
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling POM–peptide complexes: molecular insights into metal oxide nanoparticle–protein interactions
Björn H.
Greijer
and
Vadim G.
Kessler
 *
*
Department of Molecular Sciences, BioCenter, Swedish University of Agricultural Sciences, Box 7015, SE-75007 Uppsala, Sweden. E-mail: vadim.kessler@slu.se
First published on 25th February 2025
Abstract
Mineral nanoparticles (NPs) play a crucial role in biological systems, exhibiting enzyme-like “nanozyme” activity in protein oxidation and hydrolysis. To study NP interactions at the molecular level, we characterized complexes of peptides with poly-oxo-metalate (POM) species, the smallest known NPs. Our findings highlight the importance of factors such as metal–oxygen bond polarity, peptide hydrophilicity, medium conditions, and structure-directing amino acids. Using single-crystal models and 2D NMR, we also explored interactions between larger NPs as nanozymes and proteins relevant for specific oxidation of amino acids and proteins.
 Björn H. Greijer | Björn H. Greijer graduated from the Master's programme in Applied Biotechnology at Uppsala University in 2019. He defended his PhD in Chemistry in September 2024 under supervision of Professor Vadim Kessler at the Swedish University of Agricultural Sciences, focusing on X-ray single crystal analysis of model systems, where polyoxometallates and oligopeptides were used to mimic nanoparticle interactions with proteins. |
 Vadim G. Kessler | Vadim G. Kessler is Professor of Inorganic Chemistry and Bionanotechnology at the Swedish University of Agricultural Sciences since 2004. His major scientific interests are in understanding of molecular mechanisms in formation of mineral nanoparticles in nature and in chemical synthesis by sol–gel process, and gaining insights into their roles in metabolic processes in plants, animals and humans. |
Introduction
Mineral nanoparticles (NPs), components of soil and ground water with at least one dimension between 1 and 100 nm,1 are ubiquitous in nature, and are present everywhere from the ocean, to soil, and to our bodies. They form naturally from weathering of minerals, and dissolved matter undergoing natural sol–gel processes.1,2 NPs have been present on Earth from the beginning, and have likely influenced the development of life in ways we cannot begin to predict. What can be observed, however, is how NPs affect our health, due either to incidental exposure or deliberate treatments.3,4 The chemical reactivity of NPs may have played an important role in the development of life itself. The redox reactivity of NPs has been highlighted as the foundation of their nanozyme action, which is particularly crucial for plant stress resistance.5 Additionally, their ability to complex with blood proteins has demonstrated significant benefits in wound healing.6,7To understand the molecular mechanisms underlying protein interactions with NPs, considerable effort has been devoted to investigating the spectroscopic characteristics of the so-called “protein corona”.8–10 Theoretical chemistry has also been employed to model possible attachment modes.11 While X-ray diffraction is the most insightful method for revealing molecular structures and interactions, its application has been hindered by challenges such as the non-uniform size of NPs and the significant disparity in X-ray reflectivity between solid inorganic matter and the less electron-dense amino acids, proteins, or peptides. Although it has been observed that NPs tend to form common dense packing motifs, the exact nature of their molecular attachments remains enigmatic.12
The smallest “mineral” metal oxide NPs—the poly-oxo-metalate species (POMs) have been proposed in our recent studies as potential models for investigation of protein interactions. We selected for this purpose Keggin POMs, which are spherical, chemically distinct species with the general formula EM12O40, E = P, As, Si, Ge; M = Mo, W (V)13 and typically have sizes just above 1 nm. The distance between opposite oxygen atoms on the surface of Keggin POMs is actually 1.04 nm (10.4 Å) for phosphomolybdates and -tungstates.14 This implies that the hydrodynamic size must be somewhat larger, about 1.2 nm. That might have been too small for the Dynamic Light Scattering (DLS) instruments in the past,15,16 used commonly for estimation of NP sizes. It falls, however, within the range of the contemporary multi-channel DLS that can distinguish the sizes from 0.3 nm to at least 15 μm.17 Application of DLS for determination of POM sizes has taken pace and the sizes of 1.2–1.3 nm have been successfully measured.18 Worth noting is that mineral NP generated by nucleation phenomena are strikingly uniform in size as proved by high-resolution Transmission Electron Microscopy (TEM) studies,19,20 or truly uniform at molecular level as proved in recent works by I. A. Weinstock et al.21,22
