Optimizing oxygen vacancy concentration and electronic transport processes in a MnxCo/CeO2 nanoreactor: regulation mechanism of the radical to non-radical pathway†
Received
24th September 2024
, Accepted 7th November 2024
First published on 21st November 2024
Abstract
Enhancing the efficiency of electron transfer and augmenting the utilization rate of peroxymonosulfate (PMS) pose challenges for advanced oxidation processes (AOPs). A high-performance bimetallic-doped catalyst (MnCo/CeO2) with an appropriate concentration of oxygen vacancies (OVs) was successfully designed using a straightforward synthesis strategy. It primarily activates PMS through non-radical pathways. Systemic characterization, experiments, and theoretical calculations have demonstrated that reasonable OVs and the Mn/Co bimetallic doping strategy effectively modulated the surface spatial electron structure and greatly improved interfacial electron transfer processes (ETP). Ultimately, MnCo/CeO2 exhibits a remarkable ciprofloxacin (CIP) removal efficiency of 93.71% (k = 0.03501 min−1) within 50 min (after 5 cycles, 89%), which is 5.03 times faster than that of traditional CeO2 (k = 0.00696 min−1), and the possible degradation pathway as well as toxicity of intermediate products were identified using LC-MS, Fukui function analysis, and toxicity evaluation. This work proposes a feasible strategy for designing bimetallic-doped metallic oxide catalysts, which have great application potential for the degradation of organic contaminants under actual harsh environmental conditions.
Environmental significance
Activating PMS can effectively remove CIP from aqueous environments, reduce its environmental pollution load, improve water quality and the ecological environment, and help achieve greener pollution control. This study presents an MnCo/CeO2/PMS reaction system that effectively degrades CIP in a short period through careful material design, optimization of reaction conditions, and cost control. The degradation mechanism of the MnCo/CeO2/PMS/CIP system and the toxicity of intermediates were comprehensively examined. Furthermore, the efficacy of this system was validated using actual water samples and various pollutants, demonstrating its potential application in actual pollution control, thereby promoting environmental protection and sustainable development.
|
1. Introduction
Peroxymonosulfate (PMS)-based advanced oxidation processes (AOPs) have proven to be extremely efficacious in removing persistent pollutants.1,2 Diverse, plentiful reactive oxygen species (ROS), including singlet oxygen, sulfate radicals, hydroxyl radicals, and superoxide radicals (1O2, ·SO4−, ·OH, and ·O2−), are primarily produced by breaking the O–O bond,3 which efficiently degrade target pollutants. Since PMS-based AOPs offer more distinct advantages than traditional Fenton reactions in the generation of ·SO4−, activated PMS has been extensively utilized in AOPs. Recently, catalytic activation has attracted significant attention owing to its straightforward operational system and the absence of additional energy supply. Since the catalysts have a crucial role in the reaction mechanism, the quest for an appropriate catalyst to activate PMS remains a hot topic.
As a highly active rare-earth oxide catalyst, CeO2 readily undergoes redox reactions between Ce4+ and Ce3+, resulting in the formation of numerous oxygen vacancies (OVs) within its structure.4 Therefore, CeO2 is widely used for the removal of organic pollutants. Excellent catalysts are also used in various areas.5,6 However, a single catalyst is not conducive to catalytic reactions, and both too high and too low concentrations of oxygen vacancies are unfavorable for contaminant removal.7 Currently, diverse approaches have been developed to enhance the performance of catalysts. These strategies include doping, morphology regulation, heterojunction construction, defect engineering, and loading.8–14 Among them, defect engineering and transition metal doping are of great interest owing to their efficiency and practicality. Not only does the incorporation of transition metal ions increases the amount of active sites available, but it also facilitates the formation of OVs.15 It is important to highlight that excessive OVs can reduce catalyst selectivity, thus negatively impacting pollutant degradation efficiency. Conversely, too low OVs concentration may result in insufficient catalytic active sites, thereby hindering the degradation process. Thus, an appropriate OV concentration is essential for the degradation efficiency of pollutants.16–19 Simultaneously, the presence of OVs augments the binding affinity of PMS molecules, thereby improving the charge transfer processes between interfaces.17,20 Additionally, it enhances the anchoring of dioxygen onto the surface of the target catalyst and meanwhile lowers the activation energy required for the self-decomposition of the PMS surface.21
In a previous study, Co(II)-doped CeO2 was used to remove refractory pollutants, resulting in positive outcomes. This study found that the decorated Co, serving as the main active site, was capable of transferring electrons to both the PMS and Ce sites.22 Moreover, CeO2 acted synergistically to enhance the electron transfer process (ETP), further enhancing the catalytic activity of Co2Ce8-300-2. In a parallel effort, Mei et al. synthesized Co–CeO2via solvothermal calcination, which generated more defects and sites on its surface compared with pure CeO2, thereby improving its adsorption performance for tetracycline.23 Despite exhibiting superior catalytic activity compared to traditional CeO2, the synergistic effects of these materials have not been fully harnessed. Therefore, constructing an efficient catalyst is crucial. Compared to single metal doping, bimetallic doping typically exhibits enhanced catalytic activity and stability. The valence and coordination environment of different metals can adjust the surface electron density, thereby enhancing the activity of the catalytic sites. Moreover, the majority of research has indicated that the primary pathways for the catalytic activation to degrade pollutants involve the free radical pathway together with the non-radical pathway (including ETP). These primary pathways comprise the organics attacking the PMS directly (adjacent transfer) and electrons transferring to the PMS via a catalyst (electrons shuttle).24 Free radicals exhibit high reactivity towards organic pollutants, while non-radical reactions can avoid free radical self-quenching and enhance the utilization rate of PMS.25 Compared with other radicals, 1O2 is a weak oxidant that demonstrates notable selectivity towards electron-rich pollutants. The combined action of both radical and non-radical routes provides a more effective approach for PMS activation compared to single pathway activation.3 Therefore, the introduction of Co and Mn can not only effectively regulate the concentration of OVs in CeO2 but also improve the surface space electronic structure of PMS to further enhance the ETP in the reaction process and greatly enhance the catalytic capacity. However, how to optimize the Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ratio and OVs concentration to enhance the catalytic activity of radical and non-radical avenues has been rarely reported.
:Mn ratio and OVs concentration to enhance the catalytic activity of radical and non-radical avenues has been rarely reported.
Herein, MnCo/CeO2 with suitable OVs was prepared using a simple hydrothermal calcination strategy. Multi-characterization confirmed that MnCo/CeO2 could successfully activate PMS to pure ciprofloxacin (CIP) under the combined effect of both free radicals and non-radicals. Furthermore, research on the catalytic mechanism unveiled that an appropriate concentration of OVs could facilitate electron migration, improve the oxidation activity, and enhance PMS utilization. Besides, the influences of different factors on the catalytic performance were investigated, encompassing PMS and CIP concentration, catalyst dosage, different temperatures, initial pH, anion types as well as natural organic matter. In addition, the activation of PMS by the MnCo/CeO2 catalyst was scrutinized via density functional theory (DFT) calculations. Meanwhile, the Fukui function was calculated using DFT to predict the attacking sites of different actives on CIP. The production of different intermediates was also demonstrated using LC-MS, the degradation routes were proposed and the toxicity was evaluated. The feasibility of CIP removal within an authentic water environment was analyzed. This work furnishes a promising foundation for ongoing research on activating PMS through bimetallic-doped metallic oxide.
2. Experimental section
2.1 Fabrication of MnxCo/CeO2
In a representative synthetic route, 6 mmol Ce(NO3)3·6H2O, 2 mmol CoCl2·6H2O, and 2x (2x = 0.5, 2, 6) mmol MnCl2·4H2O were respectively solubilized in 100 mL ultrapure water by ultrasonication and transferred to a thermostat water bath. A tan suspension was formed by rapidly adding 4 mL of NH3·H2O to the above solution. After stirring the mixture for 2 h at 40 °C, it was aged for 12 h. Then, the as-acquired ingredient (precipitation form) was washed sufficiently with the aid of ultrapure water as well as anhydrous ethanol. The product was then dried using a vacuum freeze dryer. Finally, the prepared products were heated in a tube furnace (300 °C, 2 °C min−1, 2 h). The obtained products were designated as Mn0.25Co/CeO2, MnCo/CeO2, and Mn3Co/CeO2, respectively. For other sample synthesis steps, see the supplementary information. See Text S1† for detailed information on the chemicals included in the present study.
2.2 Computational methods and data analysis
To further elucidate the mechanism of the excellent performance of MnCo/CeO2, first-principles calculations were performed. The calculation details are included (see Text S2†).
