
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg–NiP deposited green carbon channel embedded NiP panels for sustainable water splitting†
Revathy B.
Nair
a,
A. Anantha
Krishnan
b,
Aneesh Kumar
M. A.
b,
Sivaraj
Rajendran
b,
Sreehari
Harikumar
 b,
Vidhya
C.
a,
M.
Ameen Sha
c,
Thomas
Mathew
b,
Sajith
Kurian
*a and
P. S.
Arun
*b
b,
Vidhya
C.
a,
M.
Ameen Sha
c,
Thomas
Mathew
b,
Sajith
Kurian
*a and
P. S.
Arun
*b
aDepartment of Chemistry, Mar Ivanios College (Affiliated to University of Kerala), Nalanchira, Thiruvanathapuram, Kerala - 695015, India. E-mail: sajith.kurian@mic.ac.in
bDepartment of Chemistry, St. John's College (Affiliated to University of Kerala), Anchal, Kollam, Kerala - 691306, India. E-mail: arunps@stjohns.ac.in
cDepartment of Applied and Environmental Chemistry, University of Szeged, Szeged, 6720, Hungary
First published on 12th September 2024
Abstract
Ag–NiP-deposited carbon channels on NiP panels were successfully developed through lemon juice extract (Ag–CL/NiP) and citric acid (Ag–CC/NiP)-assisted methodologies. The methods involved the precise execution of electroless deposition of the advanced Ag–carbon matrix with NiP. The lemon juice-assisted method produced carbon channels with a dense concentration of Ag–NiP on the electrode surface, whereas the citric acid method resulted in a less dense deposition of Ag–NiP on the electrode surface, as observed via FE-SEM. The Ag–CL/NiP has remarkably higher electro- and photocatalytic water splitting performance due to the compact and conductive Ag–NiP connected with carbon channels. Electrochemical impedance analysis of Ag–CL/NiP revealed a low Rct of 491.3 Ω at the open circuit potential, indicating enhanced conductivity. The electrocatalytic oxygen evolution reaction (OER) overpotential of Ag–CL/NiP was 401 mV to achieve a current density of 50 mA cm−2, with a Tafel slope of 46.5 mV dec−1. The panel exhibited good stability, with a proven durability of over 1000 cycles of CV during OER. The developed panel achieved an impressive photocurrent density of ∼9.5 mA cm−2 at 1.37 V vs. RHE when subjected to light irradiation with a wavelength exceeding 420 nm. Furthermore, the Ag–CL/NiP panel demonstrated the ability to generate 17.5 mmol cm−2 of H2 over a 4-hour sunlight irradiation period. The temperature-controlled photocatalytic water splitting experiment revealed that the panel maintained its activity at temperatures as low as ∼12 °C, but with a 40% drop in efficiency compared to normal sunlight conditions.
Introduction
The global economy is turning its attention to the significance of hydrogen and its associated technologies, driven by the pressing need for cleaner fuel sources and raw materials in today's world. On a global scale, there is a growing imperative to promote and scale up the green methods of hydrogen production, from the catalytic splitting of water.1–7 Nickel (Ni)-based materials have garnered substantial attention from both researchers and industrialists as a viable substitute for noble metals in catalytic applications, particularly in photocatalytic and electrocatalytic water splitting processes. This is primarily due to their ability to achieve a high charge separation rate for photo-induced electrons and holes.8,9 NiP sites can serve as an efficient trapping site for capturing photo-induced electrons, thereby enhancing the effectiveness of water splitting.10 Further, the NiP sites not only function as electron probes at the solid–liquid interface but also reduce the energy barrier height for electron transfer during the water splitting process.11 In certain nanocomposite systems, NiP acts as an electron carrier, promoting a significant increase in the separation of interfacial charge carriers. This, further extends the lifespan of photogenerated charge carriers.12–16 These advantageous electronic and chemical properties establish NiP as a promising candidate for electrochemical water splitting processes.17,18The widespread adoption of catalysts doped with noble metals, such as Pt, Pd, Au, and Ag, represents an effective strategy for enhancing the water splitting reaction. This is primarily attributed to the formation of highly conductive Schottky heterojunctions by these noble metals, which in turn promotes the efficient generation and separation of charge carriers.19,20 Additionally, metals like Au and Ag exhibit the surface plasmon resonance (SPR) effect, further augmenting the efficiency of charge transfer kinetics of photogenerated electron–hole pairs during photocatalytic reactions.21,22 In the pursuit of highly efficient photo/electrocatalytic water splitting processes, researchers have already explored systems incorporating silver nanoparticles. It has been documented that the integration of silver nanoparticles into catalytic systems holds promise for achieving remarkable catalytic activity.23–25 Notably, when silver is introduced as a dopant into nickel-based catalyst systems, it facilitates rapid electron transfer and promotes the formation of active NiOOH intermediates. Consequently, this Ag–Ni bimetallic system accelerates the rate of electrocatalytic reactions, particularly in the context of the oxygen evolution reaction (OER).26 Through theoretical investigations, it has come to light that the interplay of multiple components, particularly Ag and Ni, leads to synergistic effects that significantly boost the electrocatalytic performance during the OER. These effects are attributed to several factors, including the reduction of interfacial charge transfer resistance, the optimal adsorption energy for OER intermediates, and the reinforcement of Ni–O bonding.27 This synergistic interaction between Ni and Ag not only influences the electrocatalytic OER but also facilitates bulk charge separation, promoting the transfer of electron density between Ag and Ni during the electrocatalytic water reduction process.28,29
Recently, there has been a growing interest in utilizing transition metals in conjunction with carbon-based materials as highly effective photo- and electrocatalysts for the water splitting reaction.30–34 Notably, the introduction of single-atom silver onto carbon in Ni-based systems has proven to enhance OER performance. This enhancement is attributed to the presence of Ag on the Ni site, which elevates the valency of Ni and subsequently reduces the adsorption energy of oxygen-containing intermediates on the active Ni sites, ultimately leading to improved OER performance.35,36 When considering carbon-based materials, graphitic carbon nitride, carbon nanotubes, and graphene have been explored as active catalysts.37–45 Their appeal lies in their ability to rapidly separate and transport photogenerated charge carriers, possess a high surface area, and exhibit a strong visible light harvesting capability. It has been reported that single-atom Ni, accompanied by a neighboring carbon atom, offers active sites for the overall water splitting reaction under visible light irradiation.46,47 Incorporating carbon-covered NiP into the catalyst system has demonstrated the capacity to selectively attract and extract photoinduced electrons for the water reduction process, primarily through the formation of heterojunctions.48–50 The distinctive morphology of the carbon component within the NiP catalyst further enhances the availability of active sites, reducing charge transfer resistance and ultimately leading to improved and stable hydrogen evolution performance.51 The combination of Ni with amorphous carbon has been documented as exhibiting remarkable performance in electrocatalytic water splitting.52,53 Furthermore, the utilization of a Ni–Ag bimetallic system supported on various carbon structures has shown promise as an effective choice for electrocatalytic oxidation and reduction processes.54,55 Recent reports have highlighted the efficacy of lemon juice-based green approaches as effective reducing agents and capping agents in the synthesis of catalytically active nanoparticles and optically active carbon quantum dots.56–61
In this context, we developed highly conductive green carbon channels by utilizing the lemon juice extract as both carbon source and reducing agent. To boost their activity, these channels are further deposited with Ag–NiP, drawing inspiration from the excellent water-splitting performance of the Ag–Ni bimetallic system. This led us to propose Ag–NiP covered green carbon channels on NiP panels for enhanced electrocatalytic OER and photocatalytic hydrogen evolution reaction (HER). For comparison, Ag–NiP decorated carbon channels using citric acid were also developed. Surprisingly, the lemon juice-assisted methodology yielded a dense Ag–NiP, covered with carbon channels on the electrode surface. The system exhibited a low overpotential of 401 mV to achieve a high current density of 50 mA cm−2, with a low Tafel slope of 46.5 mV dec−1. Further, a remarkable photocurrent of ∼9.5 mA cm−2 at 0.35 V vs. Ag/AgCl was observed when subjected to light irradiation with a wavelength >420 nm, whereas the citric acid-assisted panels generated only ∼0.43 mA cm−2. According to Marwat et al.,62 the benchmark material Sb2Se3 achieves a photocurrent density of 30 mA cm−2 at a high potential of 1.0 V vs. Ag/AgCl. In comparison, the Ag–CL/NiP panel developed using the lemon juice extract exhibits remarkably higher electrocatalytic and photocatalytic activity (Tables S1 and S2, ESI†) surpassing many recently explored catalysts. Temperature-controlled photocatalytic water splitting reactions were performed to analyze the effect of temperature on this reaction system. The Ag–CL/NiP panel retained its photocatalytic activity even at a lower temperature of ∼12 °C. The reusable Ag–CL/NiP panels demonstrated exceptional catalytic activity in both photocatalytic and electrocatalytic water splitting reactions. The Ag–CL/NiP panel exhibited good stability over 1000 CV cycles during the OER as well as five cycles of the photocatalytic HER. In the present work, we are introducing a highly stable and efficiently reusable catalytic system developed by exploring lemon juice-derived green panels which is capable of contributing to the growing demand for sustainable and clean energy production.
