
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review of filiform corrosion and its prevention on polymer-coated aluminium alloys
Erlind
Mysliu†
*,
Iman
Taji‡
and
Andreas
Erbe

Department of Materials Science and Engineering, NTNU, Norwegian University of Science and Technology, 7491 Trondheim, Norway. E-mail: erlind.mysliu@sintef.no
First published on 17th December 2024
Abstract
Filiform corrosion (FFC) is an underpaint corrosion form frequently observed on metals with a high band gap oxide, e.g., aluminium and magnesium. Macroscopically, FFC shows as filaments propagating at a metal/coating interface. Electrochemically, an anodic active region is followed by a cathodic tail and a region where solid corrosion products precipitate. Two forms of FFC are observed, (i) fast anodic undercutting at the metal/coating interface and (ii) successive pitting. This work reviews experimental evidence for FFC mechanisms especially on 3xxx and 6xxx rolled and extruded aluminium alloys and the effect of the surface composition and microstructure. Recent evidence suggested that the hydrogen evolution and related bubble detachment near the anodic region in the head are more important for coating detachment than previously envisaged. For efficient FFC protection, surface pretreatments such as anodising and the sequence of etching, desmutting and conversion coating are crucial. Industrial focus has recently been on FFC on aluminium with a large content of post-consumer (a.k.a. end-of-life) scrap.
1 Organic coatings in corrosion protection
Corrosion has huge economic consequences, costing annually a few % of the gross domestic product of industrialised countries.1 The use of organic polymer coatings is often an attractive method for corrosion protection.2–4 Coatings are pricewise attractive and possibly also the most sustainable method of corrosion protection.5 Coatings are in addition frequently used to determine the visual appearance of materials. Corrosion protection through classical passive coatings is realised by simply isolating a reactive metal from its corrosive environment.2–4 On the other hand, coatings can also fail, with the result that their protective properties deteriorate significantly, or their decorative purposes vanish.2–4Several mechanisms of coating failure exist.3 There is the possibility of cohesive failure under mechanical load and the possibility of chemical degradation of the substances constituting the coating, e.g., by photochemical or radical activation. These mechanisms depend on the specific molecular composition of the coatings.
Adhesive failure at a metal/polymer interface can occur via chiefly mechanical processes, or by corrosion driven processes. On steel including galvanised steel under wet conditions, cathodic delamination is typically the fastest mechanism.2–4,6 On the other hand, anodic undercutting is a process in which the anodic metal dissolution at a metal/polymer interface drives the disbonding process.3,6 Corrosion science typically classifies corrosion forms according to their macroscopic appearance; one form of underpaint corrosion where an anodic process is driving the failure of a metal/coating interface is FFC.
Defined for the first time as “hair-like corrosion” by Sharman in 1944,7 FFC is a type of localised corrosion process taking place mainly on coated metals such as aluminium and magnesium. On iron, FFC is possible although the most common type of underpaint corrosion is cathodic delamination.2,4 To illustrate the macroscopic appearance of FFC, an optical microscopy image of FFC on a coated AA3005 aluminium alloy is shown in Fig. 1. The threadlike filaments can grow from coating defects such as cut edges or locations in which the coating has been damaged or not perfectly deposited. In these regions, the penetration of electrolytes favors metal dissolution. Although some of these principles are the same for aluminium, iron, and magnesium, their respective FFC propagation mechanism is different.
 | ||
| Fig. 1 Optical microscopy image of FFC affecting a rolled AA3005 aluminium alloy coated with a weak poly(methyl methacrylate) transparent model coating. FFC was induced by dripping concentrated HCl in an artificially made coating defect. Samples were exposed at 82% R.H. and 40 °C in a humidity chamber for 1000 h.§ | ||
The presence of FFC as a macroscopic corrosion form is not limited to metals coated with polymer coatings. Most prominently on some magnesium and aluminium alloys, FFC as a corrosion form can also be observed when exposed to conditions of atmospheric corrosion or exposed to electrolytes. In these cases, the oxide layers on the respective metal take the role of the coating, and FFC propagates at the metal/oxide interface.
2 Filiform corrosion: phenomenology and mechanistic discussions
2.1 FFC on aluminium: general features
On aluminium alloys, a representation of the FFC process is shown in Fig. 2. Specifically for aluminium, FFC has recently been reviewed only under the special conditions of adhesive bonding.8 On the other hand, there is increasing interest in the community because of the increased availability of recycled aluminium based on PCS and the role FFC may play in coated structures of PCS-based aluminium.9–12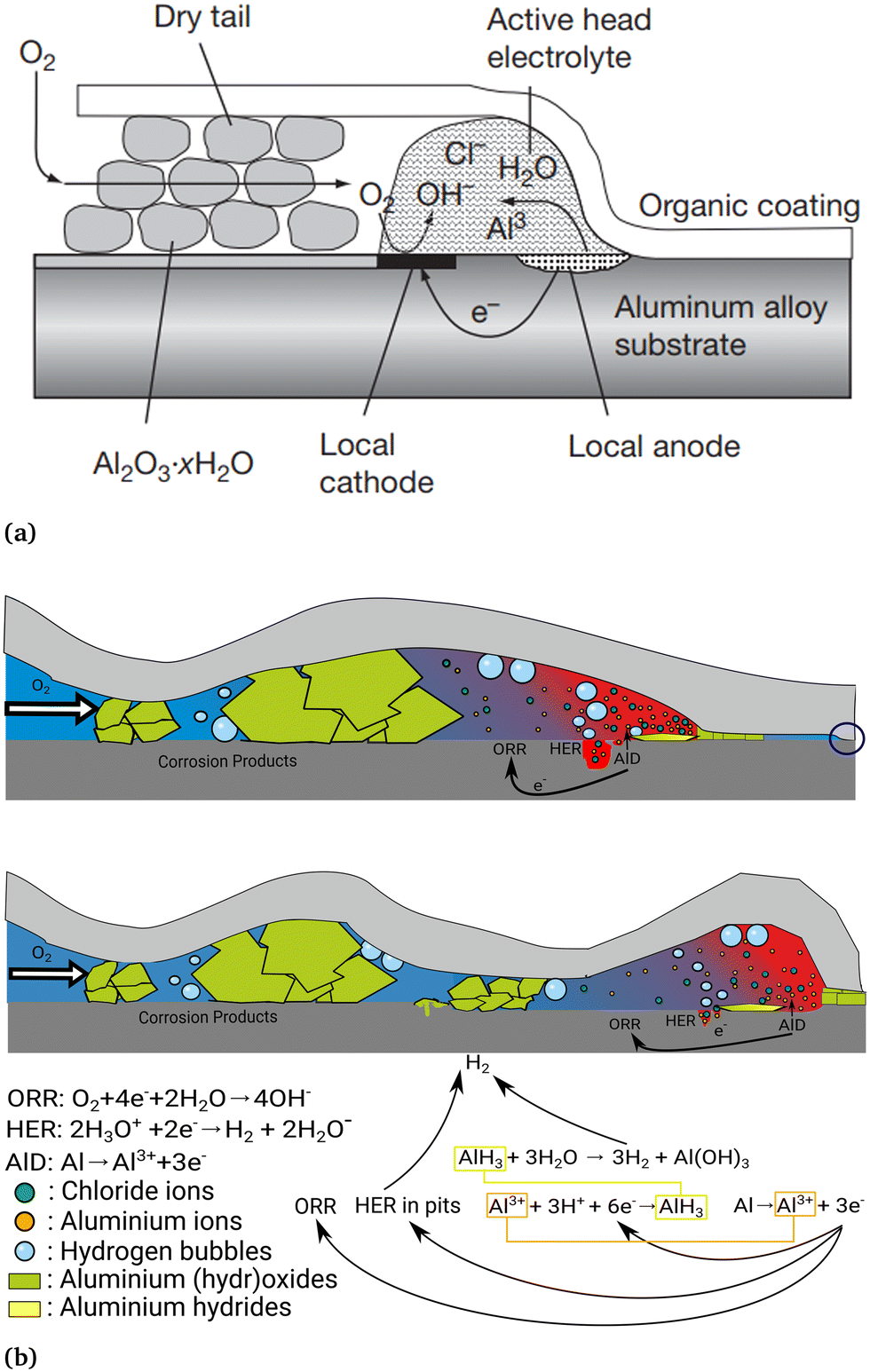 | ||
| Fig. 2 Schematic representation of the FFC mechanism on an aluminium alloy showing the different characteristic regions inside the filament: the tail of the filament where the metal dissolution takes place, the back of the head where the cathodic ORR takes place and the filament tail where the aluminium based corrosion products are deposited. (a) Schematic version, reprinted from Shreir's Corrosion, vol. 2, H.N. McMurray, G. Williams, chapter 2.14 - under film/coating corrosion, pp. 988–1004, Copyright (2010), (https://doi.org/10.1016/B978-044452787-5.00040-8) with permission from Elsevier, https://www.elsevier.com/.6 (b) A model depicting more details, including pitting and hydrogen evolution near the head.13 Adapted from ref. 13 with permission from the PCCP Owner Societies. | ||
From the current understanding of the FFC mechanism, the presence of chlorides penetrated inside a coating defect, on the one hand, deteriorates the naturally present aluminium (hydr)oxide layer and on the other hand, produces soluble species such as aluminium chlorides or oxychlorides favoring the aluminium dissolution and formation of pit-like morphologies.
Oxygen is consumed faster in the defect region far from the filament head leading to an oxygen concentration gradient inside the defect. The different oxygen concentrations lead to a difference in electrode potential; consequently, the farthest region from the coating defect acts as an anode and the closest region to the coating defect acts as a cathode. The development of a potential gradient allows the start of filament propagation. Three main regions are present inside a filament during its growth:
1. The tip of the head where an acidic environment characterized by a pH ranging from 1 to 3,14,15 with a higher concentration of chlorides (estimated to be between 0.5 and 0.96 M)14,16 compared to other parts of the filament, is the site of the anodic metal dissolution. In this region, Al3+ is generated via metal dissolution. Al3+ migrates towards the back of the head.
