
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the non-radiative losses encountered in intramolecular charge transfer compounds with benzodithiophene-4,8-dione acceptors†
Stephanie
Montanaro
 a,
Alexander J.
Gillett
b,
Patrick
Kimber
a,
Dong
Xing
a,
Sascha
Feldmann
c,
Emrys W.
Evans
b,
Stefan
Warrington
ad,
Felix
Plasser
a,
Richard H.
Friend
*b and
Iain A.
Wright
*d
a,
Alexander J.
Gillett
b,
Patrick
Kimber
a,
Dong
Xing
a,
Sascha
Feldmann
c,
Emrys W.
Evans
b,
Stefan
Warrington
ad,
Felix
Plasser
a,
Richard H.
Friend
*b and
Iain A.
Wright
*d
aDepartment of Chemistry, Loughborough University, Epinal Way, Loughborough, Leicestershire LE11 3TU, UK
bOptoelectronics Group, Cavendish Laboratory, University of Cambridge, Cambridge CB3 0HE, UK. E-mail: rhf10@cam.ac.uk
cInstitute of Chemical Engineering and Sciences, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland
dSchool of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: iain.wright@ed.ac.uk
First published on 29th July 2024
Abstract
A new yellow-emitting quadrupolar donor–π–acceptor–π–donor (D–π–A–π–D) molecule compound has been synthesised featuring benzo-[1,2-c:4,5-c′]dithiophene-4,8-dione as the acceptor. This molecule was prepared for the purpose of elucidating the origins of the very low photoluminescence quantum yield encountered in its thermally activated delayed fluorescent (TADF) red-emitting isomer which used benzo-[1,2-b:4,5-b′]dithiophene-4,8-dione as the acceptor. The molecule was designed to circumvent the energy gap law, by having a wider HOMO–LUMO gap, while retaining a comparable singlet–triplet gap but ultimately demonstrates even weaker photoluminescence than the red isomer. It shows extremely fast intersystem crossing followed by rapid non-radiative decay and no observable TADF. The electronic structure of this new molecule has been studied using cyclic voltammatery alongside steady-state and transient optical spectroscopy, with observations underpinned by computational insights. To identify whether the observations made from the experimental results might be general properties of benzodithiophene-4,8-dione containing emitters, a computational study is extended to the four isomers of benzodithiophene-4,8-dione in comparison with 9,10-anthraquinone. The results suggest that the singlet and triplet manifolds of these systems are strongly coupled via spin–orbit interactions, and explain how the relative electron-accepting strength of these quinones arises from an interplay between the resonance gains or losses of the central benzene and fused thiophene rings upon photoexcitation. This provides valuable insights into the design principles required for efficient organic light-emitting materials.
Introduction
Organic molecules with a donor–acceptor (D–A) structure often display intramolecular charge transfer (ICT) bands in their absorbance and emission spectra. By judicious choice of D and A moieties, and control of the configuration of the bonds linking them together, the location of these bands can be tuned across the visible spectrum and into the near-IR. They have been subject to intense study as many of these compounds display thermally activated delayed fluorescence (TADF) comprising the third generation of emitters for organic light emitting devices (OLEDs).1–4TADF is a triplet mediated emission mechanism, the efficiency of which is governed by the rate of reverse intersystem crossing (krISC). Reverse intersystem crossing (rISC) is a spin-forbidden process which relies upon spin–orbit coupling (SOC) and the mixing of singlet and triplet states.5,6 This is often achieved by either the use of heavy atoms, notably large halogen atoms, or by minimising the magnitude of the energy gap between the lowest excited singlet and triplet states (ΔEST). Many ICT-based TADF emitters have a dipolar D–A or quadrupolar D–A–D structure, often with “π-spacers” positioned between the D and A components. These spacers serve to modulate the extent of overlap between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) providing further control over the magnitude of ΔEST.3,7,8
Compounds 1–3 (Fig. 1) are examples of D–π–A–π–D structures featuring 3,6-di(tert-butyl)carbazole donors, 1,4-phenylene π-spacers, and quinone acceptors. We previously reported molecule 29 as a thiophene based analogue of the red TADF emitter 3.102 was designed to emit at longer wavelengths than 3 due to the deeper LUMO energy of its acceptor component benzo-[1,2-b:4,5-b′]dithiophene-4,8-dione 4 compared to anthraquinone (AQ) as used in 3.
 | ||
| Fig. 1 Chemical structures of the new ICT compound 1 and the previously reported red emitters 2 and 3 alongside the structures of 9,10-anthraquinone AQ and the four isomers of benzodithiophene-4,8-dione 4–7. | ||
Using 4 as the acceptor did indeed lead to a desirable >100 nm red-shift of the ICT emission for 2 (λmax = 664 nm) compared to 3 (λmax = 558 nm) however the photoluminescence quantum yield (PLQY, Φ) for 2 in both solution and thin film was extremely low. The size of any delayed component to the emission was also very small both in solution and thin films. A number of possible explanations can be considered for the large drop in Φ. The wider ΔEST for 2 compared to 3 may provide some explanation for its much less efficient rISC, alongside simple invocation of the energy gap law, which posits that non-radiative decay accelerates as the energy gap between the electronic states involved decreases, in this case S0 → S1.11 Subsequent computational investigation indicated that symmetry breaking in the excited state of 3 may be a factor in rendering its S1 emissive12 however, these factors notwithstanding, the magnitude of the reduction in Φ moving from AQ to a benzodithiophene seemed quite extreme. In accordance with the El-Sayed selection rule13,14AQ itself is well known to undergo very rapid ISC and also rapid non-radiative decay and therefore demonstrates negligible prompt fluorescence at ambient temperatures and only weak delayed fluorescence and phosphorescence at low temperatures.15–20 However despite this fact, 3 still functions as a relatively efficient red emitter while 2 does not. Furthermore, the transient absorption studies of 2 revealed that the rates of both ISC (kISC) and the non-radiative decay of triplet states (knr) in thin films were very fast at 1.42 × 108 s−1 and 7.18 × 104 s−1 respectively. These observations raised questions about what specific aspects of the benzodithiophene-4,8-dione are responsible for its tendency to shut down emission with such efficacy.