Experimental methods
Tailoring procedure of Ag–CL/NiP and Ag–CC/NiP panels
The silver–carbon powder was developed using AgNO3 and a carbon source by the following procedure: AgNO3 (AR grade, 1 g) was dissolved in 50 mL of DI water followed by a gentle heating at 80 °C using a water bath for 10 min. A suitable amount of unprocessed lemon juice was introduced into the solution until a black color was observed. Here, the lemon juice acts as the reducing agent as well as the carbon source. The solution was subjected to stirring at a rate of 500 rpm for a duration of 2 hours. Following this, the mixture was left undisturbed overnight at room temperature. To complete the reduction of silver ions in the solution, 0.2 mL of hydrazine (AR grade) was cautiously added drop by drop while maintaining agitation. The mixture was then subjected to stirring at 500 rpm for a period of one hour. After that, the mixture underwent filtration and several washings with DI water. The resulting residue was subjected to drying at 160 °C for 5 hours. For comparison, citric acid-derived silver–carbon was also developed by a similar procedure, utilizing citric acid (AR grade) as the carbon source instead of lemon juice. Both the developed Ag–carbon were deposited on a NiP plate by a customized electroless deposition technique to develop the electrodes.63An electroless deposition bath was prepared by dissolving nickel sulfate (30 g L−1) and succinic acid (25 g L−1) in DI water. The pH of this solution was carefully adjusted and maintained between 4.5 and 5 by using NH4OH. The temperature of the bath was maintained at 70 °C throughout the deposition process. The reducing agent sodium hypophosphite (25 g L−1) was added, along with 100 mg of as-prepared silver–carbon powder. The pre-treated Cu plates were dipped into the bath by fixing it in a vertical manner. The deposition was carried out for 90 minutes with stirring (500 rpm) and heating (70 °C). The NiP panels with silver–carbon developed using lemon juice extract and citric acid are labelled as Ag–CL/NiP and Ag–CC/NiP respectively. The schematic representation of the fabrication of Ag–CL/NiP panels is shown in Fig. 1.
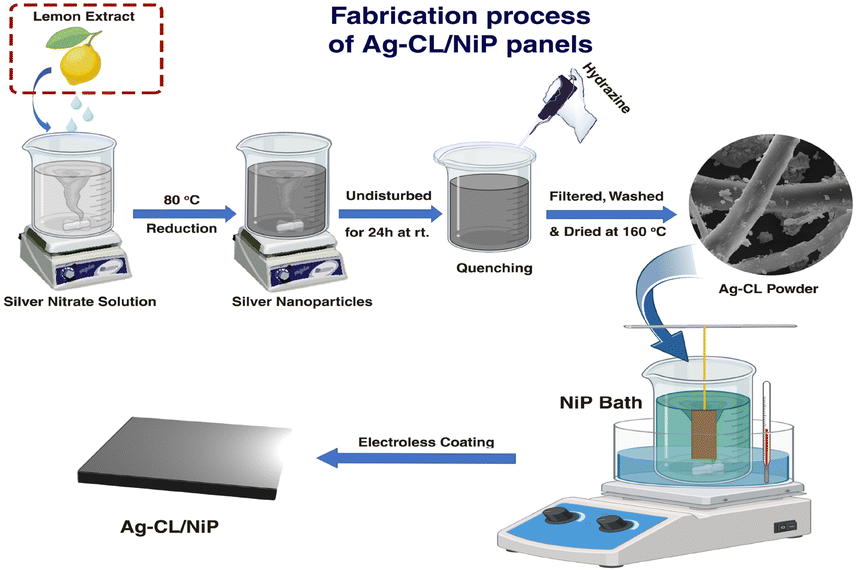 | ||
| Fig. 1 Scheme indicating the fabrication sequence leading to the formation of Ag–CL/NiP panels. | ||
Physico-chemical characterization
The developed systems were characterized using different techniques in order to unfold their material characteristics. XRD was carried out using a Bruker D8 Advance twin–twin spectrometer. UV-vis spectroscopic analysis was carried out on a Thermoscientific Nicolet iS50 - Cary 5000 high performance UV-Vis-NIR spectrophotometer and XPS analysis was performed using a Thermo Scientific ECSALAB Xi+ photoelectron spectrometer with an Al Kalpha (1486.6 eV) X-ray source. The binding energy obtained was corrected using carbon 1s (284.6 eV) as a reference. The SEM/EDAX analysis with Carl Zeiss EVO 18 research and FE-SEM/EDAX analysis with Carl Zeiss-Sigma 300 were performed.Electrochemical characterization and OER efficiency analysis
All the electrochemical characterization and OER performance analysis were carried out using an electrochemical workstation (CH16041E USA) and the IR corrected values are reported. The electrochemical characteristics and reaction kinetics of the developed panel were analyzed by electrochemical impedance spectroscopy (EIS) analysis performed in a 1 M NaOH solution at the open circuit potential (OCP) in the AC frequency range of 1 Hz to 10 kHz. Electrochemically active surface area (ECSA) was analyzed by conducting CV analysis in the non-faradaic region at different scan rates. The OER performance of the developed panels was assessed by cyclic voltammetry at a scan rate of 10 mV s−1 in 1 M NaOH. LSV analysis was conducted to calculate the OER overpotential at a scan rate of 10 mV s−1. The electrochemical stability during OER was confirmed by linear sweep voltammetric analysis before and after conducting 1000 cycles of CV. The electrochemical characteristics and OER performance were evaluated using three electrodes set up with a Pt mesh (5 cm2) as the counter electrode.Photocatalytic water splitting analysis
The photocatalytic water splitting efficiencies of Ag–CL/NiP and Ag–CC/NiP were investigated simultaneously under natural sunlight. A surface area of 2 cm2 on the developed panel was exposed to sunlight. The prepared panels were placed in 40 mL of water–methanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture in an airtight borosilicate round bottom flask (70 mL). Then N2 gas was purged through the mixture for 45 minutes to provide an inert atmosphere during irradiation. The variation of temperature of the reaction medium throughout the reaction was monitored. The volume of hydrogen gas generated was periodically monitored by using a PerkinElmer gas chromatograph Clarus 590 with a thermal conductivity detector. The reusability of the panel was monitored under exactly the same conditions for 5 cycles. The temperature dependence of the photocatalytic activity of the catalyst was monitored at temperatures starting from ∼0 °C under natural sunlight, and hydrogen evolution was quantified to analyze the effect of temperature.
:1) mixture in an airtight borosilicate round bottom flask (70 mL). Then N2 gas was purged through the mixture for 45 minutes to provide an inert atmosphere during irradiation. The variation of temperature of the reaction medium throughout the reaction was monitored. The volume of hydrogen gas generated was periodically monitored by using a PerkinElmer gas chromatograph Clarus 590 with a thermal conductivity detector. The reusability of the panel was monitored under exactly the same conditions for 5 cycles. The temperature dependence of the photocatalytic activity of the catalyst was monitored at temperatures starting from ∼0 °C under natural sunlight, and hydrogen evolution was quantified to analyze the effect of temperature.