2. The back of the head where the oxygen supplied by diffusion through the tail is reduced to hydroxide ions. The pH can range from neutral to alkaline.15
3. The tail of the filament filled (or partially filled) with mainly aluminium-based corrosion products, but also carbonates and chlorides.13,17,18
The acidic environment at the tip of the head caused by the partial hydrolysis of aluminium ions,
| [Al(H2O)x]3+ ⇌ [Al(OH)(H2O)x−1]2+ + H+ ⇌ … | (1) |
The active head of the filament has recently been investigated in situ by Raman spectroscopy, and spectral features consistent with the presence of aluminium hydrides were found.12 This result leads to a number of questions about the involvement of hydrogen, generated either via electrochemical or non-electrochemical processes (as for example hydrolysis of aluminium hydrides), in the early stages of FFC. For similar stages in cathodic delamination, the limited availability of water was argued to lead to start–stop cycles of the progression of the disbonding process.22 Formation of aluminium hydride particles during alkaline etching was already reported previously.23,24 Hydride was also discussed as an important species to explain the corrosion potential of aluminium.25
A feature of FFC which merits discussion is the fact that despite the fact that there is an anodic region around the head, FFC usually does not propagate deep into the material. Obviously, the spatiotemporal correlation of anodic and cathodic sites does not enable sustained growth of stable pits into the material. With increasing separation of the anodic region from the corresponding cathode, resistance increases locally, leading to a permanent or temporary stop of the propagation into the material. Mechanisms of permanent, or at least sustained, deactivation of the filament will be discussed in section 2.5. Locally, the main question is whether and where novel cathodic areas become available. The surface contains IMPs, and these particles can become active cathodes. In other words, there is the competition between propagation into the material's bulk and along the interface; in the latter case, novel cathodic areas and solvents are more readily available. A consequence is the jumping movement during FFC, where IMPs have been shown to play a role, as shown schematically based on experimental data in Fig. 3.26,27 The more recent results of the role of cathodic processes in the active head are in line with this interpretation.13,19 Importantly, it is not given that pits will always deactivate. Initiation of other forms of localised corrosion has been observed in relation to FFC.28
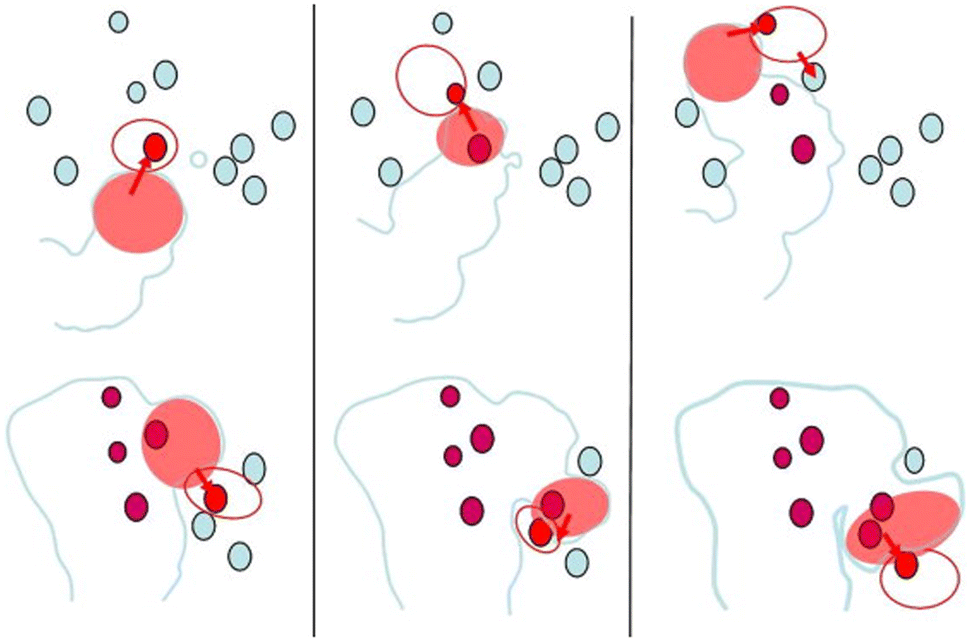 | ||
| Fig. 3 Schematic illustration of the “jumps” observed during the propagation of the active head by in situ SKPFM. The IMPs nearest to the active filament are shown in red. After incorporation into the filament, they are shown in ruby. The top sequence shows the propagation of the filament in the first 4.5 h and the bottom sequence shows the further propagation imaged in the following 17.5 h of an experiment on AA2017-T4. Reprinted from Electrochimica Acta, vol 56, Ceylan Senöz, Michael Rohwerder, “Scanning Kelvin probe force microscopy for the in situ observation of the direct interaction between active head and intermetallic particles in filiform corrosion on aluminium alloy”, 9588–9595, Copyright (2011), with permission from Elsevier.26 | ||
2.2 FFC on other material systems: magnesium and steel
For what concerns magnesium, most of the time, the term FFC has been used to indicate the type of corrosion that takes place when magnesium alloys are immersed in aqueous chloride-containing solutions.29–31 At least on some alloys, FFC also occurs when under conditions of atmospheric corrosion. The same macroscopic corrosion form is obtained as the native oxide layer can take the role of a coating. On magnesium, the HER has been noted to drive a cathodic delamination type of process; the dominance of anodically driven FFC over cathodic delamination strongly depended on the salt used for initiation.32 Detailed recent reviews on the role of FFC in magnesium and its alloys are available elsewhere;31,33–35 FFC on magnesium is thus excluded from this review unless necessary. Recent insights include specific differences between specific ions in the FFC kinetics and mechanism.36,37Different models have been proposed regarding the mechanism of FFC on iron and steel. One suggestion is that of anodic disbondment where the oxygen penetrating from the filament tail is reduced in the head where the anodic metal dissolution takes place as well.38 However, there is no general consensus regarding the location of the anode and cathode when analysing the studies over several decades. One of the first models was proposed in the 1980s.39 In this model, a small cathodic region is present in the front of the propagating filament. In this region, hydroxide ions are produced as a consequence of the ORR. This region tends to grow until it merges with the filament leading edge, i.e., the site of the anodic process. The positive ions generated by the metal dissolution tend to migrate towards the cathodic region. What was initially the cathodic region becomes in this way the new anodic edge of the filament during propagation.39 This model would be consistent with the “jumping” movement of FFC on iron.40
Ruggeri and Beck contested the previous model by arguing that if the cathodic region directs the FFC from an intact area in front of the filament head, the ion transport must occur either through the coating or an ∼35 nm thin aqueous layer present at the metal/coating interface.41 Furthermore, no experimental evidence of coating disbondment in front of the filament was observed at that time. However, experimental evidence for the existence of a cathodic area in front of the propagating filament was given by Maeda et al. based on the observation of an alkaline region at the rim of the head of a filament which was argued to stem from cathodic activity.42 The existence of cathodic regions near the head on steel was later confirmed by SKPFM.40 Cathodic activity was observed in a region extending outside the filament front suggesting the possibility of cathodic disbondment. Furthermore, a recent study on cathodic delamination shows the possibility of cation insertion at the delamination front favoured by the presence of negative charges as induced by oxygen adsorption in the same region.43 During preparation of this work, a comprehensive review on the mechanism of FFC on steel became available.44
2.3 Role of O2 supply: aluminium vs. steel
One of the most accepted hypotheses regarding the way of oxygen supply to the cathodic region is that O2 is provided by diffusion through the porous corrosion products deposited in the filament tail instead of diffusing through the coating. Experiments conducted by covering the organic coating with metallic foil to hinder possible oxygen diffusion from the head show that FFC propagation rates remain unaltered.45 However, in the mechanism of underpaint corrosion proposed for steel it is usually assumed that oxygen diffuses through the coating so the reason why for aluminium with similar coatings this shouldn't be the case is unclear.The effect of the oxygen source on the filament life time can be illustrated by a virtual experiment (Fig. 4). The left-side square in the geometry (Fig. 4a) represents the defect and a filament with a length of 100 μm. On the Al surface, three reactions may take place: Al dissolution, ORR and HER. All reactions obey the Butler–Volmer equation but may become transport-controlled. The electrolyte is considered to be saturated with oxygen as initial conditions. The top part of the defect is blocked and no oxygen can enter via the defect. However, from the polymeric coating on top of the filament, it is possible for oxygen to diffuse in. For 50 μm of the polymeric coating, the highest possible amount of oxygen flux for any polymer listed in the Polymer Handbook is calculated to be 1.2 × 10−6 mol m−2 s−1.46Fig. 4b shows the resulting Al dissolution current density as a function of time. During the early stages of the simulation, the filament shows active behaviour, but during the time the current density of Al dissolution decays to zero, the filament is no longer active. Deactivation of the filament here occurred due to lack of oxygen driving the cathodic reaction as vital part of a corrosion process. The amount of oxygen coming from the polymer in this case is not high enough to maintain the dissolution.
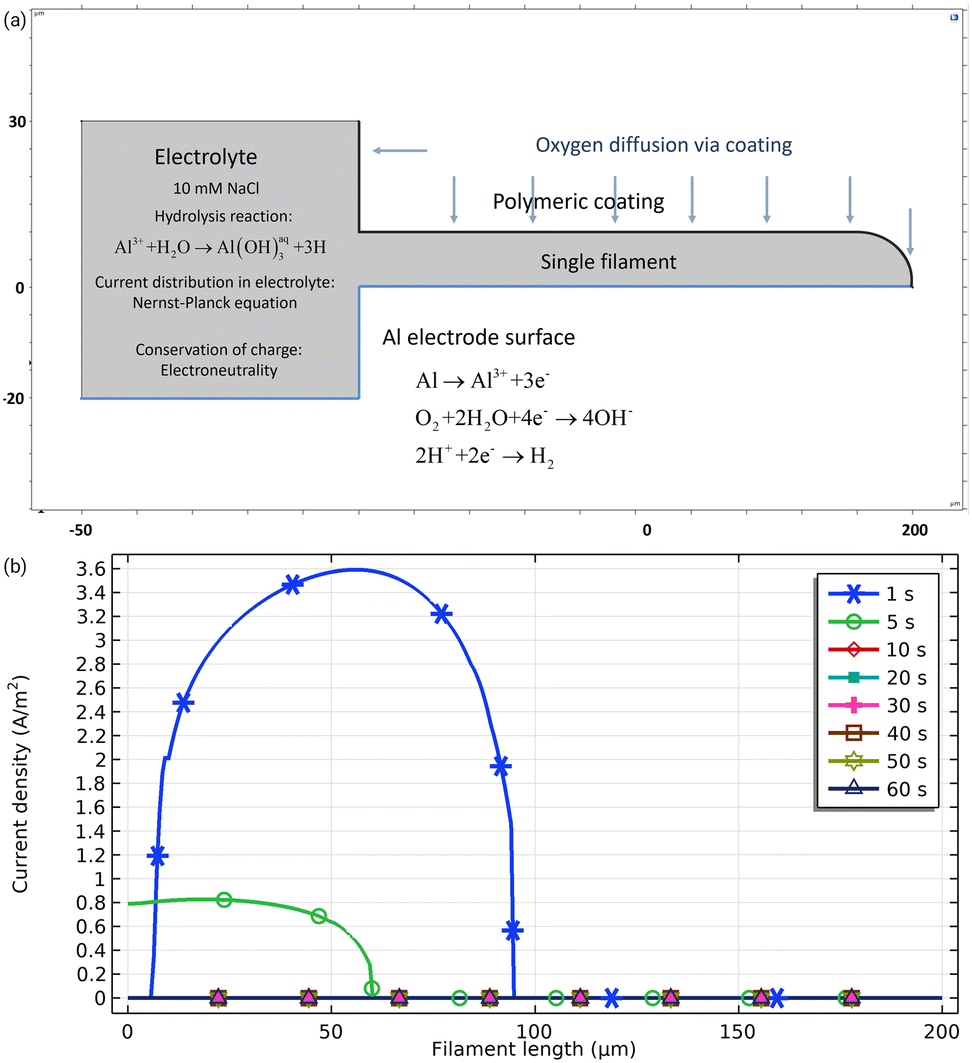 | ||
| Fig. 4 Computational experiment investigating the role of the oxygen consumption in underpaint corrosion. For the 2D geometry shown in (a), with numbers indicating dimensions in μm, the tertiary current distribution was obtained by solving the Nernst–Planck equation using the finite element method through COMSOL Multiphysics. The electrolyte is 10 mM NaCl. Convection was neglected. Electroneutrality was enforced for conservation of charge. (b) Al dissolution current density for different times after start.§ | ||
Differences between iron and aluminium could stem from the difference in the electronic structure of the oxides present on aluminium and iron. In the case of iron—which is usually covered with a low band-gap semiconducting oxide—electron transfer through the oxide is possible. Moreover, the presence of mixed oxides constituted by different valence states of iron (Fe2+ and Fe3+) facilitates both cathodic and anodic reactions as the ratio Fe2+/Fe3+ is potential dependent. If oxygen diffuses to the filament tip of the head through the polymer, the ORR is possible and can be accompanied by the anodic reaction of the iron oxide oxidation. More and more Fe2+ ions are converted to Fe3+ until the thermodynamic driving force for the electron transfer becomes so low that the ORR rate becomes negligible. In this scenario, the filament head would be polarised positively with respect to the filament tail as it is the case for cathodic delamination.4,47
On the other hand, the high band-gap oxide present on aluminium hinders the electron transfer and the ORR in a region in front of the filament tip becomes very slow. The sluggish kinetics of the ORR on the aluminium oxide in front of the tip of the filament favours instead the formation of a semi-occluded cell with a low concentration of oxygen at the tip of the head and a higher concentration of oxygen at the back of the head. Such a distribution of reactive species leads to a negative polarization of the tip of the head with respect to the back.48 Thus, for FFC on iron, the interface has an opposite polarisation compared to the polarisation found in the case of FFC on aluminium. The potential distribution profile along the filament and the different mechanisms could thus be attributed to the different semiconducting properties of the surface oxides of the different metals. For cathodic delamination, a recent study highlighted the importance of cation insertion into the intact interface,49 a step which could also play a role in FFC, and then related differences between metals to differences in chemical interactions between different oxides.
