
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent lanthanide-doped calcium phosphate from oyster shell waste: an example of bright recycling†
Paula
Puentedura-Navarro
a,
Raquel
Fernández-Penas
 a,
Francisco Javier
Acebedo-Martínez
a,
Carla
Triunfo
b,
Jorge F.
Fernández-Sánchez
c,
Antonia
Follenzi
d,
Francesca
Oltolina
*d,
Giuseppe
Falini
b and
Jaime
Gómez-Morales
*a
a,
Francisco Javier
Acebedo-Martínez
a,
Carla
Triunfo
b,
Jorge F.
Fernández-Sánchez
c,
Antonia
Follenzi
d,
Francesca
Oltolina
*d,
Giuseppe
Falini
b and
Jaime
Gómez-Morales
*a
aLaboratorio de Estudios Cristalográficos, IACT (CSIC), Avda. Las Palmeras, no 4, 18100 Armilla, Spain. E-mail: jaime.gomez@csic.es
bDepartment of Chemistry “Giacomo Ciamician”, University of Bologna, via F. Selmi 2, 40126 Bologna, Italy
cDepartment of Analytical Chemistry, Faculty of Sciences, University of Granada, Avda. Fuentenueva s/n, 18071 Granada, Spain
dDipartimento di Scienze della Salute, Università del Piemonte Orientale, “A. Avogadro” Via Solaroli 17, 28100 Novara, Italy. E-mail: francesca.oltolina@med.uniupo.it
First published on 21st January 2025
Abstract
This research explores the transformation of biogenic CaCO3 microparticles (Ø < 45 μm) from oyster shell waste into luminescent Eu- or Tb-doped calcium phosphate (apatites), using a sustainable “one-step” and eco-friendly method. The full transformation was achieved at 200 °C via a dissolution–precipitation mechanism. Precipitates were composed of Eu- or Tb-doped apatite particles, with average sizes L = 163 ± 7 nm and anisometric shapes for the former, and 41 ± 8 nm and more isometric shapes for the latter. Alongside these, particles of either EuPO4·nH2O or TbPO4·nH2O (rhabdophane) were present. The physicochemical and electrokinetic analysis revealed the A- and B-carbonate substitutions and labile CO32− species in the apatite particles, and ζ-potentials approaching zero in the aqueous suspensions at physiological pH levels, indicating a tendency for particle aggregation. The luminescence properties, such as relative luminescent intensity and luminescence lifetimes, were dependent on the lanthanide content and the presence of the rhabdophane phase. The Ap–Ln samples demonstrated cytocompatibility, with cell viability exceeding 85% when incubated with murine pancreatic endothelial cells (MS1) and murine mesenchymal stem cells (m17.ASC), regardless of the lanthanide type or the particle dosage used (ranging from 0.1 to 100 μg mL−1).
1. Introduction
Mollusc shells are described as mineral/organic hybrid composites containing CaCO3 crystals embedded in an organic matrix (OM) formed mainly of proteins and polysaccharides (1–5 wt%).1 These shells, which are mostly generated in canning industries or seafood restaurants, are a waste product of the marine aquaculture and fishing sectors. In 2018, the production of molluscs in the world, mainly bivalves, was of 17.7 million tons, which represented 2.6 kg per capita.2 Clams and oysters are the major bivalve species, with 38% and 33% of the overall production, while scallops and mussels represent 17% and 13%, respectively.3,4 The shells account for 65–90 wt% of live weight depending on the species which, considering only the bivalves, represent over 10 million tons per year.5 This waste is usually dumped in the sea6 or disposed of in landfills, causing unpleasant smell and health issues due to the microbial decomposition.7,8 Their removal involves the use of incineration or burial,9 thus representing an environmental and economic issue as well as a loss of potentially usable biomaterials. Repurposing this waste into new materials contributes to alleviating these problems while also presenting an opportunity for shell valorization, and then stimulation of the circular blue bioeconomy.10CaCO3 from seashell waste has been used by different researchers as a calcium supplement for livestock feeding,11,12 liming agents for treating the soil acidity in agricultural exploitations,13 aggregate substitutes in concrete preparation,14,15 or as absorbents in water treatment,16 CO2-capture,17 or catalysts in biodiesel production.18 Triunfo et al. showed that calcite and aragonite single crystals recovered from waste mussel shells of the species Mytilus galloprovincialis were effective substrates in removing anionic organic dyes from water, thus finding applications in water remediation.19 Basile et al. showed that for stearate-functionalized biogenic CaCO3 crystals recovered from oyster shells of the species Crassostrea gigas, when used as a filler of an ethylene vinyl-acetate polymer employed in the fabrication of shoe soles, the obtained polymeric material showed better mechanical performance than the one made with geogenic CaCO3.20 Several studies have used biogenic CaCO3 (bCC) waste for the preparation of calcium phosphate (CaP) nano or microparticles of the apatite phase (Ap). Eggshells,21 shells from mussels,22 clams,23 oysters,24 snails,25 cockles26 as well as coral skeletons27 and cuttlefish bones,28 have been used as biominerals sources. Since Ap is the major inorganic compound of bones and teeth, synthetic Ap is employed largely for biomedical applications such as coatings for dental implants, drug delivery, bone filling, substitution, replacement, and regeneration in orthopedics, and when doped with lanthanides, it can be used as luminescent probes in bioimaging applications.29–35 In this regard, these materials combine the biocompatibility and osteoconductivity of Ap with unique optical properties.35 Lanthanide ions (Ln) such as europium (Eu3+) and terbium (Tb3+), and others have been successfully incorporated into Ap structures, resulting in materials with tunable light emission.36 The concentration of dopants, particularly Tb3+, influences the morphology, structure, and photoluminescence properties of Ap.37 These novel materials have shown good biocompatibility in cell viability tests, suggesting their potential for biological system imaging and other biomedical applications.32–34,37,38 The development of rare earth-doped Ap luminescent probes from bCC represents an innovative approach for repurposing biomineral waste since the apatite market including nano-, micron- and larger sizes, is continuously growing and is expected to reach around USD 3086.05 million by 2027.39
The methods used to prepare Ap particles from bCC typically comprise two main steps. Firstly, bCC is calcined at temperatures ranging from 900 to 1200 °C, leading to the formation of calcium oxide (CaO) and carbon dioxide (CO2). The second step involves neutralization with a phosphorus reagent, usually phosphoric acid (H3PO4).40–43 Alternatively, a three-step procedure has been employed: this involves calcination to CaO and CO2, followed by carbonation to produce calcium carbonate (CaCO3) particles, and then a hydrothermal reaction with ammonium hydrogen phosphate ((NH4)2HPO4) to yield apatite nanoparticles (Ap NPs).44 These multi-step procedures, involving calcination of bCC, fully destroy the intracrystalline organic matrix. Additionally, they are neither ecologically or energy sustainable; that is, they need extremely high temperatures and emit a substantial amount of CO2 into the atmosphere, which accounts for around 44% of the bCC. A sustainable “one-step” and eco-friendly hydrothermal method, recently developed by our team, has allowed the full transformation of bCC particles (Ø < 45 μm) into Ap submicron-nanoparticles (L < 200 nm) under mild hydrothermal conditions (T < 200 °C) in experiments lasting 7 days.45,46 In this research, we will investigate the “one-step” hydrothermal transformation of bCC into luminescent Eu- and Tb-doped Ap nano-submicron particles (Ap–Eu and Ap–Tb), and characterize their physicochemical, luminescent, and biocompatibility properties.
