DOI:
10.1039/D4TA05025H
(Paper)
J. Mater. Chem. A, 2024,
12, 27340-27354
A regenerative dual-functional platform combining dendritic silica and anthraquinone amide: advancing seawater lithium detection and recovery with biosensing capabilities†
Received
19th July 2024
, Accepted 5th September 2024
First published on 6th September 2024
Abstract
Lithium is a vital energy storage material in high demand, but its excess in water bodies poses environmental risks, necessitating rigorous monitoring and remediation. Currently, there are no reports on the simultaneous detection and extraction of lithium from pure aqueous systems. Addressing this gap, a novel functionalized material named MDNS@DAAH has been developed. This material integrates dendritic silica as a substrate with an anthraquinone-based amide, a lithium-specific chelating fluorophore. MDNS@DAAH exhibits selective, turn-on green emission in the presence of lithium ions amidst common oceanic elements like magnesium, sodium, and potassium in pure aqueous settings, with a remarkably low detection limit of 4.9 nM. The turn-on emission in the presence of lithium ions arises from the disruption of the hydrogen-bonding-assisted PET process that leads to the fluorescence recovery of the anthraquinone system supported by DFT studies. Moreover, it demonstrates an exceptional adsorption capacity for lithium, surpassing established adsorbents with a high value of 97.8 mg g−1, along with rapid kinetics and recyclability over multiple cycles. As a fluorescent probe, it enables the biosensing of lithium in the living organism Artemia salina through fluorescence imaging. Additionally, this material is effective for quantifying and extracting lithium from seawater and sea bittern (91.6% recovery). This study showcases a single platform with dual functions for lithium recovery from seawater, offering benefits in recycling, biosensing, and quantification in real seawater environments.
1. Introduction
Lithium is a crucial element in energy storage materials and is extensively used in batteries, medicines, lubricants, and ceramics.1 The growing demand for electric vehicles relies on lithium-ion batteries (LIBs), which provide a balanced weight, capacity, and charging/discharging efficiency. Lithium carbonate is widely used as a mood stabilizer in the treatment of bipolar disorders, such as manic depression, and psychiatric conditions, such as schizoaffective disorder. Lithium oxide is used in special glasses and ceramics, which improves their thermal shock resistance. Lithium-based high-temperature lubricating greases are known for their stability and water resistance. Lithium is also used in nuclear fusion reactions and as a coolant in some types of nuclear reactors. Therefore, its global demand is increasing exponentially for the growing market for lithium commodities. However, major lithium resources are limited and confined to only a few countries in the form of salt lake brine or mineral ores.2 The lack of sustainable supply sources has prompted researchers to exploit unusual resources like sea brine, geothermal brine, and recycling of LIBs (lithium-ion batteries).3 Therefore, lithium mapping or detection in these dilute supply sources is a basic requirement before extraction. While lithium is essential for electronic consumer products, its improper disposal leads to the leaching of lithium into waterbodies and drinking water. Lithium has clinical applications for certain mental disorders,4 but excess lithium can be toxic to the central nervous system and the kidneys.5 The present lithium detection and quantification method largely relies on ICP (inductively coupled plasma) testing.6 However, this technique's cost and prolonged sample preparation and testing time are demerits. Other detection methods involve spectroscopic and electrochemical methods.7–17 Among various detection methods, fluorescence detection is widely used due to its high sensitivity, fast measurement time, and ease of use. The design of such a sensory system consists of a lithium-selective receptor18–20 attached to a fluorophore for optical signaling.7,8 Most of the probes consist of small molecule receptors like crown ethers and their derivatives,9–12 calixarenes,13 metallacrowns, etc.14 However, most of these probes are not water soluble and require organic solvents for detection studies,9–11,13 which is inappropriate for biological or aquatic environmental samples. Also, most probes show blue luminescence, which is not suitable for biological samples and suffers from recycling issues.
Apart from lithium sensing, lithium extraction from dilute supply sources like seawater (∼0.2 ppm) is equally important to meet the global demand. Nevertheless, capturing lithium from an aqueous system poses a well-acknowledged challenge owing to its high hydration energy and the existence of alkali and alkaline earth metal ions.21 The common methods for lithium separation involve precipitation,22 adsorption,23–29 solvent extraction,6,21,30–34 electrodeposition,35–37 and membrane separation.38–43 The adsorption technique has garnered increased interest because of its straightforward operation, minimal environmental impact, and cost-effectiveness. These adsorbents include inorganic ion sieves, composite materials, and ionophore-appended functional materials. However, most of these adsorbents often fail in dilute solutions and exhibit low selectivity and poor adsorption capacity.
The examination of the aforementioned literature indicates that lithium recognition and separation commonly rely on ion-sieve materials and small molecule receptors. However, these small molecules pose challenges in terms of removal and recyclability for extraction processes. Moreover, the utilization of these receptors for creating hybrid materials that serve the dual purposes of lithium sensing and removal is infrequent but highly desirable. Lately, there has been an increasing fascination with functional materials comprising organic–inorganic hybrids, to simultaneously achieve sensing and adsorption capabilities.44–50 While the concept of a single platform for environmental monitoring and remediation using such materials is desirable, it is relatively scarce for lithium in existing literature. Hybrid materials featuring an analyte-specific chelating probe attached to a mesoporous matrix offer the capability for dual investigations in aqueous suspension, a feat not achievable with simple organic probes alone. Despite the demonstrated success of these materials in dealing with transitional and heavy metal ions, there is currently no reported instance of their application in the simultaneous detection and extraction of lithium.
In this study, our primary objective is to create a functional material with the capability to concurrently sense and extract lithium ions from a pure aqueous system using a unified platform. We employed a modified synthesis approach to produce dendritic mesoporous silica as the substrate, and anthraquinone amide was selected as the lithium-selective chelating fluorophore. The choice of dendritic silica was based on its favorable aqueous dispersibility and high surface area featuring nanochannels suitable for substantial organic loading.51,52 Likewise, anthraquinone amide was chosen for its strong lithium binding affinity and selectivity.6,53 Our group recently demonstrated that anthraquinone-based amides exhibit strong lithium binding and extraction capabilities.53 However, these small molecule receptors face challenges in lithium extraction using biphasic solvent systems, which require large volumes of volatile organic solvents. Handling weak brine (low-concentration lithium) under biphasic solvent extraction conditions demands significant amounts of organic solvents and effort. In contrast, the adsorption process is advantageous for dealing with weak brine. Therefore, our objective was to create functional materials derived from anthraquinone amides for lithium extraction using a coordination-driven adsorption method. To enhance selectivity, the open-end amide terminals were covalently attached to a silica surface, closing the open ends to form a macrocyclic-like system that improves selectivity. The resultant green-emitting material (MDNS@DAAH) was utilized for both lithium-ion sensing and adsorption studies in an aqueous medium while in dispersion. This material demonstrates a rare selective turn-on green emission with lithium ions in the presence of other major cations commonly found in seawater. The green emission with turn-on fluorescence offers advantages over blue-emitting materials for biological samples. We examined the potential application of this probe material for lithium biosensing in the living organism Artemia salina using fluorescence imaging. Moreover, we investigated the material's ability to strip lithium ions and regenerate to showcase its recyclability across multiple cycles. Additionally, the material accurately quantifies lithium in seawater and shows a high extraction efficiency (∼90%) from sea bittern, a notably challenging task. The evaluation of the material's performance in lithium-ion sensing, adsorption, recyclability, and practical applications underscores its superior capabilities compared to those reported in existing literature.
2. Experimental section
The materials and methods with additional experimental procedures are available in the ESI† of this manuscript.
2.1 Synthesis of dimethyl 2,2′-((9,10-dioxo-9,10-dihydroanthracene-1,8-diyl)bis(oxy))diacetate
In a typical procedure, 3.7 g (15.45 mmol) of 1,8-dihydroxyanthraquinone was refluxed with 6.39 g (465.29 mmol) of K2CO3 in an 80 mL solution of acetonitrile for an hour and then allowed to cool to room temperature. Next, 5.12 mL (46.16 mmol) of ethyl bromoacetate was added to the above solution and was refluxed for another 48 hours. After the reaction, the solvent was evaporated by rotary evaporation, resulting in a yellow crude product. This product was purified by washing it with methanol several times, leading to the recovery of a pure product (5.734 g, yield: 90%). The pure product was then characterized by 1H, 13C NMR, and LC-MS techniques (Fig. S1–S3†). 1H NMR (600 MHz, CDCl3 solvent): δ (ppm): 7.91 (d, 2H), 7.61 (t, 2H), 7.22 (d, 2H), 4.85 (s, 4H), 4.25 (q, 4H), 1.28 (t, 6H). 13C NMR (δ, ppm): 14.23, 61.57, 67.23, 120.78, 121.13, 125.09, 133.90, 134.96, 157.95, 168.66, 182.08, 183.52.
2.2 Synthesis of 2,2′–((9,10-dihydroanthracene-1,8-diyl)bis(oxy))diacetohydrazide
The above synthesized diester (2.5 g, 6.06 mmol) was refluxed in ethanol with hydrazine hydrate (1.132 mL, 36.373 mmol) for 6 h to obtain orange-coloured precipitates. Afterward, the solvent was rotary evaporated and the crude product was washed several times with chloroform and methanol and dried in a desiccator under a vacuum to afford the pure DAAH ligand (1.909 g, yield: 88%). The synthesized ligand was well characterized using 1H, 13C NMR, and LC-MS (Fig. S4–S6†). 1H NMR (600 MHz, DMSO-d6), δ (ppm): 4.55 (s, 4H), 4.79 (s, 4H), 9.31 (m, 2H), 7.81 (m, 4H), 7.55 (d, 2H). 13C NMR (600 MHz), δ (ppm): 68.04, 119.88, 120.74, 121.16, 134.60, 135.57, 156.98, 157.63, 183.38.
2.3 Synthesis of mesoporous dendritic nano-silica (MDNS)
A biphasic (aqueous and organic) emulsion method was followed for the synthesis of MDNS, as illustrated in Scheme 1 and Fig. S7.† Initially, 2.5 g (7.35 mmol) of BDAC (benzyl dimethyl dodecyl ammonium chloride) was dissolved in 150 mL Milli-Q water at 600 rpm for 20 min, affording micellar froth. To this solution, 2.8 g of urea (46.62 mmol) was added and the reaction mixture was stirred for another 30 min. To this aqueous phase, the organic phase consisting of a mixture of tetramethyl orthosilicate (TMOS) (8 ml, 54.2 mmol) in 50 mL of trimethylbenzene (TMB) was added slowly for 45 min at 600 rpm. Afterward, the mineralizing agent isopropanol (IPA) was added to the above reaction mixture and refluxed for 12 h, affording white colloidal precipitates. The obtained white precipitates were centrifuged (8000 rpm) and further washed with methanol and water 3 times each and finally dried overnight in an oven at 75 °C, affording pure white MDNS (6.12 g).
