
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A lot to unpack: a decade in high Z′ crystal structures†
Paul G.
Waddell
 *
*
School of Natural and Environmental Sciences, Newcastle University, Bedson Building, Newcastle upon Tyne, NE1 7RU, UK. E-mail: paul.waddell@ncl.ac.uk
First published on 13th January 2025
Abstract
Crystal structures that form with more than one molecule in the asymmetric unit (Z′ > 1) are a fascinating and important, if overlooked, aspect of crystal engineering. With the recent publication of the results of the ‘seventh blind test of crystal structure prediction’ the challenges that these structures present and the questions they provoke for the prediction and design of crystalline solids are brought sharply into focus. This article documents developments in the study of high Z′ structures over the last ten years and shines a spotlight on the most extreme and intriguing examples from recent publications. The lessons learned from these studies will inform future crystal engineering and design efforts as strides are made to work around the computational expense inherent in the prediction of structures with large asymmetric units.
 Paul Waddell | Paul Waddell obtained his PhD from the University of St Andrews under the supervision of Alex Slawin and Derek Woollins. He then worked as a post-doctoral research associate with Jacqui Cole at the University of Cambridge, firstly as part of an NSERC Discovery grant with the University of New Brunswick and latterly in conjunction with the Australian Nuclear Science and Technology Organisation (ANSTO). Since 2014 he has been responsible for the chemical crystallography service at Newcastle University. Their research interests include polymorphism, phase transitions and supramolecular chemistry. He was recently involved in developing RODIN with the Cambridge Crystallographic Database Centre, providing access to raw diffraction data for education and outreach. In 2024 his work on high Z′ structures won the CrystEngComm prize at the British Crystallographic Association spring meeting. |
Introduction
Homomolecular crystal structures that crystallise with more than one molecule in the asymmetric unit (Z′ > 1) have been puzzling crystallographers since the advent of crystal structure determination and instances of high values of Z′ continue to perplex and astound to this day.1 Structures where Z′ > 1 account for ca. 10% of entries in the Cambridge Crystallographic Database2 (CSD, version 5.45, Update 2, Jun 2024) and, as a general trend, the more molecules in the asymmetric unit, the scarcer the examples of structures become (Table 1), though even values of Z′ are more common than odd.3 Instances where the value of Z′ is extremely high (Z′ ≥ 12) are a true crystallographic rarity, with only 129 such structures reported in the CSD.‡| Z′ value | Number of hits |
|---|---|
| >1 | 134![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 084 084 |
| ≥4 | 9289 |
| ≥8 | 630 |
| ≥12 | 129 |
| ≥16 | 53 |
| ≥20 | 16 |
This phenomenon has consistently proven to be hard to anticipate and there are strong indications that such structures can inform our understanding of the processes of nucleation and crystal growth.4,5 As the rational design of crystalline materials requires control over various aspects of supramolecular chemistry there is much to be learned from structures of this kind rather than their being mere curiosities.
Indeed, the study of high Z′ structures goes hand in hand with polymorphism and all that that means to a great many fields in chemistry and that of pharmaceuticals in particular, where the financial ramifications can be significant.
Central to the formation of these structures are the concepts of ‘frustration’, where intermolecular interactions of similar energies are in competition, and ‘awkwardness’, molecules with shapes that preclude ordered packing.6 The propensity of structures with high Z′ values to exhibit approximate symmetry adds another layer of complexity to the problem.7 Most high Z′ structures can be rationalised in terms of translational modulations, approximate symmetry consistent with the crystallographic symmetry or confined to 2D layers or 1D rods, or a combination of both.
As specific molecular and structural features correlate with instances of high Z′, it is an aspect of crystal engineering that is hard to avoid and needs to be considered and embraced as part of the design process. The ability to predict instances of high Z′ and approximate symmetry will surely lead to better design protocols.
At this point in time, our ability to predict structures with multiple molecules in the asymmetric unit is still somewhat limited. In the latest iteration of the blind test for crystal structure prediction, the largest Z′ value of any of the structures in the test was 3.8,9 In this case, only six of the twenty eight groups involved in the test predicted structures with Z′ = 3. This is mostly due to a convention within crystal structure prediction (CSP) as considering structures with Z′ > 2 is computationally expensive. There have been examples where structures with Z′ = 4 have been successfully predicted10 but in general, high Z′ structures still represent a challenge for CSP algorithms. Without the ability to address this, it is possible that a great many potential polymorphs and metastable structures may be overlooked.11
In this work, recent advances in the field of high Z′ structures are detailed in the context of their potential impact on crystal engineering, solid-state design and CSP. Examples of homomolecular crystal structures with Z′ ≥ 12 published since 2018 are reviewed and their true frequency in recent literature assessed. The nature of the relationship between CSP and structures with Z′ > 2 is also expanded upon with some commentary on what can be done to ensure structures of this kind are taken into account in the future.
Recent advances
A comprehensive review of structures with high Z′ values was published in 2015 by the Steeds and has proven to have been a truly seminal work on the topic.12 In the following ten years, further strides have been made in terms of our understanding of the formation and frequency of these structures and it may behove us to highlight a few of these in this article. Details of all structures referred to are available in the ESI.†In the time since the Steeds' review there have been a number of surveys of high Z′ structures that have highlighted the structural features that are prevalent in instances of high Z′ and can potentially be exploited to improve the probability of producing structures of this kind. In particular, the work of Brock in identifying the ‘organising principles’ of organic crystal structures with high Z′ has been pivotal.3 Here, a survey of a curated list of organic crystal structures in the CSD that exhibit asymmetric units comprising 5 or more molecules, revealed a series of structural features that are common among this type of structure. In terms of the molecular species involved, somewhat counterintuitively, it was found that there is often negligible conformational variation between the symmetry independent molecules in the asymmetric unit. In terms of their supramolecular chemistry, the structures were commonly found to crystallise as layered structures and strong intermolecular interactions such as classical hydrogen bonds were also frequently observed.
Specific aspects of the symmetry of the structures were also found to be prevalent where a high Z′ is observed as the structures had a tendency to crystallise in Sohncke space groups and quite often with apparent approximate symmetry. An interesting observation was also that the combination of coincident features such as the ones described could lead to doubling or tripling of the value of Z′ and such was often the case where extreme values were observed. This may also account for the distribution of odd and even values of Z′ in the CSD.
With all these factors being identified as portents of high Z′ structures, it is clear to see how they could be applied to design protocols for structures with Z′ > 1. An example from our own work demonstrates the rational design of a shikimate amide structure with Z′ > 1 (BOHYIK),13 leveraging the features identified by Brock to improve the probability of success.
A similar survey was carried out by Taylor et al. with this work focusing specifically on intermolecular interaction motifs observed in high Z′ organic crystal structures.14 Though analyses of this kind focusing on specific interaction motifs have been previously reported, their work applied a much more comprehensive algorithmic approach to assess the frequency of all such motifs.
The study identified the motifs most likely to occur in both centrosymmetric and non-centrosymmetric structures with Z′ > 1 and that these are the motifs that tend to occur between symmetry-independent molecules. Hydrogen bonds of the type OH⋯O and edge-to-face π-interactions were found to be more prevalent in the centrosymmetric structures, while the non-centrosymmetric structures tended to form ring motifs, weak hydrogen bonds and π⋯π interactions.
Most interestingly, the results of the analysis suggest a causative link between the motifs identified as prevalent and the formation of high Z′ structures. The association between instances of high Z′ and Sohncke space groups, pseudosymmetry and strong intermolecular interactions was also observed, reflecting the ‘organising principles’ outlined by Brock.
The phenomenon of approximate symmetry was specifically highlighted by the observation that there is correlation between ‘inversion favouring’ interactions and non-centrosymmetric structures with Z′ = 2, in which two homomers often mimic an inversion relationship.15 Pinpointing these interactions and identifying the molecules between which they are likely to occur will surely prove incredibly useful when designing or applying CSP to high Z′ structures.