3. Results and discussion
3.1 Morphology and microstructure of MnxCo/CeO2
MnxCo/CeO2 was synthesized through a typical hydrothermal calcination method. As shown in Fig. S1,† the XRD spectra confirmed the successful preparation of MnxCo/CeO2. The catalyst samples' diffraction peaks were precisely matched with those of CeO2 (PDF: 43-1002). However, the diffraction intensities of the sample facets varied significantly with increasing amounts of Mn:Co, attributed to the presence of defects. The diffraction peak of MnCo/CeO2 became wider, which is attributed to its lower crystallinity.26 Additionally, all samples exhibited stalactite-like morphologies, and the surface particles become progressively smaller and more tightly packed with the increase in the Mn/Co content, thereby providing more adsorption sites (Fig. S2†). The morphology and structure of all the catalysts were similar, suggesting that the incorporation of Mn and Co failed to alter the morphology of CeO2. Simultaneously, TEM characterization was performed to further indicate the microstructure of the samples using the MnCo/CeO2 catalyst as the representative material. This is shown in Fig. 1(a). The TEM results obtained were consistent with the SEM results, confirming that the sample's surface was comprised of numerous fine particles. In the meantime, the HRTEM images presented in Fig. 1(b) revealed the presence of OVs near the (111) facets of CeO2 (0.307 nm) due to the incompleteness of the crystal structure (red dashed line). Moreover, a ringlike SAED pattern diagram confirmed the polycrystalline nature of the synthesized catalyst.27 The mapping images of MnCo/CeO2 shown in Fig. S3† discerned that in the samples, Co, Ce, Mn, and O were uniformly allocated. This observation further confirmed that the MnCo/CeO2 catalyst was successfully synthesized.7 Moreover, Fig. S4† demonstrated the porosity of MnCo/CeO2, and it can be seen that this sample exhibited type II adsorption–desorption isotherms. The pore distribution was primarily focused on 2.79 nm, and MnCo/CeO2 exhibited an encouraging surface area of 187.4 m2 g−1, which is ready to provide more adsorption sites and active centers. Fig. S5† displayed the contact angles of all the samples, indicating that MnCo/CeO2 (18.703°) was of superior hydrophilicity compared to other catalysts, which facilitated mass transfer during catalysis.28
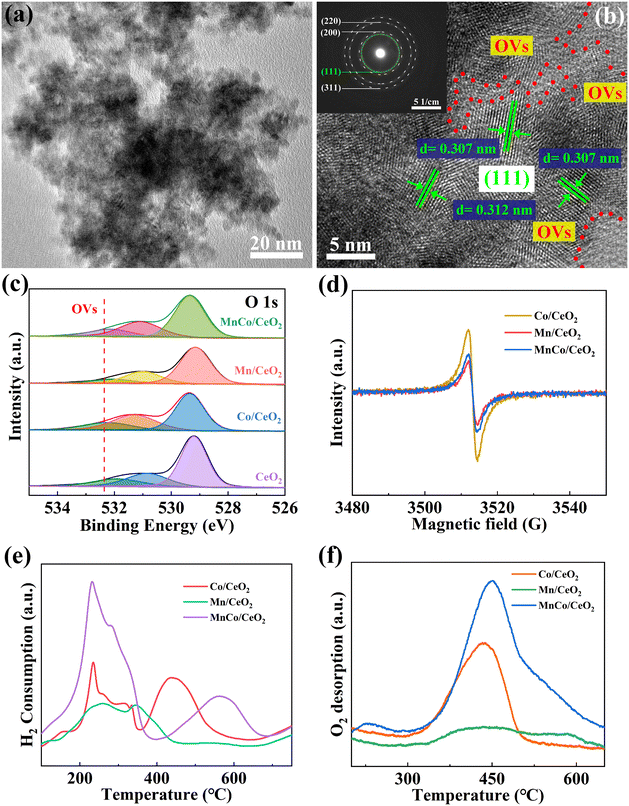 |
| | Fig. 1 (a) TEM and (b) HRTEM (SAED image in the inset) of MnCo/CeO2. (c) O 1s XPS spectra, (d) ESR patterns, (e) H2-TPR, and (f) O2-TPD patterns of target samples. | |
3.2 Analysis of oxygen vacancies
The microscopic chemical environments as well as the bond structures of CeO2, Co/CeO2, Mn/CeO2, and MnCo/CeO2 were analyzed via the XPS pathway, calibrated with the C 1s spectrum (binding energy of 284.8 eV). As demonstrated in Fig. S6(a),† Ce, Co or/and Mn, and O were identified within the survey spectra of CeO2, Co/CeO2, Mn/CeO2, and MnCo/CeO2, which indicated that the nanomaterials were effectively prepared. Furthermore, elemental high-resolution spectra were used so as to analyze the OVs and electron-transfer directions. The Ce 3d spectrum was fitted according to eight peaks (Fig. S6b†), corresponding to Ce 3d5/2 and Ce3d3/2, respectively. Among them, u′′′, u′′ and u belong to Ce4+ 3d3/2, v′′′, v′′ as well as v peaks belong to Ce4+ 3d5/2, and the u′ peak pertains to the signal of Ce3+ 3d3/2, while the v′ peak pertains to Ce3+ 3d5/2.29 As depicted in Fig. S7,† the proportion of Ce3+ decreased when compared to pure CeO2. This was primarily attributed to the doping of Co and Mn atoms, leading to a reduction in the concentration of OVs concentration in both Co/CeO2 and Mn/CeO2.30 However, the bimetallic doping strategy can enhance the formation of Ce3+ compared to single metal doping, attributed to the synergistic effect, which alters the electronic states on the catalyst surface and transforms some high valence states of Ce4+ into low valence states of Ce3+. The presence of Ce3+ indicated the formation of OVs.31,32 The O 1s fine-structure spectrum is shown in Fig. 1(c), in which the peaks observed at 529.35 eV (representative lattice oxygen), 531.1 eV (indicator-adsorbed surface hydroxy groups), together with 532.15 eV (OVs) in MnCo/CeO2 could be assigned, respectively.33 It was evident that the presence of Co or Mn in CeO2 altered the content of OVs. Co/CeO2 exhibited the largest integral area at OVs, whereas Mn/CeO2 displayed the lowest area. This result shows that an appropriate concentration of OVs facilitates electron migration. This situation is favorable in the process of activating PMS, which can improve the activation efficiency of PMS and further enhance the degradation performance of the target pollutants. As we know, it has been reported that high-resolution spectroscopy can be utilized to investigate the direction of electron migration more precisely and accurately.34 The peaks located at 796.7 eV (Co2+)/794.95 eV (Co3+) and 781.4 eV (Co2+)/779.8 eV (Co3+) were an indicator of the Co 2p1/2 and Co 2p3/2 states in Co/CeO2 (Fig. S6c†).35 It implies that electrons flow from MnCo/CeO2 to Co/CeO2. Intriguingly, the corresponding peaks in MnCo/CeO2 exhibited a shift towards a lower binding energy. Additionally, the high-resolution spectrum concerning Mn 2p is depicted in Fig. S6(d).† The deconvoluted Mn 2p3/2 spectrum was resolved into three distinct peaks at 640.85 eV (Mn2+), 642.65 eV (Mn3+), and 645.90 eV (Mn4+), respectively.36,37 At the same time, the blue shift for Mn/CeO2 was observed. It means that electrons flow from Mn/CeO2 to MnCo/CeO2. The aforementioned phenomena indicate that the electrons in CeO2 flow to Co, while CeO2 gets electrons from Mn. ICP-OES also showed that the cobalt and manganese doping in MnCo/CeO2 was 6.21 wt% and 7.51 wt% (in Table S1†), respectively. The results presented herein demonstrate the effective doping of Co and Mn in CeO2.38 XPS analysis revealed that the close coexistence of Mn, Co, Ce, and O could enhance the electron and oxygen transfer efficiency, thereby ameliorating CIP degradation.
In addition, the OVs in Co/CeO2, Mn/CeO2, and MnCo/CeO2 were further characterized through ESR. Fig. 1(d) reveals that Co/CeO2 exhibits the strongest signal peak, indicating the highest concentration of OVs. Conversely, Mn/CeO2 displays the weakest signal peak and lowest concentration of OVs. The signal peak of MnCo/CeO2 is located between Co/CeO2 and Mn/CeO2, indicating that an appropriate concentration of OVs can enhance the electron transport rate of MnCo/CeO2. This indicates that the catalyst is conducive to improving the activation efficiency of PMS and enhancing the purification performance for pollutants. It was also consistent with the results from the XPS characterization results.
OV is a common factor that directly impacts the catalytic activity of materials. Additionally, the high redox capacity of the catalyst can effectively transfer oxygen to the metal nano-catalyst, thus influencing its catalytic performance. Typically, the catalytic redox performance, surface oxygen species, and active oxygen content of CeO2-based materials were characterized using H2-TPR and O2-TPD. Based on Fig. 1(e), it is evident that CeO2-based catalytic materials exhibit two primary reduction peaks within the temperature range of 300–700 °C. The peak of reduction observed in the H2-TPR curve of Co/CeO2 at 235 °C/258 °C was primarily attributed to the reduction of CeO2 and Mn/Co oxides.39 The second peak at 345 °C for Co/CeO2 or 438 °C for Mn/CeO2 can be attributed to the spillover of hydrogen from the Co or Mn species to the adjacent CeO2.40 It was widely acknowledged that the result of O2-TPD reflects the total OVs concentration (TOVs).34 In order to identify changes in the OVs concentration, the peak areas of Co/CeO2, Mn/CeO2, and MnCo/CeO2 were calculated according to the corresponding O2-TPD plots, as portrayed in Fig. 1(f) together with Fig. S8.† According to Fig. S8,† it is evident that MnCo/CeO2 exhibits the highest concentration of TOVs, which may be a result of the synergistic effect between Co and Mn, leading to the partial oxidation and generation of both surface OVs and corresponding interface OVs. Due to the bimetallic doping of Mn and Co, it leads to a more disordered lattice structure and increased defects. As a result, the MnCo/CeO2 catalyst reflects higher TOVs concentration than Co/CeO2 and Mn/CeO2, which also has a significant impact on its catalytic performance. These findings concur with the results of XRD characterization. However, in the whole of the MnCo/CeO2/PMS reaction system, only the surface or near-surface OVs plays a major role, which further proves that MnCo/CeO2 with appropriate OVs concentration is more conducive to activating PMS, enhancing ETP, and ultimately improving its catalytic performance.
