
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModifying bacterial cellulose dispersions with deep eutectic solvent and pectin to tune the properties of open-celled foams†
Hareesh
Iyer
,
Aban
Mandal
,
Michael
Holden
and
Eleftheria
Roumeli
 *
*
Department of Materials Science and Engineering, University of Washington, Roberts Hall, Seattle, Washington, USA. E-mail: eroumeli@uw.edu; Tel: +(206)616-2832
First published on 26th December 2024
Abstract
The overconsumption of plastics has led to a significant micro/nanoplastics pollution problem, driving an urgent need for sustainable alternatives. Synthetic polymer foams such as expanded polystyrene (EPS), polyethylene (PE), and polyurethane (PU), contribute significantly to plastic waste, often ending up in landfills after short service lives. In this article, we present a comprehensive investigation of bacterial cellulose (BC)/pectin composite foams, focusing on how modifications to the biopolymer network and macromolecular interactions influence colloidal and solid-state properties. By treating BC with a citric acid-based deep eutectic solvent (DES), we enhance its colloidal stability, achieving a zeta potential 81.2% more negative, and improve the compressive strength of the resulting foams by 23.8%. Introducing pectin further transforms the structure of the BC network, and significantly alters its electrostatic and rheological properties. The zeta potential reaches absolute values as high as 30.3 mV at 80% pectin, while the recoverability increases and the storage and loss moduli decrease with increasing pectin concentration. Small-angle X-ray scattering (SAXS) reveals modifications in the network structure that provide insight into the substantial changes in the morphological and mechanical properties of the foams. The resulting binary biopolymer foams demonstrate strength and stiffness rivaling those of synthetic polymer foams of similar density. Overall, we demonstrate the critical role of colloidal interactions in tuning the mechanical properties of binary biopolymer solid foams, and highlight the potential of this sustainable and biodegradable system to address pressing environmental issues caused by plastic waste.
1 Introduction
Synthetic polymer foams such as expanded polystyrene (EPS), polyethylene (PE), and polyurethane (PU), provide inexpensive, lightweight, and versatile solutions for a variety of applications, ranging from protective parts for transportation, and padding in shoes and seats, to food packaging and flotation devices. However, the extensive utilization of petroleum-based polymers has had a detrimental effect on the environment due to their reliance on petroleum, non-biodegradable nature, and the energy-intensive processes involved in their production.1–3 According to the US Environmental Protection Agency (EPA), the production of petroleum-based plastic packaging materials in the USA reached 14.5 million tons in 2018, constituting 5% of municipal solid waste. Of this, only 13.6% was recycled, while 16.9% was incinerated for energy recovery. The remaining 69.5%, or 10 million tons, ended up in landfills.4 Many of these products break down into microplastics, which are pervasive in essentially every environment on Earth due to their abundance and small size.5 As consumers have grown increasingly concerned about microplastics, manufacturers have placed an increased importance on having sustainable and circular economies for their products’ lifecycles.6 Recent research has focused on developing bioplastics that utilize renewable resources, such as algae and cellulose, as well as sustainable processes and end-of-life scenarios.7–9Cellulose is the most abundant natural polymer in the world, found in the cell walls of algae and plants, and can even be synthesized by certain strains of bacteria.10,11 Compared to plant-derived cellulose, bacterial cellulose (BC) offers several distinct advantages, such as a significantly higher aspect ratio of the fibers, and higher crystallinity, that contribute to it having a higher strength and stiffness.11,12 Moreover, BC can be produced at commercially viable volumes with high purity, reducing the need for energy-intensive extraction methods such as Kraft pulping, which is used for wood-derived cellulose.12
To improve the strength of cellulosic materials, chemical treatments have been used as a way to create nanofibers and nanocrystals, or to simply defibrillate the cellulose fibers.13,14 Deep eutectic solvents (DES) offer a promising, environmentally-friendly alternative to conventional acid hydrolysis treatments of cellulose, avoiding the use of harsh acids.7,15,16 Prior literature has suggested that using various DES treatments on cellulose can nano-fibrillate BC and increase the absolute zeta potential, allowing for more colloidally stable suspensions. Some literature has shown that DES can also be used to add functional groups to the cellulose.7,17,18
For example, Henschen et al., demonstrated that esterification and acid hydrolysis of cellulose with oxalic acid dihydrate resulted in improved mechanical properties of cellulose films due to the nano-fibrillation of cellulose fibers.19 Similarly, Nguyen et al. developed an adsorbent foam for the removal of wastewater dyes based on the crosslinking of BC using citric acid.20 DES has also been used with cellulose from both plants and bacteria as a more sustainable solvent for producing a variety of foams and aerogels.21–25 Farooq et al. extracted microfibers from Firmiana simplex bark and used a DES to make aerogels that provided more than 90% purification efficiencies for air pollutants.21 Similarly, Laitinen et al. used DES to defibrillate kraft pulp before a silylation process that allowed for the creation of hydrophobic aerogels with the ability to absorb marine diesel from water at capacities of up to 142.9 grams of diesel per gram of aerogel.23 Long et al. also used DES and carbon nanofibers (CNFs), along with carbonation, to produce an aerogel filter with a volumetric absorption capability for organic pollutants and oils from water of 74–95%.24 For a sensing application, Panikar et al. used DES to fabricate gold nanoparticles on a cellulose nanocrystal (CNC) scaffold. This composite was able to detect pesticides in rice and tea extracts and Methylene Blue in fish muscle extract, although sensitivity was dependent on nanoparticle morphology.22 Recently, PVA, palm wax, and cellulose foams were created using DES by Li et al., with the DES treatment allowing for a high cellulose yield from the cotton fibers that were the feedstock for the study.25 They found that the combination of these materials resulted in a hydrophobic foam that may be an option for packaging materials in the future. Based on these studies, DES is typically used to treat cellulosic materials before other processes, and the resultant aerogels have largely been studied for filtering applications. Only one of these studies25 focused on properties relevant to packaging applications.
Another approach to alter the properties of cellulosic materials is to use similarly sustainable additives. Pectin, for instance, is a critical component in the cell walls of terrestrial plants, acting as a compliant matrix for the stiff load-bearing cellulose fibers. Industrially, it is often used in food as a gelling agent, giving structure and recoverability to food products. It can also be used as a food stabilizer, helping to keep fruit components, such as pulp fibers, from separating out in a mix. The ability for pectin to improve recoverability and aid colloidal stability, as well as its natural presence in plant cell walls, makes it a promising candidate as an additive to cellulosic materials. Cellulose and pectin is therefore a material system that has attracted significant interest, and has been well-studied in both colloidal and solid states. Examples include cellulose/pectin hydrogels and colloids,26,27 films,28 and foams.29 BC specifically has also been studied with pectin, typically in the form of pellicles or films with the pectin added in the culture medium in an effort to better understand natural polymerization reactions or biopolymer functions in plant cells30,31 or to create composite films with compelling mechanical properties.32
Here, we present how citric acid DES-treatment and pectin incorporation transforms the BC network structure in the colloidal state and subsequently, how those changes influence the morphology and mechanical properties of the resulting solid foams. First, we investigate the effects of treating BC dispersions with a citric acid and choline chloride DES, and how that treatment alters electrostatic, rheological, colloidal, morphological, and mechanical properties of the BC dispersions and the foams made from lyophilizing them. We then add pectin to the BC dispersions and study how the pectin changes those properties. We highlight how the changes to the BC-based materials and their colloidal properties affect the properties of the resulting freeze-dried foams. Finally, we compare the BC/pectin composites with commercial foams such as EPS, PE, and PU, which are commonly used in packaging applications. We show that biomatter-based foams can be competitive with petroleum-derived foams for such applications, providing a potential path toward sustainability for the packaging sector.
