
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational analysis of biomineral proteins interacting with inorganic minerals using dispersive mineral particles for solution NMR
Kei
Futagawa
,
Yugo
Kato
and
Michio
Suzuki
 *
*
Department of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan. E-mail: amichio@g.ecc.u-tokyo.ac.jp
First published on 24th February 2025
Abstract
Minerals formed by living organisms are referred to as biominerals, and the process of their formation is known as biomineralization. These biominerals result from organic–inorganic interactions involving biomineral proteins and minerals. However, the detailed molecular mechanisms underlying these interactions remain unclear due to the lack of established methods for analyzing the conformations of biomineral proteins. Existing conformational analysis techniques, such as X-ray crystallography and cryo-electron microscopy, are insufficient for studying these proteins, as they cannot reveal protein structures on solid surfaces. Solution nuclear magnetic resonance spectroscopy offers high-resolution capabilities for determining protein structures but requires solubilized proteins that can move and rotate freely in liquid solutions. Dispersive mineral particles provide a solid surface for proteins while allowing rotation in solution without inducing anisotropic effects. This novel conformational analysis method using dispersive mineral particles presents a promising approach to elucidate the molecular mechanisms of various biomineral proteins. Furthermore, this insight may enable the synthesis of valuable and eco-friendly mineral materials by mimicking biomineralization processes.
1 Introduction
1.1 Mineral synthesis via organic–inorganic interaction
Living organisms, including animals, plants, and microorganisms, are primarily composed of organic matrices. However, inorganic elements also play critical roles in maintaining biological functions. Many inorganic elements exist in bodily fluids as ions. Conversely, living organisms accumulate these inorganic elements and utilize them to form hard tissues by converting inorganic ions into solid-state minerals.1 Minerals formed by living organisms are referred to as biominerals, and the processes leading to their formation are known as biomineralization.2,3 Typical examples of biominerals include calcium carbonate (CaCO3) in the exoskeletons of mollusks and shells, and otoliths;4–7 calcium phosphate in the bones and teeth of vertebrates;8,9 calcium oxalate in the stems and leaves of plants;10 and hydrated amorphous silica accumulated in rice.11 Additionally, certain microorganisms synthesize minerals such as magnetite and gold nanoparticles for detoxification and metabolic purposes.12–15 Biominerals are found across diverse life forms, from prokaryotes to higher organisms, where they support structural integrity, provide protection from external threats, and store inorganic substances. Organic molecules, such as proteins associated with biominerals, regulate crystal polymorphs and other properties to fulfill specific biological purposes.16The pathways of crystallization through organic–inorganic interactions are presented in Fig. 1. Crystals generally form via the addition and attachment of individual ions, atoms, or molecules, resulting in the growth of bulk crystals through classical pathways. Alternatively, minerals that are unstable under standard temperature and pressure, such as amorphous phases and nanoparticles, can be synthesized with the involvement of organic substances like proteins. These unstable nanoparticles, stabilized by organic matrices, serve as an essential initial phase for controlling crystal polymorphs, morphology, orientation, and defects during crystal growth in non-classical pathways. Minerals synthesized via organic–inorganic interactions exhibit exceptional toughness and stiffness due to their fine microstructures. Despite these advancements, elucidating the molecular conformation and reactions involved in organic–inorganic interactions during this transitional process remains challenging. A novel method utilizing solution nuclear magnetic resonance (NMR) with dispersive minerals offers a promising approach to clarify these interactions in the non-classical pathway. This conformational information could pave the way for the development of environmentally friendly techniques to synthesize valuable materials under standard conditions, avoiding toxic byproducts. Consequently, mechanisms governing diverse crystal formation processes under inorganic conditions have garnered significant research interest.
 | ||
| Fig. 1 Classical and non-classical pathways to crystallization. One ion or molecule directly becomes bulk crystal in the classical pathway. On the other hand, non-classical pathways use the intermediate process with clusters or nanoparticles. The biological process of crystallization in biomineralization is non-classical pathway. | ||
1.2 Conformational analysis methods of protein in the mineral surface
Conformational analyses of proteins are crucial for elucidating the detailed molecular mechanisms of their reactions and interactions with ligands. However, analyzing the conformation of proteins involved in organic–inorganic interactions remains challenging for several reasons.To investigate the conformational changes of biomineral proteins on solid surfaces, mixture samples of minerals and proteins in solution are required. Conventional methods for analyzing these samples face several limitations. X-ray crystallography, a widely used technique, determines atomic-level protein structures by analyzing diffraction patterns from crystallized protein samples.17–19 However, it is not feasible to prepare protein crystals in the presence of minerals because mineral crystals grow much faster than protein crystals. This rapid growth results in the precipitation of bulk mineral crystals, which hinders protein crystallization. Furthermore, X-ray crystallography is a static conformational analysis method and is suitable only for proteins with fixed or ordered three-dimensional structures. Cryo-electron microscopy, a state-of-the-art technique, enables high-resolution visualization of protein complexes in near-native states without requiring protein crystallization.20–22 However, in mixture samples, scattered light from minerals interferes with the production of high-resolution images, making this technique unsuitable for analyzing biomineral proteins involved in organic–inorganic interactions.
Given these challenges, we chose to analyze the conformational changes of biomineral proteins using NMR.23 Solid-state NMR can detect signals from proteins in solid mineral samples. However, it lacks sufficient resolution to assign detailed proton, carbon, and nitrogen signals from individual amino acids, which are essential for determining atomic distances and performing detailed conformational analyses. Conversely, solution NMR offers high-resolution capabilities for analyzing the three-dimensional structures of proteins but requires that proteins and minerals remain uniformly dispersed in solution. Unfortunately, bulk minerals are unable to rotate or disperse in solution, leading to mineral precipitation and causing anisotropic effects in the NMR spectrum. Consequently, both conventional solid-state NMR and solution NMR methods fail to achieve high-resolution spectra for conformational analysis. To address these challenges, we designed the following experimental system. Proteins are mixed with dispersive minerals in solution.24–26 Unlike bulk minerals, dispersive minerals can rotate and disperse within the solution, thereby reducing the anisotropic effects on the spectrum and producing sharper signals.27 The addition of dispersive mineral particles to the solution enables the analysis of conformational changes in biomineral proteins on solid surfaces using solution NMR,27 offering a novel method for studying organic–inorganic interactions. This Highlight covers three main topics: the interaction between aragonite and the biomineral protein Pif 80 in the pearl oyster, the interaction of amorphous calcium carbonate (ACC) with paramyosin, and the interaction of gold nanoparticles with DP-1, a protein found in microorganisms.
DQF-COSY (double quantum filtered correlation spectroscopy) and TOCSY (total correlation spectroscopy) detect correlations between protons within the same amino acid. HSQC (heteronuclear single quantum correlation) and HMBC (heteronuclear multiple bond coherence) detect correlations between protons and carbons. ROESY (rotating-frame Overhauser effect spectroscopy) is used to identify spatial relationships between protons within a molecule.
The method to analyze the conformational changes of biomineral proteins is presented in (Fig. 2). First, all 1H and 13C signals in solution NMR are attributed to individual amino acids using 2D-NMR spectra (DQF-COSY, TOCSY, HSQC, and HMBC). Next, the sequence of the amino acids is identified based on HMBC and ROESY data. The obtained NMR data are processed using NMRPipe to refine the topology of each 2D-NMR spectrum, and ROESY spectra are analyzed with SPARKY.28,29 Using SPARKY, assignments are made for all ROESY signals, and peak intensities are converted into distance constraints. Based on 1H chemical shift assignments from SPARKY, peptide conformations are computed using CYANA (combined assignment and dynamics algorithm).30–32
