
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D modelling of pulmonary arterial stenosis and endothelial dysfunction in CTEPH†
Salina Nicoleaua,
Ylenia Roger Vallebc,
Olga Tura-Ceidebcd,
Chloe H. Armourae,
Joan Albert Barberàbc,
Thomas. A. J. McKinnonf,
Deepa Gopalang and
Beata Wojciak-Stothard *a
*a
aNational Heart and Lung Institute, Imperial College London, ICTEM Building, Hammersmith Campus, Du Cane Road, London W12 0NN, UK. E-mail: b.wojciak-stothard@imperial.ac.uk; Tel: +44 (0)207 594 6821
bDepartment of Pulmonary Medicine, Hospital Clínic-Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, 08036 Barcelona, Spain
cBiomedical Research Networking Centre on Respiratory Diseases (CIBERES), Madrid, Spain
dTranslational Research Group on Cardiovascular Respiratory Diseases (CAREs), Institut d'Investigació Biomèdica de Girona (IDIBGI-CERCA), Parc Hospitalari Martí i Julià, Edifici M2, 17190 Salt, Spain
eDepartment of Chemical Engineering, Imperial College London, UK
fDepartment of Immunology and Inflammation, Imperial College London, UK
gDepartment of Radiology Imperial College NHS Trust London, UK
First published on 16th July 2025
Abstract
Chronic thromboembolic pulmonary hypertension (CTEPH) arises from progressive thrombotic occlusion of pulmonary arteries, involving vessel blockage by unresolved thrombi and small vessel arteriopathy. This disrupts blood flow, increases lung pressure, and alters vessel geometry, contributing to endothelial dysfunction. However, the mechanisms remain unclear. To study these interactions, we developed microfluidic 3D models of pulmonary arteries with 30–80% stenosis using CTPAs from CTEPH and acute pulmonary embolism (APE) patients, in silico flow simulations, 3D printing, and soft lithography. Unlike standard circular channels, we designed semi-circular channels enclosed by a coverslip, which computational modelling confirmed closely mimics real vessel flow dynamics. Human pulmonary artery endothelial cells (HPAECs) cultured in 30–80% stenosis channels exhibited increased expression of pro-inflammatory, pro-thrombotic, and pro-angiogenic genes, with responses varying by stenosis severity and location. Cells in post-stenotic dilatation regions (60–80% stenosis) lost alignment and junctional integrity due to disturbed flow. The transcriptional profile of HPAECs from 80% stenosis channels closely resembled that of CTEPH pulmonary endarterectomy specimens. Platelet adhesion, dependent on von Willebrand factor (VWF), varied with stenosis severity and flow rate. Low perfusion rates increased adhesion in stenotic regions, while higher flow rates promoted adhesion post-stenosis. Our patient data-based stenosis models provide a robust platform for studying the effects of vascular geometry on blood flow, endothelial responses, and platelet aggregation, advancing research on CTEPH, pulmonary embolism, and other diseases associated with vascular occlusion.
Introduction
The pulmonary endothelium plays a vital role in regulating vascular homeostasis, including vascular repair, inflammation, and thrombosis. Endothelial cells respond to changes in blood flow, particularly wall shear stress, flow patterns, and pressure.1 Dysregulation of these responses has been linked to several cardiovascular disorders.2Chronic thromboembolic pulmonary hypertension (CTEPH) is a rare, late complication of acute pulmonary embolism (APE).3 It occurs due to the incomplete resolution of pulmonary emboli, which, along with the progressive narrowing of the arteriolar lumen caused by vascular wall remodelling, restricts blood flow and increases pulmonary arterial pressure, ultimately leading to right heart failure.
Pathological changes in wall shear stress and flow patterns, which influence thrombus formation and contribute to pulmonary endothelial dysfunction, have been implicated in CTEPH. However, the underlying mechanisms remain poorly understood. Recent advances in 4D flow MRI analysis have enabled the non-invasive characterization of blood flow patterns in CTEPH4,5 Computational fluid dynamics (CFD) modelling has identified shear stress gradients and recirculating flow patterns at and downstream of vascular stenosis, likely creating pro-thrombotic environments.6
In the last decade, development of 3D printing methods based on CT angiograms opened up prospects of producing microfluidic devices recreating patient-specific vascular geometry.7 Most research however focused on modelling flow in tubular stenotic channels in coronary vascular disease8 whilst research on the effects of pulmonary arterial stenosis remains limited.9
Patient blood-derived endothelial cells, also called late outgrowth endothelial colony forming cells (ECFCs)10,11 and endothelial cells obtained from the pulmonary endarterectomies (PEA),12,13 are often used as surrogates for CTEPH endothelium, providing insights into the disease pathobiology. Both cell types exhibit increased expression of inflammatory markers, cell adhesion molecules, and evidence of mitochondrial dysfunction, oxidative stress, and altered fatty acid metabolism12–14 and therefore can be used for validation of results produced by the in vitro modelling. They also display a pro-thrombotic phenotype, characterised by elevated levels of von Willebrand factor (VWF), plasminogen activator inhibitor-1 (PAI-1), and tissue factor (TF), alongside reduced levels of anti-thrombotic factors such as thrombomodulin (TM).12,14–17 ECs from PEA samples further show diminished angiogenic capacity compared to healthy ECs.14,18
In this study, we present easy-to-fabricate 3D-printed semi-circular microfluidic models of pulmonary vascular stenosis, based on vascular geometries observed in CTEPH lungs. We explore how these arterial geometries, altered by varying degrees of stenosis, affect pressure, flow dynamics, endothelial morphology, gene expression, and platelet aggregation.
Methods
Detailed description of experimental procedures is provided in ESI† Methods.Patient data and ethical approvals
Computed tomography pulmonary angiograms (CTPAs) were obtained from CTEPH, acute pulmonary embolism (APE), chronic thromboembolic disease (CTED) and non-PE patients attending the Hammersmith Hospital (Imperial College Healthcare NHS Trust, London UK). The scans used for modelling were completely anonymised data collected as a part of a retrospective study that was approved by the Institutional Review Board [IRAS project ID 280472] at Imperial College London and the need for informed consent was waived.CTPAs were identified from the Radiology records using the Radiology Information System (RIS). The search was carried out by Dr Deepa Gopalan, cardiovascular radiologist at the Hammersmith Hospital.
All experiments involving patient-derived cells were conducted in accordance with the 1964 Declaration of Helsinki. The study protocol was granted by the Hospital Clínic of Barcelona ethics committee (HCB/2018/0837 and HCB/2018/0434), and all the subjects provided written informed consent.
Segmentation of pulmonary arterial geometries
Segmentations and 3D reconstructions of pulmonary vascular tree were performed with Mimics 23.0 (Materialise, 2021). CTPA scans were imported as Digital Imaging and Communications in Medicine (DICOM) files. Pulmonary arterial diameters and lengths were measured by fitting a centerline through the reconstructed 3D vascular tree. Further details are provided in ESI† Methods, with illustrations of this process shown in Fig. S1–S4.†Design of semi-circular channels
Fully circular and semi-circular channels recreating the ring-like stenoses and average dimensions observed in CTPA scans from CTEPH patients obtained from the Hammersmith Hospital (London, UK) (n = 7) and the literature19–22 were designed in Fusion 360 (AutoDesk). CT-scan- and literature-derived measurements of pulmonary arterial stenosis are provided in Tables S1 and S2,† respectively.The semi-circular and fully circular channels aimed to recreate a Strahler order 13 (ref. 22) subsegmental pulmonary arteries and were both 28 mm in length and 4 mm width, with initial heights of 2 mm and 4 mm, respectively. Both models contained the following 4 regions: pre-stenosis (8.5 mm length), stenosis (5 mm length), post-stenotic dilatation (2 mm length) and a downstream region (12.5 mm length) tapering to 1 mm and 2 mm diameter in the semi- and fully circular model, respectively, to model the natural tapering of a pulmonary artery. Concentric stenosis degrees of 30%, 60% and 80% were applied, and a straight channel (0% stenosis) was used as control. In both fully and semi-circular channels, the post-stenotic dilatation region had a width of 4 mm in the 30% and 60% stenosis models, while the same region had a width of 3 mm in the 80% stenosis model. The moulds' dimensions were 36 mm × 20 mm × 7 mm (L × W × H). A 2 mm gap was left between the ends of the channels and the walls of the moulds.
3D-printing of the semi-circular moulds
Moulds with semi-circular channels were designed in Fusion 360 (AutoDesk) and printed in Model V2 resin (FormLabs), using a Form3 SLA 3D-printer (FormLabs).The moulds were activated with trichloro(chloromethyl)silane (Sigma-Aldrich, cat. no. 254436) before polydimethylsilane (PDMS) casting. PDMS was prepared by mixing 10 parts of elastomer to 1 part of curing agent (SYLGARD® 184, Dow Corning, Midland, MI, USA). The mixture was then slowly poured onto the moulds and left to cure for 24 h at 65 °C. The cured PDMS devices were peeled off the moulds and bonded to either glass coverslips (Ibidi, cat. no. 10812) for platelet adhesion assays, or polymer coverslips (ibiTreat coverslips, Ibidi, cat. no. 10812) for endothelial flow assays using PDMS and set to cure at 65 °C for 12 hours. Blunt needles (16G × 10 mm, tailored made from Coopers Needle Works, Ltd) were then inserted into the inlet and outlet regions bonded to the devices using PDMS. The edges of the bonded devices were sealed with another layer of PDMS to strengthen bonding to the coverslips, and the devices were left to cure again at 65 °C for 12 hours. Completed models were flushed with absolute ethanol and left under UV light to sterilize for 4 hours.