We now present the synthesis of 1 and aspects of its electrochemistry, time-resolved spectroscopy and the results of computational experiments. 1 is an isomer of 2 that uses benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (5) as the acceptor. 5 is a weaker acceptor than 421 therefore blue-shifted absorbance and emission in comparison with 2 and 3 can be anticipated, and so circumventing losses attributed to the energy gap law. By using the same donor and π-spacer moieties any observed changes in rate constants, Φ or other photophysical characteristics can be ascribed to as great an extent as possible to the chemical makeup of the acceptor moiety 5. Preliminary TDDFT (ESI†) also predicted a ΔEST for 1 which is comparable to that of 2 therefore a similarly low proportion of TADF might be expected. While additional differences in TADF behaviour may be expected to arise from the precise locations of the donor moieties on the acceptors,22–24 for example whether they are bound across (as in 2 and 3) or adjacent to the quinone core (as in 1), in the wider context of organic electronic materials 5 is an interesting subject for study as this acceptor is ubiquitous in high-performance, low band gap polymers used in bulk heterojunction (BHJ) organic solar cells, notably including PM625 and the PBDB-T family of polymers26–28 among others.29,30 In all of these polymers the quinone is configured analogously to 1 with respect to the polymer backbone. 6 has also been employed as the acceptor in polymers31–37 in a similar manner to 5, while 7 has received greater attention in medicinal chemistry.38–41 We therefore extend the computational aspects of this work to the four isomers of benzodithiophene-[4,8]-dione 4–7 presented in comparison with AQ with a view to learning more about the properties of these heterocycles, and inform how they might best be used in the design of future materials. Calculations have been performed on both the ground and monoanion states of the quinones to better capture the nature of their behaviour when used as acceptors in ICT systems.
Results and discussion
Synthetic procedures
The synthesis of 1 is analogous to that reported for 2 and is presented in Scheme 1 (synthetic details can be found in the ESI†). 1,3-Dibromo-4H,8H-benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (8)42,43 was subjected to two-fold Suzuki–Miyaura cross-coupling with 4-(3,6-di-tert-butylcarbazol-9-yl)phenyl boronic acid (9)44,45 employing Pd(PPh3)4 as the catalyst and K2CO3 as the base in a mixture of tetrahydrofuran (THF) and water at a temperature of 55 °C. After workup, purification was achieved using column chromatography to produce bright yellow compound 1 in 97% yield. | ||
| Scheme 1 Synthesis of compound 1. | ||
Electrochemistry
Cyclic voltammetry of 1 was performed to obtain oxidation and reduction potentials (Fig. 2 and Table 1, the electrochemical properties of 2 are provided to aid comparison). The oxidation potentials of 1 and 2 are very close giving rise to near identical HOMO levels. This is to be expected due to the common D–π moieties. The first reduction potential of 2 is significantly less negative than that of 1 indicating a higher LUMO energy and confirming the weaker accepting power of quinone 5 compared to 4. In contrast with the cyclic voltammetry of 2 there are two reduction processes observed for 1, a quasi-reversible reduction at Ered1 = −1.79 V and an irreversible reduction at Ered2 = −2.17 V indicating sequential formation of a radical anion then dianion over the quinone. The small peaks present at −1.24 V and −0.81 V are only observed upon reversing the polarity after any reduction of 1 has occurred so are assigned to deposits of degradation products formed on the electrode surface. | ||
| Fig. 2 Cyclic voltammetry for compound 1 in 1,2-dichlorobenzene solution (ca. 10−5 M), in the presence of 10−1 M (n-Bu4N)(PF6) as a supporting electrolyte measured at a scan rate of 100 mV s−1. | ||
| E ox (V) | E red1 (V) | E red2 (V) | HOMOa (eV) | LUMOa (eV) | E g (eV) | |
|---|---|---|---|---|---|---|
| a HOMO and LUMO levels estimated from the onset of the first oxidation and reduction waves respectively and are references to the HOMO of ferrocene at −5.10 eV.46 b Electrochemical HOMO–LUMO gap. c Taken from ref. 9. qr, quasi-reversible; ir, irreversible. | ||||||
| 1 | +0.67 | −1.79qr | −2.17ir | −5.77 | −3.31 | 2.46 |
| 2 | +0.74 | −1.15 | — | −5.69 | −4.10 | 1.59 |
The unsubstituted quinones 4 and 5 were previously shown to undergo two sequential reductions in a similar fashion to 1 albeit reversibly.9 We suggest that the absence of a second reduction wave in 2 arises from the presence of cross-conjugation at the carbonyl groups. In neutral 2 no linear conjugated pathway exists between the terminal D–π fragments, however upon reduction cross-conjugation is destroyed and the resulting radical anion is able to delocalise across the π-system.47,48 This has served to increase the barrier towards further reduction. In 1 the quinone moiety is entirely pendant to the π-conjugated backbone therefore radical anion (and dianion) states of 1 remain localised over the quinone and so it behaves very similarly to the unfunctionalised acceptor 5.
