrossRef CAS.
- K. J. Harris, G. Munoz, V. Woo, S. Sauvé and A. A. Rand, Environ. Sci. Technol., 2022, 56, 14594–14604 CrossRef CAS PubMed.
- K. W. Pütz, S. Namazkar, M. Plassmann and J. P. Benskin, Environ. Sci.: Processes Impacts, 2022, 24, 1697–1707 RSC.
- D. Lay, I. Keyte, S. Tiscione, J. Kreissig, Check Your Tech: A Guide to PFAS in Electronics, 2023 Search PubMed.
- J. Glüge, R. London, I. T. Cousins, J. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, C. A. Ng, S. Patton, X. Trier, Z. Wang and M. Scheringer, Environ. Sci. Technol., 2022, 56, 6232–6242 CrossRef PubMed.
- Fracking with “Forever Chemicals”, https://psr.org/resources/fracking-with-forever-chemicals/.
- US-EPA, 2023, TSCA Section 8(a) (&) Reporting and Recordkeeping Requirements for Perfluoroalkyl and Polyfluoroalkyl Substances, https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/tsca-section-8a7-reporting-and-recordkeeping.
| This journal is © The Royal Society of Chemistry 2024 |