2 Experimental section
2.1 Materials
12 oz. kombucha SCOBYs were purchased from joshuatreekombucha.com. Gallon black tea bags were purchased from Lipton. Pectin from citrus peel, choline chloride, citric acid from citrus peel, and sodium hydroxide were purchased from Sigma-Aldrich.2.2 Methods
BC dispersions were then made by blending 75 g of the purified BC pellicle with 200 ml of DI water for 3 minutes. 10 g of the dispersion was moisture analyzed using a VWR M-Series moisture analyzer to determine water content.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of citric acid and choline chloride to 80 °C while constantly stirring until a homogenous liquid formed. The BC dispersion and DES were combined in a 1:15 ratio of BC:DES by mass. Approximately 1 L of boiling DI water was added, the beaker was covered with aluminum foil, and the mixture was heated to 105 °C for 2 hours on a hot plate with a magnetic stir bar. Once the DES treatment was done, the solution was cooled by adding cold DI water to the beaker. The solution was then vacuum filtered using a Whatman #2 filter. The resulting film was separated from the filter paper and blended for 3 minutes in DI water. This process of vacuum filtering and washing was repeated once more. The resulting dispersion was then moisture analyzed before being centrifuged down to 1 wt% of BC in water.
:1 molar ratio of citric acid and choline chloride to 80 °C while constantly stirring until a homogenous liquid formed. The BC dispersion and DES were combined in a 1:15 ratio of BC:DES by mass. Approximately 1 L of boiling DI water was added, the beaker was covered with aluminum foil, and the mixture was heated to 105 °C for 2 hours on a hot plate with a magnetic stir bar. Once the DES treatment was done, the solution was cooled by adding cold DI water to the beaker. The solution was then vacuum filtered using a Whatman #2 filter. The resulting film was separated from the filter paper and blended for 3 minutes in DI water. This process of vacuum filtering and washing was repeated once more. The resulting dispersion was then moisture analyzed before being centrifuged down to 1 wt% of BC in water.
For power law fitting, the equation I(q) = Aq−n was fit to the data with q ranging from 0.004 to 0.08 Å−1.
The experimental data was fit using SASview35 with a shape-independent model eqn (1) containing a two-stage Gauss-Lorentz gel model, taking into account the contribution from the interference between fibers in the network of physical gels.36–38
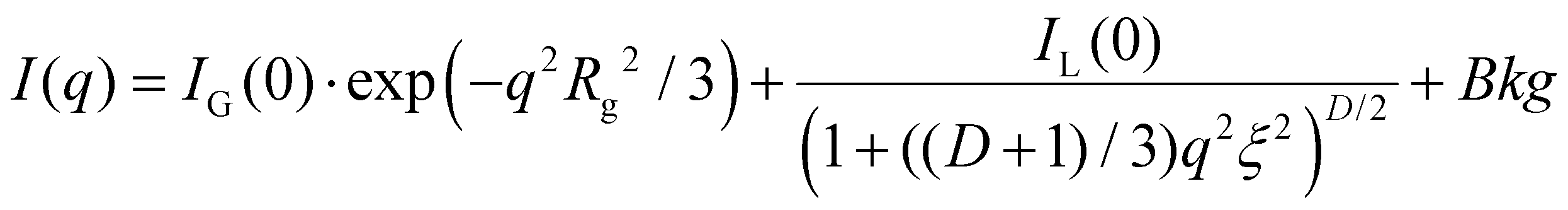 | (1) |
I(q) is the intensity in terms of the scattering vector, q. IG and IL are the scaling parameters for the Guinier and Lorentz portions of the equation, respectively, determining the relative contributions of each. Rg is the static correlation length, also referred to as the agglomerate size or entangled region size, which accounts for static accumulations of polymer pinned down by junction points in the polymer network.36,37 D is the fractal dimension, which reflects the interactions of fractal objects with their solvent.39ξ is the dynamic correlation length, often referred to simply as the correlation length, which describes fluctuations in the position of the polymer chains and is related to the average distance between polymers.40,41 Finally, Bkg is the scattering due to the background material. In this study, the background sample was DI water.
3 Results
3.1 Effects of DES treatment on bacterial cellulose
To promote defibrillation and improve the stability of BC in suspension (Fig. 1a), we treated the BC with a DES comprised of citric acid and choline chloride, under conditions similar to Xia et al., who used oxalic acid instead of citric acid7 (see Methods).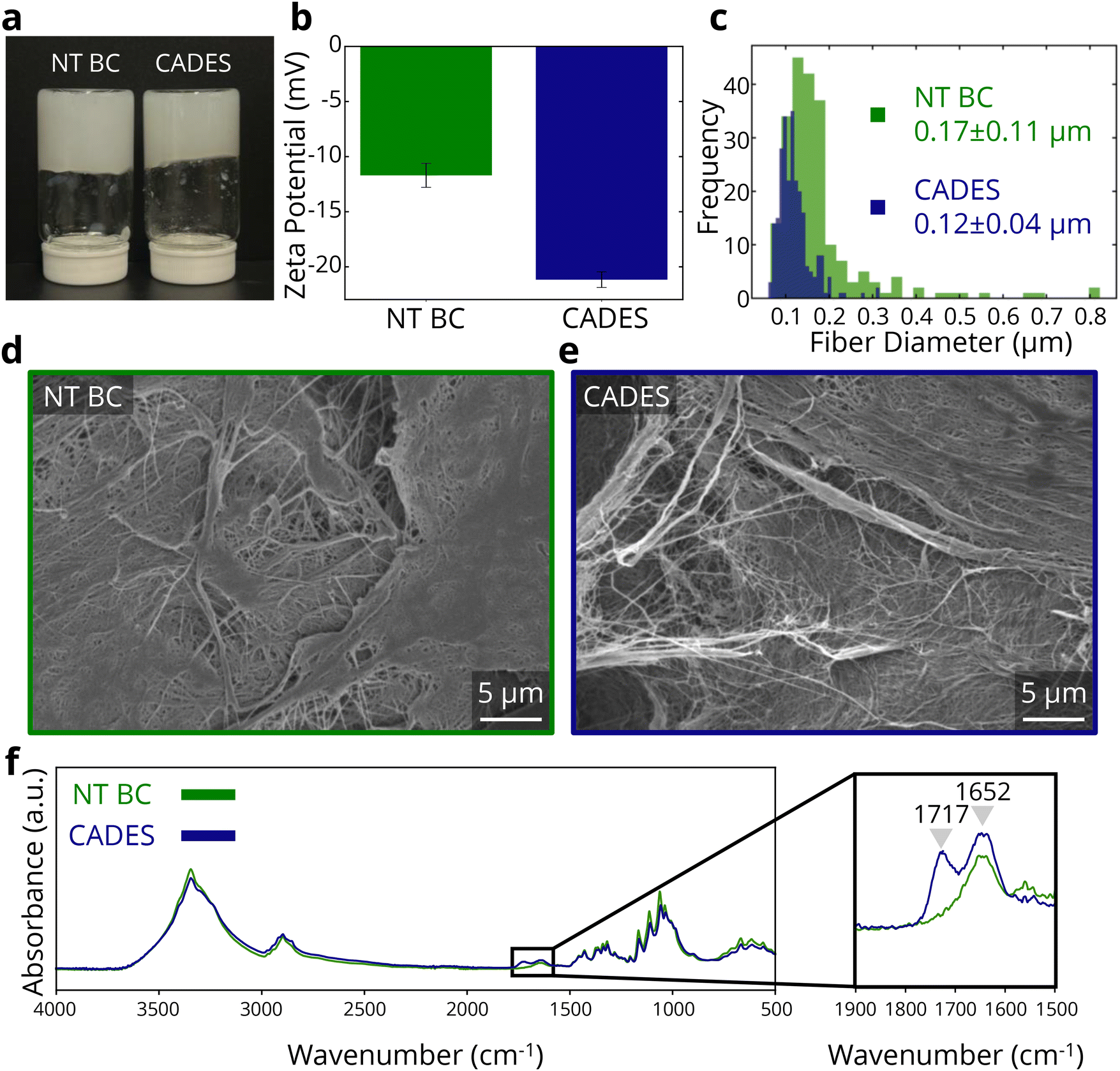 | ||
| Fig. 1 Colloidal and chemical effects of DES treatment on bacterial cellulose. (a) Non-treated and CADES BC dispersions. (b) Zeta potential of non-treated and CADES BC. (c) Comparison of fiber diameters of non-treated and CADES BC. SEM of non-treated BC (d) and CADES BC (e) films. (f) FTIR spectra of non-treated and CADES BC films. | ||
In our study, considering that the foams are produced by freeze-drying, we anticipate that the final behavior of the foams will be influenced both by how the fibers are frozen in their solution, and by the DES treatment. We hypothesized that the defibrillation and increased stability of these fibers, achieved through the DES treatment, would improve mechanical properties by creating more homogenous dispersions, and therefore more homogenous foams.