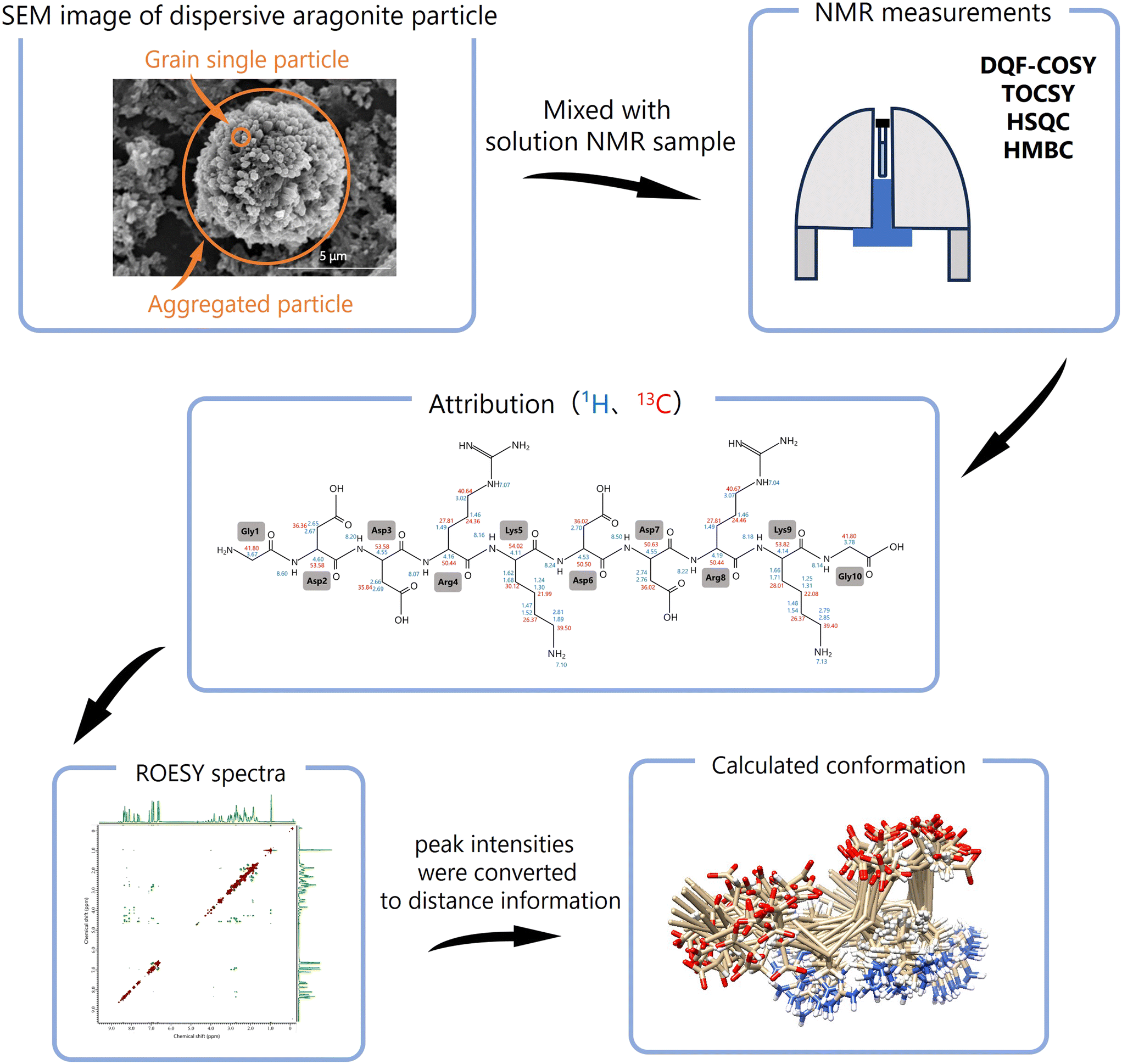 | ||
| Fig. 2 Flow of conformational analysis by solution NMR using dispersive mineral particles. The amino acid residues are attributed by 1H, 13C, DQF-COSY, TOCSY, HSQC, and HMBC. Based on 1H chemical shift attribution of ROESY, the peak intensities were converted to distance information between residues and 3D conformations of peptides were calculated and computed by using CYANA.30–32 | ||
2 Conformational analyses of biomineral proteins from the shell of the pearl oyster, Pinctada fucata
2.1 Biomineralization of invertebrates
Shellfish and crustaceans primarily consist of CaCO3 in their shells and exoskeletons, providing structural protection. Since CaCO3 is a naturally abundant substance on Earth and widely used by various organisms as a biomineral, extensive research has been conducted on its biomineralization. The general mechanisms underlying the localization and functions of organic matrices in shell biomineralization are presented in (Fig. 3(a)). The insoluble frameworks, composed of polysaccharides such as chitin (a β-polymer of N-acetyl-D-glucosamine), create scaffolds that provide space for the precipitation of inorganic minerals. Insoluble matrix proteins, which possess hydrophobic regions for protein–chitin interactions and hydrophilic regions for binding inorganic elements, mediate connections between the organic scaffolds and inorganic minerals. These proteins regulate nucleation, crystal polymorphism, and crystal orientation. Meanwhile, soluble matrix proteins, which contain only hydrophilic regions, regulate the formation and growth of inorganic minerals. During crystal growth, insoluble matrix proteins form organic frameworks as intercrystallite matrices, while soluble matrix proteins are incorporated into the CaCO3 crystals as intracrystalline organic matrices. This dynamic interplay between organic and inorganic components results in the formation of biominerals with complex structures through organic–inorganic interactions.33 | ||
| Fig. 3 (a) Schematic diagram of organic–inorganic interaction in biominerals. (b) Crystal structures of each CaCO3 polymorph. (c) Picture of shell and shell microstructures of Pinctada fucata. M indicates the myostracum, P indicates the prismatic layer, N indicates the nacreous layer, and A indicates the adductor muscle. | ||
2.2 Calcium carbonate
CaCO3 is a white powder widely used in various industries, including paint, pigments, paper, plastic fillers, tablet base materials, and chalk. It is the most abundant biomineral on Earth, forming a significant part of the planet's biomass.34,35 Carbonate sediments, accounting for 60–80% of the chemical carbon on Earth (approximately 40 million gigatons), are produced through the biomineralization processes of living organisms. These sediments play a crucial role in regulating atmospheric concentrations of carbon dioxide and bicarbonate.36,37 CaCO3 primarily exists in three crystal polymorphs: calcite, aragonite, and vaterite. Calcite, the most thermodynamically stable polymorph at room temperature and pressure,38–40 forms a hexagonal crystal system (Fig. 3(b)).41 Aragonite, a metastable orthorhombic crystal system, forms under high-pressure conditions or in the presence of magnesium ions (Fig. 3(b)).42–44 Vaterite, the most unstable polymorph of CaCO3, belongs to a hexagonal crystal system. Despite extensive research, no unified interpretation of its crystal structure has been established (Fig. 3(b)).45–47 In contrast, the predominant polymorphs found in biominerals are calcite and aragonite. Due to its instability, vaterite is rarely found in natural minerals. However, it has been identified in freshwater pearl oyster shells and fish otoliths.48–51 These crystal polymorphs are meticulously controlled within the microstructures of biominerals, largely owing to their biological functions.CaCO3 also exists in an amorphous form, referred to as ACC. ACC is highly unstable and can be synthesized in vitro under conditions of low temperature, high pressure, and high pH. Nevertheless, ACC rapidly undergoes a phase transition to more stable forms such as calcite and aragonite.38–40 In some biominerals, ACC serves as a precursor or a stable constituent alongside calcite or aragonite.38–40 For instance, ACC is present during the initial stages of calcification in the bone fragments of sea urchin larvae and the shells of bivalve larvae.52 Additionally, ACC is stabilized in the exoskeletons of crustaceans, where it plays a key role during molting, being dissolved and subsequently restructured.52–55
The synthesis of ACC under inorganic conditions has been extensively studied. Methods involving phosphate compounds, such as phosphorylated amino acids and sodium dihydrogen phosphate, have been reported.55–58 Other studies suggest that magnesium, silicate, and dimethyl carbonate also contribute to the stabilization of ACC.38,59,60
2.3 Mother of pearl, Pinctada fucata
The Japanese pearl oyster, Pinctada fucata (P. fucata), is widely used in pearl aquaculture in Japan. Its shell, a biomineral primarily composed of CaCO3, consists of mineralized microstructures, including the nacreous and prismatic layers (Fig. 3(c)). The shell is formed by a soft tissue called the mantle. The extrapallial fluid, located in the space between the mantle and shell, is separated from the surrounding seawater. The shell interacts with the mantle, which supplies the periostracum and calcified microstructures with inorganic ions and organic matrices through the extrapallial fluid. The conditions within this fluid—including the concentration of organic molecules, trace elements, and pH—regulate the polymorphs and shapes of CaCO3 crystals, determining the shell's microstructures.The shell of P. fucata consists of two primary layers: the outer prismatic layer and the inner nacreous layer (Fig. 3(c)). The outer prismatic layer is composed of calcite crystal columns.61,62 Each columnar crystal is encapsulated by an organic framework of materials such as chitin and biomineral proteins. The crystallographic c-axis of these crystals is oriented parallel to the shell surface.63–65 The inner nacreous layer comprises aragonite crystal tablets (Fig. 3(c)). In this layer, the growth direction of the aragonite crystals aligns with the c-axis of the tablets, which are also oriented parallel to the shell surface.63 The nacreous layer's stacked structure produces a unique rainbow-like luster due to the interference of light reflected from its layers.62,66
In addition to the prismatic and nacreous layers, the shell also contains a microstructure known as the myostracum, which attaches to the adductor muscle (Fig. 3(c)). The myostracum consists of elongated prismatic aragonite crystals aligned along the c-axis, further contributing to the shell's structural integrity.26
2.4 Biomineral proteins in P. fucata shell
Biomineral proteins are essential for biomineral formation, prompting extensive research into extracting these proteins from shells and identifying their sequences. Shell demineralization is achieved using agents such as EDTA (ethylenediaminetetraacetic acid) or acids to isolate biomineral proteins.4,67,68 As a result, proteins can be categorized as soluble or insoluble in EDTA and acid. To extract insoluble proteins, denaturing agents like urea and SDS (sodium dodecyl sulfate) are employed, as these proteins are cross-linked with insoluble organic matrices such as chitin. Previous studies have identified unique biomineral proteins from the prismatic and nacreous layers of P. fucata shells, shedding light on their specific roles.Several key biomineral proteins have been identified in P. fucata. MSI31, found in the prismatic layer,69 contains Gly, Asp, and Glu-rich regions that may interact with Ca2+ ions. Prismalin-14, also located in the prismatic layer,70,71 has a Gly- and Tyr-rich N-terminal domain and an Asp-rich C-terminal domain. Nacrein, found in the nacreous layer,72 includes a Gly-X-Gln repeat region that binds calcium ions. Pif, another biomineral protein in the nacreous layer,73 has a C-terminal region (Pif 80) associated with aragonite crystallization and contains an Asp-Asp-Arg-Lys (DDRK) repeat region (Fig. 4(a)). Paramyosin, the primary component of the adductor muscle in P. fucata, includes many Glu residues and a low-complexity region near its C-terminal, featuring an Glu-Glu-Ala-Glu (EEAE) repeat region (Fig. 4(c)).26 These biomineral proteins commonly contain low-complexity regions, such as repeat sequences and regions with unbalanced amino acid compositions. Proteins with such regions are categorized as intrinsically disordered proteins (IDPs) due to their lack of a fixed or ordered three-dimensional structure.74–76 Consequently, most biomineral proteins in P. fucata lack a stable three dimentional structure (Fig. 4(b)), making it challenging to determine their conformations and functions using conventional analytical methods.