Steady-state photophysics
In toluene solution (Fig. 3 and Tables 2), 1 exhibits a low energy absorbance at 403 nm which is assigned to ICT between the carbazole donors and the quinone acceptor. The onset of this band corresponds to an optical HOMO–LUMO gap of 2.58 eV which agrees well with the results from voltammetry. Excitation of the ICT band gives rise to a yellow emission at 519 nm in the photoluminescence (PL) spectrum. This emission is blue-shifted by 145 nm compared to 2 (also shown in Fig. 3) and 39 nm with respect to 3 which is indicative of the weaker accepting strength of 5. The total PLQY of 1 is Φ = 0.023 which notably lower than both 2 and 3. As compound 1 displays a significantly blue-shifted emission compared to 2 the energy gap law can be excluded as a major contributing factor to this low PLQY. Minimal sensitivity of the PLQY of the solution to oxygen was observed with a fluorescence PLQY of ΦF = 0.019 indicating that any delayed component of the ICT emission is vanishingly small in solution.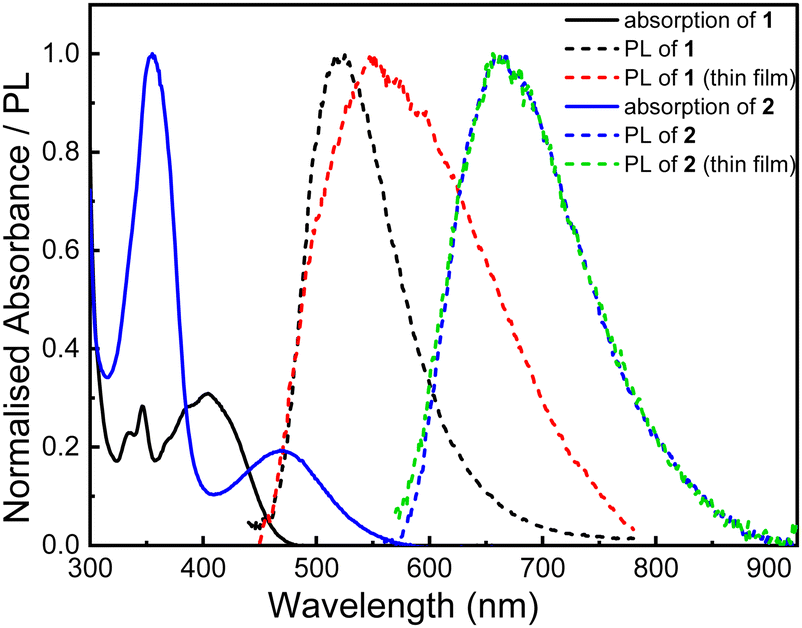 | ||
| Fig. 3 Steady-state solution absorption and emission and thin film (2 wt% in poly(styrene) PL spectra of 1 (excitation wavelength, λex = 400 nm) and 2 (λex = 520 nm. Reproduced from ref. 9). | ||
| λ abs (nm) | λ em (nm) | E g,opt (eV) | Φ (soln) | Φ F (soln) | Φ (film) | Φ F (film) | |
|---|---|---|---|---|---|---|---|
| a Optical energy gap calculated from the onset of the lowest energy absorbance. b Excitation wavelength λex = 405 nm. c Excitation wavelength λex = 520 nm. d Taken from ref. 9. | |||||||
| 1 | 346, 403 | 519 | 2.58 | 0.023b | 0.019b | 0.051b | 0.027b |
| 2 | 355, 468 | 664 | 2.14 | 0.095c | 0.084c | 0.085c | 0.064c |
The PL was also measured of a drop cast thin film (2 wt% of 1 in poly(styrene) (PS)). The profile of the emission band is broad and poorly resolved in comparison to the solution state measurement. This suggests that more than one triplet-mediated emission mechanism and/or more than one emitter is at play, with emission from aggregates of 1 a possible explanation. A larger proportion of the emission of the film is delayed with the PLQY increasing from 2.7% to 5.1% in the absence of oxygen indicating that some triplet mediated processes are at play.
Time-resolved photophysics
The evolution of the broad spectral shape of the ICT emission became apparent in ns-broadband transient photoluminescence (trPL) of the film (Fig. 4) which revealed that the immediate emission matches well to that of the solution spectrum before the band red-shifts rapidly over the first few ns. No measurable delayed fluorescence was observed. The timescale of the emission substantiates that it likely arises from radiative singlet decay of aggregates of 1 in the film.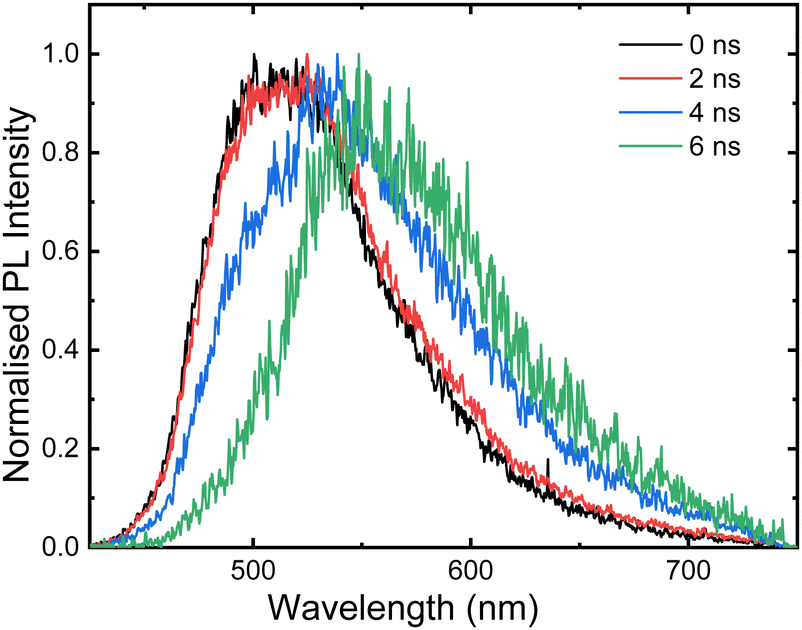 | ||
| Fig. 4 Transient photoluminescence spectroscopy of a thin film of 1 (2 wt% in poly(styrene), λex = 400 nm). | ||