Comparing SAXS data for untreated BC and DES-treated BC, we see significantly different behavior induced by the DES treatment (see ESI Table 1†). The size of the entangled region (Rg) for untreated BC is measured as ∼255 Å and ∼3279 Å for DES-treated BC, an order of magnitude difference. This may be explained by the defibrillation of the cellulose fibers due to DES treatment, leading to more overlapping fibers at each point of contact. The DES treatment shows the opposite effect on mesh size (ξ), where the mesh size of the DES-treated BC (220 Å) is an order of magnitude smaller than that of the non-treated BC (4717 Å). Therefore, the defibrillated DES-treated BC forms a network of entanglements that result in a smaller mesh size, compared to the non-treated BC which forms a significantly looser mesh structure.
To clarify the reasons for the measured differences in zeta potential and SAXS, we conducted SEM imaging (Fig. 1d and e) to evaluate changes in fibrillation due to the DES treatment. As shown in the SEM images, the DES treated samples consist of finer cellulose fibers which are also more distinct compared to the untreated BC sample surface. Fiber size analysis (Fig. 1c) reveals an average fiber diameter of 0.17 ± 0.11 μm for the untreated BC sample, while the DES-treated sample had an average fiber diameter of 0.12 ± 0.04 μm. The reduced fiber diameter as well as smaller deviation of fiber sizes indicates that the DES treatment conditions enabled the defibrillation of BC.
Next, turning to FTIR spectra (Fig. 1f) to investigate possible molecular bonding changes due to the DES treatment, we observed a new peak at 1717 cm−1, in the DES-treated BC. This peak indicates the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds,46 which are not present in pure cellulose but are found in citric acid. This suggests that cellulose carboxylation occurred through the DES treatment. However, batch-to-batch variation led to inconsistent detection of the carbonyl peak (ESI Fig. 1†), indicating that the functionalization efficiency was inconsistent. Nonetheless, the DES treatment enabled defibrillation in all the studied samples, supporting the higher absolute zeta potential of the DES-treated BC at all cases in the used reaction conditions.
O bonds,46 which are not present in pure cellulose but are found in citric acid. This suggests that cellulose carboxylation occurred through the DES treatment. However, batch-to-batch variation led to inconsistent detection of the carbonyl peak (ESI Fig. 1†), indicating that the functionalization efficiency was inconsistent. Nonetheless, the DES treatment enabled defibrillation in all the studied samples, supporting the higher absolute zeta potential of the DES-treated BC at all cases in the used reaction conditions.
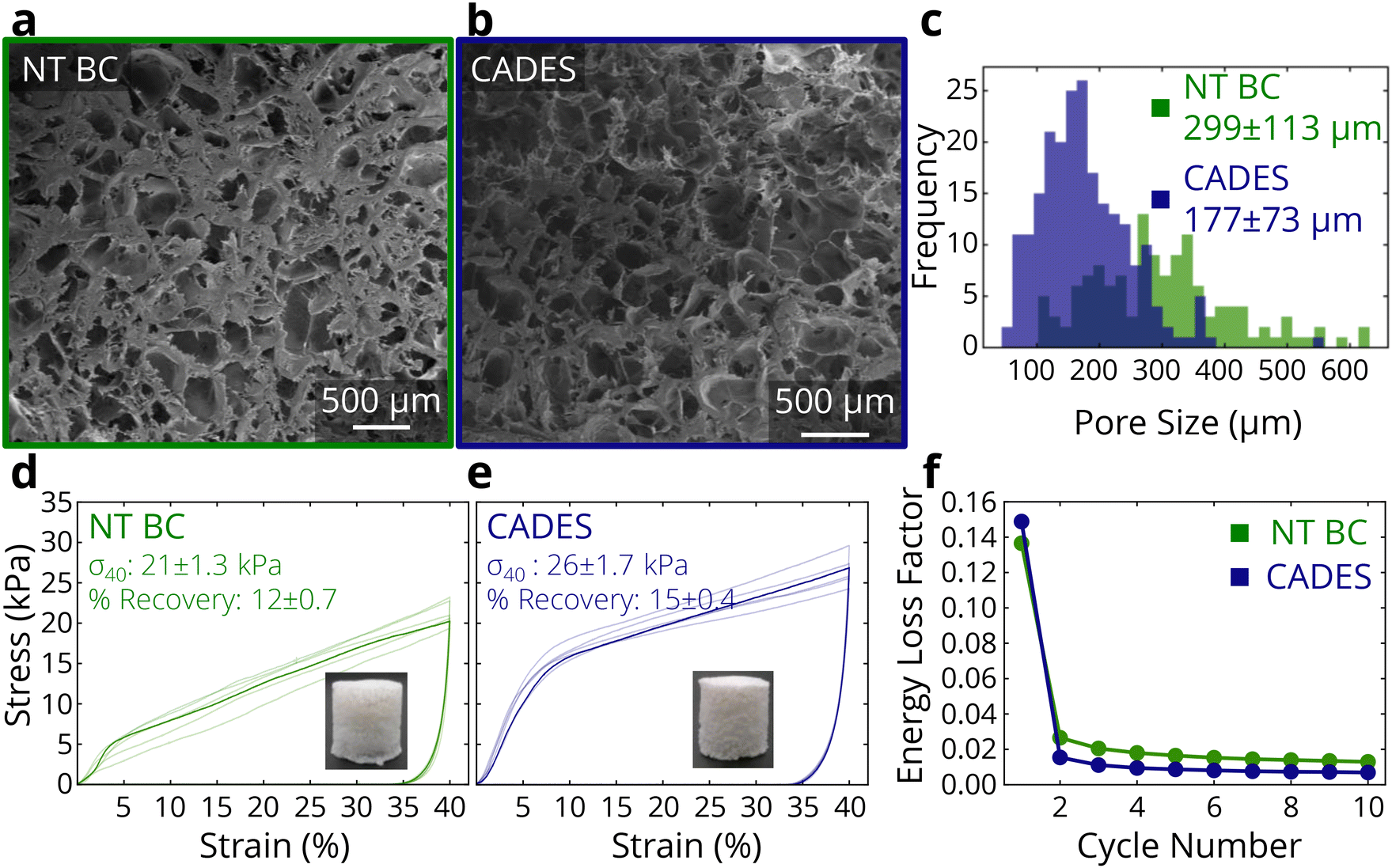 | ||
| Fig. 2 Morphological and mechanical effects of DES treatment on BC foams. SEM of non-treated BC (a) and CADES BC (b) foams. (c) Distribution of pore sizes for non-treated and CADES BC foams. Compression tests of non-treated (d) and CADES (e) BC foams. (f) Energy loss factors from cyclic testing of non-treated and CADES BC foams. | ||
To examine how the colloidal properties translate to solid materials, compression tests on lyophilized samples were carried out. In tests where samples were compressed to 40% strain and then unloaded, the DES-treated foams showed a 146% higher loading modulus, and a 25% higher compressive strength at 40% strain (Fig. 2d and e). The treatment also reduced the variability between samples, as the measured stress–strain responses show smaller differences compared to the non-treated BC. The recoverability of the foam changed negligibly between the non-treated and treated BC.