Characterization of the wall shear stress (WSS) and flow patterns using computational fluid dynamics (CFD) analyses
All models were meshed in ICEM CFD (Ansys, Inc.). Final meshes were exported to CFX-PRE (Ansys, Inc.) for pre-processing. All simulations were carried out in a laminar flow module and without thermal exchange. Water was used to simulate cell medium, using a dynamic viscosity of 1 mPa s−1, a molar mass of 0.01802 kg mol−1 and a constant density of 992 kg m−3. Blood was defined as a liquid with a density of 1060 kg m−3, a dynamic viscosity of 4 mPa s−1 and a molecular weight of 65 kg mol−1. Inlet boundary conditions for water were adjusted to obtain similar hemodynamic conditions to those observed when simulations were run with blood. Outlet pressure was determined using a derivation of the Hagen–Poiseuille's equation,23 where μ is the dynamic viscosity, Q the inlet flow rate, Ltube and Dtube the length and inner diameter of the tubing used for flow perfusion, respectively.23For semi-circular models, inlet velocities of 60.2 mm s−1 (corresponding to a flow rate of 0.5 L h−1) and 20 mm s−1 (corresponding to a flow rate of 0.17 L h−1) were used for water and blood, respectively. For fully circular models, inlet velocities of 50 mm s−1 (corresponding to a flow rate of 2.26 L h−1) and 15 mm s−1 (corresponding to a flow rate of 0.68 L h−1) were used for water and blood, respectively.

The inlet velocity values were chosen to achieve a WSS of 1 dyn cm−2 in the straight, control channels, which corresponds to the average WSS seen in the large pulmonary arteries of healthy individuals.24–26
Tubing inner diameter was set to 2.79 mm and 1.6 mm for the semi-circular models simulated with water and blood, respectively. A tubing inner diameter value of 2 mm was used for the fully circular models simulated with water and blood. Outlet pressures of 0.097 kPa and 1.16 kPa were used for the semi-circular models simulated with water and blood, respectively. For fully circular models, outlet pressures of 1.6 kPa and 1.92 kPa were used for water and blood, respectively. Inlet and outlet boundary parameters are summarised in Table S3.† A no-slip boundary condition was applied at the walls of the models. All simulations were run on the CFX Solver Manager (Ansys, Inc.).
Post-processing
All results files were post-processed and analysed in CFD POST (Ansys, Inc.). Contours around the walls of the models were created to display WSS and pressure values. Velocity streamlines (50 data points), corresponding to the line tangential to the instantaneous velocity direction, were displayed from the inlet to show the flow patterns within the channels.A polyline was created across the length of the channels to extract the exact WSS values. The polyline was derived from the intersection between the default domain (i.e. the main body of the model) and YZ plane of the models. WSS, pressure and velocity values were then plotted against the Y axis of the models and extracted as a Microsoft Excel file for downstream analyses.
In the semi-circular channels, creating a polyline allowed the characterisation of WSS, pressure and velocity values at the top, curved wall of the models (referred to here as “top”), as well as the flat surface of the channel cross-section (named here as “bottom”).
Mesh convergence analyses
A mesh sensitivity test was carried out to determine which mesh size to use for the final simulations. In the mesh sensitivity test, all simulations were performed with water, using an inlet velocity of 157 mm s−1 (1.31 L h−1) and an outlet pressure of 2.94 kPa. Part element sizes used for each mesh in the convergence analysis are provided in Table S4;† total number of elements for each mesh in Table S5;† mean WSS and % difference in WSS between meshes in each part of straight and stenotic channels in Tables S6–S9.† Details of mesh convergence analysis are provided in ESI† Methods.Blood sample preparation
Venous blood was taken from antecubital veins of consenting, healthy donors. Samples were collected in 50 mL sterile falcon tubes and treated with 10% v/v acid citrate dextrose (ACD) (Sigma-Aldrich; cat. no.: C3821-50ML). Blood samples were used within 1 h of collection. 3,3-Dihexyloxacarbocyanine iodide (DiOC6) was added to the samples to stain for platelets.Platelet adhesion and aggregation assays in collagen-coated channels
Sterilised pulmonary artery models were coated with collagen type 1 (ChronoPAR, P/N 385; 120 μL per channel, 200 μg mL−1) and left at room temperature for 2 hours. The channels were then blocked with 0.4% bovine serum albumin (in PBS, w/v). Citrated and DiOC6-labelled blood was perfused through the coated channels using an Ibidi pump system for 4 minutes at a flow rate of 0.17 L h−1 (2.8 mL min−1) or 0.50 L h−1 (8.3 mL min−1). Fluorescent platelet aggregates were visualised with a fluorescent microscope (EVOS M5000, Invitrogen, Thermo Scientific). Platelet surface coverage was analysed with ImageJ (FIJI).Platelet adhesion and aggregation assays in endothelialised channels
Sterilised pulmonary artery models were coated with fibronectin (10 μl mL−1 in PBS; EMD Millipore Corp, USA, 341631), seeded with human pulmonary arterial endothelial cells (HPAECs; PromoCell, C-12241) and then perfused with endothelial cell growth medium 2 (ECGM-2, PromoCell) for 6 hours or incubated overnight at 37 °C under static conditions, to be used as static controls. The channels left under flow were disconnected from the flow system and perfused with citrated, DiOC6-labelled whole blood at a flow rate of 0.17 L h−1 (2.8 mL min−1) for 4 min each, using a syringe pump (Fisherbrand). The channels were then examined under a fluorescence microscope (Olympus IX70). Platelet surface coverage was analysed with ImageJ (FIJI). In some assays, a polyclonal anti-VWF antibody was used to block VWF function.Cell culture
HPAECs (PromoCell, C-12241) were cultured in endothelial cell growth medium 2 (ECGM2; PromoCell, Germany C-22111) as in ref. 27 and used between passages 5–8.The arterial models were sterilised prior to cell seeding under ultraviolet light for 1 h. Additionally, the channels were flushed with 70% ethanol and then washed 4 times with sterile PBS. After that, the channels were coated with fibronectin (10 μg mL−1 in PBS, EMD Millipore Corp, USA; 341631). HPAECs, resuspended in ECGM-2 (7.0–8.0 × 105 cells per mL), were introduced into the channels (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–120000 cells per device) and incubated for 24 h to allow for complete cell adherence and monolayer formation.
000–120000 cells per device) and incubated for 24 h to allow for complete cell adherence and monolayer formation.
To allow adaptation to flow, the cells were initially exposed to a low flow rate of 0.01 L h−1 (equivalent to a shear stress of 0.01 dyn cm−2 at the inlet) for 1 h. The flow was then gradually increased by 0.1 L h−1 every hour, to reach the final flow rate of 0.5 L h−1. After that, fresh medium was added to reservoirs and the cells were left under flow for 6 h or 24 h at 37 °C. After the experiments, the devices were disconnected from the flow system and the tubing was flushed 1× with 0.2% sodium hydroxide (Sigma-Aldrich, cat. no. S5881) in PBS (w/v), followed by 1× with distilled water. The tubing was autoclaved every 2 uses.
Patient-derived endothelial colony forming cells
Late outgrowth endothelial colony forming cells (ECFCs) were isolated from systemic blood from CTEPH patients (n = 4) and control healthy individuals (n = 3) and cultured as previously described.14 Donor characteristics are summarized in Table S10.† Endothelial identity of ECFCs was confirmed as previously described.14Culture of pulmonary endarterectomy endothelial (PEA) cells
PEA cells from 2 CTEPH patients were extracted from fresh PEA lung tissue samples, taken from the right inferior lobe and left superior lobe, respectively. Briefly, the tissues were first cut into small pieces and added to the wells of a 6-well plate coated with 0.2% gelatin (Sigma; cat.: G1890). 2 mL of ECGM-2 medium (PromoCell, Germany; cat.: C-2211) supplemented with 10% HyClone FBS (Cytiva; cat.: SH30071.03IH25-40) and 1% penicillin–streptomycin (100 μg mL−1, Gibco, 15140-122) was added to each well. The following day, 1 mL of medium was replaced with fresh medium, and the procedure repeated for the next 3 days. The medium was then fully replaced with fresh medium every 2 days, without removing the pieces of tissue, for the next 6 days. After this, medium was changed, and the pieces of tissue slowly removed over a week. Full ECGM-2 supplemented with 10% HyClone FBS and 1% penicillin–streptomycin was used until colony formation.PEA cells were then passaged and cultured in 0.2% gelatin (Sigma; cat.: G1890)-coated 75 cm2 tissue culture flasks (Sarstedt; cat.: C-22011), in full ECGM-2 medium (PromoCell, Germany; cat.: C-2211) with antibiotics until 80% confluency. Endothelial identity of the PEA cells was confirmed according to previously described methods.14 Patient characteristics are summarized in Table S10.†
Flow system
The channels were perfused using a closed-circuit system driven by a REGLO ICC 4 channel 12 Roller Pump (Cole-Parmer, UK, cat. no. ISM4412). The pump was connected to two medium reservoirs acting as capacitators28 placed before and after the inlet and outlet of the PDMS device, respectively. The reservoirs had a total capacity of 2 mL, with two hose barb connectors to connect the tubing to both the pump and the device's inlet or outlet. The tops of the reservoirs have a screw thread to fit commercially available microcentrifuge tube caps lined with a silicone O-ring (Greiner Bio-One, 12 mm screw cap; cat. no.: 366380). Two small extensions were added to the bottom of the reservoir to secure them to a baseplate.The peristaltic pump was connected to each reservoir using Ismatec® pump tubing, 3-stop, PharMed® BPT tubing, 2.76 mm ID (Cole-Parmer, UK, cat. no. WZ-95714-48) and Ismatec® pump tubing, PharMed® BPT extension tubing, 2.76 mm ID; 100 ft (Cole-Parmer, UK, cat. no. WZ-95809-48). Short pieces of Ismatec® pump tubing, PharMed® BPT extension tubing, 1.14 mm ID; 100 ft (Cole-Parmer, UK, cat. no. WZ-95809-30) were used to connect the end of the 2.79 mm ID tubing to the barbs of each reservoir. The same 1.14 mm ID tubing was used to connect the inlet and outlet of the devices to the lower barbs of the reservoir.