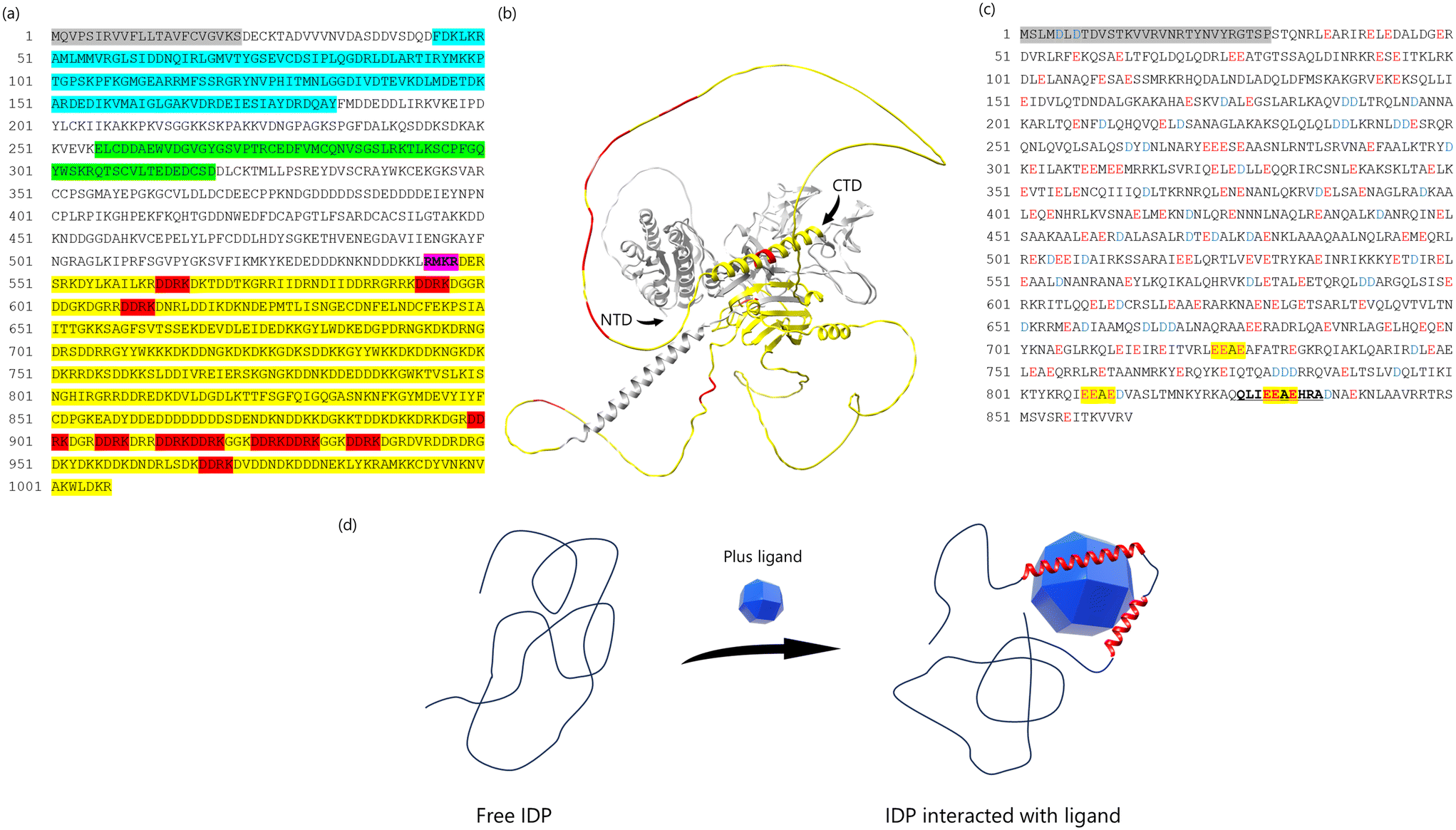 | ||
| Fig. 4 (a) The complete amino acid sequence of Pif. Pif 97 consists of 525 amino acids (from 23 to 547 amino acid). Pif 80 consists of 460 amino acids (from 548 to 1007 amino acid). Gray box is a signal peptide (from 1 to 22 amino acid). Blue box indicates the VWA domain. The green box is the chitin-binding domain. Purple box is RMKR (a Kex2-like proteinase cleavage site). Yellow box is the aragonite-binding domain (Pif 80). Red box indicates the sequence of Asp-Asp-Arg-Lys (DDRK). (b) The prediction of 3D structure of Pif using AlphaFold3.75 The yellow parts indicates Pif 80, the red parts indicates sequence of DDRK, NTD indicates N-terminal domain, CTD indicates C-terminal domain. (c) The amino acid sequence of paramyosin-3. Glu (E) indicated in red, peptide analyzing conformations are indicated in bold and underlined and EEAE sequence in yellow highlighter. (d) Schematic image of IDP binding ligand. The IDP does not form the fixed 3D structure in the solution, because of no typical secondary structure such as beta-sheet or alpha-helix. When the ligand binds to the IDP structure, the interaction between IDP and ligand decreases the flexibility of IDP structure and induce the 3D structure. We assumed that the biomineral proteins containing IDP structure employ this system. | ||
However, we decided to analyze the conformational changes of biomineral proteins with low-complexity regions for the following reason: IDPs exist in a denatured state, with their main chain adopting a highly dynamic and disordered conformation when unbound. However, IDPs can undergo conformational changes and interact with ligands in a manner that aligns with the ligand's conformation (Fig. 4(d)). Biomineral proteins with low-complexity regions often play a key role in regulating the crystallization of shells. We hypothesized that the low-complexity regions of biomineral proteins may partially adopt a three-dimensional structure upon interacting with the solid surface of CaCO3, thereby controlling shell crystallization. To test this hypothesis, specific sequences, such as repeat regions, were selected as peptides for analyzing conformational changes using solution NMR and dispersive CaCO3 particles.
2.5 Synthesis of dispersive calcium carbonate particles
The synthesis of dispersive CaCO3 particles is critical for studying the conformational changes of biomineral proteins on solid mineral surfaces. Moreover, the synthesis must occur under strictly inorganic conditions to prevent contamination by organic substances, which could interfere with solution NMR measurements. We developed a method for synthesizing dispersive aragonite particle and ACC (Fig. 5 and 6).24,58,62 These CaCO3 polymorphs were characterized and stabilized using techniques such as Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD), scanning electron microscopy (SEM) and X-ray absorption fine structure (XAFS). Based on these analyses, the dispersive particles were confirmed to be suitable for use in solution NMR measurements. | ||
| Fig. 5 (a) Schematic diagram of synthesis of dispersive CaCO3 particles. The synthesized dispersive CaCO3 particles were washed three times with ultrapure water and ultrasonicated before mixing into the NMR sample. (b)–(f) Measurement of dispersive aragonite particles. (b) SEM, (c) the histogram indicated the size and distribution of dispersive aragonite particles. (d) XRD, (e) FT-IR, and (f) XAFS. Signals derived from aragonite indicate red in (e) and (f). Reproduced with permission from ref. 24. Copyright 2023, American Chemical Society. | ||
 | ||
| Fig. 6 (a) and (b) Ca K-edge XANES spectra of (a) Pi-ACC powder and (b) Mg-ACC powder. Signals derived from ACC indicate red in (a) and (b). The peak shoulder in the range 4044.8–4045.3 eV and the maximum absorption of 4048.3 eV that are unique to ACC was detected. The features of these spectra are identical to each other. (c) and (d) SEM images of CaCO3 from Pi-ACC solution. Low magnification image (c) and high magnification image (d). (e) The histogram indicated the size and distribution of dispersive Pi-ACC particles. Reproduced with permission from ref. 58. Copyright 2020, Oxford Academic. | ||
2.6 Conformational and functional analysis of DDRK-repeat region in Pif 80
Pif, an acidic matrix protein, is essential for the formation of aragonite crystal tablets in the nacreous layer of P. fucata. The aragonite crystals grow with their c-axis aligned parallel to the shell surface, as demonstrated by knockdown assays.73 Pif is initially synthesized as a single translation product and then cleaved by a Kex2-like protease at the RMKR site (Kex2-like proteinase cleavage site), yielding two fragments: Pif 97 and Pif 80.77 Pif 97 contains von Willebrand A (VWA) and chitin-binding domains, which are involved in interactions with organic matrices. Pif 80, on the other hand, features low-complexity regions rich in both acidic and basic amino acids and exhibits aragonite-binding activity.78,79Previous studies reported that acidic and basic amino acids can interact with Ca2+ and CO32 ions, influencing the formation of CaCO3. Notably,3,80–82 Pif 80 contains a unique Asp-Asp-Arg-Lys (DDRK) repeat region. However, the molecular mechanisms underlying the organic–inorganic interactions in the DDRK repeat region remain unclear. To investigate the role of acidic and basic amino acids in Pif 80, we directly analyzed the interactions between the low-complexity region and CaCO3 using solution NMR with dispersive aragonite particles.
We hypothesized that the low-complexity region of biomineral proteins might partially adopt a three-dimensional structure on the solid CaCO3 surface, thereby influencing shell crystallization. To test this hypothesis, we synthesized peptides and mutants containing two DDRK repeat regions. The peptide sequences and their abbreviations were as follows: GDDRKDDRKG (GDR), GEERKEERKG (GER), GAARKAARKG (GAR), and GDDAADDAAG (GDA). NMR samples were denoted as “peptide name/sample condition”; for example, a sample with dispersive aragonite particles incorporated into GDR was labelled GDR/Ara.
The calculated conformations of the peptide are presented in (Fig. 7(a)–(d)). When focusing on the main chain, the peptide-only structures, in the absence of dispersive aragonite particles and Ca2+, were folded and rounded. This characteristic was particularly evident in the GER/water and GDA/water systems. By contrast, the incorporation of dispersive aragonite particles caused the main chains of the peptides to elongate, a feature that was particularly evident in the GDR/Ara and GAR/Ara systems.
 | ||
| Fig. 7 (a)–(d) Conformations of each peptide calculated using CYANA. Overlay of 20 best-matched 3D structures of each peptide (left); the main chain is labeled “ball and stick”, and all side chains are labeled (center). The surface was displayed as “coulombic surface coloring” (right). (a) GDR, (b) GER, (c) GAR, and (d) GDA. Reproduced with permission from ref. 24. Copyright 2023, American Chemical Society. | ||
Focusing on the side chain, in GDR/Ara, the four Asp Hβ residues were aligned in the same plane and sandwiched between Arg and Lys. In GER/Ara, the four Glu Hγ side chains corresponding to Asp Hβ in GDR/Ara were arranged in the same plane, even if a slight misalignment was present, and sandwiched between Arg and Lys. This conformational feature of acidic amino acids in the side chain was not observed in GDA/Ara. In GAR/Ara, the four Ala Hβ residues corresponding to Asp Hβ in GDR were arranged in the same plane and sandwiched between Arg and Lys. These results suggested that it does not matter whether the side chains are Asp, Glu, or Ala for alignment in the same plane. On the other hand, Arg and Lys are structurally indispensable for aligning the side chains of the sandwiched amino acid residues in the same plane.