3.3 MnCo/CeO2 catalyst activates PMS to eliminate CIP
CIP was identified as the target pollutant for the purpose of evaluating the performance of MnCo/CeO2 catalytic material in activating PMS. In the absence of PMS, CIP was primarily purified through adsorption. It is mainly due to the porous structure of MnCo/CeO2, which has an outstanding specific surface area. The results in Fig. 2(a) indicate that PMS has a weak ability to degrade 10 mg L−1 CIP alone, with only approximately 13% degradation observed. MnCo/CeO2 demonstrated superior degradation efficiency towards 10 mg L−1 CIP in 50 min, with a degradation rate of 93.71%. Additionally, on the basis of pseudo-first-order kinetics (Fig. 2b), MnCo/CeO2 exhibits the maximum apparent rate constant (k = 0.03501 min−1 5.03 times faster than CeO2). The above results further confirmed its outstanding catalytic activity. In the meantime, appropriate OVs were conducive to the construction of an electron transfer channel. Additionally, excessive or insufficient Mn doping can impede the degradation rate of MnxCo/CeO2 composites. This may be because when Mn is excessive, Co and some Mn occupy the position on the CeO2 surface, while some Mn enters the CeO2 lattice.22 Due to the shielding effect, it was not conducive to contact with PMS but rather, it inhibited the degradation activity.
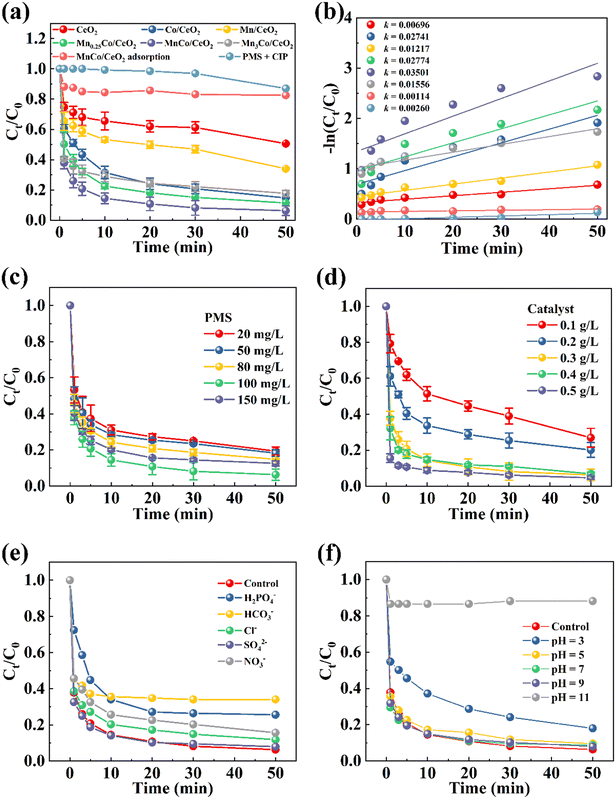 |
| | Fig. 2 (a) Purification performance of the synthesized catalysts for CIP, (b) the corresponding degradation kinetics plots (experimental conditions: [CIP] = 10 mg L−1, [PMS] = 100 mg L−1, and [catalyst] = 300 mg L−1), and the role of diverse factors in CIP decontamination: (c) PMS concentration, (d) catalyst quantity, (e) inorganic anions, and (f) pH. | |
3.4 Different factors impacting CIP degradation via MnCo/CeO2
Typically, multifarious experimental parameters will influence the decontamination effect during PMS activation. The following factors were primarily investigated in this study: PMS concentration, CIP concentration, catalyst dosage, inorganic anions, humic acid (HA) concentration, initial pH value, temperature, and different matrix water bodies. As revealed in Fig. 2(c), the degradation rate regarding CIP did not exhibit an increase with the excessive addition of PMS, which is attributed to its self-scavenging effect. Along with the PMS concentration elevated to 100 mg L−1, the catalysis performance of MnCo/CeO2 was gradually enhanced. However, the severe self-quenching of the active species led to a decrease in the PMS utilization efficiency.41 Consequently, augmenting the dosage of PMS does not significantly impact the catalytic activity. Moreover, the experimental study on CIP degradation at different initial concentrations revealed a decrease in the removal rate from 93.6% to 57.4% upon the CIP concentration, reaching 30 mg L−1 (Fig. S9a†). This is due to the occupation of active sites on MnCo/CeO2 by substantial pollutants. Besides, varying dosages of MnCo/CeO2 can also affect the overall efficiency of CIP. In Fig. 2(d), when utilizing a specific MnCo/CeO2 concentration (0.1–0.5 g L−1), the corresponding removal efficiencies varied from 73.06% to 95.34%. Significantly, the MnCo/CeO2/PMS system demonstrated remarkable removal efficiency at high catalyst concentrations. This was because a greater quantity of catalysts would offer more active sites, prompting PMS to generate more ROS for degrading CIP.42
The MnCo/CeO2/PMS system can effectively degrade 10 mg L−1 of CIP in the context of multiple environmental-relevant anions (i.e., H2PO4−, HCO3−, Cl−, SO42−, and NO3−) in Fig. 2(e). Particularly, HCO3− in water typically raises the pH, creating an alkaline environment that impacts the degradation of CIP. Additionally, it may react with the catalyst or target, lowering the reaction rate and thus inhibiting CIP degradation. The existence of HA in natural water can impact the purification of pollutants. To explore this impact on CIP degradation, HA was added and analyzed (Fig. S9b†). The findings revealed that the performance of CIP degradation decreases with increasing HA concentration. This is mainly because HA has a strong π–π accumulation ability and is facilely anchored onto the catalyst surface, thereby obstructing the interplay between MnCo/CeO2 and PMS or CIP by competing active sites.43 Furthermore, it carries a negative charge in water that affects the degree of CIP adsorption and dispersion on MnCo/CeO2 surfaces. The role of the initial pH value in CIP clean-up has also been examined in Fig. 2(f). Under acidic conditions, when pH = 3, H+ will make HSO5− positively charged, thus hindering the interaction with the positively-charged oxide surface and resulting in insufficient catalytic oxidation efficiency.44 Within the range of pH 5 to 9, CIP can be effectively degraded, and the rate of degradation increases with increasing pH value. The optimal catalytic effect of MnCo/CeO2 was observed under neutral and weakly basic conditions. This is primarily due to Mn2+ and Co2+ effectively activating PMS under neutral conditions, leading to increased ·SO4− production and accelerated degradation reaction rates. Additionally, in weak alkaline conditions, PMS can be activated through alkali activation to generate ·SO4− as well as interact with OH− to generate ·OH.45 However, the degradation rate of CIP was only 11.9% under strongly alkaline conditions (pH = 11). A large amount of OH− forms polyhydroxy surface complexes, which attach to the surface of MnCo/CeO2, thus impeding the static interaction between the material and PMS. Eventually, it will limit the catalytic oxidation ability. Moreover, ·SO4− reacts with OH− to form ·OH, and the lifetime of ·SO4− (10–30 μs) was longer in aqueous solution than that of ·OH (<1 μs). This phenomenon was not conducive to CIP degradation. In addition, PMS may undergo alkaline catalytic hydrolysis, which would consume PMS, inhibit ROS production and finally thereby inhibit the purification of CIP. Additionally, the elimination speed of CIP in aqueous solution was found to be accelerated with an increase in temperature, as illustrated in Fig. S9(c).† The MnCo/CeO2 catalyst can also purify CIP in water with different natural substrates. However, when multiple natural substances co-exist in the natural background water body, the degradation efficiency of CIP may be inhibited to some extent (Fig. S9d†).
At the same time, the universality of MnCo/CeO2 in PMS activation was further studied, and other refractory pollutants were tested, including CTC, OTC, TC, and RhB. The results demonstrated the exceptional catalytic performance of MnCo/CeO2 in degrading organic pollutants through PMS activation (Fig. S9e†). The leaching behavior of metal ions in solution following the MnCo/CeO2 catalyst reaction was investigated using ICP-OES. As presented in Table S1,† the leaching concentrations of Co and Mn for the MnCo/CeO2 catalyst were found to be 0.55 mg L−1 and 0.26 mg L−1, respectively. This suggests that the reaction system will not contribute to environmental pollution, thereby promoting the protection of both the environment and human health. Moreover, the catalytic prowess of MnCo/CeO2/PMS was substantiated through comparative analysis with others. Compared to the materials mentioned in Table S2,† the MnCo/CeO2 catalyst in this study exhibits superior catalytic performance under higher PMS concentrations or the same conditions. Therefore, MnCo/CeO2 was a more effective catalyst.
3.5 Degradation avenues and toxicity estimation of intermediates
The mechanism of ROS attacking CIP in the MnCo/CeO2/PMS reaction system has been further elucidated through DFT calculations.46,47 DFT calculations were performed to obtain the Fukui indices for nucleophilic attack (f+), electrophilic attack (f−), together with radical attack (f0), as determined from the charge distribution (natural population analysis (NPA)) of the CIP molecule.48 The results of the Fukui function are shown in Table 1. Atom 15N displays the highest f− value (0.0913), and its f0 (0.0566) is also relatively high. This suggested that 15N was more susceptible to attack by radicals and electrophilic reagents. Simultaneously, the atoms 14C (f− at 0.0408, f0 at 0.0393) and 24N (f− at 0.0663, f0 at 0.0364) with a more positive value of Fukui index were prone to being targeted by ·SO4−, 1O2, ·OH, and ·O2−. Despite having high Fukui index values, 3C (f− at 0.0559, f0 at 0.0319) and 13O (f− at 0.0704, f0 at 0.0804) were resistant to radical attack because of their saturated sites and steric hindrance effect.48 Manifestly, the atoms 4C, 6C, 7C, 8C, 9C, and 21O were the most likely reactive sites for nucleophilic attack due to their highly positive values f+.