Furthermore, we conducted cyclic compression tests in which all samples were compressed to 20% strain and unloaded 10 times in succession (representative stress–strain curves shown in ESI Fig. 3†). The DES-treated samples showed higher compressive strength at 20% strain and more energy absorption in the first compression (1.44 kJ m−3 for CADES-BC compared to 1.26 kJ m−3 for non-treated BC). From the cyclic tests we can calculate the loss coefficient which characterizes a material's ability to dissipate energy in a loading-unloading cycle, and is defined as:
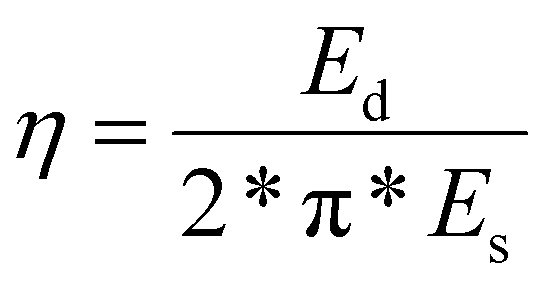 | (2) |
Overall, the DES treatment of BC leads to a higher absolute zeta potential, fiber defibrillation, and reduced agglomeration. This causes a more homogeneous foam and higher stiffness and strength, as well as the ability to store more energy during cyclic compression than non-treated BC.
3.2 Effects of pectin in BC foams
After understanding the properties of pure BC foams with and without DES treatment, we aimed to expand the attainable properties, specifically to improve the recoverability of the foams. A way to achieve a modular mechanical property change is to create a binary network with another polymer. Given the higher colloidal stability, defibrillation, strength and stiffness of the produced foams obtained after DES treatment, we chose that as the primary polymer network in our composite system. Pectin's molecular structure, as in nature, allows for a strong interacting network with the strong and stiff cellulose fibers, creating a hydrogen bond-dominated network where the softer pectin chains enable more extended deformation under loading.As described in the Methods section (2.2), we introduced water-dissolved pectin at a 1 wt% concentration into the 1 wt% CADES-BC gel in the required ratios to achieve a dry material of the desired concentrations of pectin and BC. The produced colloids were all homogenous (Fig. 3a inset) as expected, with no phase separation or precipitation noted. The binary network colloids were subsequently lyophilized to produce composite foams.
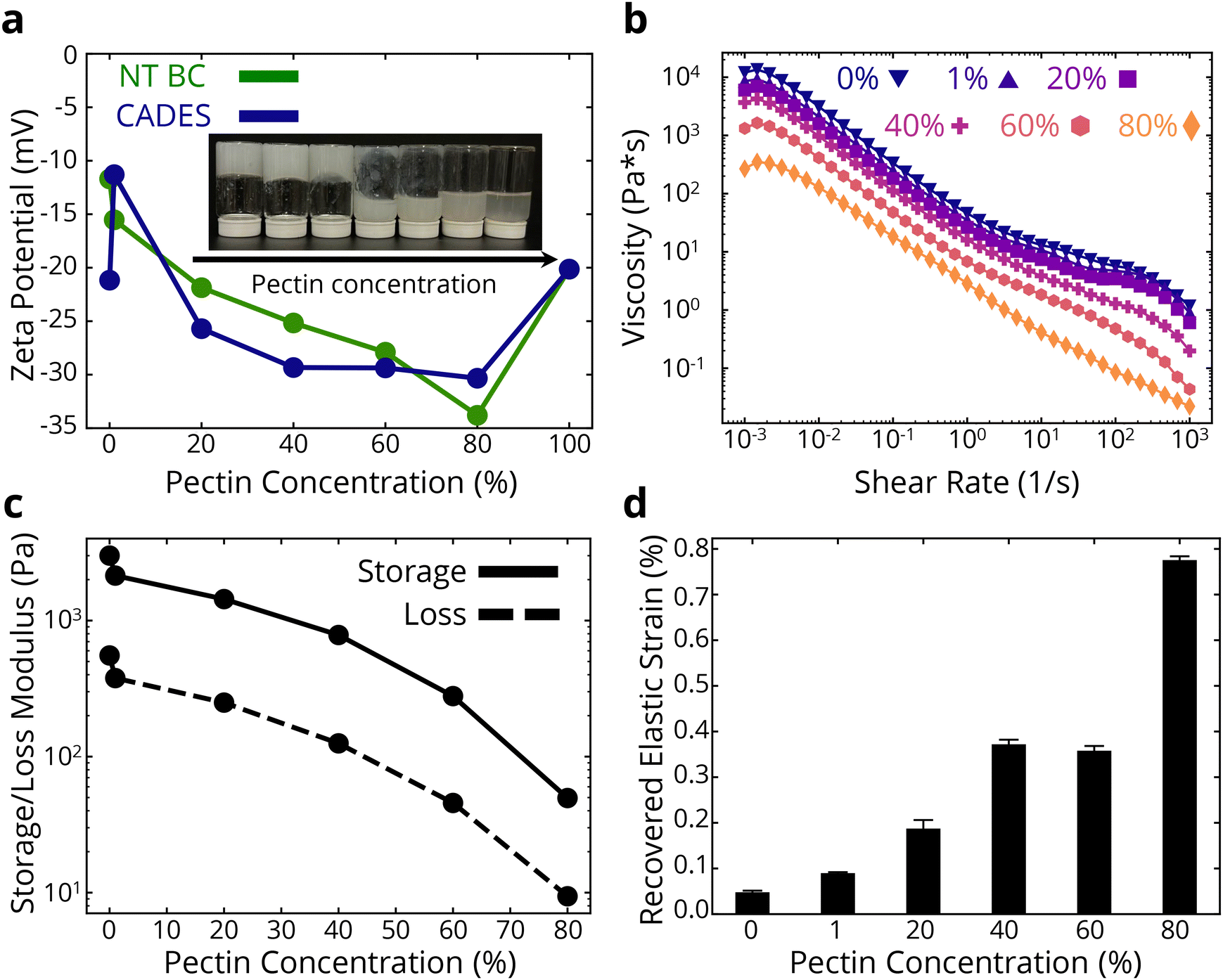 | ||
| Fig. 3 Effects of adding pectin on colloidal properties. (a) Zeta potential measurements of non-treated and CADES BC dispersions with varying amounts of pectin added. Viscosity measurements (b) and storage/loss moduli at ∼5 rad s−1 (c) of CADES BC/pectin dispersions. (d) Recovered elastic strain percentage of CADES BC/pectin dispersions. | ||
To better understand the entanglements in the colloidal system, SAXS was performed on the samples. Using a power law fit, we can get an idea of the type of network formed by the BC-pectin gels (Fig. 4a and ESI Table 2†). Note that the power is approximately 2 for all BC/pectin composites, indicating the presence of an entangled system.53 By using a combination of the Guinier and Lorentz models (see section 2.2), we can investigate the agglomerate size and correlation lengths of the colloidal systems. In eqn (1), Rg represents the agglomerate size, or the size of an entangled region pinned down by junctions.36,37ξ is associated with the thermodynamic fluctuations of polymer chains37 and is related to the average distance between polymers due to the screening of excluded volume interactions.40,41 In Fig. 4b and c, we see that the agglomerate size generally decreases with pectin concentration, while the correlation length generally increases with pectin concentration. This suggests that pectin molecules shield cellulose chains from each other, reducing the amount and size of cellulose agglomerates and increasing the size of the mesh in the cellulose-pectin matrix. Fig. 4d shows that the reptation tube diameter increases with pectin concentration. The reptation tube indicates the path along which a polymer chain's center of mass moves due to Brownian motion. Recognizing limitations of using reptation theory in semiflexible polymers54 such as BC and pectin, we can still get useful insights. We first calculated the tube diameter using the relationship d∝ξ2Rg−1.54 The aforementioned shielding due to pectin molecules may also explain the increase in reptation tube diameter, as this shielding would increase the excluded volume of the cellulose polymers, giving more space for each polymer to move. Additionally, Fig. 4e shows that the storage modulus, taken at the LVR limit from the amplitude sweeps, decreases with reptation tube diameter. With the motion of polymer chains confined to a tube-like region, a larger tube diameter would result in increased chain mobility, leading to a decrease in storage modulus.