In the GDR/Ca ion and GER/Ca ion samples, the acidic and basic residue side chains were clustered on the same side, and these peptides formed a folded and rounded structure, indicating that free Ca2+ in the solution promoted electrostatic interactions between acidic and basic residues and induced the folded structure of the peptides. On the other hand, Ca2+ in aragonite was fixed in the crystal. Fixed Ca2+ may promote the elongated and aligned structure of the peptides.
These results of conformational analysis using ROESY indicated that the main GDR chain was stretched, and the acidic residue side chains in GDR were located on the same plane, suggesting that GDR/Ara may be the most suitable structure to adhere to the aragonite surface.
We also measured STD (saturation transfer difference) to clarify which amino acid residue mainly interacts with aragonite. The STD method is commonly used to detect correlations between ligands and receptors.83,84 In this study, H2O hydrated on the aragonite surface was used as the fixed receptor, and a peptide was used as the ligand.85,86 The STD results of GDR/Ara indicated that Asp Hβ was the residue with the highest STD0 value, suggesting that Asp Hβ is closest to aragonite and binds to it (Fig. 8(a) and (b)).
 | ||
| Fig. 8 (a) STD amplification factors plotted against saturation time (STD buildup curves) for well-separated peaks. (b) Bar graph of the relative initial rate of the Astd buildup curve, STD0, for the Hβ protons of each residue. The STD0 value for the Asp Hβ is set to 100%. (c) Schematic diagram of the 3D structure of GDR on the aragonite surface. The trapezoid has four side chains with acidic residues at the top. The upper side was 6.9 Å, the lower side was 20.3 Å, and the height was 3.5 Å. The positions of Ca2+ were corresponding to the corners in the trapezoid. Reproduced with permission from ref. 24. Copyright 2023, American Chemical Society. | ||
To investigate the effect of peptides on CaCO3 crystallization, we conducted in vitro crystallization experiments as follows. When Ca2+ and CO32− concentrations become supersaturated, spherical shapes of ACC and vaterite initially form. These phases eventually dissolve, leading to the precipitation of rhombohedral calcite.87 Peptides were added to this system to evaluate their interaction with CaCO3 by comparing the duration for which ACC and vaterite were retained. ACC and vaterite exhibited spherical morphologies, whereas calcite displayed a characteristic rhombohedral shape.58,87 The crystalline polymorphism of CaCO3 was assessed based on the observed morphologies using SEM.
Among the peptides tested, GDR maintained ACC for the longest duration. Conformational analysis revealed that the acidic side chains of GDR aligned in the same plane, facilitating interaction with the CaCO3 surface. In contrast, ACC retention time for GDA was shorter than that of GDR, suggesting that basic side chains also contribute to stronger interactions with CaCO3. GER retained ACC for a shorter time than both GDR and GDA, indicating that Asp interacts more strongly with CaCO3 than Glu.
Based on these experimental results, the GDR/Ara structure appeared to be particularly well-suited for adhesion to the CaCO3 surface and interaction with aragonite. The distance between the carboxyl groups of the four Asp side chains in GDR was measured to understand this interaction further. The four Asp side chains formed a trapezoid with a 6.9 Å upper side, 20.3 Å lower side, and 3.5 Å height (Fig. 8(c)). This trapezoidal arrangement closely resembled the spacing of Ca2+ ions in the {001} plane of aragonite, which has a trapezoid shape with a 5 Å upper base, 20 Å lower base, and 4.3 Å height. These results suggest that the spatial arrangement of Asp side chains in GDR aligns with the Ca2+ positions in aragonite, facilitating binding to aragonite crystals during the crystallization process.
From our results, we observed that the acidic side chains of GDR closely matched the arrangement of Ca2+ ions on the {001} plane of aragonite. Fig. 8(c) provides a schematic diagram of the GDR structure on the aragonite surface, showing that Asp Hβ residues aligned in the same plane may interact directly with the {001} plane of aragonite. The DDRK repeat sequences found in Pif play a crucial role in forming organic frameworks and mediating interactions between organic frameworks and aragonite.73 Since the {001} plane is the primary surface of aragonite tablets in the nacreous layer, the DDRK sequence likely binds to this plane via Asp residues, enhancing organic–inorganic interactions.
This binding of the carboxyl group in the DDRK sequence to the {001} plane of aragonite regulates crystal growth along the c-axis, facilitating the formation of aragonite tablets in the nacreous layer.
2.7 Conformational and functional analysis of amorphous calcium carbonate-binding protein paramyosin in P. fucata
ACC serves as a critical precursor phase for the formation of aragonite crystals in the shells of P. fucata. To identify proteins involved in ACC binding within the inner aragonite layer (nacreous layer and myostracum) shell extracts were subjected to ACC-binding experiments. Semiquantitative analyses using liquid chromatography-mass spectrometry (LC-MS/MS) identified paramyosin, a major component of the adductor muscle, as strongly associated with and specifically incorporated into ACC.Western blotting confirmed that paramyosin is present in the myostracum, a microstructure in the shell attached to the adductor muscle (Fig. 3(c)). Since aragonite is the main component of the myostracum, we synthesized aragonite crystals in vitro in the presence of paramyosin. These experiments revealed that paramyosin promotes the formation of prism-shaped aragonite crystals elongated along the c-axis, which resembles the morphology of prism aragonite crystals in the myostracum (Fig. 9(a)). From these findings, paramyosin was identified as an ACC-binding protein that influences the formation and morphology of prism-shaped aragonite crystals. Paramyosin contains numerous Glu residues and a low-complexity region near its C-terminal, including the Glu-Glu-Ala-Glu (EEAE) repeat region (Fig. 4(c)). We hypothesized that the EEAE repeat region in paramyosin may undergo conformational changes upon interacting with the ACC surface, thereby influencing CaCO3 crystal formation in a manner similar to Pif 80. Previous studies have shown that acidic amino acids strongly interact with Ca2+, affecting CaCO3 crystal formation.3,80,81 However, differences between Asp and Glu in their roles in CaCO3 crystal formation remain unclear. To address this, we analyzed the conformational changes of the EEAE peptide (QLIEEAEHRA) and DDAD peptide (QLIDDADHRA) (Fig. 4(c)) using solution NMR with ACC, applying the same methodology as for the DDRK repeat region. The small size of ACC, with a diameter of approximately 100 nm, allows it to rotate and disperse freely in solution, minimizing anisotropy effects and producing sharp NMR signals.27,50,60
 | ||
| Fig. 9 (a) (i), (ii) SEM and (iii) TEM images of CaCO3 crystals after 24 h under aragonite-forming conditions with 50 μg mL−1 of paramyosin added. (iv) Electron diffraction pattern of (iii). (b) The most likely conformation of each peptide (left). The surface is displayed as “coulombic surface coloring” (center). Overlay of 20 best-matched conformation of each peptide (right). The white box above shows the conformations only backbone. Conformation shows that the main chain is labeled “ball and stick”, and all side chains are labeled. (i) EEAE and (ii) DDAD. (c) STD amplification factors plotted against saturation time (STD buildup curves) for well-separated peaks. (d) STD0 of the 1Hβ of each residue. (i) EEAE; (ii) DDAD. Reproduced with permission from ref. 26. Copyright 2024, American Chemical Society. | ||
Based on 1H chemical shift attributions, peptide conformations were calculated using CYANA-3.98.15 with ROESY peaks.31 The results are presented in (Fig. 9(b)). Both EEAE/water and DDAD/water structures exhibited folded and rounded main chains in the absence of ACC. However, incorporation of ACC elongated the entire molecule. Examining the side chains, the EEAE/ACC structure revealed that the three Glu side chains and the imidazole group of His8 were aligned in the same plane, a feature not observed in DDAD/ACC. These results suggest that EEAE/ACC forms a structure more suited for attachment to the ACC surface. The longer side chain of Glu in EEAE likely facilitates flexible conformational changes during ACC binding, enabling alignment of carboxy and imidazole groups for optimal interaction with the ACC surface (Fig. 9(b)).
To identify the amino acid residues primarily interacting with ACC, we performed STD measurements. The residue with the highest STD0 value—indicating the greatest degree of saturation transfer—was Ala6 Hβ in the EEAE peptide and Asp Hβ in the DDAD peptide (Fig. 9(c)). Relative saturation mobility was computed, normalizing the STD0 values of Ala6 in the EEAE peptide and Asp in the DDAD peptide to 100% (Fig. 9(d)).
In the EEAE peptide, Glu Hβ and Ala10 Hβ exhibited the next highest relative values at 51.4% and 49.7%, respectively (Fig. 9(d)). These results suggest that if the carboxyl group of Glu binds to Ca2+ ions on the ACC surface, Ala6 may be positioned on the opposite side of the ACC particle. However, STD measurements primarily reflect the proximity of residues to hydrated water molecules associated with ACC. Thus, when the carboxyl group of Glu binds to Ca2+ and penetrates into the deeper interior of ACC, Ala6 is likely the residue closest to the hydrated water molecules on the ACC surface. In the DDAD peptide, Asp Hβ and Ala Hβ exhibited the highest relative values, at 100% and 81.2%, respectively. These findings indicate that the Asp side chain, due to its shorter length and lack of flexibility compared to Glu, binds to Ca2+ ions on the surface of ACC rather than penetrating deeper into the ACC interior. Consequently, Asp remains closer to the hydrated water molecules on the ACC surface.