Table 1 Fukui index and NPA charge distribution of CIP
Notes: molecular structure of CIP  . . |

|
Rooted in DFT calculation and LC-MS analysis, potential CIP degradation pathways and intermediate products are proposed in Fig. S10 and S11.† CIP was purified mainly through the cleavage of the piperazine ring and stepwise oxidation, defluorination, cyclopropane ring cleavage, as well as hydroxylation.49 The most important pathway (I) was induced by the oxidation and cleavage of the piperazine ring. This was due to the high activity and f0 values of the 15N sites in piperazine. It was regarded as the most reactive site for ·SO4− and/or ·OH attacks.50 CIP underwent a ring-opening reaction via radical attack and was oxidized stepwise P1 and P2 to form P3 (m/z = 360.51) with a dialdehyde derivative structure. Subsequently, P6 was formed by piperazine ring cleavage and oxidation. The defluorination of P6 can generate P8 (m/z = 274.47). In pathway II, the elimination of CIP proceeds via the pathway CIP → P9 (m/z = 321.20) → P10 (m/z = 323.49) → P11 (m/z = 322.56) herein. CIP is also amenable to direct defluorination. The intermediate P12 (m/z = 331.89) was a product after defluorination and hydroxylation in pathway III, and P8 (m/z = 263) was produced by decarboxylation. Furthermore, P15 was formed through hydroxylation, oxidation, and defluorination in pathway IV. Meanwhile, intermediate P15 can lose its cyclopropyl group to generate P16 (m/z = 274.52).51 Ultimately, the intermediate was further oxidized to CO2, H2O, and others.
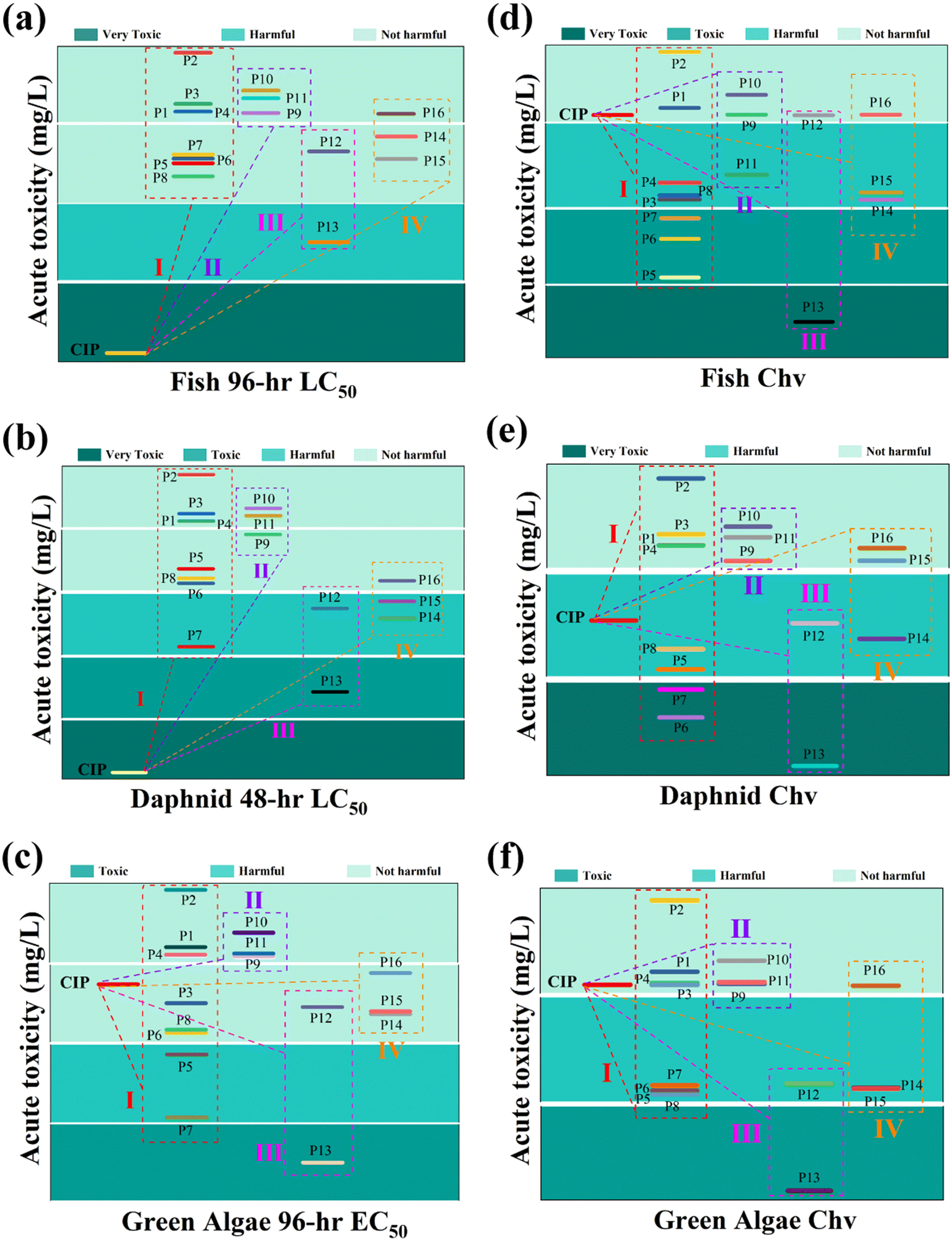
In addition, ECOSAR (v1.11) predictive model devised by U.S. EPA was leveraged to appraise and scrutinize the ecotoxicity.52,53 Acute toxicity was determined by LC50 and/or EC50, while chronic toxicity was expressed using ChV (Text S3†). The results, presented in Table S3,† classified toxicity into four levels based on different values: not harmful, harmful, toxic, and very toxic.54,55 Different intermediates exhibited diverse toxicity levels to different organisms. Fig. 3(a and b) revealed that the LC50 values of fish and daphnid acute toxicity values for all CIP intermediates were lower than those for CIP itself. Fig. 3c revealed that these intermediates had little effect on green algae. On the whole, compared with the parent CIP, the intermediate products had less effect on the acute toxicity of aquatic organisms, but some intermediates had potential chronic toxicity, as shown in Fig. 3(d–f). Toxicity calculation found that this system effectively degraded contaminants to small molecules of less toxic or not harmful category, which dramatically declined the ecological and environmental risks associated with the substrates and intermediates. The findings indicated that the MnCo/CeO2/PMS system was a green and efficient method for pollutant degradation.
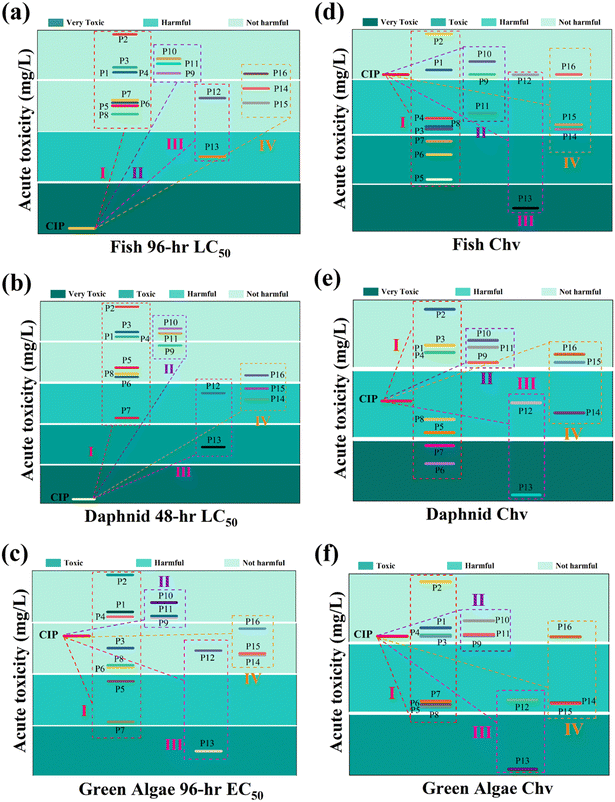 |
| | Fig. 3 Acute and chronic toxicity of (a and d) fish, (b and e) daphnid, (c and f) green algae of CIP and its degradation products. | |
3.6 Reactive species
The radicals in the MnCo/CeO2/PMS reaction system have been identified through trapping experiments. Among them, free radical scavengers have the functions of delaying, inhibiting, and blocking active oxygen species. Furfuryl alcohol, methanol, p-benzoquinone, together with tert-butanol (FFA, MeOH, PBQ, and TBA) were regarded as ROS scavengers. ·SO4− and ·OH can be efficiently quenched with MeOH (k = 1.6–7.7 × 107, k = 9.7 × 108 mol−1 s−1 for ·SO4−and ·OH, respectively) and TBA (k = 4.0–9.1 × 105 and 3.8–7.6 × 108 mol−1 s−1 for ·SO4− and ·OH, respectively).22 In the activity experiment of the MnCo/CeO2/PMS system, as presented in Fig. 4(a), MeOH with varying concentration gradients was introduced. The decomposition efficiency of CIP was found to be about 77–78%, indicating a certain degree of inhibition in the catalytic activity of this system. When adding 1:1000 of PMS:TBA (Fig. 4b), 10 mg L−1 CIP can be purified by 74.8% in 50 min. Comparison between MeOH and TBA revealed that the degradation performance of ·SO4− on the target pollutants was slightly greater than that of ·OH. However, none of them showed obvious inhibition, manifesting that ·SO4− together with ·OH could not be the primary active species in the reaction system. In contrast, the decontamination rate gradually diminished to 34.4% as the PMS/PBQ molar ratio increased from 1:0 to 1:50, indicating an enhanced inhibition effect and a significant contribution of ·O2− to the catalytic activity, as reflected in Fig. 4(c). However, when the PMS/FFA ratio was 1:50, the catalytic activity was significantly inhibited, indicating that 1O2 was the primary active oxygen species in Fig. 4(d). This means that in the MnCo/CeO2/PMS reaction system, non-radicals dominate. At the same time, it is also demonstrated that the reaction process dominated by free radicals was changed. The synergistic effect of an appropriate OV concentration and doping strategy facilitates the conversion of radicals to non-radicals. Additionally, ESR was exploited to confirm the existence of active oxygen species in the MnCo/CeO2 system. As depicted in Fig. 4(e), DMPO is able to capture ·OH/·SO4− to create adducts named DMPO–·OH/DMPO–·SO4−. Meanwhile, the obvious ESR signal allocated to DMPO–·O2− as well as TEMP–1O2 adducts were also recorded. These findings provide evidence for the prevalence of ·OH, ·SO4−, ·O2− together with 1O2 in the MnCo/CeO2/PMS process.