A graphic illustration of the potential distribution profile inside a filament during FFC on aluminium as calculated by numerically solving the Laplace equation¶ is shown in Fig. 5. The obtained profile is very similar to experimental profiles measured with SKP.17Fig. 5 is meant to give an indication of a possible potential distribution; the real potential distribution can depend on many factors such as sample composition, atmosphere composition and the surface pretreatment.17 In some cases, a lack of oxygen induced by saturating the atmosphere with argon leads to a flattening of the potential in the tail. Only the tip of the head remains polarised, indicating that the ORR takes place mainly in the tail region.17 In other cases, however, the filament becomes completely inactive when oxygen is removed.48 Furthermore, the potential values are also indicative. Potential values different from each other have been measured with SKP in different studies,40,48,51 evidencing one more time the complexity of the local environment present in the filament.
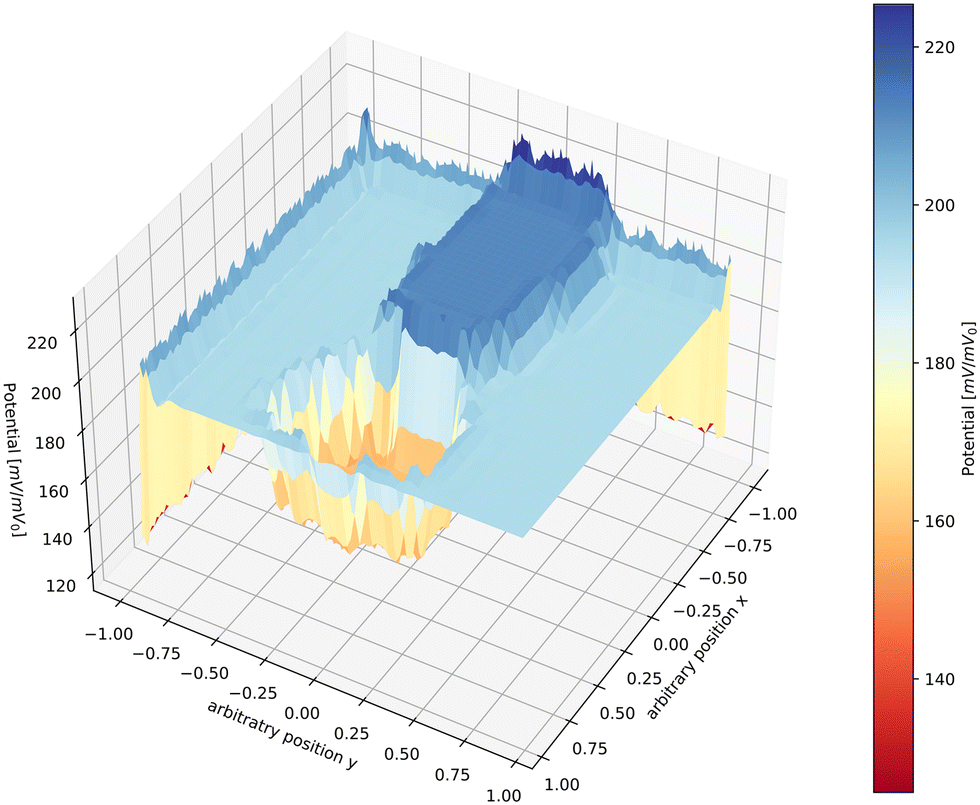 | ||
| Fig. 5 Computed potential distribution profile during FFC as calculated by solving the Laplace equation. The head of the filament has a potential that is more negative than the tail. The tail of the filament has a potential that is higher than the head and the matrix itself, indicating passivation of the tail region because of the precipitation of corrosion products.§ | ||
Following the previous considerations, the electrochemical mechanism in the case of aluminium would be more similar to what is found in the case of pitting or crevice corrosion. Both pitting and crevice corrosion are characterised by a self-accelerating propagation mechanism. The formation of an electrochemical cell is caused by the difference in oxygen concentration between the deepest part of the pit and the region close to the surface. The deepest part of the pit becomes the region where the anodic reaction of metal dissolution takes place and the walls of the pit (and eventually part of the top surface depending on the electrolyte resistance) become the region where the cathodic ORR takes place. The electric field formed in the pit induces ion migration of negatively charged ions such as Cl– towards the tip of the pit. Chloride ions undermine the oxide layer which usually passivates aluminium leading to an electrochemically more active tip of the pit.52,53 Furthermore the hydrolysis of aluminium ions, reaction (1), leads to a decrease in pH. The local acidification of the interface increases the probability of dissolution of the aluminium surface oxide, hence the process is self-accelerating. Consequently, also the acid–base properties of the surface affect pitting initiation.54 As long as the oxide dissolution and aluminium dissolution are faster than the oxide formation, the pit will continue propagating. The relationship between FFC susceptibility and pitting susceptibility has been highlighted different times.14,55–58 The correlation could be attributed to the analogous potential distribution observed in the head of an active filament and a propagating pit.
2.4 Filament environment
The composition of the inner part of the filament was studied by IR spectroscopy to characterise the formation of different chemical species both in the head and tail regions of the filament as well as to elucidate the possible propagation mechanism.17,50 The formation of complex carbonate-based polymeric species forming in the filament tail has been observed. The formation of these carbonate-based polymeric structures could incorporate chloride ions contributing to the deactivation of the filament.13,17 Furthermore, the presence of aluminium hydroxychloride was found whose hygroscopic properties determine the influence of climatic parameters such as temperature and relative humidity and the effect of wet–dry transitions on FFC.50 Notably, exact humidity has a very strong effect on the severity of the attack even within the limits of industrial testing standards.59For mill-finished non-anodised samples, the inner part of the filament tail is not always completely filled with corrosion products and there are areas still active after the passage of the corrosion front. The movement of corrosion products can take place inside the tail. This movement could be expected especially if the corrosion products are not well adherent to the surface and if the formation of gas bubbles contributes to convective motions.
The formation of hydrogen bubbles has been reported a few times in the literature but its effect on the FFC propagation rate has traditionally been neglected.14,15 However, the production of bubbles can generate convective motions which can facilitate the mixing of the acidic solution characterising the filament head and less acidic solution characterising the filament back of the head. The formation of a neutral environment resulting from the mixing of solutions with high and low pH favours the precipitation of aluminium (hydr)oxides. Indeed, more recent studies suggested a much stronger role of hydrogen evolution in FFC on magnesium alloys than previously thought.32,37 The observation of aluminium hydrides on aluminium gives the possibility for both chemical and electrochemical steps for hydrogen evolution.13 The presence of oxidation products favours the passivation of the surface and contributes to the deactivation of the filament.
The complexity of the filament's inner environment could also be responsible for the different Volta potentials measured during different times after the initiation of the filament propagation,17,27,50,60 although an effect of the technical parameters when measuring the surface potential with SKP cannot be excluded. The formation, dissolution and movement of complex species, the possibility of pit formation and the formation of gas bubbles due to the HER make the definition of polarisation regions not so obvious.13,40 The presence of pits in the filament head would for example change the potential distribution and electric field, influencing thus the direction of the ion migration. As a result, two different characteristic morphologies have been observed for FFC on steel, where mass transfer has been used to explain differences.40 The successive pitting, i.e., cyclic start of pitting initiation and stop of pitting propagation, has been used to explain a wave-like pattern observed after FFC.13
When it comes to understanding of mechanistic aspects of FFC, modern techniques may also unravel novel features. For example, acoustic emission is becoming an increasingly useful in situ technique, which can distinguish phases of the coating detachment such as tensile phases and shearing phases.61
2.5 Filament deactivation
Although not completely understood, the mechanism of filament deactivation could be similar to the deactivation mechanism of pitting. A lack of chloride ions in the head of the filament could be a reason,13,50 as chloride is needed to undermine the passive layer. The impossibility of oxygen diffusion through the filament tail leading to the zeroing of the electrochemical gradient between the anode and cathode can also be an alternative. Finally, also an increased concentration of Al3+ ions in the tip of the head would lead to a reduced potential gradient between the anode and cathode causing the deactivation of the macroscale electrochemical cell. A combination of the three cannot be excluded.SKP measurements showed that the Volta potential of the tail region becomes higher after the passage of the corrosion front indicating precipitation of corrosion products and possible surface passivation.17,47,48 The passivity of the tail region is the probable cause of the fact that the crossing of two filaments has never been observed. The possibility of an active area in a region in front of the visible filament head has been proposed and could be an explanation for the direction change of some filaments well before being close to the intersection with the tail of other filaments.62
An example of a computed surface potential distribution after filament deactivation is shown in Fig. 6. This figure shows that the potential difference between the tip of the head and the back of the head which is responsible for the filament propagation is no longer present (i.e., close to 0). The possible presence of an active region present in front of the visible filament head has been proposed,62 and the low potential region shown in Fig. 6 could not be exactly corresponding to the visible filament head but extend further in the apparently inactive surface in front.
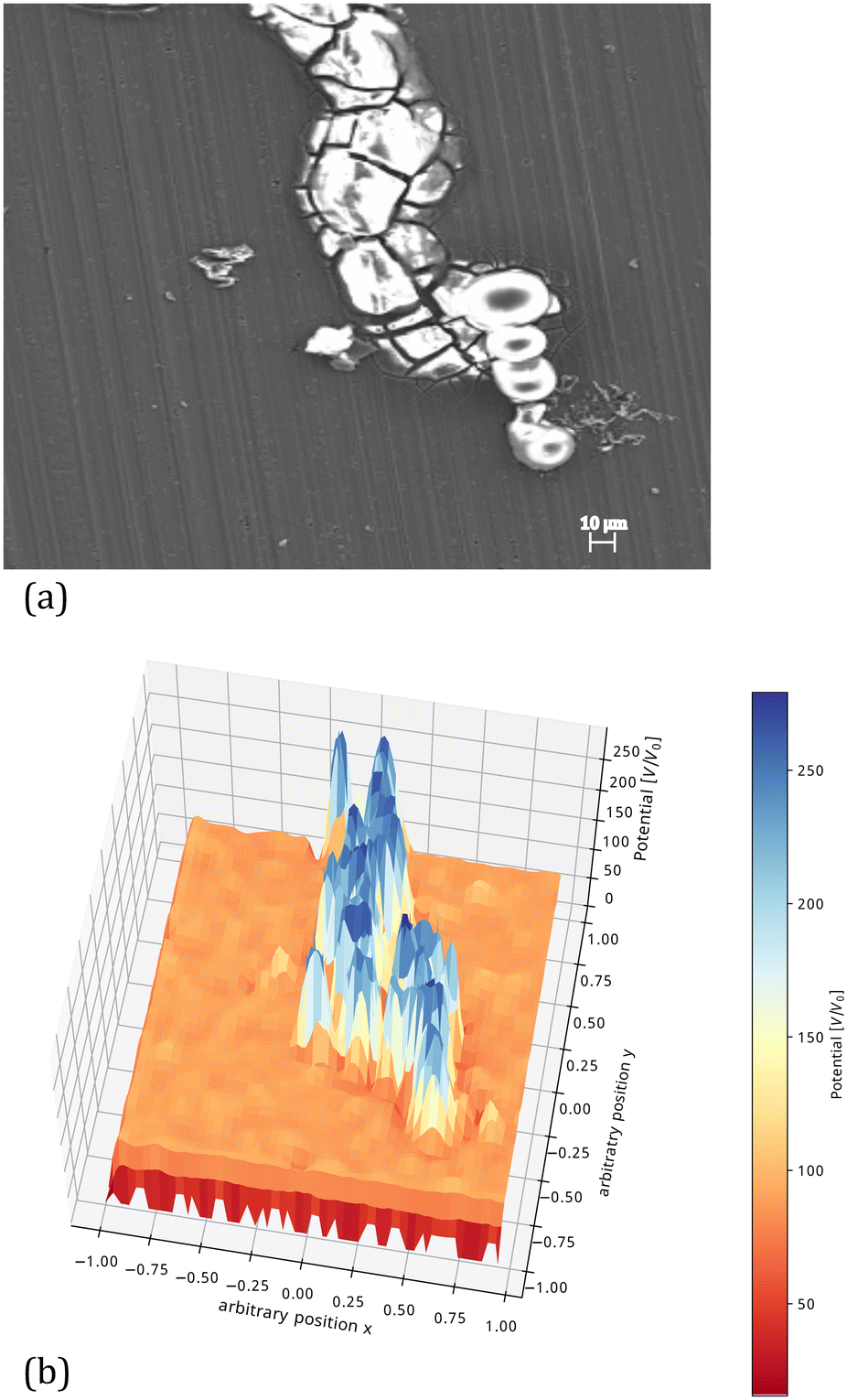 | ||
| Fig. 6 (a) SEM image of an inactive filament after coating removal. The white regions show the presence of corrosion products. (b) Computed potential distribution profile along an inactive filament calculated in the same way as that for Fig. 5.§ | ||
2.6 Differences between different alloy classes and microstructures
Filaments tend to grow following the rolling or extrusion lines. In these regions, the possible precipitation of IMPs along the elongated GBs usually offers higher potential difference between the first few hundreds of nm of the surface and the base alloy for the filament propagation.15,51 Despite the usually simplified version of the filament morphology and inner composition shown in textbooks,6,63 the morphology and especially the environment inside the filament head and tail are rather complex. Morphologies also depend on the surface treatment.Different surface treatments lead to different types of attack. On AA6111, mechanical treatment before coating with a weak polymer model coating could be used to induce different mechanisms of FFC.64 For hot-AC anodised 3005 aluminium alloys, the propagation of the filament takes place following a successive pitting type of mechanism.65 In this case, the attack is not uniformly distributed and the movement of the solution contained in the inner part of the filament diffuses towards the front through under-surface canals in the oxide layer.