2. Experimental
2.1. Reagents and preparation of raw materials
The bCC powder was prepared from oyster shell of the species Crassostrea gigas (Mg-calcite, ∼5% at Mg) provided by F.lli Terzi (Palosco, BG, Italy). The shells were first washed with tap water, then immersed in a 5% v/v hypochlorite solution for 24 hours to remove the surface organic residues. Afterwards, they were washed again with tap water, deionized water, and then air-dried. The bleached shell was crushed by a hammer mill and the resulting bCC powder sieved at Ø < 45 μm. Potassium phosphate monobasic (KH2PO4, ACS reagent, 99% purity), europium(III) chloride hexahydrate (EuCl3·6H2O, ACS Reagent, 99.9% purity), and terbium(III) chloride anhydrous (TbCl3, 99.9% pure, trace metals) were provided by Sigma-Aldrich (St Louis, MO, USA). All solutions and CaCO3 suspensions were prepared using deionized Milli-Q water (0.22 μS, 25 °C, Millipore, Burlington, MA, USA).2.2. Hydrothermal synthesis and particle characterization
Based on previous results,45,46 the experiments for the current study have been performed within the temperature range 100 °C ≤ T ≤ 200 °C (Δ20 °C) in an oven with circulating forced air for 7 days. The experiments were carried out in an aluminum box provided with a rack of PTFE tubes inside a PTFE-coated aluminum cap to close the box hermetically. The PTFE tubes were filled with 0.5 g of bCC powder suspended in 8 mL of a KH2PO4 solution of concentration 375 mmol L−1 and either 10 or 20 mmol L−1 Eu3+ and Tb3+. The percentage fill of the tubes was 70% to avoid an overpressure. After the trials were completed, the precipitates were washed with deionized water through centrifugation at 9000 rpm for 20 minutes, repeated four times, and then freeze-dried overnight at −50 °C under a vacuum of 3 mbar.Particle characterization was performed using various techniques: X-ray diffraction (XRD) patterns were acquired with an X-ray powder diffractometer Bruker D8 Advance Vario Series II (Bruker AXS, Bruker GmbH, Karlsruhe, Germany). The equipment is provided with a CuKα1 radiation (1.5406 Å) generator. Crystallite size of more pure samples was determined using software TOPAS 7.0 (Coelho Software, Brisbane, Australia) considering the instrumental contribution from a measurement of Si standard (NIST). Fourier transform infrared spectra (FTIR) were recorded with a Hyperion 3000 (Bruker, Massachusetts, USA) instrument in transmittance mode within the wavenumber range from 4000 cm−1 to 400 cm−1. The instrument is equipped with an attenuated total reflectance (ATR) accessory of diamond crystal. We conducted additional measurements to determine the degree of carbonation by preparing pellets composed of approximately 1 mg of sample mixed with about 100 mg of anhydrous KBr. These mixtures were then pressed with a hydraulic pump at a pressure of 10 tons. For the measurements, pure KBr disks were used to record the background.
Complementary spectroscopic characterization was performed by Raman spectroscopy. Spectra were recorded with a LabRAMHR spectrometer (Jobin-Yvon, Horiba, Tokyo, Japan) equipped with a laser diode emitting at a wavelength of 532 nm. Crystal size distribution (CSD) was determined by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS analyzer (Malvern Instruments Ltd., Malvern, UK). The measurements were done at room temperature in disposable polystyrene vials containing aqueous particulate suspensions (∼0.5 mg mL−1). For ζ-potential versus pH measurements using the same instrument, the pH of the suspensions was adjusted with HCl and NaOH solutions (0.25 and 0.1 M, respectively) as titration agents, without adding any additional electrolyte. For the bCC particles, the CSD measurements were performed by laser diffraction with a Mastersizer 2000 (Malvern, UK) particle size analyzer coupled to a Hydro 2000SM fully automated large-volume wet sample dispersion unit.
Scanning electron microscopy (SEM) analysis was conducted using a variable pressure Zeiss SUPRA40VP scanning electron microscope (VPSEM), which was equipped with a large X-Max 50 mm area detector for energy dispersive X-ray spectroscopy (EDX) microanalysis. Samples were deposited on conventional stubs and carbon-sputtered, prior observation. Element composition was determined averaging 7–15 measurements in different particles of each sample. Transmission electron microscopy (TEM) imaging was performed with a Libra 120 Plus TEM instrument (EELS) at 80 kV (Carl Zeiss, Jena, Germany). Particles were dispersed in ethanol (99.8% v/v) and then deposited on copper microgrids coated with a FORMVAR carbon film. High-resolution TEM (HRTEM) analysis was done with a TITAN G2 60-300 FEI Instrument (FEI, Hillsboro, OR, USA) operating at 300 kV. The instrument is equipped with EDX Super X detector to perform microanalysis and a STEM type HAADF.
Elemental analysis of Eu and Tb was also carried out by inductively coupled plasma mass spectroscopy (ICP-MS) using a Perkin Elmer NexION 300D ICP mass spectrometer (Perkin Elmer), while for Ca and P we used a Perkin Elmer ICP-OES OPTIMA 8300 spectrometer (Perkin Elmer). The pH of solutions was measured by using a Sension++ CAT pH electrode connected to a Hach Sension+ pH-meter with an accuracy ≥0.02 pH.
2.3. Luminescence spectroscopy
The luminescence properties of the solid Ap–Eu and Ap–Tb samples such as luminescence spectra, luminescence lifetime (τ) and the relative luminescence intensities (RLI) were recorded with a Cary Eclipse Varian fluorescence spectrophotometer (Varian Australia, Mulgrave, Australia), using a front surface accessory. The spectra for Tb-containing particles were recorded using the following instrumental parameters: λexc= 372 nm, λem= 543 nm, slit-widthexc/em = 10/10 nm, delay time (td) = 0.120 μs, gate time (tg) = 5 ms and detector voltage = 580 V. The instrumental parameters for recording the spectra of Eu-containing particles were λexc = 395 nm, λem = 616 nm, slit-widthexc/em = 10/10 nm, delay time (td) = 120 μs, gate time (tg) = 5 ms and detector voltage = 530 V.Luminescence lifetimes (τ) were measured using the following conditions: a) For Ap–Tb particles: λexc/em = 372/543 nm, slit-widthexc/em = 20/20 nm, td = 0.1 μs, tg = 0.120 ms and detector voltage = 800 V; and b) for Ap–Eu particles: λexc/em = 395/616 nm, slit-widthexc/em = 10/10 nm, td = 0.1 μs, tg = 0.120 ms and detector voltage = 700 V.
2.4. Cytocompatibility
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio after being incubated under usual conditions (37 °C, 5% CO2). Every three days, the medium was changed.
:10 ratio after being incubated under usual conditions (37 °C, 5% CO2). Every three days, the medium was changed.
3. Results
3.1. Compositional, crystallographic and physico-chemical characteristics of precipitates
The evolution of the transformation of bCC with the temperature in the presence of 10 mM lanthanide, characterized by XRD, FTIR and Raman spectroscopy, is shown in Fig. 1a–i and S1a–f (see ESI† material). From bottom to top, it is shown the characterization results of the reference samples bCC and Ap blank, and then of the samples obtained at each temperature. The XRD pattern of bCC shows the most intense reflections of CaCO3 (C, calcite, PDF 01-086-2339) at 2θ = 23.06°(012) and at 2θ = 29.4°(104). The pattern of Ap-blank is characterized by the reflections at 2θ = 25.8°(002), 28.05°(102), 28.9°(210) and at 31.75°, 32.12°, 32.88°, and 33.9°, corresponding to the reflections (211), (112), (300) and (202), respectively (PDF 01-089-5631). In both Ap–Eu and Ap–Tb samples prepared at 160 °C a small C(104) reflection remains, while a new reflection at around 29.8° (marked with *) is visible. The latter corresponds to the (200) reflection of EuPO4·nH2O,48 (PDF 20-1044) and TbPO4·nH2O,49 (PDF 20-1244), both rhabdophane phase, and is slightly higher in intensity in the Ap–Tb sample. | ||
| Fig. 1 Conversion of bCC powder to Ap–Eu and Ap–Tb particles at 100, 160 and 200 °C for 7 days analyzed by XRD (a, d and g), FTIR spectroscopy (b, e and h) and Raman spectroscopy (c, f and i). | ||
At 200 °C, both the C(104) of bCC and that of the LnPO4(200) are negligible, indicating the almost complete transformation to the Ap phase. Nonetheless, when increasing the Eu3+ and Tb3+ concentrations to 20 mM at 200 °C the LnPO4(200) reflection appeared again, being also visible the (100) and (101) reflections of these phases (Fig. S2a–d†). A small amount of rhabdophane was also found when using the citrate-based thermal decomplexing method to prepare Ap–Eu at long maturation times (7 days or more) or at shorter maturation periods (96 h) with higher Eu3+ concentrations33 as well as when preparing Ap–Tb.34
The analysis by FTIR (Fig. 1b, e, h, and S3†) and Raman (Fig. 1c, f, i, and S4†) of these samples allow us to complement and confirm the XRD results. Hence, in the FTIR spectra bCC displays the CO32− signal at ∼1415 cm−1 (ν3) and two bands at 875 cm−1 (ν2) and 710 cm−1 (ν4) while Ap (blank) displays the vibrational modes of PO43− at ∼1020 cm−1 (ν3), and at 560 and 604 cm−1 (ν4). The CO3 band at 875 cm−1 is commonly observed in CO3-doped Ap. At 200 °C (Fig. 1h), the bands of calcite disappeared, while a small remnant at ∼1419, ∼1449 cm−1 and 875 cm−1 correspond to the CO3 groups of the Ap. A deconvolution of the latter band reveals three sub-bands (Fig. S5†). For Ap–Eu they are located at 880, 873 and 869 cm−1 while for Ap–Tb these bands are found at 880, 873, and 866 cm−1. These sub-bands are attributed to A-type (CO32− replacing OH−), B-type (CO32− replacing PO43−) and labile CO32− species located at the surface of the particles.50,51 The degree of carbonation of these apatite samples, estimated with the method that compares the intensity of ν2CO3 band with those of the ν1–ν3PO4 ones,51 was 2.2 ± 0.2 wt% for the Ap blank, 3.1 ± 0.1 wt%. for Ap–Eu and 3.5 ± 0.2 wt% for Ap–Tb. The Raman spectra (Fig. 1c, f, i, and S4†) display the characteristic Ap band at approximately 962 cm−1, corresponding to υ1(PO4).52 Additionally, the spectra show the mode υ1(CO3) of calcite at around 1087 cm−1,53 as the most representative signal. This band above 100 °C (Fig. 1f) is insignificant.