Flow rate measurements
Flow rate measurements were performed using a Sensirion SLF3S-1300F flow sensor (Mouser electronics, cat. no. 403-SLF3S-1300F). The flow circuit was primed with distilled water (as per the manufacturer's instructions) prior to testing, by connecting the inlet and outlet tubing together using a blunt needle and filling in the outlet reservoir until the inlet reservoir became full. After priming, the devices were connected to the reservoirs via tubing and perfused with distilled water for 1 min at a flow rate of 0.5 L h−1 (8.63 mL min−1). The flow sensor was then placed between the outlet of the channel and the reservoir, and connected via USB cable to a laptop, on which waveforms measurements were performed using the Sensirion Control Center software (Sensirion). Data was acquired for 1 min at a flow rate of 0.5 L h−1 and analyzed using the Sensirion Data Viewer software (Sensirion).The shear rate in the selected regions of the channel can be determined using the formula below:23
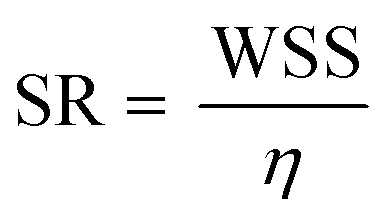
Introduction of flow reservoirs acting as capacitators, effectively reduced flow pulsatility, in agreement with previous reports.28
Viability assay
The LIVE/DEAD™ Viability/Cytotoxicity kit (ThermoFisher, cat. no. L3224) was used to perform cell survival analyses. The cells were imaged under an inverted fluorescent microscope (Olympus IX70).Immunostaining and cell imaging
HPAECs were fixed with 4% paraformaldehyde (PFA; Sigma Aldrich, UK) (w/v) in PBS for 20 min and permeabilised with 0.5% Triton X-100 (Sigma Aldrich, UK) (v/v) in PBS for 5 min. The cells were washed three times with PBS between each step. HPAECs were then blocked in 2% BSA (A9418, Sigma Aldrich, UK) (w/v) in PBS for 30 min and incubated with Alexa Fluor 488-conjugated mouse monoclonal anti-VE-cadherin antibody (eBioscience, cat. no. 53-1449-42, 1:100) and TRITC-phalloidin in 1% BSA (w/v) in PBS for 2 h at room temperature in the dark. The channels were washed three times with PBS, mounted with DAPI in Vectashield (Vector Laboratories) and imaged under an inverted fluorescent microscope (Olympus IX70) or a confocal microscope (Leica Stellaris 8).
Cell coverage and cell alignment analyses were carried out in ImageJ (FIJI). Cells were considered aligned to flow if the angle between nuclear long axis and flow direction was below 30°.
Flow exposure of patient-derived endothelial colony forming cells
60% stenosis channels were coated with 0.2% gelatin (Sigma; cat.: G1890) for 10 min at 37 °C and left to air-dry. ECFCs (7.0–8.0 × 105 cells per mL; 100000–120000 cells per device) were introduced into the channels and incubated for 24 h to allow for complete cell adherence and monolayer formation. The cells were then exposed to flow for 6 h or were fixed and stained for markers of interests.
RNA extraction, reverse transcription and qPCR
To isolate cells from different parts of the microfluidic channels, regions of interest (ROIs) termed “upstream”, “stenosis”, “post-stenotic dilatation”, and “downstream” were marked with a pen at the bottom side of the thin polymer coverslips. Culture medium was removed and the PDMS chamber was peeled off the coverslips. ROIs with cells still attached were carefully cut out with sterile scissors and transferred directly into DNA/RNA-free tubes filled with 500 μL TRIzolTM Reagent (Thermo Scientific; cat.: 15596026) and incubated for 5 min at RT. After 2–3 gentle shakes to dislodge cells, plastic cutouts were removed from the tubes with sterile tweezers and the remaining cell suspension was vortexed for 1 min and placed on ice or stored at −80 °C for future use. Samples were kept on ice until all ROIs were collected.A detailed description of experimental procedures is provided in ESI† Methods. Tables S11–S15† provide details of qPCR reverse transcription mix, RT cycling conditions, list of primers, components of qPCR reactions and amplification and cycling conditions.
Statistical analysis
All graphs and statistical analyses were performed using either GraphPad Prism software 9 (GraphPad Software, USA) or Rstudio (RStudio Inc, version 1.2.5042).All experiments were performed in at least four biological replicates, with 3 technical replicates performed per experiment, unless stated otherwise. All data were tested for normal distribution using the Shapiro–Wilk test. An unpaired Student's t test was used to analyse the normally distributed data from two sample groups, while a one- or two-way ANOVA test was used to analyse three or more sample groups as appropriate.
Statistical significance was accepted as p < 0.05. All error bars are representative of mean (±SEM).
Results
Wall shear stress, flow patterns and pressure within semi-circular models
Most vascular models are designed with a circular cross-section, to resemble natural vascular geometry. However, visualising and accessing cells growing within the selected regions of interest present a significant challenge. To address this limitation, we designed semi-circular vascular channels, where the top half of a cylindrical channel is bound to a thin, tissue culture compatible plastic coverslip. We introduced 4 degrees of vascular stenosis ranging from 30–80%, most commonly found in CTEPH and APE lungs. Device design and fabrication are illustrated in Fig. 1.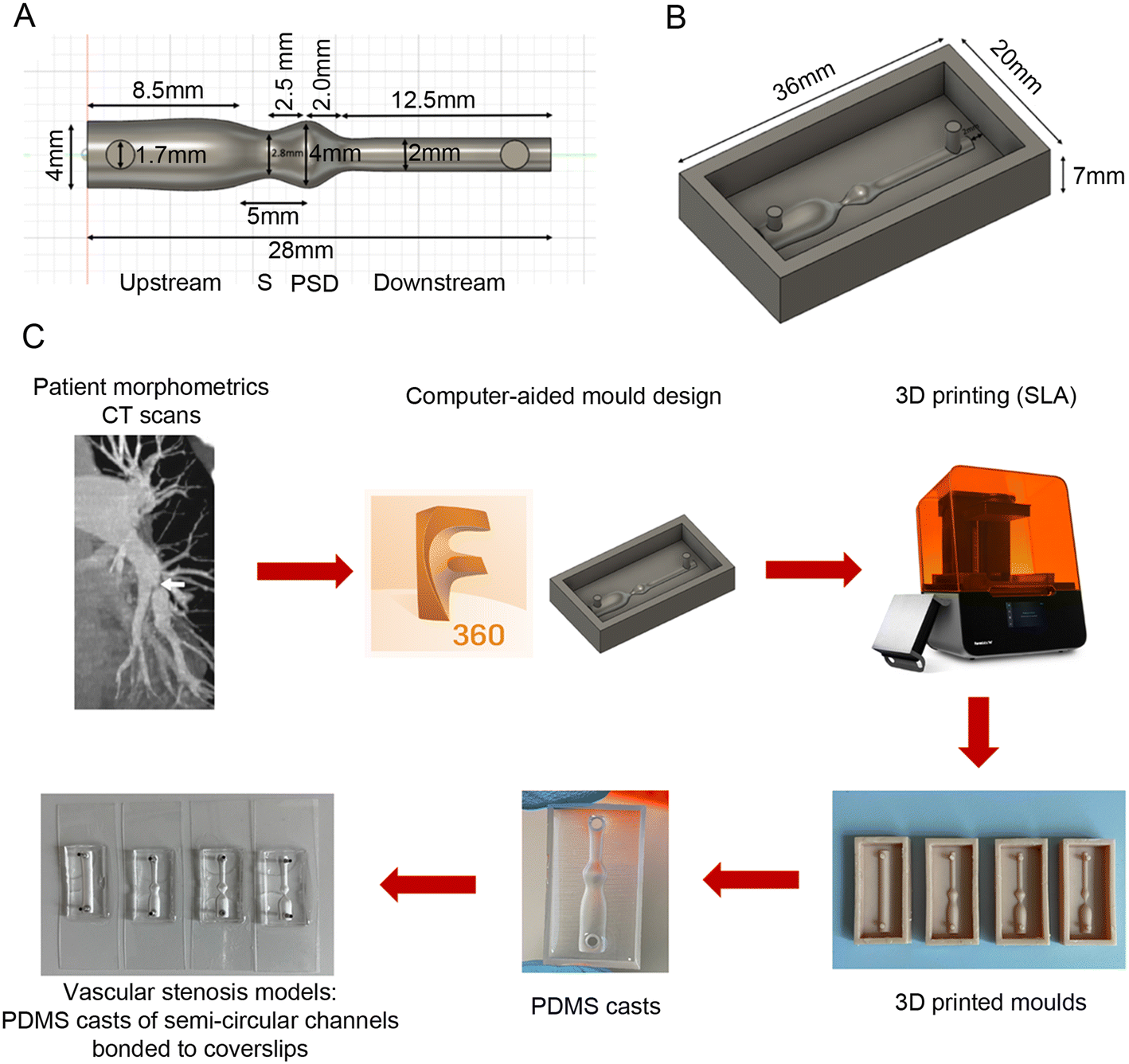 | ||
| Fig. 1 Design and fabrication of pulmonary vascular stenosis models. (A) Design of a 30% stenosis channel, and (B) 3D-printed mould for an 80% stenosis channel created using Fusion 360 (Autodesk). (C) Fabrication process of semi-circular models of vascular stenosis. Computed tomography (CT) scans from patients with CTEPH were used to determine the most commonly found degrees of stenosis. Moulds of stenotic channels were designed using Fusion 360 (Autodesk) and printed with a stereolithography (SLA) 3D printer (Form3, FormLabs). Polydimethylsiloxane (PDMS) was then cast onto the moulds to produce semi-circular replicas of control (straight) and stenotic vessels. Finally, the chambers were bonded to polymer coverslips (ibiTreat coverslips, Ibidi), with blunt needles inserted into the inlets and outlets to serve as connectors for tubing used in flow perfusion experiments. | ||
To distinguish between the stenotic channel model and the specific areas of stenosis, we refer to channels without stenosis as “straight controls”, channels with 30% stenosis as “30% models”, channels with 60% stenosis as “60% models”, and channels with 80% stenosis as “80% models”. Different areas within these channels are referred to as “upstream”, “stenosis”, “post-stenotic dilatation (PSD)” and “downstream” (Fig. 1A).