Transient absorption (TA) spectroscopy was performed on the PS thin film. As shown in Fig. 5(a), at short-timescales (0.2–0.3 ps) the TA shows two photo-induced absorption (PIA) bands that can be assigned to the singlet exciton: the first with a maximum at 570 nm and a shoulder at 670 nm, and the second in the near-infrared (NIR) at 950 nm. At wavelengths >1000 nm a third band evolves over a few hundred ps with concomitant quenching of the singlet PIAs (clearly observed in the normalized TA spectrum Fig. 5(b)). This is attributed to rapid triplet exciton formation and implies that ISC is occurring on a similar timescale to singlet emission. This contrasts with 2 for which a weak delayed component was clearly observed for all the prominent features of the TA spectra, indicating that limited rISC was occurring. The band at 570 nm fits well to a biexponential decay (Fig. 5(c) and (d)) with lifetimes of τ1 = 8.6 ps and τp = 131 ps and decays more rapidly than the NIR band. This is an observation which was also made for 2. Existing TA studies on D–A dyads of AQ in solution also have an ultrafast τ1 assigned to triplet–triplet absorbance of the CT band,49,50 however in the doped film measured here τ1 is clearly associated with the singlet CT state which gives rise to the weak prompt emission. This more likely arises from structural relaxation of the emitter in the solid-state host.51 The measured τp is comparable to those obtained for other CT-based AQ systems.49,50,52,53
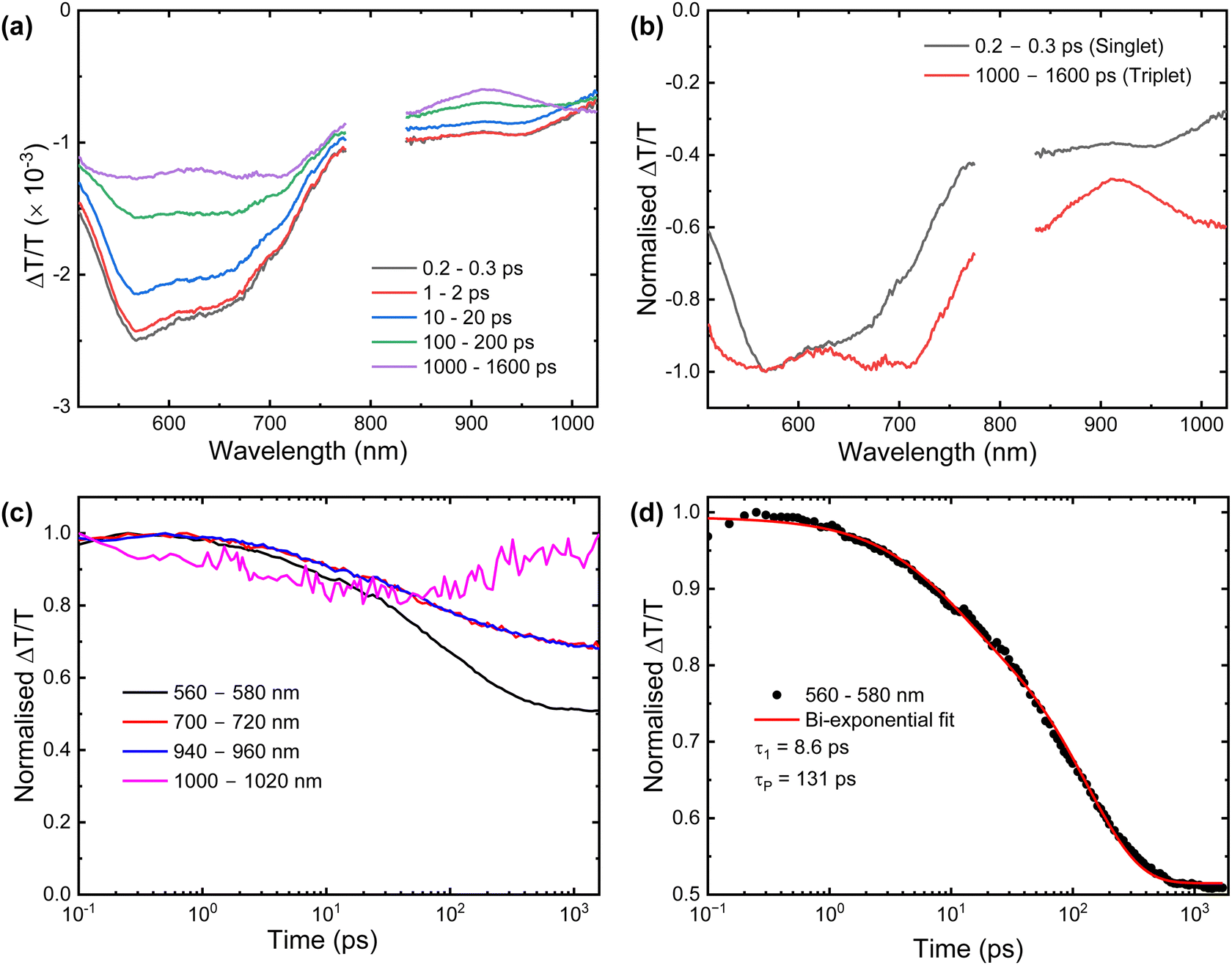 | ||
| Fig. 5 (a) Short timescale evolution of the TA spectra of a thin film of 1. (b) Normalised spectra of the singlet and triplet states. (c) Normalised kinetics taken from the wavelength regions of 560–580 nm, 700–720 nm, 940–960 nm, and 1000–1020 nm. (d) The normalised kinetics taken from the wavelength region of 560–580 nm fitted to a biexponential decay function, which is represented by the solid line. Emission data were collected upon excitation with 400 nm pulses ca. 100 fs long, at 1 kHz repetition rate). | ||
Considering the longer timescale TA (1 ns–300 μs) in Fig. 6(a), there is no further evolution in the TA spectra, and all PIA features decay together over microsecond timescales with a decay lifetime (τd) of 13.4 μs (Fig. 6(b)). This indicates that once ISC is completed on sub-ns timescales, there is no further evolution of the excited state population. The PLQY of the film does increase slightly in the absence of oxygen, suggesting a small amount of very weak delayed fluorescence may be present, but the largely mono-exponential decay indicates that non-radiative triplet deactivation dominates. We note that the value obtained for τd is comparable to that of 2 which may suggest that similar non-radiative triplet decay pathways are active for both molecules.
 | ||
| Fig. 6 (a) Long time TA spectra of a thin film of 1 doped at 2 wt% in poly(styrene), time taken at different points. The film was excited at λex = 355 nm, with a fluence of 63 μJ cm−2. (b) The normalised kinetics taken from the wavelength regions of 550–600 nm, 860–880 nm, and 980–1000 nm. All three kinetics fitted a single monoexponential decay function, which is represented by the solid line. | ||
The important rate constants are summarised in Table 3. For 1, the absence of rISC and the timescale of the decay processes involved is consistent with the hypothesis that ISC has already happened by 1 ns, following this the rate of non-radiative decay outcompetes that of any triplet mediated emission. The rapid ISC in 1 is therefore acting as a major limiting factor on the PLQY of the compound.