These results suggest that the EEAE peptide is more likely to interact with ACC than the DDAD peptide. The longer and more flexible Glu side chains in the EEAE peptide allow for conformational adjustments, aligning carboxyl groups for electrostatic interactions with Ca2+ ions. This flexibility enables the carboxyl groups of Glu to penetrate deeply into the ACC structure and bind to Ca2+ ions in the interior (Fig. 10(a)). In contrast, the shorter and less flexible Asp side chains in the DDAD peptide limit its ability to adjust conformation, restricting its interactions to Ca2+ ions on the ACC surface. As a result, Asp remains closer to the hydrated water molecules on the ACC surface (Fig. 10(a)).
 | ||
| Fig. 10 (a) Model of peptide binding to ACC: (i) EEAE; (ii) DDAD. (b) Model of myostracum formation. Reproduced with permission from ref. 26. Copyright 2024, American Chemical Society. | ||
Based on our findings, we propose the following model for myostracum formation presented in (Fig. 10(b)). In the area where the adductor muscle attaches to the nacreous layer, both calcification and muscle cell destruction occur. Cell destruction results in the release of paramyosin and intracellular proteases, leading to paramyosin degradation. The degraded paramyosin forms a scaffold on the nacreous layer, facilitating myostracum formation and promoting the growth of columnar aragonite crystals. This study demonstrated that paramyosin, a Glu-rich muscle protein with a repeating EEAE sequence, stabilizes ACC from within CaCO3 and promotes the growth of columnar aragonite crystals, highlighting its critical role in myostracum biomineralization.
3 Conformational analyses of the protein involved in the synthesis of gold nanoparticles
3.1 Synthesis of gold nanoparticles by Collimonas sp. strain D-25
Gold nanoparticles (AuNPs) are widely utilized in fields such as electronics, sensors, probes, catalysts, electronic wiring, and medical materials.13,88,89 Compared to traditional chemical and physical synthesis methods, microbial synthesis of AuNPs offers an environmentally friendly alternative. In this study, we focused on a microorganism, Collimonas sp. strain D-25 (D-25), isolated from Japanese soil. D-25 demonstrated the ability to reduce gold ions under both aerobic and anaerobic conditions. When D-25 bacterial cells were washed with sterile water and incubated in a gold chloride solution, the solution turned purple, indicating the synthesis of AuNPs, a characteristic color (Fig. 11(a)).90 TEM (transmission electron microscopy) and EDS (energy dispersive X-ray spectroscopy) analyses confirmed the formation of AuNPs with an average particle size of 25.7 nm around the bacterial cells (Fig. 11(b)). | ||
| Fig. 11 (a) and (b) AuNPs synthesized using D-25. (a) TEM image of bio-synthesized AuNPs and bacteria D-25. Insert photo indicates AuNPs solution. (b) EDS spectrum of biosynthesized AuNPs. Insert histogram indicates particle size and distribution of AuNPs. (c) TEM images of AuNPs synthesized after incubated using rDP-1. Insert photo indicates AuNPs solution. (d) TEM images of synthesized AuNPs using GBP. Insert photo indicates AuNPs solution. Reproduced with permission from ref. 90. Copyright 2024, Chemistry Europe. | ||
To identify substances contributing to AuNP synthesis by D-25, we examined the ultrasonic disruption solution of D-25. Bacterial suspensions were subjected to ultrasonic irradiation, and the resulting supernatant was mixed with gold chloride solution. After incubation, AuNPs with an average particle size of 20.3 nm were synthesized.
The protein components in the ultrasonic disruption solution were analyzed via SDS-PAGE (polyacrylamide gel electrophoresis). A protein band around 20 kDa disappeared after 5 hours of reaction, suggesting that this protein was involved in the synthesis of AuNPs.
LC-MS/MS analysis revealed that a protein with a molecular weight of 20.7 kDa had an amino acid sequence with no homology to any known proteins in the database. This novel protein was designated as DP-1.
Recombinant DP-1 (rDP-1) was expressed using Escherichia coli BL21 (DE3) and purified through nickel affinity chromatography. The size of AuNPs synthesized was dependent on the concentration of rDP-1. At a concentration of 100 μg mL−1 of rDP-1, AuNPs with an average particle size of 13.6 nm were produced. Observations of gray contrasts around the AuNPs indicated that rDP-1 formed a protein complex, acting as an organic membrane enveloping the entire surface of the AuNPs (Fig. 11(c)). In contrast, AuNPs synthesized without any protein aggregated and precipitated. These findings confirm that DP-1 plays a critical role in the stabilization and dispersion of AuNPs.
3.2 Structural analyses of DP-1, a protein with the ability to bind gold nanoparticles
DP-1 regulates both the size and stability of AuNPs.90 However, the detailed molecular mechanisms governing the interaction between DP-1 and AuNPs remain unclear. To address this, we aimed to identify the specific region of DP-1 responsible for AuNP binding and analyze its conformational changes using solution NMR.Peptide fragments generated by trypsin digestion were screened for their AuNP-binding capabilities to identify the key Au-binding domain of DP-1. A fragment consisting of 16 amino acid residues (GHAATPEQYGVVTANK) was identified as the gold-binding peptide (GBP) with the highest AuNP-dispersive activity (Fig. 11(d)).
Conformational changes of GBP on the AuNP surface were analyzed by solution NMR using AuNPs, similar to the approach used with dispersive CaCO3 particles. The conformational analysis revealed that the main chain of GBP's latter segment (Tyr9 to Lys16) was elongated upon interaction with AuNPs, while side chains of negatively charged residues in the peptide's initial segment (Gly1 to Gln8) such as Thr5, Glu7, and Gln8, were exposed on the AuNP surface (Fig. 12(a) and (b)). These results suggest that DP-1 may interact with AuNPs primarily through electrostatic interactions involving negatively charged residues such as Thr5, Glu7, and Gln8 in the first part of the peptide. Furthermore, hydrophobic residues in the latter part of the peptide, including Val11, Val12, and Ala14, likely contribute to the self-assembly of peptides via hydrophobic interactions. These self-assembly forms an organic membrane on the AuNPs surface. Self-assembling peptides have a stability on the solid surface; therefore, individual peptides are expected to form a specific conformation and a uniform molecular complex91 The schematic diagram of interaction between the DP-1 and AuNPs is as follows (Fig. 12(c)). Many molecules of DP-1 molecules bind to the AuNP surface, forming a protein corona. This corona stabilizes AuNPs by maintaining dispersion through spatial and charge interactions, preventing aggregation and ensuring uniform particle distribution.
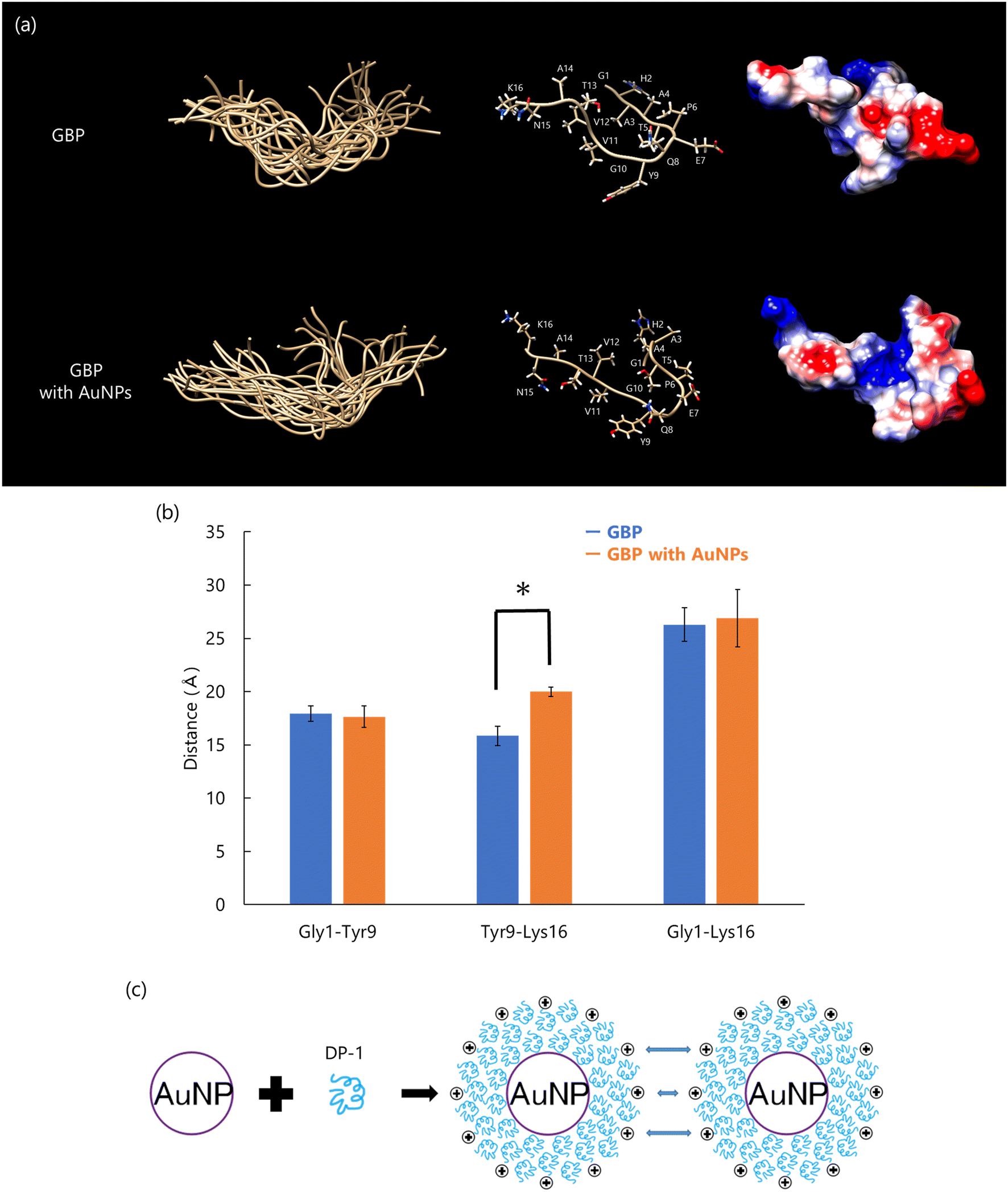 | ||
| Fig. 12 (a) Conformation of peptide calculated using CYANA. Overlay of 20 best-matched 3D structures of each peptide (left); the main chain is labeled “ball and stick”, and all side chains are labeled (center). The surface was displayed as “coulombic surface coloring” (right). (b) The mean distance between Gly1, Tyr9, and Lys16 of Cα; error bars show S.D. (n = 20), *p < 0.01, t-test, GBP (blue) vs. GBP with AuNPs (orange). (c) Interaction mechanism of DP-1 with AuNPs. Reproduced with permission from ref. 25. Copyright 2024, Chemistry Europe. | ||
4 Conclusions and prospects
Conformational analysis of proteins is critical for understanding detailed molecular mechanisms, such as reaction pathways and ligand interactions. However, conformational analysis of proteins involved in organic–inorganic interactions has historically been challenging due to the difficulty in preparing mixed samples of minerals and proteins. Solution NMR with dispersive minerals provides a unique opportunity to elucidate the conformational changes of biomineral proteins on mineral surfaces.Dispersive minerals synthesized under inorganic conditions are essential for obtaining the sharp signals required for solution NMR analysis. However, conventional methods often result in bulk CaCO3 crystals that precipitate in solution, limiting their applicability for conformational studies. To address this issue, we developed improved methods to synthesize dispersive CaCO3 particles that can rotate and disperse in solution, including ACC and aragonite particles synthesized under room temperature and pressure conditions.24,58 These advancements enabled the first detailed conformational analyses of biomineral proteins, such as Pif 80 and paramyosin, on CaCO3 surfaces.