Results and discussion
Deciphering the physico-chemical characteristics of the Ag–CL/Ni–P panels
The crystallographic characteristics of the Ag–carbon powder Ag–CL and Ag–CC, NiP and Ag–CL/NiP panels were examined using XRD, and the results are shown in Fig. 2(a). The XRD pattern evidenced the formation of crystalline Ag with a cubic structure (JCPDS65-2871) in the case of both lemon juice and citric acid assisted methods. The diffraction pattern had sharp peaks at 2θ values of 38.2°, 44.4°, and 64.6°, which can be ascribed to the features from the (111), (200), and (220) lattice planes of cubic Ag (JCPDS65-2871). The most intense plane of Ag, (111), has high density atoms and a flat, two dimensional surface making it more suitable for catalytic reactions.64 Ag can form an effective Schottky junction with NiP which can also enhance the photocatalytic water splitting performance.64 Both the planes of NiP, (111) at around 44° and (200) around 52°, were observed in the Ag–CL/NiP panel. The efficiency of various planes with respect to electrocatalytic activity may vary with the reaction conditions, media and materials.65 The intense peaks of Ag were also retained after the incorporation of the Ag–carbon powder in the NiP panels. These results revealed that Ag with a cubic crystal structure is incorporated along with NiP in the Ag–CL/NiP panel via the electroless coating.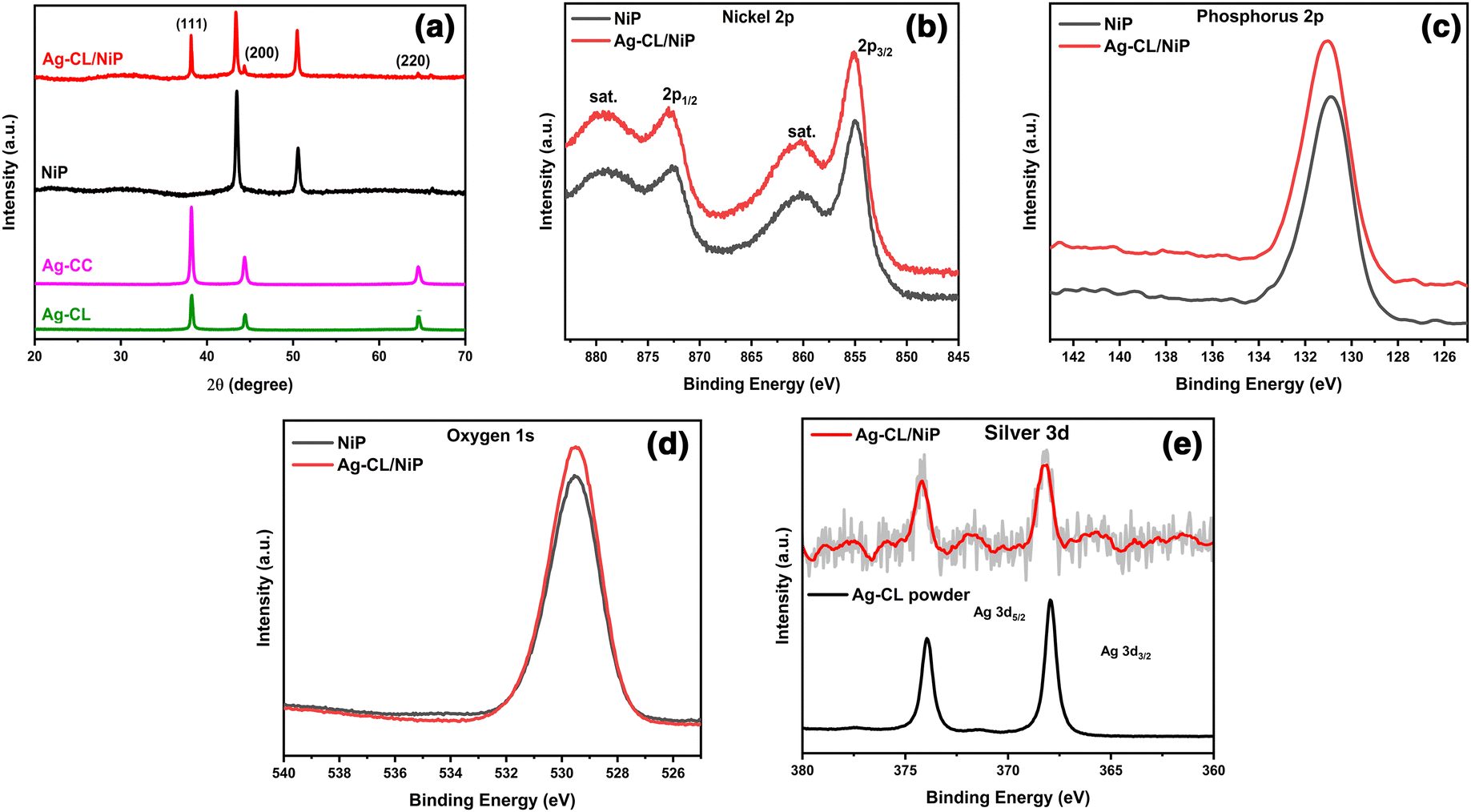 | ||
| Fig. 2 (a) XRD pattern of Ag–CL powder, Ag–CC powder, NiP, and Ag–CL/NiP. (b)–(e) XPS spectra showing Ni2p, P2p, O1s and Ag3d, respectively, of NiP and Ag–CL/NiP. | ||
The chemical states of elements in the systems were analysed using XPS and the results are shown in Fig. 2(b–e). In the case of NiP, the predominant peak for Ni 2p3/2 was observed at 854.94 eV, and the coupling constant value was determined to be 17.46 eV, consistent with the Ni2+ oxidation state.66 Conversely, in the Ag–CL/NiP system, the major peak for Ni 2p3/2 was recorded at 855.34 eV, with a coupling constant value of 17.71 eV, further confirming the presence of the Ni2+ oxidation state in both NiP and Ag–CL/NiP.66 The phosphorus 2p3/2 peaks in both NiP and Ag–CL/NiP were detected at 130.3 eV, Fig. 2(d), providing evidence for the existence of elemental phosphorus (2p3/2).67 Also, the Ag 3d5/2 core level feature was identified at 368.2 eV, accompanied by a spin–orbit coupling constant of 6 eV, indicative of the Ag0 oxidation state.68 The binding energy of Ni 2p3/2 positively shifted by 0.4 eV when the Ag–carbon powder was loaded into NiP. While the binding energy of Ag 3d5/2 negatively shifted by 0.8 eV, there was no significant shift in the binding energy of phosphorus when Ag–carbon was loaded onto NiP. There was a discernible shift in the Ag 3d5/2 peak to 367.4 eV in the case of Ag–CL/NiP, potentially suggesting electron transfer from NiP to silver.69
The elemental composition of NiP was analyzed using EDAX and is shown in Fig. S2 of the ESI.† The EDAX analysis shows that the NiP panel consists solely of nickel and phosphorus, and the corresponding EDAX mapping is shown in Fig. S1(e and f) of the ESI.† In contrast, the Ag–CL/NiP panel exhibited a more complex composition, including Ag, Ni, phosphorus, and carbon, as seen in Fig. 3(c). EDAX mapping of Ag–CL/NiP is shown in Fig. 4(d–h). There were no indications of any other impurities, demonstrating dense coverage over the NiP surface by the developed Ag–NiP covered carbon channels.