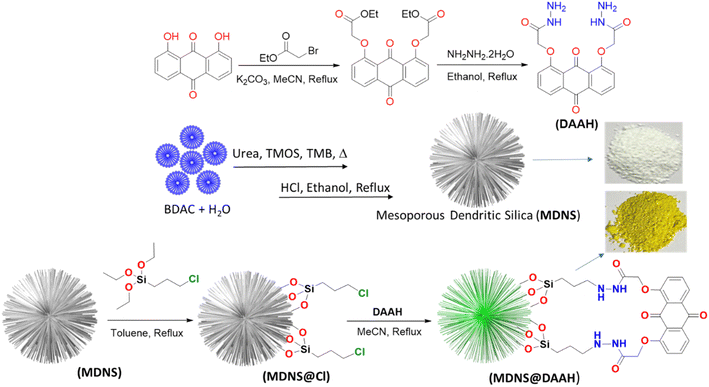 |
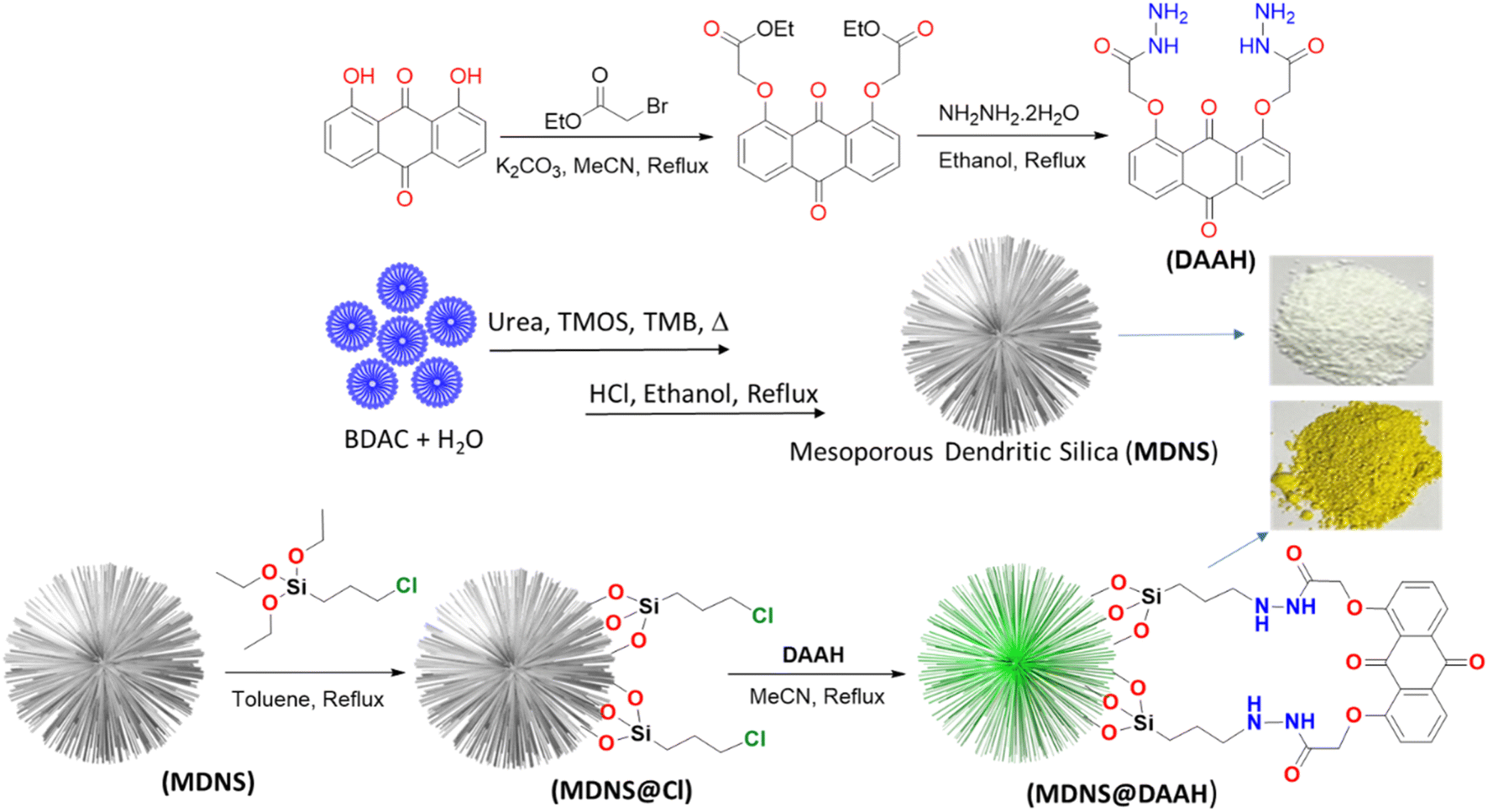
| | Scheme 1 The synthetic depiction of the anthraquinone-based ligand (DAAH) and the mesoporous dendritic nano-silica (MDNS), along with MDNS@Cl and the final material MDNS@DAAH. | |
2.4 Synthesis of MDNS@Cl
For the synthesis of MDNS@Cl, typically 3 g of the above synthesized MDNS material was refluxed with 3-CPTES (6.5 mL, 38.06 mmol) in 100 mL toluene at 110 °C for 24 h. After the reaction times, the precipitates were collected through centrifugation, washed with toluene and methanol 3 times, and finally dried in an oven at 60 °C for 5 h, affording 2.18 g of the MDNS@Cl product.
2.5 Synthesis of the final material MDNS@DAAH
For the synthesis of the final material MDNS@DAAH, 2 g of the synthesized material MDNS@Cl was refluxed with the synthesized DAAH ligand (1.8 g, 4.68 mmol) in 150 ml dry acetonitrile at 82 °C for 24 h under argon. Afterwards, the material was collected through centrifugation and subjected to washing with ethanol and chloroform 3 times and dried in an oven at 55 °C for 5 h, affording 2.21 g of the final material MDNS@DAAH. The final material was then thoroughly characterized by various analytical techniques.
3. Results and discussion
3.1 Synthesis and surface characterization
As per the objective of the proposed work, an anthraquinone-based amide was synthesized to serve as the chelating fluorescence tag and dendritic nano-silica as the substrate, as depicted in Scheme 1 and detailed in the Experimental section. The starting material 1,8-dihydroxyanthraquinone undergoes a nucleophilic substitution reaction with 2-bromoethyl acetate to produce the ester, which subsequently reacts with hydrazine hydrate to produce the amide ligand (DAAH) (Fig. S1–S6†). The free amine site of the hydrazine is covalently functionalized with the dendritic silica. Mesoporous dendritic nano-silica (MDNS) was created using a biphasic emulsion system, employing tetramethyl orthosilicate (TMOS) as the silica source, urea for hydrolysis, BDAC (benzyl dimethyl dodecyl ammonium chloride) as a template, and trimethyl benzene/H2O as the processing medium. Here, TMOS is used as a silica source, which is known to undergo rapid hydrolysis compared to conventional tetraethyl orthosilicate (TEOS). Nevertheless, control over the hydrolysis rate was achieved through the selection of the emulsion system and optimal surfactant concentrations, resulting in a dendritic structure with a high surface area matrix. The step-wise formation of the material is illustrated in Fig. S7†. Initially, the template (BDAC) was dissolved in water to form a micellar solution, followed by the addition of the hydrolyzing agent urea. The organic phase, containing TMOS in TMB, was then added to this aqueous solution. As TMOS crosses the phase boundary into the aqueous phase, it undergoes hydrolysis, followed by condensation reactions, leading to the formation of oligomers. These oligomers subsequently develop into branched structures, resulting in the creation of mesoporous dendritic nanosilica (MDNS). This MDNS material was further modified by chemically reacting it with 3-chloropropyltriethoxysilane (CPTES) to form the intermediate MDNS@Cl and finally with the ligand DAAH to produce the MDNS@DAAH material.
The morphological, structural, and chemical information was obtained using SEM, TEM, PXRD, TGA, XPS, BET, and FTIR techniques. The FeSEM images of the synthesized mesoporous dendritic nano-silica (MDNS) material exhibited 120–150 nm-sized particles with a regular and ordered spherical morphology (Fig. 1). The ordered and regular spherical morphology remains nearly unaltered during the chemical processing with the CPTES group to form the MDNS@Cl material and subsequently with the DAAH ligand to form the final material MDNS@DAAH. The TEM and HR-TEM images of the synthesized material revealed a dense and fibrous 3-D morphology with a particle size of 120–150 nm as observed in the SEM images (Fig. 2).
 |
| | Fig. 1 (A–C) FeSEM images of the synthesized materials MDNS and MDNS@Cl, and the final material MDNS@DAAH. (D–F) A magnified FeSEM view of the MDNS, MDNS@Cl and MDNS@DAAH materials. | |
 |
| | Fig. 2 (A–C) TEM images of the synthesized materials MDNS and MDNS@Cl and the final material MDNS@DAAH. (D–F) HR-TEM images of the MDNS, MDNS@Cl, and MDNS@DAAH materials. | |
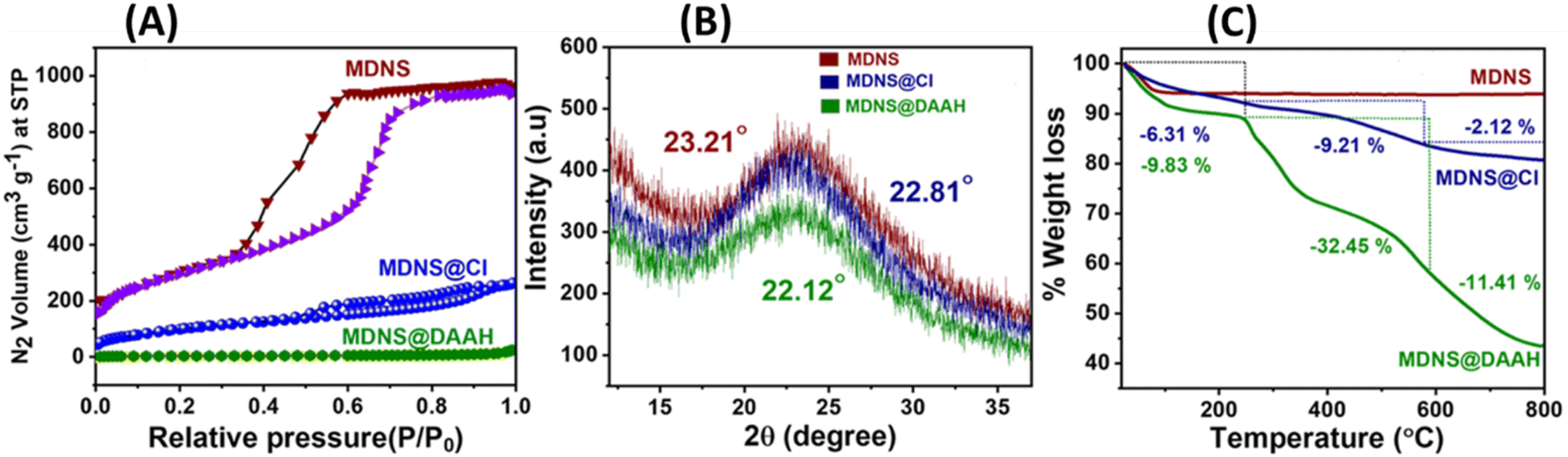
The BET measurements provided insights into the surface area, pore size, pore volume, and characteristics of the materials (Fig. 3A and Table 1). The initial material MDNS exhibited a substantial surface area of 1240 m2 g−1, characterized by mesoporous pore channels measuring 5.8 nm and a notable pore volume of 1.6 cm3 g−1 (Fig. S8†). Upon covalent attachment of the CPTES group and the loading of the DAAH ligand, the surface area, pore size, and pore volume reduce significantly hinting towards successful covalent attachment of the CPTES group and the loading of the DAAH ligand to form the final material MDNS@DAAH. Also, the order of capillary condensation reduces during the chemical processing, which also signifies the attachment of the functional groups. The PXRD pattern of MDNS exhibited an amorphous nature having a broad band at 23.21°, which remains largely unaffected throughout the chemical processing (Fig. 3B). However, a decrease in the intensity of the bands was observed, which can be anticipated due to the functionalization process. Thermogravimetric analysis (TGA) of the materials was also performed to derive information regarding stability, ligand loading, organic content, and the nature of functionalities (Fig. 3C). The starting material MDNS showed high thermal stability and no organic content loss was observed, which is an important feature of silica-based materials. The intermediate material MDNS@Cl showed a weight loss of 6.31% in the region of 0–250 °C, which can be ascribed to the surface adsorbed water molecules, which increased slightly to 9.83% in MDNS@DAAH possibly due to a larger number of electronegative atoms and the presence of NH functionality that can promote the surface adsorption of water molecules. The second region from 250–580 °C signifies the loss of organic functionalities, which is also a measure of organic content in the materials. The final material MDNS@DAAH showed a weight loss of 32.45% as compared to 9.21% in the MDNS@Cl material. The significant organic presence within the final material confirms the successful incorporation of the DAAH ligand, forming the final material and indicating a substantial level of functionalization. This is possibly due to the presence of dense fibrous pore channels, which allow the radial accessibility of the pore channels, which is ideal for effective ligand immobilization. The final phase from 580–800 °C corresponds to the destruction phase, where dihydroxylation of surface silanol groups was observed.