The EDX chemical composition of the Ap–Eu and Ap–Tb samples (Table 1), shows the presence of Eu, Tb, Ca, P, and other minor or trace elements such as Na, K, Ni, Si, and Mg doping the samples, the latter becoming from the raw material bCC. The elemental composition of Eu, Tb, Ca and P of some representative samples determined by ICP is shown in Table S1.† The atomic percentages of Eu doping in the samples fall between 1.1 and 2.5 atom% and those of Tb between 1.4 and 1.7 atom%. They increase with the doping concentration when using Eu3+ and slightly decrease when incorporating Tb3+, even though the latter are rather similar. The high S.D. that affects the concentration of each element within the analysed crystals indicates significant heterogeneity in its distribution. Regarding the (Ca + M + Ln)/P ratios, they increase when the doping concentration of both lanthanides increase, reaching a value around 1.67 (the ratio of stoichiometric hydroxyapatite) at 200 °C, thus reflecting the substitution of Ca with M and Ln in the apatite crystals.
| Sample | T (°C), CLn(mM) | Eu | Tb | Ca | P | (Ca + Ln)/P | (Ca + M + Ln)/P |
|---|---|---|---|---|---|---|---|
| EDX (7–15 spectra per sample). M = Na (0–0.5 atom%), K (0.4–2.0 atom%), Mg (0.5–1.2 atom%), Ni and Si (0.1–0.3 atm%). Average ± S.D. Lanthanide (Ln = Eu and Tb9). | |||||||
| Ap–Eu | 160, 10 | 1.1 ± 0.6 | 17.9 ± 5.8 | 12.6 ± 2.3 | 1.47 ± 0.16 | 1.56 ± 0.17 | |
| Ap–Eu | 200, 10 | 1.9 ± 1.6 | — | 18.9 ± 7.5 | 14.1 ± 3.4 | 1.38 ± 0.32 | 1.47 ± 0.32 |
| Ap–Eu | 160, 20 | 2.5 ± 2.1 | — | 20.7 ± 9.9 | 15.4 ± 2.8 | 1.48 ± 0.30 | 1.58 ± 0.26 |
| Ap–Eu | 200, 20 | 2.1 ± 1.8 | — | 20.4 ± 7.8 | 14.2 ± 2.5 | 1.52 ± 0.93 | 1.67 ± 0.94 |
| Ap–Tb | 160, 10 | — | 1.5 ± 1.3 | 19.2 ± 7.5 | 14.8 ± 2.8 | 1.38 ± 0.35 | 1.48 ± 0.35 |
| Ap–Tb | 200,10 | — | 1.7 ± 1.1 | 15.9 ± 4.6 | 13.4 ± 2.7 | 1.29 ± 0.13 | 1.44 ± 0.11 |
| Ap–Tb | 160, 20 | — | 1.4 ± 0.7 | 17.3 ± 4.0 | 13.1 ± 1.0 | 1.42 ± 0.15 | 1.54 ± 0.09 |
| Ap–Tb | 200, 20 | — | 1.4 ± 1.3 | 21.9 ± 8.8 | 14.6 ± 2.6 | 1.56 ± 0.27 | 1.69 ± 0.25 |
3.2. Morphology, microstructure, crystal size distribution and electrokinetic properties
The morphological features of the Eu- and Tb-doped samples are different, as shown in Fig. 2. | ||
| Fig. 2 VPSEM micrographs of Ap–Eu and Ap–Tb samples prepared at 160 °C in the presence of 10 mM of Eu3+ (a) and 10 mM Tb3+ (d), at 200 °C in the presence of 10 mM Eu3+ (b) and 10 mM Tb3+ (e), and at 200 °C in the presence of 20 mM Eu3+ (c) and 20 mM Tb3+ (f). | ||
While Ap–Eu samples display crystals arranged to form flower-like morphologies and elongated needle-like crystals (Fig. 2a–c), those Ap–Tb present more isometric crystals (almost spherical) and platelets (Fig. 2d–f). According to XRD results, only samples prepared at 200 °C, 10 mM lanthanide (Fig. 2b and e), were composed of Ap–Ln. Their average sizes determined with ImageJ were L = 163 ± 7 nm for Ap–Eu and 41 ± 8 nm for Ap–Tb. In contrast, the crystallite sizes determined from XRD measurements using the Software TOPAS 7.0 were found to be 53.9 nm for Ap–Eu and 51.2 nm for Ap–Tb. The crystallite size for Ap–Eu is around one third of the average particle size, suggesting that the Ap–Eu particles are polycrystalline. In comparison, the crystallite size for Ap–Tb particles is similar to the average particle size, suggesting that Ap–Tb particles are monocrystalline.
A more detailed analysis of these samples by TEM and HRTEM (Fig. 3) was performed and compared to the Ap blank prepared at 200 °C. This shows that Ap blank is formed of needle-like Ap crystals (Fig. 3a). The SAED pattern (inset) shows rings corresponding to its planes (002), (300) and (321). The HR-TEM image (Fig. 2b) of one of the elongated apatite crystals displays lattice fringes with a d-spacing of 8.211 Å, corresponding to its (100) plane. The inset shows the fast Fourier transform (FFT) of this image displaying the (100) plane. The TEM micrographs of the Ap–Eu at 200 °C (Fig. 3c and d) shows big aggregates of long needles, whose SAED pattern (inset) display the (002) and (102) planes of Ap, and small aggregates of nanocrystals whose SAED pattern (inset) shows the representative planes (104), (214), (1 0 10), and (0 2 10) of the whitlockite phase. However, the amount of this phase is negligible since it is hardly observable in the XRD pattern. Fig. 3e shows the micrograph of the Ap–Tb sample displaying nanorods and some needles. The SAED pattern (inset) displays rings, some of them corresponding to planes (002), (301), and (402) as more representative of the Ap phase, while Fig. 3f–h show EDX mappings of Ca, P and Tb of the sample of Fig. 3e.