In addition to these surveys, strides have also been made through various systematic studies of specific structures and compounds. A very interesting observation was made by Martins et al. in their analysis of packing polymorphs of an organic benzothiazole.16 Their work compared the structures of three polymorphs of 2-(thiophen-2-yl)-1,3-benzothiazole (CAMBAV02/03 and EGULOK), all with Z′ ≥ 4. These rigid, planar molecules exhibit little variation in conformation, as might be expected, yet are observed to pack very differently. Satisfyingly, when these structures were compared along with a known form with Z′ = 2 (CAMBAV),17 it was found that the value of Z′ increases as the strength of the intermolecular interactions decrease. Given the potential biomedical applications of this compound and the importance of polymorphism in the field of pharmaceuticals, this trend will prove an important consideration and shows that the Z′ value of a structure cannot be ignored in this context.18
It appears however, that when Z′ ≥ 1 polymorphs of more conformationally flexible molecules are observed, the same trend in interaction energies may not apply. Chopra et al. reported a pair of polymorphs of (Z)-2-fluoro-N′-phenyl benzamidamide (SUXLII/01), a molecule with several degrees of rotational freedom, with Z′ values of 2 and 3.19 In this instance, the interaction energies were found to be incredibly close (isoenergetic) and the packing so similar that the two structures could also be described as ‘quasi-isostructural’.
Work on a dipeptide by Otekani et al. is heavily implied to provide an answer as to why high Z′ structures form.20 This revelation stems from their study of Boc-L-methionyl glycine methyl ester (MGP; MARJEY/01/02/03), which was observed to form a structure with Z′ = 8 at 100 K but presented as a structure with Z′ = 4 at 160 K. Both crystallised in the space group P1. Analysis of the structure revealed that dynamic disorder exacerbated by the higher temperature effectively averages the positions of pairs of molecules, thereby reducing the asymmetric unit from 8 to 4 (Fig. 1).
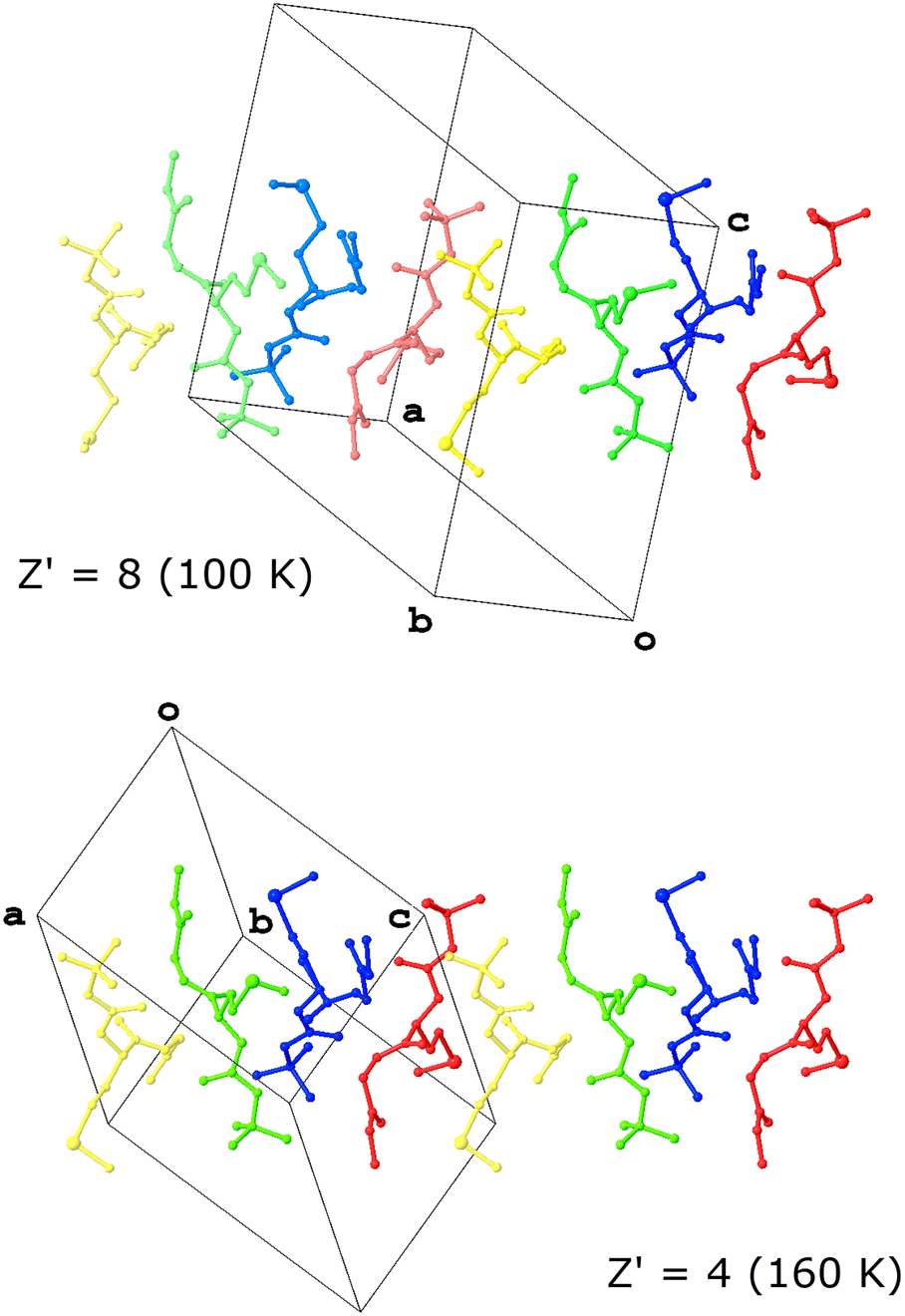 | ||
| Fig. 1 A comparative view of the two high Z′ polymorphs of MGP (CSD Refcodes: MARJEY (top) and MARJEY01 (bottom)). At 160 K, pairs of molecules rendered in the same colour are crystallographically-equivalent and those of similar shades at 100 K correspond to those pairs after the phase transition. | ||
In an effort to understand the kinetic stability of the high Z′ structure observed at low temperature, the authors compared it to a predicted structure with Z′ = 2 generated using two of the conformers observed in the real structure. Interestingly, the lattice energy calculated for the predicted structure was lower than that of the measured structure, potentially putting paid to the notion that a large Z′ is a response to frustration. However, the authors pointed out that the calculation was performed at 0 K and without taking entropy into account, hence suggesting another factor in the formation of high Z′ structures may lie in the entropy term.
Working with a different dipeptide molecule, the group of van Smaalen et al. have suggested a new approach for describing some instances of high Z′.21 Noting that a number of these structures form as a result of translational modulations, they applied the superspace method, more commonly used to describe incommensurately modulated structures, to a case of commensurate modulation. In their work, it was found that the structure of glycyl-L-valine, previously reported as having Z′ = 7 in conventional 3D space (WEVWOK),22 could be considered as Z′ = 1 when described in (3 + 1)-dimensional superspace (WEVWOK01/02). Through this analysis they were able to relate this structure to a new, high temperature phase of glycyl-L-valine and more effectively rationalise the phase transition between the two. Adoption of this rather elegant structural model where translational modulations are manifest, presented alongside the conventional description, literally adds an extra dimension to the analysis of high Z′ structures.
From a crystal engineering perspective, some work has been undertaken to apply the lessons learned from these and earlier studies and use them to design specific systems that exploit the features that promote high Z′ structures. In addition to the shikimate ester study mentioned above,13 which we will not elaborate on further so as not to appear self-indulgent, another example of this is the work of Borbone and Centore et al. into reversible crystal–crystal phase transitions.23 Acknowledging the link between polymorphism and high Z′ structures, the authors designed molecules with strong hydrogen bonding potential, that also promote frustration of one of the hydrogen bond donors. Encouraging frustration in this way creates solid state structures where various packing motifs of similar energies are possible and hence the chance of observing multiple polymorphs is increased.