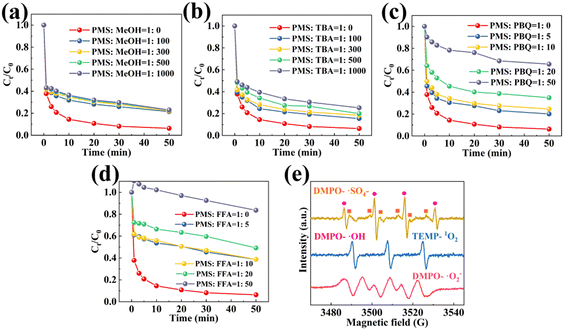 |
| | Fig. 4 Decontamination efficiency of CIP with the involvement of various scavengers using MnCo/CeO2: (a) MeOH, (b) TBA, (c) PBQ, and (d) FFA; (e) ESR spectra of the MnCo/CeO2/PMS system. | |
3.7 Theoretical calculations based on DFT
At the same time, the electron interaction between PMS and MnCo/CeO2 was further clarified by the DFT calculation.22 According to XRD and HRTEM images, it can be judged that the main exposed surface of MnCo/CeO2 was the (111) lattice plane, on which PMS was adsorbed.56 Therefore, DFT chose the (111) lattice plane for the calculation. The calculations disclose that the adsorption energies (Eads) of PMS onto the surface of CeO2, Co/CeO2, Mn/CeO2, and MnCo/CeO2 are −1.914, −1.909, −1.966, and −2.094 eV, respectively (Fig. 5a). It was confirmed that the adsorption energy onto the MnCo/CeO2 surface was more negative than that on other catalyst surfaces, enhancing the PMS capture and subsequent activation.57 Mn and Co doping can effectively modulate the electronic structure regarding CeO2 and promote transfer as well as distribution concerning the electrons. The introduction of Mn and Co may increase the unoccupied electron states on the CeO2 surface, which helps to enhance the adsorption interaction with PMS molecules. In addition, the adsorption energy of CeO2 (−1.914 eV) for PMS was higher than that of traditional CeO2 (−1.385 eV), which means that OVs were also active sites, which is beneficial for interfacial electron transfer,58 in Fig. 5(a) as well as Fig. S12(a and b).† Moreover, the bond length for O–O in PMS (lO–O) on the corresponding catalyst surface was 1.457 Å, 1.527 Å, 1.561 Å, 1.542 Å, and 1.610 Å, respectively, as shown in Fig. 5(b). This phenomenon showed that when Mn and Co are co-doped, the lO–O was the longest, it was easier to break, and the activation effect was the best. It is worth noting that in the MnCo/CeO2 system, PMS ultimately formed ·OH and ·SO42− with the extension of the O–O bond.58 To better comprehend the activation of PMS on the MnCo/CeO2 surface, the conveyance of charges between the MnCo/CeO2 catalyst surface and PMS was also investigated. The calculated average charge transfer Δq, CeO2, Co/CeO2, Mn/CeO2, and MnCo/CeO2 on PMS substrates was −2.350e, −1.030e, −2.062e, and −0.445e, respectively (Fig. S12b–d†). This showed that bimetallic doping and appropriate concentration of OVs can promote electron transportation from the catalyst to PMS, and abundant active sites facilitate the stable adsorption of PMS on its surface, which makes it easier to activate PMS and increase the production of ROS.59 In addition, by analyzing the 3 main positions at which the Ce atoms were substituted, it can be found, as in Fig. 5(c and d), at atomic number 11, 28, and 34. When doped with Co, partial electrons were transferred from Co/CeO2 to PMS. When doped with Mn, the PMS releases electrons, and electrons are transferred from PMS to Mn/CeO2. When Co and Mn were doped, electrons were transferred from the target catalyst to the PMS moiety in the MnCo/CeO2/PMS reaction system, ultimately activating the PMS. These calculations indicated that MnCo/CeO2/PMS with longer lO–O, lower adsorption energies, and appropriate OVs were more effective than other catalysts in adsorbing and decomposing PMS. To enhance the understanding of the relationship between the structure and activity, the corresponding function was established. As shown in Fig. 5(e), the fitting results demonstrate that the relationship between the pseudo-first-order kinetic constant k and the O–O bond length in PMS fits well with the ExpGrol function (R2 = 0.95458). As doping transitions from Mn or Co doping to bimetallic doping with both Mn and Co, the k value progressively increases, indicating that lO–O can be effectively adjusted by altering the type of doped metal. In bimetallic doping, where the lO–O length gets longer and is more prone to fracture, MnCo/CeO2 exhibits the best catalytic performance.
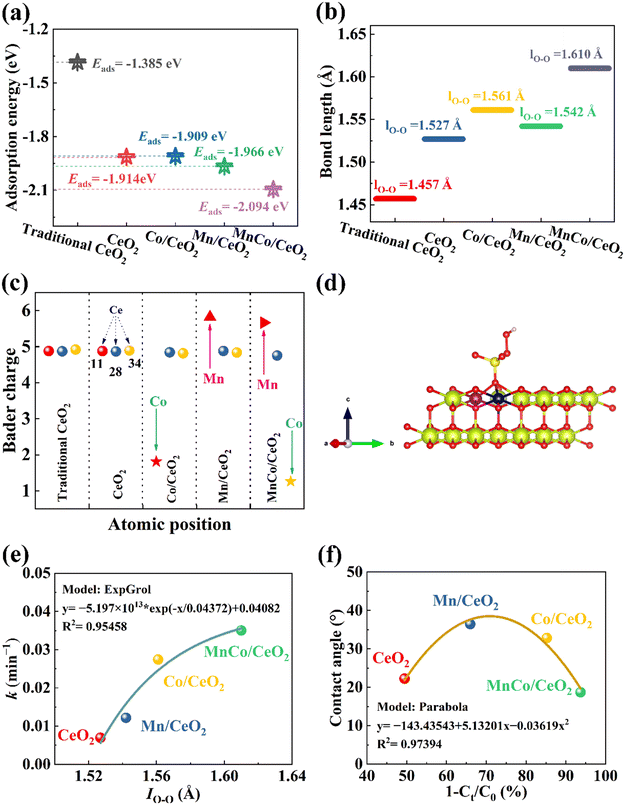 |
| | Fig. 5 (a) Adsorption energy (Eads). (b) Bond length for O–O in PMS on the surface of the catalyst. (c) Bader charge at atomic numbers 11, 28 and 34 in the structure of the synthetic catalyst. (d) Optimized structures of PMS captured by the MnCo/CeO2(111) substrate with OVs. The O, Ce, S, H, Co, and Mn elements are marked in red, green, yellow, pink, blue, and purple, respectively. (e) Correlation function model: k and the bond length for O–O in PMS, (f) contact angle and degradation activity. | |
3.8 Proposed reaction mechanism for stimulating PMS to eliminate CIP
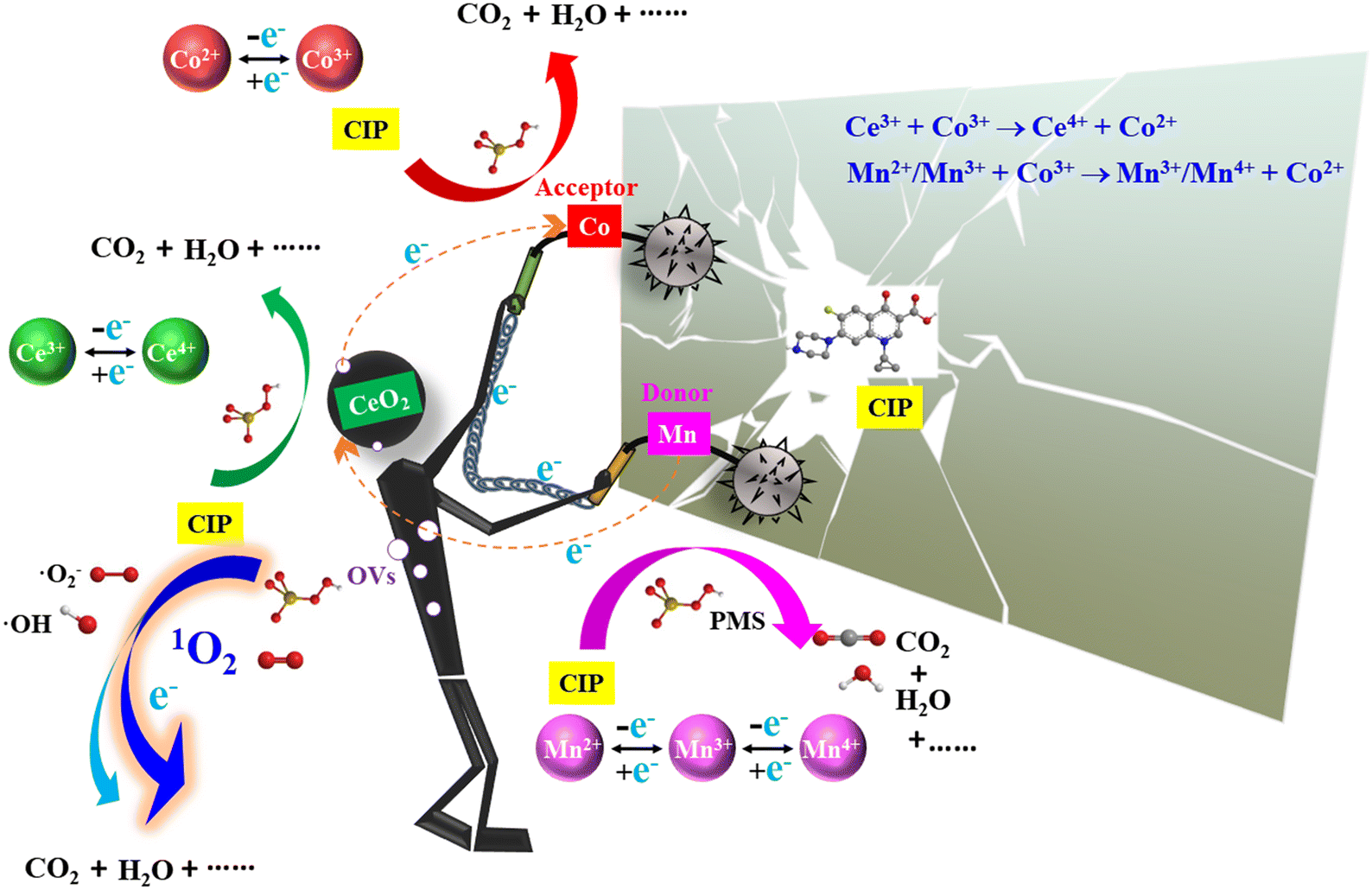
Rooted in the preceding analysis, as illustrated in Fig. 6, the mechanism for PMS stimulation and CIP degradation under the participation of appropriate OVs was proposed. Firstly, the strong hydrophilicity of the catalyst was supportive of the mass transfer process between PMS and target pollutant. In addition, by constructing the function between the degradation rate of each catalyst and the corresponding contact angle, it is found that the structure–activity relationship between the two follows the Paola function, with a correlation coefficient of R2 = 0.97394. It can be observed from Fig. 5(f) that the hydrophilic and hydrophobic properties of the catalyst can be effectively optimized by adjusting the doping amount, which further enhances the mass-transfer process. The special stalactite morphology was convenient for providing more catalytic and adsorption sites at the same time, which can effectively adsorb abundant substrates and intermediates.