In addition to biomineral proteins, we also analyzed the conformational changes of GBP in interaction with AuNPs. These findings highlight the versatility of solution NMR with dispersive particles for studying organic–inorganic interactions, providing new insights into the molecular mechanisms underlying biomineralization and nanoparticle stabilization.
Our findings suggest that common conformational changes in organic–inorganic interactions follow a general pattern (Fig. 13). Biomineral proteins without minerals do not adopt a fixed or ordered three-dimensional structure. However, when interacting with the solid surface of minerals, these proteins undergo specific conformational changes to facilitate organic–inorganic interactions. These results demonstrate that conformational analysis using solution NMR with dispersive minerals can elucidate organic–inorganic interactions at the molecular level.
 | ||
| Fig. 13 Schematic diagram of biomineral protein on the mineral surfaces. The side chains of amino acid sequences from biomineral proteins in solution cause the random structure because of lack of stabilization of minerals. On the solid surface of minerals, the amino acid sequences of biomineral proteins are extended to cover the solid surface and likely to make the tight binding of minerals. These results suggested the fundamental mechanism of biomineral proteins on the solid minerals. | ||
If dispersive minerals can be synthesized under inorganic conditions, solution NMR enables the elucidation of protein conformations on mineral surfaces. However, the complexity of preparation methods for dispersive mineral particles depends on the type of mineral and its polymorph. Despite these challenges, solution NMR offers a promising avenue for clarifying organic–inorganic interactions at the molecular level.
There are certain limitations to the current conformational analysis method using solution NMR. Our study utilized 2D-NMR spectra with non-labeled peptides, which can only determine the conformation of peptides with up to 20 amino acid residues. In contrast, solution NMR with 3D-NMR measurements can resolve the structure of proteins up to 300 amino acids in length by using 13C/15N-labeled recombinant proteins. However, acquiring the spectra for 3D-NMR requires significant time and effort. Additionally, NMR sample solutions are typically maintained at a pH of up to 7, which is unsuitable for biominerals like CaCO3 that dissolve under these conditions. Therefore, the synthesis of dispersive minerals that remain stable and suspended in solution at pH 7 for extended periods is necessary for conducting 3D-NMR measurements.
The use of 13C/15N-labeled recombinant proteins is both costly and labor-intensive compared to non-labeled peptides. Our approach using non-labeled peptides for solution NMR offers a cost-effective and easily preparable alternative with conventional instruments. To systematically clarify the organic–inorganic interactions between biomineral proteins and minerals, various features of minerals can be synthesized using recombinant proteins, partial peptides, and high-molecular-weight organic compounds that mimic biomineral proteins. These engineered minerals have significant material value due to their customizable properties, including crystal polymorph, size, morphology, defect density, and trace element composition. Furthermore, these synthesis methods are environmentally friendly, as they enable mineral production at room temperature and atmospheric pressure without the use of organic solvents.92,93 Consequently, our studies may contribute to the advancement of mineral synthesis that is both high-value and eco-friendly.
Data availability
No original data is provided in the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was partially funded by two Grants-in-Aid for Scientific Research B and A (JP19H03045, JP21H02284 and JP23H00339) and Scientific Research on Innovative Areas IBmS: JSPS KAKENHI (JP19H05771). This study was partially based on results obtained from project JPNP22100161-0, which was subsidized by the New Energy and Industrial Technology Development Organization (NEDO). This study was partially funded by the Environment Research and Technology Development Fund (grant number CN2201) of the Environmental Restoration and Conservation Agency of Japan. This study was supported by a Sasakawa Scientific Research Grant from the Japan Science Society to K. F. (2024-4008). This work was supported by JST SPRING, Grant Number JPMJSP2108.References
- S. Mann, Molecular Recognition in Biomineralization, Nature, 1988, 332(6160), 119–124, DOI:10.1038/332119a0.
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, 2001 Search PubMed.
- S. Weiner and L. Addadi, Acidic Macromolecules of Mineralized Tissues: The Controllers of Crystal Formation, Trends Biochem. Sci., 1991, 16(7), 252–256, DOI:10.1016/0968-0004(91)90098-g.
- M. Suzuki and H. Nagasawa, Mollusk Shell Structures and Their Formation Mechanism, Can. J. Zool., 2013, 91(6), 349–366, DOI:10.1139/cjz-2012-0333.
- G. Falini, S. Albeck, S. Weiner and L. Addadi, Control of Aragonite or Calcite Polymorphism by Mollusk Shell Macromolecules, Science, 1996, 271(5245), 67–69, DOI:10.1126/science.271.5245.67.
- N. Tsutsui, K. Ishii, Y. Takagi, T. Watanabe and H. Nagasawa, Cloning and Expression of a cDNA Encoding an Insoluble Matrix Protein in the Gastroliths of a Crayfish, Procambarus Clarkii, J. Zool., 1999, 16(4), 619–628, DOI:10.2108/zsj.16.619.
- E. Murayama, A. Okuno, T. Ohira, Y. Takagi and H. Nagasawa, Molecular Cloning and Expression of an Otolith Matrix Protein cDNA from the Rainbow Trout, Oncorhynchus Mykiss, Comp. Biochem. Physiol., Part B:Biochem. Mol. Biol., 2000, 126(4), 511–520, DOI:10.1016/S0305-0491(00)00223-6.
- B. Wopenka and J. D. Pasteris, A Mineralogical Perspective on the Apatite in Bone, Mater. Sci. Eng., C, 2005, 25(2), 131–143, DOI:10.1016/j.msec.2005.01.008.
- M. Iijima, H. Tohda and Y. Moriwaki, Growth and Structure of Lamellar Mixed Crystals of Octacalcium Phosphate and Apatite in a Model System of Enamel Formation, J. Cryst. Growth, 1992, 116(3), 319–326, DOI:10.1016/0022-0248(92)90639-Z.
- U. Matsushima, A. Hilger, W. Graf, S. Zabler, I. Manke, M. Dawson, G. Choinka and W. B. Herppich, Calcium Oxalate Crystal Distribution in Rose Peduncles: Non-Invasive Analysis by Synchrotron X-Ray Micro-Tomography, Postharvest Biol. Technol., 2012, 72, 27–34, DOI:10.1016/j.postharvbio.2012.04.013.
- J. F. Ma, Role of Silicon in Enhancing the Resistance of Plants to Biotic and Abiotic Stresses, Soil Sci. Plant Nutr., 2004, 50(1), 11–18, DOI:10.1080/00380768.2004.10408447.
- M. E. Compeán-Jasso, F. Ruiz, J. R. Martínez and A. Herrera-Gómez, Magnetic Properties of Magnetite Nanoparticles Synthesized by Forced Hydrolysis, Mater. Lett., 2008, 62(27), 4248–4250, DOI:10.1016/j.matlet.2008.06.053.
- R. A. Sperling, P. R. Gil, F. Zhang, M. Zanella and W. J. Parak, Biological Applications of Gold Nanoparticles, Chem. Soc. Rev., 2008, 37(9), 1896–1908, 10.1039/B712170A.
- C. Daruich De Souza, B. Ribeiro Nogueira and M. E. C. M. Rostelato, Review of the Methodologies Used in the Synthesis Gold Nanoparticles by Chemical Reduction, J. Alloys Compd., 2019, 798, 714–740, DOI:10.1016/j.jallcom.2019.05.153.
- Y. Kato and M. Suzuki, Synthesis of Metal Nanoparticles by Microorganisms, Crystals, 2020, 10(7), 589, DOI:10.3390/cryst10070589.
- J. Moradian-Oldak, F. Frolow, L. Addadi, S. Weiner and L. N. Johnson, Interactions between Acidic Matrix Macromolecules and Calcium Phosphate Ester Crystals: Relevance to Carbonate Apatite Formation in Biomineralization, Proc. R. Soc. London, Ser. B, 1997, 247(1318), 47–55, DOI:10.1098/rspb.1992.0008.
- J. D. Bernal and D. Crowfoot, X-Ray Photographs of Crystalline Pepsin, Nature, 1934, 133(3369), 794–795, DOI:10.1038/133794b0.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff and D. C. Phillips, A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis, Nature, 1958, 181(4610), 662–666, DOI:10.1038/181662a0.
- M. F. Perutz, M. G. Rossmann, A. F. Cullis, H. Muirhead, G. Will and A. C. T. North, Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis, Nature, 1960, 185(4711), 416–422, DOI:10.1038/185416a0.
- J. Dubochet, M. Adrian and J.-J. Chang, et al., Cryo-Electron Microscopy of Viruses, Q. Rev. Biophys., 1988, 21(2), 129–228 CrossRef CAS PubMed.