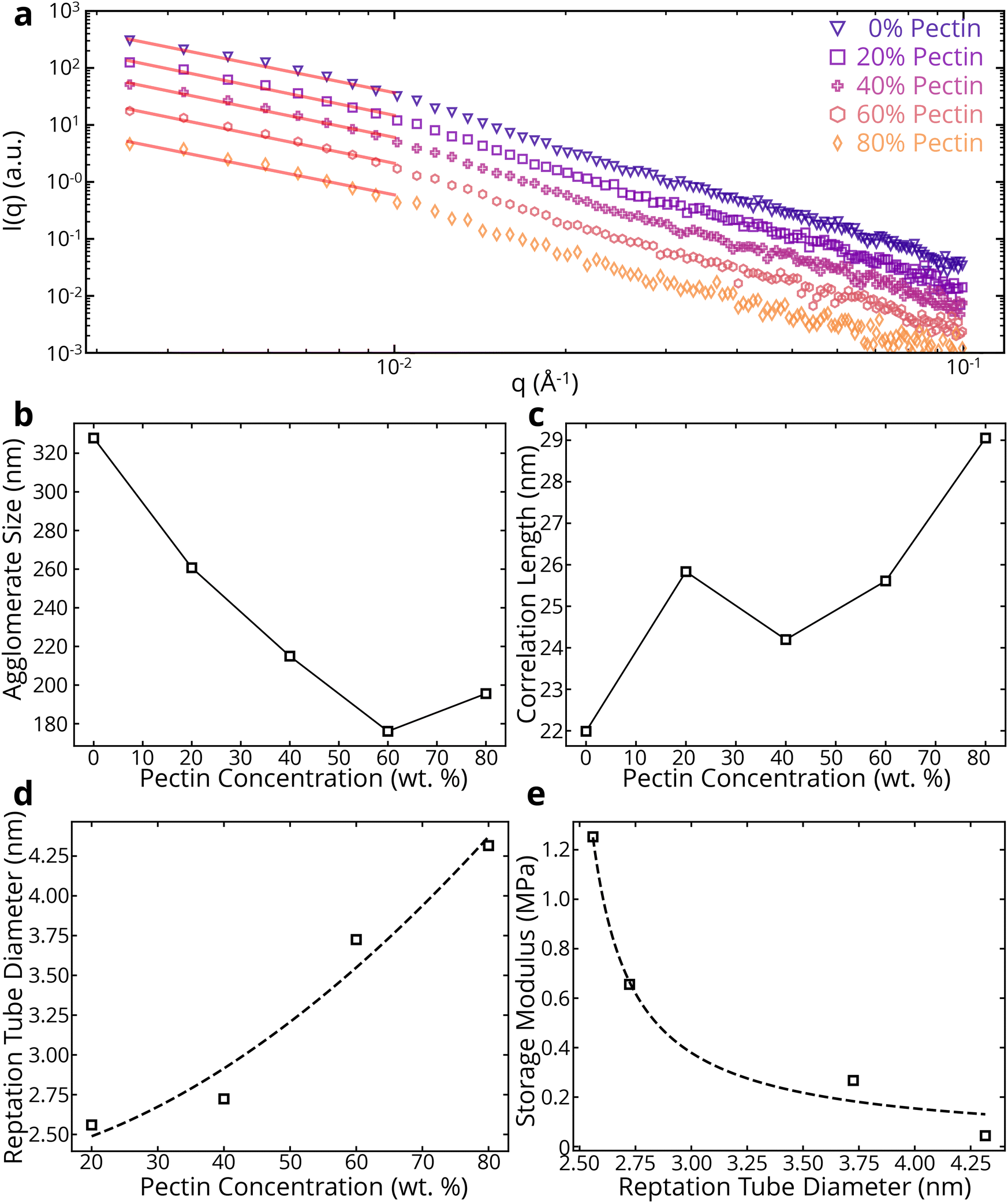 | ||
| Fig. 4 Effects of adding pectin on network properties via SAXS. (a) SAXS data for CADES BC/pectin composites with a power-law fit in the low-q region. (b) Agglomerate size, (c) correlation length, (d) and reptation tube diameter of CADES BC/pectin composites. (e) Effect of reptation tube diameter on storage modulus of the colloids. | ||
Rheology was used to further analyze the colloidal behavior of the BC-pectin gels. The flow curves in Fig. 3b show that increasing pectin concentration reduces the viscosity of the gel at all shear rates. We see smaller effects for lower concentrations of pectin, where the reduction in viscosity at a shear rate of 1 s−1 is ∼43% when adding 20 wt% pectin. At higher pectin concentrations, the viscosity reductions are greater, such as going from 60 wt% to 80 wt% causing a ∼58% reduction in viscosity at 1 s−1. A reduction in both the storage and loss moduli is observed as pectin concentration is increased (Fig. 3c). Again, we see smaller effects of pectin at lower concentrations, where the reduction in storage modulus is ∼53% when adding 1 wt% pectin. At higher pectin concentrations, the effects are increased, showing a ∼83% reduction in storage modulus when going from 60 to 80 wt% pectin. Amplitude sweep curves for the dispersions are shown in ESI Fig. 4.† These curves show clear viscoelastic behavior, and the storage modulus is higher than the loss modulus for all composites that have some amount of BC in them, until a crossover strain at which point the loss modulus dominates. The gels have a non-brittle fracture, as indicated by the crossover points being in the negative slope portion of the curves. This is consistent with observations of the behaviors of the gels; they have a clear but weak network structure, and the relatively low viscosity allows for flow under gravity at higher pectin concentrations, as shown in the inset of Fig. 3a. Combined, as expected, we see that introducing pectin in the stiff cellulose network leads to an electrostatically stable binary network with reduced rigidity and stiffness. We note that in this study, we used deionized water as our dispersion medium. A means to improve the network strength would be to provide cations in the system which would promote ionic crosslinking in the pectin chains.55,56
To analyze the time-dependent behavior of our binary networks, we conducted creep tests, as shown in ESI Fig. 5 and 3d.† Constant stress was applied for 10 minutes and released for another 10 minutes, allowing us to measure the recoverable strain of the gel network. To characterize the ability for the foams to elastically recover after the release of stress, the following equation was used to first model the non-elastic recovery:
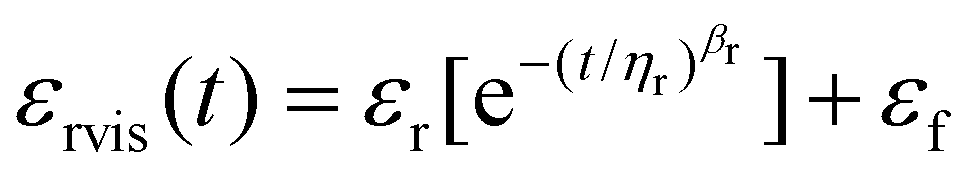 | (3) |
| εrecov = εrvis + εe | (4) |
Increasing the pectin content increased the recovered elastic strain by ∼87% at 1 wt% pectin and over 16× at 80 wt% pectin. The recovered viscoelastic strain, εr, also increased with pectin concentration (ESI Fig. 6†). The increasing recoverable elastic and viscoelastic strain in the presence of pectin could be due to the improved elastic fiber mobility in the network, afforded by the presence of pectin macromolecules.