 |
| | Fig. 6 Probable mechanisms for pollutant decontamination in the MnCo/CeO2 system. | |
The catalytic oxidation process is an ETP in essence. This work disclosed that the electron transfer between Mn and Co was facilitated in MnCo/CeO2. The activation mechanism of PMS using Ce, Mn, and Co components can be described by eqn (1)–(6). Low-valent Mn and Co can react with PMS via ETP to form ·SO4− (eqn (1), (3) and (5)). Subsequently, the activation of PMS can lead to the reduction of high-valent Mn and Co (eqn (2), (4) and (6)), resulting in the formation of ·SO5−. Compared with ·SO4− (E0 at 2.5–3.1 V), ·SO5− (E0 at 1.1 V) had a much lower reduction potential,60 was a weak ROS, and had a poor ability to degrade CIP. Thus, the purification efficiency of CIP was unsatisfactory in the Mn/CeO2/PMS and Co/CeO2/PMS system. Mn4+/Mn3+ with E0 = 0.95 V was obviously lower than that of HSO5−/·SO5− (E0 at 1.1 V).42 Considering that the potential regarding Co3+/Co2+ (E0 at 1.81 V) is superior to that of Ce3+/Ce2+ (E0 at 1.44 V), Mn3+/Mn2+ (E0 at 1.51 V), and Mn4+/Mn3+, it is thermodynamically beneficial to reduce Co3+via Mn and Ce components.42 Therefore, the regeneration of Co3+ can speed up ROS formation and facilitate CIP degradation. The reduction from Co3+ to Co2+ was accomplished by Mn and Ce components (eqn (7) and (8)) in the MnCo/CeO2/PMS system. This phenomenon manifested the cooperative interaction effect of Ce, Mn, and Co. Specifically, within this reaction system, the Mn component acts as the electron donor, while the Co component functions as the electron acceptor. CeO2 with appropriate OVs functions serves as a mediator for electron transportation, thereby the boosting electron shift efficiency and enhancing the removal rate of CIP. Also, a fast electron transportation conduit was formed between the investigated CIP and PMS due to the presence of OVs in the catalyst. Some CIP and its intermediates were enriched on the surface of the sample. ·SO4− can be partially transformed into ·OH through eqn (S1) and (S2).† ·O2− can be generated not only via the decomposition of PMS (eqn (S3) and (S4)†) but also through the reaction OH− and ·SO4− forming ·OH, which subsequently evolved into ·O2−, as described in eqn (S5) and (S6).† In the meantime, 1O2 was produced via the conversion of ·O2− (eqn (S7)†) and ·SO5− (eqn (S8)†). PMS self-decomposition induces the production of 1O2 through eqn (S9)† as well. MnCo/CeO2 boosts the effectiveness of PMS activation via a non-radical mechanism, thereby facilitating the purification of pollutants from water with improved efficacy. Meanwhile, theoretical calculations also proved that electrons were transported from the MnCo/CeO2 to PMS. Ultimately, bimetallic doping and suitable OVs effectively improved the surface electronic structure of PMS and strengthened the ETP in reaction systems, thereby facilitating the decontamination of CIP through the combined action of ·O2− and 1O2.
| | | Ce3+ + HSO5− → Ce4+ + ·SO4− + OH− | (1) |
| | | Ce4+ + HSO5− → Ce3+ + ·SO5− + H+ | (2) |
| | | Mn2+/Mn3+ + HSO5− → Mn3+/Mn4+ + ·SO4− + OH− | (3) |
| | | Mn4+/Mn3+ + HSO5− → Mn3+/Mn2+ + ·SO5− + H+ | (4) |
| | | Co2+ + HSO5− → Co3+ + ·SO4− + OH− | (5) |
| | | Co3+ + HSO5− → Co2+ + ·SO5− + H+ | (6) |
| | | Mn2+/Mn3+ + Co3+ → Mn3+/Mn4+ + Co2+ | (7) |
| | | Ce3+ + Co3+ → Ce4+ + Co2+ | (8) |
Finally, the appropriate concentration of OVs promotes electron migration, facilitates free radical together with non-radical processes, and enhances PMS activation efficiency. In addition to altering the electronic states on the catalyst surface, Mn and Co bimetallic doping also created numerous dangling bonds by introducing appropriate OVs. These dangling bonds disrupted surface charge equilibrium regarding CeO2, enhancing both the oxidation activity of the sample and the adsorption energy of PMS. Therefore, CIP was effectively removed.
3.9 Reusability and stability of the MnCo/CeO2 catalyst
The durability and stability of the sample were studied through cyclic testing. To prolong the lifetime of the catalyst, thermal annealing treatment is adopted to eliminate the intermediates adsorbed on the catalyst and restore the active sites.61 MnCo/CeO2 can eliminate about 89% of CIP after 5 cycles in Fig. 7(a), indicating that the catalyst had excellent stability after thermal annealing treatment. Furthermore, there was no remarkable change in the XRD spectra of MnCo/CeO2 before and after cycling (Fig. 7b). In conclusion, bimetallic-doped CeO2 catalysts effectively improved the performance of PMS for CIP degradation and enhanced the reusability and stability of the reaction system.
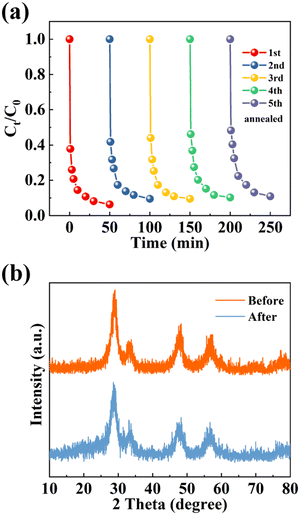 |
| | Fig. 7 (a) Recyclability of MnCo/CeO2 for 5 cycles. (b) XRD results of MnCo/CeO2 before and after repeated cycles. | |
4. Conclusions
The present work reports a Mn and Co bimetallic-doped CeO2 (MnCo/CeO2) nanoreactor with tunable OVs. The study provided a comprehensive understanding of the PMS activation mechanism, highlighting the cooperation with ·O2−, 1O2, and ETP. The target contaminant in this water treatment model, CIP, can be efficiently eliminated within 50 min. It is worth noting that the reaction system exhibits a wide operational pH range (pH 3–9). Furthermore, the potential influences of temperature and anions such as Cl−, SO42−, and NO3− are deemed negligible owing to the selective oxidation of CIP by 1O2. Even after continuous operation for 250 min, the system maintained a substantial removal efficiency of 89% for CIP in aqueous solutions. Importantly, the synthesis of MnCo/CeO2 nanoreactors relies on readily available and low-cost raw materials, thus facilitating scalable production. This work not only establishes a blueprint for the future design of CeO2-based nanoreactors but also provides a promising approach for purifying water pollutants.
Data availability
The data are available upon reasonable request.
Author contributions
Hailan Qin: methodology, validation, visualization, writing – original draft. Jiahao Wang: data curation, methodology. Siyuan Di: data curation, writing – review & editing. Yunkang Liu: data curation. Pin Chen: data curation, validation. Min Liu: validation. Qiuyue Zhang: validation. Shukui Zhu: funding acquisition, methodology, project administration, resources, supervision, validation, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the grants from the National Key Research and Development Program of China (Grants No. 2023YFC3707701 and 2023YFC3706505), the Environmental Protection Department of Hubei Province (No. 2017HB04), and State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences (No. GKZ24Y653).