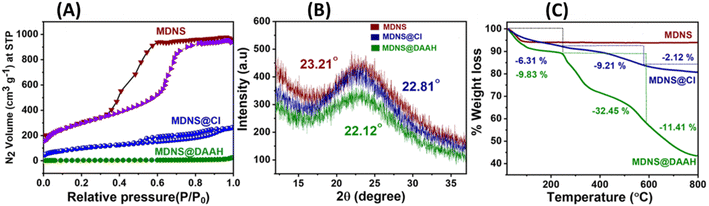 |
| | Fig. 3 (A) Surface area profiles of the MDNS, MDNS@Cl and MDNS@DAAH materials. (B) High-angle PXRD measurements of the materials. (C) TGA profiles of the materials MDNS, MDNS@Cl and MDNS@DAAH. | |
Table 1
S
BET parameters of the synthesized materials
| Material |
Surface area (m2 g−1) |
Pore size (nm) |
Pore volume (cm3 g−1) |
| MDNS |
1240 |
5.8 |
1.6111 |
| MDNS@Cl |
210.21 |
3.4 |
0.9231 |
| MDNS@DAAH |
49.21 |
2.3 |
0.054 |
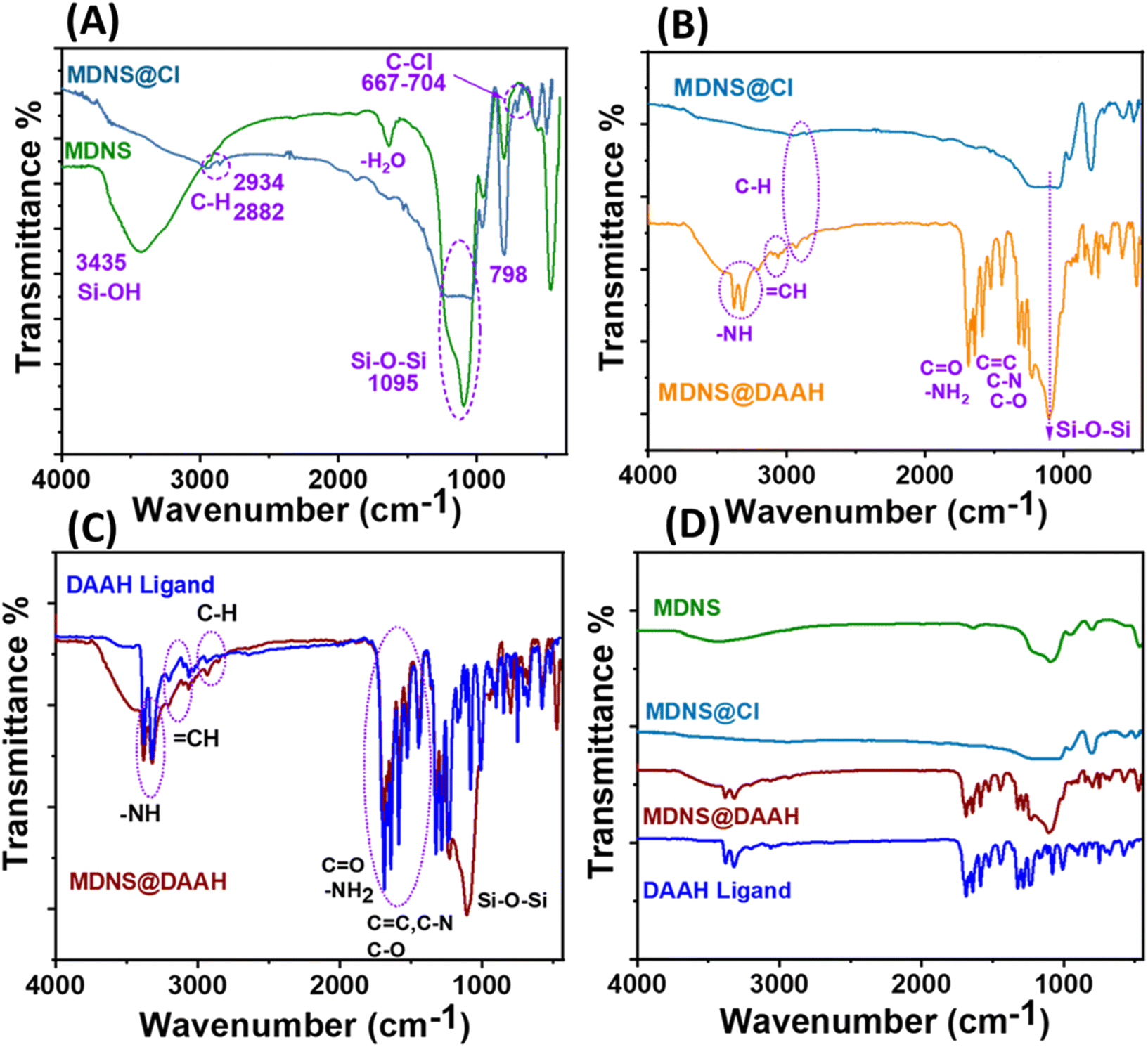
The information regarding surface functionalization was also evaluated from the FT-IR spectral analysis of the synthesized materials (Fig. 4). The spectrum of dendritic silica showed characteristic peaks at 3435, 1095, and 798 cm−1 corresponding to Si–OH and Si–O–Si stretching and bending vibrations. Covalent functionalization with the CPTES group was ensured with peaks at 2934, 2882, and 667–704 cm−1 corresponding to the alkyl stretching (C–H) of the CPTES group and C–Cl stretching (Fig. 4A). Other peaks at 3388–3314, 3100, 1705, 1730, 1685, 1620, 1585, 1590, 1378, and 1210 cm−1 in the FT-IR spectrum of MDNS@Cl and the final material MDNS@DAAH were clearly distinguished upon closer observation (Fig. 4B). The peaks located at around 3388–3314 and 3100 cm−1 correspond to the –NH and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH stretching vibrations, while the peaks at 1705, 1730, 1685, and 1620 cm−1 correspond to carbonyl and symmetric and asymmetric amide carbonyl group and NH stretching vibrations. The CC, C–N, and C–O bonds were observed at peaks of 1585, 1590, 1378, and 1210 cm−1 respectively. Additionally, all peaks characteristic of the DAAH ligand were present in the final material, serving as evidence of the successful synthesis of the product (Fig. 4C and D).
CH stretching vibrations, while the peaks at 1705, 1730, 1685, and 1620 cm−1 correspond to carbonyl and symmetric and asymmetric amide carbonyl group and NH stretching vibrations. The CC, C–N, and C–O bonds were observed at peaks of 1585, 1590, 1378, and 1210 cm−1 respectively. Additionally, all peaks characteristic of the DAAH ligand were present in the final material, serving as evidence of the successful synthesis of the product (Fig. 4C and D).
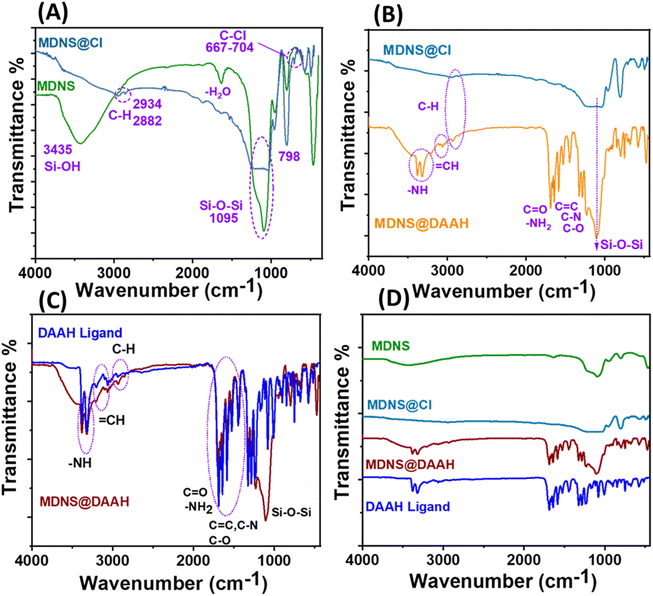 |
| | Fig. 4 (A) FT-IR spectrum of MDNS and MDNS@Cl. (B) FT-IR spectrum of MDNS@Cl and MDNS@DAAH. (C) Comparison of the FT-IR spectrum of the synthesized Ligand DAAH and the final material MDNS@DAAH. (D) Full FT-IR profile of the synthesized ligand and materials. | |
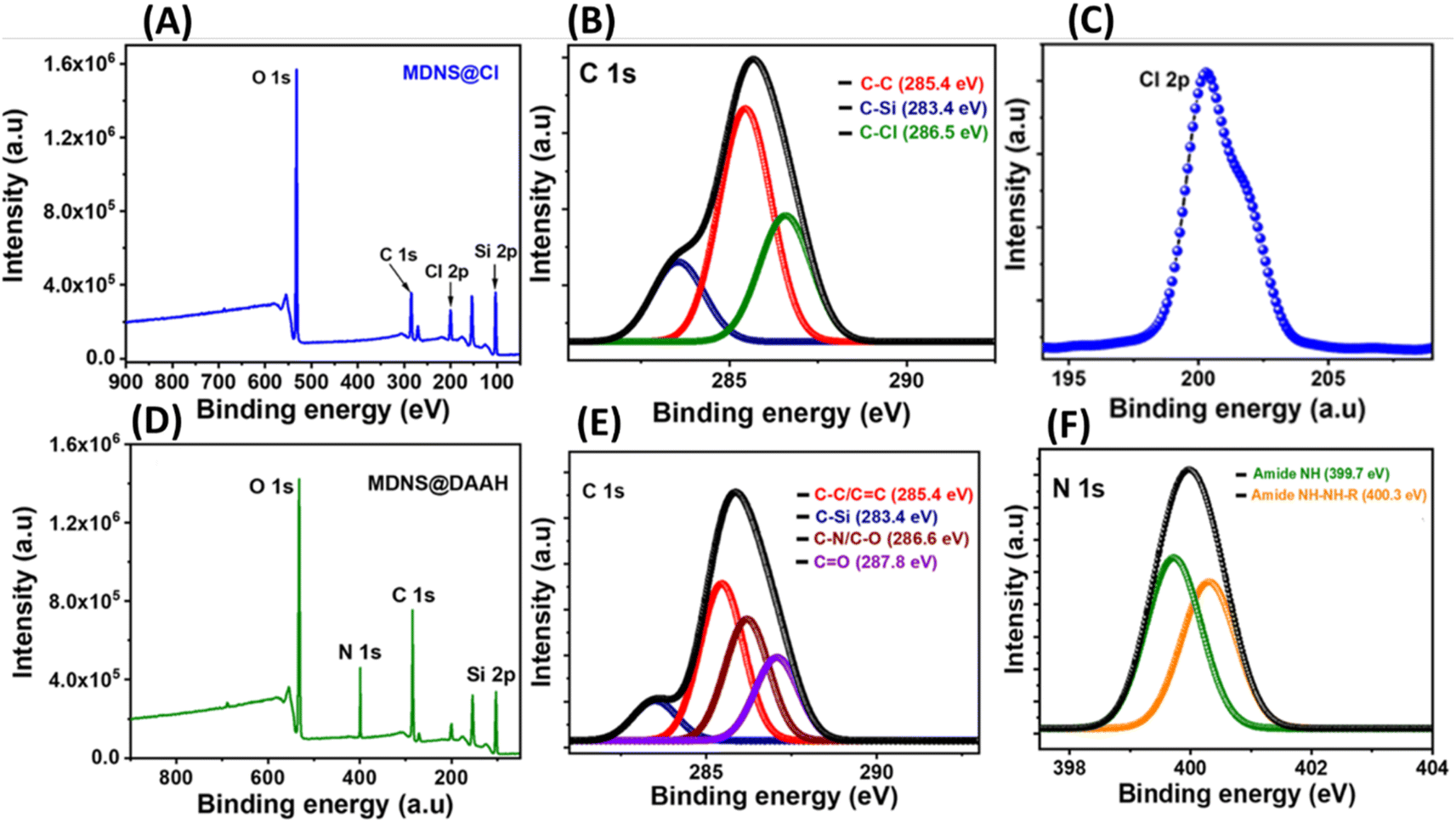
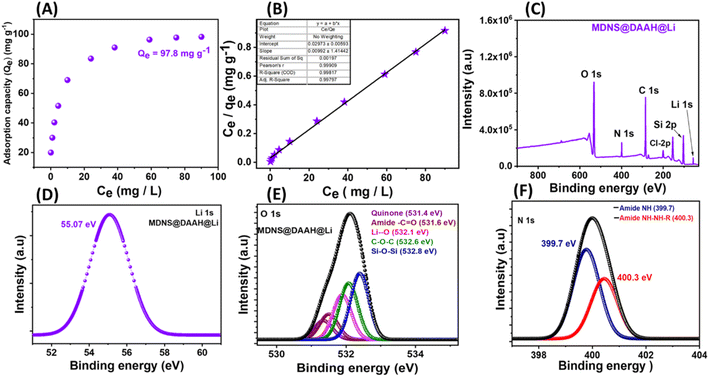
Surface states in the materials were explored using X-ray photoelectron spectroscopy (XPS) (Fig. 5). The synthesis of dendritic silica was confirmed by analyzing the full-scan XPS spectrum of the starting material (MDNS), which disclosed signals corresponding to silicon (Si) and oxygen (O) elements (Fig. S9†). The covalent attachment of the CPTES group to the MDNS@Cl material was established by the presence of Cl 2p (at 200.01 eV) and C 1s peaks in the full-scan XPS spectrum (Fig. 5A and C). Additionally, the C 1s spectra were deconvoluted into three peaks (285.4, 283.4, and 286.5 eV), representing C–C, C–Si, and C–Cl linkages, confirming the successful attachment of the 3-CPTES group to form the MDNS@Cl material (Fig. 5B). The XPS spectra of the final material (MDNS@DAAH) exhibited signals for O 1s, N 1s, C 1s, and Si 2p in the full-scan spectrum, confirming the immobilization of the DAAH ligand (Fig. 5D). Furthermore, the deconvolution of the C 1s spectra showed four peaks at 285.4, 283.4, 286.6, and 287.8 eV, which correspond to the C–C/CC, C–Si, C–N/C–O, and CO functionalities (Fig. 5E). The deconvoluted N 1s spectra show two peaks at 399.7 and 400.3 eV for amide NH and alkyl NH present in the material (Fig. 5F). Oxygen linkages were confirmed by the O 1s spectra, which show four peaks upon deconvolution at 531.2, 531.6, 532.4, and 532.84 eV, corresponding to quinone, amide (–CO), C–O–C, and Si–O–Si linkages (Fig. S10 and S11†). This provides evidence for the effective loading of the DAAH ligand to form the final material MDNS@DAAH.