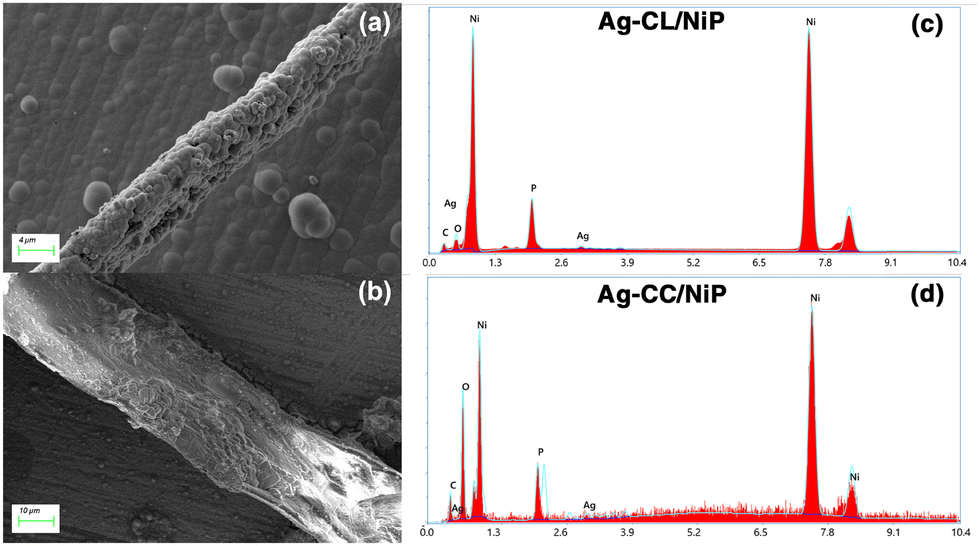 | ||
| Fig. 3 FE-SEM image of Ag–NiP deposited carbon channels on the surface of the NiP panel: (a) Ag–CL/NiP and (b) Ag–CC/NiP. EDAX spectra of (c) Ag–CL/NiP and (d) Ag–CC/NiP. | ||
 | ||
| Fig. 4 (a)–(c) FE-SEM images of Ag–CL/NiP at different magnifications. (d)–(h) EDAX mapping of the Ag–CL/NiP panel. | ||
The morphological characteristics of Ag–CL/NiP and Ag–CC/NiP were analyzed using FE-SEM and are shown in Fig. 3(a and b). The FE-SEM analysis revealed the formation of channel-like structures on the NiP panels' surface, which were densely covered with Ag–NiP in Ag–CL/NiP. EDAX spectroscopy confirmed that these channels consist of nickel, phosphorus, carbon, and silver, as shown in Fig. 3(c and d). The surface carbon concentration was notably higher in Ag–CC/NiP than in Ag–CL/NiP. This suggests that the lemon juice-assisted method generated carbon channels densely covered with Ag–NiP. In addition to that, the FE-SEM images and EDAX mapping shown in Fig. 4(a–d) and Fig. S3(a–h) of the ESI† also support this observation. The morphological characteristics of the Ag–carbon powder developed using lemon juice extract were also assessed through SEM analysis, and the corresponding images are presented in Fig. S1(c and d) of the ESI.† The SEM images and EDAX mapping show the presence of agglomerated particles intertwined with channel-like structures. Elemental mapping (Fig. S1(g and h) of the ESI†) confirms that these agglomerated and channel-like structures are primarily composed of a carbon framework, with silver particles adorning the carbon surfaces.
Electrochemical characteristics and active surface area analysis
EIS analysis was employed to assess the electrical conductivity of the fabricated Ag–CL/NiP and Ag–CC/NiP and the results were compared with the EIS results of NiP panel. The corresponding Nyquist plots at their open circuit potential are shown in Fig. 5(a). The diameter of the semicircle in the Nyquist plot corresponding to the developed Ag–CL/NiP panel was notably smaller when compared to that of the Ag–CC/NiP and NiP panels in Fig. 5(a. inset). This observation strongly supports the idea of enhancement of conductivity of the Ag–CL/NiP panel, signifying a prolonged lifetime for electron–hole pairs, a characteristic that augments photo- and electrocatalytic activity.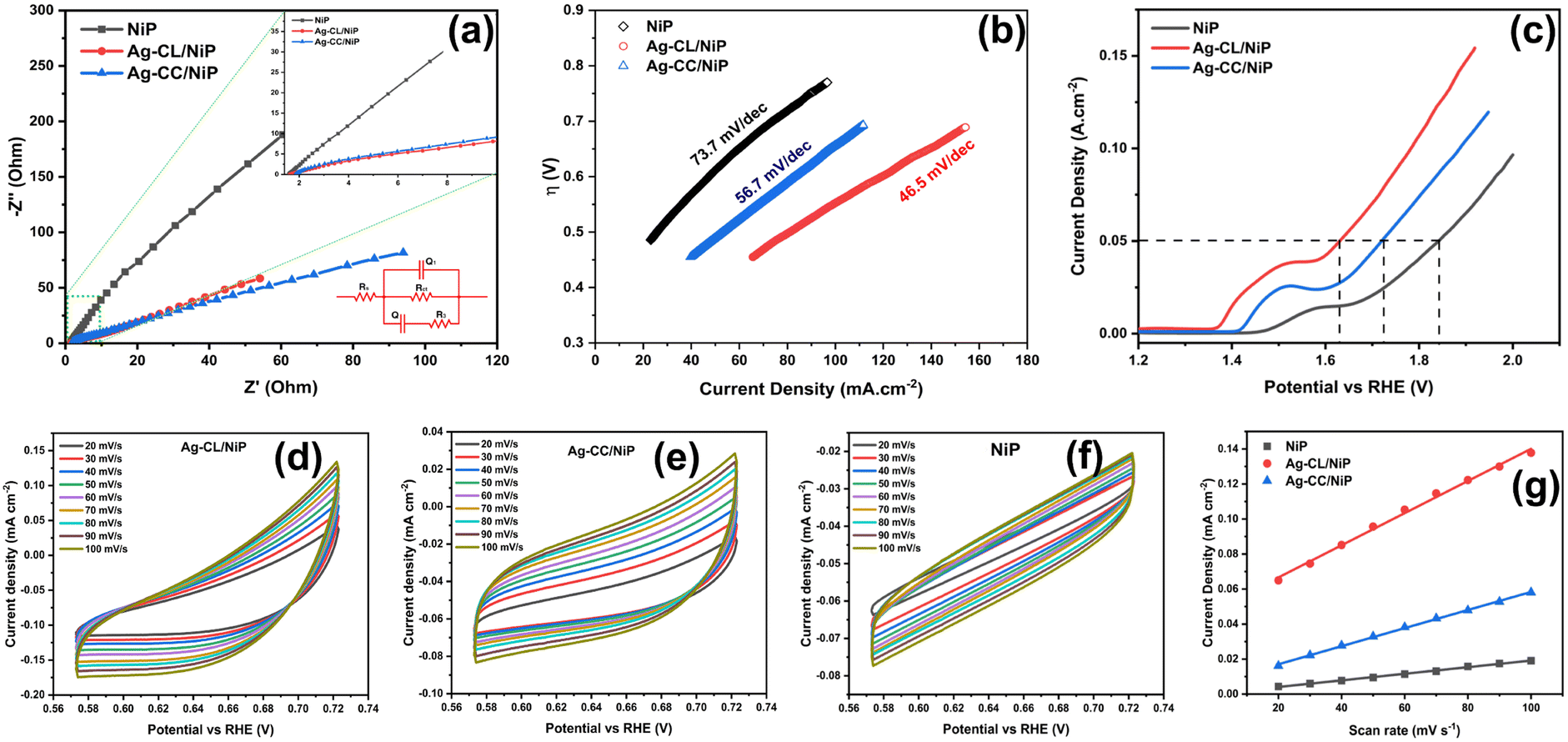 | ||
| Fig. 5 (a) Nyquist plot and equivalent circuit. (b) Tafel curves. (c) LSV curves. Analysis of the electrochemically active surface area (ECSA): CV curves of (d) Ag–CL/NiP, (e) Ag–CC/NiP and (f) NiP. (g) Determination of Cdl using the plot of double layer charging current density variation vs. scan rate by a linear regression of Ag–CL/NiP, Ag–CC/NiP and NiP. | ||
The Nyquist plots, along with the corresponding equivalent circuit used for fitting, are depicted in Fig. 5(a). Notably, the charge transfer resistance (Rct) value for the Ag–CL/NiP panel measured at OCP as 491.3 Ω stands in stark contrast to the Rct values obtained for the Ag–CC/NiP and NiP panels using the same circuit, which were 598.6 Ω and 55744 Ω respectively. Also, the analysis involved the determination of the constant phase element (Q) to establish the electrochemical performance. It is worth noting that the Q value for the developed Ag–CL/NiP panel was 2.87 × 10−3 F cm−2 sα−1, whereas for Ag–CC/NiP, it was lower at 2.49 × 10−3 F cm−2 sα−1. These findings revealed that in Ag–CL/NiP, there was an improvement in the electrochemically active surface area, consequently enhancing the electrochemical performance of the panel.70,71
To confirm this observation, the ECSA of the panels was determined using the double layer capacitance method, following the approach described by Anantharaj et al.72,73 CV analyses of Ag–CL/NiP and Ag–CC/NiP were conducted within the non-faradaic region at various scan rates, and the corresponding graphs are shown in Fig. 5(d–g), and the resulting current density was plotted against the scan rate. ECSA was calculated using the following equation:
| ECSA = Cdl/CS |
The ECSA value determined for the developed Ag–CL/NiP panel (11.81 cm2) was higher than that of the Ag–CC/NiP panel (6.72 cm2). These findings clearly establish that the electrocatalytically active surface area of the developed Ag–CL/NiP panels surpasses that of Ag–CC/NiP by a factor of two. These results reveal that the dense covering of carbon channels with Ag–NiP on the NiP surface enhances the electrochemically active surface area. Therefore, this dense Ag–NiP covering plays a crucial role in the electrochemical activity of Ag–CL/NiP.