References
- X. Liu, P. Shao, S. Gao, Z. Bai and J. Tian, Benzoquinone-assisted heterogeneous activation of PMS on Fe3S4 via formation of active complexes to mediate electron transfer towards enhanced bisphenol A degradation, Water Res., 2022, 226, 119218 CrossRef CAS.
- U. von Gunten, Oxidation Processes in Water Treatment: Are We on Track?, Environ. Sci. Technol., 2018, 52, 5062–5075 CrossRef CAS PubMed.
- M. Kohantorabi, G. Moussavi and S. Giannakis, A review of the innovations in metal- and carbon-based catalysts explored for heterogeneous peroxymonosulfate (PMS) activation, with focus on radical vs. non-radical degradation pathways of organic contaminants, Chem. Eng. J., 2021, 411, 127957 CrossRef CAS.
- Gh. Eshaq, S. Wang, H. Sun and M. Sillanpaa, Superior performance of FeVO4@CeO2 uniform core-shell nanostructures in heterogeneous Fenton-sonophotocatalytic degradation of 4-nitrophenol, J. Hazard. Mater., 2020, 382, 121059 CrossRef CAS.
- Y. Xie, L. Yu, L. Chen, C. Chen, L. Wang, F. Liu, Y. Liao, P. Zhang, T. Chen, Y. Yuan, Y. Lu, B. Huang, H. Yang, S. Wang, S. Wang, L. Ma, F. Luo, Y. Liu, B. Hu, H. Wang, D. Pan, W. Zhu, N. Wang, Z. Wang, L. Mao, S. Ma and X. Wang, Recent progress of radionuclides separation by porous materials, Sci. China: Chem., 2024, 67, 3515–3577 CrossRef CAS.
- P. Chen, S. Di, W. Xie, Z. Li and S. Zhu, One-step synthesis of triazine-based covalent organic frameworks at room temperature for efficient photodegradation of bisphenol A under visible light irradiation, Front. Mater. Sci., 2023, 17, 230661 CrossRef.
- H. Qin, J. Sun, D. Xia, H. Xu, Q. Yu, Y. Zheng and Y. Shi, Boosting nonradical process in BiOI/BiOCl heterostructure by interface oxygen vacancies, Chem. Eng. J., 2022, 435, 134847 CrossRef CAS.
- H. Liu, P. Sun, M. Feng, H. Liu, S. Yang, L. Wang and Z. Wang, Nitrogen and sulfur co-doped CNT-COOH as an efficient metal-free catalyst for the degradation of UV filter BP-4 based on sulfate radicals, Appl. Catal., B, 2016, 187, 1–10 CrossRef CAS.
- J. Lim and M. R. Hoffmann, Peroxymonosulfate (PMS) activation on cobalt-doped TiO2 nanotubes: degradation of organics under dark and solar light irradiation conditions, Environ. Sci.: Nano, 2020, 7, 1602–1611 RSC.
- P. Chen, S. Di, X. Qiu and S. Zhu, One-step synthesis of F-TiO2/g-C3N4 heterojunction as highly efficient visible-light-active catalysts for tetrabromobisphenol A and sulfamethazine degradation, Appl. Surf. Sci., 2022, 587, 152889 CrossRef CAS.
- Z. Jiang, X. Zhang, S. Guo, Y. Zheng, J. Wang, T. Wen and X. Wang, Recent advances and perspectives of emerging two-dimensional transition metal carbide/nitride-based materials for organic pollutant photocatalysis, Mater. Chem. Front., 2023, 7, 4658–4682 RSC.
- W. Bai, H. Lu, Y. Liu, X. Yuan, Y. Ai, L. Wang and Z. Chen, Crystallinity regulation-induced organic degradation on ultra-thin 2D Co3O4/SiO2 nanosheets: the critical trigger of oxygen vacancies, Environ. Sci.: Nano, 2024, 11, 2507–2520 RSC.
- Z. Zhu, J. Ye, X. Tang, Z. Chen, J. Yang, P. Huo, Y. H. Ng and J. Crittenden, Vacancy-rich CoSx@LDH@Co-NC catalytic membrane for antibiotic degradation with mechanistic insights, Environ. Sci. Technol., 2023, 57, 16131–16140 CrossRef CAS.
- X. Zhang, M. Feng, R. Qu, H. Liu, L. Wang and Z. Wang, Catalytic degradation of diethyl phthalate in aqueous solution by persulfate activated
with nano-scaled magnetic CuFe2O4/MWCNTs, Chem. Eng. J., 2016, 301, 1–11 CrossRef CAS.
- R. Yu, J. Zhao, Z. Zhao and F. Cui, Copper substituted zinc ferrite with abundant oxygen vacancies for enhanced ciprofloxacin degradation via peroxymonosulfate activation, J. Hazard. Mater., 2020, 390, 121998 CrossRef CAS PubMed.
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, Insights into the influence of CeO2 crystal facet on CO2 hydrogenation to methanol over Pd/CeO2 catalysts, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- J. Yang, M. Zhang, M. Chen, Y. Zhou and M. Zhu, Oxygen Vacancies in piezoelectric ZnO twin-mesocrystal to improve peroxymonosulfate utilization efficiency via piezo-activation for antibiotic ornidazole removal, Adv. Mater., 2023, 35, 2209885 CrossRef CAS PubMed.
- L. Li, Q. Diao, Z. Liu, G. Zhu, C. Huang, G. Shi, X. Huang, J. Zhang and M. Jiao, Facile synthesis of oxygen vacancies mediated Co3O4 by Mn doping for high-performance ethanol sensing, Vacuum, 2024, 224, 113202 CrossRef CAS.
- M. Yang, T. Xu, X. Jin, Q. Shen and C. Sun, Oxygen vacancies enriched Bi2WO6 for enhanced decabromodiphenyl ether photodegradation via C-Br bond activation, Appl. Surf. Sci., 2022, 581, 152439 CrossRef CAS.
- C. Su, X. Duan, J. Miao, Y. Zhong, W. Zhou, S. Wang and Z. Shao, Mixed conducting perovskite materials as superior catalysts for fast aqueous-phase advanced oxidation: A mechanistic study, ACS Catal., 2017, 7, 388–397 CrossRef CAS.
- P. Gao, X. Tian, Y. Nie, C. Yang, Z. Zhou and Y. Wang, Promoted peroxymonosulfate activation into singlet oxygen over perovskite for ofloxacin degradation by controlling the oxygen defect concentration, Chem. Eng. J., 2019, 359, 828–839 CrossRef CAS.
- S. Di, J. Wang, Y. Zhai, P. Chen, T. Ning, C. Shi, H. Yang, Y. Bao, Q. Gao and S. Zhu, Efficient activation of peroxymonosulfate mediated by Co(II)-CeO2 as a novel heterogeneous catalyst for the degradation of refractory organic contaminants: Degradation pathway, mechanism and toxicity assessment, J. Hazard. Mater., 2022, 435, 129013 CrossRef CAS PubMed.
- Y. Mei, Y. Zhang, J. Li, X. Deng, Y. Yang, Q. Yang, B. Jiang, B. Xin, T. Yao and J. Wu, Synthesis of Co-doped CeO2 nanoflower: Enhanced adsorption and degradation performance toward tetracycline in Fenton-like reaction, J. Alloys Compd., 2022, 904, 163879 CrossRef CAS.
- Z.-J. Xiao, X.-C. Feng, H.-T. Shi, B.-Q. Zhou, W.-Q. Wang and N.-Q. Ren, Why the cooperation of radical and non-radical pathways in PMS system leads to a higher efficiency than a single pathway in tetracycline degradation, J. Hazard. Mater., 2022, 424, 127247 CrossRef CAS PubMed.
- S. Ye, G. Zeng, X. Tan, H. Wu, J. Liang, B. Song, N. Tang, P. Zhang, Y. Yang, Q. Chen and X. Li, Nitrogen-doped biochar fiber with graphitization from Boehmeria nivea for promoted peroxymonosulfate activation and non-radical degradation pathways with enhancing electron transfer, Appl. Catal., B, 2020, 269, 118850 CrossRef CAS.
- C. Chen, T. Jiang, J. Hou, T. Zhang, G. Zhang, Y. Zhang and X. Wang, Oxygen vacancies induced narrow band gap of BiOCl for efficient visible-light catalytic performance from double radicals, J. Mater. Sci. Technol., 2022, 114, 240–248 CrossRef CAS.
- A. Wang, Z. Zheng, H. Wang, Y. Chen, C. Luo, D. Liang, B. Hu, R. Qiu and K. Yan, 3D hierarchical H2-reduced Mn-doped CeO2 microflowers assembled from nanotubes as a high-performance Fenton-like photocatalyst for tetracycline antibiotics degradation, Appl. Catal., B, 2020, 277, 119171 CrossRef CAS.
- H. Li, J. Shang, H. Zhu, Z. Yang, Z. Ai and L. Zhang, Oxygen vacancy structure associated photocatalytic water oxidation of BiOCl, ACS Catal., 2016, 6, 8276–8285 CrossRef CAS.
- J. Wu, J. Gao, S. Lian, J. Li, K. Sun, S. Zhao, Y. D. Kim, Y. Ren, M. Zhang, Q. Liu, Z. Liu and Z. Peng, Engineering the oxygen vacancies enables Ni single-atom catalyst for stable and efficient C-H activation, Appl. Catal., B, 2022, 314, 121516 CrossRef CAS.
- B. Nematollahi, M. Rezaei and E. N. Lay, Preparation of highly active and stable NiO–CeO2 nanocatalysts for CO selective methanation, Int. J. Hydrogen Energy, 2015, 40, 8539–8547 CrossRef CAS.