| τ p (ns) | τ d (μs) | k p (s−1) | k d (s−1) |
k
Sr
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (s−1)
(s−1) |
k ISC (s−1) | |
|---|---|---|---|---|---|---|
| a Equations for the radiative singlet decay rate (kSr) and the rate of intersystem crossing (kISC) are in the ESI. b Taken from ref. 9. | ||||||
| 1 | 0.131 | 13.4 | 7.63 × 109 | 7.46 × 104 | 2.06 × 108 | 7.43 × 109 |
| 2 | 6.6 | 13.6 | 1.52 × 108 | 7.35 × 104 | 9.70 × 106 | 1.42 × 108 |
Computational analysis
An analysis of the excitation energies of push–pull systems 1–3 is illustrated in Fig. 7 (S0–S4 and T1–T5 for every molecule). Generally, we find that 2 has lowest singlet and triplet excitation energies with S1 at 2.88 eV and T1 at 2.11 eV, while 1 presents the highest S1 (3.442 eV) and 3 has the highest T1 (2.900 eV). This is in good agreement with ref. 12 where the excitation energies of 2 are lower than 3. Also, molecules 1–3 show some similarities to the corresponding BDT cores 5, 4 and AQ, where 4 has lower excitation energies than the others.
 | ||
| Fig. 7 Excitation energies of the singlet and triplet excited states of molecules 1–3. SOC values above 30 cm−1 are shown as solid lines, above 10 cm−1 as dotted lines. | ||
SOC values are shown in Fig. 7 as solid and dotted lines connecting the individual states. All SOC values between excited singlet and triplet states are found to be below 30 cm−1 (shown as dotted lines). Nonetheless, all three molecules do show SOC from S2 to a triplet state (T5 for 1 and 2, T3 for 3), indicating the occurrence of ISC. In addition, all the molecules present one or two notable SOC channels from the triplet manifold to S0, at least one of which strong (>30 cm−1).
Concerning the electronic structure of the benzodithiophene-4,8-dione acceptors themselves. An analysis of the excitation energies of the lowest lying singlet and triplet states of AQ and molecules 4–7 is shown in Fig. 8. In this figure, nπ* states are shown in red, ππ* states in black. Fig. 8 highlights some similarities but also that the energies and state ordering are quite divergent between the different molecules which explains their different photophysical properties. One finds that the S1 is always of nπ* character whereas the character for T1 varies, being ππ* for 4 and 7 and nπ* for 5 and 6. The highest S1 (3.45 eV) as well as T1 (2.96 eV) are found for 5, closely followed by AQ with its S1 at 3.37 eV and T1 at 2.90 eV. Conversely, the lowest S1 (2.99 eV) and T1 (2.47 eV) are found for 7. SOC in Fig. 8 is represented via lines connecting the singlet and triplet states (solid lines for SOC >30 cm−1, dotted lines for SOC >10 cm−1). The SOC nicely illustrates El-Sayed's rules13,14 highlighting that SOC is only significant when one of the states is of nπ* character and the other one either of ππ* character or the closed-shell ground state. Furthermore, one finds a reduction in the number of non-vanishing terms for the centrosymmetric systems (AQ, 4, 5) owing to symmetry selection rules noting that only states of the same parity can be coupled by SOC. Nonetheless, all molecules show reasonable SOC from S1 into the triplet manifold to a state of similar energy, highlighting that quick ISC to the triplets can be expected. Furthermore, all molecules possess at least one triplet that is strongly coupled to S0 providing a non-radiative deactivation channel. This deactivation occurs from T1 in the case of AQ, 5, and 6 whereas it would involve higher excited states in the case of 4 and 7, indicating that this channel is more accessible in the former class of molecules.
 | ||
| Fig. 8 Excitation energies of the singlet and triplet nπ* (red) and ππ* (black) excited states of the cores of the molecules studied. SOC values above 30 cm−1 are shown as solid lines, above 10 cm−1 as dotted lines. | ||
To provide more information on the differences between the molecules, we present an analysis of the resonance structures involved in the neutral and reduced states (Fig. 9) focusing on the involvement of aromatic sextets following ref. 61. As mentioned in the introduction, the formally reduced state has been selected as a model for the acceptors in the ICT compounds after photoexcitation.
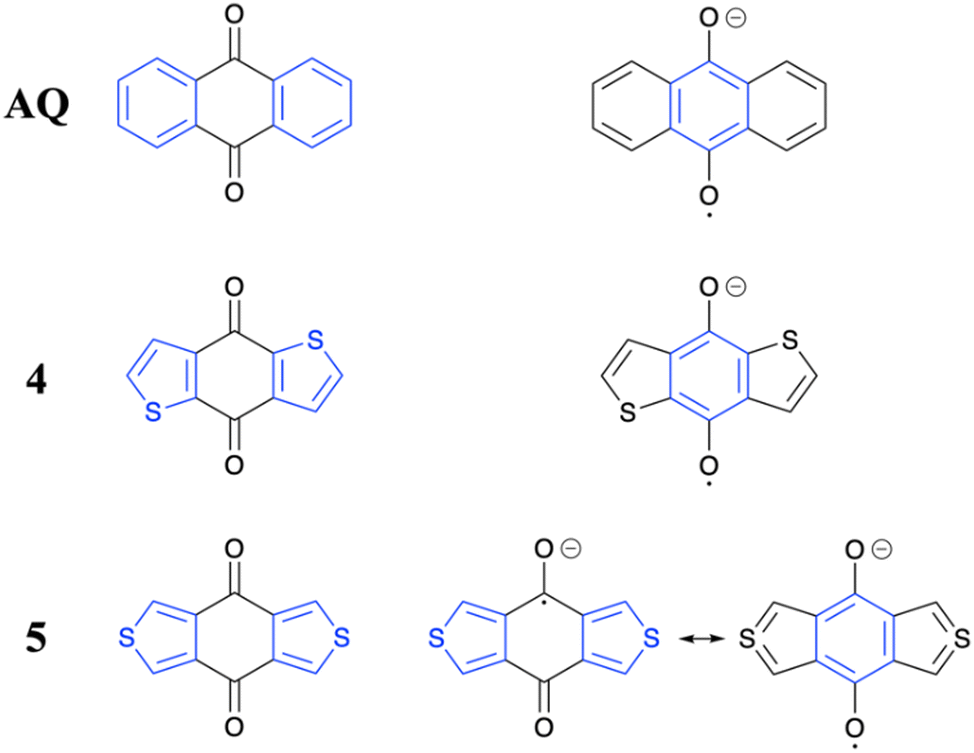 | ||
| Fig. 9 Analysis of available resonance structures for selected molecules in the neutral ground state (left) and reduced state (right). Aromatic sextets are shown in blue. | ||
Starting with AQ, we find that it has a neutral ground state featuring two aromatic sextets. Moving to the reduced state, we place a radical on one oxygen atom and the negative charge on the other. At the centre, this leaves a π-system resembling anthracene where, crucially, one of the sextets is destroyed. This loss of aromaticity explains the relatively weak electron accepting ability of AQ. Analogous resonance structures can be drawn for 4 and 7. However, the crucial difference in this case is that the relatively weak aromaticity of two thiophene rings is traded against one strongly aromatic benzenoid sextet providing a more favourable resonance structure, nicely explaining the lower LUMO energies of 4 as well as 7. An entirely different situation is present in the case of 5. In this case it is not possible to draw a resonance structure with an aromatic sextet at the central ring unless one invokes tetravalent sulfur. We show both possibilities on the lower right in Fig. 9. To the left we show the case where the radical anion is localized on one CO group. To the right we show the case where tetravalent sulfur is invoked (producing a resonance structure resembling the SO2 molecule). Both options are clearly unfavourable when compared to 4 above. These more unfavourable resonance structures explain the higher LUMO and, therefore, higher S1 and T1 energies of this molecule. Finally, quinone 6 occupies a middle ground with one side resembling 4/7 and the other resembling 7.