 | ||
| Fig. 3 TEM micrographs of Ap blank (a), Ap–Eu (c and d) and Ap–Tb (e) particles prepared at 200 °C in presence of 10 mM of lanthanide. Insets show the indexed SAED patterns corresponding to Ap (a), small particles of whitlockite and long apatite–Eu crystals (c), and apatite–Tb nanoparticles (e). HR-TEM image of (a) Ap-blank crystals displaying the lattice fringes corresponding to d-spacing of the (100) plane (b). Inset shows the corresponding FFT images displaying the (100) plane. EDX element mapping analysis of Ca (f), P (g) and Tb (h) of the sample shown in micrograph (e). | ||
The characterization of the CSD and ζ-potential versus pH for suspensions of the Ap–Ln particles at physiological pHs allow us to evaluate their possible uses as luminescent nanocarriers in nanomedicine or as luminescent osteoinductive materials. The tendency for these particles to either disperse or aggregate in physiological fluids (pH ∼7.4 in the blood), or within the tumor microenvironment (pH < 6.5), is influenced by their size and surface charge.33,54 The CSD in volume of Ap-blank, Ap–Eu and Ap–Tb prepared at 200 °C with 10 mM lanthanide is presented in Fig. 4a.
 | ||
| Fig. 4 Crystal size distribution in volume (a) and ζ-potential vs. pH (b) of the samples Ap blank (orange), Ap–Eu (red) and Ap–Tb (green line) prepared at 200 °C in presence of 10 mM lanthanide. | ||
It is evident that two peaks characterize the CSD of Ap blank: one around 44 nm, which represents the size of individual particles, and another around 1280 nm dominated by aggregation. In contrast, the CSD for Ap–Eu and Ap–Tb samples is primarily dominated by aggregation, reaching sizes up to approximately 5000 nm. Notably, for Ap–Tb, the smaller particles remain within the nanometric range, as confirmed by SEM images.
To evaluate the impact of Eu3+ and Tb3+ doping on the aggregation/dispersion behavior of Ap, we compared the curves of ζ-potential versus pH, as shown in Fig. 4b. For Ap blank, the ζ-potential decreases across nearly the entire pH range, indicating increasing adsorption of hydroxide ions (OH–) on the surface of the particles. At pH values relevant to physiological conditions (between 6 and 8), the ζ-potential decreases to values between −7.5 mV and −12.5 mV, which are still too low to achieve significant dispersion of the particles. In contrast, when doped the particles with Eu3+ and Tb3+, the ζ-potential in this pH range approaches zero, suggesting compensation of the surface charge. This condition indicates a high propensity for particle aggregation. While dispersion of colloidal particles is essential when used as nanocarriers for chemotherapeutic or anti-inflammatory drugs,32,55 it is not a critical feature for their application as osteoinductive materials.
3.3. Luminescent properties
The luminescence properties in the solid phase of Ap–Eu and Ap–Tb materials are shown in Fig. 5a and b, respectively. For Ap–Eu particles the sensitized luminescence excitation wavelengths are 320, 360, 380, 393, 466 and 537 nm, and emission wavelengths are 592, 616 and 700 nm; while for Ap–Tb particles the wavelengths are 320, 353 and 373 nm for excitation and 490, 545, 590 and 624 nm for emission.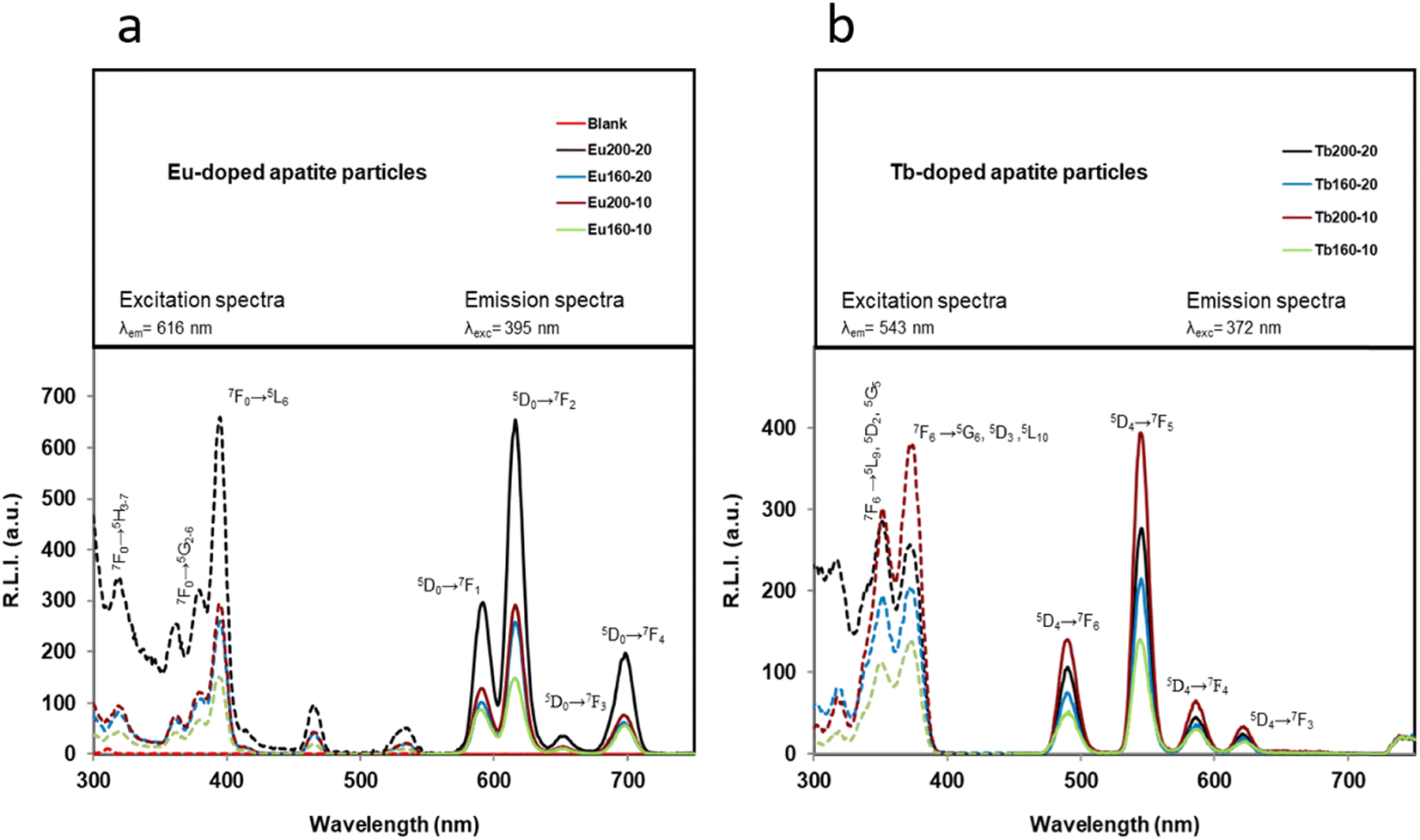 | ||
| Fig. 5 (a) Uncorrected excitation (dashed lines) and emission (solid lines) spectra of different Eu-doped particles. Slit-widthexc/em = 10/10 nm, td = 120 μs, tg = 5 ms and voltage detector = 530 V. (b) Uncorrected excitation (dashed lines) and emission (solid lines) spectra of the Tb-doped particles. Slit-widthsexc/em = 10/10 nm, td = 120 μs, tg = 5 ms and voltage detector = 580 V. | ||
These wavelengths are in concordance with those obtained in the literature for other lanthanides-doped particles.33,56 It is well known that Eu- and Tb-doped materials can be excited at the charge transfer band (CTB) which is centred around 230 nm (see Fig. S6†), but a larger excitation wavelength is preferred because the same emission spectrum is obtained and the biological applicability is increased.32 In fact, 395 nm (corresponding to the Eu3+ 7F0→5L6 transition) and 372 nm (corresponding to the Tb3+ 7F6→5G6, 5D3 transition) were selected as excitation wavelengths. Concerning the emission wavelength, those producing the highest relative luminescence intensity (RLI) are 616 and 543 nm. These wavelengths correspond to the hypersensitive transition without inversion center (5D4→7F5 for Ap–Eu and 5D4→7F5 for Ap–Tb, respectively), and are in concordance with other Eu/Tb-doped materials.57