- Y. Ma, Z. Tian, W. Zhai and Y. Qu, Insights on catalytic mechanism of CeO2 as multiple nanozymes, Nano Res., 2022, 15, 10328–10342 CrossRef CAS.
- W. Liu, J. Zhou and J. Yao, Shuttle-like CeO2/g-C3N4 composite combined with persulfate for the enhanced photocatalytic degradation of norfloxacin under visible light, Ecotoxicol. Environ. Saf., 2020, 190, 110062 CrossRef CAS.
- H. Qin, Y. Zhang, S. He, Z. Guan, Y. Shi, X. Xie, D. Xia, D. Li and H. Xu, Increasing the migration and separation efficiencies of photogenerated carriers in CQDs/BiOCl through the point discharge effect, Appl. Surf. Sci., 2021, 562, 150214 CrossRef CAS.
- C. Deng, H. Xu, H. Qin, D. Xia, D. Li, Q. Yu, D. Chen, Y. Zheng and Y. Wang, Enhancing the separation efficiency of photo-induced carriers in a Bi2S3/BiOCl heterostructure by cooperative influence of oxygen vacancies and the interfacial electric field, New J. Chem., 2022, 46, 9195–9206 RSC.
- C. Jin, M. Wang, Z. Li, J. Kang, Y. Zhao, J. Han and Z. Wu, Two dimensional Co3O4/g-C3N4 Z-scheme heterojunction: Mechanism insight into enhanced peroxymonosulfate-mediated visible light photocatalytic performance, Chem. Eng. J., 2020, 398, 125569 CrossRef CAS.
- J. Fan, Q. Wang, W. Yan, J. Chen, X. Zhou and H. Xie, Mn3O4-g-C3N4 composite to activate peroxymonosulfate for organic pollutants degradation: Electron transfer and structure-dependence, J. Hazard. Mater., 2022, 434, 128818 CrossRef CAS.
- J. Xu, Y. Wang, J. Wan and L. Wang, Facile synthesis of carbon-doped CoMn2O4/Mn3O4 composite catalyst to activate peroxymonosulfate for ciprofloxacin degradation, Sep. Purif. Technol., 2022, 287, 120576 CrossRef CAS.
- B. Han, Q. Pan, Y. Chen, D. Liu, C. Zhou, K. Xia and Q. Gao, High-entropy perovskite oxide washcoated porous alumina ceramic as a superb catalyst for activating peroxymonosulfate to eliminate refractory organic pollutants, Chem. Eng. J., 2023, 455, 140828 CrossRef CAS.
- C. Wang, Y. Li, H. Liu, Z. Hu, X. Hao, H. Jia, J. Chen and C. Lu, Constructing a high concentration CuO/CeO2 interface for complete oxidation of toluene: The fantastic application of spatial confinement strategy, J. Rare Earths, 2023, 41, 850–861 CrossRef CAS.
- M. Wu, M. Miao, W. Li, X. Zhang, L. Zhang, T. Zhen, Y. Fu, J. Jin and L. Yuan, Metal-organic framework-derived one-dimensional Pd@CeO2 catalysts with enhanced activity for methane oxidation, Fuel, 2023, 331, 125575 CrossRef CAS.
- X. Zhou, Y. Tang, X. Xu, X. Zhou, G. Zhao, M. Zhou, G. Wan and G. Wang, CoP/C hollow hybrids inducing abundant active interfaces and fast electron transfers to activate peroxymonosulfates for bisphenol A degradation, Mater. Today Nano, 2021, 14, 100116 CrossRef CAS.
- D. Yu, J. He, T. Xie, J. Yang, J. Wang, J. Xie, H. Shi, Z. Gao, B. Xiang and D. D. Dionysiou, Boosting catalytic activity of SrCoO2.52 perovskite by Mn atom implantation for advanced peroxymonosulfate activation, J. Hazard. Mater., 2023, 442, 130085 CrossRef CAS.
- H. Fu, P. Zhao, S. Xu, G. Cheng, Z. Li, Y. Li, K. Li and S. Ma, Fabrication of Fe3O4 and graphitized porous biochar composites for activating peroxymonosulfate to degrade p-hydroxybenzoic acid: Insights on the mechanism, Chem. Eng. J., 2019, 375, 121980 CrossRef CAS.
- S. Yu, Q. Zhang, X. Sun, S. Chen, J. Tang, J.-J. Zhu and Y. Dang, Doping Sb into CuFe2O4 improved the catalytic performance in the electrochemically enhanced homogeneous peroxymonosulfate-heterogeneous catalytic system for the degradation of ciprofloxacin, J. Environ. Chem. Eng., 2022, 10, 108335 CrossRef CAS.
- X. Luo, L. Bai, J. Xing, X. Zhu, D. Xu, B. Xie, Z. Gan, G. Li and H. Liang, Ordered mesoporous cobalt containing perovskite as a high-performance heterogeneous catalyst in activation of peroxymonosulfate, ACS Appl. Mater. Interfaces, 2019, 11, 35720–35728 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Y. Liu, H. Liu, Z. Li, H. Qin, S. Di, P. Chen, M. Liu, Q. Zhang, M. Sillanpää and S. Zhu, N-doped CuFe-MOF-919 derivatives in activation of peroxymonosulfate for enhanced degradation of organic pollutants: 1O2 dominated non-radical pathway, Sep. Purif. Technol., 2024, 345, 127324 CrossRef CAS.
- C. Liu, S. Mao, M. Shi, F. Wang, M. Xia, Q. Chen and X. Ju, Peroxymonosulfate activation through 2D/2D Z-scheme CoAl-LDH/BiOBr photocatalyst under visible light for ciprofloxacin degradation, J. Hazard. Mater., 2021, 420, 126613 CrossRef CAS PubMed.
- W. Li, S. Li, Y. Tang, X. Yang, W. Zhang, X. Zhang, H. Chai and Y. Huang, Highly efficient activation of peroxymonosulfate by cobalt sulfide hollow nanospheres for fast ciprofloxacin degradation, J. Hazard. Mater., 2020, 389, 121856 CrossRef CAS PubMed.
- C. Jiang, Y. Ji, Y. Shi, J. Chen and T. Cai, Sulfate radical-based oxidation of fluoroquinolone antibiotics: Kinetics, mechanisms and effects of natural water matrices, Water Res., 2016, 106, 507–517 CrossRef CAS PubMed.
- A. Huang, D. Zhi, H. Tang, L. Jiang, S. Luo and Y. Zhou, Effect of Fe2+, Mn2+ catalysts on the performance of electro-Fenton degradation of antibiotic ciprofloxacin, and expanding the utilizing of acid mine drainage, Sci. Total Environ., 2020, 720, 137560 CrossRef CAS.
-
O. US EPA, EPI Suite™-Estimation Program Interface, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface.
-
O. US EPA, Ecological Structure Activity Relationships (ECOSAR) Predictive Model, https://www.epa.gov/tsca-screening-tools/ecological-structure-activity-relationships-ecosar-predictive-model.
- Y. Jia, S. K. Khanal, L. Yin, L. Sun and H. Lu, Influence of ibuprofen and its biotransformation products on different biological sludge systems and ecosystem, Environ. Int., 2021, 146, 106265 CrossRef CAS.
- P. Shi, X. Yue, X. Teng, R. Qu, A. Rady, S. Maodaa, A. A. Allam, Z. Wang and Z. Huo, Degradation of butylated hydroxyanisole by the combined use of peroxymonosulfate and Ferrate(VI): Reaction kinetics, Mechanism and Toxicity Evaluation, Toxics, 2024, 12, 54 CrossRef CAS PubMed.
- J. Zhang, Y. Ma, Y. Sun, L. Wang, L. Wang, Z. Wang, B. Zhao, J. Gao and M. Xu, Ultrahigh-flux 2D CoO@MoS2 composite membrane activated peroxymonosulfate through enhanced electron transfer for rapid degradation of refractory benzotriazole, Chem. Eng. J., 2023, 471, 144837 CrossRef CAS.
- S. Guo, H. Wang, W. Yang, H. Fida, L. You and K. Zhou, Scalable synthesis of Ca-doped α-Fe2O3 with abundant oxygen vacancies for enhanced degradation of organic pollutants through peroxymonosulfate activation, Appl. Catal., B, 2020, 262, 118250 CrossRef CAS.
- X.-L. Wu, S. Liu, Y. Li, M. Yan, H. Lin, J. Chen, S. Liu, S. Wang and X. Duan, Directional and ultrafast charge transfer in oxygen-vacancy-rich ZnO@single-atom cobalt core-shell junction for photo-Fenton-like reaction, Angew. Chem., Int. Ed., 2023, 62, e202305639 CrossRef CAS PubMed.
- S. Mao, P. Zhao, Y. Wu, C. Liu, M. Xia and F. Wang, Promoting charge migration of Co(OH)2/g-C3N4 by hydroxylation for improved PMS activation: Catalyst design, DFT calculation and mechanism analysis, Chem. Eng. J., 2023, 451, 138503 CrossRef CAS.
- X. Zhou, C. Luo, M. Luo, Q. Wang, J. Wang, Z. Liao, Z. Chen and Z. Chen, Understanding the synergetic effect from foreign metals in bimetallic oxides for PMS activation: A common strategy to increase the stoichiometric efficiency of oxidants, Chem. Eng. J., 2020, 381, 122587 CrossRef CAS.
- N. Du, Y. Liu, Q. Li, W. Miao, D. Wang and S. Mao, Peroxydisulfate activation by atomically-dispersed Fe-Nx on N-doped carbon: Mechanism of singlet oxygen evolution for nonradical degradation of aqueous contaminants, Chem. Eng. J., 2021, 413, 127545 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*


 .
.