NP in the nature are usually strongly surface charged due to well-known double electric layer phenomenon. For example, nano TiO2 in a pH-neutral aqueous medium bear charge between 0.30 and 0.05 C m−2.23 POMs are also negatively charged and stable in acidic media, though some POM analogs, such as [Al13(μ3-OH)6(μ-OH)18(H2O)24]15+ (ref. 24) and [Fe(O4){Fe(OH)2(OH2)}12]7+ (ref. 25) aggregates, carry a positive charge and are thus stable in basic environments. With the charge density of about 1 atomic unit per nm2, corresponding to 0.16 C m−2 (the average for TiO2 in water!) POMs serve as effective models for visualizing the nature of chemical bonding at the surface. What is also extremely important to notice is that kinetics of the surface reactions for both Lindquist and Keggin POMs has recently been demonstrated to display reaction speeds in the range typical for (micro) heterogeneous processes, confirming their validity as NP models in reaction mechanism studies.26
General trends in, in the first hand, heteroatom substituted POM bonding to real protein structures have been summarized by Parac-Vogt et al. in a recent review, highlighting the role of charge interactions and hydrogen bonding.27
In this study, we systematically varied key parameters influencing NP–peptide bonding, such as the polarity of M–O bonds, peptide hydrophilicity/hydrophobicity, the acidity and salinity of the medium, and the presence of structure-directing functions such as phenyl alanine and tyrosine amino acids in the ligand structure. We also utilized POMs as structural models to investigate the mechanisms of NP so-called nanozyme action. This innovative approach offers valuable insights into the molecular dynamics and bonding characteristics of NP–protein systems important for the growing field of biopolymer-NP and biopolymer-POM hybrid materials design.28 The present highlight article, however, is only reporting principal observations from systematic exploratory studies and makes no claims on crystal engineering in purposeful application of those.
Effect of the M–O bond polarity
At the outset of our studies, we investigated the complexation of phosphomolybdic acid and phosphotungstic acid with a simple dipeptide, GlyGly (bis-glycine). When combined in a POM-to-peptide ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, reflecting formal charge balance, the resulting complexes—while identical in composition—exhibited markedly different structures. These structural differences appear to stem from variations in the nature of the interactions between the POM and the peptide, driven by differences in the polarity of the metal–oxygen bonds.
:3, reflecting formal charge balance, the resulting complexes—while identical in composition—exhibited markedly different structures. These structural differences appear to stem from variations in the nature of the interactions between the POM and the peptide, driven by differences in the polarity of the metal–oxygen bonds.
For [PMo12O40]3− species, the electrostatic interactions dominated, with the negatively charged POM anion interacting strongly and uniformly with the protonated peptide cations. Hydrogen bonding primarily occurred between peptide molecules themselves or as a complement to the electrostatic interactions between the ammonium –NH3+ groups and the POM. In contrast, for [PW12O40]3−, while charge interactions were still significant, they were strongly enhanced by hydrogen bonding. This is likely due to the higher polarity of the W–O bonds, which readily interacted with the amide protons of the peptide (see Fig. 1).29 The difference is particularly evident in the molecular packing. For Mo-POM, the protonated peptides form pairs that are predominantly hydrogen-bonded to each other. In contrast, for W-POM, individual protonated peptide cations are primarily hydrogen-bonded directly to the POM anions.
 | ||
| Fig. 1 Asymmetric units and packing arrangements in the structures of (HGlyGly)3[PMo12O40](H2O)4 (A and B) and (HGlyGly)3[PW12O40](H2O)4 (C and D) respectively.29 | ||
Influence of thermal pre-treatment of solutions
It turned out that while all crystallizations occurred at room temperature, the structure of the products was distinctly dependent on the thermal pre-history of the solutions. Intermediate heating was leading to a less dense structure incorporating more of the water molecules per structural unit. Composition of the resulting materials was (HGlyGly)3[PMo12O40](H2O)10 and (HGlyGly)3[PW12O40](H2O)6 respectively (Fig. 2).29 | ||
| Fig. 2 Crystal structure fragments visualizing differences in packing between the structures of the (HGlyGly)3[PMo12O40](H2O)10 (A) and (HGlyGly)3[PW12O40](H2O)6 (B).29 | ||
In the packing, which gains higher symmetry, monoclinic instead of triclinic, we can clearly see the persistent trend for hydrogen bonding between peptide cations in the Mo-POM and more pronounced H-bonding to POM anions in the W-POM. The reason of gaining more water and higher symmetry is apparently that higher mobility of the POMs in the intermediate heated up medium results in gaining a secondary solvation sphere that is then preserved on cooling, and kept on transition into the solid crystal structure.