Overall, the above analyses of the BC-pectin colloids show that the binary network is more stable in water than either of the pure component gels (as shown by the more negative zeta potentials of the composite gels), and that the presence of pectin affords an improvement in the network's recoverability at the expense of gel stiffness. The presence of pectin appears to create a weaker network, but due to electrostatic repulsion there may be less fiber entanglements, allowing for fiber rearrangements during loading. Next, we will examine how these colloidal properties translate to solid properties of the lyophilized foams.
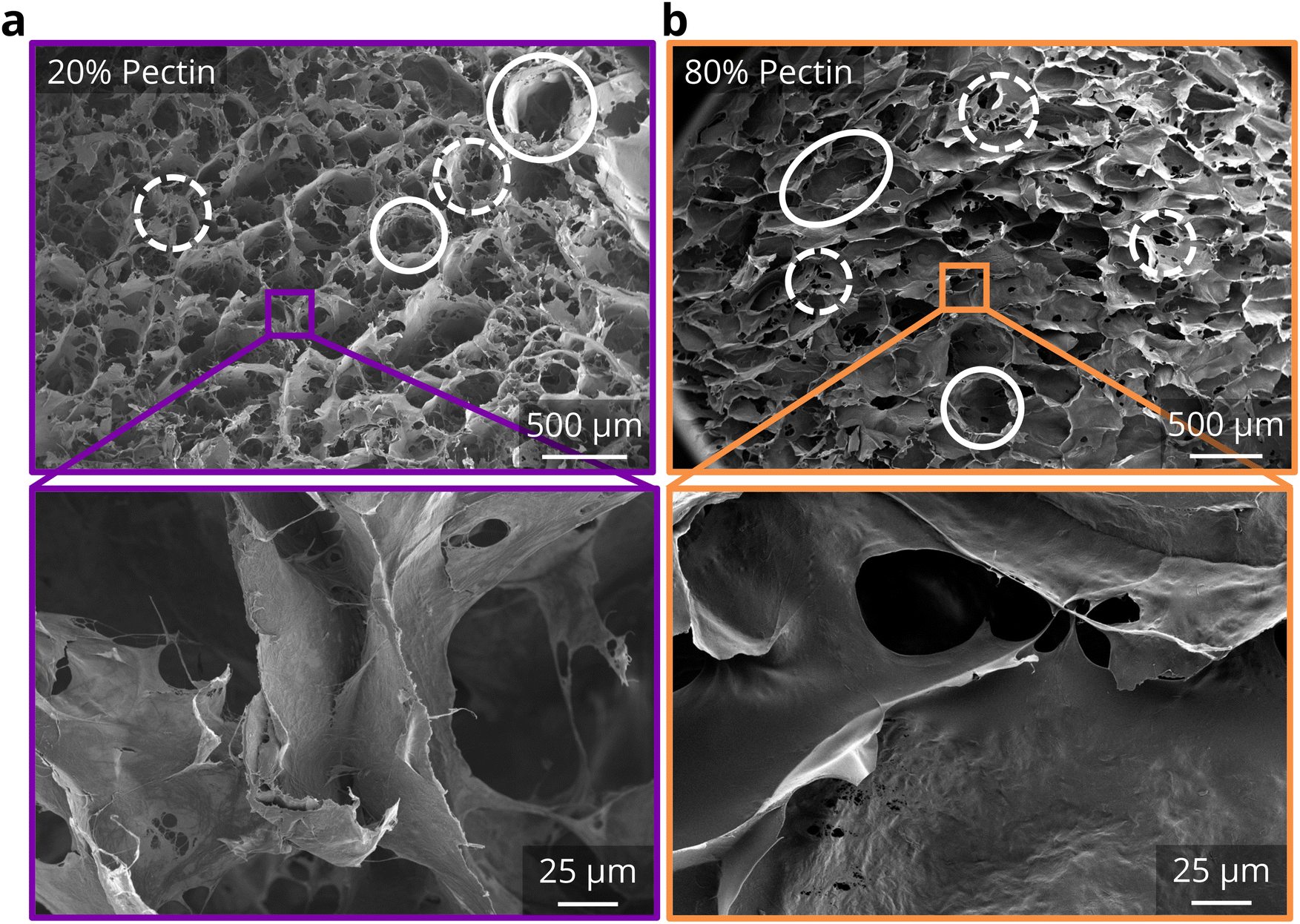 | ||
| Fig. 5 Effects of adding pectin on morphological properties. SEM images of a CADES BC foam with (a) 20% pectin and (b) 80% pectin at 30× (top row) and 500× (bottom row) magnifications. Solid circles indicate cells in the foams, dashed circles indicate holes in the matrix. | ||
Indeed, compression tests show a wide property space for BC-pectin foams depending on pectin concentration. Using the 40% compression testing (Fig. 6a and ESI Fig. 8†), we see that compressive strength values at 40% strain increased by approximately 26% and the loading modulus increased by about 10% at 20 wt% pectin concentrations. Thus, a synergistic effect was observed for the strength and modulus of the binary network foams with the lowest tested pectin content. However, strength decreased after 20 wt%, dropping down to 7.6 kPa at 100 wt% pectin (an approximately 72% decrease compared to pure CADES-BC). On the other hand, the recovery of the foams increased significantly with pectin concentration (Fig. 6b). At 20 wt% pectin, where compressive strength was the highest, the foams exhibited 26% recovery, compared to 15% recovery for pure CADES-BC (a ∼73% increase). The recovery percentage increases steadily, getting to almost 40% recovery at 90 wt% pectin. Pure pectin foams show a recovery of about 68%, indicating the presence of even 10 wt% BC significantly decreases the recoverability of BC-pectin composite foams. The increasing recoverability with increasing pectin concentration matches with what we saw in the rheological creep tests (section 3.2.1), where the increase in pectin increased the recovered strain of the gels. The data suggest that the presence of pectin adds elastic energy absorption pathways in both the gel and solid composites.
 | ||
| Fig. 6 Effects of adding pectin on mechanical properties. Compressive strength (a) and recovery percentage (b) from 40%-strain compression testing. (c) Stress decay ratio, and (d) energy loss factor of CADES BC/pectin composites. (e) Comparison of CADES BC/pectin composites with synthetic polymer foams by unloading modulus and density. | ||
Overall, the lowest amount of pectin probed in our experiments enabled a synergistic improvement in the compressive strength and modulus of the foam, exceeding the properties of pure CADES-BC, while also enabling an improvement of the recoverability. The addition of pectin reduces the amount of irrecoverable strain in the foams, and it creates a fiber-in-matrix morphology that improves the load transfer between BC fibers.
To further examine the effects of pectin on BC foams, we conducted dynamic mechanical tests. Stress relaxation tests (Fig. 6c) show an increase in the stress decay, which indicates how much energy is dissipated while the compressive strain is held constant. Stress decay is calculated as the ratio of the stress lost during the relaxation period to the stress at the start of the relaxation period, as shown below:
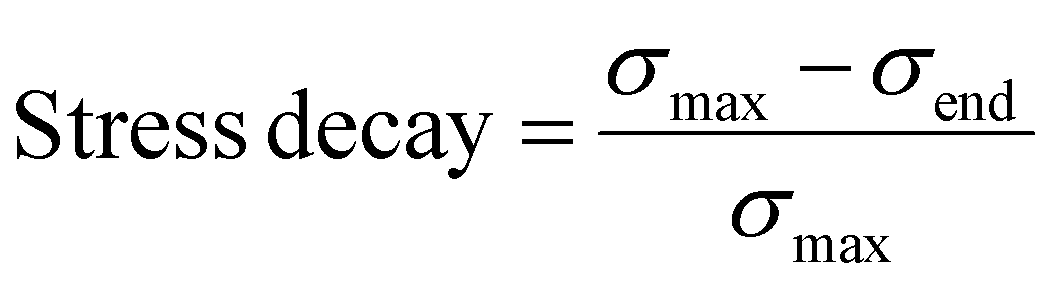 | (5) |
An increase in stress decay shows an increase in the ability for the matrix to accommodate the stress being applied to it, demonstrating an increase in the chain mobility of the binary system. Pure pectin samples have a significantly higher stress decay, and therefore higher chain mobility, than pure BC foams. As pectin concentration increases, the overall chain mobility in the foam also increases.