Fig. 7a and b show an example of the successive pitting FFC morphology where round-shaped features are present and can be attributed to the formation of hydrogen bubbles or corrosion products that are able to lift part of the surface oxide. For mill-finished non-anodised samples of the same alloy, the attack is rather different as shown in Fig. 7c; the whole inner part of the filament is superficially attacked. However, in the center of the filament's body (area between the orange lines in Fig. 7c) the attack is deeper with respect to the sides (area between the orange and green lines on either side of the filament centre in Fig. 7c). The deeper attack is probably a result of the higher exposure time to the aggressive solution characterising the inner part of the filament during the propagation process.13 Furthermore, the passage of the corrosion front leaves behind a porous surface with a morphology that hints to dissolution of the matrix as a result of galvanic coupling with nobler IMPs (Fig. 7d). Fig. 7c evidences the presence of round shaped thin canals (some of them shown by the red arrows). The formation of these features has been ascribed to matrix dissolution due to the presence of cathodic IMPs.66,67 However, from these pictures, there doesn't seem to be a co-location of IMPs and these features. In addition, the circumference of these features is much higher than that of IMPs so their formation mechanism remains unexplored.
 | ||
| Fig. 7 (a and b): Example of FFC on a hot-AC anodised AA3005 aluminium alloy showing the surface resulting after a successive pitting-type of FFC propagation mechanism. Picture (b) is a higher magnification of the area delimited by the red rectangle in picture (a). Coating and corrosion products have been removed before taking the pictures. (c and d): Example of FFC on a non-anodised AA3005 aluminium alloy after removal of coating and corrosion products. Picture (d) is a higher magnification of a part of a different filament with respect to the one shown in (c). The red arrows in (c) indicate selected canals formed during FFC; the area between the orange boundaries presents the centre of the filament, whereas the areas between the orange and green lines on each side of the filament centre present the sides of the filament. In all the cases, FFC was induced by making an artificial defect on the coated samples and keeping the defect in contact with HCl (37%) for 1 minute before putting the samples in a humidity chamber at 40 °C and 86% relative humidity. Exposure was stopped after initiation of FFC was observed as described in ref. 13.§ | ||
3 Role of the microstructure and alloy composition in FFC on aluminium
The surface structure and composition play a pivotal role in FFC susceptibility. During thermo-mechanical processes such as extrusion or rolling, the high shear stress to which the near-surface region is subjected induces the formation of a NSDL.68–73 This layer is characterized by nano-sized grains that can extend for a few μm below the top surface. Hot and cold rolling processes can cause enough preferential surface deformation to lead to the precipitation of a high number of fine secondary IMPs. For example, for 3005 aluminium alloys, these are predominately α-(Mn,Fe)3SiAl12.72 The combination of a high density of noble Fe-rich IMPs with a reduced amount of Mn present in solid solution with respect to the underlying bulk material increases the electrochemical activity of the surface and thus the FFC susceptibility. Microgalvanic coupling between these particles and the matrix is essential and FFC does not occur on aluminium alloys in their absence.74For 6xxx aluminium alloys, analogous considerations can be made. The thermo-mechanical history and the sample composition influence the surface structure, composition and thus the FFC susceptibility. Because of the dissolution of the precipitate distribution formed during hot-extrusion, the effect of pre-extrusion heat treatments has a negligible effect on the FFC susceptibility. A post-extrusion heat treatment can however lead to the formation of a high density of fine or intermediately sized precipitates which increase the corrosion attack.56 Fe-rich noble particles in 6060 alloys are predominantly α-AlFe and MnSi phase IMPs.75 The high shear stress to which the surface is subjected during extrusion leads to (i) formation of an electrochemically active NSDL, (ii) surface enrichment of alloying elements and impurities, and (iii) precipitation of Fe-rich noble particles; although a certain susceptibility remains,76 the control of the bulk concentration of certain elements such as Si, Cu, and Pb,63 and the removal of the NSDL and Fe-rich particles improve the FFC resistance of the family of 6xxx aluminium alloys.77
With the different compositions of the different alloy classes, NSDLs will naturally differ, not only between alloy classes, but also between production processes. Table 1 gives an overview over the most important trends. Generally, the presence of a NSDL increases the susceptibility towards FFC to different degrees, implying that its removal improves FFC resistance. Naturally, the exact nature and composition depend not only on the alloy class but also on the nature of the thermomechanical processing. It is also clear that not for all possible combinations of NSDLs and coatings, it is meaningful to systematically investigate FFC susceptibility.
| Alloy | NSDL characteristics | Effect on FFC | Key ref. |
|---|---|---|---|
| a Removal of IMPs (“pure aluminium”) will remove susceptibility towards FFC.74 | |||
| 1xxx | No systematic study availablea | ||
| 2xxx | Ultrafine grains, enrichment of Mg, O, and Li | — | 78–81 |
| 3xxx | Two types of NSDLs. Fine grains decorated with oxide particles (after hot rolling) or fine grains without oxide particles (after hot rolling + cold rolling) | Severely increased susceptibility | 70, 72, 82 |
| 4xxx | No data available | ||
| 5xxx | Two types of NSDLs. Fine grains decorated with oxide particles (after hot rolling) or fine grains without oxide particles (after hot rolling + cold rolling) | Moderately increased susceptibility | 65, 71 |
| 6xxx | For an AA6111 single layer with ultrafine grains (50–150 nm diameter) induced by mechanical grinding. Enrichment of Cu, Mg, and silicon at the GBs. For AA6082, an inner layer (nm-sized grains) and an outer layer (μm-sized grains) were observed after mechanical grinding. Mg and Cu segregate at the GBs | Moderately increased susceptibility, possible combination with IGC | 64, 83, 84 |
| 7xxx | The NSDL induced by hot-rolling has a bi-layered structure with the bottom layer containing a significantly higher concentration of precipitates as Cu and Zn. A one layer structure is observed if the NSDL is induced by machining instead | Largely increased susceptibility | 85–88 |
| 8xxx | Amorphous and nanocrystalline NSDLs characterised by nanosized grains (5–50 nm) with IMPs (α-particles) and oxides decorating the GBs | Severely increased susceptibility | 89 |
Different efficient methods for selectively removing the noble element rich particles from the top surface were proposed—especially for 6060 aluminium alloys—two decades ago.74 However, in some cases, the noble particle removal does not have a strong effect on FFC since the process could still be sustained by the emergence of new IMPs when the surface is exposed to an aggressive environment as the inner part of the filament. For this reason, alternative solutions, such as chemical CCs, which can have different mechanisms of corrosion protection, have been explored.90–92
After hot and cold rolling, two different types of NSDLs can be formed.82 The first one, produced during hot rolling, is characterized by very fine grains decorated with magnesium and aluminium oxide particles, and the second type, produced during cold rolling, is characterized by a similar structure but without oxide particles. The precipitation of another type of fine dispersoid containing Mn and Fe has also been observed.93,94 In this last case, the more noble precipitated particles act cathodically and are galvanically coupled with the nano-grain bodies favoring the dissolution of the latter. However, other surface treatments such as machining induce only the formation of one type of NSDL.86
On extruded alloys, the NSDL has been less studied compared to rolled products. Nevertheless, the presence of an active layer with a detrimental effect on the FFC resistance has been demonstrated.77 The deformed microstructure can consist of sub-micron sized grains with oxide particles decorating GBs.95 The NSDL on extruded alloys is however thinner and easier to remove than the NSDL present on rolled alloys, e.g., ref. 96. It is good industrial practice to tune the production parameters such that the formation of the NSDL is minimised. For this reason, in many cases the quality of a pretreatment is judged on the basis of the amount of material that is removed by etching after extrusion. The recommended amount varies from 1 to 2 g m−2 based on the application region of a certain product.97,98
OCP transients associated with the preferential dissolution of the NSDL with respect to the base substrate and the unstable polarization method confirm an increased activity of the top part of the surface.69,99 OCP and pitting potential tend to become more positive towards the bulk.100 In general, an increased amount of IMPs combined with a reduced amount of noble elements in solid solution enhances the electrochemical activity of the NSDL, thus providing an additional driving force for the corrosion process and increasing the FFC susceptibility.14 Even though the NSDL can be removed by alkaline etching and acidic desmutting, post-production processes such as machining, grinding, and abrasion can also lead to the formation of a NSDL affecting in a negative way the FFC resistance.88,101,102 Formation of heterogeneous electrochemically active zones induced by friction stir welding is also associated with the formation of a NSDL.79
The surface activation mechanism induced by some elements is different from the commonly known galvanic coupling. Pioneering studies demonstrated that certain trace elements such as Pb and Sn can enrich at the surface and precipitate along the GBs of the NSDL during thermal treatments.103,104 For some elements, such as Pb and Sn, the activation mechanism is analogous to the “mercury drop” dissolution.105 Liquid Hg or Ga in contact with the bare aluminium surface prevents the reformation of protective aluminium oxide by hindering the passage of oxygen and water. The oxidation of the base substrate occurs first by the dissolution of aluminium in the Hg drop and then by oxidizing it at the drop/air interface. In the case of activation due to the presence of Pb and Sn, the presence of chloride ions is required.105 The surface activation induced by the presence of nano-sized grains and precipitation of noble or heavy elements in this region leads to a fast FFC propagation that is usually limited to the NSDL itself. The NSDL acts as a sacrificial anode, cathodically protecting the underlying base substrate.86
Surface activation can be obtained by heat treatment of AA3005 aluminium alloys which leads to segregation of noble elements to the surface or the depletion of, e.g., Mn from solid solution to secondary phase particles.106 An example of a polarisation curve of an alloy with surface activation induced by heat treatment and the effect of noble element concentration is shown in Fig. 8. Activation can be observed in both cases (i.e., heat treatment or noble element concentration) from an increase in anodic current with increasing temperature or noble element concentration (Fig. 8 left and right, respectively). During FFC, the passage of the corrosion front consumes the NSDL. When this active surface layer is consumed and converted into corrosion products, the possible passage of a second corrosion front has been suggested.60 In this last case, the previously formed corrosion products are either dissolved or mechanically broken and a deeper attack characterized by pitting or IGC takes place.94
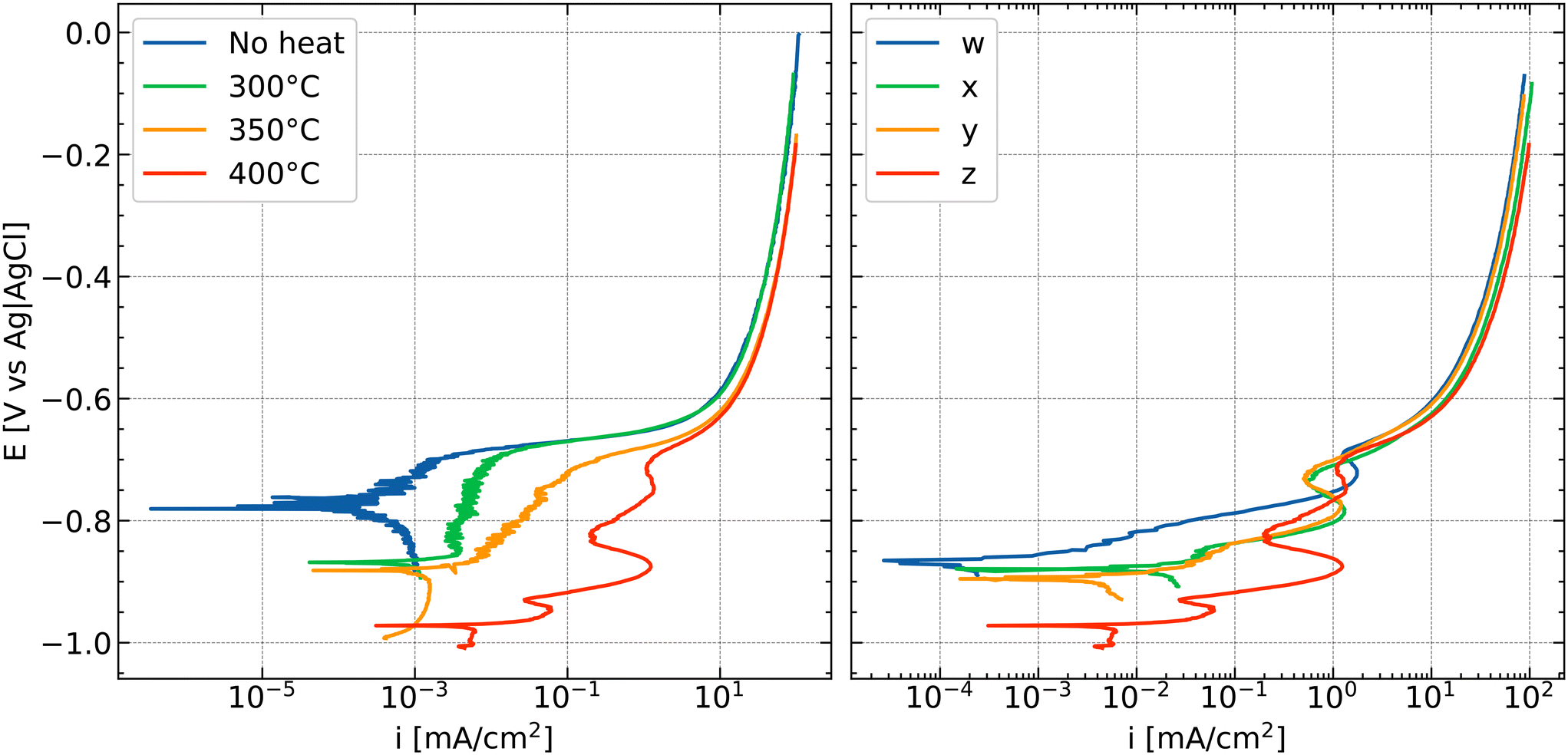 | ||
| Fig. 8 Effect of annealing temperature (left) and amount of elements (right) on anodic polarization curves of AW3005 aluminium alloys (Si 0.48–0.5; Fe 0.55–0.59; Mg 0.34–0.35 wt%; Cu, Mn, Zn varied, see below; balance Al) due to segregation of noble elements, as studied systematically.105,107 Tests were performed in a 0.86 M NaCl solution exposed to air. The numbers in the legend in the right figure represent the amount of elements in the alloy where w < x ⋍ y < z (increasing Cu and Mn concentration; w – Cu 0.15, Mn 1.04, Zn 0.09 wt%; x – Cu 0.15, Mn 1.39, Zn 0.08 wt%; y – Cu 0.22, Mn 1.08, Zn 0.14 wt%; z – Cu 0.22, Mn 1.39, Zn 0.22 wt%). Samples on the right (1 to 4) were all annealed at 400 °C. The surface activation increases with increasing the annealing temperature (left; for sample z) or the amount of alloying elements (right).§ | ||