To assess whether the semi-circular channels can replicate changes in flow dynamics caused by vascular stenosis as effectively as the fully circular channels, we compared velocity streamlines, WSS, and pressure distributions in both channel types, using simulations with water and blood. The inlet velocity was chosen to achieve a WSS of 0.1 Pa (1 dyne per cm2) in the straight, control channel, which corresponds to a physiological WSS in large pulmonary arteries.25,26
Flow characteristics in semi-circular channels simulated with water and blood are provided in Fig. S5 and S6† and flow characteristics in fully circular channels in Fig. S11 and S12.†
All models showed flow acceleration at the stenosis apex, followed by flow deceleration. Flow turbulence was noted in the PSD area in 60% and 80% models simulated with water (Fig. 2 and S5 and S11†).
 | ||
| Fig. 2 Wall shear stress and velocity streamlines in circular and semi-circular channels simulated with water. Velocity streamlines are shown in (A) semi-circular and (B) fully circular straight channel, whilst (C) and (D) show corresponding wall shear stress (WSS) values, as indicated. (E) Shows velocity streamlines in semi-circular and (F) in fully circular 80% stenosis models, respectively. (G and H) Show WSS changes in semi-circular and fully circular 80% stenosis models, as indicated. All simulations were performed in CFX (Ansys, Inc.), using an inlet velocity of 60.2 mm s−1 (0.5 L h−1) and outlet pressure of 0.097 kPa for the semi-circular channels, and an inlet velocity of 50 mm s−1 (2.26 L h−1) and outlet pressure of 1.60 kPa for the fully circular channels. | ||
WSS followed a similar pattern in all models and simulations, increasing at the stenosis apex and declining at PSD. Maximal WSS was observed at the stenosis apex in the 60% and 80% models. Predicted WSS values in upstream, stenosis, PSD and downstream areas in 30–80% models simulated with water and blood are provided in Tables S7–S9.† A buildup of pressure was observed at the upstream of stenosis, with the highest increase in 80% model (Fig. S9, S10, S15 and S16†).
WSS profiles at the curved top and flat bottom sides of the semi-circular channels showed similar pattern of changes, with only small variations observed in PSD of 60% and 80% models simulated with water (Fig. 3 and S17–S20†).
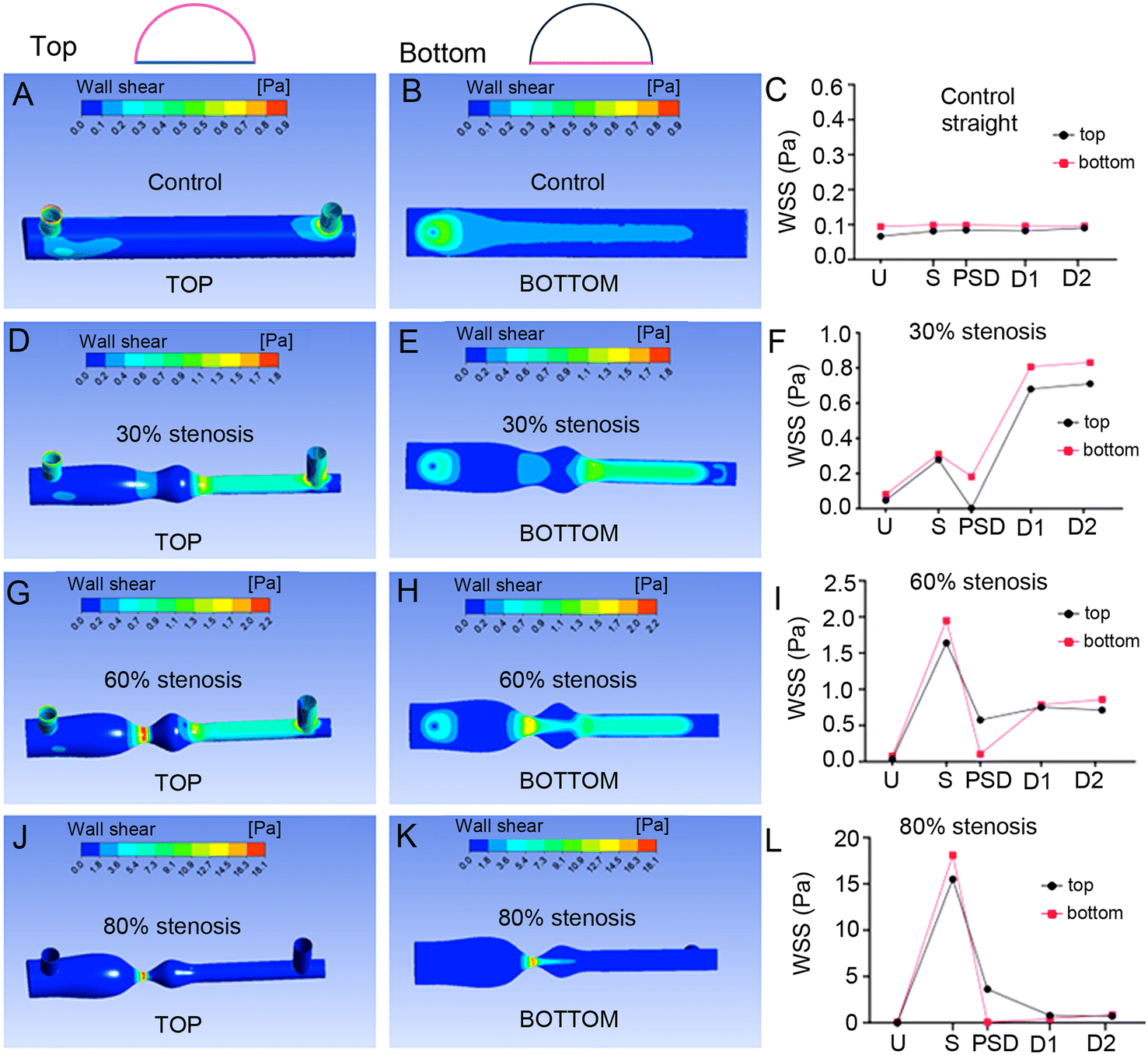 | ||
| Fig. 3 Wall shear stress in semi-circular channels simulated with water. Computational fluid dynamics (CFD) results showing wall shear stress (WSS) at the top and bottom of the semi-circular in the control (A–C), 30% stenosis (D–F), 60% stenosis (G–I) and 80% stenosis (J–L) models. Colour scales at the top of the images in (A)–(H) represent a WSS range between 0.0–2.0 Pa (0.0–20 dyn cm−2), while in (J and K) the scale ranges from 0.0–18.1 Pa (0.0–181 dyn cm−2). In (C, F, I and L) the x axis indicates the different regions of the channel: upstream (U; 7 mm), stenosis (S; 11 mm), post-stenotic dilatation (PSD; 13.5 mm), downstream 1 (D1; 18 mm) and downstream 2 (D2; 22.5 mm). All simulations were performed in CFX (Ansys, Inc.), using an inlet velocity of 60.2 mm s−1 (0.5 L h−1) and outlet pressure of 0.097 kPa. | ||
In summary, in silico analyses demonstrated that semi-circular channels can effectively replicate the pattern of WSS and pressures seen in fully circular channels. They also showed that predicted magnitude and pattern of mechanical forces acting at the curved roof of the channel closely resemble those seen at the bottom coverslip, justifying further use of this surface for cell culture and studies of cellular responses to stenosis.