Electrocatalytic oxygen evolution
The kinetics of OER catalysis and OER efficiency of both Ag–CL/NiP and Ag–CC/NiP panels were evaluated using a three-electrode system, as outlined in the experimental section. To analyze the OER activity of the developed panels, Tafel (Fig. 5(b)) and LSV (Fig. 5(c)) analyses were performed, and the overpotential at a current density of 50 mA cm−2 was assessed. The Ag–CL/NiP panel exhibited an overpotential of 401 mV, which is markedly less than that of both Ag–CC/Ni–P (491 mV) and NiP (614 mV) to achieve the same current density. The corresponding Tafel analysis results also indicated that the Ag–CL/NiP panel possessed a Tafel slope value of 46.5 mV dec−1, which was less than that of both Ag–CA/Ni–P (56.7 mV dec−1) and Ni–P (73.7 mV dec−1) panels. These findings confirm a substantial improvement in the kinetics of OER catalysis attributable to the incorporation of Ag–NiP covered carbon channels within the NiP panel. The low charge transfer resistance observed at the Ag–CL/NiP electrode surface–electrolyte interface and the high Cdl values suggest that this panel can be employed effectively for electrocatalytic OER.The reversible characteristics of Ag–CL/NiP panels can be observed from CV, as shown in Fig. S4 of the ESI,† providing clear evidence of the superior OER efficiency of the developed panels. The voltammetric response observed before 1.4 V (vs. RHE) was due to Ni oxidation (as detailed in the mechanism of OER) and the oxygen evolution reaction which started alongside the Ni oxidation reaction led the current density peak to reach 10 mA cm−2 (Fig. S7 of ESI†).
The reversible OER activity is clearly illustrated through CV analysis, as presented in Fig. S4 of ESI.† The Ag–CL/NiP panels exhibit remarkable reversible OER performance, despite the OER process being inherently sluggish and requiring a high overpotential due to its demanding 4-electron process. The reversible nature can be seen by looking into the ΔE value (ΔE = Ered. − Eox.). The ΔE value for Ag–CL/NiP was 0.29 V, compared to 0.22 V for Ag–CC/NiP. The peak current (ip) value of 63 mA cm−2 for Ag–CL/NiP was significantly higher than that of Ag–CC/NiP (42 mA cm−2) and NiP (6.9 mA cm−2). The overall electrochemical stability was confirmed by LSV analysis conducted before and after 1000 cycles of CV (Fig. S5 and S6 of ESI†). Only a slight shift in the LSV curve was observed after 1000 cycles of CV, indicating that the developed Ag–CL/NiP panel has exceptionally high electrocatalytic OER efficiency and stability. The results of electrocatalytic OER performance obtained for Ag–CL/NiP are compared with recent literature reports in Table S1 of ESI.† These comparisons reveal that Ag–CL/NiP possesses superior electrocatalytic OER efficiency compared to recently reported systems.
Electrocatalytic OER mechanism
Drawing insights from the OER test results and a thorough examination of the developed panel's characters, a plausible mechanism for the OER process is proposed. The Ag–CL/NiP catalyst displays heightened OER activity in alkaline environments, which can be attributed to the dual-site adsorbate evolution (AEM) electron transfer mechanism. Within the Ag–CL/NiP structure, phosphorus naturally has an inclination to abstract oxygen or hydroxide species on its surface or bond covalently with O, resulting in the formation of phosphorus oxide or hydroxide.74,75 The presence of Ag and Ni enhances the metal sites' ability to absorb OOH* intermediates, facilitating a dual-site mechanism that promotes the OER process.76–78 Similarly, the XPS data of Ag–CL/NiP show a higher binding energy Ni 2p3/2 peak (855.34 eV) compared to that in NiP (854.94 eV), indicating that the Ni2+ species are in a less electron dense state. This state is conducive to electrostatic interactions with hydroxide ions, facilitating their rapid conversion into nickel hydroxide. The reactions that might happen during OER are as follows:79| Ni(OH)2 + OH− → Ni(OH)3 + e− |
| Ni(OH)3 → NiOOH + H2O |
The emergence of NiOOH is an intermediate step within the widely acknowledged OER mechanism.80 In this context, the rapid formation of NiOOH is facilitated by the electrostatic interaction between the higher oxidation states of nickel and OH− ions, effectively amplifying OER activity.81–83 Another pivotal factor contributing to excellent OER activity is the presence of an ample number of adsorption sites for OH− during the OER process. In this particular system, apart from the Ni adsorption sites, Ag also serves as an OH adsorption site, thereby enhancing OER activity through a dual site adsorption mechanism.84
During the adsorption process, [O*] and OH* (* denotes the adsorption site) intermediates exhibit a tendency to bind to carbon sites, while the OOH* intermediate exhibits a preference for adsorption on nickel ion sites.85 Incorporating the heteroatom Ag into the carbon ribbon within NiP introduces additional active sites for the adsorption of OOH*, enhancing the dual site mechanism. Furthermore, Ag can act as sites for hydrogen interaction, facilitating the accumulation of protons from OOH* intermediates and the efficient release of O2 molecules. This hydrogen abstraction from OOH* and the creation of new active sites consequently reduce the system's overpotential. The OER process with Ag–CL/NiP achieved a low overpotential of 401 mV at 50 mA cm−2, signifying the superior performance of this panel in comparison to both Ag–CC/NiP and NiP panels. The presence of the carbon channel and the heteroatom Ag serves as an electron transport facilitator, expediting ion transport and leading to a significant reduction in the Rct value, which measured at 491.3 Ω.
Photocatalytic water splitting performance
The photocatalytic performance of the developed panel was assessed by exposing a 2 cm2 area of the panel, as mentioned in the experimental section. Over a 4-hour irradiation period, the Ag–CL/NiP panel demonstrated the ability to generate 17.5 mmol cm−2 of H2 (Fig. 6(c)), while Ag–CC/NiP yielded only 1.2 mmol cm−2 of H2. This superior photocatalytic efficiency can be attributed to the very low interfacial charge transfer resistance and the higher double layer capacitance resulting from the presence of the highly conductive Ag–NiP covered carbon channels on the surface of NiP. | ||
| Fig. 6 Photocurrent measurement: (a) single step of CA and (b) multistep of CA. (c) Comparison of photocatalytic water splitting analysis of Ag–CL/NiP and Ag–CC/NiP. (d) Temperature dependence of photocatalytic water splitting performance of Ag–CL/NiP. (e) Reusability analysis of Ag–CL/NiP. | ||
To validate this observation, a photocurrent measurement was conducted using chronoamperometric analysis at 1.37 V vs. RHE, utilizing light with a wavelength exceeding 420 nm. Fig. 6(a and b) illustrates that the Ag–CL/NiP panel consistently generated a high current density of ∼9.5 mA cm−2, whether in a single-step or multi-step procedure, when exposed to light at 420 nm and 1.37 V vs. RHE. In similar analysis conditions, Ag–CC/NiP panel only managed to produce a current density of 0.43 mA cm−2. According to Marwat et al.,62 the benchmark materials Sb2Se3 and TiO2−NR/CdS–NiOx86 achieved a photocurrent density of 30 mA cm−2 at a high potential of 1.0 V vs. Ag/AgCl. Here, the Ag–CL/NiP panel only needs a potential of 0.35 V vs. Ag/AgCl to provide a photocurrent of ∼9.5 mA cm−2.
These findings indicate that, under light irradiation, a substantial number of electron–hole pairs are generated and efficiently transported to the surface sites due to the exceptional conductivity of Ag–CL/NiP. In contrast, the significantly lower current observed for Ag–CC/NiP underscores the crucial role played by the Ag–NiP covered carbon channels. Moreover, the method of deposition and the use of unprocessed lemon juice as a carbon source are identified as the primary contributing factors to these exceptional qualities in the Ag–CL/NiP panel.