Effect of the peptide hydrophilicity/hydrophobicity
To investigate the influence of hydrophilicity and hydrophobicity on POM–peptide interactions, we synthesized both Mo-POM and W-POM complexes with oligopeptides of varying lengths, such as GlyGlyGly and GlyGlyGlyGly. A notable finding was the need to lower the pH to below 1.0 by adding a strong acid to facilitate complex formation, as the peptides exhibited poor solubility at higher pH levels. The resulting structures were predominantly shaped by hydrogen bonding in both Mo-POM and W-POM systems, although the number of hydrogen bonds per POM unit was significantly higher for W-POM, as shown in Fig. 3.30 | ||
| Fig. 3 Asymmetric units in the molecular structures of (HGly4)2(H3O)PMo12O40(H2O)9 (A) and (HGly4)1.33(H3O)1.67PW12O40(H2O) (B).30 | ||
For the more hydrophobic GlyGlyGlyGly peptide, the compositions of the complexes differed markedly between Mo-POM and W-POM: (HGly4)2(H3O)PMo12O40(H2O)9 and (HGly4)1.33(H3O)1.67PW12O40(H2O), respectively. The Mo-POM complex was more hydrated, with weaker hydrogen bonding between the peptide and the POM. In contrast, the W-POM complex contained minimal crystalline water but exhibited strong hydrogen bonding interactions between the POM and the peptide. In this case, the peptide was effectively “wrapped” around the POM, forming robust peptide-NH-POM hydrogen bonds.30
Effects of salinity and acidity of media
Nanoparticles (NPs) can enter the body through either the respiratory system or the gastrointestinal tract. The latter environment is characterized by high acidity and significant salinity. To explore the influence of these factors, we examined the formation of complexes between poly-oxo-metalates (POMs) and the GlyGly peptide under varying conditions.31Our initial experiments focused on the effect of the POM-to-peptide ratio, considering the intrinsic acidity of POMs as strong acids. For W-POM, the only isolable product was the previously reported (HGlyGly)3[PW12O40](H2O)4 complex. In contrast, for Mo-POM, at low pH and POM-to-peptide ratios greater than 1:1, a new crystalline phase of (HGlyGly)3[PMo12O40](H2O)4 was observed, with a structure analogous to that of the W-POM complex.
These findings suggest that increased acidity facilitates the formation of hydrogen bonds, enabling the Mo-POM complex to adopt a structure similar to the H-bond-dominated W-POM complex.