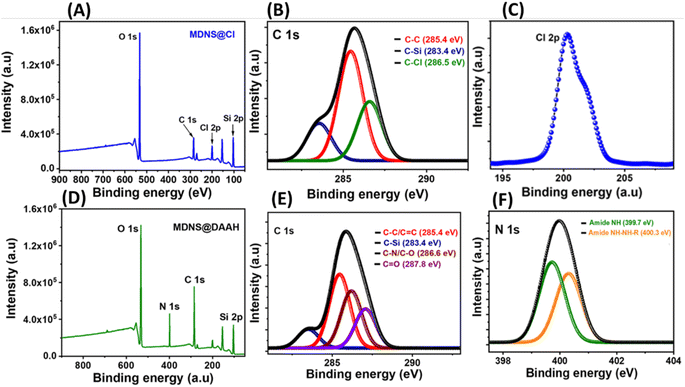 |
| | Fig. 5 (A and D) The XPS full scan spectrum of the 3-CPTES functionalized material MDNS@Cl and the final material MDNS@DAAH. (B and E) The deconvoluted C 1s XPS spectra of MDNS@Cl and the final material MDNS@DAAH. (C and F) The deconvoluted Cl-2p spectra of the MDNS@Cl material and the N 1s spectra of the final material MDNS@DAAH. | |
3.2 Optical selectivity studies
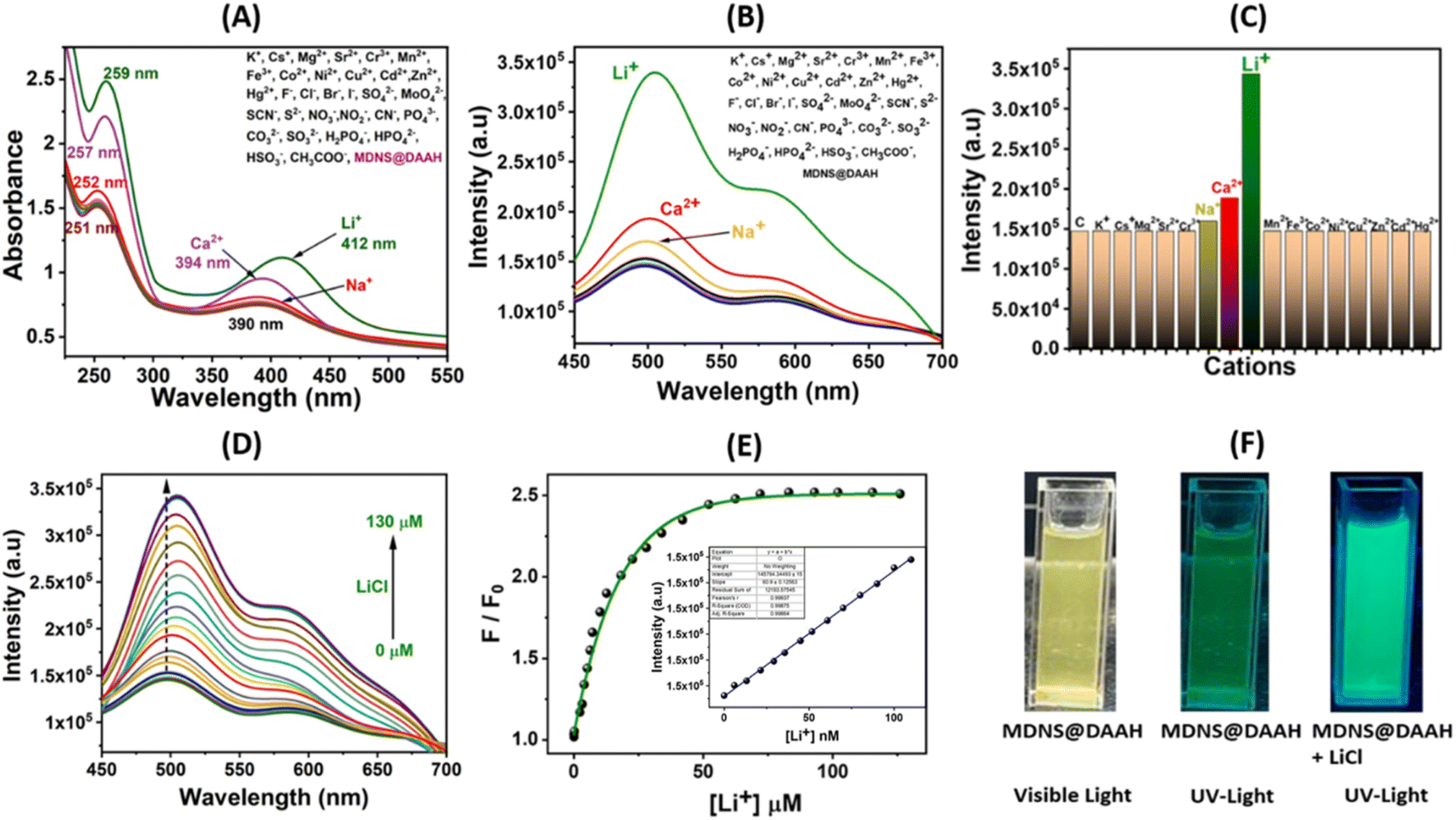
To examine the optical characteristics of the final MDNS@DAAH material, UV-Vis and fluorescence studies were conducted in a neutral aqueous buffer medium (HEPES buffer, pH ∼7.2) while in dispersion (5 mg/50 mL). Various cations and anions were present during these studies (Fig. 6). The UV-Vis spectra revealed two distinct bands at 253 and 390 nm, likely corresponding to π–π* and n–π* transitions. Upon adding Li+ and Ca2+, the 390 nm band exhibited a slight blue shift, while the 253 nm band showed increased intensity (Fig. 6A). These spectral alterations were not observed with other cations and anions under identical conditions. Similarly, the fluorescence emission studies were conducted in dispersion with different cations and anions (Fig. 6B). The material displayed a strong fluorescence emission band at 510 nm upon excitation at 390 nm, resulting in a significant Stokes shift advantageous for analytical studies. Studies on selectivity demonstrated an absolute preference for Li+ ions, exhibiting fluorescence enhancement (turn-on), whereas no notable intensity changes were observed with other cations and anions. A minor intensity enhancement was noted with Ca2+ and Na+ ions, but it remained substantially high with Li+ ions (Fig. 6C). Interference studies and response time studies were performed with other coexisting ions in real tap water and aqueous systems (Fig. S12A and B†). The material exhibited a rapid response to lithium ions (15 s) and a fluorescence response similar to control experiments. Further investigation on selectivity reveals that Ca2+ and Na+ showed no change in fluorescence response at micromolar concentrations (Fig. S12C and D†). The fluorescence response for these ions was only observed at millimolar concentrations and the fluorescence intensity saturates within a small millimolar concentration (5 mM to 35 mM for Ca2+ and 2 mM to 10 mM for Na+). To ascertain lithium-ion detection parameters, fluorescence titration was conducted by gradually adding Li+ ions until reaching a saturated concentration. The material displayed a fluorescence enhancement behaviour, and a non-linear fitting analysis yielded a large enhancement constant (Kb) of 2.85 × 105 M−1.51 The limit of detection (LOD) was estimated by the 3σ method to be 4.9 nM,46 while the limit of quantification (LOQ) based on the 10σ method48 and linear range (LR) values were found to be 14.42 nM and 14.42–90 nM, respectively (Fig. S13A†). To elucidate the binding sites and affinity of the probe material, a Scatchard plot (Fig. S13B†) was employed using the formula log{(F0 − F)/F} = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kb + nlog[Q].48 In this equation, F0 and F represent the fluorescence intensity of the probe material before and after the introduction of Li+ ions, respectively, and [Q] is the concentration of [Li+]. The linear plot of log{(F0 − F)/F} versus log[Q] revealed a binding constant (Kb) of 1.21 × 105 M−1 with a number of binding sites (n) of approximately 1.88, in good agreement with the two binding pockets provided by the ligand (Fig. S13B†). These results demonstrate the selective and strong binding of lithium ions, leading to selective lithium recognition.
Kb + nlog[Q].48 In this equation, F0 and F represent the fluorescence intensity of the probe material before and after the introduction of Li+ ions, respectively, and [Q] is the concentration of [Li+]. The linear plot of log{(F0 − F)/F} versus log[Q] revealed a binding constant (Kb) of 1.21 × 105 M−1 with a number of binding sites (n) of approximately 1.88, in good agreement with the two binding pockets provided by the ligand (Fig. S13B†). These results demonstrate the selective and strong binding of lithium ions, leading to selective lithium recognition.