This conclusion is further supported by cyclic testing, as shown in Fig. 6d. All materials show a non-linear dependence of loss coefficient, with the first compression causing the greatest energy loss and subsequent compressions showing very small drops in energy loss. Practically, after the second compression cycle all foams approach a steady state response. With the exception of the 60 wt% pectin concentration, there is a steady increase in the energy loss coefficient, meaning there is an increase in damping ability, as pectin concentration is increased.
Overall, in the binary biopolymer foams we observed that the strong and stiff BC fibers are immersed in a compliant pectin matrix. Mechanical testing consistently showed that pectin improves the flexibility of the network, allowing for more recoverability and energy absorption under compressive load. The lowest tested pectin concentration (20 wt%) enabled small improvements in strength, modulus, recoverability, and energy absorption, relative to pure BC fiber foams, but at higher pectin concentration, the recoverability and loss coefficients were further improved at the expense of network strength and stiffness.
Finally, to contextualize our findings, we compare the quasi-static compression performance (to a 40% compression) of our binary network foams against commercially available EPS, PE, and PU foams with similar apparent density. In Fig. 6e we report the measured unloading moduli versus density for all tested materials (representative stress–strain curves at ESI Fig. 9†). The BC/pectin foams fall within and exceed the range of moduli of the synthetic foams. Our foams have unloading moduli ranging from 0.2 to 2.6 MPa at densities of 0.012 ± 0.001 g cm−3 on average. Synthetic commercial foams have unloading moduli ranging from 0.3 to 0.8 MPa at densities ranging from 0.007 g cm−3 to 0.030 g cm−3. When comparing strength, we similarly see that the BC/pectin foams are on the same order of magnitude in terms of strength (ESI Fig 10†). Our foams have strengths ranging from 7.6 kPa to 34.2 kPa, while the synthetic foams range from 16.3 kPa to 49.8 kPa. However, if we compare the extent of recovery of the foams after unloading, we see that the recoverability of the synthetic polymer foams far exceeds that of our BC/pectin foams, showing the significant gap in mechanical properties that still exist (ESI Fig 11†). Our foams have recoverabilities ranging from 15% to 68%, while commercial foams have recoverabilities ranging from 54% to 92%. One stark difference between the tested synthetic foams and our biopolymer foams, besides the chemical structures, is that the synthetic foams are all closed-cell foam structures. Our biopolymer foams are all open-celled foam structures, which causes a significant change in the mechanical behavior of the foams. With the current preparation method that applies lyophilization on the colloids to produce the foams, it is not possible to achieve a closed-cell structure with which to compare to the synthetic foams.
4 Conclusion
In this article, we investigated the effects of DES treatment on BC, and the incorporation of pectin to enhance the properties of BC foams. The DES treatment with citric acid and choline chloride successfully promoted defibrillation, leading to finer and more stable cellulose fibers, as evidenced by SEM and zeta potential measurements. This treatment also introduced functional groups, as indicated by FTIR spectra, though with inconsistent efficiency. The resulting DES-treated BC foams exhibited improved mechanical properties, including higher loading modulus and compressive strength, compared to non-treated BC foams. However, upon cyclic compressive loading the CADES-BC foams had slightly lower loss coefficient values, potentially indicating a lesser ability to dampen and disperse energy.Incorporating pectin into the CADES-BC foams created a binary network that further modified the mechanical and colloidal properties. Pectin, which is acidic in solution, increased the absolute zeta potential of BC/pectin composite dispersions, which is the opposite of what is usually expected for cellulose in acidic environments. Additionally, the absolute zeta potential of the composites was higher than that of either of the constituent components, suggesting a synergistic interplay in the binary network. The incorporation of pectin also reduced the viscosity and the storage/loss moduli, with higher pectin concentrations leading to higher decreases. Rheological creep tests showed that an increase in pectin increased the colloid's ability to recover strain after a constant stress was removed, demonstrating the pectin's ability to improve the fiber mobility in the network.
Morphological analysis revealed a transition from a fibrillar to a fiber-in-matrix structure upon introduction of pectin in the BC foams, improving flexibility and recoverability of the foams. Strength at 40% strain was also found to increase up to 20 wt% pectin but decreased at higher concentrations. Recovery of the foams consistently increased as pectin concentration increased, and the ability of the foams to dissipate energy under constant load increased at higher pectin concentrations as well.
Overall, our findings indicate that DES treatment and pectin incorporation are effective strategies for tuning the properties of BC foams. The treated and composite foams exhibited promising mechanical performance, with potential applications in areas requiring lightweight, high-strength, and energy-absorbing materials. We also showed that these biomatter-based foams can be competitive with petroleum-derived foams, and therefore can provide a path forward for sustainability in packaging materials. Future work includes optimizing the functionalization process and exploring the potential for closed-cell foam structures to further enhance the mechanical properties.
Author contributions
Hareesh Iyer: conceptualization, methodology, software, formal analysis, investigation, data curation, writing, visualization, supervision. Aban Mandal: conceptualization, methodology, software, formal analysis, investigation. Michael Holden: conceptualization, methodology, formal analysis, investigation, writing. Eleftheria Roumeli: conceptualization, resources, writing – review and editing, supervision, project administration, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation Award #2332640. This material is based in part upon work supported by the state of Washington through the University of Washington Clean Energy Institute. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement no. 654000. Part of this work was conducted at the Washington Clean Energy Testbeds, a facility operated by the University of Washington Clean Energy Institute. The authors acknowledge the use of facilities and instrumentation supported by the U.S. National Science Foundation through the UW Molecular Engineering Materials Center (MEM-C), a Materials Research Science and Engineering Center (DMR-2308979).References
- S. P. Bangar and W. S. Whiteside, Int. J. Biol. Macromol., 2021, 185, 849–860 CrossRef CAS PubMed.
- B. Kunwar, H. N. Cheng, S. R. Chandrashekaran and B. K. Sharma, Renewable Sustainable Energy Rev., 2016, 54, 421–428 CrossRef CAS.
- W. Zhang, Y. Zhang, J. Cao and W. Jiang, Int. J. Biol. Macromol., 2021, 166, 288–296 CrossRef CAS PubMed.
- O. US EPA, Containers and Packaging:Product-Specific Data, 2017, https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/containers-and-packaging-product-specific.
- Z. Pan, Q. Liu, X. Sun, W. Li, Q. Zou, S. Cai and H. Lin, Gondwana Res., 2022, 108, 31–40 CrossRef CAS.
- D. R. Enarevba and K. R. Haapala, Procedia CIRP, 2023, 116, 654–659 CrossRef.
- Q. Xia, C. Chen, Y. Yao, J. Li, S. He, Y. Zhou, T. Li, X. Pan, Y. Yao and L. Hu, Nat. Sustainability, 2021, 4, 627–635 CrossRef.
- J. L. Fredricks, H. Iyer, R. McDonald, J. Hsu, A. M. Jimenez and E. Roumeli, J. Polym. Sci., 2021, 59, 2878–2894 CrossRef CAS.
- H. Iyer, P. Grandgeorge, A. M. Jimenez, I. R. Campbell, M. Parker, M. Holden, M. Venkatesh, M. Nelsen, B. Nguyen and E. Roumeli, Adv. Funct. Mater., 2023, 33, 2302067 CrossRef CAS.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- S. Wang, F. Jiang, X. Xu, Y. Kuang, K. Fu, E. Hitz and L. Hu, Adv. Mater., 2017, 29, 1702498 CrossRef PubMed.
- Y. Lu, M. Mehling, S. Huan, L. Bai and O. J. Rojas, Chem. Soc. Rev., 2024, 53, 7363–7391 RSC.
- S. Liu, Z.-X. Low, Z. Xie and H. Wang, Adv. Mater. Technol., 2021, 6, 2001180 CrossRef CAS.