When the POM-to-peptide ratio was maintained at 1:3 and the acidity and concentration of Na+(aq) or other highly charged cations were increased, complexes with lower peptide content were formed. These new structures incorporated sodium ions, which played a role in directing structural organization. Sodium ions were observed to form coordination fragments by being solvated by the peptide molecules (see Fig. 4 and 5).31
 | ||
| Fig. 4 Molecular (A) and crystal (B) structures of the Na(HGlyGly)2[PMo12O40]·8H2O compound.31 | ||
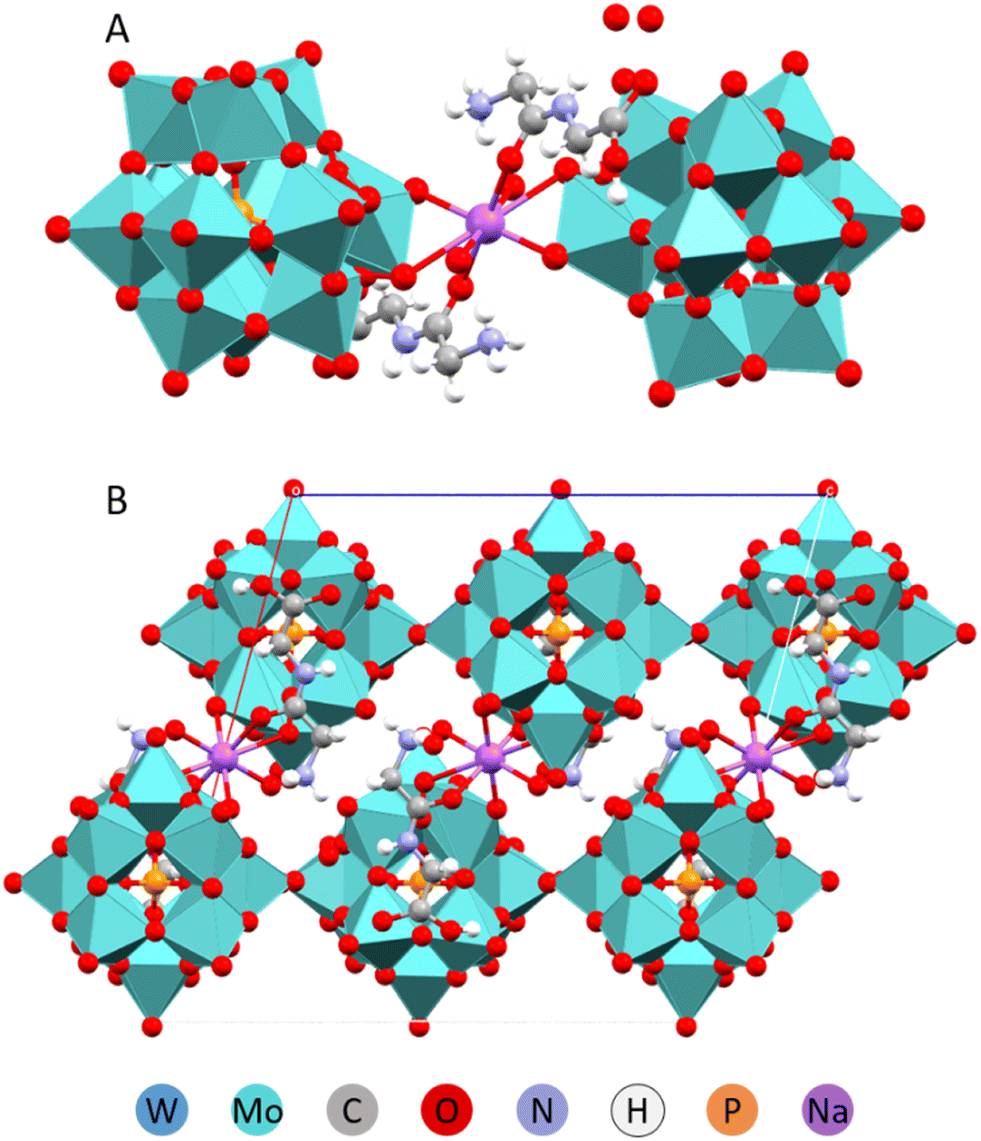 | ||
| Fig. 5 Molecular (A) and crystal (B) structures of the Na(HGlyGly)(H3O)[PMo12O40]·3H2O compound.31 | ||
Cumulative effects of acidity and salinity on Mo-POM GlyGly peptide complexes are summarized in Fig. 6.
 | ||
| Fig. 6 Comparative diagram reflecting the cumulative effects of acidity and salinity on the formation of GlyGly-Mo-POM complexes.31 | ||
This variety was not observed for similar experiments with W-POM. The reason may be the relatively weaker interactions between peptides and Mo-POM as compared to those with W-POM, as discussed above. Weaker interactions may allow for more “options” with similar Gibbs free energy.
Effects of structure-directing ligands
Peptides naturally form anisotropic structures due to their inherent asymmetry and chirality. Amino acids such as phenylalanine (Phe) and tyrosine (Tyr) play a key role in determining these structures. Their aromatic rings are not only sterically demanding but also facilitate self-assembly through π-stacking interactions. A special role is also assigned to oligo alanine (Ala) fragments that contribute to structure formation via manifestation of hydrophobic interactions or hindering the hydrophilic ones. We have investigated therefore complex formation between Keggin W-POM and such peptides as Phe-Ala, Ala-Phe, Ala-Ala, Ala-Ala-Ala, and just the amino acid Tyr.32In structures containing Phe and Tyr, the peptide appeared to be the driving force behind structure formation, forcing the POM to arrange into columns or planes. In case of the Phe-Ala peptide, where the bulky and π-stacking prone phenyl ring was located at the end opposite to the carboxylic acid group, the hydrogen bonding was apparently competing with the ring stacking (Fig. 7). The resulting complex structure had phenyl rings parallel to each other but shifted in space. The chemical composition was 2 protonated peptide cations per single POM anion.