Indeed, the compound itself was observed in six forms (KOSSIY/01/02/03/04/05), a number of which had Z′ > 1 with the largest value being Z′ = 6. Single-crystal-to-single-crystal (SCSC) transitions were also observed between many of them and, as transitions of this kind are hard to anticipate, this work provides insight into the future design of similar systems with a range of potential applications. Indeed, the authors suggest that CSP may be used to predict sets of polymorph structures with common supercells, thereby identifying systems with potential SCSC transitions.
Some extreme cases
In addition to penning what is widely considered to be the definitive work on the subject of high Z′ structures,12 the Steeds also created a dedicated website, https://zprime.co.uk/, to collate and disseminate information on the topic.24 This included a comprehensive list of all structures in the CSD with Z′ > 4. Unfortunately, this list was last updated in 2018, and so it may be worthwhile to review a little of what has been reported since. A search of the CSD returns 632 structures with Z′ > 4 deposited since 2018. Of course, not all of these will be novel structures and there may be some that either have a misassigned Z′ value or have a large reported Z′ value as the result of being solved in the wrong space group; such structures are listed on https://zprime.co.uk/ but with explanatory notes.There are clearly more structures than can be discussed in this article but, as our own interest was piqued by our work on the structure of methyl shikimate ester with Z′ = 12 (CSD Refcode: BOHYUW),13 this section will detail recent examples of other structures with extreme Z′ values (Z′ ≥ 12). Each structure will be referred to by its CSD Refcode where appropriate.
The https://zprime.co.uk/ website lists three structures with Z′ ≥ 12 from 2018 and the CSD returns 56 further structures deposited since then. As our previous work involved homomolecular crystal structures, our focus will be on those comprising one chemical species and will not concern, co-crystals, salts or polymeric structures. Our definition of co-crystal includes solvates, hydrates and any model in which a solvent mask such as SQUEEZE25 has been applied. Quite often examples of co-crystals have a formal Z′ value lower than that reported. For example, WIYKUO26 has a reported Z′ of 12, however as it is a hemi-hydrate the formal value is 6. These examples are often analysed through the lens of their Z′′ (Z double prime) value, typically interpreted as the number of species in the asymmetric unit,27 though alternative designations such as Zr and Z* may perhaps be more useful.3,28
Kryptoracemates, such as the structure of OTOGOW,29 are also technically co-crystals comprising two enantiomers and hence two different molecules and therefore the formal Z′ is half that reported by convention in the CSD. As OTOGOW has a reported Z′ of 16 but is formally 8 it will not be considered as having an extreme Z′ value.
In addition, redeterminations of known structures with extreme Z′ that are already featured on the https://zprime.co.uk/ website's list, such as LUXYOU03,30 first reported in 2015,31 will not be included. Likewise, structures that were first deposited with the CSD as CSD Communications later to be included in a journal article generating another entry are counted only once. Structures with R1 > 0.075 were omitted to preclude refinements where reliability could not be guaranteed.
With these search parameters in place, there are 15 homomolecular crystal structures with Z′ ≥ 12 with no errors and for which 3D coordinates are available to be considered. As part of this survey, the ADDSYM routine of the program PLATON32 was run for each of these structures to check for potential missed symmetry. It is important to note that structural interpretations may vary and the purpose of this review is not to ‘correct’ any of the structure determinations discussed. There are, however, a few that are somewhat ambiguous and should be regarded as needing further investigation rather than a being simply right or wrong.
Polymorphism studies
A number of the structures with extreme Z′ were reported as part of a study into the polymorphism of a particular compound. This is unsurprising as a concerted exploration of polymorphic space is more likely to turn up this kind of anomaly. In the following examples, there is analysis of the structure in terms of its extreme Z′ value in the original article.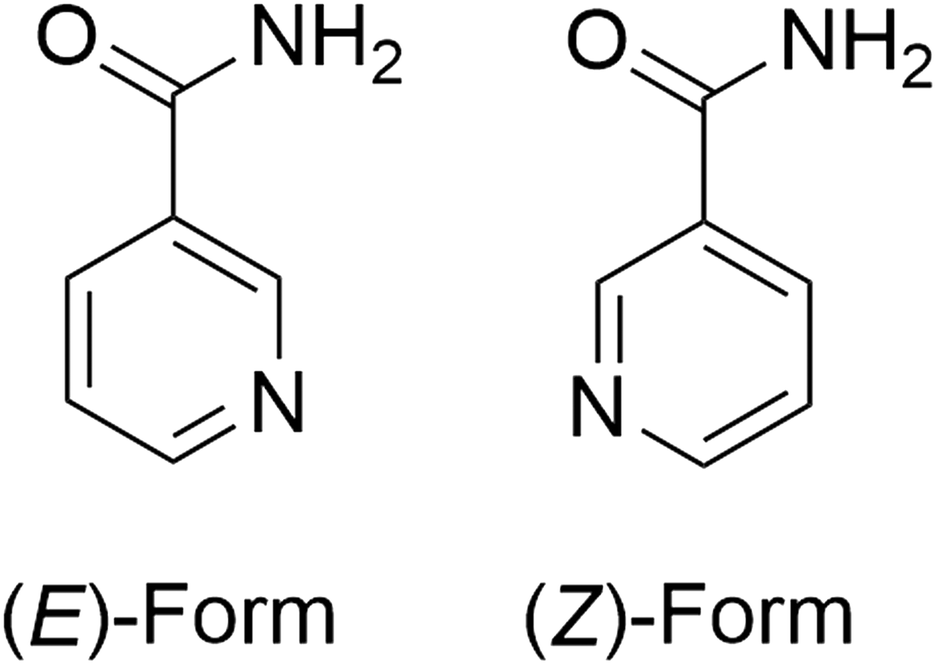 | ||
| Fig. 2 The two conformers of nicotinamide observed in NICOAM12. Cis and trans designations relate to the relationship of the amide nitrogen to the pyridyl nitrogen. | ||
The large Z′ value appears to be the result of a combination of translational modulations, most noticeable in the [001] direction, approximate 2-fold symmetry and the 7:3 ratio of (E)-form to (Z)-form conformers, similar to BOYHUW where there are two distinct conformers but the remainder of the molecule is otherwise rigid.
This example brings into focus the general observation that high Z′ occurs in molecules with strong intermolecular interactions and little conformational flexibility. By way of contrast, if one considers the most polymorphic molecule known, 5-methyl-2-((nitrophenyl)amino)-3-thiophenecarbonitrile (ROY; Refcode family QAXMEH),34 one would think that there would be a greater chance of observing a structure with high Z′ among its 14 polymorphs and yet the highest reported value is Z′ = 2 (QAXMEH57).35 This can be attributed to the variation in the twist angle of this molecule and the lack of strong structure directing hydrogen bonds.
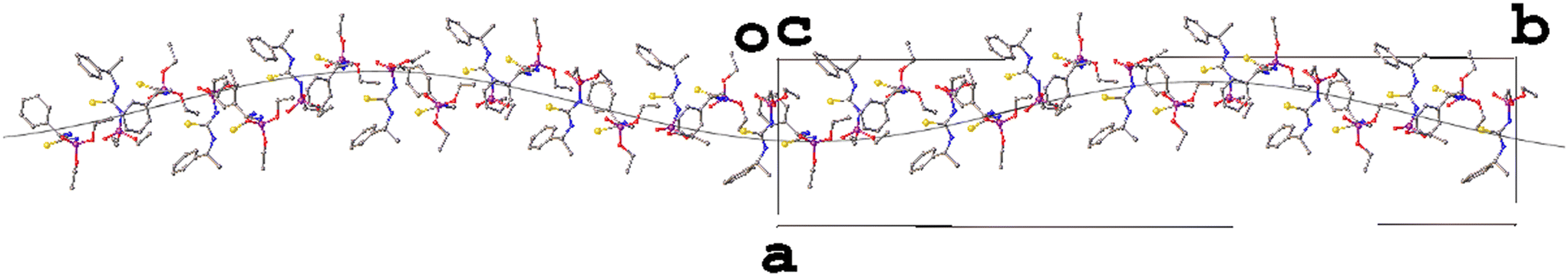 | ||
| Fig. 3 The helical motif in the structure of AZOMAG04 showing the wave-like formation comprised of adjacent asymmetric units in the [010] direction. | ||
The wave-like modulation brings to mind the work van Smaalen21 and a cursory analysis suggests it is possible that by the same superspace approach this structure could be interpreted as Z′ = 4 with a commensurate modulation.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), was found to have Z′ = 12.37 The large asymmetric unit in this case was attributed to competition between lateral chalcogen bonds and pancake-bonding between the 1,2,3-dithiazole rings, the latter of which are perturbed by the presence of methyl groups on the molecule, favouring the former; a classic case of frustration.