Excursion: polarisation curves of anodically activated alloys
The anodic polarisation curves in Fig. 8 show typical examples of anodic activation. The blue curve in the left panel (no heat treatment) presents a typical anodic polarisation curve of an aluminium alloy.67–69 At a potential above the corrosion potential of ≈−0.78 V vs. Ag/AgCl/KCl(sat.), an increase in current is observed with a lot of small variations in current that may indicate metastable pitting. Above a critical potential, here ≈−0.68 V vs. Ag/AgCl/KCl(sat.), the current rapidly increases; this potential is typically identified as the critical pitting potential. At current densities above 10 mA cm−2, the increase of current with potential flattens which is usually caused by a combination of transport limitation—such as transport of dissolved species into the bulk of the electrolyte—and the effect of ohmic potential drop.After different heat treatments, the effect of anodic activation becomes apparent. In general, the corrosion potential has moved to more negative values after anodic activation. Second, the current densities in the anodic branch before reaching the pitting potential have been increased. Third, the anodic polarisation curves show “wiggles”, i.e., maxima and minima, as the red curve in the left panel of Fig. 8 illustrates as a prototypical example. Such features always indicate changes in the surface composition, resulting in a modified electrochemical response as anodic polarisation progresses. In these cases here, corrosion processes proceed at the aluminium|oxide interface, changing the active area and dissolution conditions inside the active area during recording of the polarisation curve.
Interestingly, in some cases,78,80 on AA2098-T351, the electrochemical activity of precorroded samples or samples with a NSDL can be lower than the activity of the base alloy. Both the HER and ORR can be more pronounced when corrosion products or a NSDL is not present. In the presence of corrosion products (precorroded surfaces) or in the presence of a NSDL, the surfaces can be less susceptible to severe localised corrosion.78,80 These observations seem to be partly in contradiction with what was discussed above. However, the presence of non-noble elements such as Mg in the deformed layer could contribute to the protection by galvanic coupling to the surrounding matrix.80 The system under consideration would be in the first phase of the process mentioned above and discussed in ref. 60 in which the localised corrosion process on samples that contain a NSDL or deposited corrosion products takes place after the dissolution or mechanical removal of the corrosion products. However, similarities between coated and uncoated surfaces must be considered carefully since the surface composition and electrocatalytic properties of IMPs can vary substantially between coated and uncoated systems due to the formation of unique local environments.19
4 Surface treatments are crucial to prevent FFC
Aluminium and its alloys have a stable, naturally formed oxide layer which covers the surface and guarantees good corrosion resistance. The oxide layer thickness ranges from 2–4 nm and has a high band gap (6–7 eV).108,109 However, at acidic and alkaline pH, i.e., <4 and >8, depending also on the Al3+ concentration and the presence of specific anions or possibly complex formers, the solubility of the oxide is highly increased.Alkaline etching is one of the most common pretreatments used both at the industrial scale and in lab research in order to prepare aluminium surfaces for further surface pretreatment steps such as conversion coating, anodising or painting. In combination with its impact on the further surface pretreatments, alkaline etching has a direct impact on FFC. Susceptibility to FFC can be reduced by the removal of the NSDL. When the surface active layer is removed by alkaline etching in NaOH, the resistance towards FFC increases and the morphology of the attack changes.68 When the NSDL is present, the filaments are wide and they tend to spread faster, the lifting of the coating is little, and the attack is limited to the NSDL similar to what is shown in Fig. 7 (bottom), where the NSDL was not removed before FFC testing. In contrast, after alkaline etching, FFC propagation is much slower and characterized by repeated blistering along the filament length with a more pronounced lifting of the coating.65
Although FFC resistance is improved by alkaline etching, a surface enrichment of elements such as Cu, Zn, Mn, Cr and Fe takes place during this step.12,110–114 Aluminium is highly soluble at high pH and its selective dissolution occurs during alkaline etching resulting in an increased relative concentration of elements with a nobility higher than the matrix.115 When less-noble elements are present in the alloy, these will selectively dissolve. In the case of Li for example, its preferential dissolution and incorporation into the aluminium oxide layer can also improve the Al corrosion resistance.116
In general, surface enrichment of noble elements is not desirable, which is why an acidic oxidising desmutting step is used to remove the remains of alkaline etching. The remaining IMPs with high electrocatalytic activity towards the ORR and HER enhance cathodic activity. Based on mixed potential theory,38,117,118 a faster kinetics for the HER and ORR leads to an increase in corrosion current, thus having a negative effect on localised corrosion.119 In practice, the situation is rather complex. The presence of noble elements at the metal/oxide interface and in the oxide layer is not the only important factor when it comes to FFC resistance or in more general to localised corrosion resistance. In fact, the presence of a smut layer as a result of alkaline etching can modify the interaction of the metallic surface with the CCs successively applied.120,121 The enrichment of noble elements such as Cu could on the one hand have a negative effect for the reasons mentioned above; however, on the other hand, it could also have an overall positive impact on FFC resistance if it guarantees the formation of a more homogeneous and uniform conversion layer.121,122 Furthermore, an optimal etching time can reduce the noble element surface enrichment,123 and could also be of fundamental importance in order to guarantee the formation of a highly protective anodic layer in the case of anodised samples.124
Adhesion properties of organic coatings can also be improved by an alkaline etching step. The interaction between hydrolysed organosilane polymers and oxidised aluminium surfaces of comparable roughness increases with increasing the hydroxylation of the aluminium surface.125 The degree of hydroxylation can affect the performance of both Zr and Ce-based CCs.120,126
Despite many decades of research dedicated to understanding and improving the alkaline etching process, there are still new challenges, new applications and fundamental concepts pictured only in the last few years. Challenges related to the preferential grain etching when an amount of Zn higher than 0.03 wt% is present in AA6060 aluminium alloys could limit the ambition of increasing the amount of PCS-based recycled aluminium used for alloy production.9,127,128 Fundamental understanding of the deposition mechanism and structure of the layer formed after alkaline etching shed light onto its effect on the structure of CCs,121,122,129,130 its effect on the structure and corrosion protection properties of anodic layers,124,131,132 and its effect on the enrichment of noble elements.123 In addition to the dissolution of the matrix, there is also significant dissolution of alloy elements, including noble alloy elements, during alkaline etching.12,133–135 Alkaline etching has also an effect on the efficacy of CeIII acetate used as a corrosion inhibitor in NaCl solution136 and on the deposition of CCs.122,129,131,137,138 Importantly, after applying CCs, it is not given that the cathodic activity of a surface is directly correlated to the susceptibility towards FFC.122
The behaviour of different particles and the time dependent cathodic activity of their surface during alkaline etching have also only recently been explored.113,114 Detailed mechanistic studies on the dissolution of aluminium in alkaline solution performed using a rotating cylinder electrode have been reported more than 30 years ago,139 and more details have been added through the years by employing more recently developed techniques such as atomic emission spectroscopy coupled with electrochemistry,140 dynamic electrochemical impedance spectroscopy (dEIS),141 or detailed analysis of classical electrochemical measurement combined with solution analysis.12
5 FFC of PCS-based aluminium alloys
New technologies of scrap separation make it possible to obtain aluminium with controlled composition using more than 70–80% of PCS.142,143 However, the concentration of elements such as Cu, Zn, Ni, Fe and Mn can sometimes be slightly higher than in comparative primary alloys.144 Importantly, the alloy element content can vary to a larger degree than for primary alloys. Even in the absence of a deformed layer, in some cases recycled products may have a slightly higher FFC susceptibility.122 On recycled AA5050 aluminium alloys, controlling the Fe impurities can help in increasing the FFC resistance.106 Some of the most common alloys contain elements such as Cu and Mn. Atomistic simulations and experimental data have shown that aluminium forms a more stable and protective oxide when Mn is present as an alloying element.145 In other cases, an increased amount of Mn in solid solution diminishes the potential difference between matrix and noble IMPs. The reduced potential difference reduces the probability of localised corrosion initiation.146 However, although on a microscopic scale, the increased amount of impurities increases the heterogeneity and thus changes the electrochemical properties of the surface. For certain recycled alloys, an increased amount of elements such as Pb and Cu does not necessarily result in an increased FFC susceptibility.147On 6xxx alloys, however, an effect of elements such as Cu, Si and Pb has been observed. Some sources recommend to control the concentration of these elements so that Cu <0.02 wt%, Si >0.55 wt% and Pb <0.022 wt%,63 but some of the rationale behind these numbers remains unclear and may depend on other alloy elements. There are indications that in the range of 0 to 0.1 wt%, Cu has a slightly negative effect on the FFC resistance of 6060 powder coated aluminium alloys.11 The best FFC protection is achieved for samples which have the lowest Cu surface enrichment. However, the performance difference between samples coated at different plants can be higher than the performance difference due to the increased Cu content (Fig. 9).11
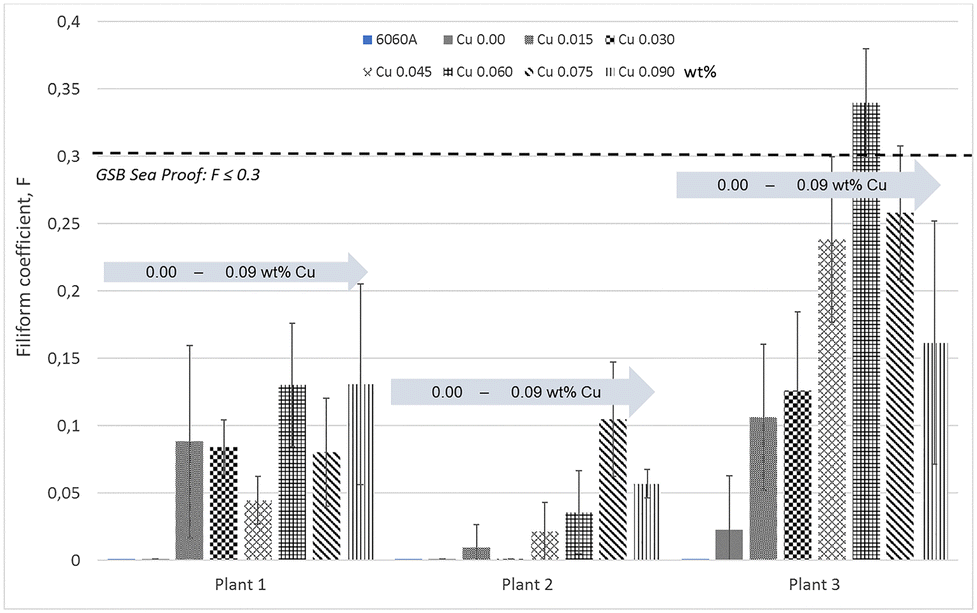 | ||
| Fig. 9 Results from industrial tests in which samples from the same batch of AA6060 have been pretreated and powder coated in different plants, and subsequently FFC-tested by the same methodology.11 The same set of samples (the same composition and thermomechanical history) containing increasing amounts of Cu (from 0 to 0.09 wt%) were pretreated and FFC-tested at three different plants. The bar charts show that the variability in performance between plants is in most of the cases higher than the variability in performance due to sample composition. Data from: Halseid et al., “The effect of trace additions of copper and pretreatment conditions on the filiform corrosion of powder-coated aluminium alloy AA6060”, Surface and Interface Analysis, 51, (2019), 1225–1230, © 2019 John Wiley & Sons, Ltd. | ||
In the study in ref. 11, the copper content in 6060 aluminium alloys was systematically varied, samples from the same batches were pretreated and powder coated at different plants, and then FFC tested with exactly the same methodology in the same chamber. Results indicate a large variability between plants; whereas some of the coatings show a performance almost independent of the Cu-content, others show an increase with the Cu content up to a certain level, and a decrease again at the highest Cu levels. The Cu surface concentration can be leveled down by a desmutting step in a concentrated nitric acid solution which can lead to a similar surface enrichment regardless of the Cu concentration in the bulk alloy.11 Consequently, details of the pretreatment process are more important than the exact alloy composition.