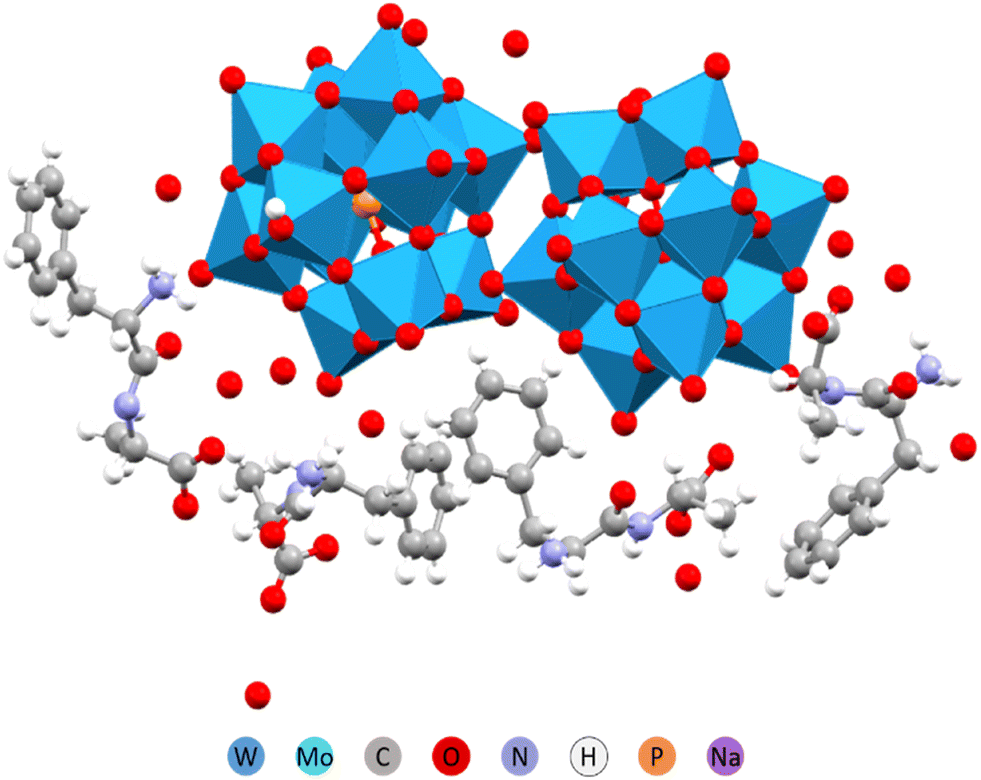 | ||
| Fig. 7 Molecular structure of the (H3O)2(HPhe-Ala)4[PW12O40]2·11H2O.32 | ||
In case of more sterically encumbered Ala-Phe peptide, with the ring shielding the carboxylic acid group, there was observed formation of two compounds, different in both the structure and chemical composition, but both featuring domination of π-stacking over the hydrogen bonding (with benzene rings of Phe fragment situated exactly above each other).
The product crystallizing quickly on solution drying in air, contained only two peptide cations per POM. It was bearing a lot of interstitial water, featuring the (HAla-Phe)2[HPW12O40]·4H2O composition (Fig. 8A). Slow crystallization in solution offered unexpectedly a water-free product with 3:1 peptide-to-POM ratio (Fig. 8B), giving a (HAla-Phe)3[PW12O40] composition.
 | ||
| Fig. 8 Molecular structures of the (HAla-Phe)2[HPW12O40]·4H2O (A) and (HAla-Phe)3[PW12O40] (B).32 | ||
When the same 3:1 peptide to POM ratio was used in synthesis, the structure of POM complex with Tyr is rather principally different from that of Phe containing peptides, most probably, because in this case only a single amino acid was present and no sterical hindrance contributed. The composition is 2:1 peptide-to-POM, (HTyr)2[HPW12O40]·4H2O (Fig. 9), analogous to that of the Phe-Ala derivative. However, both the amino acid cations and the POM ligands are located in infinite chains.
 | ||
| Fig. 9 Molecular (A) and crystal (B) structures of the (HTyr)2[HPW12O40]·4H2O.33 | ||
The oligo alanine derivatives produced in part rather unexpected results. The di-alanine structure resembled the diglycine structures, where the POM units were arranged in close packing of spheres, with the peptide cations filling in the gaps.
On an attempt to produce tri-alanine derivatives, the only product that crystallized was the derivative of amino acid alanine, resulting apparently from hydrolysis of the original tri-peptide.