HPAEC culture in stenotic channels
Changes in the channel geometry did not significantly affect HPAEC attachment, though some cell loss was observed in PSD in the 80% model, compared with straight controls (88.8% ± 2.1) (Fig. 4A). | ||
| Fig. 4 HPAECs surface coverage and orientation in semi-circular channels after flow perfusion. (A) HPAECs surface coverage in upstream, stenosis, PSD and downstream (Downstr) areas in semi-circular channels in 30%, 60%, and 80% stenosis models after 6 h flow at 0.5 L h−1. Image analysis was carried out with ImageJ (FIJI). N = 3–5 biological repeats. *P < 0.05, comparison with surface coverage in straight channel control; unpaired t-test. (B) HPAECs alignment. Alignment was measured by scoring % of nuclei oriented within 30 degrees of the flow direction in static controls (representing random orientation), straight channels under flow (control flow) and different areas of 30%, 60% and 80% stenosis models, as indicated. Image analysis was carried out with ImageJ (FIJI). N = 3–6 biological repeats. ###P < 0.01, comparison with flow control; *P < 0.05, **P < 0.01, ***P < 0.00, comparisons, as indicated; unpaired t-test. Values are shown as mean ± SEM. (C) Representative images of HPAECs in static culture and different areas of 80% stenosis model under flow, with predicted mean WSS values. In confocal microscopy images nuclei are (blue), F-actin (red) and VE-cadherin (green). White arrows indicate flow direction. Bar = 50 μm. | ||
The cells aligned within the flow direction, with the highest degree of cell alignment observed at 60% and 80% stenosis (Fig. 4B and C). Cell alignment and endothelial junctional integrity were reduced in PSD (Fig. 4B and C).
Transcriptional responses of HPAECs to changes in vascular geometry
Expression of marker genes of thrombosis, inflammation, angiogenesis, proliferation, and stimulation by flow (named here as “flow responses”) was measured in static and flow-stimulated HPAECs.HPAECs in 30% models showed increased expression of inflammatory markers (VCAM1, ICAM1, SELE, NFKB1, CCL5) across all regions of the channel, compared with static controls. Expression of tissue factor (TF), a key initiator of coagulation, was upregulated at stenosis, PSD, and downstream, whilst flow-response genes (KLF4, KLF6, PLXND1) and angiogenesis marker genes were upregulated in PSD and downstream. Expression of proliferation markers (CDC45, BRD4, CDKN1B) remained largely unchanged (Fig. 5).
 | ||
| Fig. 5 Transcriptional profile of HPAECs cultured in 30% and 60% stenosis models. (A) Heatmap showing fold change of expression of marker genes of thrombosis, inflammation, angiogenesis, proliferation, and stimulation by flow (named here as “flow responses”) in 30% model under flow, relative HPAECs cultured in the straight, control channel, under static conditions (static control); p < 0.05. (B) Summary diagram illustrating direction of changes in gene expression in HPAECs cultured in the 30% stenosis model under flow (0.5 L h−1, 6 h). (C) Heatmap showing fold changes in marker gene expression in 60% stenosis model. (D) Summary diagram illustrating direction of changes in gene expression in 60% model. Heatmaps in (A) and (C) were generated using R 4.3.3 (The R Project for Statistical Computing, 2024). PSD: post-stenotic dilatation. Diagrams in (B) and (D) were made using PowerPoint (Microsoft) and Fusion360 (Autodesk). | ||
60% stenosis models showed upregulation of inflammatory responses in all regions of the channel. Expression of angiogenic, thrombotic and EMT marker genes as well as flow-response genes was markedly upregulated in PSD and downstream regions. Overall, 60% stenosis induced a more widespread endothelial inflammatory, thrombotic, and tissue repair responses, compared with the 30% model (Fig. 5).
HPAECs cultured in the 80% stenosis model exhibited signs of endothelial damage, characterised by a significant (p < 0.05) increase in the expression of apoptosis markers, alongside an enhanced inflammatory, pro-thrombotic, pro-angiogenic, and pro-proliferative response observed throughout the entire length of the channel. Similar to the 60% stenosis model, the expression of EMT markers was elevated in both the PSD and downstream PSD regions (Fig. 6A and B).
 | ||
| Fig. 6 Transcriptional profile of 80% stenosis HPAECs and PEA endothelial cells. (A) Heatmap showing fold change of expression of marker genes of thrombosis, inflammation, angiogenesis, proliferation, and stimulation by flow (named here as “flow responses”) in 80% model under flow (0.5 L h−1, 6 h), relative HPAECs cultured in the straight, control channel, under static conditions (static control). p < 0.05. (B) Direction of changes in gene expression in HPAECs cultured in the 80% stenosis model under flow (0.5 L h−1, 6 h). (C) Isolation of endothelial cells from pulmonary endarterectomy (PEA) specimens. (D) Expression of selected gene markers of inflammation, thrombosis, angiogenesis, proliferation and apoptosis, in PEA endothelial cells from 2 donors. *P < 0.05, **P < 0.001, ***P < 0.001, comparison with static HPAEC control, n = 4. Data normalised to β2 microglobulin (B2M). Heatmap in (A) was generated using R 4.3.3 (The R Project for Statistical Computing, 2024). Diagram (B) was made using PowerPoint (Microsoft) and Fusion 360 (Autodesk). | ||
Pulmonary endarterectomy (PEA) endothelial cells were isolated from the intimal layer attached to the organised clot, and their endothelial cell identity was confirmed by increased expression of marker genes NOS3, PECAM1, CDH5 and VWF.14 CTEPH-derived PEA endothelial cells displayed an overall pro-thrombotic, pro-proliferative, pro-angiogenic, and pro-inflammatory phenotype, with some evidence of endothelial-to-mesenchymal transition. This phenotype was consistent with the endothelial profile observed in organised thrombi in situ13,29,30 and corresponded to the phenotype of HPAECs in 60% and 80% stenosis models (Fig. 6).
In contrast to HPAECs, transcriptional responses of blood-derived ECFCs to vascular stenosis showed high donor-dependent variability (Fig. S21†). Though the changes in marker gene expression did not reach significance, a trend towards increased apoptosis, proliferation, and inflammation was noted (Fig. S21†). The origin of these blood-derived cells is poorly understood,10 which may explain differences in their response to mechanical stimulation compared to arterial endothelial cells.
Platelet adhesion and aggregation in collagen-coated channels
To evaluate an impact of vessel geometry on platelet behaviour we studied platelet adhesion in stenosis models perfused with citrated whole blood at low (0.17 L h−1) and high (0.5 L h−1) flow. Under low flow conditions, platelets preferentially adhered at the stenosis apex in the 80% model (88.9 ± 4.5% surface coverage, P < 0.001), compared with straight controls (28.4 ± 2.7% surface coverage, P < 0.001) (Fig. 7A and B). In 30–60% stenosis models, platelet adhesion was relatively uniform across all regions, with a small increase observed within stenosis and downstream PSD (Fig. 7A and B). | ||
| Fig. 7 Platelet adhesion in collagen-coated stenotic channels perfused with blood. (A) Platelet adhesion (% surface coverage) in collagen-coated semi-circular channels with 30–80% stenosis, perfused with citrated whole blood for 4 min at 0.17 L h−1 (low flow) and (B) corresponding fluorescence microscopy images. (C) Platelet adhesion at high flow of 0.5 L h−1 and (D) corresponding images of platelets in channels perfused at 0.5 L h−1. In (A and C) the left Y axis shows% of surface coverage and the right Y axis indicates the wall shear stress (WSS; lines) at various regions of the channels (upstream, stenosis, PSD and downstream), as indicated. N = 3–5 biological repeats. Unpaired t-tests were performed to compare two individual groups. **P < 0.01, ***P < 0.001, compared to straight control. Bars show means ± SEM. In (B and D) platelets were labelled with 3,3-dihexyloxacarbocyanine iodide (DiOC6) (green fluorescence). Scale bar = 500 μm. | ||
At high blood perfusion rate (0.5 L h−1), in the 80% model platelet adhesion was lowest at stenosis (13.8 ± 1.2% surface coverage, P < 0.001) and highest at PSD (72.9 ± 1.5% surface coverage, P < 0.001), where flow re-circulation occurred (Fig. 7C and D). In the 60% model, platelet adhesion was increased in all regions of the channel, with highest values observed within stenosis (61.1 ± 2.3% coverage), PSD (65.5 ± 1.6% coverage), and downstream regions (62.9 ± 1.5% coverage). In the 30% model, platelet surface coverage was highest in the downstream region (43.8 ± 0.9%) (Fig. 7C and D).