), was found to have Z′ = 12.37 The large asymmetric unit in this case was attributed to competition between lateral chalcogen bonds and pancake-bonding between the 1,2,3-dithiazole rings, the latter of which are perturbed by the presence of methyl groups on the molecule, favouring the former; a classic case of frustration.
In addition to being comprised of rigid molecules and exhibiting strong intermolecular interactions, AYADOW01 is an interesting example of multiple factors doubling and tripling the Z′ value to the point where it reaches an extreme value. As is expected for thiazyl rings they form pancake-bonded dimers and, as they are rigid and sterically-hindered by the methyl groups, they cannot stack directly one on top of the other are hence not related by translation symmetry (Z′ = 2). These dimers arrange into hexamers (three dimers) through chalcogen bonding (Fig. 4), which would have approximate 3-fold symmetry but for the orientation of one of the dimers and the perturbations resulting from the positions of the methyl groups (Z′ = (2 × 3) = 6). The competition between the various interdimer contacts in the [011] direction and the awkward shape of the hexamer unit result in an asymmetric unit formed of two hexamers (Z′ = (6 × 2) = 12).
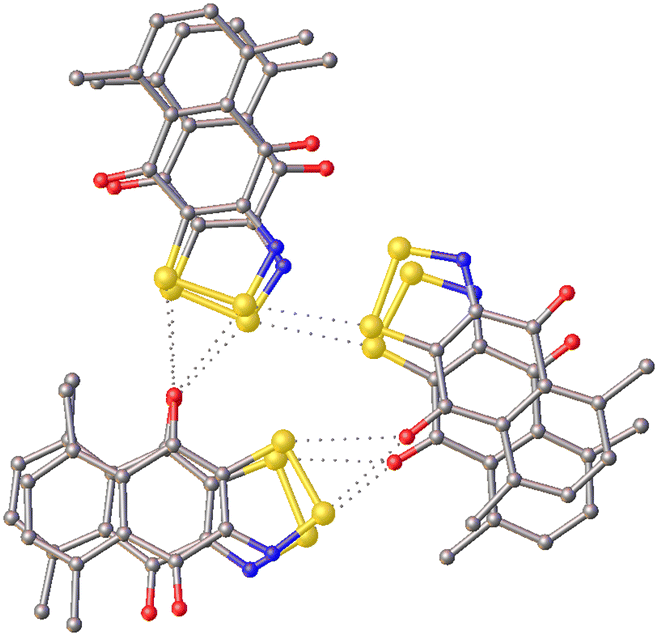 | ||
| Fig. 4 One of the chalcogen and pancake-bonded hexamer units in the structure of AYADOW01. | ||
The article includes data for a structure with Z′ = 16 (DAKCEX08, P) but the authors are wary to classify it as a polymorph in its own right as it appears very similar to form VII (DAKCEX07) of ATPH. In fact, two such structures, forms VIIa (DAKCEX08) and VIIb (DAKCEX09), are included and referred to as being different ‘versions’ of a single, layered structure. Each of these forms produces a similar powder diffractogram and diffuse scattering is observed in the direction of the layers, a common indicator of layer defects.39
Though PLATON was unable to identify any alternative space groups for the form VII versions, an examination of the structures reveals that they are almost identical in terms of their packing (Fig. 5). It seems likely that they are indeed the same structure, but that the diffuse scattering may have made unit cell determination difficult. There do not appear to be rational relationships between the various unit cell parameters so it is hard to confirm whether these structures are truly the same or not without access to the raw diffraction data. Despite this, an assessment of form VIIa with its extreme Z′ value can perhaps be made. As the packing is identical in each structure and forms VII and VIIb have been determined in the space group P21/c, the glide plane and screw axis symmetries must be present in the structure of form VIIa, which was determined in P, and this is indeed the case (Fig. 6).
 | ||
| Fig. 5 The packing in the structures of forms VII (left, DAKCEX07, P21/c, Z′ = 4), VIIa (centre, DAKCEX08, P, Z′ = 16) and VIIb (right, DAKCEX09, P21/c, Z′ = 8) of ATPH. | ||
 | ||
| Fig. 6 Packing diagram of form VIIa of ATPH (DAKCEX08) showing the presence of glide planes (magenta) and screw axes (red). | ||
It would seem that this may have been a case where symmetry was missed, which could be due to the presence of diffuse scattering. The structure DAKCEX08 does constitute one of the hits in the CSD for structures with Z′ ≥ 12 but it should perhaps be considered somewhat tentatively.
) is one of only 4 in the CSD to have a Z′ of 13. In this case the prime number value results from the asymmetric unit comprising 6 dimer units formed by As⋯Br contacts and one lone molecule, which is revealed to be half a dimer modelled across a centre of inversion when the full contents of the unit cell are generated. The structure is very closely related to that of its chloro-analogue (KEFSUM) which also crystallises with a prime number value of Z′: 17, making it unique among structures deposited in the CSD.41 Both structures are characterised by translational modulations and exhibit negligible conformational variation owing to the rigidity of the molecules.
On the other hand, it is possible that this structure may be incommensurately modulated. As the structure of the chloro-analogue was determined to be incommensurately modulated in 2013 (ref. 42) and the structures are so similar, the same could be true of LUBVUC. Determining whether the modulations in this structure are commensurate or incommensurate would require access to the raw data.
The δ-P4 polymorph (LOFRAD/LOFRAD01, P212121) was crystallised serendipitously as a twinned orthorhombic crystal with tetragonal metric symmetry and no evidence of incommensurate modulation in the diffraction pattern. Upon solution, the asymmetric unit was found to contain 29 crystallographically-independent molecules, the largest prime number Z′ value ever observed.
The structure was comparable to that of α-Mn, which has four independent atoms in the asymmetric unit, but with the distribution of P4 tetrahedra exhibiting different degrees of distortion resulting in the 29 molecules observed in the asymmetric unit of δ-P4.
Synthetic studies
Some structures with an extreme Z′ value are reported as part of articles detailing organic syntheses. In these cases, the crystal structure data are often included solely to aid the characterisation of the target molecules and the origins (or often times, existence!) of the large asymmetric unit is tangential and hence not discussed. As a structural analysis of these examples is lacking, they are worth investigating further as part of this review. Five of the high quality, homomolecular crystals structures with Z′ ≥ 12 submitted to the CSD since the last update of https://zprime.co.uk/ fall into this category and are discussed here.Considering both polymorphs, there is very little conformational variation despite the rotatable bonds present and the twist angles between the rings remain essentially the same for all 12 independent molecules of OYACEX01 and the single molecule of OYACEX. The difference between the two structures becomes clear when the packing is considered. Where the molecules in OYACEX directly stack in the [100] direction through π⋯π interactions, the asymmetric unit of OYACEX01 can be defined as forming a helical cluster. Here, three pairs of molecules are related by an approximate three-fold rotation (Fig. 7), perpendicular to the 21 screw axis is the [010] direction.