- R. Henderson, J. M. Baldwin, T. A. Ceska, F. Zemlin, E. Beckmann and K. H. Downing, Model for the Structure of Bacteriorhodopsin Based on High-Resolution Electron Cryo-Microscopy, J. Mol. Biol., 1990, 213(4), 899–929, DOI:10.1016/S0022-2836(05)80271-2.
- J. Frank, Three-Dimensional Electron Microscopy of Macromolecular Assemblies: Visualization of Biological Molecules in Their Native State, Oxford University Press, 2006 Search PubMed.
- K. Wüthrich, Protein Structure Determination in Solution by NMR Spectroscopy, J. Biol. Chem., 1990, 265(36), 22059–22062, DOI:10.1016/S0021-9258(18)45665-7.
- K. Futagawa, T. Morioka, K. Furihata, H. Watanabe, Y. Ito, T. Ikeya, A. Hokura, K. Nagata and M. Suzuki, Structural and Functional Analyses of the Acidic and Basic Amino Acid Repeat Sequence (DDRK) in Pif 80 from Pinctada Fucata on the Aragonite Crystal Surface Using NMR, Cryst. Growth Des., 2023, 23(7), 5264–5278, DOI:10.1021/acs.cgd.3c00467.
- K. Futagawa, D. Tang, Y. Kato, K. Nagata and M. Suzuki, Structural Analyses of DP-1, a Protein with the Ability To Bind Gold Nanoparticles, by Using Nuclear Magnetic Resonance Spectroscopy, ChemBioChem, 2024, 25(1), e202300554, DOI:10.1002/cbic.202300554.
- K. Futagawa, H. Ikeda, L. Negishi, H. Kurumizaka, A. Yamamoto, K. Furihata, Y. Ito, T. Ikeya, K. Nagata, D. Funabara and M. Suzuki, Structural and Functional Analysis of the Amorphous Calcium Carbonate-Binding Protein Paramyosin in the Shell of the Pearl Oyster, Pinctada Fucata, Langmuir, 2024, 40(16), 8373–8392, DOI:10.1021/acs.langmuir.3c03820.
- R. Babou-Kammoe, S. Hamoudi, F. Larachi and K. Belkacemi, Synthesis of CaCO3 Nanoparticles by Controlled Precipitation of Saturated Carbonate and Calcium Nitrate Aqueous Solutions, Can. J. Chem. Eng., 2012, 90(1), 26–33, DOI:10.1002/cjce.20673.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes, J. Biomol. NMR, 1995, 6(3), 277–293, DOI:10.1007/BF00197809.
- W. Lee, M. Tonelli and J. L. Markley, NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy, Bioinformatics, 2015, 31(8), 1325–1327, DOI:10.1093/bioinformatics/btu830.
- P. Güntert, Automated NMR Structure Calculation With CYANA, in Protein NMR Techniques, ed. A. K. Downing, Humana Press, Totowa, NJ, 2004, pp. 353–378, DOI:10.1385/1-59259-809-9:353.
- T. Ikeya, T. Terauchi, P. Güntert and M. Kainosho, Evaluation of Stereo-Array Isotope Labeling (SAIL) Patterns for Automated Structural Analysis of Proteins with CYANA, Magn. Reson. Chem., 2006, 44(S1), S152–S157, DOI:10.1002/mrc.1815.
- P. Güntert and L. Buchner, Combined Automated NOE Assignment and Structure Calculation with CYANA, J. Biomol. NMR, 2015, 62(4), 453–471, DOI:10.1007/s10858-015-9924-9.
- S. Weiner and L. Addadi, Crystallization Pathways in Biomineralization, Annu. Rev. Mater. Res., 2011, 41(1), 21–40, DOI:10.1146/annurev-matsci-062910-095803.
- J. D. Milliman, C. P. Summerhayes and H. T. Barretto, Oceanography and Suspended Matter off the Amazon River February-March 1973, J. Sediment. Res., 1975, 45(1), 189–206, DOI:10.1306/212F6D02-2B24-11D7-8648000102C1865D.
- V. J. Fabry, Aragonite Production by Pteropod Molluscs in the Subarctic Pacific, Deep-Sea Res., Part A, 1989, 36(11), 1735–1751, DOI:10.1016/0198-0149(89)90069-1.
- D. Gebauer, A. Völkel and H. Cölfen, Stable Prenucleation Calcium Carbonate Clusters, Science, 2008, 322(5909), 1819–1822, DOI:10.1126/science.1164271.
- R. E. Zeebe, J. C. Zachos, K. Caldeira and T. Tyrrell, Carbon Emissions and Acidification, Science, 2008, 321(5885), 51–52, DOI:10.1126/science.1159124.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, The Role of Magnesium in Stabilising Amorphous Calcium Carbonate and Controlling Calcite Morphologies, J. Cryst. Growth, 2003, 254(1), 206–218, DOI:10.1016/S0022-0248(03)01153-9.
- L. Addadi, S. Raz and S. Weiner, Taking Advantage of Disorder: Amorphous Calcium Carbonate and Its Roles in Biomineralization, Adv. Mater., 2003, 15(12), 959–970, DOI:10.1002/adma.200300381.
- M. Faatz, F. Gröhn and G. Wegner, Amorphous Calcium Carbonate: Synthesis and Potential Intermediate in Biomineralization, Adv. Mater., 2004, 16(12), 996–1000, DOI:10.1002/adma.200306565.
- Y. Kitano, N. Kanamori and A. Tokuyama, Effects of Organic Matter on Solubilities and Crystal Form of Carbonates, Am. Zool., 1969, 9(3), 681–688, DOI:10.1093/icb/9.3.681.
- G. J. F. MacDonald, Experimental Determination of Calcite-Aragonite Equilibrium Relations at Elevated Temperatures and Pressures*, Am. Mineral., 1956, 41(9–10), 744–756 CAS.
- R. A. Berner, The Role of Magnesium in the Crystal Growth of Calcite and Aragonite from Sea Water, Geochim. Cosmochim. Acta, 1975, 39(4), 489–504, DOI:10.1016/0016-7037(75)90102-7.
- M. Boon, W. D. A. Rickard, A. L. Rohl and F. Jones, Stabilization of Aragonite: Role of Mg2+ and Other Impurity Ions, Cryst. Growth Des., 2020, 20(8), 5006–5017, DOI:10.1021/acs.cgd.0c00152.
- B.-G. Mao, D.-Q. Chu, A.-X. Wang, L.-M. Wang, H.-M. Sun, Z.-Y. Zhang and X.-Z. Yang, Fabrication of Flowerlike Vaterite Calcium Carbonate Crystal Aggregates by Self-Assembly in Water/Ethanol Mixtures, Eur. J. Inorg. Chem., 2013, 2013(35), 5958–5963, DOI:10.1002/ejic.201300892.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, The Kinetics and Mechanisms of Amorphous Calcium Carbonate (ACC) Crystallization to Calcite, via Vaterite, Nanoscale, 2011, 3(1), 265–271, 10.1039/C0NR00589D.
- T. Okumura, G. Takahashi, M. Suzuki and T. Kogure, Stacking Structure of Vaterite Revealed by Atomic Imaging and Diffraction Analysis, Chem. – Eur. J., 2024, 30(52), e202401557, DOI:10.1002/chem.202401557.
- L. Qiao and Q. L. Feng, Study on Twin Stacking Faults in Vaterite Tablets of Freshwater Lacklustre Pearls, J. Cryst. Growth, 2007, 304(1), 253–256, DOI:10.1016/j.jcrysgro.2007.02.001.
- A. L. Soldati, D. E. Jacob, U. Wehrmeister and W. Hofmeister, Structural Characterization and Chemical Composition of Aragonite and Vaterite in Freshwater Cultured Pearls, Mineral. Mag., 2008, 72(2), 579–592, DOI:10.1180/minmag.2008.072.2.579.
- B. M. Jessop, J. C. Shiao, Y. Iizuka and W. N. Tzeng, Prevalence and Intensity of Occurrence of Vaterite Inclusions in Aragonite Otoliths of American Eels Anguilla Rostrata, Aquat. Biol., 2008, 2(2), 171–178, DOI:10.3354/ab00047.
- G. Takahashi, T. Okumura, T. Nagaya, M. Suzuki and T. Kogure, Mineralogical Aspects of Asteriscus of Goldfish (Carassius Auratus) Consisting of Vaterite, J. Mineral. Petrol. Sci., 2024, 119(1) DOI:10.2465/jmps.231206.
- Y. Weiss, E. P. Simoncelli and E. H. Adelson, Motion Illusions as Optimal Percepts, Nat. Neurosci., 2002, 5(6), 598–604, DOI:10.1038/nn0602-858.
- G. Luquet and F. Marin, Biomineralisations in Crustaceans: Storage Strategies, C. R. - Palevol., 2004, 3(6), 515–534, DOI:10.1016/j.crpv.2004.07.015.
- M. Ueno, Calcium Transport in Crayfish Gastrolith Disc: Morphology of Gastrolith Disc and Ultrahistochemical Demonstration of Calcium, J. Exp. Zool., 1980, 213(2), 161–171, DOI:10.1002/jez.1402130202.
- A. Akiva-Tal, S. Kababya, Y. S. Balazs, L. Glazer, A. Berman, A. Sagi and A. Schmidt, In Situ Molecular NMR Picture of Bioavailable Calcium Stabilized as Amorphous CaCO3 Biomineral in Crayfish Gastroliths, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(36), 14763–14768, DOI:10.1073/pnas.1102608108.
- S. Akhtar, J. E. Barralet, D. Farrar and A. J. Wright, Influence of Phosphate Ions on the Stability of Amorphous Calcium Carbonate, Key Eng. Mater., 2006, 309–311, 179–182, DOI:10.4028/www.scientific.net/KEM.309-311.179.
- S. Bentov, S. Weil, L. Glazer, A. Sagi and A. Berman, Stabilization of Amorphous Calcium Carbonate by Phosphate Rich Organic Matrix Proteins and by Single Phosphoamino Acids, J. Struct. Biol., 2010, 171(2), 207–215, DOI:10.1016/j.jsb.2010.04.007.