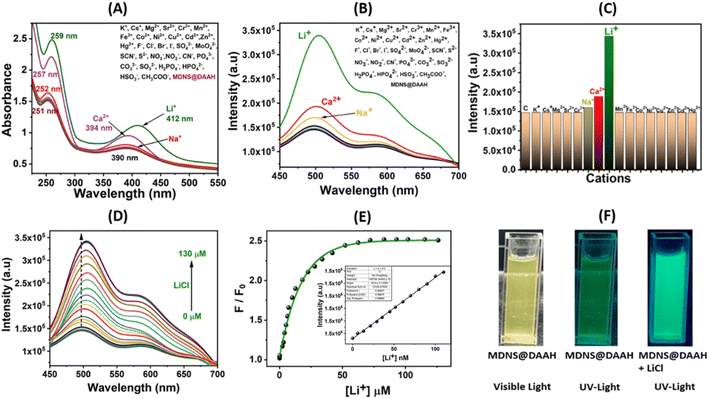 |
| | Fig. 6 (A and B) Investigation of UV-Vis and fluorescence selectivity in the presence of diverse cations and anions with 50 mM concentration. (C) Evaluation of relative fluorescence emission intensity for the MDNS@DAAH material with cations. (D) The fluorescence titration profile with LiCl up to a saturation concentration. (E) The non-linear fitting to analyze titration data and LOD. plot in the inset. (F) The color change in the material suspension under UV light in the absence and presence of Li+ ions. | |
3.3 DFT studies and mechanism
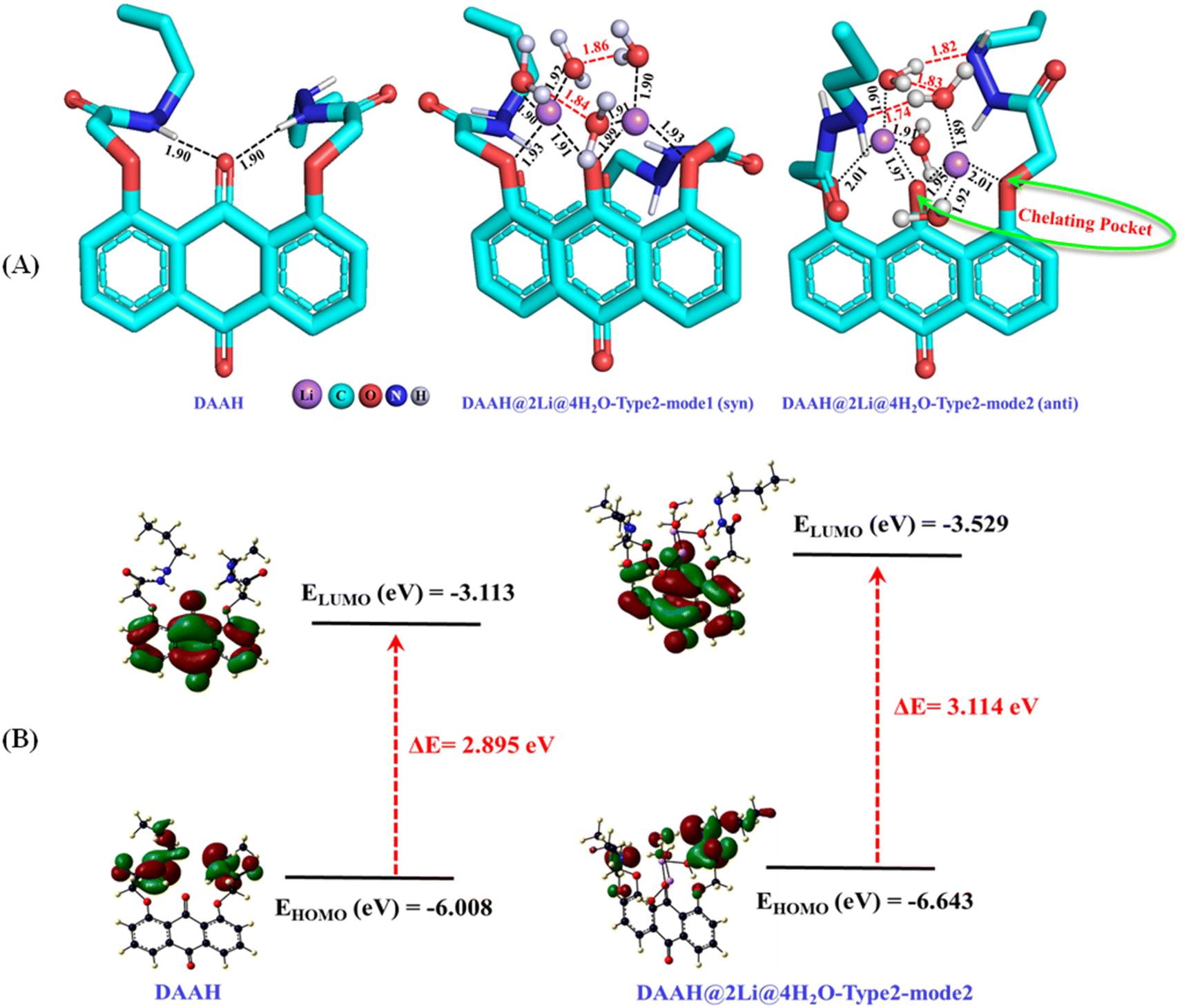
To comprehend the binding mechanism of the ligand within the lithium-ion, various potential binding modes were examined using DFT calculations at the B3LYP-D3/Def2-SVP level of theory in an aqueous medium. The optimized geometry of the DAAH ligand reveals two hydrogen bonding interactions involving the amide NH and the central carbonyl oxygen of the anthraquinone motif (Fig. 7A). The ligand component was further analyzed to explore the potential modes of binding with Li+ ions in an aqueous solvent. Initially, the possible binding sites were examined with one lithium chloride. Considering the three binding sites: type-1 (binding through amide carbonyl oxygen), type-2 (binding through interior carbonyl oxygen of the anthracene moiety, chelating pocket), and type-3 (binding through exterior carbonyl oxygen of the anthraquinone motif), as shown in Table S1† (entries 1–3, respectively), it was found that type-2 binding was energetically more favourable. The interior pocket provided by the anthraquinone ligand is energetically preferred for the binding as two oxygen atoms are involved. The calculations performed with the explicit water molecule replacing the chloride ion showed higher binding energy of lithium ions in type-2 binding mode compared to the binding energy with Cl− ions (entries 4–6 for the binding mode of type-1, 2, and 3, respectively, Table S1†). The possibility of binding a second Li+ ion in type-1 and type-2 modes was also examined with Cl− ions and H2O molecules, showing that type-2 binding in both cases is energetically more favourable compared to that of the former binding mode, involving two oxygen atoms (entries 7 and 8 for Li–Cl and 9 and 10 for Li–H2O, Table S1†). Further, the calculations have been performed with the type-2 binding mode exploiting the tetra-, penta-, and hexa-coordination of lithium by adding chloride anions and water molecules. The calculated results reveal that the penta- and hexa-coordination were not feasible for the binding of Li+ ions with the DAAH ligand molecule. The interaction of Li+ ions with the DAAH amide carbonyl oxygen or the amide NH group was also not observed. The tetra-coordinated lithium complex with one chloride and one water molecule (entries 11–13, Table S1†) and with two water molecules have further been examined (entries 14 and 15, Table S1†). The tetra-coordinated lithium complex with the arrangements of Cl−, H2O, and the auxiliary amide arms in the same directions was considered as syn–syn. The arrangement of Cl− and H2O in opposite direction and the auxiliary amide arms in the same direction as that of the tetra coordinated Li-complex was considered as anti–syn, while, the Cl−, H2O, and auxiliary amide arms in opposite directions was termed anti–anti (entries 11–13, Table S1†). The calculated results suggest that the tetra-coordinated complexes with two water molecules are energetically more favourable compared to those with one Cl− and H2O (entries 14 and 15, Table S1†). In these complexes, the anthraquinone carbonyl oxygen and side-arm ethereal oxygen create a chelating pocket for lithium-ion binding (Fig. 7A). The calculated tetrahedral complexes with two H2O molecules further reveal that the structure with the auxiliary amide arms in the opposite direction (anti) is more stable (∼4.5 kcal mol−1) than the structure with amide arms in the same direction (syn) due to additional hydrogen bonding interactions (Fig. 7A).
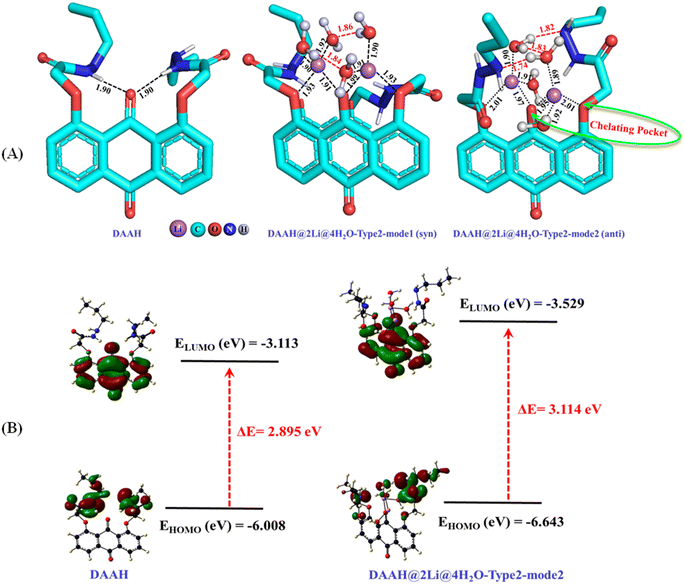 |
| | Fig. 7 (A) B3LYP-D3/Def2-SVP optimized geometries of the DAAH ligand motif and its lithium complexes (red lines represent the hydrogen bonding interactions; bond distances are given in Angstrom (Å)). (B) HOMO–LUMO energy gaps for the DAAH part and the optimized lithium complex. | |
We have also examined the HOMO and LUMO energy levels for the DAAH and the DAAH@2Li@4H2O-type 2-mode 2 complex at the same level of theory (Fig. 7B). For DAAH, the highest occupied molecular orbital (HOMO) at −6.008 eV is entirely localized on the amide side arms of the anthraquinone motif, whereas the lowest unoccupied molecular orbital (LUMO) at −3.113 eV is localized entirely on the accepting group, i.e., the anthraquinone ring (Fig. 7B). Similarly, in the lithium complex, the HOMO at −6.643 eV is localized on the amide side arms of the ligand, and two H2O molecules are involved in the hydrogen bonding, while the LUMO at −3.529 eV is exclusively on the anthraquinone ring. The HOMO–LUMO energy gaps (ΔE) for DAAH and the complex are 2.895 eV and 3.114 eV, respectively.
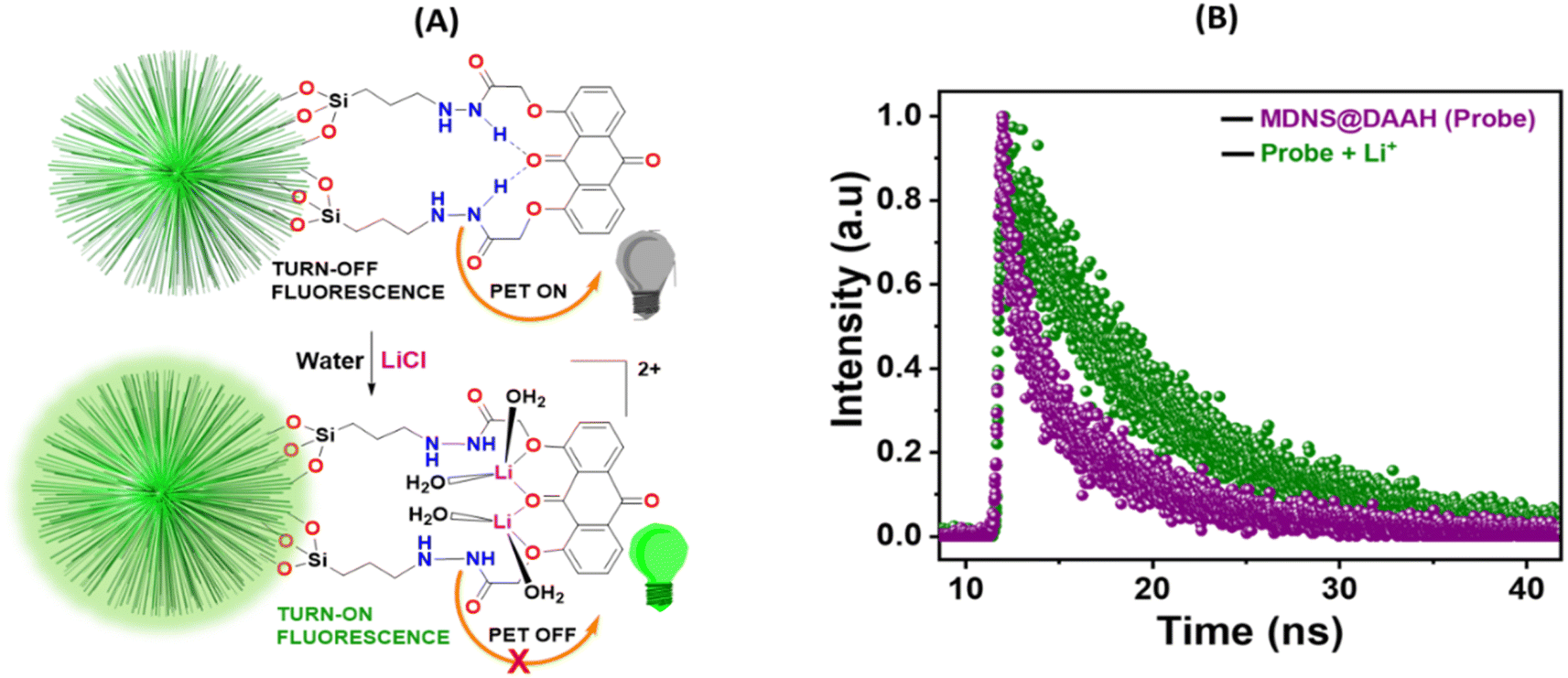
The smaller HOMO–LUMO energy gap in DAAH facilitates effective photoelectron transfer (PET), where the hydrogen bonding between the amide NH and the carbonyl oxygen of the anthraquinone motif brings them closer together and expedites PET, resulting in weak fluorescence (turn-off) of the material (Fig. 8A). However, in the presence of lithium ions, the turn-on fluorescence is attributed to the chelation-enhanced fluorescence (CHEF) effect due to lithium complexation. Lithium complexation disrupts the hydrogen bond between the amide NH and carbonyl oxygen, interrupting the PET process and restoring the fluorescence (turn-on fluorescence) of the DAAH ligand. The turn-on emission in the presence of lithium ions was further assessed using fluorescence lifetime measurements (Fig. 8B). The fluorescence lifetime of the MDNS@DAAH material exhibited a τ value of 2.8 ns (χ2 = 1.02), which significantly increased to 3.6 ns (χ2 = 1.1) in the presence of lithium ions. This observation confirms the turn-on behaviour of the material in the presence of Li+ ions, supported by UV-Vis and fluorescence changes.