The temperature-dependent photocatalytic activity of the Ag–CL/NiP panel was investigated under natural sunlight, with temperatures ranging from approximately 0 °C. The resulting hydrogen evolution was quantified to elucidate the effects of temperature on the catalytic process, and the results are shown in Fig. 6(d). Notably, gas production from water splitting with this system becomes feasible only above a critical temperature threshold of ∼12 °C, where significant gas evolution initiates. At this temperature, the water splitting process was carried out for 4 hours, yielding a gas production rate of 10.1 mmol cm−2. However, under natural sunlight, without temperature control, the system generated 17.1 mmol cm−2 of gas after 4 hours, with the solution temperature varying between ∼33 °C and ∼44 °C throughout the experiment. The temperature fluctuations over time are depicted in Fig. S8 of ESI.† These results demonstrated that when temperature was controlled to ∼12 °C, there was ∼40% decrement in hydrogen evolution performance which underlines the material's catalytic potential in water splitting under low temperature. The apparent quantum yield was decreased from 1.8 × 10−2 % to 1.04 × 10−2 % when the temperature was reduced from RT to ∼12 °C, as shown in the ESI.†
The assessment of reusability demonstrated that the catalytic performance of the Ag–CL/NiP panel remains nearly unchanged even after undergoing five cycles of continuous water splitting, and the results are shown in Fig. 6(e). Any slight variations in activity can be attributed to fluctuations in solar intensity. Importantly, these panels can be easily reused without any additional treatment. This reusability feature makes these panels readily suitable for integration into industrial water splitting processes. The results of the photocatalytic water splitting performance obtained in this study were compared to those of notable systems reported in the literature, as shown in Table S2 of ESI.† These comparisons reveal that the current system exhibits superior photocatalytic efficiency.
Electron transfer mechanism in photocatalysis
A plausible electron transfer mechanism in Ag–CL/NiP is demonstrated with the SPR effect of noble Ag nanoparticles. Besides the SPR effect of Ag, a Schottky barrier and synergism are developed between Ag and NiP on the green carbon channels. The UV-visible diffuse reflectance spectra shown in Fig. 7(a) clearly demonstrate that the synthesized Ag–CL and Ag–CC powder particles have characteristic absorption in the UV-visible region. It is also evident that the synthesized NiP exhibited a characteristic absorption in the UV-visible region; however, in the Ag–CL/NiP, the intensity of the absorption band in the UV-visible region significantly increased due to Ag–CL powder incorporation. These results imply that Ag–CL/NiP is able to absorb in the entire UV-visible region of the spectra. Moreover, XPS analysis provided insights into the valence band of the developed panels, as depicted in Fig. 7(b). The valence band for NiP was 1.6 eV, while for Ag–CL/NiP it was 1.2 eV. A significant reduction in the valence band position was observed in the case of Ag–CL/NiP. This shift can be attributed to the effective overlap of Ni and Ag orbitals facilitated by the carbon channels derived from lemon juice, as shown in Fig. 7(b). This observation can be attributed to the synergistic interaction between Ni and Ag.87 | ||
| Fig. 7 (a) UV-visible diffuse reflectance spectra of NiP, Ag–CC/NiP, Ag–CL/NiP, Ag–CC powder and Ag–CL powder. (b) Valence band analysis using XPS. (c) Mott–Schottky plot used to calculate the conduction band. | ||
The determination of the conduction band position for the panels was carried out through Mott–Schottky analysis under dark conditions at 1 kHz (Fig. 7(c)). Specifically, for NiP, the conduction band was identified at −0.43 eV, while for Ag–CL/NiP, it was positioned at −0.5 eV. This shift is attributed to the Fermi level equilibration between Ag and NiP, facilitating a smooth electron transfer from Ag to NiP.88 The calculated band gap values for Ag–CL/NiP and NiP were found to be 1.7 eV and 2.03 eV, respectively. These findings indicate a substantial reduction in the bandgap upon incorporating the Ag–CL powder into the NiP panel. This decrease can be attributed to the robust overlap between the orbitals of Ni and Ag. As reported by XPS data, the binding energy of Ni 2p3/2 shifted to the positive region by 0.6 eV, while the binding energy of Ag shifted to the negative region by 0.8 eV. These results point to the migration of electrons from Ni to Ag alongside the migration of electrons from nickel to phosphorus indicating a synergic effect between Ni and Ag. A plausible mechanism for Ag–CL/NiP is depicted in Fig. 8.
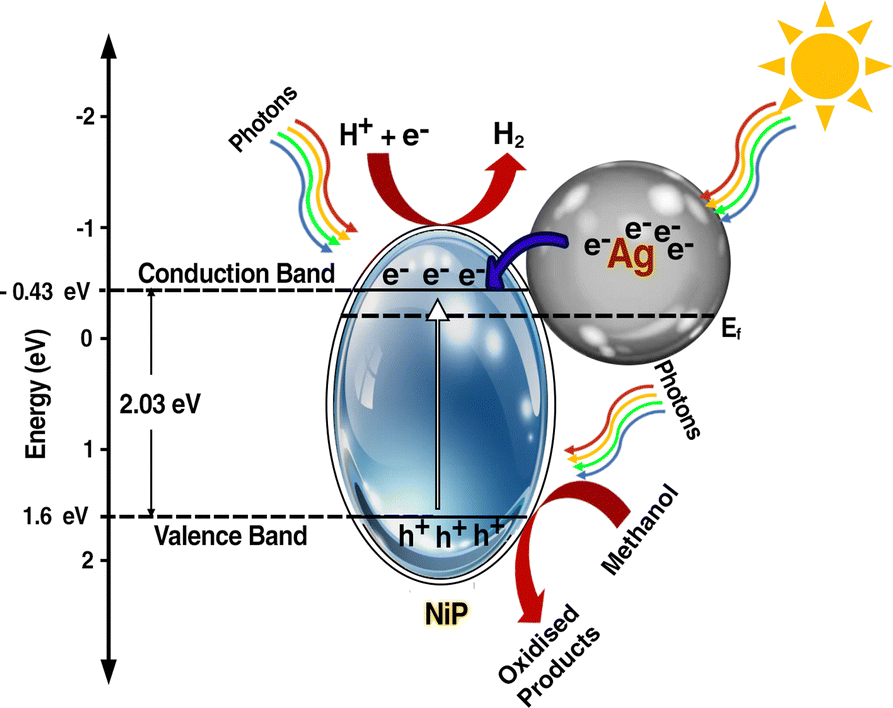 | ||
| Fig. 8 Plausible photocatalytic water splitting mechanism of Ag–CL/Ni–P. | ||
Under the irradiation of sunlight, electron excitation takes place simultaneously in both electron rich Ag and in NiP in Ag–CL/NiP. Electron shuttling occurs through the conductive carbon channels. Due to the SPR effect of Ag, the electron–hole generation is enhanced. The photogenerated electron migrates from the CB of Ag to the CB of NiP through the green carbon channel which also facilitates the smooth electron transfer process. Ag forms a Schottky barrier at the interface, which prevents the electron and hole recombination by generating an energy barrier that the electrons must overcome to recombine with holes, thus prolonging the lifetime of charge carriers. However, hydrogen reduction cannot occur at the surface of Ag due to the weak bond strength between Ag and H (Ag–H).87 Therefore, at the CB position of Ni–P, H+ ions are reduced to generate H2 gas. The holes are gathered on the VB of NiP and oxidize the methanol to useful organic compounds.