An indication of a negative effect of an increased amount of Cu was recently reported;10 however, also in this case, the variation in corrosion susceptibility due to trace element concentration was lower than the variation between coating plants or different pretreatment processes highlighting one more time the importance of the latter. Moreover, the type of powder coating could influence whether alloys with higher noble element concentration will be more susceptible to FFC. Overall, the role of Cu remains controversial. Although in some instances, an increased Cu concentration in the alloy leads to an increased susceptibility to FFC which can be attributed to the galvanic coupling of Cu-based noble IMPs and the aluminium matrix, in other cases Cu has no effect on the FFC susceptibility,147 or it can also be beneficial.148
Temperature also has an impact on FFC and the rate of the filament propagation increases in the range of temperatures between 25 and 50 °C. Samples with large amounts of noble elements plated on the surface, or the propensity for re-plating noble elements, have the highest increase in the filament propagation rate with temperature.16 The increase in the propagation rate depends on the kinetics of the cathodic reactions such as the HER and ORR. The presence of surface features with higher electrocatalytic activity towards the ORR will increase the total corrosion current based on the mixed potential theory,38,117,118 and consequently the FFC propagation rate. The effect of Cu is, however, not completely clear since both beneficial149 and negative150,151 effects have been reported.
In summary, in certain cases, the effect of impurities and trace elements has no major impact on the FFC performance,147 in other cases, an effect can be observed and the removal of noble surface precipitates is not sufficient to reduce the susceptibility to FFC due to the exposure of new noble IMPs during the filament propagation,74 while in other cases, the effect of the surface pretreatment overcomes the effect of the sample composition.10,11 For these reasons, in addition to considering the sample composition, a suitable surface pretreatment must be designed in order to obtain a high resistance towards FFC and possibly increase the amount of PCS used for aluminium production. The large variety of data reported motivates a deep look into mechanistic aspects of both FFC and pretreatment.
6 Conclusion
Filaments pointing to the corrosion form of FFC can be observed on a variety of different materials in a variety of different situations. On aluminium, FFC can be observed under coatings, but also in the absence of organic coatings.There is a general consensus that the active head in FFC is anodic, and the cathodic reaction tails behind; this sequence distinguishes FFC from cathodic delamination. There is an interaction of the anodic head with cathodic IMPs. The limited amount of water in the confined space under the coating gives room for unusual reaction mechanisms and processes which are more difficult to observe in free solution.
On aluminium, two types of filaments can be observed, (i) filaments as a result of a fast anodic undercutting process that can be induced, e.g., by surface segregation of low-melting point metals such as Pb or Sn at the oxide|metal interface, and (ii) filaments that undergo start–stop cycles by successive pitting. Mechanism (i) is rather fast but can be easily avoided by appropriate heat- and surface treatment. On the other hand, mechanism (ii) needs more careful tuning of surface treatment.
Due to the buried nature of the coating|oxide|metal interface which transforms during FFC, there are still a number of open questions. The role of hydrogen evolution as a driving force is not very well explored. Reasons for the development of corrosion morphologies are still to be understood in detail. Particularly challenging is an understanding of the processes during interaction in the oxide layers, including native oxides and CCs, with anionic species such as chloride during the FFC process, and the effect such an interaction has on the FFC kinetics.
Appropriate pretreatment significantly decreases the propensity of aluminium towards FFC. For PCS-based recycled aluminium with its potentially larger composition windows compared to primary produced alloys, more detailed tuning of the pretreatment parameters may be needed. However, there is no evidence for increased propensity of PCS-based recycled aluminium towards FFC as long as correct pretreatment is applied.
List of abbreviations
| AA | Aluminium alloy |
| CC | Conversion coating |
| EOL | End-of-life |
| FFC | Filiform corrosion |
| GB | Grain boundary |
| HER | Hydrogen evolution reaction |
| IGC | Intergranular corrosion |
| IMP | Intermetallic particle |
| NSDL | Near-surface deformed layer |
| OCP | Open circuit potential |
| ORR | Oxygen reduction reaction |
| PCS | Post-consumer scrap |
| SKP(FM) | Scanning Kelvin probe (force microscopy) |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
Related research to this work at the author's lab was co-funded by aluminium companies Speira and Hydro.Acknowledgements
We thank Otto Lunder, Jan Halvor Nordlien, Jan Tore Buvik Gundersen, Mattijs ten Kate, Malgorzata Halseid and Kemal Nisancioglu for helpful discussions. Yukihiro Takahashi is acknowledged for making ref. 42 accessible to us. This work was supported by the Research Council of Norway (NFR) through Innovation Project for the Industrial Sector CORAL (grant number 309875), in collaboration with industrial partners Hydro and Speira.References
- G. Koch, J. Varney, N. Thompson, O. Moghissi, M. Gould and J. Payer, International Measures of Prevention, Application, and Economics of Corrosion Technologies Study, NACE International, Houston, TX, USA, 2016 Search PubMed.
- O. Ø. Knudsen and A. Forsgren, in Corrosion Control Through Organic Coatings, CRC Press, Boca Rayton, USA, 2nd edn, 2017, ch. Protection Mechanisms of Organic Coatings Search PubMed.
- G. Grundmeier and A. Simões, in Encyclopedia of Electrochemistry, ed. A. Bard, M. Stratmann and G. Frankel, Wiley-VCH, Weinheim, Germany, 2007, vol 4, ch. Corrosion Protection by Organic Coatings, pp. 500–566 Search PubMed.
- C. D. Fernández-Solis, A. Vimalanandan, A. Altin, J. S. Mondragón-Ochoa, K. Kreth, P. Keil and A. Erbe, Soft Matter at Aqueous Interfaces, Springer, Cham, Switzerland, 2016, vol 917, pp. 29–70 Search PubMed.
- J. Tang, 19th Asia Pacific Corrosion Control Conference, Guangzhou, China, 2023.
- H. N. McMurray and G. Williams, in Shreir's Corrosion, Elsevier, Oxford, 2010, ch. 2.14 - Under Film/Coating Corrosion, pp. 988–1004 Search PubMed.
- C. F. Sharman, Nature, 1944, 153, 621–622 CrossRef CAS.
- F. Cavezza, M. Boehm, H. Terryn and T. Hauffman, Metals, 2020, 10, 730 CrossRef.
- A. Lutz, L. Malet, J. Dille, L. H. de Almeida, L. Lapeire, K. Verbeken, S. Godet, H. Terryn and I. De Graeve, J. Alloys Compd., 2019, 794, 435–442 CrossRef CAS.
- A. Lutz, M. C. Halseid and I. D. Graeve, Mater. Corros., 2022, 73, 1575–1585 CrossRef CAS.
- M. Halseid, J. T. B. Gundersen, Ø. Bauger and T. Hentschel, Surf. Interface Anal., 2019, 51, 1225–1230 CrossRef CAS.
- E. Mysliu, K. S. Storli, E. Kjørsvik, O. Lunder and A. Erbe, J. Electrochem. Soc., 2023, 170, 011503 CrossRef CAS.
- E. Mysliu, O. Lunder and A. Erbe, Phys. Chem. Chem. Phys., 2023, 25, 11845–11857 RSC.
- J. M. C. Mol, B. R. W. Hinton, D. H. Van Der Weijde, J. H. W. De Wit and S. Van Der Zwaag, Corros. Sci., 2000, 35, 1629–1639 CAS.
- A. Bautista, Prog. Org. Coat., 1996, 28, 49–58 CrossRef CAS.
- D. A. Little, M. A. Jakab and J. R. Scully, Corrosion, 2006, 62, 300–315 CrossRef CAS.
- N. Le Bozec, D. Persson, A. Nazarov and D. Thierry, J. Electrochem. Soc., 2002, 149, B403 CrossRef CAS.
- R. Szymanski, D. Jamieson, A. Hughes, A. Mol, S. van der Zwaag and C. Ryan, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 190, 365–369 CrossRef CAS.
- A. Almalla, O. Ozcan and J. Witt, Adv. Eng. Mater., 2022, 24, 2101342 CrossRef CAS.
- C. Gu, J. Hu and X. Zhong, Prog. Org. Coat., 2020, 147, 105774 CrossRef CAS.
- G. Williams and H. N. McMurray, J. Electrochem. Soc., 2003, 150, B380 CrossRef CAS.
- D. Iqbal, A. Sarfraz, M. Stratmann and A. Erbe, Chem. Commun., 2015, 51, 16041–16044 RSC.
- S. Adhikari, J. Lee and K. R. Hebert, J. Electrochem. Soc., 2008, 155, C16 CrossRef CAS.
- S. Adhikari and K. R. Hebert, J. Electrochem. Soc., 2008, 155, C189 CrossRef CAS.
- G. G. Perrault, J. Electrochem. Soc., 1979, 126, 199–204 CrossRef CAS.
- C. Senöz and M. Rohwerder, Electrochim. Acta, 2011, 56, 9588–9595 CrossRef.
- C. Senöz, S. Borodin, M. Stratmann and M. Rohwerder, Corros. Sci., 2012, 58, 307–314 CrossRef.