Fig. 10 shows an analysis of aromaticity properties for 4–6 using the VIST (visualization of chemical shielding tensors) plot technique.62 as implemented in TheoDORE 3.1.63 VIST plots were based on NICS (nucleus independent chemical shift) values64 computed at the PBE0/def2-SVP level65 using Gaussian 09.66 Within the VIST plots, shielded (aromatic) contributions are shown in blue whereas deshielded contributions are shown in red. In their neutral ground states, the VIST plots of all three molecules look very similar with pronounced aromaticity (ca. −22 ppm) for the thiophene rings and a slightly deshielded central ring. Note, however, that the deshielding of the central ring should be associated with residual contributions from the outer rings rather than actual anti-aromaticity within the central ring. Reducing 4 to 4− leaves the outer rings more or less unaltered but produces a notable shielded (aromatic) contribution on the central ring as explained in Fig. 9.
 | ||
| Fig. 10 Analysis of neutral and anionic state for molecules 4, 5 and 6 where the VIST plots for neutral state (left) and anionic state (middle) with natural difference orbitals (NDOs) for electron attachment (right). EEA for electron affinity. See ESI,† Fig. S3–S7 show similar analysis of the monocation, monoanion and T1 excited states for all of the quinones AQ and 4–7. | ||
Interestingly, upon reduction of 5 one finds a similar enhancement of aromaticity for the central ring but in this case also the outer rings become more aromatic. We associate the enhanced aromaticity of the central ring and thiophene rings with the two respective resonance structure shown in Fig. 9. It is at first counterintuitive that the enhanced aromaticity of 5 goes along with strongly reduced electron affinity of 1.27 eV compared to 2.02 eV in 4. However, this fact can be explained by considering that the enhanced aromaticity comes at the cost of reduced charge delocalization and/or the formation of tetravalent sulfur as indicated in Fig. 7. Finally, an analysis of 6 is presented at the bottom of Fig. 8. As expected, this molecule occupies a middle ground between 4 and 5 where the strongly nonsymmetric shape of the VIST plot of the anion is particularly noteworthy.
Natural difference orbitals (NDOs)67 are shown to the right in Fig. 10. These highlight that the extra electron attaches predominantly to the central benzene ring along with the two oxygen atoms. It is also noteworthy that the dominant NDO exhibits one nodal plane on the benzene ring, resembling the HOMO of benzene. In line with ref. 68, this can be interpreted in the sense that upon reduction the central ring becomes more like benzene and, hence, more aromatic. The NDOs also provide a clear depiction of the symmetry breaking seen for 6 where the electron is strongly pulled to the left side.
Conclusions
We have presented the synthesis and properties of the yellow ICT compound 1 which uses a benzo[1,2-c:4,5-c′]dithiophene-4,8-dione as the acceptor component, in order to help understand why thiophene analogues of anthraquinone can have such a negative influence on emissive properties. Our observations suggest a combination of factors are at play. The thiophene quinones seem to behave much like anthraquinone itself in terms of triplet harvesting. ISC is both permitted and extremely fast so triplet quantum yields are high. While we have previously demonstrated that symmetry breaking has then rendered the AQ molecule emissive, this has not been observed to date in any emitters that use the thiophene quinones. Instead, they undergo non-radiative decay from the triplet manifold. The presence of heavy sulphur atoms in these acceptors improves ISC and is also likely to be a major contributing factor. Our computational results have revealed a powerful interplay between the aromaticity gains and losses of the 5- and 6-membered rings of each of the isomeric quinones 4–7 after accepting an electron, which has contextualised their relative electron accepting ability and individual electronic structures.We note that aspects of the excited state structures of the molecules presented here such as degeneracy of triplet states have been shown to have a commanding influence on other functional properties such as organic room temperature phosphorescence and singlet oxygen generation in non-quinone based ICT systems.69
The question remains of why acceptors such as 5 can have such a destructive presence in ICT emitters, but are among the best performing acceptor moieties used in high performance thiophene-based conjugated polymers for BHJ and other photovoltaic devices. Firstly, the CT between chains or with discrete acceptor molecules in the BHJ is extremely fast, occurring on the picosecond timescale.70–73 This is very much faster than the rates of ISC we have measured for these acceptors. Secondly, existing computational results generally depict the LUMO of polymers containing these quinones as being delocalised along the conjugated backbone to a much greater extent than it lies directly over the quinone moiety.25,26,30,37 The interplay between oligomers or polymers of thiophene with ring-fused electroactive moieties such as tetrathiafulvalenes74–78 or transition metal dithiolenes79–81 have previously been studied and are known to be complex. The results presented here suggest that the quinone acceptors in polymers such as PM6 are essentially pendant, and have more limited influence on excited state properties than is observed in small molecules with very defined donor and acceptor regimes such as 1 and 2.