Fig. 6a compares the RLI of Ap–Eu and Ap–Tb prepared at 160 and 200 °C with 10 and 20 mM lanthanide. At 200 °C, where the amounts of impurities bCC and LnPO4 are negligible, as described in section 3.1, it is observed that an increase in the content of Eu3+ or Tb3+ doping the material leads to an increase of the RLI (see Table 1); the sample Ap–Eu 200, 10 (1.9 Eu3+ atom%) provides a lower RLI than Ap–Eu 200,20 (2.1 Eu3+ atom%) and the same for Tb, the sample Ap–Tb 200, 10 (1.7 Tb3+ atom%) shows a higher RLI than Ap–Tb 200,20 (1.4 Tb3+ atom%). Similar results are shown at 160 °C, the sample Ap–Eu 160,10 (1.1 Eu3+ atom%) provides a lower RLI than the sample Ap–Eu 160,20 (2.5 Eu3+ atom%), and Ap–Tb 160,10 (1.5 Tb 3+ atom%) shows a higher RLI than Ap–Tb 160,20 (1.4 Tb3+ atom%). Therefore, we can conclude that the luminescence emission intensity is affected by the lanthanide content, as was found in Ap–Eu and Ap–Tb prepared by the citrate-based thermal decomplexing method.33,34
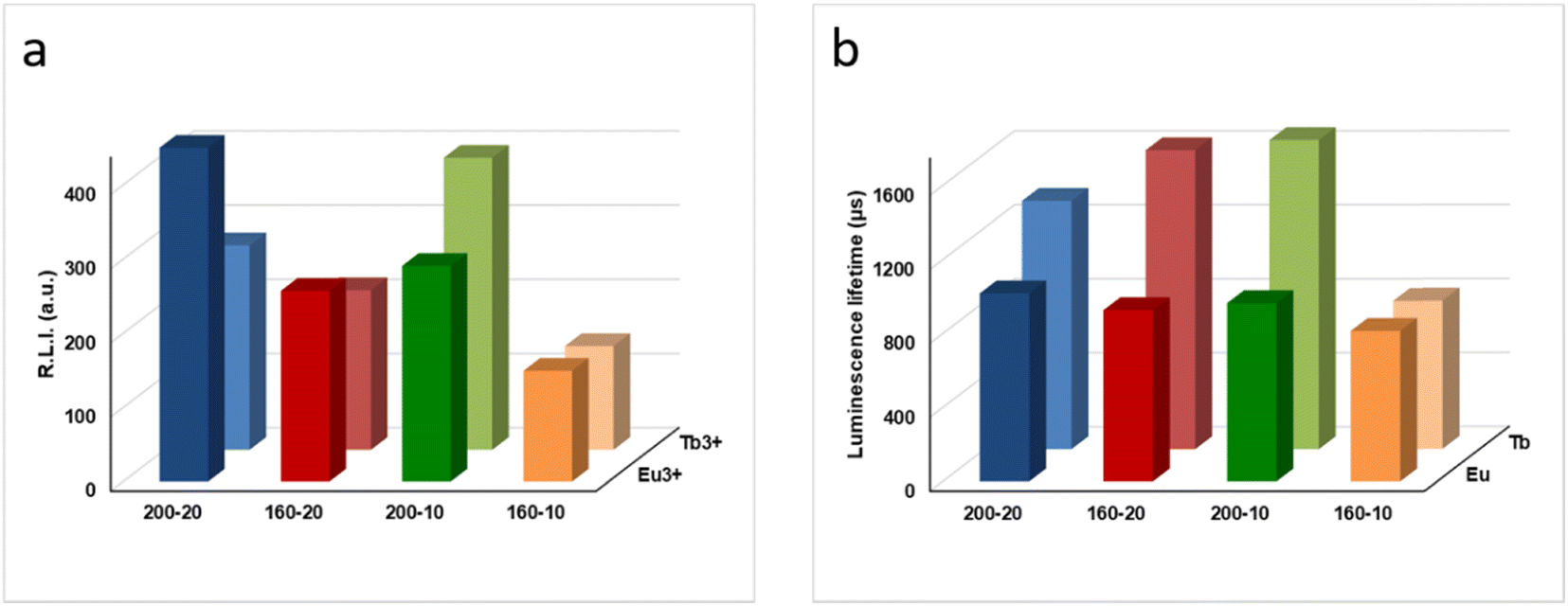 | ||
| Fig. 6 (a) Relative luminescence intensity of the Ap–Eu and Ap–Tb samples. Slit-widthexc/em = 10/10 nm, td = 120 μs, tg = 5 ms. For Ap–Eu particles, λexc/em = 395/616 nm and voltage detector = 530 V; for Ap–Tb particles λexc/em = 372/543 nm and voltage detector = 580 V. (b) Luminescence lifetime comparison between materials doped with Eu and Tb. For Ap–Eu particles λexc/em = 395/616 nm, slit-widthexc/em = 10/10 nm, and detector voltage = 700 V. For Ap–Tb particles λexc/em = 372/543 nm slit-widthexc/em = 20/20 nm, and detector voltage = 800 V. | ||
However, if RLI at 160 and 200 °C are compared, there is no consistency with the lanthanide content. This is due to the presence of the impurities at 160 °C that are not present at 200 °C, as described above and in the literature.58 The presence of the EuPO4·nH2O and TbPO4·nH2O (rhabdophane) phases impurifying the samples provides a decrease on the RLI and for this reason the RLI of Ap–Ln at 160 °C are always lower than those at 200 °C. Therefore, we can state that the presence of the EuPO4·nH2O and TbPO4·nH2O (rhabdophane) phases impurifying the samples are also important from the point of view of the RLI.
Fig. S7 and S8† show the luminescence lifetimes (τ) of Ap–Eu and Ap–Tb materials, respectively. The decay profile for each sample was analysed as a single exponential component (RLI = A × e − t/τ + C). A comparison of τ of the Ap–Eu samples (see Fig. 6b) reveals that neither the Eu3+ concentration nor the temperature significantly affects τ because it is due to the presence of Eu3+. Only, the Ap–Eu sample prepared at 160 °C, 10 mM Eu3+ shows a slightly lower τ. However, it can be considered that the lifetimes of all Ap–Eu materials are practically the same, regardless of the amount of Eu3+ atom% they contain and the presence of impurities.
Nevertheless, the τ of the Ap–Tb sample prepared at 160 °C with 10 mM Tb3+ shows a much lower luminescence lifetime than the other Tb-doped materials. This effect was previously observed.34 The decrease of τ has been attributed to the presence of bCC, rhabdophane, and undoped Ap impurifying the sample. Therefore, we can conclude that the luminescence lifetimes of the Tb-containing samples depend on the presence of impurities in the sample, while the lifetimes of the Eu-containing samples do not.
Regarding the luminescence observed by naked eyes (see Fig. 7) under UV light the Ap–Eu materials exhibit red luminescence emission, increasing the color intensity with the temperature and Eu3+ content. The sample Ap–Eu 160 °C, 10 mM, with the lower amount of Eu3+, emits pink colour, a mixture of red and blue colors. Ap–Tb materials, on their side, emit a greenish luminescence while materials free of lanthanides provides a strong blue emission. Interestingly the samples Ap–Tb 200 °C, 10 mM and Ap–Tb 160 °C, 10 mM emit in violet, a mixture of green and blue, indicating that part of the Ap is undoped. No luminescence is detected when the materials are exposed to white light.
 | ||
| Fig. 7 Pictures of the materials exposed to white (sun) or UV lights. | ||
3.4. Cytocompatibility of Ap–Eu and Ap–Tb particles
Apatite is a widely used material for biomedical applications and the introduction of partial metal doping may significantly impact its biological properties, which needs to be investigated. To assess the cytotoxicity of the particles, an MTT assay was performed after 3 days of incubation with two murine cell lines: MS-1 (pancreatic endothelial cells) and m17.ASC (mesenchymal stem cells).Fig. 8 illustrates the cell viability results, with the chemotherapeutic drug doxorubicin (reported as Doxo) serving as internal control. All treatments are given at doses of 0.1, 1, 10 and 100 μg mL−1 and compared to untreated cells (CTRL–). The particles demonstrated excellent cytocompatibility in both cell lines, with viability consistently above 85% for all materials and concentrations tested, surpassing the limit established by ISO 10993-5:2009.59 As expected, cell viability in the presence of Doxo dropped below 50% at all doses, confirming the cytocompatibility of the materials.