The same structure, (HAla)H5[PW12O40]2·4H2O, involving L-alanine was obtained using D,L-racemic amino acid mixture. It resembled a metal–organic framework (MOF), in that the structure was highly porous and made up of POMs linked with alanine via apparently extremely strong hydrogen bonding (Fig. 10).32
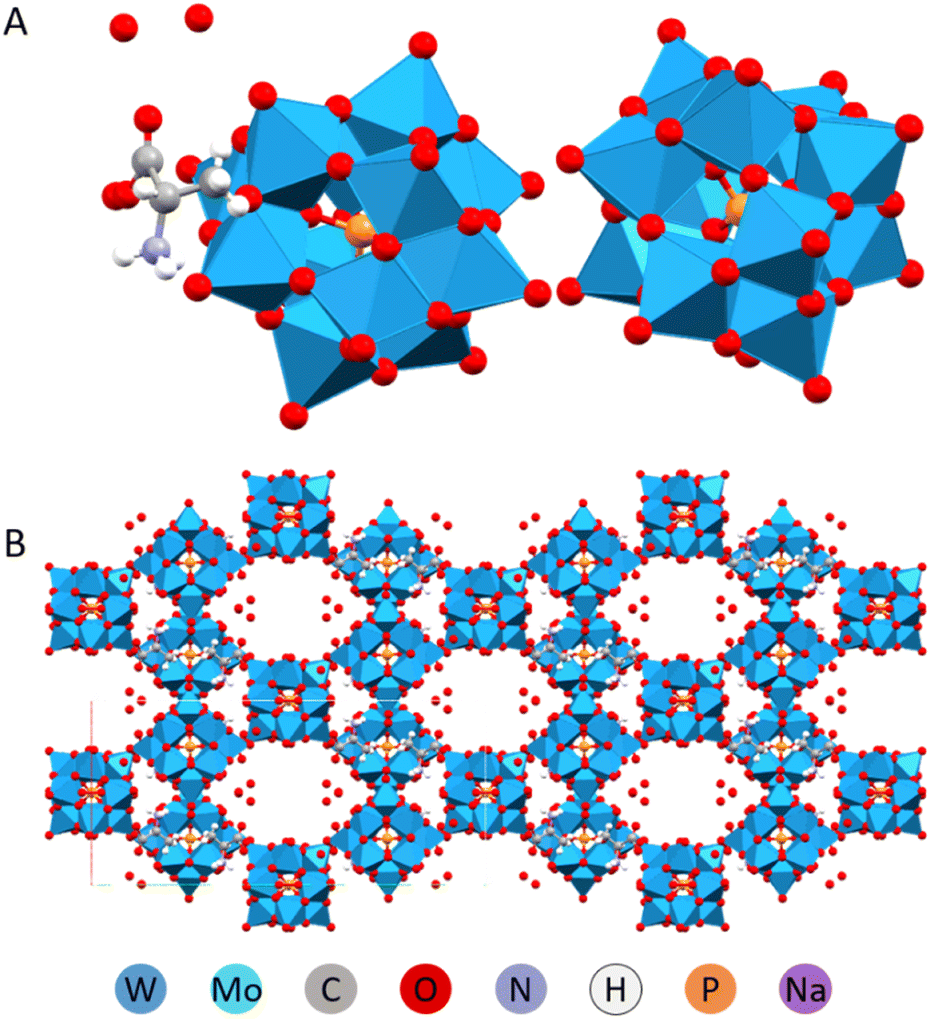 | ||
| Fig. 10 Molecular (A) and crystal (B) structure of the (HAla)H5[PW12O40]2·4H2O.33 | ||
Probing redox reaction mechanisms using POM models
Among the principally interesting problems that can potentially be clarified via structural studies of POM complexes is the challenge in understanding of molecular mechanisms in oxidation catalysis performed by nanoparticles of oxide minerals, nanozymes.33,34 General understanding is that NP are generating reactive oxygen species (ROS), especially when subjected to illumination by UV or visible light.An alternative pathway may actually be direct catalytic transformation on the surface of NP, associated with simultaneous proton and electron transfer or even cascade of the electron-and-proton transfer events. In the recent study of Mo-POM and W-POM complexes with Trp, we observed emergence of colour associated with charge transfer along with short hydrogen bond supported contacts between the imidazole ring and POM, responsible apparently for the electron transfer and between the ammonium –NH3+ entity and the POM, opening for the proton transfer (Fig. 11).21 Interestingly, it was the same Trp molecule that was involved in both, demonstrating how the redox cascade, leading to formation of plant hormone compounds can occurs with the aid of oxidative mineral NP.
 | ||
| Fig. 11 A fragment of the crystal structure of (HTRP)3PW12O40·5H2O with indication of strong interactions responsible for electron and proton transfer.34 | ||
It is interesting to note that the observed oxidation of Trp amino acid by both CeO2 and Keggin POMs in the presence of dissolved oxygen was resulting in a single specific primary product, 3-hydroxopyrroloindole carboxylic acid (PIC), demonstrating nanozyme character of this process.35 Following electron-and-proton transfer cascade sequence was proposed to explain the reaction mechanism (Scheme 1).