Therefore, we can summarise that while quinones 4–7 have many appealing characteristics the results to date suggest that these compounds should be utilised with due consideration. More detailed predictive calculations than might otherwise be expected should be applied prior to embarking on synthesis.
Data availability
Computational research data for this article including molecular geometries and input/output files are available via a separate repository, DOI: 10.17028/rd.lboro.25773591. Other data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. and P. K. thank Loughborough University for PhD Studentships. R. H. F. thanks the EPSRC for support. A. J. G. thanks the Leverhulme Trust for an Early Career Fellowship (ECF-2022-445). I. A. W. and S. W. thank the EPSRC (EP/T028688/2) for funding.References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC.
- T. Zhang, Y. Xiao, H. Wang, S. Kong, R. Huang, V. K.-M. Au, T. Yu and W. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301896 CrossRef CAS PubMed.
- M. K. Etherington, Front. Chem., 2020, 8, 716 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926 RSC.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707 CrossRef CAS PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- S. Montanaro, A. J. Gillett, S. Feldmann, E. W. Evans, F. Plasser, R. H. Friend and I. A. Wright, Phys. Chem. Chem. Phys., 2019, 21, 10580 RSC.
- Q. S. Zhang, H. Kuwabara, W. J. Potscavage, S. P. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1969, 18, 145 CrossRef.
- P. Kimber, P. Goddard, I. A. Wright and F. Plasser, Phys. Chem. Chem. Phys., 2021, 23, 26135 RSC.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834 CrossRef CAS.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8 CrossRef CAS.
- P. Nag, S. V. K. Isukapalli, A. Nath and S. R. Vennapusa, J. Phys. Chem. A, 2002, 126, 3680 CrossRef PubMed.
- K. Gollnick, S. Held, D. O. Mártire and S. E. Braslavsky, J. Photochem. Photobiol., A, 1992, 69, 155 CrossRef CAS.
- A. A. Lamola and G. S. Hammond, J. Chem. Phys., 1965, 43, 2129 CrossRef CAS.
- S. A. Carlson and D. M. Hercules, J. Am. Chem. Soc., 1971, 22, 5611 CrossRef.
- C. A. Parker and C. G. Hatchard, Analyst, 1962, 87, 664 RSC.
- V. Ya Oginets, J. Appl. Spectrosc., 1985, 43, 849 CrossRef.
- K. Kobayashi, C. L. Gajurel, K. Umemoto and Y. Mazaki, Bull. Chem. Soc. Jpn., 1992, 65, 2168 CrossRef CAS.
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal, J. V. Gražulevičius and A. P. Monkman, J. Mater. Chem. C, 2019, 7, 9184 RSC.
- S. Kothavale, W. J. Chung and J. Y. Lee, J. Mater. Chem. C, 2022, 10, 6043 RSC.
- F.-M. Xie, J.-X. Zhou, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2020, 8, 9476 RSC.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655 CrossRef CAS PubMed.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. Tan and J. Hou, Macromolecules, 2012, 45, 9611 CrossRef CAS.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed.
- Z. Zheng, H. Yao, L. Ye, Y. Xu, S. Zhang and J. Hou, Mater. Today, 2020, 35, 115 CrossRef CAS.
- X. Xu, K. Feng, L. Yu, H. Yan, R. Li and Q. Peng, ACS Energy Lett., 2020, 5, 2434 CrossRef CAS.
- N. G. Yang, G. P. Kini, H. S. Lee, J. Y. Kim and D. K. Moon, Dyes Pigm., 2024, 224, 112045 CrossRef CAS.
- J. Cao, W. Zhang, Z. Xiao, L. Liao, W. Zhu, Q. Zuo and L. Ding, Macromolecules, 2012, 45, 1710 CrossRef CAS.
- H. Bin, L. Xiao, Y. Liu, P. Shen and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1929 CrossRef CAS.
- G. Zhang, J. Guo, J. Zhang, W. Li, X. Wang, H. Lu and L. Qiu, Dyes Pigm., 2016, 126, 20 CrossRef CAS.
- X. Zhao, L. Qian, J. Cao, S. Yan and L. Ding, Polym. Chem., 2016, 7, 1226 RSC.
- K.-M. Lu, W.-M. Li, P.-Y. Lin, K.-T. Liu and C.-Y. Liu, Adv. Synth. Catal., 2017, 359, 3805 CrossRef CAS.
- P. Chao, H. Chen, Y. Zhu, H. Lai, D. Mo, N. Zheng, X. Chang, H. Meng and F. He, Adv. Mater., 2020, 32, 1907059 CrossRef CAS PubMed.
- M. Jeong, J. Oh, Y. Cho, B. Lee, S. Jeong, S. M. Lee, S.-H. Kang and C. Yang, Adv. Funct. Mater., 2021, 31, 2102371 CrossRef CAS.
- Y.-F. Wen, K.-H. Lee, P.-T. Huang, M.-H. Chen, W.-C. Shin, L.-J. Huang, M.-H. Hsu, C.-J. Chen and S.-C. Kuo, Bioorg. Med. Chem. Lett., 2007, 17, 2908 CrossRef CAS PubMed.
- C.-J. Chen, Y.-F. Wen, P.-T. Huang, M.-H. Hsu, K.-H. Lee, M.-H. Chen, W.-C. Shin, L.-J. Huang and S.-C. Kuo, Leuk. Res., 2009, 33, 1664 CrossRef CAS PubMed.
- J.-S. Yang, C.-A. Lin, C.-C. Lu, Y.-F. Wen, F.-J. Tsai and S.-C. Tsai, Oncol. Rep., 2017, 37, 1786 CrossRef CAS PubMed.