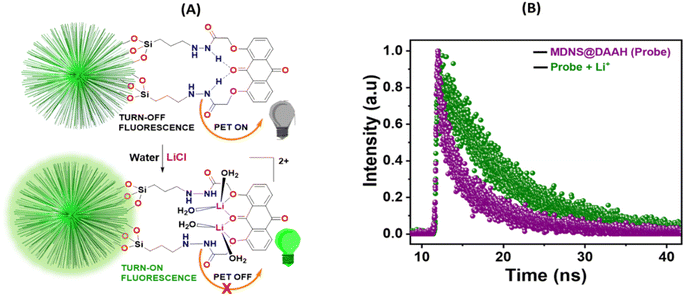 |
| | Fig. 8 (A) PET-based turn-on-off mechanism for fluorescence enhancement in MDNS@DAAH with lithium-ion binding. (B) Lifetime measurements of the material MDNS@DAAH before and after the addition of Li+. | |
3.4 Adsorption studies
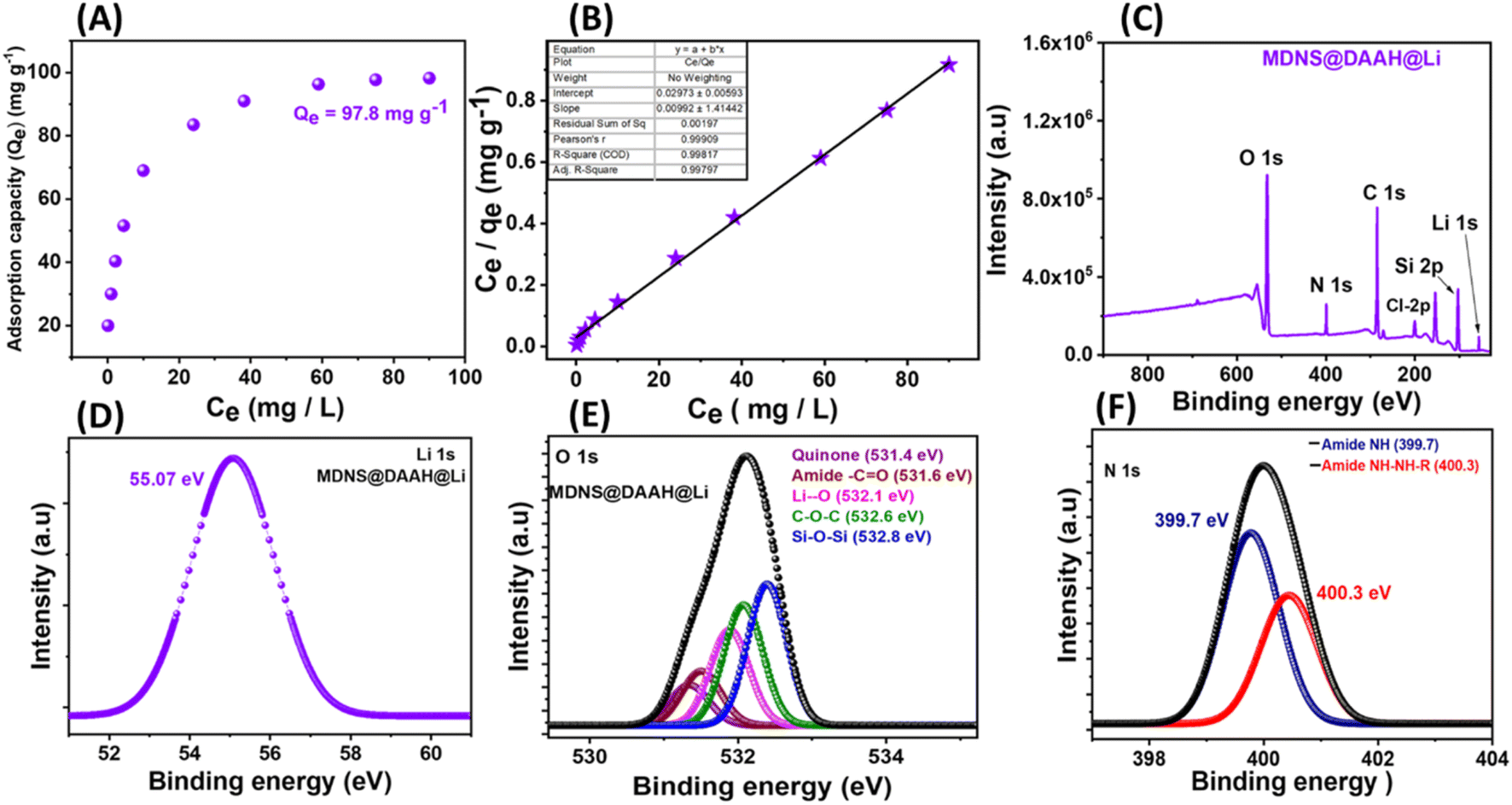
The material's potential for lithium extraction was assessed through batch adsorption studies. Typically, 5 mg of the material was exposed to treatment in a LiCl aqueous solution with concentrations extending from 5 to 100 ppm, with agitation until equilibrium was reached (2 hours). After the treatments, the sample tubes were centrifuged and filtered through a 0.24 μm syringe filter; afterward, the lithium concentrations in the filtrate were measured using ICP-MS analysis. Qe, the equilibrium adsorption capacity, was determined using the formula Qe = (Ci − Ce)V/W, where Qe is the amount of metal ion adsorption at equilibrium, Ci and Ce refer to the initial and final concentrations of Li+ ions, V refers to the volume in liters, and W denotes the weight of the adsorbent in grams (g). The maximum adsorption capacity, Qe, for lithium ions derived from the adsorption isotherm plot was 97.8 mg g−1, quite high compared to the reported literature (Fig. 9A). The adsorption behaviour was analyzed using adsorption isotherm models: Langmuir and Freundlich (Fig. 9B, S14 and Table S3†). The Langmuir fitting was identified as the most suitable, exhibiting a good correlation factor close to unity, suggesting the monolayer adsorption behavior of the analyte. Additionally, a competitive adsorption experiment demonstrated a selective uptake of lithium with 92.34% extraction efficiency (Table S4†). This selectivity was further supported by the XPS full scan spectrum of the lithium-adsorbed material (Fig. 9C). Following the competitive adsorption experiment, the material underwent washing with water and evaluation using XPS spectroscopy, revealing the presence of Li 1s at 55 eV (Fig. 9D). Also, a detailed analysis of core–shell O 1s deconvoluted spectra indicated an approximately 0.2 eV shift in binding energy and also the appearance of a new peak at 532.1 eV for Li–O suggesting an interaction of lithium coordination (Fig. 9E). To assess the effect of Ca2+ and Na+ on adsorption capacity, batch adsorption experiments conducted in the presence of a 1000-fold excess of these metal ions showed a 3–4% decrease in adsorption capacity (Fig. S15†), indicating minimal interference.
 |
| | Fig. 9 (A) The equilibrium adsorption capacity (Qe) plot of the material towards the Li+ ions. (B) The Langmuir adsorption isotherm plot towards the Li+ ions. (C) The full scan spectrum of the material after the Li+ adsorption from the competitive experiment showing the presence of lithium. (D) The Li 1s XPS spectra of the final material after adsorption. (E) The core–shell O 1s XPS spectra of the final material after lithium adsorption. (F) The core–shell N 1s XPS spectra of the material after lithium adsorption. | |
3.5 Adsorption kinetics and thermodynamics
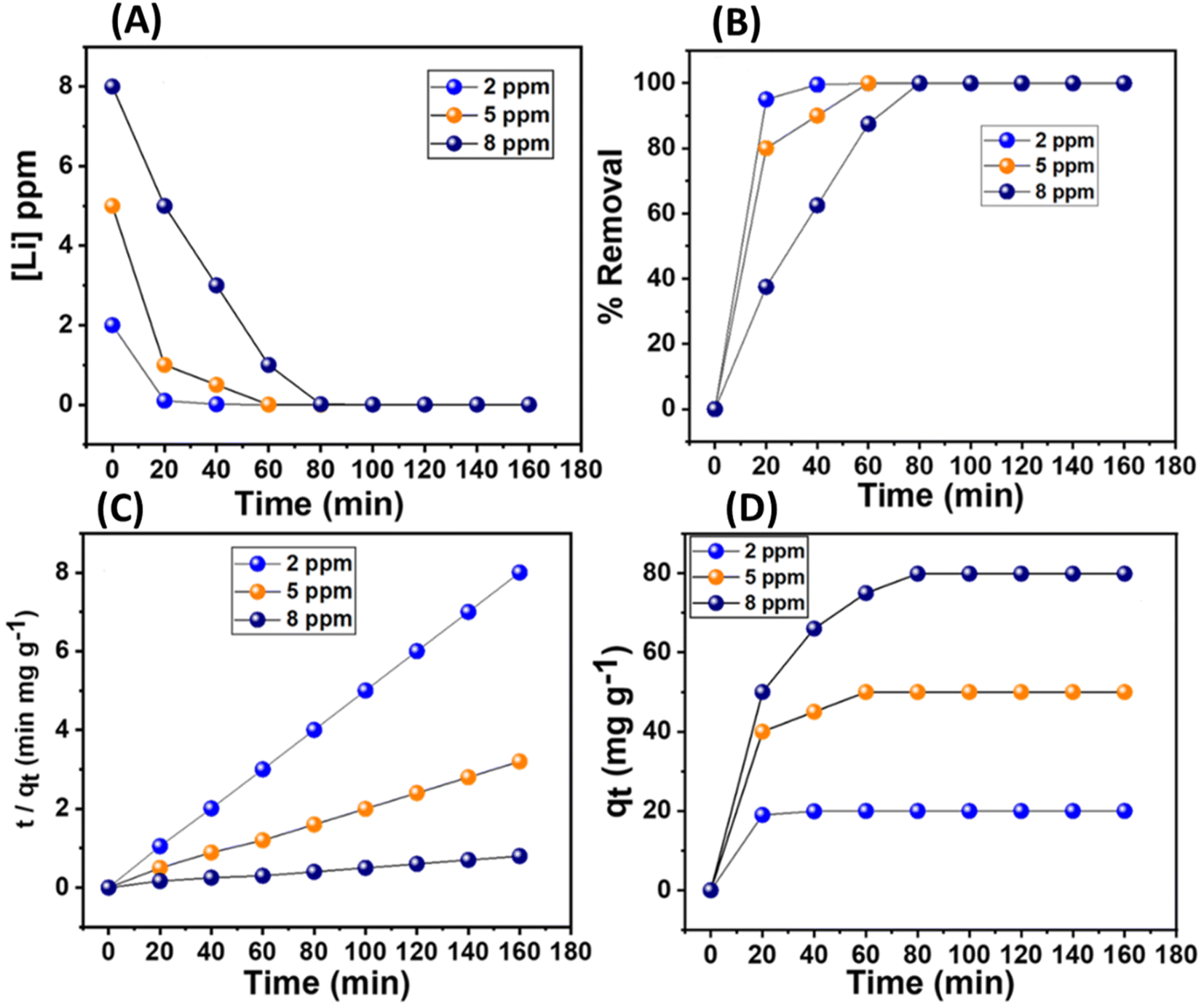
The adsorption rate kinetics were performed by treating 5 mg of the material in varied concentrations of lithium ions (2, 5, and 8 ppm). It was observed that lithium removal rates of ≥90 were attained within 20, 40, and 60 minutes for concentrations of 2, 5, and 8 ppm, respectively (Fig. 10A and B). The lithium adsorption values corresponding to this set of concentrations were about 20, 50, and 79 mg g−1, respectively (Fig. 10C). The kinetic models for the adsorption rate represented by the equations, namely pseudo-first-order and pseudo-second-order, were applied: ln(qe – qt) = ln(qe) – (k1)t and t/qt = 1/(k2qe2) + t/qe. Here, qe is the adsorption capacity at equilibrium, qt is the adsorption capacity at time t, and k1 and k2 are the respective rate constants for pseudo-first-order and pseudo-second-order kinetics. The first and second-order rate kinetic models were examined and compared elaborately, which revealed that the pseudo-second-order model very closely fitted the experimental data, and the correlation factor, R2, was close to unity (Table 2 and Fig. S16†). Additionally, the plots of t/qtvs. t exhibited a linear relationship, further suggesting chemisorption, primarily governed by the complexation reaction of lithium in this context (Fig. 10D).