- H. Iwahashi, A. Araki, C. Numako, A. Hokura and M. Suzuki, Identification of Amorphous CaCO3 in Aqueous Solution Using XANES Analysis, Chem. Lett., 2020, 49(8), 982–985, DOI:10.1246/cl.200328.
- J. Jiang, M.-R. Gao, Y.-H. Qiu and S.-H. Yu, Gram-Scale, Low-Cost, Rapid Synthesis of Highly Stable Mg–ACC Nanoparticles and Their Long-Term Preservation, Nanoscale, 2010, 2(11), 2358–2361, 10.1039/C0NR00443J.
- A. Gal, S. Weiner and L. Addadi, The Stabilizing Effect of Silicate on Biogenic and Synthetic Amorphous Calcium Carbonate, J. Am. Chem. Soc., 2010, 132(38), 13208–13211, DOI:10.1021/ja106883c.
- A. Lutts, J. Grandjean, C. Grégoire, A. Lutts, J. Grandjean and C. Grégoire, X-Ray Diffraction Patterns from the Prisms of Mollusk Shells, Arch. Physiol. Biochem., 1960, 68(5), 829–831, DOI:10.3109/13813456009075173.
- P. U. P. A. Gilbert, A. Young and S. N. Coppersmith, Measurement of C-Axis Angular Orientation in Calcite (CaCO3) Nanocrystals Using X-Ray Absorption Spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(28), 11350–11355, DOI:10.1073/pnas.1107917108.
- D. Chateigner, C. Hedegaard and H.-R. Wenk, Mollusc Shell Microstructures and Crystallographic Textures, J. Struct. Geol., 2000, 22(11), 1723–1735, DOI:10.1016/S0191-8141(00)00088-2.
- H. Mutvei, The Nacreous Layer inMytilus, Nucula, andUnio (Bivalvia), Calcif. Tissue Res., 1977, 24(1), 11–18, DOI:10.1007/BF02223291.
- G. A. Kahler, R. L. Sass and F. M. Fisher, The Fine Structure and Crystallography of the Hinge Ligament ofSpisula Solidissima (Mollusca: Bivalvia: Mactridae), J. Comp. Physiol., B, 1976, 109(2), 209–220, DOI:10.1007/BF00689419.
- P. Stempflé, O. Pantalé, M. Rousseau, E. Lopez and X. Bourrat, Mechanical Properties of the Elemental Nanocomponents of Nacre Structure, Mater. Sci. Eng., C, 2010, 30(5), 715–721, DOI:10.1016/j.msec.2010.03.003.
- M. Suzuki, Structural and Functional Analyses of Organic Molecules Regulating Biomineralization, Biosci., Biotechnol., Biochem., 2020, 84(8), 1529–1540, DOI:10.1080/09168451.2020.1762068.
- H. Kintsu, R. Nishimura, L. Negishi, I. Kuriyama, Y. Tsuchihashi, L. Zhu, K. Nagata and M. Suzuki, Identification of Methionine-Rich Insoluble Proteins in the Shell of the Pearl Oyster, Pinctada Fucata, Sci. Rep., 2020, 10(1), 18335, DOI:10.1038/s41598-020-75444-4.
- S. Sudo, T. Fujikawa, T. Nagakura, T. Ohkubo, K. Sakaguchi, M. Tanaka, K. Nakashima and T. Takahashi, Structures of Mollusc Shell Framework Proteins, Nature, 1997, 387(6633), 563–564, DOI:10.1038/42391.
- M. Suzuki, E. Murayama, H. Inoue, N. Ozaki, H. Tohse, T. Kogure and H. Nagasawa, Characterization of Prismalin-14, a Novel Matrix Protein from the Prismatic Layer of the Japanese Pearl Oyster (Pinctada Fucata), Biochem. J., 2004, 382(1), 205–213, DOI:10.1042/BJ20040319.
- M. Suzuki and H. Nagasawa, The Structure–Function Relationship Analysis of Prismalin-14 from the Prismatic Layer of the Japanese Pearl Oyster, Pinctada Fucata, FEBS J., 2007, 274(19), 5158–5166, DOI:10.1111/j.1742-4658.2007.06036.x.
- H. Miyamoto, T. Miyashita, M. Okushima, S. Nakano, T. Morita and A. Matsushiro, A Carbonic Anhydrase from the Nacreous Layer in Oyster Pearls, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(18), 9657–9660, DOI:10.1073/pnas.93.18.9657.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, An Acidic Matrix Protein, Pif, Is a Key Macromolecule for Nacre Formation, Science, 2009, 325(5946), 1388–1390, DOI:10.1126/science.1173793.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly Accurate Protein Structure Prediction with AlphaFold, Nature, 2021, 596(7873), 583–589, DOI:10.1038/s41586-021-03819-2.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3, Nature, 2024, 630(8016), 493–500, DOI:10.1038/s41586-024-07487-w.
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Sequence Complexity of Disordered Protein, Proteins: Struct., Funct., Bioinf., 2001, 42(1), 38–48, DOI:10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3.
- T. L. Burgess and R. B. Kelly, Constitutive and Regulated Secretion of Proteins, Annu. Rev. Cell Biol., 1987, 3(1), 243–293, DOI:10.1146/annurev.cb.03.110187.001331.
- M. Suzuki, T. Kogure and H. Nagasawa, Studies on the Chemical Structures of Organic Matrices and Their Functions in the Biomineralization Processes of Molluscan Shells, AGri-Biosci. Monogr., 2017, 7(2), 25–39, DOI:10.5047/agbm.2017.00702.0025.
- M. Suzuki, A. Iwashima, M. Kimura, T. Kogure and H. Nagasawa, The Molecular Evolution of the Pif Family Proteins in Various Species of Mollusks, Mar. Biotechnol., 2013, 15(2), 145–158, DOI:10.1007/s10126-012-9471-2.
- L. Addadi and S. Weiner, Interactions between Acidic Proteins and Crystals: Stereochemical Requirements in Biomineralization, Proc. Natl. Acad. Sci. U. S. A., 1985, 82(12), 4110–4114, DOI:10.1073/pnas.82.12.4110.
- S. Weiner and L. Hood, Soluble Protein of the Organic Matrix of Mollusk Shells: A Potential Template for Shell Formation, Science, 1975, 190(4218), 987–989, DOI:10.1126/science.1188379.
- J. Liang, J. Xie, J. Gao, C.-Q. Xu, Y. Yan, G.-C. Jia, L. Xiang, L.-P. Xie and R.-Q. Zhang, Identification and Characterization of the Lysine-Rich Matrix Protein Family in Pinctada Fucata: Indicative of Roles in Shell Formation, Mar. Biotechnol., 2016, 18(6), 645–658, DOI:10.1007/s10126-016-9724-6.
- J. Klein, R. Meinecke, M. Mayer and B. Meyer, Detecting Binding Affinity to Immobilized Receptor Proteins in Compound Libraries by HR-MAS STD NMR, J. Am. Chem. Soc., 1999, 121(22), 5336–5337, DOI:10.1021/ja990706x.
- M. Mayer and B. Meyer, Group Epitope Mapping by Saturation Transfer Difference NMR To Identify Segments of a Ligand in Direct Contact with a Protein Receptor, J. Am. Chem. Soc., 2001, 123(25), 6108–6117, DOI:10.1021/ja0100120.
- Y. Suzuki, H. Shindo and T. Asakura, Structure and Dynamic Properties of a Ti-Binding Peptide Bound to TiO2 Nanoparticles As Accessed by 1H NMR Spectroscopy, J. Phys. Chem. B, 2016, 120(20), 4600–4607, DOI:10.1021/acs.jpcb.6b03260.
- Y. Suzuki and H. Shindo, Binding Sites and Structure of Peptides Bound to SiO2 Nanoparticles Studied by Solution NMR Spectroscopy, Polym. J., 2018, 50(10), 989–996, DOI:10.1038/s41428-018-0084-0.
- M. Suzuki, H. Nagasawa and T. Kogure, Synthesis and Structure of Hollow Calcite Particles, Cryst. Growth Des., 2006, 6(9), 2004–2006, DOI:10.1021/cg0602921.
- J. J. Storhoff, A. A. Lazarides, R. C. Mucic, C. A. Mirkin, R. L. Letsinger and G. C. Schatz, What Controls the Optical Properties of DNA-Linked Gold Nanoparticle Assemblies?, J. Am. Chem. Soc., 2000, 122(19), 4640–4650, DOI:10.1021/ja993825l.
- J. Zhang, L. Mou and X. Jiang, Surface Chemistry of Gold Nanoparticles for Health-Related Applications, Chem. Sci., 2020, 11(4), 923–936, 10.1039/C9SC06497D.
- D. Tang, Y. Kato, D. Zhang, L. Negishi, H. Kurumizaka, T. Hirata, M. Nakakido, K. Tsumoto, F. Shuji, S. Tsuguyuki, T. Okumura, K. Nagata and M. Suzuki, Dispersion Function of a Protein, DP-1, Identified in Collimonas Sp. D-25, for the Synthesis of Gold Nanoparticles, ChemBioChem, 2023, 24(14) DOI:10.1002/cbic.202300221.
- D. Khatayevich, C. R. So, Y. Hayamizu, C. Gresswell and M. Sarikaya, Controlling the Surface Chemistry of Graphite by Engineered Self-Assembled Peptides, Langmuir, 2012, 28(23), 8589–8593, DOI:10.1021/la300268d.
- Y. Namikawa, K. Moriyasu, K. Yasumoto, S. Katsumata and M. Suzuki, Carbonic Anhydrase Activity Identified in the Powdered Nacreous Layer of Pinctada Fucata, Process Biochem., 2023, 128, 22–29, DOI:10.1016/j.procbio.2023.02.007.
- Y. Namikawa and M. Suzuki, Atmospheric CO2 Sequestration in Seawater Enhanced by Molluscan Shell Powders, Environ. Sci. Technol., 2024, 58(5), 2404–2412, DOI:10.1021/acs.est.3c09273.
| This journal is © The Royal Society of Chemistry 2025 |