- J.-O. Nilsson, Proceedings of Eurocorr 2019, Madrid, Spain, 2019, p. 222366.
- O. Lunder, J. E. Lein, S. M. Hesjevik, T. K. Aune and K. Nişancioǧlu, Mater. Corros., 1994, 45, 331–340 CrossRef CAS.
- P. Schmutz, V. Guillaumin, R. S. Lillard, J. A. Lillard and G. S. Frankel, J. Electrochem. Soc., 2003, 150, B99 CrossRef CAS.
- M. Esmaily, J. Svensson, S. Fajardo, N. Birbilis, G. Frankel, S. Virtanen, R. Arrabal, S. Thomas and L. Johansson, Prog. Mater. Sci., 2017, 89, 92–193 CrossRef CAS.
- G. Williams, C. Kousis, N. McMurray and P. Keil, npj Mater. Degrad., 2019, 3, 41 CrossRef.
- E. Ghali, W. Dietzel and K.-U. Kainer, J. Mater. Eng. Perform., 2013, 22, 2875–2891 CrossRef CAS.
- L. Wang, J. He, J. Yu, S. Arthanari, H. Lee, H. Zhang, L. Lu, G. Huang, B. Xing, H. Wang and K.-S. Shin, Materials, 2022, 15, 6197 CrossRef CAS PubMed.
- A. Atrens, G.-L. Song, Z. Shi, A. Soltan, S. Johnston and M. Dargusch, Encyclopedia of Interfacial Chemistry, Elsevier, Oxford, 2018, pp. 515–534 Search PubMed.
- C. Kousis, N. McMurray, P. Keil and G. Williams, Corrosion, 2020, 77, 156–167 CrossRef PubMed.
- C. Kousis, P. Keil, N. M. Hamilton and G. Williams, Corros. Sci., 2022, 206, 110477 CrossRef CAS.
- P. Pedeferri, Corrosione e protezione dei materiali metallici, Polipress, Milano, Italy, 2010 Search PubMed.
- W. Funke, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 343–347 CrossRef CAS.
- P. P. Leblanc and G. S. Frankel, J. Electrochem. Soc., 2004, 151, B105 CrossRef CAS.
- R. T. Ruggeri and T. R. Beck, Corrosion, 1983, 39, 452–465 CrossRef CAS.
- S. Maeda, T. Hayashi, K. Yamamoto and T. Tanaka, Proc. 56th Met. Finish. Conf. Jpn., 1977, pp. 102–103.
- J. M. Prabhakar, P. Kerger, A. de Vooys and M. Rohwerder, Corros. Sci., 2022, 199, 110185 CrossRef CAS.
- A. Cristoforetti, S. Rossi, F. Deflorian and M. Fedel, Prog. Org. Coat., 2024, 192, 108469 Search PubMed.
- H. Lenderik, PhD thesis, Delft University of Technology, Delft, The Netherlands, 2018 Search PubMed.
- J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer handbook, Wiley, New York, 1999, vol 89 Search PubMed.
- G. Grundmeier, W. Schmidt and M. Stratmann, Electrochim. Acta, 2000, 45, 2515–2533 CrossRef CAS.
- W. Schmidt and M. Stratmann, Corros. Sci., 1998, 40, 1441–1443 CrossRef CAS.
- N. Khayatan and M. Rohwerder, Corros. Sci., 2022, 202, 110311 CrossRef CAS.
- N. Le Bozec, D. Persson and D. Thierry, J. Electrochem. Soc., 2004, 151, B440 CrossRef CAS.
- G. Williams, H. N. McMurray, D. Hayman and P. C. Morgan, PhysChemComm, 2001, 4, 26–31 RSC.
- K. Nişancioǧlu and H. Holtan, Mater. Corros., 1979, 30, 105–113 CrossRef.
- K. Nisancioglu and H. Holtan, Corros. Sci., 1978, 18, 1011–1023 CrossRef CAS.
- M. A. Petrunin, L. B. Maksaeva and T. A. Yurasova, Corros. Rev., 2023, 41, 515–535 CrossRef CAS.
- V. Poulain, J. P. Petitjean, E. Dumont and B. Dugnoille, Electrochim. Acta, 1996, 41, 1223–1231 CrossRef CAS.
- J. M. C. Mol, J. van de Langkruis, J. H. W. de Wit and S. van der Zwaag, Corros. Sci., 2005, 47, 2711–2730 CrossRef CAS.
- M. Fedel, C. Zanella, L. Ferrari and F. Deflorian, Electrochim. Acta, 2021, 381, 138288 CrossRef CAS.
- E. Mysliu, T. Holm, M. ten Cate and A. Erbe, ChemElectroChem, 2023, 10, e202300374 CrossRef CAS.
- M. Asendorf and P. Plagemann, JOT J. fuer Oberflaechentechnik, 2020, 60, 7–9 Search PubMed.
- H. N. McMurray, A. J. Coleman, G. Williams, A. Afseth and G. M. Scamans, J. Electrochem. Soc., 2007, 154, C339 CrossRef CAS.
- C. Abarkane, A. Florez-Tapia, J. Odriozola, A. Artetxe, M. Lekka, E. García-Lecina, H.-J. Grande and J. Vega, Corros. Sci., 2023, 214, 110964 CrossRef CAS.
- J. L. Delplancke, S. Berger, X. Lefèbvre, D. Maetens, A. Pourbaix and N. Heymans, Prog. Org. Coat., 2001, 43, 64–74 CrossRef CAS.
- C. Vargel, Corrosion of Aluminium, Elsevier, Amsterdam, 2nd edn, 2020, pp. 247–265 Search PubMed.
- H. McMurray, A. Holder, G. Williams, G. Scamans and A. Coleman, Electrochim. Acta, 2010, 55, 7843–7852 CrossRef CAS.
- X. Zhou, G. E. Thompson and G. M. Scamans, Corros. Sci., 2003, 45, 1767–1777 CrossRef CAS.
- J. V. Kloet, W. Schmidt, A. W. Hassel and M. Stratmann, Electrochim. Acta, 2003, 48, 1211–1222 CrossRef.
- H. Leth-Olsen, PhD thesis, NTNU, Norwegian University of Science and Technology, Trondheim, Norway, 1996 Search PubMed.
- H. Leth-Olsen and K. Nisancioglu, Corros. Sci., 1998, 40, 1179–1194 CrossRef CAS.
- H. Leth-Olsen, A. Afseth and K. Nisancioglu, Corros. Sci., 1998, 40, 1195–1214 CrossRef CAS.
- A. Afseth, J. H. Nordlien, G. M. Scamans and K. Nisancioglu, Corros. Sci., 2001, 43, 2093–2109 CrossRef CAS.
- A. Afseth, J. H. Nordlien, G. M. Scamans and K. Nisancioglu, Corros. Sci., 2001, 43, 2359–2377 CrossRef CAS.
- A. Afseth, J. H. Nordlien, G. M. Scamans and K. Nisancioglu, Corros. Sci., 2002, 44, 2491–2506 CrossRef CAS.
- A. Afseth, J. H. Nordlien, G. M. Scamans and K. Nisancioglu, Corros. Sci., 2002, 44, 2529–2542 CrossRef CAS.
- O. Lunder, K. F. Heen and K. Nisancioglu, Corrosion, 2004, 60, 622–631 CrossRef CAS.
- L. F. Mondolfo, Aluminum Alloys, Butterworth-Heinemann, 1976, pp. 661–663 Search PubMed.
- O. Lunder, B. Olsen and K. Nisancioglu, Int. J. Adhes. Adhes., 2002, 22, 143–150 CrossRef CAS.
- J. H. Nordlien, J. Defrancq, W. Züst, M. Benmalek and R. Stuckart, Mater. Corros., 2000, 51, 473–480 CrossRef CAS.
- R. M. P. da Silva, M. X. Milagre, L. A. de Oliveira, U. Donatus, R. A. Antunes and I. Costa, Surf. Interface Anal., 2019, 51, 982–992 CrossRef CAS.
- R. M. P. da Silva, J. Izquierdo, M. X. Milagre, J. V. de S. Araujo, R. A. Antunes, R. M. Souto and I. Costa, Electrochim. Acta, 2022, 427, 140873 CrossRef CAS.
- R. M. P. da Silva, M. X. Milagre, J. Izquierdo, A. M. Betancor-Abreu, L. A. de Oliveira, J. V. de S. Araujo, R. A. Antunes, R. M. Souto and I. Costa, Mater. Charact., 2022, 191, 112130 CrossRef CAS.
- U. Donatus, R. E. Klumpp, N. V. V. Mogili, R. Altobelli Antunes, M. X. Milagre and I. Costa, Surf. Interface Anal., 2019, 51, 275–289 CrossRef CAS.
- X. Zhou, Y. Liu, G. E. Thompson, G. M. Scamans, P. Skeldon and J. A. Hunter, Metall. Mater. Trans. A, 2011, 42, 1373–1385 CrossRef CAS.
- Y. Liu, X. Zhou, G. Thompson, T. Hashimoto, G. Scamans and A. Afseth, Acta Mater., 2007, 55, 353–360 CrossRef CAS.
- X. Meng, Y. Li, Y. Lv, Z. Dong and X. Zhang, Corros. Sci., 2024, 230, 111907 CrossRef CAS.
- E. Liu, Q. Pan, B. Liu, J. Ye and W. Wang, Materials, 2023, 16, 1–18 Search PubMed.
- B. Liu, X. Zhang, X. Zhou, T. Hashimoto and J. Wang, Corros. Sci., 2017, 126, 265–271 CrossRef CAS.
- B. Liu, X. Zhou and X. Zhang, Mater. Charact., 2017, 123, 91–98 CrossRef CAS.
- B. Liu, X. r. Zhou and X. x. Zhang, Trans. Nonferrous Met. Soc. China, 2020, 30, 2056–2066 CrossRef CAS.
- T. Thomé, S. Martinez, C. Czechowski, L. Thomé, J. P. Debove and S. Benchabane, Surf. Interface Anal., 2014, 46, 157–163 CrossRef.
- F. Mansfeld, S. Lin, S. Kim and H. Shih, Corrosion, 1989, 45, 615–630 CrossRef CAS.
- A. J. Davenport, H. S. Isaacs and M. W. Kendig, Corros. Sci., 1991, 32, 653–663 CrossRef CAS.
- H. A. Katzman, G. M. Malouf, R. Bauer and G. W. Stupian, Appl. Surf. Sci., 1979, 2, 416–432 CrossRef CAS.
- A. Cassell, G. E. Thompson, X. Zhou, A. Afseth, H. Dunlop, M.-A. Kulas, L. Peguet and G. Scamans, Surf. Interface Anal., 2013, 45, 1604–1609 CrossRef CAS.
- Y. Liu, T. Hashimoto, X. Zhou, G. E. Thompson, G. M. Scamans, W. M. Rainforth and J. A. Hunter, Surf. Interface Anal., 2013, 45, 1553–1557 CrossRef CAS.
- J. Wang, M. Clinch, G. Scamans, C. Pargeter and X. Zhou, Aluminium Surface Science & Technology Symposium, 2023.
- F. Andreatta, A. Lanzutti, S. Maschio and L. Fedrizzi, Surf. Interface Anal., 2019, 51, 1240–1250 CrossRef CAS.
- GSB International, International Quality Regulations for the Coating of Aluminium Building Components, 2017, https://gsb-international.de/en/quality-regulations/, accessed 2023-07-03.
- Qualicoat, Qualicoat Specifications 2021, Qualicoat technical report, 2021.
- M. Huisert, PhD thesis, Delft University of Technology, Delft, The Netherlands, 2001 Search PubMed.
- P. Premendra, J. H. W. De Wit, L. Katgerman and H. Terryn, Innovative Pre-Treatment Techniques to Prevent Corrosion of Metallic Surfaces, Woodhead Publishing, 2007, pp. 71–82 Search PubMed.
- M. D. Goodall, S. Pawar, M. Curioni, S. Morsch, M. G. Unthank, S. R. Gibbon and X. Zhou, Corros. Eng., Sci. Technol., 2022, 57, 97–104 CrossRef CAS.
- S. S. Wang, D. Huber, J. D. Poplawsky, H. Colijn and G. S. Frankel, Materialia, 2021, 16, 101065 CrossRef CAS.