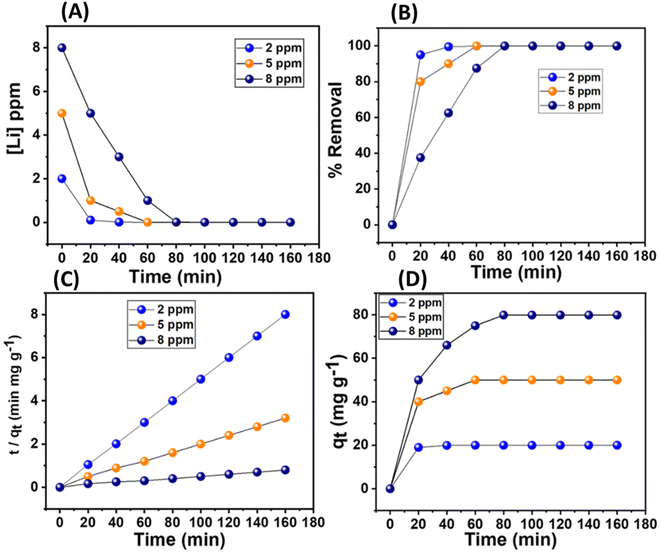 |
| | Fig. 10 (A) The plot of concentration changes of Li+ with time in lithium chloride aqueous solution (2, 5, and 8 ppm). (B) Lithium removal efficiency (%) of the MDNS@DAAH material with time. (C and D) The linear plot for pseudo-second-order kinetics and the sorption plot (qt) for specific Li+ concentrations. | |
Table 2 The adsorption kinetic parameters towards LiCl aq. solutions
| Pseudo-second-order kinetic parameters |
Analyte |
q
e (experimental) (mg g−1) |
k
2 (g mg−1 min−1) |
q
e (calculated) (mg g−1) |
R
2
|
| Li+ (2 ppm) |
19.99 |
1.02 |
20.08 |
0.99 |
| Li+ (5 ppm) |
49.99 |
0.97 |
50.99 |
0.98 |
| Li+ (8 ppm) |
79.90 |
0.95 |
80.12 |
0.99 |
| Pseudo-first-order kinetic parameters |
Analyte |
q
e (experimental) (mg g−1) |
k
1 (min−1) |
q
e (calculated) (mg g−1) |
R
2
|
| Li+ (2 ppm) |
19.99 |
0.061 |
16.39 |
0.95 |
| Li+ (5 ppm) |
49.99 |
0.016 |
62.50 |
0.93 |
| Li+ (8 ppm) |
79.90 |
0.015 |
66.66 |
0.97 |
To investigate the thermodynamic aspects of adsorption at various temperatures, specifically 298, 308, 318, 328, and 333 K, a study was conducted using 5 mg of adsorbent in an 8 ppm lithium-ion solution, with all other variables held constant. It was observed that the adsorption rate of Li+ ions enhanced with temperature, rising from 88 mg g−1 at 298 K to 125 mg g−1 at 333 K. Consequently, increasing the temperature results in increased adsorption of Li+ ions. Further evaluation of the thermodynamic parameters was made using Van't Hoff's equation [log(qe/Ce) = ΔS/2.303R − ΔH/2.303RT] and the Gibbs free energy equation (ΔG = ΔH − TΔS). In these expressions, ΔS is the change in entropy, whereas ΔH is the change in enthalpy, R is the universal gas constant (8.314 J mol−1 K−1), qe/Ce is the adsorption affinity, and ΔG stands for the Gibbs free energy. The values of the enthalpy change, ΔH, and entropy change, ΔS, were evaluated from the slopes and intercept of the plot log(qe/Ce) versus 1/T. The change in enthalpy was found to be 105.30 kJ mol−1, while that obtained for the change in entropy was 0.375 kJ mol−1 (Table 3 and Fig. S17†). Also, the ΔG values at different temperatures were calculated. Positive and significant enthalpy and entropy values suggest a strong interaction between the material and Li+ ions, confirming a chemisorption adsorption process that intensifies with increasing temperature. Additionally, the negative ΔG values advocate for a spontaneous and favorable adsorption process.
Table 3 The thermodynamic parameters towards the adsorption of Li+ ions
| Analyte |
ΔH (kJ mol−1) |
ΔS (kJ K−1 mol−1) |
ΔG (kJ mol−1) |
| Temperature (K) |
298 |
308 |
318 |
328 |
333 |
| Li+ |
105.30 |
0.375 |
−6.5 |
−10.25 |
−14.0 |
−17.7 |
−19.6 |
3.6 Recyclability and regeneration studies
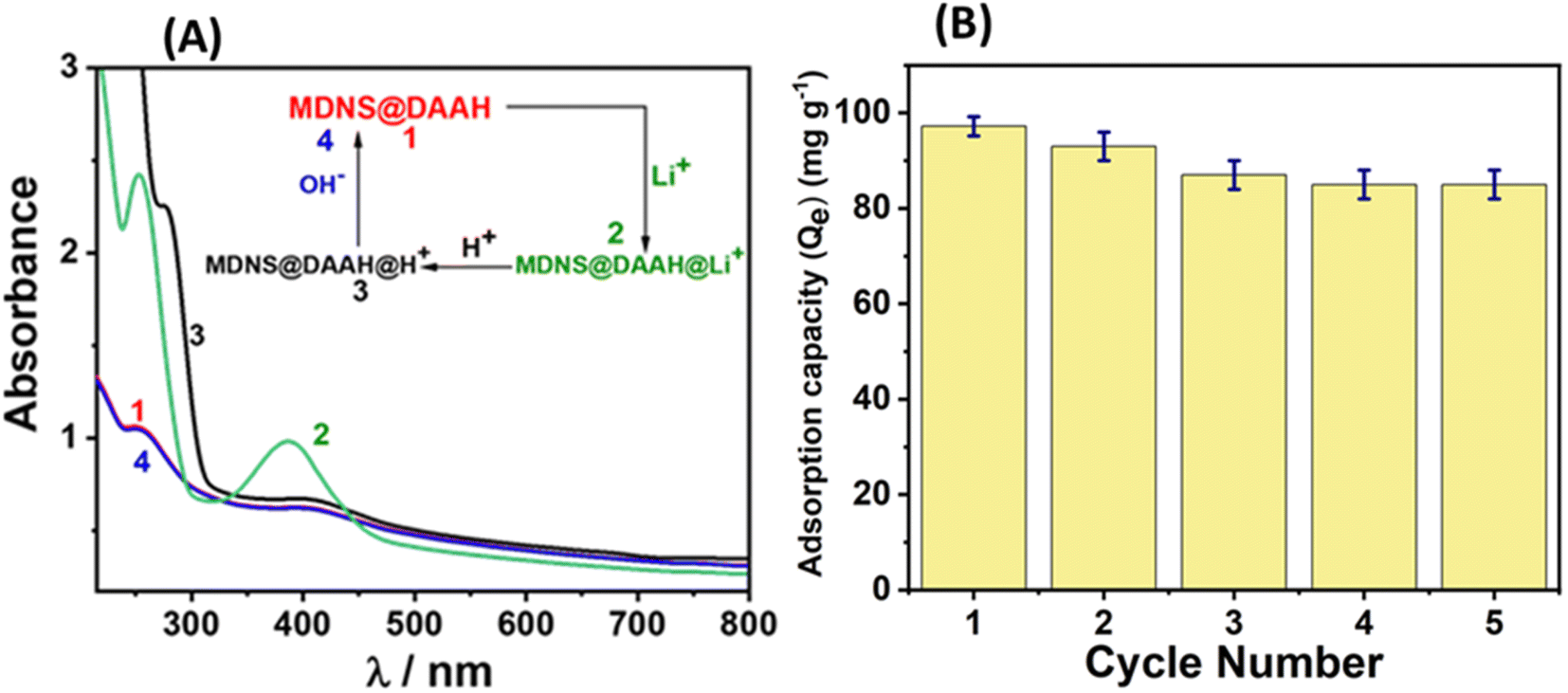
The material's reusability holds significance for economic, environmental, and sustainability considerations. Consequently, the recyclability of the material for lithium adsorption and desorption cycles was investigated. At the outset, the reversibility of lithium adsorption and desorption was evaluated using UV-Vis and zeta potential studies (Fig. 11 and S18†). The UV-Vis spectra of the material displayed spectral alterations following lithium binding, as illustrated in Fig. 11A. Upon treating the lithium-adsorbed material with 0.5 N HCl, pronounced UV-Vis spectral changes occurred, with a significant increase in the absorbance band around 300 nm, likely attributed to the protonation of the NH moiety present in the material. The subsequent treatment of this protonated material with 0.5 N NaOH solution restored the UV-Vis spectrum to its original appearance, indicating that lithium adsorption and desorption can be achieved through straightforward acid-base treatments. Following these reversibility studies, sorption experiments were conducted for 5 cycles, and the adsorption capacity was monitored (Fig. 11B). The results indicated that the material maintained a stable adsorption capacity after the 2nd cycle, with minimal decrease in efficiency. The regenerated material after the 5th cycle underwent comprehensive characterization using various techniques, confirming its structural and functional integrity, resembling that of the original material (Fig. S19†). The assessment of a material's performance in lithium-ion sensing, adsorption, recyclability, and practical applications highlights its superior capabilities in comparison to those documented in the current literature (Table S5†).
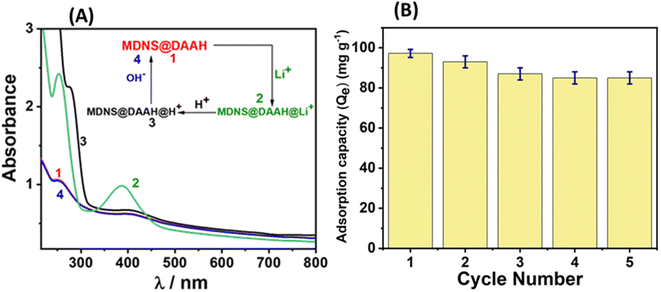 |
| | Fig. 11 (A) The regeneration studies of the material towards Li+ ions. (B) The adsorption capacity of the material towards Li+ up to 5 cycles. | |
3.7
In vivo biosensing applications
Lithium finds clinical applications in managing bipolar disorder, and an excess of lithium poses toxicity risks to living organisms. Consequently, the biosensing of lithium within living organisms becomes crucial. The lithium-biosensing capability of this material was assessed through fluorescence imaging in Artemia salina, serving as the animal model (details are provided in the Experimental section). Initially, without material treatment, fluorescence imaging of Artemia was established following ref. 54 (Fig. 12A and B). Subsequently, upon material treatment, in the brine containing Artemia, fluorescence imaging revealed a faint green staining of the gastrointestinal (GI) tract, indicating material ingestion by Artemia (Fig. 12C and D). Following the treatment of these material-ingested Artemia with a lithium chloride solution, fluorescence imaging showed vivid green staining of the GI tract under similar measurement conditions (Fig. 12E and F). These observations support the fluorescence emission behavior (turn-on) of the material in the presence of lithium. Furthermore, toxicity studies indicated no adverse effects of the material on Artemia during the experiments (Table S6†). These findings underscore the material's biosensing potential for lithium ions in animal models, offering utility in real-time monitoring applications.
 |
| | Fig. 12 (A) Bright-field image of Artemia nauplii before the material feeding. (B) Artemia nauplii under a UV filter (negative control). (C and D) Bright-field image of Artemia nauplii after material feeding and weak green staining of the GI tract of Artemia nauplii under a UV filter after material feeding. (E and F) Bright green staining of the GI tract of Artemia nauplii under a UV filter after treatment with the probe material MDNS@DAAH with lithium chloride solution. | |
3.8 Lithium quantification in seawater as the real sample
The developed material was employed for the quantification of lithium in actual seawater using the fluorescence method, utilizing seawater with a density of 3 °Bé. In this experiment, 5 mg of the material was dispersed in seawater, and fluorescence emission was measured after spiking known concentrations of lithium ions (15, 30, and 45 μM). The fluorescence calibration plot derived from the fluorescence titration experiment was utilized to determine the lithium concentration (Fig. S20 and Table S7†). The results obtained indicate recovery percentages ranging from approximately 102% to 106%, with a relative standard deviation (RSD) value below 3%. The slight excess in recovery percentage may be attributed to the interference of calcium in seawater. These findings underscore the potential application of the material in detecting lithium in real environmental samples.
3.9 Lithium extraction from sea bittern as the real sample
To extract lithium, sea bittern55 was chosen as the real sample, offering several advantages. Sea bittern is the residual brine remaining after the production of common salt through solar evaporation. During the successive evaporation of brine and the separation of various salts to yield common salt, the lithium concentration in the bittern increases.53 Additionally, with a higher bittern density, a significant portion of calcium is eliminated in the form of gypsum (CaSO4), providing an advantage for our system. As we observed interference of calcium and its quantity is much higher than the lithium concentration, we chose the extraction operation from a bittern of higher density. A sea bittern sample was obtained from the Experimental Salt Farm (ESF) of CSIR-CSMCRI, Bhavnagar, India, for conducting the adsorption experiments. The sea bittern with a measurement of 33.13 °Bé (specific gravity: 1.296) underwent analysis for major ionic components, revealing Ca2+ (not found), Mg2+ 7.78%, Na+ 3.36%, K+ 2.13%, and 5.6 ppm of lithium. A 50 mL portion of this bittern was treated with 10 mg of the material and agitated for 6 hours in a shaker. The treated bittern, syringe-filtered for analysis, exhibited a selective lithium extraction of 91.6%, contrasting with the negligible uptake of other cations (Na+ ∼2%, K+ ∼1.5%, and Mg ∼3%). The experiment was scaled up using 4 L of sea bittern and 0.5 g of the material, with lithium being desorbed using an aqueous HCl solution. Treating the aqueous solution with a saturated Na2CO3 solution under hot conditions resulted in the precipitation of Li2CO3, which was characterized by powder XRD. The purity of Li2CO3 was approximately 98%, as confirmed by ICP-MS analysis (detailed in ESI, Fig. S21†).