Wall shear stress values in different regions of the semi-circular channels perfused with whole blood at 0.17 L h−1 and 0.50 L h−1 are shown in Fig. 7A and C and Tables S6–S9.† Platelet adhesion and thrombus formation were prevented in the presence of polyclonal anti-VWF antibodies31 (Fig. 8).
 | ||
| Fig. 8 Platelet adhesion in collagen-coated channels with 80% stenosis perfused with citrated whole blood at 0.17 L h−1 and 0.50 L h−1. (A) Images of platelet adhesion and aggregation in control (straight) semi-circular channels after perfusion with citrated whole blood for 4 min at 0.17 L h−1 and (B) 0.50 L h−1. (C) and (D) Show platelet adhesion within the region of 80% stenosis at low and high flow, as indicated. (E) Platelet adhesion within 80% stenosis at low flow (0.17 L h−1). (F) Platelet adhesion in 80% stenosis channel in the presence of anti-vWF antibodies. Images were taken under a fluorescence microscope (EVOS M5000, Invitrogen, Thermo Scientific) at the 4 min time point. Platelets were labelled with 3,3-dihexyloxacarbocyanine iodide (DiOC6) (green fluorescence). Arrows indicate the direction of flow. In (A–D) scale bar = 1 mm, in (E and F) bar = 500 μm. | ||
Platelet adhesion and aggregation in endothelium-coated channels
In the presence of endothelium, platelet adhesion was relatively low in 30% and 60% models, compared with straight control. Significantly increased platelet adhesion was observed in all regions of the 80% model, particularly within the PSD and downstream areas (Fig. 9). | ||
| Fig. 9 Platelet adhesion in endothelium-coated semi-circular channels perfused with citrated whole blood at 0.17 L h−1. Platelet adhesion in semi-circular channels seeded with human pulmonary artery endothelial cells (HPAECs) and perfused with citrated whole blood for 4 min at 0.17 L h−1. N = 3 biological repeats. Endothelial channels were exposed to flow for 6 hours before blood perfusion. *P < 0.05; **P < 0.01, compared to flow control; ∼P < 0.05, comparison, as indicated; unpaired t-test. Bars show means ± SEM. | ||
Discussion
In this study, we employed CTPA, 3D printing and soft lithography to produce CTEPH-based microfluidic models of pulmonary arterial stenosis. The semi-circular models of arterial channels were bonded to biocompatible polymer coverslips to facilitate microscopic analysis and site-specific analysis of cell morphology and gene expression.Vascular channels with circular cross-section are typically fabricated using hydrogels or PDMS. However, the poor optical properties of these materials, along with light refraction and distortion within tubular environments, complicate data acquisition. To address this, we designed semi-circular models featuring a thin plastic coverslip enclosing a PDMS semi-circular channel. This coverslip, optimized for high-resolution microscopy and tissue culture, can be easily detached, allowing different regions of the stenotic channel, along with attached cells, to be excised and analysed.
Flow simulations with water and blood demonstrated close similarities in flow dynamics in upstream, stenosis, PSD, and downstream regions of semi-circular and circular channels. Additionally, WSS values and patterns at the coverslip closely matched those at the channel roof. This suggests that, under the conditions described in this study, cells grown on a coverslip positioned at the bottom of a semicircular channel can serve as reliable proxies for those grown on the curved walls of the circular channel.
Variations in flow patterns, notably WSS gradients and recirculation of flow, identified by CFD simulations at stenosis and PSD in 60–80% stenosis models, align with the published data.6,7,32,33
Changes in flow patterns, characterised by flow acceleration at the stenosis apex, and flow disturbances (deceleration and recirculation) in PSD shown here are likely to account for the loss of HPAEC alignment, compromised junctional integrity and increased platelet adhesion. Disturbed flow is known to induce cytoskeletal remodelling by activating RhoA, which increases actomyosin contractility and traction forces, disrupting cell–cell adhesion.34,35
Changes in channel geometry led to significant alterations in the endothelial cell transcriptome. Specifically, there was a marked increase in the expression of pro-thrombotic, pro-inflammatory, pro-angiogenic, and pro-proliferative marker genes in regions of high WSS at the stenosis and flow recirculation at the PSD, with expression levels increasing in proportion to the degree of stenosis. These changes indicate heightened potential for in situ thrombosis, as well as the activation of endothelial repair mechanisms, including increased cell proliferation and angiogenesis.
In 80% stenosis channels coated with HPAECs, platelet adhesion was prominent at stenosis, PSD and downstream, whilst in collagen-coated channels perfused at the same flow rate (0.17 L h−1), platelet adhesion occurred predominantly at stenosis. These differences may be explained by combined effects of endothelium-derived factors and shear micro-gradients.36 Expression of tissue factor (TF), plasminogen activator inhibitor-1 (PAI-1) and thrombomodulin (THBD) in HPAECs was significantly elevated at PSD, whilst VWF expression was low at stenosis but was elevated in all other regions of the channel. TF is a key initiator of the coagulation cascade, while PAI-1 promotes thrombus growth and stabilisation by inhibiting the degradation of fibrin; increased endothelial expression of both proteins may therefore create a pro-thrombotic environment in PSD. This aligns with published data showing that reduced wall shear stress (WSS) at the stenosis outlet increases VWF secretion and platelet aggregation in human umbilical vein endothelial cells (HUVECs).31 In the 80% stenosis model, platelet aggregation was completely inhibited by anti-VWF antibodies, indicating that VWF activation, typically occurring at shear rates above 1000 s−1,37 was required for this response. Investigations into the involvement of mechanosensing vWF-binding platelet receptors such as glycoprotein Ib (GPIb), glycoprotein IIb/IIIa (GPIIb/IIIa), and glycoprotein VI (GPVI),38 will be important for developing future therapeutic strategies.
At a low perfusion rate (0.17 L h−1) in collagen-coated 80% stenotic channels, platelets preferentially aggregated at the stenosis. However, at a higher flow rate (0.50 L h−1), platelet aggregation occurred at the PSD. This may be explained by the greater shear deceleration observed during the transition from stenosis to PSD under high flow compared to low flow (13740 s−1 at the stenosis and 367.5 s−1 at the PSD under high flow, versus 6250 s−1 at the stenosis and 250 s−1 at the PSD under low flow).
HPAECs cultured within the stenosis models displayed a pro-angiogenic profile, particularly in regions of high WSS. This may be a mark of an early endothelial attempt to resolve vascular occlusion in response to acute embolism. In addition to the activation of endothelial repair, areas with high WSS and/or disturbed flow also showed increased expression of apoptotic markers such as BAK1 and CASP9, as well as markers of inflammation, including the master regulator of inflammatory gene activation, transcription factor NFkB, and NFkB-regulated genes, including CCL5,39 SELE,40 ICAM1,41 VCAM1 (ref. 42) and TF.43 HPAECs cultured in 60% and 80% models showed increased levels of FN1 expression, which may promote endothelial-to-mesenchymal transition, one of the key features of vascular remodelling.44 Interestingly, increased FN1 expression was also seen in PEA endothelial cells.
Vessel-on-a-chip models have primarily been used to study thrombosis in the systemic circulation, particularly in the context of atherosclerosis or deep vein thrombosis.45 These models often employ human umbilical vein endothelial cells (HUVECs) and are designed to mimic the high-pressure, high-shear environment of systemic arteries (mean arterial pressure ∼93 mmHg; shear stress ∼1–2 Pa in arterioles), which is roughly 10 times greater than in pulmonary arteries. Thrombosis, influenced by both haemodynamics and endothelial cell type, is vascular bed-specific.46 Our model incorporates pulmonary-specific conditions to more accurately replicate the in vivo environment associated with lung thrombotic events, such as acute pulmonary embolism. Interestingly, comparative analysis of microfluidic models reveals key similarities in the response of systemic and pulmonary endothelial cells to stenosis. For example, platelet aggregation in vWF/fibrinogen- or HUVEC-coated rectangular microchannels increases downstream of stenotic atherosclerotic plaque geometries.31 This parallels our findings of a post-stenotic pro-apoptotic, pro-thrombotic, and pro-inflammatory endothelial phenotype.
Certain methodological considerations should be noted. While blood is a non-Newtonian fluid, many studies have demonstrated that blood behaves as a Newtonian fluid at high shear rates, such as those observed in stenotic vessels.47,48 Shear rates are seen to drop post stenosis (to a minimum value of 175 s−1 in our simulations) as the velocity of the blood reduces significantly when the geometry expands. Yet previous work on microfluidic stenotic geometries has also presented results confirming the Newtonian model to be a valid assumption at shear rates as low as 50 s−1.49 Thus, to reduce computational expense, the assumption of a Newtonian model was made. As future work moves towards patient-specific geometries, a non-Newtonian model such as the Quemada model,50 which allows for patient-specific hematocrit to be input, can be included.
Additionally, as stated in the methodology, the inlet flowrates were set to yield 0.1 Pa in straight controls, to match healthy subject shear stress values. As the primary focus of this study was on degree of stenosis inlet flowrates were kept constant between all models. This allowed for conclusions to be drawn solely on geometric variation. As our study focused on modelling a Strahler order 13 pulmonary artery, in vivo comparative data is not available. Thus, the suitability of our inlet flow conditions was also assessed by comparing main pulmonary artery flow parameters between CTEPH and healthy subjects: 4D-flow MRI51 and RHC52 literature report cardiac index of 1.5 and 2.5 L min−1 m−2, respectively, in CTEPH patients while in healthy subjects a value of 2.7 L min−1 m−2, measured from MRI, has been reported.53 Thus, given there are no significant order of magnitude differences, the constant flow condition between the straight and stenoses geometries was deemed appropriate. Our predicted WSS values were in line with values seen in other literature studies of CTEPH simulations.6,54 Future work however should also look to investigate the influence of potentially reduced cardiac output in CTEPH.