Conclusions
Ag–NiP deposited green carbon channel embedded NiP panels were developed using lemon juice extract and precise execution of electroless NiP coating for sustainable electro- and photocatalytic water splitting processes. The lemon juice-assisted methodology yielded a compact Ag–NiP, with green carbon channels on the electrode surface. The Ag–CL/NiP displayed reversible OER kinetics with a low overpotential of 401 mV to achieve a current density of 50 mA cm−2. It displayed a Tafel slope of 46.5 mV dec−1 and followed the dual-site adsorbate evolution mechanism. The Ag–CL/NiP achieved a photocurrent density of ∼9.5 mA cm−2 at a lower voltage of 0.35 V vs. Ag/AgCl when subjected to light irradiation with a wavelength >420 nm. Under 4 hours of sunlight irradiation, the Ag–CL/NiP panel achieved a hydrogen production rate of 17.5 mmol cm−2, demonstrating its impressive capacity for solar-driven H2 generation. Temperature-controlled photocatalytic water splitting was conducted and the system exhibited a reasonable performance (∼40% lower compared to that under normal sunlight conditions) at a lower temperature of 12 °C. These easily reusable high-performance Ag–CL/NiP panels are promising for industrial water splitting processes for carbon-neutral hydrogen production.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. All authors contributed equally.Data availability
The data that support the finding of this study are available from the corresponding author, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge DST, New Delhi, for the facility support under DST-FIST programme in St. John's College, Anchal, Kerala, India. R. B. N, A. A. K and S. H. thank the University of Kerala for research fellowships. S. R. and V. C. thank UGC for research fellowships. We gratefully acknowledge CLIF, Kerala University, STIC Kochi and Material Characterization FESEM Lab, Bannari Amman Institute of Technology for providing material analysis data.References
- B. Tam, O. Babacan, A. Kafizas and J. Nelson, Energy Environ. Sci., 2024, 17, 1677–1694 RSC.
- L. Zhao, Z. Lv, Y. Shi, S. Zhou, Y. Liu, J. Han, Q. Zhang, J. Lai and L. Wang, Energy Environ. Sci., 2024, 17, 770–779 RSC.
- E. Boivin, S. Saitzek, F. Fauth, M. Huvé, P. Roussel and H. Kabbour, Chem. Mater., 2022, 35, 447–456 CrossRef.
- C. Avcıoǧlu, S. Avcıoǧlu, M. F. Bekheet and A. Gurlo, ACS Appl. Energy Mater., 2023, 6, 1134–1154 CrossRef.
- H. Jiao, C. Wang, L. Xiong and J. Tang, Acc. Mater. Res., 2022, 3, 1206–1219 CrossRef CAS PubMed.
- R. V. Digraskar, B. B. Mulik, P. S. Walke, A. V. Ghule and B. R. Sathe, Appl. Surf. Sci., 2017, 412, 475–481 CrossRef CAS.
- H. Gunaseelan, A. V. Munde, R. Patel and B. R. Sathe, Mater. Today Sustainability, 2023, 22, 100371 CrossRef.
- X. Yan, M. Xia, H. Liu, B. Zhang, C. Chang, L. Wang and G. Yang, Nat. Commun., 2023, 14, 1–11 Search PubMed.
- Y. Zhao, W. Xue, W. Sun, H. Chen, X. Li, X. Zu, S. Li and X. Xiang, Int. J. Hydrogen Energy, 2023, 48, 31161–31171 CrossRef CAS.
- Y. Xiao, Z. Wang, L. Li, Q. Gu, M. Xu, L. Zhu and X. Fu, Int. J. Hydrogen Energy, 2023, 48, 15460–15472 CrossRef CAS.
- G. Jia, M. Sun, Y. Wang, X. Cui, B. Huang and J. C. Yu, Adv. Funct. Mater., 2023, 33, 2212051 CrossRef CAS.
- A. Chouat, D. T. Nguyen, S. Mohan and T. O. Do, ACS Appl. Nano Mater., 2022, 5, 13078–13089 CrossRef CAS.
- W. Ma, D. Zheng, B. Xiao, Y. Xian, Q. Zhang, S. Wang, J. Liu, P. Wang and X. Hu, J. Environ. Chem. Eng., 2022, 10, 107822 CrossRef CAS.
- Y. Wu, Y. Li, L. Zhang and Z. Jin, ChemCatChem, 2022, 14, e202101656 CrossRef CAS.
- L. Wei, M. A. S. Adamson and J. Vela, ChemNanoMat, 2020, 6, 1179–1185 CrossRef CAS.
- F. Li, C. Liu, H. Lin, Y. Sun, H. Yu, S. Xue, J. Cao, X. Jia and S. Chen, J. Colloid Interface Sci., 2023, 640, 329–337 CrossRef CAS PubMed.
- M. Wang, L. Zhou, M. Zhang, W. Song, W. Zhong, X. Wang and Y. Tang, ChemistrySelect, 2022, 7, e202202462 CrossRef CAS.
- J. Nie, J. Shi, T. Huang, M. Y. Xie, Z. Y. Ouyang, M. H. Xian, G. F. Huang, H. Wan, W. Hu and W. Q. Huang, Adv. Funct. Mater., 2024, 34, 2314172 CrossRef CAS.
- T. N. Q. Trang, T. B. Phan, N. D. Nam and V. T. H. Thu, ACS Appl. Mater. Interfaces, 2020, 12, 12195–12206 CrossRef CAS.
- L. Li, Q. Zhang, X. Wang, J. Zhang, H. Gu and W. L. Dai, J. Phys. Chem. C, 2021, 125, 10964–10973 CrossRef CAS.
- D. Gelija, C. Loka, M. Goddati, N. H. Bak, J. Lee and M. D. Kim, ACS Appl. Mater. Interfaces, 2023, 15, 34883–34894 CrossRef CAS.
- H. Qiu, A. Yamamoto and H. Yoshida, ACS Catal., 2023, 13, 3618–3626 CrossRef CAS.
- F. Naaz and T. Ahmad, Langmuir, 2023, 39, 9300–9314 CrossRef CAS.
- R. K. Sharma, S. Yadav, S. Dutta, H. B. Kale, I. R. Warkad, R. Zboril, R. S. Varma and M. B. Gawande, Chem. Soc. Rev., 2021, 50, 11293–11380 RSC.
- D. Li, Y. Qin, J. Liu, H. Zhao, Z. Sun, G. Chen, D. Y. Wu, Y. Su, S. Ding and C. Xiao, Adv. Funct. Mater., 2022, 32, 2107056 CrossRef CAS.
- H. Zhang, H. Tang, Q. Weng, Q. Wei, M. Duan, X. Bo, F. Fu and L. Zan, J. Solid State Chem., 2022, 316, 123556 CrossRef CAS.
- F. Wang, Y. Zhang, J. Zhang, W. Yuan, Y. Li, J. Mao, C. Liu, C. Chen, H. Liu and S. Zheng, ACS Sustainable Chem. Eng., 2022, 10, 5976–5985 CrossRef CAS.
- T. Xu, J. Wang, M. Wang, Y. Xue, J. Liu, N. Cai, W. Chen, F. Huang, X. Li and F. Yu, New J. Chem., 2021, 45, 13286–13292 RSC.
- Y. Hu, Y. Wu, J. Feng, H. Huang, C. Zhang, Q. Qian, T. Fang, J. Xu, P. Wang, Z. Li and Z. Zou, J. Mater. Chem. A, 2018, 6, 2568–2576 RSC.
- A. T. Hoang, A. Pandey, W. H. Chen, S. F. Ahmed, S. Nižetić, K. H. Ng, Z. Said, X. Q. Duong, Ü. Ağbulut, H. Hadiyanto and X. P. Nguyen, ACS Sustainable Chem. Eng., 2023, 11, 1221–1252 CrossRef CAS.
- S. S. Narwade, B. B. Mulik, S. M. Mali and B. R. Sathe, Appl. Surf. Sci., 2017, 396, 939–944 CrossRef CAS.
- R. V. Digraskar, V. S. Sapner, S. M. Mali, S. S. Narwade, A. V. Ghule and B. R. Sathe, ACS Omega, 2019, 4, 7650–7657 CrossRef CAS.
- S. S. Narwade, S. M. Mali, V. S. Sapner and B. R. Sathe, ACS Appl. Nano Mater., 2020, 3, 12288–12296 CrossRef CAS.
- B. Li, Z. Tian, L. Li, Y. H. Wang, Y. Si, H. Wan, J. Shi, G. F. Huang, W. Hu, A. Pan and W. Q. Huang, ACS Nano, 2023, 17, 3465–3482 CrossRef CAS PubMed.
- W. He, R. Zhang, H. Liu, Q. Hao, Y. Li, X. Zheng, C. Liu, J. Zhang and H. L. Xin, Small, 2023, 19, 2301610 CrossRef CAS PubMed.
- S. Lee, M. R. Ashwin Kishore, D. Kim, H. Kang, J. Chun, L. S. Oh, J. H. Park, H. J. Kim, J. S. Yoo and E. Lim, ACS Appl. Energy Mater., 2022, 5, 14658–14668 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- X. Liu, W. Yang, L. Chen, Z. Liu, L. Long, S. Wang, C. Liu, S. Dong and J. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 4463–4472 CrossRef CAS.