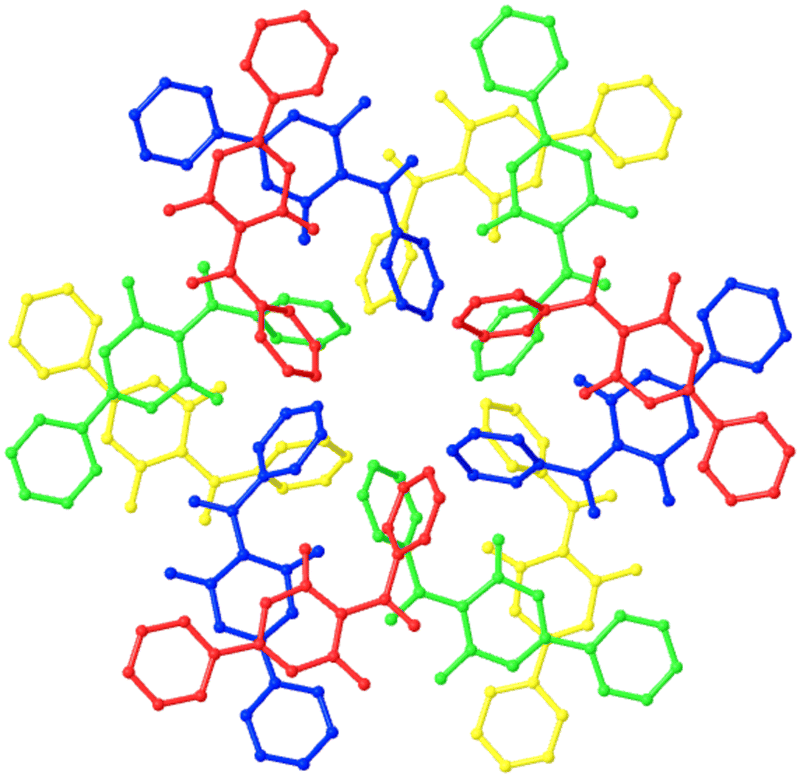 | ||
| Fig. 7 The twelve independent molecules in the asymmetric unit of OYACEX01 arranged to highlight the approximate three-fold axis with each successive group of three molecules along the axis rendered in a different colour. | ||
The structure of OYACEX01, which crystallises in the space group P21/n, is best described as a distorted P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c structure with wave-like layers in the (100) plane. This rather elegantly rationalises the Z′ value of 12. As the approximate cell is a quarter of the P21/n cell, there would be 12 molecules in the Pc cell and as the multiplicity of the general position in the Pc space group is 12, the approximate cell would have Z′ = 1.
c structure with wave-like layers in the (100) plane. This rather elegantly rationalises the Z′ value of 12. As the approximate cell is a quarter of the P21/n cell, there would be 12 molecules in the Pc cell and as the multiplicity of the general position in the Pc space group is 12, the approximate cell would have Z′ = 1.
However, in this case the packing in the two structures is virtually identical with only slight conformational perturbations differentiating the two. Additionally, the unit cell dimensions differ only in the length of one axis (a in EKAXEV and c in EKAXEV01), which is almost exactly four times longer in EKAXEV01.
PLATON highlights ‘pseudo translations’ without suggesting a new unit cell, but without the raw diffraction data it is hard to confirm whether or not these are the same structure. In addition, both datasets were collected at similar temperatures, which would seem to make a phase transition less likely. Either way it seems there is a question mark next to this particular occurrence of extreme Z′.
The molecules in IQEQUT form dimer units comprised of π⋯π interactions which in turn form tetramers through hydrogen bonding, which are arranged in layers of equivalent tetramers coplanar with (100). The hydrogen bonding between these tetramer units seem to perturb the π⋯π interactions within them producing a pronounced translational modulation in the [101] direction. This can be seen most clearly by viewing the three tetramers comprising the asymmetric unit along the [100] direction (Fig. 8).
 | ||
| Fig. 8 The asymmetric unit of IQEQUT viewed down the crystallographic [100] directions. | ||
This view clearly demonstrates the conformationally variation between all three tetramers manifest in the very different shapes they adopt. It is immediately obvious that they cannot be related by crystallographic symmetry and that this is the root of the extreme value of Z′ for this structure.
).49 The origins of the extreme Z′ value are once again evident in the asymmetric unit. In this example, there is an approximate two-fold rotation axis along the [111] direction which relates layers in (10), with the symmetry broken by the orientation of some of the ethoxy groups and phenyl rings. This is not due to any conformational variation, as the molecules do not differ in this regard, but rather their relative positions throughout the crystal structure. These perturbations result in slightly different packing environments for each of the 12 molecules in the asymmetric unit.
).50 The large value of Z′ seems to result from slight perturbations of the long alkyl substituents on this molecule resulting in a translational modulation in the [![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 52] direction. However, the reliability of this determination is brought into question as the refinement does not converge and PLATON suggests a cell a third of the size, which would give Z′ = 4. There is a likelihood that this structure may actually be incommensurately modulated.
52] direction. However, the reliability of this determination is brought into question as the refinement does not converge and PLATON suggests a cell a third of the size, which would give Z′ = 4. There is a likelihood that this structure may actually be incommensurately modulated.
Structures in the space group P1
Of the 15 structures with Z′ ≥ 12 curated for this review, 3 were reported as crystallising in the space group P1. As structures in P1 account for only 1% of data in the CSD, and even though high Z′ structures are more likely to crystallise in Sohncke space groups of which P1 is one, it seems unusual that our, albeit small, subset should boast 20% in this space group. As P1 is distinguished by a complete lack of symmetry within the unit cell any crystal structure can be described in this space group if symmetry other than translation is ignored. For this reason, these structures require special attention when considering the true incidence of extreme Z′ structures in the CSD.The unit cell and space group suggested by PLATON are seen in a second database entry (CIDHAB01),52 which appears to be a redetermination by the same author, possibly using the same data, of the original CIDHAB structure. This structure has Z′ = 1 and seems likely to be in the correct space group. The first entry remains in the CSD however and this and other structures like it artificially inflate the number of high Z′ structures in the CSD.
:1 d.r. diastereoselectivity, its chirality makes its determination in the P1 space group more plausible.
The large asymmetric unit for this structure can be rationalised by considering the intermolecular interactions and packing. The structure forms in layers coplanar to (001) with the molecules arranged in hydrogen bonded tetramers around a central ring motif. The hydrogen bonds within this ring are observed to either form outward, with the bond vector directed away from its parent molecule, or inward, with the bond vector directed across its face. These two orientations result in either edge-to-face or face-to-face π-interactions between the molecules in the tetramer and, ultimately, three different tetramer motifs. Looking at the four tetramers (Fig. 9), it is clear that even the two that exhibit the same motif are significantly different, different enough to not be related by any crystallographic symmetry. It would seem that there is some competition between the different π-interactions and subtle variations in the packing environment around each tetramer favours one over the other.
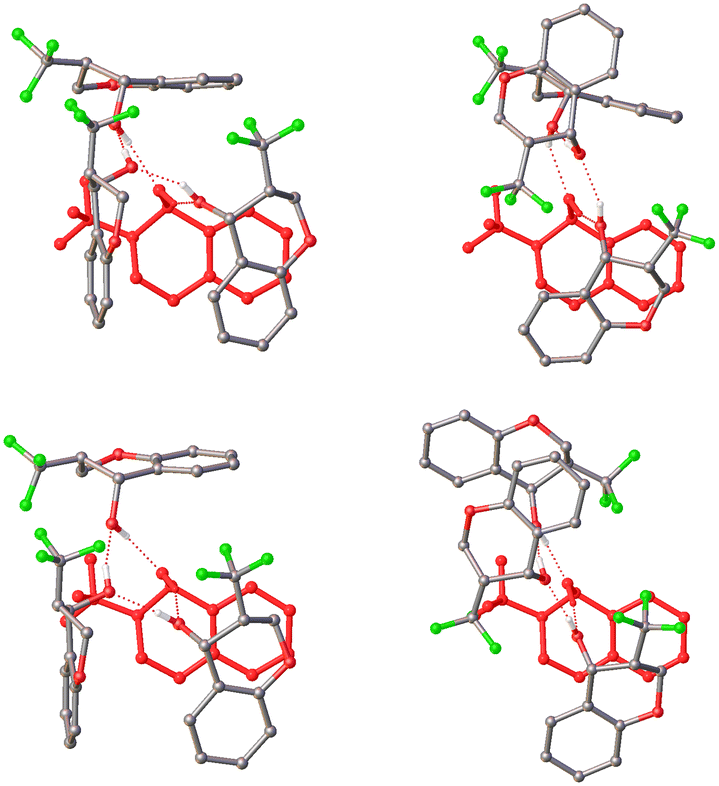 | ||
| Fig. 9 The four tetramers comprising the asymmetric unit of KEJZEI. Each tetramer has been orientated to allow one molecule (rendered in red) involved in π⋯π interactions and with an inward-facing hydrogen bond to be used as a reference. | ||
Furthermore, the ring motif and π-interactions observed in this structure are consistent with the findings of Taylor et al. when considering the expected interaction motifs for a non-centrosymmetric structure of this kind.14
Though the article is concerned primarily with the synthesis and stereochemistry of the molecules, some rather convincing explanation for the large asymmetric unit is provided by the authors. The ‘suspiciously’ hexagonal metric symmetry noted in the article can be rationalised by the ca. 60° angle between each tetramer as they propagate with the translational modulation in the [001] direction.