- K. Missoum, M. N. Belgacem and J. Bras, Materials, 2013, 6, 1745–1766 CrossRef PubMed.
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290 RSC.
- W. Liu, H. Du, K. Liu, H. Liu, H. Xie, C. Si, B. Pang and X. Zhang, Carbohydr. Polym., 2021, 267, 118220 CrossRef CAS PubMed.
- W. Yu, C. Wang, Y. Yi, W. Zhou, H. Wang, Y. Yang and Z. Tan, Cellulose, 2019, 26, 3069–3082 CrossRef CAS.
- W. Yu, C. Wang, Y. Yi, H. Wang, Y. Yang, L. Zeng and Z. Tan, Cellulose, 2021, 28, 175–188 CrossRef CAS.
- J. Henschen, D. Li and M. Ek, Carbohydr. Polym., 2019, 213, 208–216 CrossRef CAS PubMed.
- H. T. Nguyen, F. A. Ngwabebhoh, N. Saha, T. Saha and P. Saha, Int. J. Biol. Macromol., 2022, 222, 77–89 CrossRef CAS PubMed.
- A. Farooq, H. Yang, Z. Ding, F. Bu, M. Guo, W. Sun, Z. Wang and M. Tian, Int. J. Biol. Macromol., 2024, 275, 133629 CrossRef CAS PubMed.
- S. S. Panikar, K. C. Sekhar Reddy, A. L. Gonzalez, G. Ramírez-García, A. G. Rodríguez, M. A. M. Sosa, P. Salas and J. D. Mota-Morales, Anal. Chem., 2022, 94, 16470–16480 CrossRef CAS PubMed.
- O. Laitinen, T. Suopajärvi, M. Österberg and H. Liimatainen, ACS Appl. Mater. Interfaces, 2017, 9, 25029–25037 CrossRef CAS PubMed.
- S. Long, Y. Feng, Y. Liu, L. Zheng, L. Gan, J. Liu, X. Zeng and M. Long, Sep. Purif. Technol., 2021, 254, 117577 CrossRef CAS.
- P. Li, M. Zhou, X. Zhou, X. Li, Y. Wang and B. Zhou, Int. J. Biol. Macromol., 2024, 277, 133879 CrossRef CAS PubMed.
- W. Chen, S. Yuan, J. Shen, Y. Chen and Y. Xiao, Front. Bioeng. Biotechnol., 2021, 8 DOI:10.3389/fbioe.2020.627351.
- G. Agoda-Tandjawa, S. Durand, C. Gaillard, C. Garnier and J. L. Doublier, Carbohydr. Polym., 2012, 90, 1081–1091 CrossRef CAS PubMed.
- S. Ye, Z. Zhu, Y. Wen, C. Su, L. Jiang, S. He and W. Shao, Polymers, 2019, 11, 57 CrossRef PubMed.
- S. Groult, S. Buwalda and T. Budtova, Biomater. Adv., 2022, 135, 212732 CrossRef CAS PubMed.
- J.-P. Touzel, B. Chabbert, B. Monties, P. Debeire and B. Cathala, J. Agric. Food Chem., 2003, 51, 981–986 CrossRef CAS PubMed.
- E. Chanliaud and M. J. Gidley, Plant J., 1999, 20, 25–35 CrossRef CAS PubMed.
- M. S. Dayal and J. M. Catchmark, Carbohydr. Polym., 2016, 144, 447–453 CrossRef CAS PubMed.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, S. Camarero-Espinosa, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts, J. A. Shatkin, J. Simonsen, K. Stinson-Bagby, N. Wanasekara and J. Youngblood, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- J. B. Hopkins, J. Appl. Crystallogr., 2024, 57, 194–208 CrossRef CAS PubMed.
- SasView, SasView, https://sasview.github.io/.
- S. Mallam, F. Horkay, A. M. Hecht, A. R. Rennie and E. Geissler, Macromolecules, 1991, 24, 543–548 CrossRef CAS.
- C. A. Maestri, M. Abrami, S. Hazan, E. Chisté, Y. Golan, J. Rohrer, A. Bernkop-Schnürch, M. Grassi, M. Scarpa and P. Bettotti, Sci. Rep., 2017, 7, 11129 CrossRef CAS PubMed.
- M. Shibayama, T. Tanaka and C. C. Han, J. Chem. Phys., 1992, 97, 6842–6854 CrossRef CAS.
- D. Johansen, J. Trewhella and D. P. Goldenberg, Protein Sci., 2011, 20, 1955 CrossRef CAS PubMed.
- S. Morozova, E. Hitimana, S. Dhakal, K. G. Wilcox and D. Estrin, J. Appl. Phys., 2021, 129, 071101 CrossRef CAS.
- D. C. Schoenmakers, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2018, 9, 2172 CrossRef PubMed.
- G. N. Balistreri, I. R. Campbell, X. Li, J. Amorim, S. Zhang, E. Nance and E. Roumeli, RSC Appl. Polym., 2024, 2, 172–183 RSC.
- N. E.-A. El-Naggar, A. B. A. Mohammed and S. E. El-Malkey, Sci. Rep., 2023, 13, 51 CrossRef CAS PubMed.
- A. Mandal, K. Liao, H. Iyer, J. Lin, X. Li, S. Zhang and E. Roumeli, Mol. Syst. Des. Eng., 2024, 9, 1036–1050 RSC.
- T. Benselfelt, N. Kummer, M. Nordenström, A. B. Fall, G. Nyström and L. Wågberg, ChemSusChem, 2023, 16, e202201955 CrossRef CAS PubMed.
- H. Wang, J. Li, X. Zeng, X. Tang, Y. Sun, T. Lei and L. Lin, Cellulose, 2020, 27, 1301–1314 CrossRef CAS.
- J. Li, T. Song, H. Xiu, R. Cheng, X. Yang, Q. Liu, X. Zhang, E. Kozliak and Y. Ji, Wood Sci. Technol., 2019, 53, 837–854 CrossRef CAS.
- N. Lavoine and L. Bergström, J. Mater. Chem. A, 2017, 5, 16105–16117 RSC.
- P. Munier, K. Gordeyeva, L. Bergström and A. B. Fall, Biomacromolecules, 2016, 17, 1875–1881 CrossRef CAS PubMed.
- H. Lu, X. Wang, T. Zhang, Z. Cheng and Q. Fang, Materials, 2009, 2, 958–977 CrossRef CAS.
- Y. Zhang and Y. Zhao, Materialia, 2019, 6, 100286 CrossRef CAS.
- J. Zhang, R. J. Perez and E. J. Lavernia, J. Mater. Sci., 1993, 28, 2395–2404 CrossRef CAS.
- V. Guccini, S. Yu, M. Agthe, K. Gordeyeva, Y. Trushkina, A. Fall, C. Schütz and G. Salazar-Alvarez, Nanoscale, 2018, 10, 23157–23163 RSC.
- P. Lang and E. Frey, Nat. Commun., 2018, 9, 494 CrossRef PubMed.
- Z. Aydin and J. Akbugǎ, Int. J. Pharm., 1996, 137, 133–136 CrossRef CAS.
- P. Sriamornsak, Silpakorn University International Journal, 2003, 3, 206–228 Search PubMed.
- F. Daver, M. Kajtaz, M. Brandt and R. A. Shanks, Polymers, 2016, 8, 437 CrossRef PubMed.
- K. S. Fancey, J. Mater. Sci., 2005, 40, 4827–4831 CrossRef CAS.
- K. S. Fancey, J. Polym. Eng., 2001, 21, 489–510 CAS.
- C. R. Vithanage, M. J. Grimson, B. G. Smith and P. R. Wills, J. Phys.:Conf. Ser., 2011, 286, 012008 CrossRef.
- R. Palin and A. Geitmann, BioSystems, 2012, 109, 397–402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00348a |
| This journal is © The Royal Society of Chemistry 2025 |