Experimentally, cells in this study were exposed to acute flow (6 hours), the shortest practical in vitro time-point to observe meaningful endothelial adaptation to flow, especially in terms of cytoskeletal changes/alignment and gene/protein expression.55,56 This exposure time was sufficient to demonstrate significant differences in endothelial response to changes in vascular geometry. The pro-thrombotic, pro-proliferative, pro-angiogenic, and pro-inflammatory phenotype, observed in HPAECs in 60% and 80% stenosis models is consistent with the endothelial profile observed in organised thrombi in situ13,29,30 and corresponds to the phenotype of pulmonary endarterectomy (PEA) endothelial cells, isolated from the intimal layer attached to the organised clot. The development of CTEPH is associated with long-term exposure to disturbed flow and, in this regard, our model is more likely to replicate early events following pulmonary embolism (PE). Prolonging the exposure to disturbed flow in combination with hypoxia and inflammatory stimuli, may better simulate the conditions leading to vascular pathology in CTEPH.18,57
Another limitation of our model is that it does not accurately replicate the physicochemical properties of the basement membrane, instead relying on a much more rigid, collagen-coated plastic coverslip. Endothelial mechanosensing is influenced by the stiffness, stretch, and composition of the substratum.58,59 To address this, future designs should consider alternative materials, such as hydrogels or PDMS,47 which, despite their less favourable optical properties as compared with glass or plastic, may better reproduce the mechanical forces acting on the lung endothelium.
There is currently no well-established in vivo animal model that fully captures the complexity of CTEPH, which hinders the development of effective therapeutic strategies. However, the innovative use of in vitro personalized systems, such as those being developed in this project, holds significant potential to advance personalized medicine, enabling disease prediction and optimized therapeutic selection – an approach that will be further enhanced by artificial intelligence advancements.
Conclusions
To summarise, we present an easily producible easy to fabricate microfluidic model of pulmonary vascular stenosis, suitable for studying effects of local changes of flow haemodynamics on endothelial morphology and function in vitro. This model may help in understanding the complex pathophysiology underlying development of chronic thromboembolic pulmonary hypertension in a subset of individuals following acute pulmonary embolism.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
SN: performing experiments, data analysis, manuscript writing; OTC, YRV and JAB: provision of study resources, data validation; TAJMcK and DG: data collection and analysis, critical evaluation of manuscript; CHA: data acquisition and analysis; BWS: study conceptualization, management, securing resources and manuscript writing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
SN PhD project was funded by Imperial College President's Scholarship Award; CA was funded by the British Heart Foundation Fellowship FS/IPBSRF/24/27116; OTC was supported by funding from the Miguel Servet type grant from the Institute of Health Carlos III (CPII22/00006), co-funded by the European Social Fund (“Investing in your future”), and by the Institute of Health Carlos III (PI21/01212; and PI18/00960). Additional co-funding was provided by the Fondo Europeo de Desarrollo Regional (FEDER), under the initiative “Una manera de hacer Europa”.References
- J. Ando and K. Yamamoto, Effects of shear stress and stretch on endothelial function, Antioxid. Redox Signaling, 2011, 15(5), 1389–1403 CrossRef CAS PubMed.
- M. O'Rourke, Mechanical principles in arterial disease, Hypertension, 1995, 26(1), 2–9 CrossRef PubMed.
- J. Pepke-Zaba, P. Jansa, N. H. Kim, R. Naeije and G. Simonneau, Chronic thromboembolic pulmonary hypertension: role of medical therapy, Eur. Respir. J., 2013, 41(4), 985–990 CrossRef CAS PubMed.
- G. Reiter, U. Reiter, G. Kovacs, H. Olschewski and M. Fuchsjager, Blood flow vortices along the main pulmonary artery measured with MR imaging for diagnosis of pulmonary hypertension, Radiology, 2015, 275(1), 71–79 CrossRef PubMed.
- M. Kawakubo, H. Akamine, Y. Yamasaki, A. Takemura, K. Abe and K. Hosokawa, et al., Three-dimensional phase contrast magnetic resonance imaging validated to assess pulmonary artery flow in patients with chronic thromboembolic pulmonary hypertension, Radiol. Phys. Technol., 2017, 10(2), 249–255 CrossRef PubMed.
- M. Spazzapan, P. Sastry, J. Dunning, D. Nordsletten and A. de Vecchi, The Use of Biophysical Flow Models in the Surgical Management of Patients Affected by Chronic Thromboembolic Pulmonary Hypertension, Front. Physiol., 2018, 9, 223 CrossRef PubMed.
- P. F. Costa, H. J. Albers, J. E. A. Linssen, H. H. T. Middelkamp, L. van der Hout and R. Passier, et al., Mimicking arterial thrombosis in a 3D-printed microfluidic in vitro vascular model based on computed tomography angiography data, Lab Chip, 2017, 17(16), 2785–2792 RSC.
- D. Cernica, I. Benedek, S. Polexa, C. Tolescu and T. Benedek, 3D Printing-A Cutting Edge Technology for Treating Post-Infarction Patients, Life, 2021, 11(9), 910 CrossRef PubMed.
- S. Aldosari, S. Jansen and Z. Sun, Patient-specific 3D printed pulmonary artery model with simulation of peripheral pulmonary embolism for developing optimal computed tomography pulmonary angiography protocols, Quant. Imaging Med. Surg., 2019, 9(1), 75–85 CrossRef PubMed.
- Y. Lin, D. J. Weisdorf, A. Solovey and R. P. Hebbel, Origins of circulating endothelial cells and endothelial outgrowth from blood, J. Clin. Invest., 2000, 105(1), 71–77 CrossRef CAS PubMed.
- O. Tura, E. M. Skinner, G. R. Barclay, K. Samuel, R. C. Gallagher and M. Brittan, et al., Late outgrowth endothelial cells resemble mature endothelial cells and are not derived from bone marrow, Stem Cells, 2013, 31(2), 338–348 CrossRef CAS PubMed.
- S. B. Nukala, O. Tura-Ceide, G. Aldini, V. Smolders, I. Blanco and V. I. Peinado, et al., Protein network analyses of pulmonary endothelial cells in chronic thromboembolic pulmonary hypertension, Sci. Rep., 2021, 11(1), 5583 CrossRef CAS PubMed.
- V. Smolders, K. Lodder, C. Rodriguez, O. Tura-Ceide, J. A. Barbera and J. W. Jukema, et al., The Inflammatory Profile of CTEPH-Derived Endothelial Cells Is a Possible Driver of Disease Progression, Cells, 2021, 10(4), 737 CrossRef CAS PubMed.
- O. Tura-Ceide, V. Smolders, N. Aventin, C. Moren, M. Guitart-Mampel and I. Blanco, et al., Derivation and characterisation of endothelial cells from patients with chronic thromboembolic pulmonary hypertension, Sci. Rep., 2021, 11(1), 18797 CrossRef CAS PubMed.
- I. M. Lang, J. J. Marsh, M. A. Olman, K. M. Moser, D. J. Loskutoff and R. R. Schleef, Expression of type 1 plasminogen activator inhibitor in chronic pulmonary thromboemboli, Circulation, 1994, 89(6), 2715–2721 CrossRef CAS PubMed.
- F. Sakamaki, S. Kyotani, N. Nagaya, N. Sato, H. Oya and N. Nakanishi, Increase in thrombomodulin concentrations after pulmonary thromboendarterectomy in chronic thromboembolic pulmonary hypertension, Chest, 2003, 124(4), 1305–1311 CrossRef PubMed.
- I. M. Lang, J. J. Marsh, M. A. Olman, K. M. Moser and R. R. Schleef, Parallel analysis of tissue-type plasminogen activator and type 1 plasminogen activator inhibitor in plasma and endothelial cells derived from patients with chronic pulmonary thromboemboli, Circulation, 1994, 90(2), 706–712 CrossRef CAS PubMed.
- S. Alias, B. Redwan, A. Panzenboeck, M. P. Winter, U. Schubert and R. Voswinckel, et al., Defective angiogenesis delays thrombus resolution: a potential pathogenetic mechanism underlying chronic thromboembolic pulmonary hypertension, Arterioscler., Thromb., Vasc. Biol., 2014, 34(4), 810–819 CrossRef CAS PubMed.
- K. M. Moser and C. M. Bloor, Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension, Chest, 1993, 103(3), 685–692 CrossRef CAS PubMed.
- T. Kawakami, A. Ogawa, K. Miyaji, H. Mizoguchi, H. Shimokawahara and T. Naito, et al., Novel Angiographic Classification of Each Vascular Lesion in Chronic Thromboembolic Pulmonary Hypertension Based on Selective Angiogram and Results of Balloon Pulmonary Angioplasty, Circ.: Cardiovasc. Interventions, 2016, 9(10), e003318 Search PubMed.
- R. Azarian, M. Wartski, M. A. Collignon, F. Parent, P. Herve and H. Sors, et al., Lung perfusion scans and hemodynamics in acute and chronic pulmonary embolism, J. Nucl. Med., 1997, 38(6), 980–983 CAS.
- T. J. Sanada, N. Tanabe, H. Ishibashi-Ueda, K. Ishida, A. Naito and S. Sakao, et al., Involvement of pulmonary arteriopathy in the development and severity of reperfusion pulmonary edema after pulmonary endarterectomy, Pulm. Circ., 2019, 9(2), 2045894019846439 Search PubMed.
- W. Huang, R. T. Yen, M. McLaurine and G. Bledsoe, Morphometry of the human pulmonary vasculature, J. Appl. Physiol., 1996, 81(5), 2123–2133 CrossRef CAS PubMed.