Overview of extreme Z′ structures
Though the structures with extreme Z′ values (Z′ ≥ 12) discussed above represent a range of different chemical fields they are almost exclusively organic. The arsenic complex LUBVUC and the white phosphorus structure LOFRAD are the exceptions, though arsenic's classification as a metalloid makes its status somewhat ambiguous and both arsenic and phosphorus are classed as ‘organic’ for the purpose of CSD searches.The best way to assess this group of structures is to see how well they adhere to the organising principles identified by Brock.3 Most of the examples exhibit negligible conformational flexibility and strong intermolecular interactions in the form of hydrogen bonds, or pancake bonds in the case of AYADOW01. Over half crystallise in Sohncke space groups and almost all exhibit some form of approximate symmetry.
It is encouraging that these structures, which were not part of Brock's earlier survey, adhere to these principles and they are clearly incredibly important in terms of predicting incidents of high or extreme Z′ structures and should be considered crucial when designing such systems.
What is clear is that the true number of crystal structures in the CSD with Z′ ≥ 12 may be a lot fewer than the 129 that a simple search implies. Considering redeterminations of the same structure, racemates, co-crystals reported with a higher Z′ than their stoichiometry suggests and instances where the symmetry may be incorrect or the quality of the data not sufficient to be certain of it, many of the search results can probably be disregarded.
However, it may well be that the true incidence of these structures is higher than reported. The propensity of crystals of this kind to form small, low quality crystals55 may mean that they are forming much more frequently than they are being analysed and/or reported. The inherent difficulty in the refinement of high Z′ structures and the high level of confidence the crystallographer must have in the result in order to publish surely contribute further to the underreporting of these data. It will be interesting to see if the number of these structures increases as our ability to probe the structure of smaller crystals becomes more reliable.
Crystal structure prediction and high Z′ structures
Though the concepts of frustration, awkwardness and the common features present in high Z′ structures can be exploited for the rational design of crystals with Z′ ≥ 1 and highly polymorphic systems, structures of this kind are often neglected when it comes to crystal structure prediction (CSP). As stated in the introduction, the recent seventh blind test of crystal structure prediction, though an overall successful endeavour, did not consider any structures with Z′ > 3.8,9 This is currently the convention within the field of CSP as exemplified by the study into the polymorphism of nicotinamide that produced NICOAM09.33 This work included CSP calculations, but capped Z′ at 2, which seems restrictive when a third of the known forms have Z′ ≥ 4.It is not that structures with high Z′ are impossible to predict. Some predicted structures, albeit those comprising very simple, small molecules such as pyridine, have been calculated.10 One might think, as structures with high Z′ typically comprise inflexible molecules with negligible conformational variation, that the rigid molecule assumption would hold for such molecules,56 that savings could be made in terms of computation by omitting steps where molecular geometry is optimised, an approach exploited by Oketani et al. in their work on MGP.20 However, it appears that, even so, the computational cost is so much greater for each extra molecule in the asymmetric unit and this is what restricts investigations in this direction. With greater consideration being given to the environmental impact of computation in recent years, it seems harder to justify this cost and hence it is unlikely that structures of this kind will be considered by CSP for the time being.
Beside the matter of computational resources, a number of other factors contribute to the potential challenges of predicting instances of high Z′. One potential issue was highlighted in the case of the quasi-isostructural polymorphism observed by Chopra et al. in their benzamidamide structures (SUXLII/01).19 As these various polymorphs were essentially isoenergetic, they would surely prove difficult to differentiate in silico and converging on an energy minimum may not be possible. This is a known limitation of CSP in general and is only exacerbated when the asymmetric unit contains many molecules.57,58
And then there is the question of approximate symmetry. A significant number of structures with high Z′ are observed to exhibit approximate symmetry59 and its prevalence must therefore be taken into account if crystal structures of this type are to be successfully predicted.
For now, it would seem that the most effective way to identify approximate symmetry is still by eye.60 Some program packages are capable of finding specific approximate symmetries, such as inversions and translations25,61–63 but can be hard to utilise and vary in terms of reliability especially where this approximate symmetry is confined to layers as is often the case for high Z′ structures.64 A method capable of finding all types of approximate symmetry is still elusive, though promising work to this end by Baggio is under development.65
With this in mind, it may make one wonder, if the identification of approximate symmetry cannot be automated, what are the chances of it being predicted? This may be a case of putting the cart before the horse and that approximate symmetry may manifest in predicted structures regardless of whether or not it is taken into consideration. In either case, there may be hope in the hypothesis that there is a relationship between high Z′ structures and a theoretical structure with Z′ = 1 but with true symmetry instead of approximate symmetry accessible through very slight conformational changes.3,57 If this can be confirmed, Zhu suggests that if the theoretical Z′ = 1 structure can be returned by CSP, it may be possible to predict the high Z′ structure using this as a starting point,66 which would potentially be much more computationally economical.
Another recent development that may lead to more efficient processes is the work of Galanakis and Tuckerman.67 Their purely mathematical approach by-passes the generation of interatomic interaction models, often the rate-determining step in the CSP process. In their article the authors assure us that this method can successfully be applied to structures with Z′ > 2 and hence this could be yet another encouraging step towards the prediction of high Z′ crystal structures.
Conclusions
Over the last ten years, crystal structures with multiple molecules in the asymmetric unit have continued to be a fruitful area of research. Much work has gone into codifying the rules that govern their formation and molecular features and intermolecular motifs that are most likely to result in high Z′ structures. These observations have been successfully applied to the rational design or new systems demonstrating the potential of this new knowledge to crystal engineering in general.Examples of structures with extreme Z′ values continue to be reported and their complexity and variety continue to astound. Each is a unique snowflake and worthy of a story of its own but the structural characteristics they share can inform crystal design and allow for a greater exploration of the polymorph space of organic molecules. Their true incidence in the CSD may be exaggerated by redetermination and misassignments, but it is also possible that they are under-reported and more common than thought.
Crystal structure prediction (CSP) continues to develop apace, but the computational cost incurred by increasing the number of molecules in the unit cell means that structures with high Z′ often fall through the cracks. There are as yet no reliable methods for identifying approximate symmetry, a feature observed in a great many high Z′ structures.
It is almost a certainty that structures with large numbers of independent molecules will continue to surprise crystallographers by appearing as if at random, and their analysis will delight and inform us for years to come. For the time being, it would seem that future studies of these fascinating structures will not be able to rely on CSP or other automated methods. In this world of machine learning and so-called AI, it may be heartening to know that there is still a place for an experienced eye and essentially performing structural analysis ‘on vibes’.
Data Availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PGW would like to thank Prof. Carolyn Brock and Dr Lily Hunnisett for useful discussions.References
- S. B. Hendricks, J. Am. Chem. Soc., 1928, 50, 1205 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- C. P. Brock, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 807 CrossRef CAS PubMed.