 | ||
| Fig. 8 Cell viability of nanoparticles in (a) MS1 and (b) m17.ASC cells. Significance levels were assessed with Dunnett's multiple comparisons test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). | ||
4. Discussion
Lanthanide-doped Ap NPs are a relatively new class of fluorophores for bioimaging and biosensing applications.32–34,60,61 Among their key features, they exhibit long luminescent lifetimes, high quantum yield, sharp emission peaks, different emission colors depending on the lanthanide ion doping the particle and good resistance to photo-bleaching caused by the environmental conditions.62 This research explores the one-step hydrothermal conversion45 of bCC to Ap–Eu and Ap–Tb particles. The objective is to transform waste material, often sent to landfills, into high-value, biocompatible luminescent apatites using a sustainable and eco-friendly method. This approach aligns with the principles of the circular economy and is relatively new in its application. To our knowledge, there have been no reports of luminescent Ap–Ln samples derived from bCC obtained from molluscs shell waste. Only recently, Ap–Eu NPs derived from eggshell were obtained by the two-step method.63Considering these factors, the evolution of the transformation of bCC with the temperature, characterized by XRD, shows that it completely disappeared from the growth medium in experiments at 200 °C. In addition to Ap–Ln, a negligible amount of whitlockite was observed by HR-TEM. Below 200 °C, the Ap–Ln phase was usually accompanied by unconverted bCC and LnPO4·nH2O (rhabdophane phase). The presence of the latter phase increases when the concentration of doping lanthanide increases to 20 mM. Only samples prepared at 200 °C with 10 mM Ln3+ were practically free of spurious phases.
It was shown that precipitation of apatite from bCC in a H2PO4-rich aqueous suspension proceeds with an increase of pH from 5.5–5.8 to around 10–10.5, and follows a dissolution–precipitation mechanism ruled by the favorable balance between apatite precipitation and calcite dissolution.45 This is particularly true at high temperatures, due to the increasing differences between the solubility products of hydroxyapatite and calcite.64,65 This difference regulates the apatite supersaturation and precipitation until the dissolution of the CaCO3.45
In this study, we have conducted a simulated experiment using the speciation software Visual MinteQ 3.1 (ref. 66) to analyze the aqueous system calcite/H2PO4−/Ln3+ as a function of pH (Fig. S9a and c†). The simulation revealed the formation of some major species including Ca2+, CO32−, CaHCO3+, HPO42−, PO43−, CaHPO4°, Ln3+, LnCO3+, LnOH2+, and OH−. Notably, in this system, when the pH exceeds 7.2–7.3, a decrease in the concentration of Ca2+ and CaHPO4° species indicates their involvement in the calcium phosphate precipitation process. Recent findings by Yang et al.67 have provided evidence that the non-classical precipitation pathway of calcium phosphate includes the formation of prenucleation clusters (PNC), specifically identified as Ca2(HPO4)32−. These clusters consist of 2 CaHPO4° and 1 additional HPO42− species. Returning to our simulation, we represented the evolution of the saturation index (SI) with 10 mM and 20 mM of Lanthanide (see Fig. S9b and d†). The results show that the solution is supersaturated for both the hydroxyapatite (reported as HAP) and the LnPO4 across the analyzed pH range. However, while below pHs 7.2–7.3, the SI for LnPO4 is higher than that for HAP, above these pHs the opposite occurs. Additionally, at 20 mM Ln3+, the SI for LnPO4 is greater than at 10 mM. This simulation illustrates that nucleation of apatite preferentially occurs followed by LnPO4, once the available CaCO3 has been depleted, leading to an increase in LnPO4 precipitation at 200 °C.
In blank precipitation experiments using KH2PO4 with a stoichiometric P/Ca ratio, the minimum temperature required for the complete transformation of bCC to Ap was found to be 160 °C.45 Nevertheless, in our study, this temperature increased to 200 °C. This change would indicate that the presence of impurities such as Eu3+ or Tb3+ slow down the dissolution of CaCO3. These impurities also affect the ionic activity product necessary to reach the critical supersaturation for the nucleation process, as they compete with Ca2+ ions for the available phosphate species. Besides the influence at the nucleation stage, we found that the morphology and average size of the Ap particles varied between the Eu–Ap and Tb–Ap samples, as well as in comparison to the Ap blank. This variation indicates that the two metals influence the crystal growth differently. In general, Eu3+ encouraged growth along the c-axis (001 direction), while Tb3+ promoted growth along the a- and b-axes.
Concerning the influence of Eu3+ and Tb3+ ions on the electrokinetic properties of the Ap, the analysis reveals that both ions similarly affect the evolution of the ζ-potential with pH, approaching it to zero at pH values of physiological interest. This shift favors particle aggregation over their dispersion. According to Somasundaran68 for sparingly soluble salts that react with the solvent, the solvent species, the lattice ions, and their complexes, can all influence the ζ-potential. In the case of aqueous suspensions of Ap–Ln within the pH range of 4 to 11, the relevant ions include Ca2+, Ln3+, H+, OH−, HCO3−/CO32−, H2PO4−, HPO42−, PO43−, as well as their charged complexes. Compared to the ζ-potential versus pH curve of undoped Ap, the ζ-potential evolution of Ap–Ln suggests that some Ln3+ ions and their positively charged complexes remain adsorbed on the outermost layers of the particle surface. This adsorption neutralizes the negatively charged surface species that contribute to the negative ζ-potential observed in the undoped apatite sample.
When comparing the samples prepared at 200 °C, 10 mM Ln3+, to those using commercial reagents by the citrate-based thermal decomplexing method,33,34 we found out key differences: i) the particles produced in the former are larger, ii) they contain impurities such as Na, K, Ni, Si, and Mg, and iii) the apatite surfaces lack the citrate coating, which otherwise is found in apatite particles (both undoped and metal-doped) obtained by the thermal decomplexing method.33,34,69 Adsorbed citrate acts by inhibiting particle growth and enhancing particle dispersion. However, the luminescence properties of samples obtained by both methods are similar, as they are primarily influenced by the lanthanide ion and content, and their biocompatibility is comparable.
Concerning the samples obtained at T < 200 °C we have demonstrated that the heterogeneity in phase composition affect the luminescent emission but does not the biocompatibility. The luminescent emission depends solely on the amount of lanthanide incorporated into the apatite (Ap) structure and on the presence of the rhabdophane phase, which can be adjusted by varying the lanthanide concentration. Even though the luminescent emission of the rhabdophane phase is lower than that of the apatitic phase, the luminescent emission of Eu-doped samples enhanced when Eu3+ concentration is increased, due to the precipitation of the new phase of EuPO4.70 In a different research, it was shown that the maximum Tb3+ doping in the apatite structure was ∼12 wt%, and the presence of additional Tb3+-containing phases was beneficial to increase the global Tb3+ content of the samples, thus increasing the luminescence intensity almost linearly up to RLI around 800.34 In contrast, the luminescence lifetimes differ between Ap–Eu and Ap–Tb and are influenced differently by the heterogeneous matrix.
In terms of biocompatibility, all samples were found to be cytocompatible when incubated with MS1 cells (murine pancreatic endothelial cells) and m17.ASC cells (murine mesenchymal stem cells), regardless of the type of lanthanide (Ln3+) or the dose of Ap–Ln used, confirming the findings described in previous studies.35
In summary, the above findings suggest that Ap–Ln, fabricated through the one-step hydrothermal method, using oyster shell bCC particles, are valuable particles that may find bioimaging applications as an implantable luminescent material.