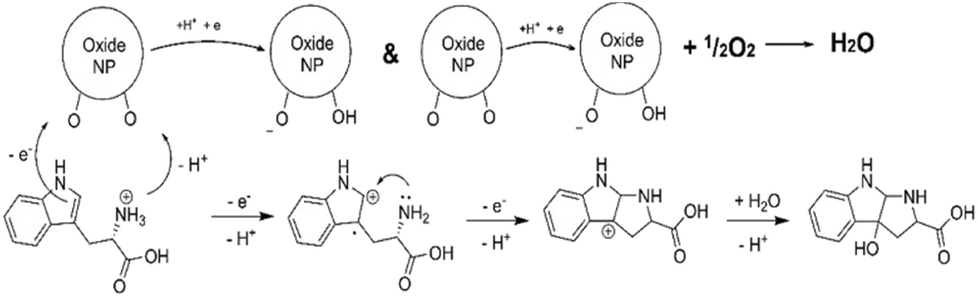 | ||
| Scheme 1 Molecular reaction mechanism in oxidation of Trp amino acid to PIC.35 | ||
Conclusions
W-POM and Mo-POM are typically thought of as analogous compounds, with nearly identical structure and sizes, along with identical formal charge, though the structures discussed here have shown that there are pronounced differences in how they interact with peptides/protein fragments. This is likely primarily due to the differing electronegativity between W and Mo; Mo is more electronegative, and thus the charge gradient of the Mo–O bond is less pronounced than for W–O, which likely leads to a lower partial charge on the Mo-bound oxygen, and, on average, weaker hydrogen bonding interactions with the ligand.How the ligand interacts with the POM and which complex structure forms depend on a number of factors. Those we have identified are temperature, pH, salinity, speed of formation, and the nature of the components; aside from the metal in the POM, the side chains of the peptides have a significant effect on the structure. POMs with diglycines as ligands typically form a hexagonal close packing motif, while larger side chains result in arrangement resembling a simple cubic packing as the result of π-stacking of the ligands.
Obtained results may help to shed light additionally on the nature of interactions in nanocomposite materials resulting in their complex structure, and perhaps aid in rational design of novel materials.
The structures reported here are intended to serve as a model of how NPs might interact with proteins. The factors identified are highly relevant for biological contexts; bodily fluids are typically quite strongly buffered towards a specific pH, and have a high salinity. Certain effects of NPs may be apparent only after a long period, e.g. if they are persistent in a certain type of tissue.
Data availability
The original data used for structure solution and refinement has been reported in publications summarized in the present Highlight article. All further details are available on request from authors.Author contributions
Both authors contributed actively to the conceptualization and writing of the manuscript. BG made major input in preparation of the figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Swedish Research Council (Vetenskapsrådet) for the support to the projects 2018-03811_VR and 2022-03971_VR, molecular mechanisms in mineral (oxide) nanoparticle interactions with proteins.Notes and references
- M. F. Hochella, S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Science, 2008, 319, 1631–1635 CrossRef CAS.
- M. F. Hochella, D. W. Mogk, J. Ranville, I. C. Allen, G. W. Luther, L. C. Marr, B. P. McGrail, M. Murayama, N. P. Qafoku, K. M. Rosso, N. Sahai, P. A. Schroeder, P. Vikesland, P. Westerhoff and Y. Yang, Science, 2019, 363, eaau8299 Search PubMed.
- H. L. Karlsson, J. Gustafsson, P. Cronholm and L. Möller, Toxicol. Lett., 2009, 188, 112–118 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, 1805368 CrossRef CAS PubMed.
- G. A. Seisenbaeva, K. Fromell, V. V. Vinogradov, A. N. Terekhov, A. V. Pakhomov, B. Nilsson, K. N. Ekdahl, V. V. Vinogradov and V. G. Kessler, Sci. Rep., 2017, 7, 15448 CrossRef PubMed.
- F. G. Svensson, V. A. Manivel, G. A. Seisenbaeva, V. G. Kessler, B. Nilsson, K. N. Ekdahl and K. Fromell, Nanomaterials, 2021, 11, 1100 CrossRef CAS.
- G. Bashiri, M. S. Padilla, K. L. Swingle, S. J. Shepherd, M. J. Mitchell and K. Wang, Lab Chip, 2023, 23, 1432–1466 RSC.