- J. W. Steed, CrystEngComm, 2003, 5, 169 RSC.
- G. R. Desiraju, CrystEngComm, 2007, 9, 91 RSC.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2008, 8(7), 2517 CrossRef CAS.
- C. P. Brock, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2022, 78, 576 CrossRef CAS PubMed.
- L. M. Hunnisett, J. Nyman, N. Francia, N. S. Abraham, C. S. Adjiman, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli, D. M. Anstine, J. E. Anthony, J. E. Arnold, F. Bahrami, M. A. Bellucci, R. M. Bhardwaj, I. Bier, J. A. Bis, A. D. Boese, D. H. Bowskill, J. Bramley, J. G. Brandenburg, D. E. Braun, P. W. V. Butler, J. Cadden, S. Carino, E. J. Chan, C. Chang, B. Cheng, S. M. Clarke, S. J. Coles, R. I. Cooper, R. Couch, R. Cuadrado, T. Darden, G. M. Day, H. Dietrich, Y. Ding, A. DiPasquale, B. Dhokale, B. P. van Eijck, M. R. J. Elsegood, D. Firaha, W. Fu, K. Fukuzawa, J. Glover, H. Goto, C. Greenwell, R. Guo, J. Harter, J. Helfferich, D. W. M. Hofmann, J. Hoja, J. Hone, R. Hong, G. Hutchison, Y. Ikabata, O. Isayev, O. Ishaque, V. Jain, Y. Jin, A. Jing, E. R. Johnson, I. Jones, K. V. J. Jose, E. A. Kabova, A. Keates, P. F. Kelly, D. Khakimov, S. Konstantinopoulos, L. N. Kuleshova, H. Li, X. Lin, A. List, C. Liu, Y. M. Liu, Z. Liu, Z.-P. Liu, J. W. Lubach, N. Marom, A. A. Maryewski, H. Matsui, A. Mattei, R. A. Mayo, J. W. Melkumov, S. Mohamed, Z. M. Abardeh, H. S. Muddana, N. Nakayama, K. S. Nayal, M. A. Neumann, R. Nikhar, S. Obata, D. O'Connor, A. R. Oganov, K. Okuwaki, A. Otero-de-la-Roza, C. C. Pantelides, S. Parkin, C. J. Pickard, L. Pilia, T. Pivina, R. Podeszwa, A. J. A. Price, L. S. Price, S. L. Price, M. R. Probert, A. Pulido, G. R. Ramteke, A. U. Rehman, S. M. Reutzel-Edens, J. Rogal, M. J. Ross, A. F. Rumson, G. Sadiq, Z. M. Saeed, A. Salimi, M. Salvalaglio, L. S. de Almada, K. Sasikumar, S. Sekharan, C. Shang, K. Shankland, K. Shinohara, B. Shi, X. Shi, A. G. Skillman, H. Song, N. Strasser, J. van de Streek, I. J. Sugden, G. Sun, K. Szalewicz, B. I. Tan, L. Tan, F. Tarczynski, C. R. Taylor, A. Tkatchenko, R. Tom, M. E. Tuckerman, Y. Utsumi, L. Vogt-Maranto, J. Weatherston, L. J. Wilkinson, R. D. Willacy, L. Wojtas, G. R. Woollam, Z. Yang, E. Yonemochi, X. Yue, Q. Zeng, Y. Zhang, T. Zhou, Y. Zhou, R. Zubatyukh and J. C. Cole, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2024, 80, 517 CrossRef PubMed.
- L. M. Hunnisett, J. Nyman, N. Francia, N. S. Abraham, C. S. Adjiman, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli, D. M. Anstine, J. E. Anthony, J. E. Arnold, F. Bahrami, M. A. Bellucci, R. M. Bhardwaj, I. Bier, J. A. Bis, A. D. Boese, D. H. Bowskill, J. Bramley, J. G. Brandenburg, D. E. Braun, P. W. V. Butler, J. Cadden, S. Carino, E. J. Chan, C. Chang, B. Cheng, S. M. Clarke, S. J. Coles, R. I. Cooper, R. Couch, R. Cuadrado, T. Darden, G. M. Day, H. Dietrich, Y. Ding, A. DiPasquale, B. Dhokale, B. P. van Eijck, M. R. J. Elsegood, D. Firaha, W. Fu, K. Fukuzawa, J. Glover, H. Goto, C. Greenwell, R. Guo, J. Harter, J. Helfferich, D. W. M. Hofmann, J. Hoja, J. Hone, R. Hong, G. Hutchison, Y. Ikabata, O. Isayev, O. Ishaque, V. Jain, Y. Jin, A. Jing, E. R. Johnson, I. Jones, K. V. J. Jose, E. A. Kabova, A. Keates, P. F. Kelly, D. Khakimov, S. Konstantinopoulos, L. N. Kuleshova, H. Li, X. Lin, A. List, C. Liu, Y. M. Liu, Z. Liu, Z.-P. Liu, J. W. Lubach, N. Marom, A. A. Maryewski, H. Matsui, A. Mattei, R. A. Mayo, J. W. Melkumov, S. Mohamed, Z. M. Abardeh, H. S. Muddana, N. Nakayama, K. S. Nayal, M. A. Neumann, R. Nikhar, S. Obata, D. O'Connor, A. R. Oganov, K. Okuwaki, A. Otero-de-la-Roza, C. C. Pantelides, S. Parkin, C. J. Pickard, L. Pilia, T. Pivina, R. Podeszwa, A. J. A. Price, L. S. Price, S. L. Price, M. R. Probert, A. Pulido, G. R. Ramteke, A. U. Rehman, S. M. Reutzel-Edens, J. Rogal, M. J. Ross, A. F. Rumson, G. Sadiq, Z. M. Saeed, A. Salimi, M. Salvalaglio, L. S. de Almada, K. Sasikumar, S. Sekharan, C. Shang, K. Shankland, K. Shinohara, B. Shi, X. Shi, A. G. Skillman, H. Song, N. Strasser, J. van de Streek, I. J. Sugden, G. Sun, K. Szalewicz, B. I. Tan, L. Tan, F. Tarczynski, C. R. Taylor, A. Tkatchenko, R. Tom, M. E. Tuckerman, Y. Utsumi, L. Vogt-Maranto, J. Weatherston, L. J. Wilkinson, R. D. Willacy, L. Wojtas, G. R. Woollam, Z. Yang, E. Yonemochi, X. Yue, Q. Zeng, Y. Zhang, T. Zhou, Y. Zhou, R. Zubatyukh and J. C. Cole, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2024, 80, 548 CrossRef PubMed.
- J. van de Streek and M. A. Neumann, CrystEngComm, 2011, 13, 7135 RSC.
- J. Bernstein, J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2008, 8(6), 2011 CrossRef CAS.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895 CrossRef CAS PubMed.
- R. Ragbirsingh, M. J. Hall, M. R. Probert and P. G. Waddell, Cryst. Growth Des., 2024, 24(3), 1429 CrossRef CAS PubMed.
- R. Taylor, J. C. Cole and C. R. Groom, Cryst. Growth Des., 2016, 16, 2988 CrossRef CAS.
- R. E. Marsh, Acta Crystallogr., Sect. B, 1999, 55, 931 CrossRef PubMed.
- M. E. Alvarenga, A. K. S. M. Valdo, L. Ribeiro, J. A. Do Nascimento Neto, D. P. De Araujo, C. M. Da Silva, Â. De Fátima and F. T. Martins, Acta Crystallogr., Sect. C:Struct. Chem., 2019, 75, 667 CrossRef CAS PubMed.
- F. Renz, Y. Baran, P. Gutlich and D. Schollmeyer, CSD Communication, 2011, CCDC 815737 Search PubMed.
- W. Clegg, Acta Crystallogr., Sect. C:Struct. Chem., 2019, 75, 833 CrossRef CAS PubMed.
- D. Dey, S. P. Thomas, M. A. Spackman and D. Chopra, Chem. Commun., 2016, 52, 2141 RSC.