- P. Bachler, N. Pinochet, J. Sotelo, G. Crelier, P. Irarrazaval and C. Tejos, et al., Assessment of normal flow patterns in the pulmonary circulation by using 4D magnetic resonance velocity mapping, Magn. Reson. Imaging, 2013, 31(2), 178–188 CrossRef PubMed.
- M. Schafer, D. D. Ivy, S. H. Abman, K. Stenmark, L. P. Browne and A. J. Barker, et al., Differences in pulmonary arterial flow hemodynamics between children and adults with pulmonary arterial hypertension as assessed by 4D-flow CMR studies, Am. J. Physiol., 2019, 316(5), H1091–H1104 CAS.
- M. Schafer, V. O. Kheyfets, J. D. Schroeder, J. Dunning, R. Shandas and J. K. Buckner, et al., Main pulmonary arterial wall shear stress correlates with invasive hemodynamics and stiffness in pulmonary hypertension, Pulm. Circ., 2016, 6(1), 37–45 CrossRef PubMed.
- A. J. Ainscough, T. J. Smith, M. Haensel, C. J. Rhodes, A. Fellows and H. J. Whitwell, et al., An organ-on-chip model of pulmonary arterial hypertension identifies a BMPR2-SOX17-prostacyclin signalling axis, Commun. Biol., 2022, 5(1), 1192 CrossRef CAS PubMed.
- P. L. Voyvodic, D. Min and A. B. Baker, A multichannel dampened flow system for studies on shear stress-mediated mechanotransduction, Lab Chip, 2012, 12(18), 3322–3330 RSC.
- R. Quarck, M. Wynants, A. Ronisz, M. R. Sepulveda, F. Wuytack and D. Van Raemdonck, et al., Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients, Respir. Res., 2012, 13(1), 27 CrossRef PubMed.
- R. Quarck, M. Wynants, E. Verbeken, B. Meyns and M. Delcroix, Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension, Eur. Respir. J., 2015, 46(2), 431–443 CrossRef CAS PubMed.
- E. Westein, A. D. van der Meer, M. J. Kuijpers, J. P. Frimat, A. van den Berg and J. W. Heemskerk, Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(4), 1357–1362 CrossRef CAS PubMed.
- N. V. Menon, C. Su, K. T. Pang, Z. J. Phua, H. M. Tay and R. Dalan, et al., Recapitulating atherogenic flow disturbances and vascular inflammation in a perfusable 3D stenosis model, Biofabrication, 2020, 12(4), 045009 CrossRef CAS PubMed.
- Y. J. Deng, J. A. Duque, C. X. Su, Y. Q. Zhou, M. Nishikawa and T. H. Xiao, et al., Understanding stenosis-induced platelet aggregation on a chip by high-speed optical imaging, Sens. Actuators, B, 2022, 356 CAS.
- B. Wojciak-Stothard and A. J. Ridley, Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases, J. Cell Biol., 2003, 161(2), 429–439 CrossRef CAS PubMed.
- T. Miyazaki, K. Honda and H. Ohata, m-Calpain antagonizes RhoA overactivation and endothelial barrier dysfunction under disturbed shear conditions, Cardiovasc. Res., 2010, 85(3), 530–541 CrossRef CAS PubMed.
- W. S. Nesbitt, E. Westein, F. J. Tovar-Lopez, E. Tolouei, A. Mitchell and J. Fu, et al., A shear gradient-dependent platelet aggregation mechanism drives thrombus formation, Nat. Med., 2009, 15(6), 665–673 CrossRef CAS PubMed.
- S. W. Schneider, S. Nuschele, A. Wixforth, C. Gorzelanny, A. Alexander-Katz and R. R. Netz, et al., Shear-induced unfolding triggers adhesion of von Willebrand factor fibers, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(19), 7899–7903 CrossRef CAS PubMed.
- Y. Chen and L. A. Ju, Biomechanical thrombosis: the dark side of force and dawn of mechano-medicine, Stroke Vasc. Neurol., 2020, 5(2), 185–197 CrossRef PubMed.
- M. I. Wickremasinghe, L. H. Thomas, C. M. O'Kane, J. Uddin and J. S. Friedland, Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis, J. Biol. Chem., 2004, 279(26), 27199–27210 CrossRef CAS PubMed.
- U. Schindler and V. R. Baichwal, Three NF-kappa B binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression, Mol. Cell. Biol., 1994, 14(9), 5820–5831 CAS.
- K. Bunting, S. Rao, K. Hardy, D. Woltring, G. S. Denyer and J. Wang, et al., Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel, J. Immunol., 2007, 178(11), 7097–7109 CrossRef CAS PubMed.
- M. F. Iademarco, J. J. McQuillan, G. D. Rosen and D. C. Dean, Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1), J. Biol. Chem., 1992, 267(23), 16323–16329 CrossRef CAS PubMed.
- Y. D. Li, B. Q. Ye, S. X. Zheng, J. T. Wang, J. G. Wang and M. Chen, et al., NF-kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis, J. Biol. Chem., 2009, 284(7), 4473–4483 CrossRef CAS PubMed.
- Y. M. Hao, H. Q. Yuan, Z. Ren, S. L. Qu, L. S. Liu and H. Dang, et al., Endothelial to mesenchymal transition in atherosclerotic vascular remodeling, Clin. Chim. Acta, 2019, 490, 34–38 CrossRef CAS PubMed.
- J. F. Ren, Z. Wang, N. Du, W. L. Cheng and L. A. Ju, Charting the course of blood flow: vessel-on-a-chip technologies in thrombosis studies, Microstructures, 2024, 4, 2024037 CrossRef.
- W. C. Aird, Vascular bed-specific thrombosis, J. Thromb. Haemostasis, 2007, 5(Suppl 1), 283–291 CrossRef CAS PubMed.
- D. N. Ku, D. P. Giddens, C. K. Zarins and S. Glagov, Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress, Arteriosclerosis, 1985, 5(3), 293–302 CAS.
- Y. I. Cho and K. R. Kensey, Effects of the non-Newtonian viscosity of blood on flows in a diseased arterial vessel. Part 1: Steady flows, Biorheology, 1991, 28(3–4), 241–262 CAS.
- Y. C. Zhao, P. Vatankhah, T. Goh, R. Michelis, K. Kyanian and Y. Zhang, et al., Hemodynamic analysis for stenosis microfluidic model of thrombosis with refined computational fluid dynamics simulation, Sci. Rep., 2021, 11(1), 6875 CrossRef CAS PubMed.
- A. Skiadopoulos, P. Neofytou and C. Housiadas, Comparison of blood rheological models in patient specific cardiovascular system simulations, J. Hydrodyn., 2017, 29(2), 293–304 CrossRef.
- W. Xu, X. Sun, X. Tao, D. Wang, Y. Zhen and X. Liu, et al., Characteristics of Right Ventricular Blood Flow in Patients With Chronic Thromboembolic Pulmonary Hypertension: An Analysis With 4-Dimensional Flow Cardiovascular Magnetic Resonance Imaging, Front. Cardiovasc. Med., 2022, 9, 900301 CrossRef PubMed.
- J. Pepke-Zaba, M. Delcroix, I. Lang, E. Mayer, P. Jansa and D. Ambroz, et al., Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry, Circulation, 2011, 124(18), 1973–1981 CrossRef PubMed.
- C. P. Cheng, R. J. Herfkens, C. A. Taylor and J. A. Feinstein, Proximal pulmonary artery blood flow characteristics in healthy subjects measured in an upright posture using MRI: the effects of exercise and age, J. Magn. Reson. Imaging, 2005, 21(6), 752–758 CrossRef PubMed.
- H. Tsubata, N. Nakanishi, K. Itatani, M. Takigami, Y. Matsubara and T. Ogo, et al., Pulmonary artery blood flow dynamics in chronic thromboembolic pulmonary hypertension, Sci. Rep., 2023, 13(1), 6490 CrossRef CAS PubMed.
- C. G. Galbraith, R. Skalak and S. Chien, Shear stress induces spatial reorganization of the endothelial cell cytoskeleton, Cell Motil. Cytoskeleton, 1998, 40(4), 317–330 CrossRef CAS PubMed.
- B. D. Johnson, K. J. Mather and J. P. Wallace, Mechanotransduction of shear in the endothelium: basic studies and clinical implications, Vasc. Med., 2011, 16(5), 365–377 Search PubMed.
- M. L. Bochenek, N. S. Rosinus, M. Lankeit, L. Hobohm, F. Bremmer and E. Schutz, et al., From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension, Thromb. Haemostasis, 2017, 117(4), 769–783 Search PubMed.
- N. Chandra Sekar, S. Aguilera Suarez, N. Nguyen, A. Lai, P. Thurgood and Y. Zhou, et al., Studying the Synergistic Effect of Substrate Stiffness and Cyclic Stretch Level on Endothelial Cells Using an Elastomeric Cell Culture Chamber, ACS Appl. Mater. Interfaces, 2023, 15(4), 4863–4872 CrossRef CAS PubMed.
- A. Lai, P. Thurgood, C. D. Cox, C. Chheang, K. Peter and A. Jaworowski, et al., Piezo1 Response to Shear Stress Is Controlled by the Components of the Extracellular Matrix, ACS Appl. Mater. Interfaces, 2022, 14(36), 40559–40568 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lc00300h |
| This journal is © The Royal Society of Chemistry 2025 |