- R. Shen, J. Xie, Y. Ding, S. Y. Liu, A. Adamski, X. Chen and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 3243–3250 CrossRef CAS.
- S. Mousavi-Salehi, S. Keshipour and F. Ahour, J. Phys. Chem. Solids, 2023, 176, 111239 CrossRef CAS.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., 2014, 126, 4461–4465 CrossRef.
- B. R. Sathe, X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 RSC.
- S. S. Narwade, S. M. Mali, R. V. Digraskar, V. S. Sapner and B. R. Sathe, Int. J. Hydrogen Energy, 2019, 44, 27001–27009 CrossRef CAS.
- V. S. Sapner, P. P. Chavan, A. V. Munde, U. S. Sayyad and B. R. Sathe, Energy Fuels, 2021, 35, 6823–6834 CrossRef CAS.
- Y. Li, Y. Wang, C. L. Dong, Y. C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huangfu, D. Zhao, L. Gu and S. Shen, Chem. Sci., 2021, 12, 3633–3643 RSC.
- L. Liu, S. Du, X. Guo, Y. Xiao, Z. Yin, N. Yang, Y. Bao, X. Zhu, S. Jin, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2022, 144, 2747–2754 CrossRef CAS PubMed.
- Y. Liu and Z. Xiang, ACS Appl. Mater. Interfaces, 2019, 11, 41313–41320 CrossRef CAS PubMed.
- G. Wang and Z. Jin, ChemistrySelect, 2019, 4, 3602–3610 CrossRef CAS.
- L. Lu, Q. Li, R. Zhang, J. Jiang, S. Zhang, J. Guo, P. Cheng and W. Shi, ACS Appl. Energy Mater., 2023, 6, 8072–8080 CrossRef CAS.
- F. Yang, S. Huang, B. Zhang, L. Hou, Y. Ding, W. Bao, C. Xu, W. Yang and Y. Li, Nanomaterials, 2019, 9, 1022 CrossRef CAS PubMed.
- S. Shanmugam, A. Sivanantham, M. Matsunaga, U. Simon and T. Osaka, Electrochim. Acta, 2019, 297, 749–754 CrossRef CAS.
- S. Sirisomboonchai, S. Li, A. Yoshida, S. Kongparakul, C. Samart, Y. Kansha, X. Hao, A. Abudula and G. Guan, Catal. Sci. Technol., 2019, 9, 4651–4658 RSC.
- J. R. N. dos Santos, I. C. B. Alves, A. L. B. Marques and E. P. Marques, Electrocatalysis, 2022, 13, 713–730 CrossRef CAS.
- S. S. Yu, T. H. Lee and T. H. Oh, Fuel, 2022, 315, 123151 CrossRef CAS.
- M. Mahiuddin and B. Ochiai, ACS Omega, 2022, 7, 35626–35634 CrossRef CAS PubMed.
- B. T. Hoan, P. D. Tam and V. H. Pham, J. Nanotechnol., 2019, 2019, 2852816 Search PubMed.
- A. Tadesse, M. Hagos, D. Ramadevi, K. Basavaiah and N. Belachew, ACS Omega, 2020, 5, 3889–3898 CrossRef CAS PubMed.
- T. K. Mondal, A. Gupta, B. K. Shaw, S. Mondal, U. K. Ghorai and S. K. Saha, RSC Adv., 2016, 6, 59927–59934 RSC.
- E. M. Schneider, A. Bärtsch, W. J. Stark and R. N. Grass, J. Chem. Educ., 2019, 96, 540–545 CrossRef CAS.
- F. Ortega, V. B. Arce and M. A. Garcia, Carbohydr. Polym., 2021, 252, 117208 CrossRef CAS PubMed.
- M. A. Marwat, M. Humayun, M. W. Afridi, H. Zhang, M. R. Abdul Karim, M. Ashtar, M. Usman, S. Waqar, H. Ullah, C. Wang and W. Luo, ACS Appl. Energy Mater., 2021, 4, 12007–12031 CrossRef CAS.
- A. Anantha Krishnan, A. K. Aneesh, R. B. Nair, R. Sivaraj, A. Lamiya, P. K. Jishnu, S. Kurian, T. Mathew, M. Ameen Sha and P. S. Arun, New J. Chem., 2022, 46, 22256–22267 RSC.
- S. S. Mani, S. Rajendran, N. Nalajala, T. Mathew and C. S. Gopinath, Energy Technol., 2022, 10, 1–12 Search PubMed.
- D. Ahmadkhaniha, F. Eriksson and C. Zanella, Coatings, 2020, 10, 1179 CrossRef CAS.
- Y. Zhang, X. Hou, X. Li, D. Li, F. Huang and Q. Wei, J. Colloid Interface Sci., 2020, 578, 805–813 CrossRef CAS PubMed.
- X. Cheng, J. Zheng, J. Li and X. Luo, ChemistrySelect, 2019, 4, 4271–4277 CrossRef CAS.
- P. Bazant, I. Kuritka, L. Munster and L. Kalina, Cellulose, 2015, 22, 1275–1293 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC.
- Y. Wei, Z. Han, T. Liu, X. Ding and Y. Gao, Chem. Commun., 2023, 59, 11572–11575 RSC.
- P. Cördoba-Torres, T. J. Mesquita and R. P. Nogueira, J. Phys. Chem. C, 2015, 119, 4136–4147 CrossRef.
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem., 2021, 133, 23235–23251 CrossRef.
- M. Mathankumar, S. Anantharaj, A. K. Nandakumar, S. Kundu and B. Subramanian, J. Mater. Chem. A, 2017, 5, 23053–23066 RSC.
- M. Ledendecker, S. Krickcalderõn, C. Papp, H. P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- Y.-H. Wang, L. Li, J. Shi, M.-Y. Xie, J. Nie, G.-F. Huang, B. Li, W. Hu, A. Pan, W.-Q. Huang, Y.-H. Wang, L. Li, J. Shi, M.-Y. Xie, J. Nie, G.-F. Huang, B. Li, W.-Q. Huang, W. Hu and A. Pan, Adv. Sci., 2023, 10, 2303321 CrossRef CAS PubMed.
- F. Song, M. M. Busch, B. Lassalle-Kaiser, C. S. Hsu, E. Petkucheva, M. Bensimon, H. M. Chen, C. Corminboeuf and X. Hu, ACS Cent. Sci., 2019, 5, 558–568 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- L. Yang, Y. Wu, F. Wu, Y. Zhao, Z. Zhuo, Z. Wang, X. Li, Y. Luo and J. Jiang, J. Mater. Chem. A, 2020, 8, 20946–20952 RSC.
- P. Chandrasekharan Meenu, P. K. Samanta, T. Yoshida, N. J. English, S. P. Datta, S. A. Singh, S. Dinda, C. Chakraborty and S. Roy, ACS Appl. Energy Mater., 2022, 5, 503–515 CrossRef CAS.
- G. F. Chen, T. Y. Ma, Z. Q. Liu, N. Li, Y. Z. Su, K. Davey and S. Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314–3323 CrossRef CAS.
- A. A. Krishnan, H. Sreehari, M. A. A. Kumar, R. B. Nair, S. Kurian, M. A. Sha and P. S. Arun, Catal. Sci. Technol., 2024 10.1039/D4CY00627E.
- S. Khatun, S. Pal and P. Roy, J. Alloys Compd., 2024, 977, 173393 CrossRef CAS.
- K. Srinivas, Y. Chen, B. Wang, B. Yu, Y. Lu, Z. Su, W. Zhang and D. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 55782–55794 CrossRef CAS PubMed.
- C. Huang, J. Zhou, D. Duan, Q. Zhou, J. Wang, B. Peng, L. Yu and Y. Yu, Chin. J. Catal., 2022, 43, 2091–2110 CrossRef CAS.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 11(1), 63–72 CrossRef.
- I. Han Yoo, S. S. Kalanur and H. Seo, Appl. Catal., B, 2019, 250, 200–212 CrossRef.
- D. Gao, W. Liu, Y. Xu, P. Wang, J. Fan and H. Yu, Appl. Catal., B, 2020, 260, 118190 CrossRef CAS.
- R. Purbia and S. Paria, Dalton Trans., 2017, 46, 890–898 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00463a |
| This journal is © The Royal Society of Chemistry 2024 |