- R. Oketani, H. Takahashi, S. Clevers, A. Oyamada, I. Hisaki, G. Coquerel, K. Yamanaka and H. Tsue, Cryst. Growth Des., 2022, 22, 2230 CrossRef CAS.
- T. Rekis, A. Schönleber, L. Noohinejad, M. Tolkiehn, C. Paulmann and S. van Smaalen, Cryst. Growth Des., 2021, 21, 2324 CrossRef CAS.
- C. H. Gorbitz, S. Agustsdottir and F. Bleken, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2007, 63, 58 CrossRef PubMed.
- E. Parisi, E. Santagata, E. Simone, F. Barbone and R. Centore, J. Am. Chem. Soc., 2024, 146, 19405 CrossRef CAS PubMed.
- Z′, https://zprime.co.uk/, (date accessed: 11/11/2024) Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- P. Zhao, B.-S. Xin, F.-M. He, L. Ye, Z.-T. Ma, J.-L. Hao, R. Shi, X.-H. He, G.-D. Yao, X.-X. Huang and S.-J. Song, New J. Chem., 2023, 47, 20890 RSC.
- Y.-C. Yao, Z. Jin, M. Hong, B. Zhu, G.-B. Ren and M.-H. Qi, Cryst. Growth Des., 2024, 24, 554 CrossRef CAS.
- K. M. Anderson, M. R. Probert, C. N. Whiteley, A. M. Rowland, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2009, 9, 1082 CrossRef CAS.
- Z. Liu, G. K. Kole, Y. P. Budiman, Y.-M. Tian, A. Friedrich, X. Luo, S. A. Westcott, U. Radius and T. B. Marder, Angew. Chem., Int. Ed., 2021, 60, 16529 CrossRef CAS PubMed.
- X. Ou, X. Li, H. Rong, L. Yu and M. Lu, Chem. Commun., 2020, 59, 9950 RSC.
- C. Brandel, Y. Cartigny, N. Couvrat, M. Erminlinda, S. Eusébio, J. Canotilho, S. Petit and G. Coquerel, Chem. Mater., 2015, 27, 6360 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- X. Li, X. Ou, B. Wang, H. Rong, B. Wang, C. Chang, B. Shi, L. Yu and M. Lu, Commun. Chem., 2020, 3, 152 CrossRef CAS PubMed.
- J. Weatherston, M. R. Probert and M. J. Hall, J. Am. Chem. Soc., 2025 Search PubMed , accepted.
- A. R. Tyler, R. Ragbirsingh, C. J. McMonagle, P. G. Waddell, S. E. Heaps, J. W. Steed, P. Thaw, M. J. Hall and M. R. Probert, Chem, 2020, 6, 1755 CAS.
- K. Ivshin, K. Metlushka, R. Zinnatullin, K. Nikitina, A. Pashagin, D. V. Zakharychev, A. Efimova, A. Kiiamov, S. Latypov and O. Kataeva, Cryst. Growth Des., 2021, 21, 5460 CrossRef CAS.
- R. A. Mayo, I. S. Morgan, D. V. Soldatov, R. Clérac and K. E. Preuss, Inorg. Chem., 2021, 60, 11338 CrossRef CAS PubMed.
- A. G. Nunez Avila, B. Deshênes-Simard, J. E. Arnold, M. Morency, D. Chartrand, T. Maris, G. Berger, G. M. Day, S. Hanessian and J. D. Wuest, J. Org. Chem., 2022, 87, 6680 CrossRef CAS PubMed.
- T. R. Welberry and T. Weber, Crystallogr. Rev., 2016, 22, 2 CrossRef CAS.
- M. Lyczko, K. Lyczko, A. Majkowska and A. Bilewicz, Molecules, 2019, 24, 3865 CrossRef CAS PubMed.
- T. T. P. Tran, D. M. C. Ould, L. C. Wilkins, D. S. Wright, R. L. Melen and J. M. Rawson, CrystEngComm, 2017, 19, 4696 RSC.
- R. C. Bakus II, D. A. Atwood, S. Parkin, C. P. Brock and V. Petricek, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2013, 69, 496 CrossRef PubMed.
- R. Herbst-Irmer, X. Wang, L. Haberstock, I. Köhne, R. Oswald, J. Behler and D. Stalke, IUCrJ, 2023, 10, 766 CrossRef CAS PubMed.
- A. A. M. Sarhan, M. Haukka, A. Baraket and A. T. A. Boraei, Tetrahedron Lett., 2020, 61, 152660 CrossRef CAS.
- T. Luo, M. Dai, S.-L. Zheng and S. L. Schreiber, Org. Lett., 2011, 13, 2834 CrossRef CAS PubMed.
- P. Messakul, W. Jaidee, C. Richardson, R. J. Anderson, B. O. Patrick, A. C. Willis, C. Muanprasat, J. Wang, X. Lei, S. Hadsadee, S. Jungsuttiwong, S. G. Pyne and S. Laphookhieo, Phytochemistry, 2020, 171, 112248 CrossRef PubMed.
- Z. Li and R. Tong, Synthesis, 2016, 48, 1630 CrossRef CAS.
- G. S. Bisht and B. Gnanaprakasam, J. Org. Chem., 2019, 84, 13516 CrossRef CAS PubMed.
- X.-Q. Zhu, H. Yuan, Q. Sun, B. Zhou, X.-Q. Han, Z.-X. Zhang, X. Lin and L.-W. Ye, Green Chem., 2018, 20, 4287 RSC.
- J. M. de Souza, I. Abdiaj, J. Chen, K. Hanson, K. T. de Oliviera and D. T. McQuade, J. Org. Chem., 2020, 85, 11822 CrossRef CAS PubMed.
- M. Fu, CSD Communication, 2018, CCDC 1838663 Search PubMed.
- M. Fu, CSD Communication, 2018, CCDC 1871767 Search PubMed.
- R. B. Betancourt, L. Bacheley, A. Karapetyan, G. Guillamot, P. Phansavath and V. Raovelomanana-Vidal, ChemCatChem, 2022, 14, 595 Search PubMed.
- S. R. Jakkaraj, V. G. Young Jr. and G. I. Georg, Tetrahedron, 2018, 74, 2769 CrossRef CAS PubMed.
- G. S. Nichol and W. Clegg, CrystEngComm, 2007, 9, 959 RSC.
- G. M. Day, in Computational Pharmaceutical Solid State Chemistry, ed. Y. A. Abramov, Wiley-VCH, Weinheim, 2016, p. 87 Search PubMed.
- D. Nyman and G. M. Day, CrystEngComm, 2015, 17, 5254 RSC.
- S. L. Price and J. G. Brandenburg, in Non-Covalent Interactions in Quantum Chemistry and Physics, ed. A. O. de la Rosa and G. A. di Labrio, Elsevier, Amsterdam, 2017, p. 333 Search PubMed.
- C. P. Brock, CrystEngComm, 2020, 22, 7371 RSC.
- C. P. Brock, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2022, 78, 576 CrossRef CAS PubMed.
- A. Gavozzetti, CrystEngComm, 2008, 10, 389 Search PubMed.
- N. V. Somov and E. V. Chuprunov, Crystallogr. Rep., 2009, 54, 727 CrossRef CAS.
- A. Collins, R. I. Cooper and D. J. Watkin, J. Appl. Crystallogr., 2006, 39, 842 CrossRef CAS.
- C. P. Brock, Cryst. Growth Des., 2024, 24, 6211 CrossRef CAS.
- R. Baggio, Acta Crystallogr., Sect. C:Struct. Chem., 2019, 75, 837 CrossRef CAS PubMed.
- Q. Zhu and S. Hattori, J. Mater. Res., 2022, 38, 19 CrossRef.
- N. Galanakis and M. E. Tuckerman, Nat. Commun., 2024, 15, 9757 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce01186d |
| ‡ Z′ (Z prime) is the number of symmetry-independent molecules in the asymmetric unit of a crystal structure. |
| This journal is © The Royal Society of Chemistry 2025 |