5. Conclusions
The preparation of Ap–Eu and Ap–Tb particles by transforming bCC microparticles (Ø < 45 μm) from oyster shell waste was successfully carried out through the one-step hydrothermal method. A complete transformation of bCC was attained at 200 °C. Precipitates were composed of Ap–Eu submicron particles, with average sizes L = 163 ± 7 nm and anisometric shapes, and Ap–Tb NPs with L = 41 ± 8 nm and more isometric shapes (nearly spherical rods). The precipitates were accompanied of LnPO4·nH2O (rhabdophane) as a spurious phase irrespective of the reaction temperature (between 100 °C and below 200 °C). Its presence increased with the concentration of the doping lanthanide. Both Ap–Ln possess A- and B-carbonate substitutions and labile carbonate ions, and the ζ-potentials approached zero at pHs of physiological interest, indicating a trend to aggregation. Ap–Eu and Ap–Tb materials emit strong sensitized luminescence at 616 and 543 nm, respectively, when they are excited at 395 and 372 nm, with an average luminescence lifetime time of 1000 and 1500 μs respectively, in concordance with those reported in the literature. Their luminescence emissions are affected by the lanthanide content of the samples, irrespective of the doping concentration, as well as the presence of the rhabdophane phases (EuPO4·nH2O and TbPO4·nH2O) whose RLI and luminescence lifetimes are lower than those of Ap–Eu and Ap–Tb. Regardless of the lanthanide ion and the dose of particles used (ranging from 0.1 to 100 μg mL−1) the Ap–Ln samples were cytocompatible (cell viability >85%) against MS1 (murine pancreatic endothelial cells) and m17.ASC (murine mesenchymal stem cells).Data availability
The authors declare that the data supporting this study's findings are available within the paper and its ESI† files. Should any raw data files be needed in another format, they are available from the corresponding author upon reasonable request.Author contributions
Conceptualization, J. G.-M., G. F., F. O.; investigation, J. F. F.-S., F. O., F. J. A.-M., A. F., R. F. P., C. T.; methodology, J. G.-M., G. F., A. F., F. O.; supervision, J. G.-M., F. O.; writing – original draft preparation, J. G.-M., F. O.; writing – review and editing, J. G.-M., F. O., A. F., G. F., J. F. F.-S.; funding acquisition, J. G.-M., F. O., A. F. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work has been performed in the framework of the project “Advanced materials using biogenic calcium carbonate from seashell waste (CASEAWA), Ref. PCI2020-112108 funded by MCIN/AEI/10.13039/501100011033 and the European Union “NextGenerationEU”/PRTR”. PCI2020-112108 is part of the ERA-NET Cofund BlueBio Programme (H2020) supported by the European Union. We also acknowledge projects GBRMat (ref. 2023-151538NB-100, MCIU/AEI, Spain) and ARCHER (MUR-M4C2 I1.2 of PNRR with ID project no. MSCA_0000008, Italy). AF was supported by Ministero della Sanità RF-2018-12366471 and Compagnia San Paolo Trapezio No 68155.References
- D. E. Jacob, A. L. Soldati, R. Wirth, J. Huth, U. Wehrmeister and W. Hofmeister, Geochim. Cosmochim. Acta, 2008, 72, 5401–5415 CrossRef CAS.
- FAOSTAT, Food and Agriculture Organization of the United Nation, Available online: https://www.fao.org/faostat/en/#home.
- J. W. M. Wijsman, K. Troost, J. Fang and A. Roncarati, Global Production of Marine Bivalves. Trends and Challenges, in Goods and Services of Marine Bivalves, ed. A. C. Smaal, J. G. Ferreira, J. Grant, J. K. Petersen and Ø. Strand, Springer International Publishing, Cham, Switzerland, 2019, pp. 7–26, ISBN 978-3-319-96776-9 Search PubMed.
- A. C. Smaal, J. G. Ferreira, J. Grant, J. K. Petersen and Ø. Strand, Goods and Services of Marine Bivalves, 2019, p. 591 Search PubMed.
- D. Summa, M. Lanzoni, G. Castaldelli, E. A. Fano and E. Tamburini, Resources, 2022, 11, 48 CrossRef.
- Y. Hou, A. Shavandi, A. Carne, A. A. Bekhit, T. B. Ng, R. C. F. Cheung and A. E. d. A. Bekhit, Crit. Rev. Environ. Sci. Technol., 2016, 46, 1047–1116 CrossRef CAS.
- K. H. Mo, U. J. Alengaram, M. Z. Jumaat, S. C. Lee, W. I. Goh and C. W. Yuen, Constr. Build. Mater., 2018, 162, 751–764 CrossRef CAS.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- D. Suteu, D. Bilba, M. Aflori, F. Doroftei, G. Lisa, M. Badeanu and T. Malutan, Clean, 2012, 40, 198–205 CAS.
- P. Morseletto, Resour., Conserv. Recycl., 2020, 153, 104553 CrossRef.
- C. McLaughlan, P. Rose and D. C. Aldridge, Environ. Manage., 2014, 54, 1102–1109 CrossRef PubMed.
- A. O. Oso, A. A. Idowu and O. T. Niameh, J. Anim. Physiol. Anim. Nutr., 2011, 95, 461–467 CrossRef CAS PubMed.
- C. H. Lee, D. K. Lee, M. A. Ali and P. J. Kim, Waste Manage., 2008, 28, 2702–2708 CrossRef CAS PubMed.
- E. I. Yang, S. T. Yi and Y. M. Leem, Cem. Concr. Res., 2005, 35, 2175–2182 CrossRef CAS.
- W. Ten Kuo, H. Y. Wang, C. Y. Shu and D. S. Su, Constr. Build. Mater., 2013, 46, 128–133 CrossRef.
- J. H. Shariffuddin, M. I. Jones and D. A. Patterson, Chem. Eng. Res. Des., 2013, 91, 1693–1704 CrossRef CAS.
- K. W. Ma and H. Teng, J. Am. Ceram. Soc., 2010, 93, 221–227 CrossRef CAS.
- J. Boro, D. Deka and A. J. Thakur, Renewable Sustainable Energy Rev., 2012, 16, 904–910 CrossRef CAS.
- C. Triunfo, S. Gärtner, C. Marchini, S. Fermani, G. Maoloni, S. Goffredo, J. Gomez Morales, H. Cölfen and G. Falini, ACS Omega, 2022, 7, 43992–43999 CrossRef CAS PubMed.
- M. L. Basile, C. Triunfo, S. Gärtner, S. Fermani, D. Laurenzi, G. Maoloni, M. Mazzon, C. Marzadori, A. Adamiano, M. Iafisco, D. Montroni, J. Gómez Morales, H. Cölfen and G. Falini, ACS Omega, 2024, 9, 11232–11242 CrossRef CAS PubMed.
- N. K. Nga, N. T. Thuy Chau and P. H. Viet, Colloids Surf., B, 2018, 172, 769–778 CrossRef CAS PubMed.
- S. Scialla, F. Carella, M. Dapporto, S. Sprio, A. Piancastelli, B. Palazzo, A. Adamiano, L. D. Esposti, M. Iafisco and C. Piccirillo, Mar. Drugs, 2020, 18, 309 CrossRef CAS PubMed.
- A. Pal, S. Maity, S. Chabri, S. Bera, A. R. Chowdhury, M. Das and A. Sinha, Biomed. Phys. Eng. Express, 2017, 3, 015010 CrossRef.
- S. Rujitanapanich, P. Kumpapan and P. Wanjanoi, Energy Procedia, 2014, 56, 112–117 CrossRef CAS.
- A. Shavandi, A. E. D. A. Bekhit, A. Ali and Z. Sun, Mater. Chem. Phys., 2015, 149–150, 607–616 CrossRef CAS.
- S. Hajar Saharudin, J. Haslinda Shariffuddin, N. Ida Amalina Ahamad Nordin and A. Ismail, Mater. Today: Proc., 2019, 19, 1208–1215 CAS.
- Y. Xu, D. Wang, L. Yang and H. Tang, Mater. Charact., 2001, 47, 83–87 CrossRef CAS.
- H. Ivankovic, E. Tkalcec, S. Orlic, G. G. Ferrer and Z. Schauperl, J. Mater. Sci.:Mater. Med., 2010, 21, 2711–2722 CrossRef CAS PubMed.
- J. Gómez-Morales, M. Iafisco, J. M. Delgado-López, S. Sarda and C. Drouet, Prog. Cryst. Growth Charact. Mater., 2013, 59, 1–46 CrossRef.
- A. Ressler, A. Žužić, I. Ivanišević, N. Kamboj and H. Ivanković, Open Ceram., 2021, 6, 100122 CrossRef CAS.
- M. Okada and T. Furuzono, Sci. Technol. Adv. Mater., 2012, 13, 064103 CrossRef CAS PubMed.
- S. M. Cano- Plá, A. D'urso, J. F. Fernández-Sánchez, D. Colangelo, D. Choquesillo-Lazarte, R. Ferracini, M. Bosetti, M. Prat and J. Gómez-Morales, Nanomaterials, 2022, 12, 562 CrossRef PubMed.