- J.-S. Yang, C.-Y. Lee, H.-C. Cho, C.-C. Lu, S.-C. Kuo, Y.-F. Wen, F.-J. Tsai, M.-R. Lee and S.-C. Tsai, Oncol. Rep., 2018, 39, 383 CAS.
- K. Shiraishi and T. Yamamoto, Polym. J., 2002, 34, 727 CrossRef CAS.
- D. W. H. MacDowell and J. C. Wisowaty, J. Org. Chem., 1972, 37, 1712 CrossRef CAS.
- E. Ishow, R. Camacho-Aguilera, J. Guérin, A. Brosseau and K. Nakatani, Adv. Funct. Mater., 2009, 19, 796 CrossRef CAS.
- W. Sun, N. Zhou, Y. Xiao, S. Wang and X. Li, Dyes Pigm., 2018, 154, 30 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- C. M. Guédon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. van der Molen, Nat. Nanotechnol., 2012, 7, 305 CrossRef PubMed.
- A. Alanazy, E. Leary, T. Kobatake, S. Sangtarash, M. Teresa González, H.-W. Jiang, G. R. Bollinger, N. Agräit, H. Sadeghi, I. Grace, S. J. Higgins, H. L. Anderson, R. J. Nichols and C. J. Lambert, Nanoscale, 2019, 11, 13720 RSC.
- H. J. van Ramesdonk, B. H. Bakker, M. M. Groeneveld, J. W. Verhoeven, B. D. Allen, J. P. Rostron and A. Harriman, J. Phys. Chem. A, 2006, 110, 13145 CrossRef CAS PubMed.
- K. Okamoto, T. Hasobe, N. V. Tkachenko, H. Lemmetyinen, P. V. Kamat and S. Fukuzumi, J. Phys. Chem. A, 2005, 109, 4662 CrossRef CAS PubMed.
- A. J. Gillett, A. Pershin, R. Pandya, S. Feldmann, A. J. Sneyd, A. M. Alvertis, E. W. Evans, T. H. Thomas, L.-S. Cui, B. H. Drummond, G. D. Scholes, Y. Olivier, A. Rao, R. H. Friend and D. Beljonne, Nat. Mater., 2022, 21, 1150 CrossRef CAS PubMed.
- H. Inoue, M. Hida, N. Nakashima and K. Yoshihara, J. Phys. Chem., 1982, 86, 3184 CrossRef CAS.
- O. F. Mohammed, D. Xiao, V. S. Batista and E. T. J. Nibbering, J. Phys. Chem. A, 2014, 118, 3090 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- T. Yanai, D. P. Yew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- B. de Souza, G. Farias, F. Neese and R. Izsák, J. Chem. Theory Comput., 2019, 15, 1896 CrossRef CAS PubMed.
- W. Zeng, O. El Bakouri, D. W. Szczepanik, H. Bronstein and H. Ottosson, Chem. Sci., 2021, 12, 6159 RSC.
- F. Plasser and F. Glöcklhofer, Eur. J. Org. Chem., 2021, 2529 CrossRef CAS PubMed.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- F. Plasser, Chemistry, 2021, 3, 532 CrossRef CAS.
- S. Paredis, T. Cardeynaels, S. Kuila, J. Deckers, M. van Landgehem, K. Vandewal, A. Danos, A. P. Monkman, B. Champagne and W. Maes, Chem. – Eur. J., 2023, e202301369 CrossRef CAS PubMed.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, J. Am. Chem. Soc., 2020, 142, 12751 CrossRef CAS PubMed.
- D. Lee, C.-M. Oh, J. Ryu, S.-Y. Jang, I.-W. Hwang and S. Cho, Mater. Adv., 2023, 4, 4868 Search PubMed.
- A. C. Jakowetz, M. L. Böhm, J. Zhang, A. Sadhanala, S. Huettner, A. A. Bakulin, A. Rao and R. H. Friend, J. Am. Chem. Soc., 2016, 138, 11672 CrossRef CAS PubMed.
- S. Shoaee, H. M. Luong, J. Song, Y. Zou, T.-Q. Nguyen and D. Neher, Adv. Mater., 2024, 36, 2302005 CrossRef CAS PubMed.
- I. A. Wright, P. J. Skabara, J. C. Forgie, A. L. Kanibolotsky, B. González, S. J. Coles, S. Gambino and I. D. W. Samuel, J. Mater. Chem., 2011, 21, 1462 RSC.
- I. A. Wright, N. J. Findlay, S. Arumugam, A. R. Inigo, A. L. Kanibolotsky, P. Zassowski, W. Domagala and P. J. Skabara, J. Mater. Chem. C, 2014, 2, 2674 RSC.
- R. Berridge, P. J. Skabara, C. Pozo-Gonzalo, A. Kanibolotsky, J. Lohr, J. J. W. McDouall, E. J. L. McInnes, J. Wolowska, C. Winder, N. S. Sariciftci, R. W. Harrington and W. Clegg, J. Phys. Chem. B, 2006, 110, 3140 CrossRef CAS PubMed.
- A. L. Kanibolotsky, L. Kanibolotskaya, S. Gordeyev, P. J. Skabara, I. McCulloch, R. Berridge, J. E. Lohr, F. Marchioni and F. Wudl, Org. Lett., 2007, 9, 1601 CrossRef CAS PubMed.
- A. L. Kanibolotsky, N. J. Findlay and P. J. Skabara, Beilstein J. Org. Chem., 2015, 11, 1749 CrossRef CAS PubMed.
- I. A. Wright, C. A. Wilson, S. J. Coles and P. J. Skabara, Dalton Trans., 2019, 48, 107 RSC.
- C. Pozo-Gonzalo, R. Berridge, P. J. Skabara, E. Cerrada, M. Laguna, S. J. Coles and M. B. Hursthouse, Chem. Commun., 2002, 2408 RSC.
- P. J. Skabara, C. Pozo-Gonzalo, N. Lardiés Miazza, M. Laguna, E. Cerrada, A. Luquin, B. González, S. J. Coles, M. B. Hursthouse, R. W. Harrington and W. Clegg, Dalton Trans., 2008, 3070 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02099e |
| This journal is © The Royal Society of Chemistry 2024 |