4. Conclusion
In brief, this study focuses on the development of a silica material functionalized with small molecules to simultaneously detect and extract lithium ions from aqueous systems. This research addresses the recycling challenges associated with small molecule receptors and achieves dual functional activity within a single platform. The functional material comprises a dendritic silica matrix and an anthraquinone-based amide ligand designed for lithium specificity. Through fluorescence methods, the resulting material exhibits selective detection of lithium over other abundant cations in seawater, such as sodium, potassium, magnesium, and calcium. In the presence of lithium ions, the material's strong binding affinity disrupts the hydrogen-bonding-assisted PET process, leading to the fluorescence recovery of the anthraquinone system supported by DFT studies. This results in rare turn-on luminescence with lithium ions with nanomolar sensitivity (limit of detection: 4.9 nM) in the green region. Additionally, the material demonstrates high lithium adsorption capacity with fast kinetics, as observed in batch adsorption studies, and can be easily recycled through simple acid–base treatments. In practical applications, this material proves effective in quantifying lithium in real seawater and extracting lithium from sea bittern (91.6% recovery). It also serves as a biosensing probe for detecting lithium in the living animal model Artemia salina through fluorescence imaging. Notably, the material possesses several advantages over existing literature reports, including superior selectivity, turn-on green emission, high adsorption capacity, dual functionality, and reliable performance with real samples.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
The manuscript was collaboratively written by all authors, and all have approved the final version.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
N. Choudhary, S. Yadav, V. Sonpal, T. R. Patel, B. Ganguly, and A. R. Paital acknowledge the Council of Scientific and Industrial Research (CSIR), Government of India, for their financial assistance. N. C., S. Y., V. S., and T. R. P. also extend their appreciation to AcSIR for supporting their PhD pursuits. Additionally, the ADCIF division of CSIR-CSMCRI is acknowledged for providing analytical facilities. A CSIR-CSMCRI communication no. 106/2024 is acknowledged.
References
- G. Martin, L. Rentsch, M. Höck and M. Bertau, Energy Storage Mater., 2017, 6, 171–179 CrossRef.
- S. H. Mohr, G. M. Mudd and D. Giurco, Minerals, 2012, 2, 65–84 CrossRef.
- C. Grosjean, P. H. Miranda, M. Perrin and P. Poggi, Renewable Sustainable Energy Rev., 2012, 16, 1735–1744 CrossRef.
- J. Mertens, Q.-W. Wang, Y. Kim, D. X. Yu, S. Pham, B. Yang, Y. Zheng, K. E. Diffenderfer, J. Zhang, S. Soltani, T. Eames, S. T. Schafer, L. Boyer, M. C. Marchetto, J. I. Nurnberger, J. R. Calabrese, K. J. Oedegaard, M. J. McCarthy, P. P. Zandi, M. Alda, C. M. Nievergelt, S. Mi, K. J. Brennand, J. R. Kelsoe, F. H. Gage and J. Yao, Nature, 2015, 527, 95–99 CrossRef CAS PubMed.
- D. H. P. Kang, M. Chen and O. A. Ogunseitan, Environ. Sci. Technol., 2013, 47, 5495–5503 CrossRef CAS.
- H. Gohil, S. Chatterjee, S. Yadav, E. Suresh and A. R. Paital, Inorg. Chem., 2019, 58, 7209–7219 CrossRef CAS PubMed.
- M. Kamenica, R. Kothur, A. Willows, B. Patel and P. Cragg, Sensors, 2017, 17, 2430 CrossRef.
- E. Villemin and O. Raccurt, Coord. Chem. Rev., 2021, 435, 213801 CrossRef CAS.
- J. Kang, E. Li, L. Cui, Q. Shao, C. Yin and F. Cheng, Sens. Actuators, B, 2021, 327, 128941 CrossRef CAS.
- H. Kim and B. Koo, RSC Adv., 2022, 12, 31976–31984 RSC.
- R. V. Hangarge, D. D. La, M. Boguslavsky, L. A. Jones, Y. S. Kim and S. V. Bhosale, ChemistrySelect, 2017, 2, 11487–11491 CrossRef CAS.
- D. Citterio, J. Takeda, M. Kosugi, H. Hisamoto, S. Sasaki, H. Komatsu and K. Suzuki, Anal. Chem., 2007, 79, 1237–1242 CrossRef CAS PubMed.
- H. Wang, L. O. Jones, T. Zhao, I. Hwang, V. M. Lynch, N. M. Khashab, G. C. Schatz, Z. A. Page and J. L. Sessler, Chem. Sci., 2023, 14, 4120–4125 RSC.
- S. Rochat, Z. Grote and K. Severin, Org. Biomol. Chem., 2009, 7, 1147–1153 RSC.
- X. Cheng, F. Xian, Z. Hu, C. Wang, X. Du, H. Zhang, S. Chen, S. Dong and G. Cui, Angew. Chem., Int. Ed., 2019, 58, 5936–5940 CrossRef CAS.
- D. Jiang, R. Xu, L. Bai, W. Wu, D. Luo, Z. Li, T. Asahi, Y. Mai, Z. Liu, Y. Yamauchi and X. Xu, Coord. Chem. Rev., 2024, 516, 215923 CrossRef CAS.
- F. Javanbakht, H. Najafi, K. Jalili and M. Salami-Kalajahi, J. Mater. Chem. A, 2023, 11, 26371 RSC.
- S.-Y. Huang, H. Wang, H. Celio, N. M. Khashab, Z. A. Page and J. L. Sessler, J. Mater. Chem. A, 2022, 10, 14788–14794 RSC.
- J. Zhang, M. Wenzel, L. Yang, R. C. Luckay and J. J. Weigand, Front. Chem. Eng., 2023, 5, 1271045 CrossRef.
- C. Xu, Q. Tran, L. Wojtas and W. Liu, J. Mater. Chem. A, 2023, 11, 12214–12222 RSC.
- Y. J. Lim, K. Goh, A. Goto, Y. Zhao and R. Wang, J. Mater. Chem. A, 2023, 11, 22551–22589 RSC.
- J. W. An, D. J. kim, K. T. Tran, M. J. Kim, T. Lim and T. Tran, Hydrometallurgy, 2012, 117, 64–70 CrossRef.
- Z. Wu, F. Weigend, D. Fenske, T. Naumann, J. M. Gottfried and S. Dehnen, J. Am. Chem. Soc., 2023, 145, 3802–3811 CrossRef CAS PubMed.
- L. Bai, R. Xu, W. Wu, C. Ma, S. Li, H. Gao, D. Luo, B. Liu, S. Melhi, Y. Zhao, Z. Liu, Y. Yamauchi and X. Xu, J. Mater. Chem. A, 2024, 12, 10676–10685 RSC.
- P. Wang, J. Dai, Y. Ma, L. Chen and J. Pan, Chem. Eng. J., 2020, 380, 122495 CrossRef CAS.
- X. Bai, J. Dai, Y. Ma, W. Bian and J. Pan, Chem. Eng. J., 2020, 380, 122386 CrossRef CAS.
- H.-J. Hong, I.-S. Park, J. Ryu, T. Ryu, B.-G. Kim and K.-S. Chung, Chem. Eng. J., 2015, 271, 71–78 CrossRef CAS.
- D. Weng, H. Duan, Y. Hou, J. Huo, L. Chen, F. Zhang and J. Wang, Prog. Nat. Sci.: Mater. Int., 2020, 30, 139–152 CrossRef CAS.
- U. Kamran, Y.-J. Heo, J. W. Lee and S.-J. Park, J. Alloys Compd., 2019, 794, 425–434 CrossRef CAS.
- B. Swain, J. Chem. Technol. Biotechnol., 2016, 91, 2549–2562 CrossRef CAS.
- J. Zhang, N. Tanjedrew, M. Wenzel, P. Royla, H. Du, S. Kiatisevi, L. F. Lindoy and J. J. Weigand, Angew. Chem., Int. Ed., 2023, 62, e202216011 CrossRef CAS PubMed.
- J. Zhang, M. Wenzel, J. Steup, G. Schaper, F. Hennersdorf, H. Du, S. Zheng, L. F. Lindoy and J. J. Weigand, Chem.–Eur. J., 2022, 28, e202103640 CrossRef CAS.
- L. Zhang, L. Li, D. Shi, X. Peng, F. Song, F. Nie and W. Han, Hydrometallurgy, 2018, 175, 35–42 CrossRef CAS.
- T. Kanagasundaram, O. Murphy, M. N. Haji and J. J. Wilson, Coord. Chem. Rev., 2024, 509, 215727 CrossRef CAS.
- M. Lee, W. Jung, H. J. Kwon and G. Lim, J. Mater. Chem. A, 2022, 10, 4621–4633 RSC.
- C. Liu, Y. Li, D. Lin, P.-C. Hsu, B. Liu, G. Yan, T. Wu, Y. Cui and S. Chu, Joule, 2020, 4, 1459–1469 CrossRef CAS.
- Y. Xiong, J. Zhou, P. Lu, J. Yin, Y. Wang and Z. Fan, Matter, 2022, 5, 1760–1791 CrossRef CAS.
- J. Hou, H. Zhang, A. W. Thornton, A. J. Hill, H. Wang and K. Konstas, Adv. Funct. Mater., 2021, 31, 2105991 CrossRef CAS.
- H. Peng and Q. Zhao, Adv. Funct. Mater., 2021, 31, 2009430 CrossRef CAS.
- Y. Guo, Y. Ying, Y. Mao, X. Peng and B. Chen, Angew. Chem., 2016, 128, 15344–15348 CrossRef.
- S. J. Warnock, R. Sujanani, E. S. Zofchak, S. Zhao, T. J. Dilenschneider, K. G. Hanson, S. Mukherjee, V. Ganesan, B. D. Freeman, M. M. Abu-Omar and C. M. Bates, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022197118 CrossRef CAS.
- H. Jo, T.-H. Le, H. Lee, J. Lee, M. Citter, S. Lee, M. Chang and H. Yoon, Chem. Eng. J., 2023, 452, 139274 CrossRef CAS.
- A. Razmjou, M. Asadnia, E. Hosseini, A. Habibnejad Korayem and V. Chen, Nat. Commun., 2019, 10, 5793 CrossRef CAS PubMed.
- D. Kou, W. Ma and S. Zhang, Adv. Funct. Mater., 2021, 31, 2007032 CrossRef CAS.
- M. Xu, T. Wang, P. Gao, L. Zhao, L. Zhou and D. Hua, J. Mater. Chem. A, 2019, 7, 11214–11222 RSC.
- S. Chatterjee and A. R. Paital, Adv. Funct. Mater., 2018, 28, 1704726 CrossRef.
- K. Wu, X. Kong, K. Xiao, Y. Wei, C. Zhu, R. Zhou, M. Si, J. Wang, Y. Zhang and L. Wen, Adv. Funct. Mater., 2019, 29, 1807953 CrossRef.
- S. Yadav, N. Choudhary and A. R. Paital, Carbon, 2023, 205, 527–539 CrossRef CAS.
- Q. Meng, X. Zhang, C. He, G. He, P. Zhou and C. Duan, Adv. Funct. Mater., 2010, 20, 1903–1909 CrossRef CAS.
- S. Yadav, N. Choudhary, V. Sonpal and A. R. Paital, Small, 2023, 20, 2307491 CrossRef PubMed.
- S. Yadav, N. Choudhary, M. Ranjan Dash and A. Ranjan Paital, Chem. Eng. J., 2022, 450, 138042 CrossRef CAS.
- V. Polshettiwar, Acc. Chem. Res., 2022, 55, 1395–1410 CrossRef CAS PubMed.
- N. Choudhary, D. Rajpurohit, A. Saha, S. Yadav, S. Tothadi, B. Ganguly and A. Ranjan Paital, Chem. Eng. J., 2023, 470, 144408 CrossRef CAS.
- M. Raju, R. R. Nair, I. H. Raval, S. Haldar and P. B. Chatterjee, Analyst, 2015, 140, 7799–7809 RSC.
- S. F. Estefan, Hydrometallurgy, 1983, 10, 39–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, additional experimental procedures, computational methodology, Fig. (S1–S17) and Tables (S1–S7). See DOI: https://doi.org/10.1039/d4ta05025h |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ac
*ac