- J. Gómez-Morales, C. Verdugo-Escamilla, R. Fernández-Penas, C. M. Parra-Milla, C. Drouet, F. Maube-Bosc, F. Oltolina, M. Prat and J. F. Fernández-Sánchez, RSC Adv., 2018, 8, 2385–2397 RSC.
- J. Gómez-Morales, R. Fernández-Penas, F. J. Acebedo-Martínez, I. Romero-Castillo, C. Verdugo-Escamilla, D. Choquesillo-Lazarte, L. D. Esposti, Y. Jiménez-Martínez, J. F. Fernández-Sánchez, M. Iafisco and H. Boulaiz, Nanomaterials, 2022, 12, 1257 CrossRef PubMed.
- I. A. Neacsu, A. E. Stoica, B. S. Vasile and E. Andronescu, Nanomaterials, 2019, 9, 239 CrossRef CAS PubMed.
- L. Dong, L. Zhang, Y. Jia, B. Shao, W. Lü, S. Zhao and H. You, CrystEngComm, 2019, 21, 6226–6237 RSC.
- A. V. Paduraru, O. Oprea, A. M. Musuc, B. S. Vasile, F. Iordache and E. Andronescu, Nanomaterials, 2021, 11, 2442 CrossRef CAS PubMed.
- N. L. Ignjatović, L. Mančić, M. Vuković, Z. Stojanović, M. G. Nikolić, S. Škapin, S. Jovanović, L. Veselinović, V. Uskoković, S. Lazić, S. Marković, M. M. Lazarević and D. P. Uskoković, Sci. Rep., 2019, 9, 1–15 CrossRef PubMed.
- Proficient Market Insights|The Research Reports & Consulting, https://www.proficientmarketinsights.com/, (accessed 8 November 2024).
- S. Santhosh and S. B. Prabu, Mater. Lett., 2013, 97, 121–124 CrossRef CAS.
- B. N. Alhussary, G. A. Taqa and A. A. Taqa, J. Appl. Vet. Sci., 2020, 5, 25–32 Search PubMed.
- K. Salma-Ancane, L. Stipniece and Z. Irbe, Ceram. Int., 2016, 42, 9504–9510 CrossRef CAS.
- D. L. Goloshchapov, A. S. Lenshin, D. V. Savchenko and P. V. Seredin, Results Phys., 2019, 13, 102158 CrossRef.
- D. F. Fitriyana, R. Ismail, Y. I. Santosa, S. Nugroho, A. J. Hakim and M. S. Al Mulqi, International Biomedical Instrumentation and Technology Conference, IBITeC, 2019, pp. 7–11 Search PubMed.
- R. Fernández-Penas, C. Verdugo-Escamilla, C. Triunfo, S. Gärtner, A. D'Urso, F. Oltolina, A. Follenzi, G. Maoloni, H. Cölfen, G. Falini and J. Gómez-Morales, J. Mater. Chem. B, 2023, 11, 7766–7777 RSC.
- A. Torres-Mansilla, P. Álvarez-Lloret, R. Fernández-Penas, A. D'Urso, P. A. Baldión, F. Oltolina, A. Follenzi and J. Gómez-Morales, Nanomaterials, 2023, 13, 2299 CrossRef CAS PubMed.
- A. Zamperone, S. Pietronave, S. Merlin, D. Colangelo, G. Ranaldo, E. Medico, F. Di Scipio, G. N. Berta, A. Follenzi and M. Prat, Stem Cells Dev., 2013, 22, 2873–2884 CrossRef CAS PubMed.
- J. Gómez-Morales, C. Verdugo-Escamilla, R. Fernández-Penas, C. Maria Parra-Milla, C. Drouet, M. Iafisco, F. Oltolina, M. Prat and J. F. Fernández-Sánchez, J. Colloid Interface Sci., 2019, 538, 174–186 CrossRef PubMed.
- J. Gómez-Morales, R. Fernández-Penas, I. Romero-Castillo, C. Verdugo-Escamilla, D. Choquesillo-Lazarte, A. D'Urso, M. Prat and J. F. Fernández-Sánchez, Nanomaterials, 2021, 11, 322 CrossRef PubMed.
- C. Rey, O. Marsan, C. Combes, C. Drouet, D. Grossin and S. Sarda, Characterization of Calcium Phosphates Using Vibrational Spectroscopies, in Advances in Calcium Phosphate Biomaterials, ed. B. Ben-Nissan, Springer Series in Biomaterials Science and Engineering, Springer, Berlin, Heidelberg, 2014, vol. 2, DOI:10.1007/978-3-642-53980-0_8.
- A. Grunenwald, C. Keyser, A. M. Sautereau, E. Crubézy, B. Ludes and C. Drouet, J. Archaeol. Sci., 2014, 49, 134–141 CrossRef CAS.
- G. B. Ramírez-Rodríguez, J. M. Delgado-López and J. Gómez-Morales, CrystEngComm, 2013, 15, 2206–2212 RSC.
- M. De La Pierre, C. Carteret, L. Maschio, E. André, R. Orlando and R. Dovesi, J. Chem. Phys., 2014, 140(16), 164509 CrossRef PubMed.
- M. Iafisco, J. M. Delgado-Lopez, E. M. Varoni, A. Tampieri, L. Rimondini, J. Gomez-Morales and M. Prat, Small, 2013, 9, 3834–3844 CrossRef CAS PubMed.
- Y. Jabalera, F. Oltolina, M. Prat, C. Jimenez-Lopez, J. F. Fernández-Sánchez, D. Choquesillo-Lazarte and J. Gómez-Morales, Nanomaterials, 2020, 10, 199 CrossRef CAS PubMed.
- I. Hemmilä, S. Dakubu, V. M. Mukkala, H. Siitari and T. Lövgren, Anal. Biochem., 1984, 137, 335–343 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2006, pp. 1–954 Search PubMed.
- P. Ivanchenko, G. Escolano-Casado, L. Mino, L. Dassi, J. F. Fernández-Sánchez, G. Martra and J. Gómez-Morales, Colloids Surf., B, 2022, 217, 112620 CrossRef CAS PubMed.
- I. International Standard, in ANSI/AAMI/ISO 10993-5:2009/(R)2014; Biological evaluation of medical devices —Part 5: Tests for in vitro cytotoxicity, 2009 Search PubMed.
- T. S. H. Perera, Y. Han, X. Lu, X. Wang, H. Dai and S. Li, J. Nanomater., 2015, 2015, 705390 CrossRef.
- A. Al-Kattan, V. Santran, P. Dufour, J. Dexpert-Ghys and C. Drouet, J. Biomater. Appl., 2014, 28(5), 697–707 CrossRef PubMed.
- A. Al-Kattan, P. Dufour, J. Dexpert-Ghys and C. Drouet, J. Phys. Chem. C, 2010, 114, 2918–2924 CrossRef CAS.
- T. K. Krishnapriya, A. Deepti, P. S. B. Chakrapani, A. S. Asha and M. K. Jayaraj, J. Fluoresc., 2021, 31, 1927–1936 CrossRef CAS PubMed.
- K. H. Prakash, R. Kumar, C. P. Ooi, P. Cheang and K. A. Khor, Langmuir, 2006, 22, 11002–11008 CrossRef CAS PubMed.
- L. N. Plummer and E. Busenberg, Geochim. Cosmochim. Acta, 1982, 46, 1011–1040 CrossRef CAS.
- J. P. Gustafsson, Visual MINTEQ 3.1, freeware chemical equilibrium model, https://vminteq.lwr.kth.se/, (accessed 23 May 2021).
- X. Yang, C. Zhang, X. Yang and Z. Xu, J. Mol. Liq., 2023, 378, 121585 CrossRef CAS.
- P. Somasundaran, J. Colloid Interface Sci., 1968, 27, 659–666 CrossRef CAS.
- J. Torrent-Burgués, J. Gómez-Morales, A. López-Macipe and R. Rodríguez-Clemente, Cryst. Res. Technol., 1999, 34, 757–762 CrossRef.
- M. F. Vega-Zerpa, S. Briceño, J. Bahamonde-Duarte, K. Vizuete, A. Debut, R. Uribe, L. J. Borrero-González and G. González, Ceram. Int., 2024 DOI:10.1016/j.ceramint.2024.07.235.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce01217h |
| This journal is © The Royal Society of Chemistry 2025 |