- Y. W. Keuong, J. H. Nordlien, S. Ono and K. Nisancioglu, J. Electrochem. Soc., 2003, 150, B547 CrossRef CAS.
- B. Graver, A. M. Pedersen and K. Nisancioglu, ECS Trans., 2009, 16, 55 CrossRef CAS.
- K. Kurt, S. Diplas, J. C. Walmsley and K. Nisancioglu, J. Electrochem. Soc., 2013, 160, C542–C552 CrossRef CAS.
- P. Premendra, W. Loven, H. Terryn, J. de Wit and L. Katgerman, Aluminium Alloys 2006 - ICAA10, 2006, pp. 687–692 Search PubMed.
- Y. Yu, O. Sævik, J. H. Nordlien and K. Nisancioglu, J. Electrochem. Soc., 2005, 152, B327 CrossRef CAS.
- E. O. Filatova and A. S. Konashuk, J. Phys. Chem. C, 2015, 119, 20755–20761 CrossRef CAS.
- T. T. Song, M. Yang, J. W. Chai, M. Callsen, J. Zhou, T. Yang, Z. Zhang, J. S. Pan, D. Z. Chi, Y. P. Feng and S. J. Wang, Sci. Rep., 2016, 6, 29221 CrossRef CAS PubMed.
- C. E. Moffitt, D. M. Wieliczka and H. K. Yasuda, Surf. Coat. Technol., 2001, 137, 188–196 CrossRef CAS.
- Y. Ma, X. Zhou, G. Thompson and P. Skeldon, Corros. Sci., 2013, 66, 292–299 CrossRef CAS.
- Z. Jin, C. Cai, T. Hashimoto, D. K. Yudie Yuan, J. Hunter and X. Zhou, J. Alloys Compd., 2020, 842, 155834 CrossRef CAS.
- Z. Jin, C. Cai, T. Hashimoto, D. K. Y. Yuan, J. Hunter and X. Zhou, Corros. Sci., 2021, 179, 109134 CrossRef CAS.
- Z. Jin, C. Cai, Y. Yuan, D. Kang, J. Hunter and X. Zhou, Mater. Charact., 2021, 171, 110768 CrossRef CAS.
- O. Gharbi, N. Birbilis and K. Ogle, J. Electrochem. Soc., 2016, 163, C240 CrossRef CAS.
- O. Gharbi, N. Birbilis and K. Ogle, Electrochim. Acta, 2017, 243, 207–219 CrossRef CAS.
- C. Wagner and W. Traud, Z. Elektrochem. Angew. Phys. Chem., 1938, 44, 391–402 CrossRef CAS.
- C. Wagner and W. Traud, Corrosion, 2006, 62, 844–855 CrossRef CAS.
- O. Lunder and K. Nisancioglu, Corrosion, 1988, 44, 414–422 CrossRef CAS.
- J. Cerezo, P. Taheri, I. Vandendael, R. Posner, K. Lill, J. H. W. de Wit, J. M. C. Mol and H. Terryn, Surf. Coat. Technol., 2014, 254, 277–283 CrossRef CAS.
- C. F. Glover, M. L. C. Lim and J. R. Scully, Corrosion, 2020, 77, 40–52 CrossRef PubMed.
- E. Mysliu, K. Sletteberg Storli, H. M. Skogøy, S. Kubowicz, I.-H. Svenum, O. Lunder and A. Erbe, Electrochim. Acta, 2024, 477, 143805 CrossRef CAS.
- S. Chankitmunkong, D. Eskin, C. Limmaneevichitr, N. Kengkla and O. Diewwanit, Metals, 2022, 12, 1–19 CrossRef.
- H. Shi, M. Yu, J. Liu, G. Rong, R. Du, J. Wang and S. Li, Corros. Sci., 2020, 169, 108642 CrossRef CAS.
- M. Kono, X. Sun, R. Li, K. C. Wong, K. A. R. Mitchell and T. Foster, Surf. Rev. Lett., 2001, 08, 43–50 CrossRef CAS.
- A. S. Hamdy, A. M. Beccaria and P. Traverso, Surf. Interface Anal., 2002, 34, 171–175 CrossRef CAS.
- A. Lutz, L. Lapeire, T. Nguyen-Minh, K. Verbeken, H. Terryn and I. De Graeve, Surf. Interface Anal., 2019, 51, 1251–1259 CrossRef CAS.
- B. Holme, N. Ljones, A. Bakken, O. Lunder, J. E. Lein, L. Vines, T. Hauge, O. Bauger and K. Nisancioglu, J. Electrochem. Soc., 2010, 157, C424 CrossRef CAS.
- M. Li, S. Zanna, A. Seyeux, F. Wiame, P. Marcus and J. Światowska, J. Electrochem. Soc., 2021, 168, 041504 CrossRef CAS.
- H. Zhu, JOM, 2014, 66, 2222–2228 CrossRef CAS.
- A. S. Hamdy, I. Doench and H. Möhwald, J. Mater. Sci., 2012, 47, 3784–3792 CrossRef CAS.
- Y. Ma, X. Zhou, G. E. Thompson, X. Zhang, C. Luo, M. Curioni and H. Liu, J. Electrochem. Soc., 2013, 160, C111–C118 CrossRef CAS.
- R. G. Buchheit, R. P. Grant, P. F. Hlava, B. Mckenzie and G. L. Zender, J. Electrochem. Soc., 1997, 144, 2621–2628 CrossRef CAS.
- R. G. Buchheit, M. A. Martinez and L. P. Montes, J. Electrochem. Soc., 2000, 147, 119–124 CrossRef CAS.
- R. Buchheit, Mater. Sci. Forum, 2000, 331-337, 1641–1646 CAS.
- I. Milošev and P. Rodič, J. Electrochem. Soc., 2022, 169, 011504 CrossRef.
- J. Qi, A. Němcová, J. Walton, X. Zhou, P. Skeldon and G. Thompson, Thin Solid Films, 2016, 616, 270–278 CrossRef CAS.
- A. Sarfraz, R. Posner, M. Lange, K. Lill and A. Erbe, J. Electrochem. Soc., 2014, 161, C509–C516 CrossRef.
- D. Chu and R. F. Savinell, Electrochim. Acta, 1991, 36, 1631–1638 CrossRef CAS.
- J. Han, V. Vivier and K. Ogle, npj Mater. Degrad., 2020, 4, 1–10 CrossRef.
- J. Wysocka, S. Krakowiak, J. Ryl and K. Darowicki, J. Electrochem. Soc., 2016, 778, 126–136 CAS.
- S. Tjøtta, L. Dardinier and B. Kurth, Alum. Extrus. Finish, 2020, pp. 18–24 Search PubMed.
- S. Tjøtta, L. Dardinier, G. Rombach and R. Scharf-Bergmann, Proceedings of the Twelfth International Aluminium Extrusion Technology Seminar & Exposition, Wauconda, IL, USA, 2022, p. MI39 Search PubMed.
- D. Raabe, D. Ponge, P. J. Uggowitzer, M. Roscher, M. Paolantonio, C. Liu, H. Antrekowitsch, E. Kozeschnik, D. Seidmann, B. Gault, F. De Geuser, A. Deschamps, C. Hutchinson, C. Liu, Z. Li, P. Prangnell, J. Robson, P. Shanthraj, S. Vakili, C. Sinclair, L. Bourgeois and S. Pogatscher, Prog. Mater. Sci., 2022, 128, 100947 CrossRef CAS.
- J. Chen, J. Xiao, J. Poplawsky, F. M. Michel, C. Deng and W. Cai, Corros. Sci., 2020, 173, 108749 CrossRef CAS.
- M. Zamin, Corrosion, 1981, 37, 627–632 CrossRef CAS.
- P. Premendra, H. Terryn, J. M. C. Mol, J. H. W. de Wit and L. Katgerman, Mater. Corros., 2009, 60, 399–406 CrossRef CAS.
- D. Arango and J. Liu, Can. Metall. Q., 2022, 63, 84–96 CrossRef.
- S. Adhikari, K. A. Unocic, Y. Zhai, G. S. Frankel, J. Zimmerman and W. Fristad, Electrochim. Acta, 2011, 56, 1912–1924 CrossRef CAS.
- F. Andreatta, A. Lanzutti, L. Paussa and L. Fedrizzi, Prog. Org. Coat., 2014, 77, 2107–2115 CrossRef CAS.
- T. Lostak, S. Krebs, A. Maljusch, T. Gothe, M. Giza, M. Kimpel, J. Flock and S. Schulz, Electrochim. Acta, 2013, 112, 14–23 CrossRef CAS.
Footnotes |
| † Present address: SINTEF Industry, 7465 Trondheim, Norway. |
| ‡ Present address: ArcelorMittal Global R&D/OCAS NV, Ghent, Belgium. |
| § As several other figures in this work, this figure illustrates a typical case recorded in our lab. Even though this is a review article, we use our own data to illustrate typical, well-known phenomena. |
¶ The discrete Laplace equation 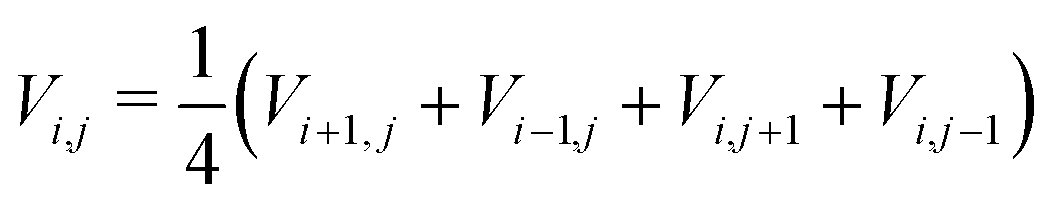 —which is a property of the true solution of the Laplace equation—was solved on a grid of points for the electrical potential V. At the boundaries along the x and y directions, the potential is set to 0. The image of the filament was drawn in gray scale using a graphic editor and imported as an array of numbers using Python. The approach assumes a potential distribution which is mirrored in corrosion product distribution. The geometry of the filament was enforced based on literature reports and own observations. The array of grayscale values was directly translated to potential values by considering all the grayscale values >95% of the maximum value present in the array fixed at a potential value H when originally imported. Similarly, all the potentials <5% of the minimum grayscale value present in the array were considered fixed at a potential value L when originally imported. At the edges, the potential was fixed to 0. Everywhere else, the initial potential was set to 0. The direction of the potential was fixed on experimental observations,17i.e., the head region was identified as having a lower potential than the tail region. The Laplace equation was solved iteratively by repeatedly setting —which is a property of the true solution of the Laplace equation—was solved on a grid of points for the electrical potential V. At the boundaries along the x and y directions, the potential is set to 0. The image of the filament was drawn in gray scale using a graphic editor and imported as an array of numbers using Python. The approach assumes a potential distribution which is mirrored in corrosion product distribution. The geometry of the filament was enforced based on literature reports and own observations. The array of grayscale values was directly translated to potential values by considering all the grayscale values >95% of the maximum value present in the array fixed at a potential value H when originally imported. Similarly, all the potentials <5% of the minimum grayscale value present in the array were considered fixed at a potential value L when originally imported. At the edges, the potential was fixed to 0. Everywhere else, the initial potential was set to 0. The direction of the potential was fixed on experimental observations,17i.e., the head region was identified as having a lower potential than the tail region. The Laplace equation was solved iteratively by repeatedly setting  except for the areas with fixed potentials which are the edges, H, and L. Because of the fact that the picture was drawn and the boundary conditions were set, the solution was forced to converge to what is represented here to illustrate the potential distribution; it does not add to experimental data which are available, e.g., in ref. 27 and 50. Furthermore, the actual values have limited physical meaning, but this method should yield an approximate picture of the potential distribution, reflected in the unit mV/mV0 where the mV0 presents an arbitrary reference potential. except for the areas with fixed potentials which are the edges, H, and L. Because of the fact that the picture was drawn and the boundary conditions were set, the solution was forced to converge to what is represented here to illustrate the potential distribution; it does not add to experimental data which are available, e.g., in ref. 27 and 50. Furthermore, the actual values have limited physical meaning, but this method should yield an approximate picture of the potential distribution, reflected in the unit mV/mV0 where the mV0 presents an arbitrary reference potential. |
| This journal is © The Royal Society of Chemistry 2025 |