- D. Costa, L. Savio and C.-M. Pradier, J. Phys. Chem. B, 2016, 120, 7039–7052 CrossRef CAS.
- S. Joshi, I. Ghosh, S. Pokhrel, L. Mädler and W. M. Nau, ACS Nano, 2012, 6, 5668–5679 CrossRef CAS PubMed.
- H. Lee, Pharmaceutics, 2021, 13, 637 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef CAS PubMed.
- J. C. Raabe, F. Jameel, M. Stein, J. Albert and M. J. Poller, Dalton Trans., 2024, 53, 454–466 RSC.
- L. Zhai and H. L. Li, Molecules, 2019, 24, 3425 CrossRef CAS.
- A. Barba-Bon, N. I. Gumerova, E. Tanuhadi, M. Ashjari, Y. Chen, A. Rompel and W. M. Nau, Adv. Mater., 2024, 36, 2309219 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 909–933 CrossRef CAS.
- https://measurlabs.com/methods/dynamic-light-scattering-dls/ .
- A. C. Venu, R. N. Din, T. Rudszuck, P. Picchetti, P. Chakraborty, A. K. Powell, S. Krämer, G. Guthausen and M. Ibrahim, Molecules, 2021, 26, 7481 CrossRef CAS.
- V. G. Kessler, G. A. Seisenbaeva, S. Håkansson and M. Unell, Angew. Chem., Int. Ed., 2008, 47, 8506–8509 CrossRef CAS PubMed.
- G. A. Seisenbaeva, G. Daniel, J. M. Nedelec and V. G. Kessler, Nanoscale, 2013, 5, 3330–3336 RSC.
- M. Baranov, Y. Duan, N. Leffler, S. Avineri, V. Ezersky and I. A. Weinstock, Chem. Commun., 2023, 59, 4364–4367 RSC.
- B. Chakraborty and I. A. Weinstock, Coord. Chem. Rev., 2019, 382, 85–102 CrossRef CAS.
- J. Perez Holmberg, E. Ahlberg, J. Bergenholtz, M. Hassellöv and Z. Abbas, J. Colloid Interface Sci., 2013, 407, 168–176 CrossRef.
- W. Wang, K. M. Wentz, S. E. Hayes, D. W. Johnson and D. A. Keszler, Inorg. Chem., 2011, 50, 4683–4685 CrossRef CAS PubMed.
- C. André Ohlin, Phys. Chem. Chem. Phys., 2020, 22, 4043–4050 RSC.
- D. Lebbie, T. Izuagie, M. Pascual-Borràs, B. Kandasamy, C. Wills, P. G. Waddell, B. R. Horrocks and R. J. Errington, Inorg. Chem., 2025, 64(5), 2379–2393 CrossRef CAS PubMed.
- S. Lentink, D. E. Salazar Marcano, M. A. Moussawi and T. N. Parac-Vogt, Angew. Chem., Int. Ed., 2023, 62, e202303817 CrossRef PubMed.
- D. E. Salazar Marcano and T. N. Parac-Vogt, Coord. Chem. Rev., 2024, 518, 216086 CrossRef CAS.
- K. M. Rominger, G. Nestor, J. E. Eriksson, G. A. Seisenbaeva and V. G. Kessler, Eur. J. Inorg. Chem., 2019, 2019, 4297–4305 CrossRef CAS.
- B. Greijer, T. De Donder, G. Nestor, J. E. Eriksson, G. A. Seisenbaeva and V. G. Kessler, Eur. J. Inorg. Chem., 2021, 2021, 54–61 CrossRef CAS.
- B. H. Greijer, G. Nestor, J. E. Eriksson, G. A. Seisenbaeva and V. G. Kessler, Dalton Trans., 2022, 51, 9511–9521 RSC.
- B. Greijer, E. Stigell, T. Guerin and V. G. Kessler, Cryst. Growth Des., 2024, 24(15), 6483–6491 CrossRef CAS.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS.
- Y. Zhang, G. Wei, W. Liu, T. Li, Y. Wang, M. Zhou, Y. Liu, X. Wang and H. Wei, Nat. Rev. Methods Primers, 2024, 4, 1–22 CrossRef.
- A. Nefedova, F. G. Svensson, A. S. Vanetsev, P. Agback, T. Agback, S. Gohil, L. Kloo, T. Tätte, A. Ivask, G. A. Seisenbaeva and V. G. Kessler, Inorg. Chem., 2024, 63, 8556–8566 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |