
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking a radioactive pertechnetate (TcO4−) treatment process with functionalized metal–organic frameworks (MOFs)
Kankan
Patra
 *ab,
Samraj
Mollick
c,
Arijit
Sengupta
bd and
Satya R.
Guchhait
ab
*ab,
Samraj
Mollick
c,
Arijit
Sengupta
bd and
Satya R.
Guchhait
ab
aNuclear Recycle Board, Bhabha Atomic Research Centre, Tarapur 401504, India. E-mail: kankan.patra2010@gmail.com
bHomi Bhabha National Institute, Anushaktinagar, Mumbai 400 094, India. E-mail: arijitbarc@gmail.com
cMultifunctional Materials & Composites (MMC) Laboratory, Department of Engineering Science, University of Oxford, Parks Road, Oxford, OX1 3PJ, UK. E-mail: samraj.mollick@eng.ox.ac.uk
dRadiochemistry Division, Bhabha Atomic Research Centre, Mumbai 400 085, India
First published on 30th January 2025
Abstract
Technetium-99 (99Tc), a troublesome radioisotope prevalent in nuclear liquid waste, poses significant environmental and human health hazards due to its long half-life, high fission yield, and fast environmental mobility. The successful mitigation of 99Tc is imperative for nuclear waste management; however, it continues to present a significant obstacle. In this comprehensive review, we explore the state-of-the-art developments in separating TcO4− ions using functionalized metal–organic framework (MOF) materials, spanning from 2010 to the present. We delve into the intricate separation mechanisms of TcO4− ions, shedding light on advanced research avenues in this field. Furthermore, we aim to provide a comprehensive understanding of the underlying receptor chemistry that is necessary for the specific targeting of pertechnetate anion-based materials. This will provide valuable insights into the molecular characteristics that are crucial for the separation of TcO4− ions from solutions containing nuclear waste. The review outlines perspectives and conclusions that pave a promising path for the comprehensive investigation of materials poised to revolutionize TcO4− separation. Finally, we provide forward-looking recommendations for future research directions, opportunities, and associated challenges, to encourage more researchers to leverage TcO4− selective materials for better management of environmental pollution.
Introduction
In the ongoing pursuit of developing alternative and sustainable renewable energy sources, nuclear power stands out as a viable option because of its cost-effectiveness and minimal greenhouse gas emissions.1 However, the effective management of radioactive waste remains a critical bottleneck, hindering the widespread adoption of nuclear energy on an industrial scale.2 The contamination of water with various radioisotopes presents a significant threat to both the environment and human health. Over recent decades, the release of radioactive materials into the environment has engendered health hazards, underscoring the urgent need for a safe, secure, and ecologically responsible radioactive waste management system to uphold the viability of nuclear energy. Of particular concern among these radionuclides is technetium-99 (99Tc), which emerges as a pivotal and pressing concern in liquid radioactive waste. Present in trace quantities in the environment, 99Tc finds its genesis in nuclear processes, particularly those involving uranium and plutonium fission.3,4 Approximately 6% of the total fission product yield comprises 99Tc, with spent nuclear fuel treatment yielding 99TcO4− ions, the stable and common oxidation state of 99Tc in aqueous solutions.5 The water solubility nature of 99TcO4− ions, particularly in the form of sodium salt, poses a challenge for environmental containment, facilitating its mobility within the Earth's crust and potential entry into the food chain. The implications of 99TcO4− ions' behavior in aqueous environments extend far beyond mere solubility.6 Their mobility within the soil and potential uptake by plants and aquatic organisms underscore the need for effective remediation strategies.6 Despite certain geochemical processes like the formation of insoluble colloids in anaerobic soils offering some level of containment, the persistent and mobile nature of 99TcO4− ions necessitates further attention to mitigate their environmental impact, particularly in the context of nuclear waste management.7 Successfully addressing these challenges associated with nuclear power is crucial for maximizing its benefits while mitigating potential environmental and health impacts. By effectively addressing these challenges, nuclear power can play a significant role in transitioning to a low-carbon energy future while ensuring environmental sustainability and public safety.In this context, anion-exchanging materials have been extensively studied for the treatment of nuclear waste streams, namely the elimination of the 99Tc species as TcO4−.8,9 The effectiveness of several materials in removing 99Tc from nuclear waste solutions has been studied. These include ion-exchange resins such as Purolite-A-520E or superLig-639, layered double hydroxides (LDHs), modified natural clays,10–12 graphene-based materials, and others.13–15 Many of these adsorbents have limitations despite these efforts, including poor radiation resistance, delayed anion-exchange kinetics, and low sorption capacity and selectivity. As a result, the environmental risks associated with 99Tc at nuclear facilities persist despite substantial study and development. With the development of advanced porous materials, such as metal–organic frameworks (MOFs), there is significant promise for environmental remediation.16–19 MOFs represent a subset of advanced porous materials formed by combining metal ions or clusters with organic struts. Their versatility spans across various domains, from gas storage and drug delivery to catalysis and environmental cleanup. This material is an enormous advance over conventional adsorbents, which do not have the tailored specificity that these advanced MOF materials possess.20 Even while this strategy could incur extra expenditure, the advantages of increased capacity and selectivity greatly outweigh these costs. Beyond just integrating functional groups, these materials can also be tailored by introducing secondary sphere interactions to improve binding, controlling pore wettability to improve sorption performance, and designing pore structures to increase charge density (Fig. 1a). These porous materials are adaptable for a variety of applications because their structure and composition can be changed at the atomic scale, and their structure and properties can be fully understood. MOFs possess promising properties, positioning them as prime contenders for tackling pollution challenges, particularly in removing contaminants like 99TcO4− from water sources, thus contributing to a more sustainable nuclear power future. Fig. 1b presents the recognition and capture of TcO4− using functionalized MOFs. Amidst the landscape of radionuclide separation research, limited attention has been directed toward the specific adsorption performance of materials targeting 99TcO4− ions. In this context, our research group presented a comprehensive analysis of MOFs utilized for the removal and remediation of radionuclides, encompassing the adsorption mechanisms of Cs, Sr, U, Tc, Pu, Am, Eu, Xe, and Kr as radionuclides.21
 | ||
| Fig. 1 (a) 99TcO4− adsorption efficiency can be increased by the following methods: designing pore architectures to boost charge density; adding secondary sphere interactions to fortify binding; and regulating pore wettability to enhance binding affinity. (b) Recognition and capture of 99TcO4− using MOFs. | ||
Very recently, our group likewise reported a comprehensive review on the management of radioactive waste, which detailed various critical aspects of radioactive metal adsorption and mechanisms of interaction.22,23 Although there exist a limited number of papers in this particular domain, there is still a lack of comprehensive reviews that specifically address the targeted removal and remediation of 99TcO4− using advanced porous materials such as functionalized MOFs.
Against this backdrop, our review aims to fill this critical gap by providing a comprehensive overview of the current state-of-the-art in 99TcO4− separating materials, with a specific focus on functionalized MOFs. By summarizing reports from 2010 to the present, we offer insights into the mechanisms of interaction and key aspects of MOFs relevant to 99TcO4− separation. We have represented different modes of interaction and sensing of 99TcO4− ions using MOFs. Fig. 2 emphasizes the potential of these materials for adsorption behaviors and metal ion separation mechanisms, and we delve into recent breakthroughs and highlight the challenges and future research directions in this burgeoning field. Through our comprehensive analysis, we aim to contribute to the advancement of nuclear waste management and environmental safety, underscoring the crucial role of functionalized MOFs in addressing the challenges posed by 99TcO4− ions in aqueous environments.
 | ||
| Fig. 2 Different mechanisms of TcO4− adsorption using functionalized MOFs. | ||
Current status TcO4− treatment
TcO4− ions are generated during the fission of uranium in the nuclear fuel cycle.24 One of the most challenging contaminants to handle at the nuclear waste management site is 99Tc, which is insoluble in storage along with uranium and plutonium. This is because of its complex chemical behavior in waste solution and its limited incorporation into a glass waste form during high-temperature vitrification processes because of its high volatility. In conventional treatment approaches, 99Tc is typically recovered from off-gas condensates generated during high-temperature vitrification processes. These processes aim to incorporate Tc-99 into a glass waste form, but its high volatility results in low concentrations of Tc-99 in the final glass product. To address this issue, current technology involves capturing Tc-99 from off-gas streams and either reintroducing it into the vitrification facility or processing it into a low-temperature waste form.Depending on the properties of the 99Tc bearing phase, the captured 99Tc can either be sent back for vitrification or encapsulated in a different waste matrix. Various methods are employed to extract and remove 99TcO4−, including ion exchange, chemical extraction, and precipitation.24–26 Among these, ion exchange has garnered significant attention due to its high recovery rate and relative ease of implementation.24 Recent efforts have focused on the use of anion-exchange resins, such as Superlig, to remove 99TcO4− from nuclear waste, particularly from Hanford tank wastes.27–30 Despite the successful removal of 99TcO4− from simulated nuclear waste streams, further research is required to enhance the selectivity and capacity of these ion-exchange systems, demonstrating the ongoing challenges in optimizing 99Tc management in nuclear waste treatment processes.
MOFs for TcO4−/RuO4− removal
In radioactive liquid legacy nuclear waste, 99Tc is one of the most hazardous and still unsolved problems for nuclear industries. Effective management of radioactive liquid waste requires the successful removal and remediation of 99TcO4− ions, which are hazardous and pose a significant challenge. MOF-based materials have demonstrated a remarkable capability for removing and remediating radionuclides, specifically oxyanions such as 99TcO4− ions.20,30,31 In recent times, MOF based materials have been employed for the adsorption of TcO4− ions from radioactive waste solution. MOFs possess advantages over other porous materials such as zeolites and activated carbons because of their hybrid nature and synthetic modularity. This enables the development of distinct materials with diverse chemical compositions, pore diameters, and thermal stability.32 Certain MOFs possess ionic frameworks, which can be attributed to the presence of extra-framework ions, ionic side chains, or a stoichiometric imbalance between metal centers and organic linkers.33 Although metal ions and functionalized organic linkers offer a wide range of structural possibilities in MOFs, their limited thermo-chemical stability when subjected to realistic ion-exchange conditions poses difficulties for their effective use in practical applications.TcO4−/RuO4− removal using MOFs through selective anion exchange
MOFs have gained significant attention for a variety of applications, including ion exchange processes and environmental remediation due to their high surface area, tunable porosity, and the ability to incorporate functional groups. One promising application of MOFs is their use as ion exchange materials for the removal of toxic or radioactive ions from water, including TcO4− ions. Certain types of i-MOFs have the ability to undergo ion exchange without altering their original structure. Ion exchange refers to the reversible replacement of ions in a material with ions of similar charge. In MOFs, the metal centers or functionalized sites on the organic linkers can be targeted for ion exchange. MOFs have tunable structures and chemical environments that can selectively bind and exchange specific ions. The high porosity of MOFs also provides a large surface area for ion exchange, improving efficiency in removing contaminants like TcO4− ions. The ion exchange mechanism in MOFs is dependent on the specific exchange sites present. The process can vary from diffusion-controlled solid-state exchange in ion-exchange resins or zeolites to solvent-mediated dissolution and re-crystallization.27 When utilizing MOFs for practical purposes, it is essential to take into account the critical aspects of selective ion exchange and basic ion exchange capabilities. MOF-based ion exchangers can achieve ion selectivity through several mechanisms, such as size exclusion and specific binding within their pores or channels. Size exclusion selectivity in MOF materials arises when ions are too large to penetrate, while preferential binding is a result of electronic interactions, such as open metal sites. MOFs typically exchange ions through van der Waals or long-range electrostatic interactions. The selectivity of the exchange is mostly dictated by the way ions are desolvated in the bulk solution and within the pores. We will cover some potential examples of MOF materials to discuss anion exchange processes for TcO4− ion removal.27,34,35Fig. 3 illustrates different processes involved in the adsorption of TcO4−/RuO4− ions using MOF based materials. | ||
| Fig. 3 MOF-based material for removal of TcO4−/RuO4−. | ||
Over the last two decades, several cationic MOFs have been developed to separate anionic pollutants.2,36–39 Since ReO4− and 99TcO4− have comparable sizes and chemical compositions, ReO4− is used in separation experiments as a substitute for 99TcO4−. Very high extraction ability and selectivity for ReO4− ions by a unique cationic MOF SLUG-21 [Ag2(4,40-bipy)2(O3SCH2CH2SO3)3·4H2O] was reported, with a sorption capacity of 602 mg g−1.40 According to them, framework flexibility plays the key role in such behavior and they have also shown high potential for oxo metal adsorption. Highly stable robust Zr-based cationic MOF for removal and remediation of ReO4− ions from an aqueous medium even in the presence of NO3− and SO42− competing anions was reported.41 The Zr-based UiO-66-NH2 MOF has demonstrated a significant advantage in its ability to adsorb ReO4− ions, with an impressive uptake capacity of 159 mg g−1. This capacity far surpasses that of other commonly used inorganic adsorbing materials, such as layered double hydroxides.37 Subsequently, to enhance the selectivity, an 8-fold interpenetrated 3D cationic MOF material (SCU-100) was reported.42 This was achieved by combining Ag+ metal as a node with a tetradentate N-donor ligand (Fig. 4). The uptake capacity for ReO4− was found to be 541 mg g−1 and 1.9 × 105 mL g−1 in an aqueous medium, respectively, which significantly surpassed that of other standard inorganic anion exchange materials. Another possible MOF, [Ag(4,4′-bipyridine)(NO3)], was reported to enhance the absorption efficiency of ReO4− ions. This MOF had a sorption capacity of 786 mg g−1 (Table 1).37
 | ||
| Fig. 4 (a) The stability of the SCU-100 MOF with respect to different pH and radiation. (b) Its sorption properties with respect to TcO4− as a function of contact time. (c) The transformation of a single crystal into another single crystal throughout the sorption process. Reproduced from ref. 37 with permission from American Chemical Society. (d) UV-vis spectra of 99TcO4− ions during the anion exchange with the SCU-102 MOF, plotted against contact time. (e) A comparison of the ReO4− sorption kinetics by Purolite A532E, SCU-102, and A530E resins. Reproduced with permission from ref. 51. | ||
| MOFs | Mechanism | Contact time | Capacity (mg g−1) | Selectivity | Ref. |
|---|---|---|---|---|---|
| UiO-66-NH2 | Ion-exchange | 24 h | 159 | Vs. NO3−, SO42−, PO43− | 36 |
| SCU-102 (Ni2(tipm)3(NO3)4) MOF | Ion-exchange | 30 min | 291 | — | 44 |
| SLUG-21 | Ion-exchange | NA | 602 | — | 40 |
| SBN | Ion-exchange | <10 min | 786 | Vs. NO3−, SO42−, CO32− | 37 |
| SCU-100 | Ion-exchange | 2 h | 541 | Vs. NO3−, CO32−, PO43− | 37 |
| MOF TJNU-216 (TJNU = Tianjin Normal University) | Ion-exchange | >6 h | 417 | — | 38 |
| SCU-101 | Ion exchange | 10 min | 217 | Vs. various anions | 37 |
| Zr6-MOF NU-1000 | Ion-exchange | — | 210 | — | 2 |
| iMOF-2C | Ion-exchange | 10 | 87 | Cl−, NO3−, Br−, SO42−, ClO4−, CO32− | 45 |
| MOF 808 | — | 5 | 85 | N.D | 46 |
| UiO-66-HCl | — | 600 | 86.8 | N.D | 47 |
| iMOF-3C | — | 5 | 73 | Cl−, NO3−, Br−, SO42−, ClO4−, CO32− | 48 |
| iMOF-1C | — | 4320 | 100 | Cl−, HCO3−, NO3−, SO42−, CO32− | 49 |
| CAU-17 | — | 600 | 20.3 | NO2−, Cl−, F−, NO3−, PO43−, SO42−, CO32−, Ac−, LAS−, B4O72−, HPO42− | 50 |
For the purpose of enhancing selectivity, a MOF (TJNU-216, TJNU = Tianjin Normal University) has been reported.51 This MOF exhibits remarkable selectivity for ReO4− and TcO4− ions in aqueous waste water, even when there are high concentrations of SO42− ions (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times) or NO3− ions (300 times). This material exhibits an impressive exchange capacity of 417 mg g−1 for these oxyanions. In order to investigate the TJNU-216 material and ion exchange of ReO4− ions, TJNU-216 crystals (30 mg, or about 0.05 mmol) were added to a 20 mL water solution containing 58 mg, or approximately 0.2 mmol, of KReO4 and it was analyzed through FT-IR, PXRD, and SEM-EDS mapping. It was observed that within one minute of anion exchange, a new strong infrared peak at 897 cm−1 emerges, as Fig. 4a illustrates, indicating the synthesis of ReO4− loaded TJNU-216. The materials' good crystallinity was shown by the SEM images and PXRD patterns (Fig. 4b). ReO4− uniformly disperses throughout the TJNU-216 crystals with the coexistence of CF3SO3.51 The transformation of a single crystal into another single crystal throughout the sorption process is described in Fig. 4c.
000 times) or NO3− ions (300 times). This material exhibits an impressive exchange capacity of 417 mg g−1 for these oxyanions. In order to investigate the TJNU-216 material and ion exchange of ReO4− ions, TJNU-216 crystals (30 mg, or about 0.05 mmol) were added to a 20 mL water solution containing 58 mg, or approximately 0.2 mmol, of KReO4 and it was analyzed through FT-IR, PXRD, and SEM-EDS mapping. It was observed that within one minute of anion exchange, a new strong infrared peak at 897 cm−1 emerges, as Fig. 4a illustrates, indicating the synthesis of ReO4− loaded TJNU-216. The materials' good crystallinity was shown by the SEM images and PXRD patterns (Fig. 4b). ReO4− uniformly disperses throughout the TJNU-216 crystals with the coexistence of CF3SO3.51 The transformation of a single crystal into another single crystal throughout the sorption process is described in Fig. 4c.
Tailoring MOFs: from Zr-based frameworks to host–guest assemblies
The role of tailoring MOFs, specifically transitioning from Zr-based frameworks to host–guest assemblies for removal of TcO4− ions, is significant in modifying the framework to optimize ion adsorption, selectivity, and capacity. By incorporating specific guest molecules and enhancing interactions between the MOFs and the target ions, the removal efficiency can be significantly improved, making these materials promising candidates for TcO4−/RuO4− remediation. In this context, an aqueous-stable Zr-MOF NU-1000 has been developed by Farha et al.2 to enhance the stability of the adsorbent in water and improve its uptake efficiency. This remarkable MOF demonstrated an impressive uptake efficiency of 210 mg g−1 and maintained a stable framework in aqueous environments. To enhance the adsorption kinetics, a Ni2(tipm)3(NO3)4 MOF (SCU-102), based on nickel, was synthesized by Shuao Wang et al. by combining Ni2+ ions as metal nodes with a hydrophobic tetradentate neutral ligand as a linker.43 The material exhibited a high distribution coefficient, fast sorption kinetics, impressive sorption capacity (291 mg g−1), and exceptional selectivity for TcO4− ions in an aqueous medium. The substance effectively removed TcO4− ions from the aqueous medium, even in the presence of SO42− and NO3− ions (Fig. 4d and e).The structural investigation confirmed that the presence of the Zr metal cluster is crucial for the separation of TcO4− in the MOF. The ligand exchange mechanism involved the replacement of hydroxyl (–OH) and water (–OH2) on the Zr nodes in the MOF by ReO4−. The structural analysis revealed that ReO4− ions form coordination bonds with the metal centres via both chelating and non-chelating (monodentate) modes. In this process, the ions replace weakly bound hydroxy groups and water molecules. Upon the adsorption of ReO4− ions within the MOF, they became immobilized and formed an insoluble solid. Furthermore, they could not be displaced by NO3− ions at a concentration 1000 times higher.
Du et al. successfully incorporated ion-exchangeable sites into MOFs using an innovative host–guest assembly approach.52 This method entails the polymerization of ionic monomers within the pore channels of a MOF host. They specifically enclosed a range of ionic liquids based on vinyl-functionalized imidazolium into the pores of MIL-101, followed by in situ polymerization. The optimized composite exhibited remarkable resilience throughout several regeneration cycles (over 30 cycles), rapid sorption kinetics (under 30 seconds), and outstanding ReO4− adsorption capabilities, even under extremely acidic conditions and simulated recycle streams. It was also able to eliminate 74% of ReO4− from a hypothetical Hanford LAW melter recycle stream. This advancement offers a versatile framework for creating high-performance composites for sorption applications, while also expanding the potential applications of MOFs in the containment of radioactive materials (Fig. 5a).52
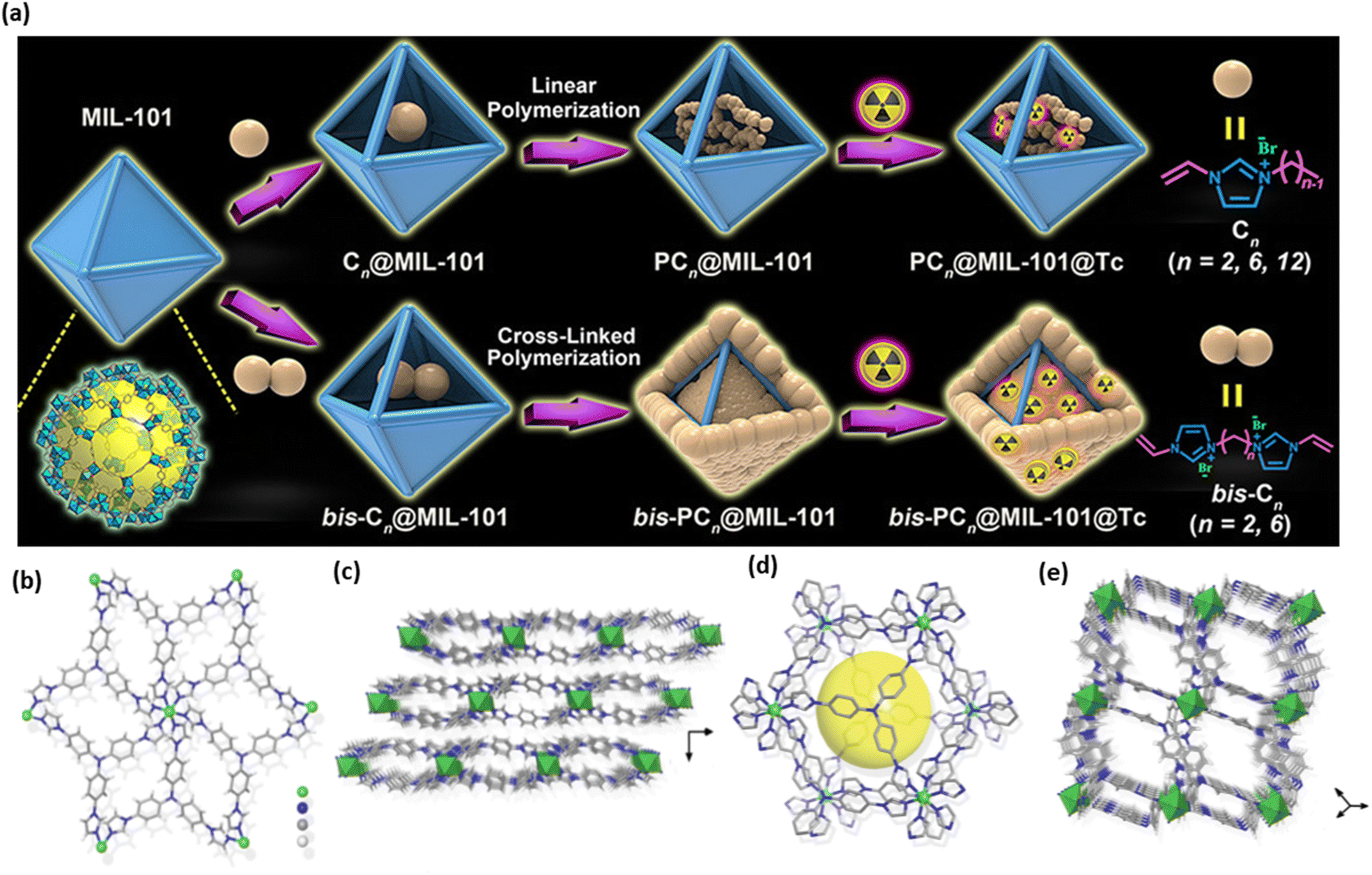 | ||
| Fig. 5 (a) Encapsulated imidazolium-based ILs in the pores of MOFs (MIL-101) was selected as the representative MOF in this design process in order to create a polymerization method for the composites of polyILs@MOFs for radionuclide sequestration. Reproduced from ref. 52 with permission from American Chemical Society. (b) The coordination environment of the Ni2+ ion with six ligands in the crystal structure of SCU-103. (c) The SCU-103 2D layer packing schematic. (d) A zoomed-in picture of the cavity created by the concave–convex 2D layers coupling face to face. (e) Porous channels are visible in the packing perspective picture of SCU-103. Reproduced from ref. 51. | ||
Overcoming stability and selectivity hurdles: stable MOF design for efficient TcO4−/RuO4− removal
The development of stable and selective MOFs for the removal of TcO4− and RuO4− ions requires overcoming key challenges related to the stability of the frameworks under environmental conditions and the need for high selectivity toward these ions. By carefully designing MOFs with tailored functional groups, redox-active centers, and appropriate pore structures, these challenges can be addressed, leading to highly efficient materials for ion removal. Further advances in MOF engineering, including hybrid materials and dynamic frameworks, could further enhance their performance in environmental and industrial applications. In this context, to improve the uptake efficiency along with selectivity, a 2D cationic MOF has been reported using Ni2+ ions, specifically [Ni(tipa)2](NO3)2 (SCU-103), and the tridentate N ligand tris[4-(1H-imidazole-1-yl)-phenyl]amine (tipa).51 It was reported that 99TcO4− was successfully separated from genuine historical nuclear waste at SRS. It also demonstrated remarkable resistance to both beta and gamma radiation. This solves the drawback of the low alkaline stability of reported cationic MOF materials. The removal and remediation of 99TcO4− ions from actual tank waste has been accomplished, and more significantly, it was highly effective for 99TcO4− separation from aqueous medium as evidenced by fast kinetics, high uptake capacity, and excellent selectivity. According to hard–soft-acid–base theory,53 the combination of high-valent metal ions (hard Lewis bases and acids) and carboxylate groups can help to form strong coordination bonds, providing frameworks with high stability in concentrated acids or even in neutral aqueous solutions.54–57Fig. 5b depicts the coordination environment of the Ni2+ ion with six ligands in the crystal structure of SCU-103. Fig. 5c shows the schematic for SCU-103's 2D layer packing. Fig. 5d shows the image of the cavity formed by concave–convex 2D layers that are coupled face to face. Porous channels were observed in the packing perspective image of SCU-103 (Fig. 5e). Nevertheless, these materials generally undergo decomposition under alkaline environments. The stability of MOFs is directly influenced by the strength of the coordination bonds and the structural disintegration caused by the competing coordination capability between organic ligands and other molecular species or anions.58,59 Due to their substantial affinity for high-valent metal ions, hydroxide (OH−) anions can easily replace carboxylate groups that are associated with high-valent metal ions. This substitution process leads to the disintegration of most of the MOF under alkaline conditions. Hence, there was a great desire for transition metal ions and organic ligands that exhibited enhanced softness in order to attain satisfactory alkaline stability. In this scenario, the coordination bonding between metal ions and linkers is sufficiently robust to resist the competition from OH− anions, resulting in a considerable reduction in the binding interaction between OH− anions and metal ions.58 MOFs composed of nitrogen heterocyclic ligands with high pKa values and transition metal ions exhibited this characteristic to a significant extent.60,61In relation to this, to improve the framework stability, by complexing 2-aminoterephthalic acid with Al(III) metal via a solvothermal technique, Chao Xue et al. synthesized an Al-based MOF, termed CAU-1MOF, to remove UO22+ and 99TcO4− ions from aqueous solution.62Fig. 6a shows the CAU-1 MOF crystal structure. The CAU-1 structure possesses a large surface area (1636.3 m2 g−1) with micropore volume (0.51 m3 g−1), rich organic functional groups (–NH2 and –OH groups), and excellent thermal stability. Additionally, batch experiments showed that the CAU-1 MOF has a high adsorption capacity for ReO4− (692.33 mg g−1) and UO22+ (648.37 mg g−1). Thermodynamic analysis revealed that the adsorption process was spontaneous and endothermic. The adsorption mechanism of ReO4− ions onto CAU-1 involved both electrostatic attraction and chelation effects, whereas the primary mechanism for UO22+ ion adsorption was the chelation effect generated by functional groups containing both nitrogen and oxygen. Therefore, the low-cost, high-capacity CAU-1 material may be seen as a workable solution for removing radioactive contaminants from aquatic environments.62
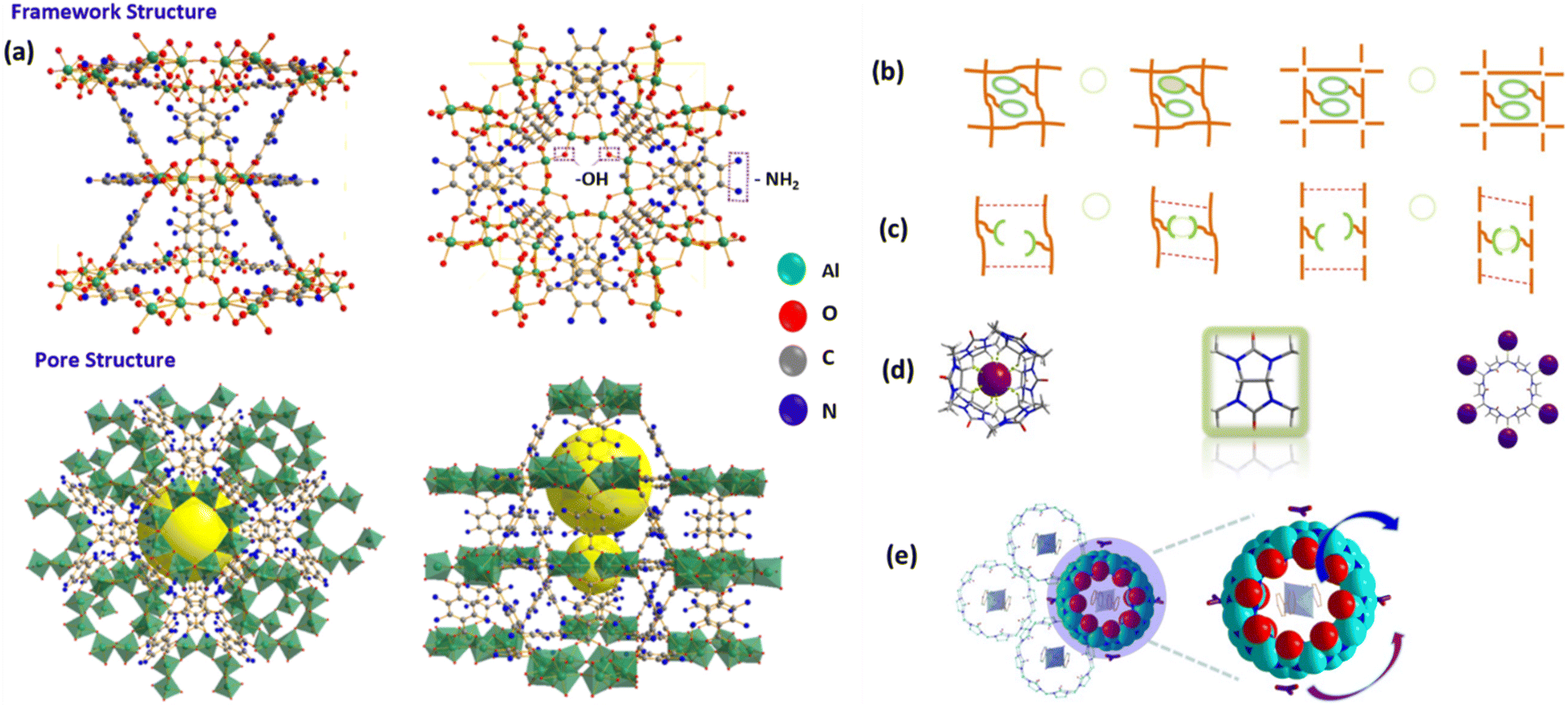 | ||
| Fig. 6 (a) CAU-1 MOF crystal structure. The figure is reproduced from ref. 62 with permission from Elsevier. (b) Modification of metal–organic sorbents with macrocyclic motifs that are capable of recognizing anions (green circles represent anion receptors, orange lines represent solid sorbent backbones, purple balls represent anions, and blue balls represent metal nodes). (c) A supramolecular construction of metal–organic sorbents or smart polymers that display anion-adaptive behaviors (green curves represent anion-recognition sites, orange lines represent solid sorbent backbones, purple circles represent anions, and blue balls represent metal nodes). (d) Two distinct ways of anion recognition using glycoluril-based macrocyclic hosts (purple balls: anions). (e) CB8 plays two roles: it encapsulates molecules to create a supramolecular network and provides contact sites for anion recognition on the exterior: full view of the CB8-based supramolecular framework is shown on the left; an expanded diagram of CB8 illustrating its interactions with the surrounding elements is shown on the right (red balls represent O atoms, light-blue balls represent C atoms, and dark blue balls represent N atoms). The figure is reproduced from ref. 63. | ||
Given this situation, to boost the framework stability, selectivity, and uptake efficiency for TcO4− ions from aqueous medium, Shi et al. reported the functionalization of SCP-IHEP-1 ([Cu((bpy)2@CB8) (H2O)4](NO3)2·18H2O), a cationic supramolecular MOF based on cucurbit[8]uril (CB8).63Fig. 6b depicts the direct alteration of metal–organic sorbents or organic polymers with macrocyclic motifs capable of detecting anions. Fig. 6c shows the supramolecular structure of metal–organic sorbents or smart polymers with anion-adaptive properties. Two independent methods of anion recognition employing glycoluril-based macrocyclic hosts (purple balls represent anions) were presented (Fig. 6d). The author of this study discusses the dual role of CB8. It acts as a container for molecules, forming a supramolecular network, while also serving as a contact point for anion recognition on its exterior (Fig. 6e). This framework was synthesized through the supramolecular collaborative assembly of CB8, 4,4′-bipyridine (bpy) and Cu(NO3)2 under hydrothermal conditions. The anion-adaptive ability of this supramolecular sorbent allows for effective recognition of TcO4−, which is comparable to the dynamic behavior of the receptor during ion recognition. The batch kinetics experiment reveals that SCP-IHEP-1 effectively eliminates ReO4− by following the pseudo-first-order model. It achieves a removal rate of over 95% after 10 minutes and 88% after 1 minute. SCP-IHEP-1 demonstrates superior ReO4− exchange kinetics compared to other cationic metal–organic materials such as SLUG, UiO-66-NH3+, and Ni(II)-based MOFs. These materials require more than 24 hours to achieve ReO4− exchange equilibrium, but SCP-IHEP-1 does it at a faster rate. When compared to SCU-101 (ref. 37) and polymeric network-type SCU-CPN-11,64 SCP-IHEP-1 demonstrates exceptional selectivity for ReO4− over competing anions (NO3− and SO42−) and has recently shown remarkable efficacy in removing ReO4−/TcO4−. Considering this, the supramolecular framework demonstrated potential for specifically adsorbing TcO4− from liquid waste, even in the presence of large quantities of competing anions (SO42− and NO3−). An examination of the crystal structure of SCP-IHEP-1-Re revealed that ReO4− was enclosed within tetrahedral pores, which were encircled by four neighboring CB8 molecules. The pores are stabilized by a cluster of C–H⋯O hydrogen bonds formed between the oxygen atoms of the anions and the CH and CH2 groups on the outer surface.63
MOFs for ReO4−/TcO4− removal: from structural design to surface functionalization
Through careful structural design of MOFs, including tuning the pore size and stability of the framework, and the surface functionalization with specific chemical groups, it is possible to create highly selective, stable, and effective materials for ion removal. The incorporation of redox-active sites, ion-exchange properties, and size-selectivity further enhances the removal efficiency, making MOFs an attractive choice for ReO4−/TcO4− removal applications. In light of this, Jie Li et al. and colleagues have proposed a method to enhance the practical uses of MOFs by transforming them into versatile materials with desirable properties.65Fig. 7a depicts the procedure used to synthesize MILP composites. A low-cost and simple-to-operate approach was employed to simultaneously extract uranium (UO22+) and rhenium (ReO4−) from water using a customized poly(ethyleneimine) (PEI) MIL-101(Cr) (MILP). The sorption performance of MILP composites was evaluated by studying the impacts of varying the amount of PEI coating, system pH, contact time, initial UO22+/ReO4− concentrations, ionic strength, and interfering ions. The maximum sorption capacities for UO22+ and ReO4− were 416.67 mg g−1 and 434.78 mg g−1, respectively, which exceeded those of the majority of adsorbents. The absorption of UO22+/ReO4− occurred spontaneously, required input of heat, and was influenced by the pH of the solution. The pseudo-second-order (PSO) kinetic equation and Freundlich isotherm equation were found to be the most suitable models for describing this process. The presence of UO22+ and high ion strength hindered the adsorption of ReO4−. The UO22+/ReO4− sorption in the MILP-3 composites was shown to be influenced by the presence of unsaturated metal sites and amino groups, as determined by X-ray photoelectron spectroscopy (XPS) analysis and batch studies. MILP-3 exhibited remarkable recycling and sorption abilities for UO22+/ReO4− in several simulated water samples. This study laid the foundation for developing a highly advanced group of candidates that may be used in many ways for treating waste water. It showed the effective removal of radioactive metal anions and cations from waste water using MILP composites. The MILP-3 material, after being exposed to ReO4−, was subjected to SEM-EDX examination. This study revealed the existence of Re throughout the material and verified the adsorption of ReO4− ions. Fig. 10b depicts the interaction between ReO4− and cavities, showcasing both the electrostatic attractions and coordination between ReO4− and amino groups. The main factors contributing to the effective adsorption of UO22+ in MIL-101(Cr) are the coordination with CUSs, the creation of N\\H⋯O hydrogen bonds, and the C\\N⋯O complexation between N-related sites and UO22+. Fig. 7b shows a graphical representation of the probable processes of UO22+ sorption onto MILP-3. In summary, the adsorption process of UO22+ or ReO4− was mostly enhanced by the presence of N-containing groups and cavities.65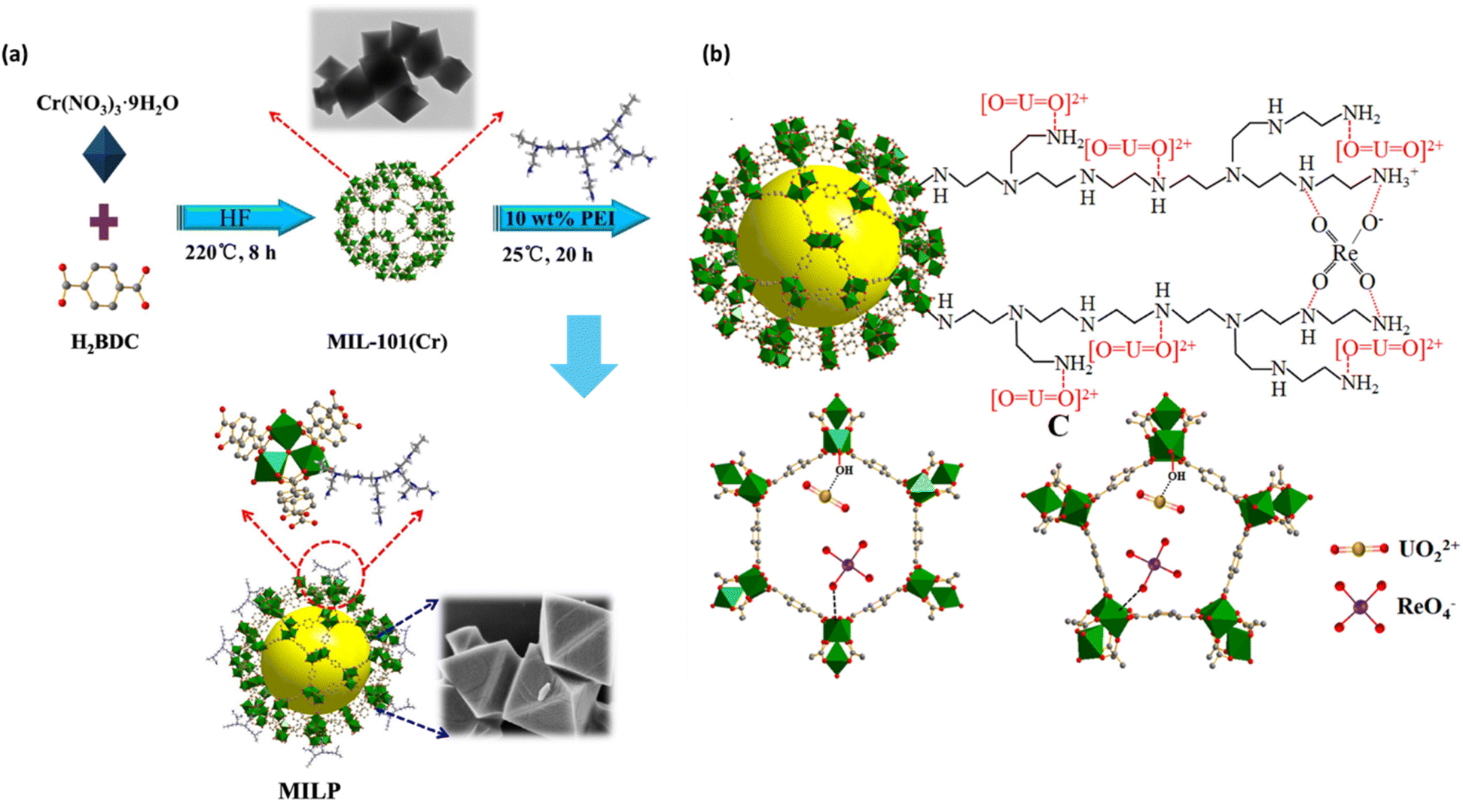 | ||
| Fig. 7 (a) The synthesis approach flowchart for MILP MOF composites. The figure is reproduced from ref. 65 with permission from Elsevier. (b) Possible host–guest interactions between UO22+ and ReO4− with the MIL-101(Cr) cavities and schematic depiction of the interaction between UO22+ and ReO4− with PEI. The figure is reproduced from ref. 65 with permission from Elsevier. | ||
To enhance the uptake efficiency and selectivity, two stable cationic MOFs have been reported recently by G. Zhang et al., Zr-tcbp-Me and Zr-tcpp-Me, Zr-tcbp-Me. Zr6O4(OH)4(tcbp)3(CH3)6, where tcbp = 4,4′,6,6′-tetracarb oxy-2,2′-bipyridine and Zr-tcpp-Me[2;Zr6O4(OH)4(H2O)4(tcpp)2(CH3)4, where tcpp = 2,3,5,6-tetrakis(4-carboxyphenyl)pyrazine] for potential use as adsorbent materials to extract 99TcO4− ions from aqueous solution.67Fig. 8a shows a simulated Zr-tcbp-Me structure with an ftw topology and a 12-connected Zr6 cluster on the c axis. A Zr-tcpp-Me structure simulated along the axis, featuring an 8-connected Zr6 cluster with an SCU topology, was presented (Fig. 8b). The ReO4− capture process is described in Fig. 8c. The pure MOF samples were subjected to an ion-exchange procedure with a saturated sodium chloride solution after methylation. Additionally, they demonstrated a high degree of selectivity and adsorption capacity for the nonradioactive ReO4− anion. The highest framework stability towards acidity was demonstrated by the MOFs Zr-tcbp-Me and Zr-tcpp-Me, which have been investigated for the removal of perrhenate from effluent water. In order to investigate the mechanism of ReO4− adsorption, FT-IR experiments were conducted to comprehend the bonding mode of ReO4− in the two MOFs. Zr-tcpp was used as a reference in this investigation, and compound 1 was chosen. Fig. 8d and e show a comparison of the Zr-tcpp and compound 1 IR spectra before and after ReO4− adsorption. The presence of a ν(ReO4−) band at 920 cm−1 in the spectra of compound 1 and Zr-tcpp indicates the adsorbed ReO4− ions. Based on the intensity of the ν(ReO4−) band, compound 1 exhibits a much higher ReO4− uptake, indicating that methylation was a useful strategy for enhancing ReO4− capture. The production of OH species by adsorbing additional protons was likely the cause of the emergence of the 3650 cm−1 band in Zr-tcpp (Fig. 11d), which balances the negative charge of ReO4− (Fig. 11e), and the positively charged N+–CH3 ions were able to stabilize the adsorbed ReO4−, which was why the ν(OH) band at 3650 cm−1 was absent. After adsorption of ReO4−, the N+–CH3 signature bands (CH3), β(CH3), and ν(C–N) become less prominent. This could be due to the compound combining with free ReO4− ions in solution, which causes the methyl groups to separate from the MOF structure. These findings suggested that the ReO4− anions in unmethylated Zr-tcpp interact with the framework through hydrogen bonding.67
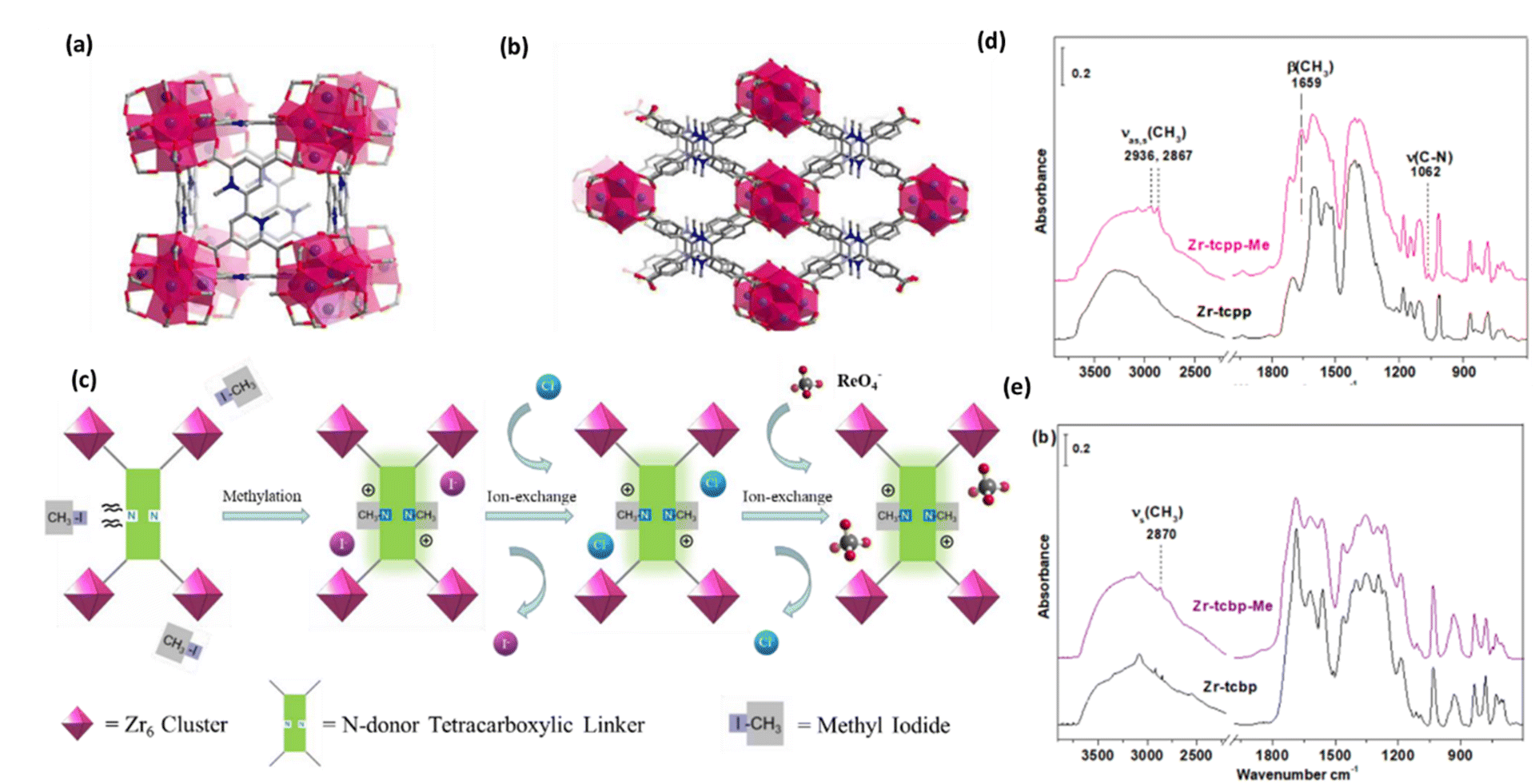 | ||
| Fig. 8 (a) A simulated Zr-tcbp-Me structure with an ftw topology and a 12-connected Zr6 cluster along the c axis. (b) A Zr-tcpp-Me structure is simulated along the axis, with an 8-connected Zr6 cluster with an SCU topology. Color scheme: N in blue; O in red; Zr in violet; C in gray. (c) ReO4− capture flow schematic. After being methylated, the pure MOF samples underwent an ion-exchange procedure using a saturated sodium chloride solution. Eventually, the cationic framework contained ReO4−. (d) The Zr-tcpp (a) and Zr-tcpp-Me. (e) FT-IR spectra of samples were obtained both before and after they were submerged in a ReO4− solution. Every spectrum is compared to that of the blank KBr pellet under vacuum (<20 mTorr). The figure is reproduced from ref. 67 with permission from the American Chemical Society. | ||
To improve the radiation stability of the MOF for TcO4− from actual waste water solution, Lin Zhu et al. synthesized SCU-101 [MOF, [Ni2(tipm)2(C2O4)](NO3)2·2H2O (SCU-101, tipm = tetrakis[4-(1-imidazolyl)phenyl]methane)], a hydrolytically stable and radiation-resistant cationic MOF with fast removal kinetics, an extraordinary distribution coefficient, and high sorption capacity towards TcO4− ions.68 This material preferentially eliminates TcO4− even in the presence of high levels of NO3− and SO42−. The presence of an excessive amount of SO42− does not significantly affect the sorption of TcO4−, even at a concentration that is 6000 times higher. The exceptional characteristics of SCU-101 facilitate the effective extraction of TcO4− from the simulant stream of Hanford's low-level waste melter off-gas scrubber. The overall structure resembles a porous 3D cationic nickel-tip expanded framework. Each Ni2+ cation has six coordinates and binds to four tipm ligands and one C2O4− anion (Fig. 9a). Fig. 9b shows three types of channels (A: ∼7 × 9 Å2, B: ∼11 × 5 Å2, and C: ∼4 × 2.5 Å2) for charge-balancing NO3− anions, as validated by ion chromatography. However, these anions could not be found in the electron density map and were utterly disordered. The tipm ligand, which connects four Ni2+ cations, works as a 4-connected node, while each [Ni2C2O4]2+ cluster acts as an 8-connected node, resulting in a binodal Flu architecture (Fig. 9c). The structure of SCU-101 was a honeycomb made up of rhombic dodecahedra (Fig. 9d). The study investigated the sorption mechanism by studying the TcO4− ion-embedded single-crystal structure of SCU-101, which is a good example of a single-crystal structure ensnaring TcO4− within a sorbent. These results were studied by density functional theory (DFT) geometry optimization, which showed that TcO4− interacts with a site formed by two tipm ligands, in agreement with the observed crystal structure (Fig. 9e–h).68
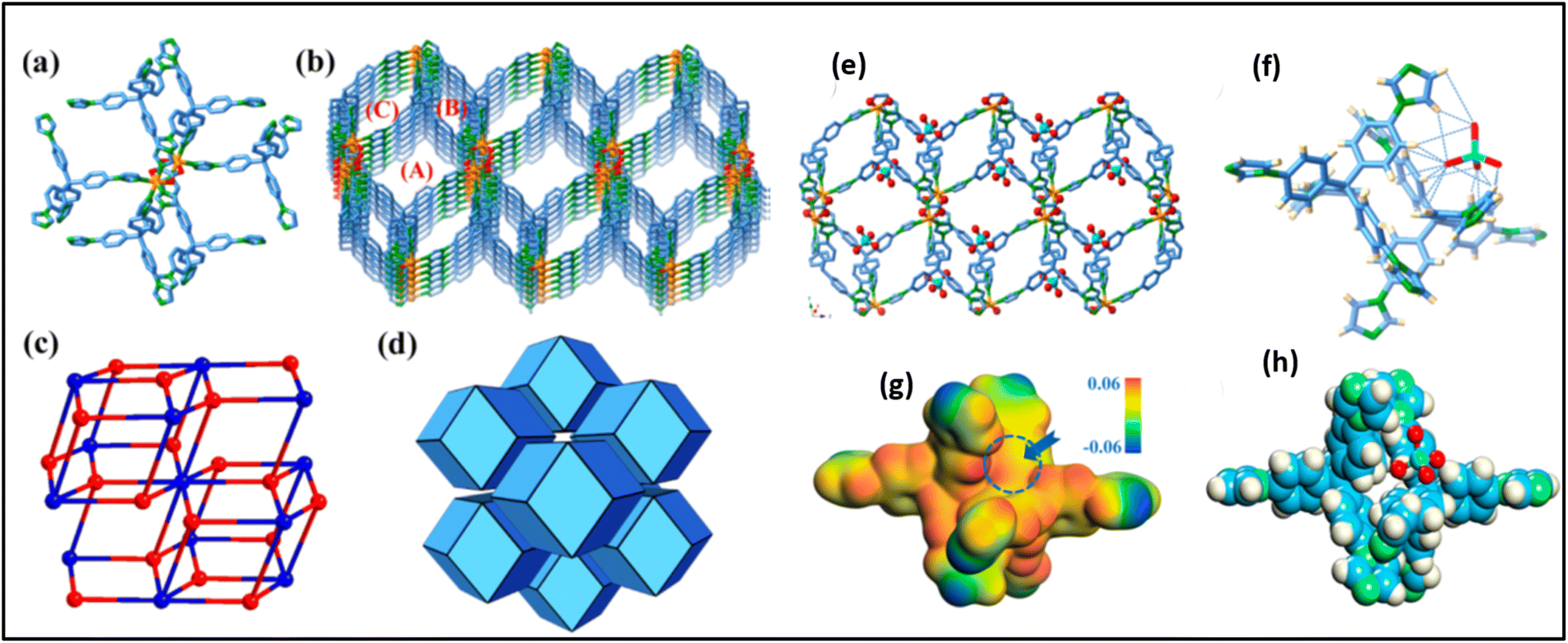 | ||
| Fig. 9 (a) The crystal structure of SCU-101. (b) Coordination environment of Ni2+ includes four tipm ligands and one oxalate group. (c) The 3D cationic framework has three types of channels. Atom colors include orange (Ni), red (O), pale blue (C), and green (N). (d) A simplified (4,8)-connected binodal Flu topology. (e) A simplified rhombic dodecahedron honeycomb structure, with TcO4− captured in type A channels. (f) Hydrogen bonds produced between TcO4− and the SCU-101 framework. (g) Electrostatic potential distribution within the incomplete framework. (h) Optimized TcO4− trapping position in the framework using theoretical calculations The figure is reproduced from ref. 68 with permission from the American Chemical Society. | ||
Conversely, Qing-Hua Hu et al. and colleagues have reported a synthetic method that is efficient in introducing halogens into a two-dimensional MOF (called Mn-MOF).69 This framework was synthesized by combining MnCl2·4H2O with a neutral nitrogen-donor ligand. It demonstrates exceptional stability in alkaline aqueous solutions, even at a concentration of 1 M NaOH. The Mn-MOF exhibits a substantial capacity of 403 mg g−1, surpassing the majority of MOF adsorbents, and demonstrates exceptional adsorption capabilities for ReO4−. Mn-MOF demonstrates exceptional selectivity towards ReO4− in the presence of highly competitive anions with high densities, such as SO42− and NO3−. This study introduced an innovative approach for the elimination of TcO4− from nuclear fuel.69 Anion exchange and cetyltrimethylammonium bromide (CTAB) functionalized MIL-101-Cr-NO3 were subsequently reported for the separation of TcO4− from groundwater.70 The parent MIL-101-Cr-F was less effective in removing ReO4− than the MIL-101-Cr that was exchanged with Cl−, I−, and CF3SO3−. The CTAB functionalized MIL-101-Cr-NO3 enhanced the ability to remove ReO4− from 39 to 139 mg g−1 in comparison to the parent framework. It also improved the reaction kinetics from around 30 to less than 10 min to reach full adsorption capacity and the selectivity for ReO4− over competing NO3−, CO32−, SO42−, and Cl−. To identify and remove ReO4−/TcO4− from radioactive wastewater, the NCU-2 was a rare case of 14-fold interpenetrated with excellent chemical stability, even at 0.5 M HNO3.71 Remarkably, the strong interaction between ReO4− and the host for the development of a non-fluorescent complex quenched the fluorescence signal of NCU-2 in the presence of ReO4−. Furthermore, NCU-2 demonstrated exceptional selectivity in detecting ReO4− even in the presence of numerous competing ions. In addition, NCU-2 demonstrated rapid sorption kinetics and a significant adsorption capacity for ReO4− (541 mg g−1), which makes it a very desirable option for waste monitoring and emergency treatment. Later on, Mei Ming et al. reported the discovery of a porous cationic Ag(I) MOF called TNU-132. This MOF combines different properties and demonstrates enhanced selectivity for capturing perrhenate/pertechnetate even in the presence of a substantial excess of 2000-fold SO42− and 300-fold NO3−.72 The anion exchange investigations of TNU-132 in the mixture of Cr2O72− and ReO4− offer a lucid elucidation of the mechanism that underlies this exceptional selectivity. To clarify, the separation method consisted of two sequential steps: the nano-sieving process and the crystalline sorbent reformation process.72,73 Shufen Gu et al. and colleagues have recently revealed that employing structural flexibility could be a viable approach to enhance the absorption of ReO4− from wastewater.66 The flexible cationic MOF that was utilized, [Co(H2O)2(TIB)][NO3]2 (also known as Co-TIB-NO3, TIB = 3,3′,5,5′-tetra(1H-imidazole-1-yl)-1,1′-biphenyl), displayed three distinct structures in response to temperature, counterions, and solvents: crystalline OP (open pore) phase, crystalline CP (closed pore) phase, and amorphous CP phase. The structural flexibility originated from a multifaceted mechanism involving the deformation of organic ligands, compression between layers, and movement of counterions from cages to channels. The material Co-TIB-NO3, which exhibits the amorphous CP phase, has been proven to have a high capacity for uptake, a quick rate of adsorption, a large distribution coefficient, excellent selectivity, and the ability to be recycled. It has demonstrated exceptional effectiveness in capturing ReO4−, showing its superior suitability for ReO4− sequestration.66 Afterward, to enhance the selectivity and adsorption kinetics of the MOF material for TcO4− ion removal and remediation from waste solution, a new chemically stable cationic MOF (iMOF-2C) with a hydrophobic core ligand strategically used to aid in the oxo-anion capture process was reported by Ghosh and colleagues.74 The compound demonstrated rapid sieving kinetics for these oxo-anions and a notable absorption capability for ReO4− (691 mg g−1) and CrO42− (476.3 mg g−1), with the latter being utilized as a stand-in counterpart for radioactive TcO4− anions. Notably, even in the presence of competing anions such as NO3−, Cl−, SO42−, ClO4−, etc., the molecule demonstrated high selectivity. Moreover, the chemical was used on a stationary phase ion column for decontamination and exhibits outstanding reusability (up to 10 cycles). After analyzing the trigonal pyramidal-shaped crystals using single crystal X-ray diffraction (XRD), it was discovered that six nitrogen atoms from six different ligand units coordinate with the core metal center to form an octahedral shape surrounding the Ni(II) center (Fig. 10b). These moieties also function as recognition sites for the incoming oxo-anions, especially those with lower charge densities (Fig. 10c). The lowest energy configuration of each anion in the primitive cell of the iMOF-2C structure was achieved from the classical simulated annealing technique (Fig. 10a and d). In this work, they have chosen a periodic model of iMOF-2C and optimized the structure with several anions (NO3−, SO42−, CrO42−, and ReO4−) by computing the corresponding binding energies using DFT calculations (Fig. 10d). It was discovered that the binding energy of the iMOF-2C-(ReO4−) structure is −300.74 kJ mol−1, which was substantially greater than the binding energy of SO42− and double that of NO3−. The experimental data of the compound iMOF-2C, which showed remarkable selectivity toward CrO42− and ReO4− over other anions, were precisely verified by the binding energy estimates.74
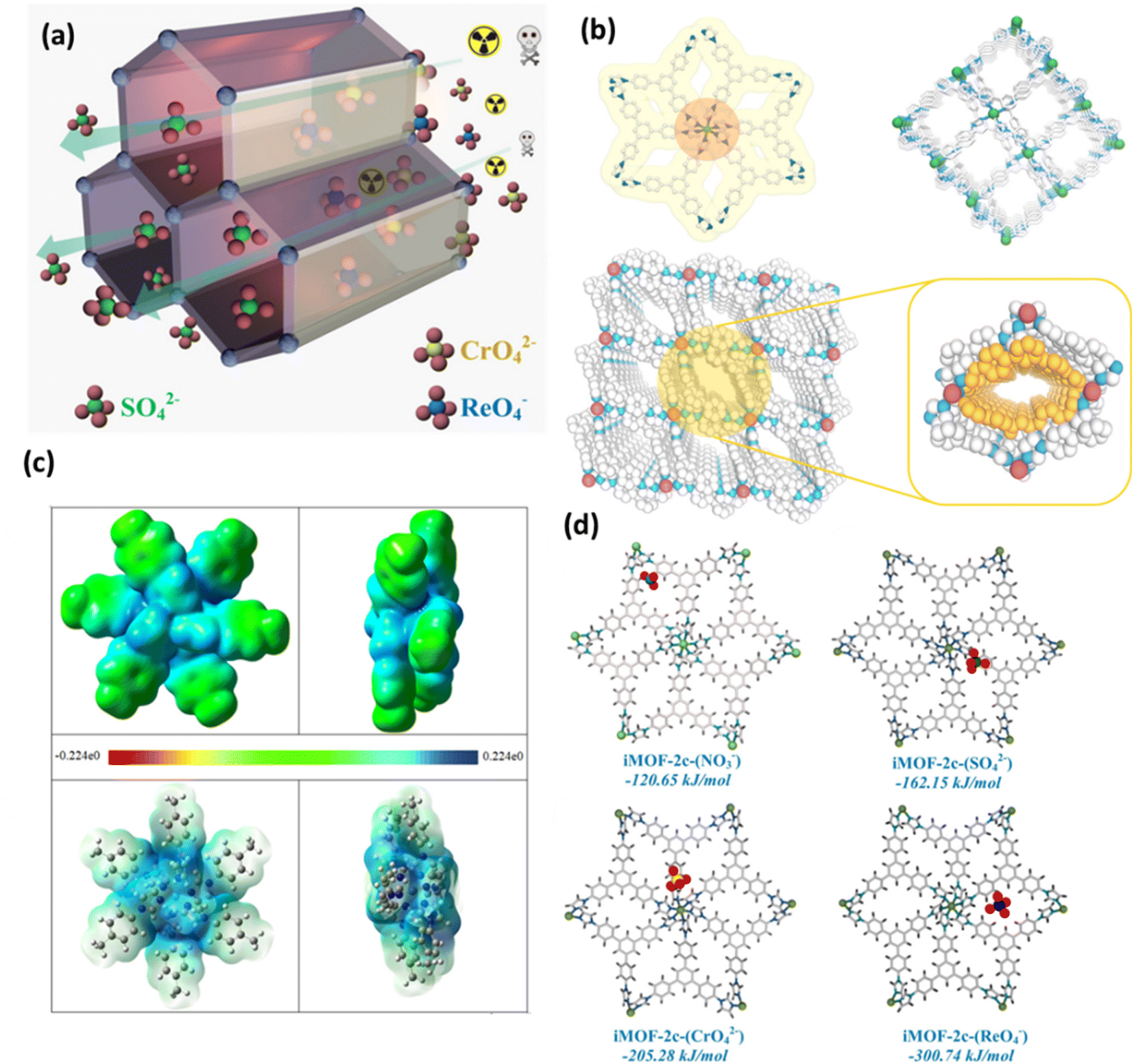 | ||
| Fig. 10 (a) Diagrammatic representation of oxo-anion capture in iMOF-2C. (b) The coordination environment in iMOF-2C (colors: gray, blue, and green; hydrogen atoms have been removed for clarity), the packing diagram of iMOF-2C, and a zoomed-in view that highlights the hydrophobic channel inside the MOF (in yellow) are shown along with a perspective view of the packing along the c-axis. (c) A section of the iMOF-2C fragment displaying the ESP isosurface (isodensity = 0.001 a.u.) at the theoretical level of (SMD)B3LYP-D3/SDD∼6-31G(d); top and side views, as well as the ESP diagram of the iMOF-2C cluster model generated by the Gaussian program. (d) The DFT-D3 approach was used to compute the binding energies of the optimized structures of iMOF-2C with various binding anions, including iMOF-2C-(NO3−), iMOF-2C-(SO42−), iMOF-2C-(CrO42−), and iMOF-2C-(ReO4−). The figure is reproduced from ref. 74 with permission from the American Chemical Society. | ||
To improve the material's stability and selectivity and particularly investigate the TcO4− ion exchange mechanism, a mesoporous cationic thorium-based MOF (SCU-8), [Th3(bptc)3O·(H2O)3]Cl·(C5H14N3Cl)·8H2O (SCU-8, H3bptc = [1,1′-biphenyl]-3,4′,5-tricarboxylicacid), was reported by Yuxiang Li et al. and team (Fig. 11).75 The main driving factors such as electrostatic interactions, hydrogen bonds, hydrophobic interactions, and van der Waals interactions immobilize PFOS anions in SCU-8. The compound crystallizes in the hexagonal space group P63/m and has a 3D open framework, according to a single-crystal X-ray diffraction study. A half thorium center, a half pbtc3− ligand, one-sixth of a μ3-O atom, and a half-coordinating water molecule were presented in the asymmetric unit. As an unconventional coordination environment best defined as a capped triangular cupola geometry, the 10-coordinate Th4+ ion adopts it (Fig. 11a–d).27
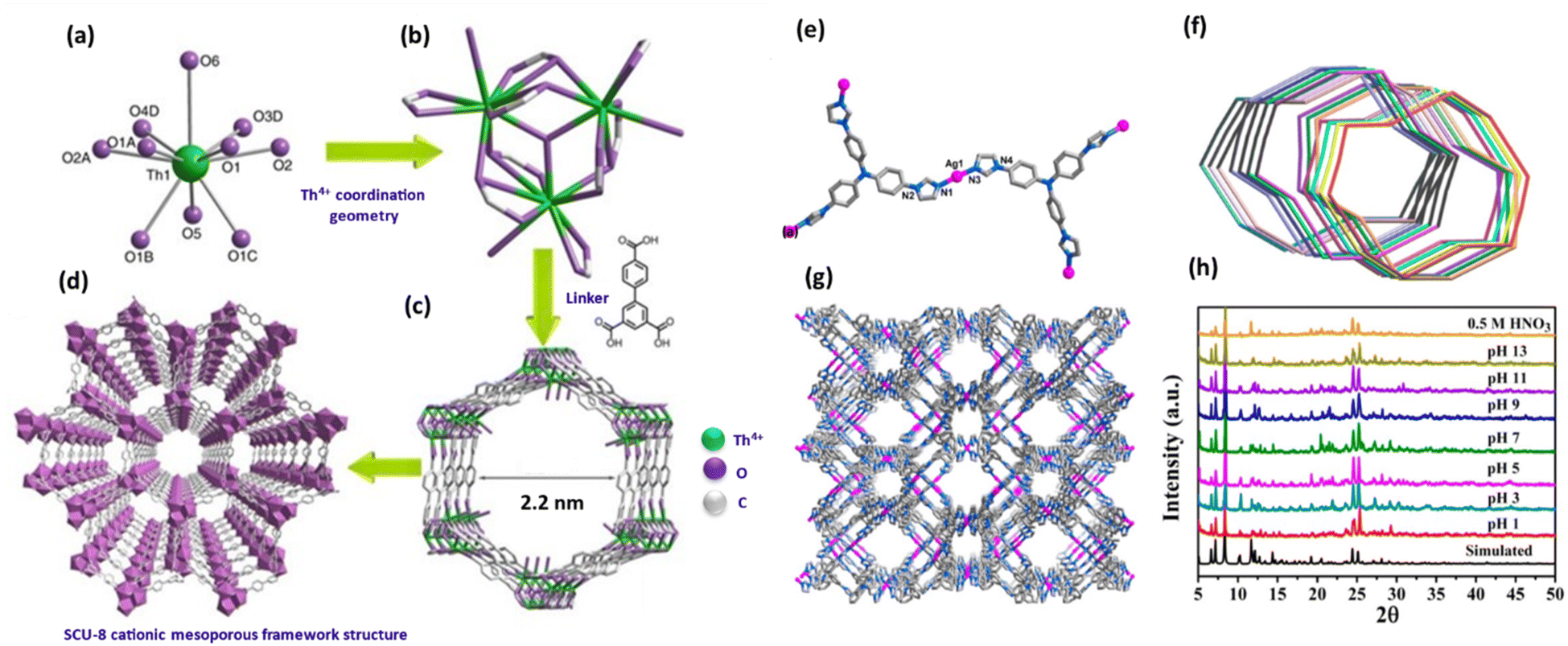 | ||
| Fig. 11 Illustrations of the SCU-8 crystal structure: (a) Th4+ coordination geometry. (b) [Th3(COO)9O(H2O)3.78]+ cationic cluster as the SBU. (c) The structure's hexagonal tubular channels. (d) A view along the c axis of the cationic mesoporous framework structure. Th = green, O = purple, and C = gray. The figure is reproduced from ref. 75. (e) The Ag+ coordination environment. (f) The 14-fold interpenetration's simplified topological structure; the various hues correspond to the various interpenetration single sets. (g) There are two kinds of NCU-2 channels and (h) NCU-2 PXRD patterns following immersion in various pH and 0.5 M HNO3 aqueous solutions. The figure is reproduced from ref. 76 with permission from the American Chemical Society. | ||
Role of luminescent MOFs in TcO4−/ReO4− recognition and removal
MOFs with luminescent properties have shown great promise due to their wide variety of multifunctional microenvironments, fast fluorescence response, low detection limit, and high sensitivity. These characteristics make them ideal for chemical identification and sensing.77–79 Luminescent MOFs represent a promising class of materials for the selective recognition and removal of TcO4− and ReO4− ions. By leveraging their luminescent properties, these materials can be used as highly sensitive sensors for detecting these ions, while also facilitating their efficient removal through adsorption, ion exchange, or redox reactions.77–79 The ability to tailor the structure and functionalization of luminescent MOFs allows for enhanced selectivity, sensitivity, and reusability, making them attractive candidates for TcO4− and ReO4− ion remediation and water purification applications. The primary concern with MOFs is their lack of stability in extreme environments, which leads to structural collapse and a decline in their sensitivity and adsorption capabilities for 99TcO4−/ReO4−. As a result, only a limited number of MOFs have been created for the purpose of detecting 99TcO4−/ReO4−, thereby hindering their practical use.2,44,80 Rapti et al. documented the exceptional sensitivity and extremely low detection limit of two Zr4+ MOFs (MOR-1 and MOR-2) as fluorescence sensors for ReO4−.81 Khan et al. revealed two luminous 2D metal–organic coordination networks that exhibit a strong sensitivity to ReO4−.82 Under harsh circumstances, both of them have poor chemical stability. It is crucial to use a new type of fluorescent MOF that has good chemical stability and specific binding efficiency for 99TcO4−/ReO4− in nuclear waste. It is an effective approach to improve structural stability, better crystal density, and guest selectivity.83–86 Afterward, Wang and colleagues synthesized two interpenetrating framework materials and showed that the higher interpenetrating network produces a more thermodynamically stable crystalline material.87 In light of this, recently Qing-Hua Hu et al. reported a method for 99TcO4−/ReO4− detection and adsorption, using tris (4-(1H-imidazole-1-yl)phenyl)amine and AgNO3, as an interpenetration strategy to synthesize a novel fluorescent MOF (known as NCU-2), which was constructed using Ag+ metal ions and a flexible ligand containing tridentate nitrogen.76Fig. 11e depicts the coordination environment of Ag+. A simplified topological structure of the 14-fold interpenetration is presented in Fig. 11f. There are two kinds of NCU-2 channels shown in Fig. 11c. NCU-2 PXRD patterns in various pH and 0.5 M HNO3 aqueous solutions are depicted in Fig. 11g. One rare example of a 14-fold interpenetrated sample with good chemical stability even at 0.5 M HNO3 is NCU-2, which is useful for removing ReO4−/TcO4− from nuclear waste. Fig. 11h shows NCU-2 PXRD patterns under various pH and 0.5 M HNO3 aqueous solutions. NCU-2 was able to detect trace quantities of ReO4− in simulated Hanford waste with a low detection limit of 66.7 nM and a wide linear range (0.2–200 μM). Moreover, NCU-2 demonstrates rapid absorption rates and a substantial ability to adsorb ReO4− (541 mg g−1), making it a potentially desirable option for waste monitoring and emergency treatment.76One common method to synthesize oxy-anion specific cationic MOFs is coordination between metal ions and nitrogen-containing ligands. This involves adding free or weakly coordinated anions like CH3COO−, NO3−, and ClO4− to the MOFs.59 Therefore, the usage of transition metal ions and nitrogen-donor ligands to produce cationic MOFs with strong sorption capacity and hydrolytic stability against anionic contaminants remains a problem.44 In actuality, triazolyl, pyridyl and imidazolyl functional groups make up the majority of nitrogen-donor ligands utilized to construct cationic MOFs.88 Pyrimidinyl can offer more binding sites than pyridyl, imidazolyl, and pyrazinyl because of its two nitrogen atoms' interaction with metal ions. Nevertheless, there are many reports indicating that cationic MOFs have not yet been created using a pyrimidyl group.89 Keeping this in mind, pyrimidyl groups were added to different organic framework materials by Kang Kang et al. to explore the superiority of pyrimidyl ligands and they synthesized cationic MOFs for the removal of radioactive oxy-anions like TcO4− (Fig. 12a).90 Three novel cationic MOF crystal structures, ZJU-X11, ZJU-X12, and ZJU-X13, were developed using AgNO3 and pyrimidyl as organic ligands. ZJU-X11 was identified as the best possible anion-exchange material and underwent extensive research and characterization, with its coordination number, sorption capacity, and selectivity compared to those of these three cationic MOFs. Hydrothermally, ZJUX11, ZJU-X12, and ZJU-X13 were synthesized by combining AgNO3 with pyrimidyl ligands in various ways. ZJU-X11 ([Ag2(L1)2]·(NO3)2·EtOH) has a colorless, long, rhombic block crystal shape (Fig. 12a).90Fig. 12b depicts the ZJU-X11 crystal optical picture, the structure of the ligand L1 in coordination with three silver ions, and the Ag+ coordination environment with one NO3− and three pyrimidyl units. Fig. 12c shows a microscopic picture of the crystals ZJU-X12, and the Ag+ coordination environment containing NO3− and pyrimidyl units. The ZJU-X11 material has shown good extraction ability of ReO4−/TcO4− ions. The findings showed that ZJU-X11 may be employed as a sorbent to remove ReO4−/TcO4− from aqueous solution without breaking the anion-exchanged framework structure.90
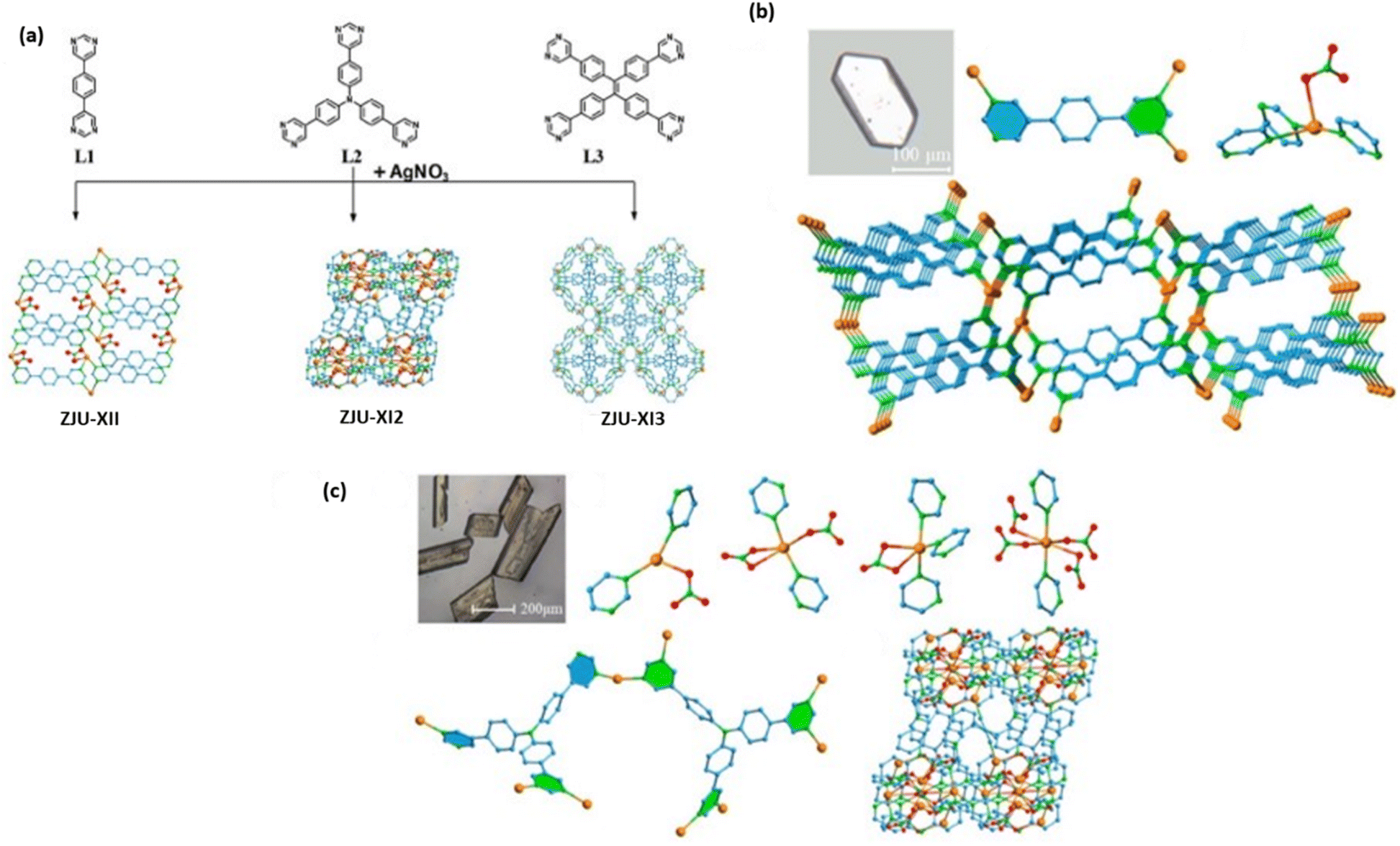 | ||
| Fig. 12 (a) Synthesized process of the cationic MOFs like ZJU-X11, ZJU-X12, and ZJU-X13, respectively. The figure is reproduced from ref. 90 with permission from the American Chemical Society. (b) ZJU-X11 crystal optical picture, structure of one ligand L1 in coordination with three silver ions, Ag+ coordination environment with one NO3− and three pyrimidyl units, and perspective packing structure of ZJU-X11 with one-dimensional channels observed along the b axis. Atom colors: Ag in orange; N in green; O in red; C in pale blue. The figure is reproduced from ref. 90 with permission from the American Chemical Society. (c) A microscopic picture of the crystals ZJU-X12 and Ag+ coordination environment containing NO3− and pyrimidyl units. Colors of atoms: Ag in orange; N in green; O in red; C in pale blue. The figure is reproduced from ref. 90 with permission from the American Chemical Society. | ||
Role of halogen bonding in MOFs for enhanced TcO4−/ReO4− capture
Halogen bonding plays a critical role in enhancing the selective capture of TcO4− and ReO4− ions in MOFs. By incorporating halogenated functional groups or metal nodes into the MOF structure, these materials exhibit strong, directional interactions with the target anions, leading to efficient ion removal. The unique combination of halogen bonding with other interactions such as electrostatics and hydrogen bonding makes these MOFs highly effective and selective for the removal of these hazardous ions. As research advances, halogen-bonded MOFs could provide a powerful solution for environmental remediation, water purification, and other industrial applications. Although sorbents have made significant progress in TcO4− removal, there has been inadequate study into the possibility of secondary contamination in cationic MOFs due to exchanged counterions such as NO3−. In order to address this, Xiao's group developed ZJU-X4, a cationic MOF that contains Cl− as counterions, which may lower the possibility of secondary contamination.91 Nickel chloride and tris(4-pyridylethynyl)triphenylamine were used in the generation of ZJU-X4. The ion-exchange kinetics of ZJU-X4 were further investigated by ion chromatography, which showed a one-to-one connection between the amount of Cl− released and the target ions absorbed. The changes in the structure of ZJU-X4 with ReO4− absorption were further confirmed by a thorough pair distribution function analysis, which also clearly demonstrated the underlying anion-exchange mechanism. ReO4− was shown to have an absorption capacity of 395 mg g−1 by ZJU-X4, and dynamic sorption tests suggested that ReO4− may be recovered and removed efficiently and reversibly using a 3 M NaCl solution. This demonstrates the potential of ZJU-X4 for real-world uses, particularly in situations where effective and reversible sorption is essential (Fig. 13a–c).91 | ||
| Fig. 13 (a) Diagram showing the anions released by the cationic MOF during ReO4− elimination. (b) An image of the dynamic sorption facility used for ReO4− based studies. (c) ZJU-X4 dynamic sorption and elution curves. The figure is reproduced from ref. 91 with permission from the American Chemical Society. (d) The Zn2+ coordination environment in NCU-3-Br. (e) A glimpse of the 3D structure that fills space. (f) A 3-connected, simplified UTP architecture. (g) NCU-3-Br PXRD patterns. The figure is reproduced from ref. 92 with permission from Elsevier. | ||
The study found that the strength of the halogen bonding (XB) interaction, specifically I > Br > Cl ≫ F, increased when the electronegativity of the halogen atoms decreased and their polarizability increased.93–95 Therefore, it is suggested that including halogen atoms in MOF structures could be a beneficial strategy to improve the adsorption abilities of MOFs for ReO4−/TcO4−. In light of this, Hu et al. Qing-Hua introduced two halogenated MOFs named NCU-3-Cl and NCU-3-Br, which were designed to adsorb ReO4−/TcO4− ions, by coordinating ZnX2 (X = Cl, Br) with ligands that bind to tris(4-(1H-imidazole-1-yl)phenyl) (Tipa).92Fig. 13d shows the Zn2+ coordination environment in NCU-3-Br. A glimpse of the 3D structure that fills space is depicted in Fig. 13e. A 3-connected, simplified UTP architecture and NCU-3-Br PXRD patterns are exhibited in Fig. 13f and g. Because of their double structures and the way the negative halogen ions work with the Zn2+ ions to spread the positive charge and thwart the OH− attack, both of them show excellent chemical stability even in 1 M NaOH solutions. NCU-3-Br (483 mg g−1) had a much better adsorption capability than NCU-3-Cl (217 mg g−1), according to the capture studies. Furthermore, NCU-3-Br demonstrated strong selectivity for ReO4− even in the presence of significant concentrations of interfering ions. The experimental and computational results suggest that halogenation plays a vital role in controlling the adsorption capacity of MOFs. The reason for this is that bromine atoms possess a relatively low electronegativity, resulting in the creation of charged σ-holes. These cavities, in turn, promote the creation of halogen bonds and enhance the stability of the positively charged framework.
Very recently, Sai Zhou et al. and colleagues reported that the benzene-rich structure of the TIB-TBPB framework improves its hydrophobicity and enables it to show exceptional thermal stability and acid–base stability.96 With a capacity of 882 mg g−1, TIB-TBPB demonstrated exceptional removal capacity for both ReO4− and TcO4−, with Kd values of 2.34 × 106 mL g−1 and 1.35 × 106 mL g−1, respectively. In addition, TIB-TBPB offers superior selectivity and recyclability, with the ability to eliminate 65.7% and 86.4% of ReO4− from 3 M HNO3 and the Hanford LAW simulated nuclear waste, respectively. In this context, Ghosh and colleagues reported that energy-efficient adsorption followed by the ion-exchange method, using a hydrolytically stable cationic MOF, known as iMOF-2C, has demonstrated preferential trapping of a selected metal-oxoanion from a mixture of other metal-oxoanionic toxic pollutants in water.45 In contaminated water systems, the cationic MOF showed rapid and selective extraction efficiency towards ReO4−.
TcO4− and ReO4− extraction mechanism in MOFs
MOFs and their composites have demonstrated high efficacy in adsorbing oxyanions like TcO4−.The majority of the fundamental mechanisms and interactions that have been reported are consolidated in Fig. 14. The potent adsorptive action of MOFs often arises from a combination of multiple techniques. The metal, clusters, and linkers can all serve as binding or interaction sites. The process of modifying the linkers with functional groups such as hydroxyl, thiol, or amide has been extensively studied as a means to enhance the adsorption efficacy and/or selectivity. The most prevalent method of metal ion adsorption by MOFs is through Lewis acid–base interaction.53 Given that metal ions act as Lewis acids, it is essential to incorporate various functional groups comprising O, S, and N elements that serve as Lewis bases into the linker backbone of MOFs. To enhance the adsorption effectiveness and selectivity of the target metal ions, the number of O-, S-, or N-containing groups in the frameworks can be increased either prior to or following synthesis. The interaction between Lewis acids and bases is crucial for the adsorption of metals onto the donor atoms of MOFs. Therefore, the pH of the solution has a substantial impact on the overall adsorption process. Adsorption is not possible at low pH values because the Lewis basic sites of linker backbones become protonated and lose their metal ion binding qualities.53 Conversely, the adsorbent's donor atoms undergo deprotonation, which enhances their ability to form complexes and adsorb the target analytes. This is achieved by increasing the pH of the aqueous samples that contain the metal ions. Further increases in pH beyond a specific threshold can lead to a reduction in the ability of sorption to effectively occur. This is because the introduction of hydroxide in alkaline solutions can result in the formation of complexes and the precipitation of various metals.37,55
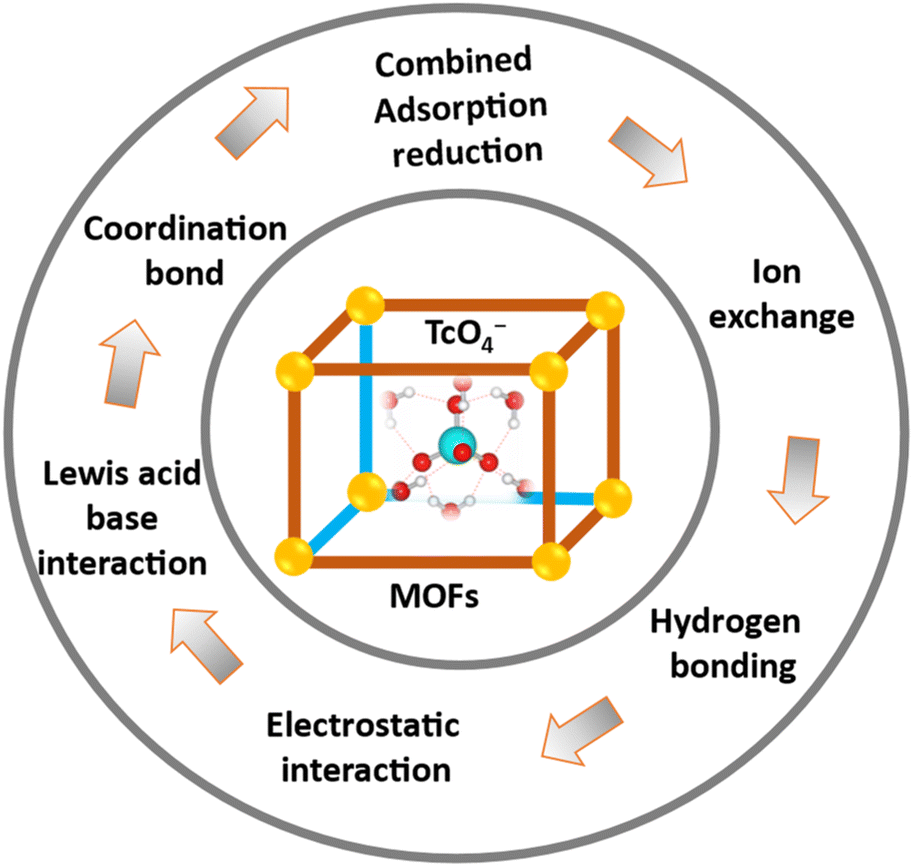 | ||
| Fig. 14 Mechanisms of TcO4−/ReO4− ion extraction with MOFs. | ||
Challenges and proposed solutions
MOF-based materials exhibit considerable benefits as adsorbents for the removal of TcO4− oxoanions, including elevated adsorption capacity, rapid adsorption kinetics, and superior selectivity. These benefits are primarily attributed to the highly ordered porous structure of the MOFs and the interactions that take place between the target TcO4− ions and sites on the MOFs, such as open metal sites and functional groups of organic ligands. Consequently, by altering the pores of MOFs and incorporating suitable functional groups, the adsorption capacity, kinetics, and selectivity for TcO4− ions can be significantly improved. Nonetheless, despite notable advancements, some substantial difficulties persist before MOF-based materials may be extensively utilized in practical water treatment applications.Key challenges
(1) Scalability of MOF synthesis: the scalability of MOF synthesis primarily relies on solvothermal methods, which require the utilization of organic solvents. These solvents not only increase production costs, particularly when costly organic ligands are necessary, but also raise environmental concerns due to waste disposal. (2) Chemical stability: numerous MOFs demonstrate restricted stability in aqueous environments, especially those with extensive surfaces. The instability is exacerbated by pH fluctuations, as extreme acidic or basic conditions might compromise the MOF structure and diminish the efficacy of functional groups in sequestering TcO4− ions. Consequently, many MOFs possess a constrained active pH range, limiting their practical utility in various aquatic environments. (3) Pore size limitations: most of the MOFs feature microporous architectures with pore dimensions smaller than TcO4− ions, thus constraining the ions' mobility inside the framework. This constrains their efficacy in collecting and sequestering these larger oxoanions. (4) Recyclability and reusability: while MOFs are theoretically reusable, their structural integrity often deteriorates during ion adsorption, resulting in the depletion of active sites and a subsequent reduction in adsorption efficacy over time. Maintaining the structural integrity of MOFs with repeated use is a significant problem for practical applications.(ii) Optimization of pore size and geometry: (a) size-selective adsorption: the pore size distribution of the MOF should be optimized to match the size of 99TcO4− (approximately 0.4–0.5 nm in diameter). This ensures that the 99TcO4− ions are preferentially adsorbed while larger or smaller ions are excluded. (b) Shape-selective sieve effects: researchers can design MOFs with pores that provide steric hindrance, allowing for selective adsorption based on the shape of the 99TcO4− ions.
(iii) Incorporation of redox-active sites: redox interactions: incorporating redox-active metal centers (e.g., transition metals like copper, iron, or manganese) into the MOF structure could facilitate electron transfer reactions, enhancing the ability of the framework to interact with pertechnetate ions. These metal centers may reduce 99TcO4− ions [Tc(VII)] to a lower oxidation state (e.g., Tc(IV)), which could be more easily captured by the MOF.
(iv) Modification of the MOF with specific metal nodes: (a) coordination chemistry tuning: the choice of metal nodes in the MOF plays a crucial role in its selectivity. The incorporation of metals with high affinity for anions or specific coordination environments (such as transition metals with specific oxidation states) could enhance the MOF's ability to selectively adsorb 99TcO4−. (b) Multivalent metal cations: using multivalent metals (e.g., Ni2+, Co2+, and Zn2+) could enable multiple interactions with the 99TcO4− anion, improving both affinity and selectivity for 99TcO4− ions.
(v) Surface charge manipulation: (a) modification of surface charge distribution: the surface charge of MOFs can be tuned by incorporating charged ligands or post-synthetic modifications. A negative charge on the MOF's surface would enhance electrostatic interactions with the negatively charged 99TcO4− anion, increasing the selectivity towards pertechnetate over other species in solution. (b) Zeta potential control: the zeta potential of the MOF can be adjusted to control the electrostatic interaction with 99TcO4−. This would ensure that 99TcO4− is preferentially adsorbed over other ions with similar charges.
(vi) Designing MOFs for kinetic selectivity: (a) kinetically selective adsorption: the author can design MOFs with highly dynamic frameworks that allow for faster adsorption kinetics specifically for 99TcO4−, which might differ from other anions. This could be achieved by optimizing the flexibility of the MOF or introducing channels that are more conducive to the rapid movement of pertechnetate ions. (b) Optimization of adsorption rates: by using MOFs with faster diffusion rates for 99TcO4− than for other species, the material can selectively adsorb pertechnetate from a mixture of anions, especially when there is a time constraint or a need for fast processing.
(vii) Post-synthetic modifications (PSMs): (a) tailoring the framework post-synthesis: the author can apply post-synthetic modification techniques (e.g., ligand exchange and functionalization with specific ligands) to tune the properties of the MOF after its initial synthesis. This allows for fine-tuning of the material's selectivity towards 99TcO4− without the need for altering the entire framework. (b) Targeted surface modifications: introducing specific chemical groups or metal clusters onto the surface of the MOF can enhance selectivity toward 99TcO4−, either through chelation, hydrogen bonding, or other specific interactions.
(viii) Competition studies and selectivity enhancement: (a) competitive adsorption studies: the author should perform adsorption studies in the presence of other ions commonly found in aqueous solutions, such as chloride, sulfate, or nitrate, to better understand the MOF's selective affinity for 99TcO4−. This will help identify the optimal conditions (e.g., pH, temperature, and ionic strength) under which the MOF shows the highest selectivity for 99TcO4−. (b) Mixed-metal MOFs: the use of mixed-metal MOFs, incorporating a combination of metals with different affinities for anions, can help increase the overall selectivity towards 99TcO4− by enhancing the binding strength for pertechnetate while reducing the adsorption of interfering ions.
(ix) Environmental and structural stability: (a) stability under operational conditions: ensure that the MOF maintains its structural integrity and selectivity in the presence of radiation or other environmental conditions (such as variations in temperature and pH) that may occur in real-world applications. (b) Radionuclide stability: given the radioactive nature of 99Tc, the MOF should be engineered for high stability against radiation, ensuring that it retains its adsorption capacity over extended periods of exposure. By combining these strategies, researchers can design MOFs with high selectivity for 99TcO4−, making them more efficient and effective for applications such as nuclear waste treatment, environmental remediation, and radiopharmaceutical purification.
Proposed solutions
(1) Eco-friendly synthesis: adopting green synthesis techniques, such as mechanical, electrochemical, or microwave-assisted approaches, may substantially diminish the environmental impact and expenses associated with MOF manufacturing.88 These approaches can eradicate dependence on detrimental organic solvents and provide more sustainable alternatives.(i) Mechanical-assisted synthesis: this method involves the use of mechanical energy (e.g., ball milling or grinding) to drive the synthesis of MOFs. By applying pressure or shear force, it facilitates chemical reactions without the need for solvents or excessive heat. This method significantly reduces the need for harmful chemicals and solvents, which are often associated with conventional synthetic routes. Furthermore, it reduces energy consumption and waste, making it an environmentally friendly and cost-effective alternative. (ii) Electrochemical synthesis: electrochemical methods use electric current to drive the synthesis of MOFs directly from metal salts and organic ligands in aqueous or non-aqueous solutions. This technique minimizes the need for organic solvents and reduces the energy required for high-temperature reactions. Electrochemical synthesis can be highly controlled, allowing for the precise formation of MOFs with tunable properties, thus enhancing efficiency and reducing environmental impact. The use of renewable energy sources for electrochemical reactions further adds to the sustainability of this method. (iii) Microwave-assisted synthesis: microwave-assisted synthesis involves using microwave radiation to rapidly heat reaction mixtures, facilitating the formation of MOFs in a much shorter time compared to traditional heating methods. This approach requires less energy and typically eliminates the need for organic solvents or toxic chemicals. By using water or green solvents, microwave-assisted synthesis provides an eco-friendlier route to MOF production. Additionally, it often leads to higher yields and better crystalline quality, making it an attractive option for large-scale synthesis.
By adopting these green synthesis techniques, the MOF production process can be made significantly more sustainable. These methods reduce the environmental impact by eliminating harmful solvents, reducing energy consumption, and minimizing waste. Additionally, they offer potential cost savings in terms of energy and raw materials, making them viable alternatives for large-scale manufacturing. The revisions emphasize the importance of these approaches for advancing both the environmental and economic sustainability of MOF-based technologies.
(2) Enhancing MOF stability: integrating functional groups specifically engineered to augment MOF stability across a wide pH range may increase their operational range. These groups would augment the structural resilience of the MOFs, especially under demanding aqueous conditions. (i) Incorporation of pH-responsive functional groups: one effective way to enhance the stability of MOFs is by incorporating functional groups that are specifically designed to respond to changes in pH. These groups could be basic (amine-based) or acidic (carboxylate- or sulfonate-based) functional groups that improve the MOF's stability across a broad pH range. By incorporating such groups into the organic linker or within the metal node, the MOF can maintain its structural integrity and functionality under acidic or basic conditions. For example, amines could be used to stabilize MOFs in acidic environments by forming hydrogen bonds or coordination interactions that help maintain the structure. Carboxylates or sulfonates may offer enhanced stability under basic conditions, as they can form strong ionic interactions with the metal centers in MOFs, preventing structural collapse. (ii) Hydrogen-bonding networks for increased stability: functional groups that can engage in hydrogen bonding can significantly contribute to the overall stability of the MOF. Functional groups such as hydroxyl (–OH), amide (–CONH2), or alcohol groups can form additional hydrogen bonds within the framework or between the organic linker and metal ions. These hydrogen bonds increase the structural resilience of MOFs in harsh aqueous environments and at extreme pH values. This approach helps to minimize the risks of framework degradation that often occur when MOFs are exposed to fluctuating pH conditions. (iii) Cross-linking and metal–organic coordination: the inclusion of functional groups that are capable of cross-linking organic linkers or forming strong coordination bonds with metal centers can further enhance the stability of MOFs. For example, introducing functional groups such as phosphonate or thiol (–SH) groups can enable additional coordination to metal centers, increasing the rigidity of the framework and preventing framework collapse, especially in the presence of water or under acidic/basic conditions. Additionally, the integration of multidentate ligands can improve the robustness of the MOF structure by enhancing metal–ligand interactions. (iv) Hydrophobic or amphiphilic functionalization: in the cases where MOFs are intended for applications in aqueous environments, incorporating hydrophobic or amphiphilic functional groups into the organic linker can help reduce the impact of water on the structural integrity of the MOF. Hydrophobic functional groups (such as fluorine-containing or alkyl groups) can help shield the metal nodes from direct exposure to water, while amphiphilic groups (such as PEGylated chains or alkylphenyl groups) may offer dual protection against both acidic and basic environments by creating a protective barrier around the MOF. (v) Metalation strategies: another promising method to enhance the stability of MOFs is through metalation—the strategic selection and incorporation of metal ions that are more resistant to degradation under extreme conditions. For example, using metals like zirconium (Zr) or yttrium (Y), which are known for their high stability under aqueous and extreme pH conditions, can strengthen the overall MOF structure. Additionally, combining these metals with pH-tolerant organic linkers with functional groups that are specifically designed for durability under diverse conditions can create a more stable MOF material. By integrating these functional groups and strategies, the MOF can be engineered to exhibit greater stability in a wide range of pH, especially under aqueous conditions that may be acidic or basic. These approaches not only enhance the structural resilience of MOFs, but also broaden their applicability in fields like water treatment and sensing, where stability in variable environments is crucial for long-term performance.
(3) Structural modification for enhanced porosity: engineering MOFs with ultrahigh porosity will optimize the use of adsorption sites, facilitating improved mobility and capture of larger-size ions such as TcO4− ions. This can be accomplished by designing bigger pore sizes or developing hybrid architectures that integrate micro- and mesoporous characteristics. (i) Designing larger pore sizes: one of the most direct ways to enhance the adsorption capacity of MOFs, particularly for larger ions like TcO4− ions, is by designing MOFs with larger pore sizes. To capture larger species, MOFs need to have pores that are not only large enough to accommodate these ions but also optimally shaped to facilitate their movement and interaction with the adsorbent. This can be achieved through several strategies: (a) selection of larger organic linkers: using organic linkers with larger molecular structures can result in expanded pore sizes within the MOF structure. For example, bipyridine, porphyrin, or pillar[5]arene-based linkers can create larger spaces for ion accommodation. (b) Tuning metal clusters or nodes: the coordination environment around the metal nodes can also influence the pore size. By using larger metal clusters or incorporating metals that form large, more spacious coordination networks (such as Zr, Ti, or U), MOFs can have expanded pores that are more suitable for trapping larger ions. (c) Pre-synthetic modification: the pore size can be increased by modifying the synthetic conditions (e.g., temperature, pressure, and solvent system) or adding template molecules that are later removed to create larger void spaces within the structure.
(ii) Hybrid architectures with micro- and meso-porosity: hybrid architectures that combine both microporosity (pores <2 nm) and mesoporosity (pores between 2 nm and 50 nm) can be an effective strategy for optimizing MOFs for large-ion capture. Such architectures offer the combined benefits of a high surface area and faster diffusion rates, which can enhance the overall efficiency of adsorption. Hybrid systems can be achieved by: (a) incorporating mesoporous materials into MOFs: by introducing mesoporous components (e.g., silica, carbon-based materials, or other nanostructured supports) into the MOF structure, researchers can create a dual porosity system that allows for both rapid transport through larger pores and selective adsorption in smaller micropores. (b) Post-synthetic modification: after synthesizing a MOF, additional treatments such as acid/base etching or templating can be used to generate mesopores within the MOF structure. For instance, selective removal of the linker or metal ions can enlarge the pores, creating a more hierarchical porosity suitable for larger ions like TcO4−. (c) Blending different pore types: another approach is blending materials with complementary porosity. For example, combining a highly microporous MOF with a mesoporous material like mesoporous silica or carbon nanotubes can result in a hybrid adsorbent with both micropores for a high surface area and mesopores for facilitating the diffusion and capture of larger ions.
(iii) Tuning pore size distribution for enhanced adsorption: the pore size distribution is crucial for enhancing the selectivity and capacity of MOFs for specific ions. By precisely engineering the pore structure, it is possible to optimize the material for capturing larger ions, such as TcO4−. This can be accomplished by: (a) controlling linker length: by carefully selecting the length of the organic linkers, the spacing between the metal nodes can be controlled, which directly affects the pore size and its distribution. Longer linkers can result in larger, more uniform pores. (b) Tailoring pore environment: the pore surface characteristics, such as hydrophobicity or charge, can be tailored by introducing functional groups that not only control the pore size but also enhance the interaction between the MOF and the adsorbate. This is particularly important for capturing ions like TcO4−, where ionic interactions play a crucial role.
(iv) Hierarchical porosity for faster diffusion and enhanced adsorption: combining hierarchical porosity (a mixture of micropores, mesopores, and even macropores) can create a system that balances the high surface area and large adsorption sites needed for effective ion capture, while allowing for faster diffusion of larger ions such as TcO4−. This can be achieved by: (a) combining different synthesis strategies: mesoporous or macroporous materials can be used as templates for MOF growth that could lead to a system with hierarchical porosity. Additionally, certain block copolymers or polymer templates can help form such hierarchical structures during MOF synthesis. (b) Creating layered or porous MOFs: layered MOFs or those with interconnected networks of pores in different size regimes (e.g., micro, meso, and macro) can offer paths for fast ion diffusion and increased capacity for ion uptake. These materials are particularly suitable for the adsorption of larger ions like TcO4− ions.
(v) To further enhance the adsorption capabilities of MOFs for larger ions like TcO4−, the integration of open metal sites (OMSs) can be beneficial. These are uncoordinated metal sites within the MOF structure that can interact directly with the adsorbed ions. Such sites provide a highly selective binding environment for larger ions, improving both the capture and the stability of the adsorbed species. The incorporation of OMS can be facilitated by: (a) using metal nodes with a low coordination number: metal centers such as Zn, Cu, or Al with low coordination numbers can leave open sites that are available for ion binding. (b) Post-synthetic modification: techniques like linker exchange or metal ion exchange can be used to introduce open metal sites into MOFs after their synthesis.
In short, by engineering larger pores, incorporating hybrid micro/mesoporous architectures, and tuning pore size distributions, MOFs can be optimized for the enhanced adsorption of larger ions like TcO4−. These strategies not only increase the porosity but also enhance the adsorption kinetics, providing a more efficient and selective adsorbent for removing large ions in applications such as environmental cleanup and radioactive ion capture. The proposed structural modifications offer promising pathways to develop highly efficient, large-capacity MOF adsorbents with tailored properties for specific applications.
(4) Developing hybrid MOF-based materials: the integration of MOFs with alternative materials can markedly enhance their recyclability and reusability. Hybrid materials may enhance longevity and inhibit the degradation of active sites, rendering them more suitable for long-term uses.
(i) Hybridization with carbon-based materials: the incorporation of carbon-based materials (such as graphene oxide (GO), carbon nanotubes (CNTs), or activated carbon) into MOFs can significantly enhance the recyclability, stability, and adsorption capacity of MOFs, particularly for pertechnetate adsorption. This integration provides the following benefits: (a) improved mechanical stability: MOFs, known for their inherent brittleness, can benefit from the mechanical strength and flexibility of carbon materials. Graphene oxide and CNTs, for example, reinforce the MOF structure, reducing the risk of structural collapse during repetitive adsorption–desorption cycles, which increases the material's long-term stability and durability. (b) Enhanced surface area and porosity: carbon materials like graphene oxide and activated carbon can contribute additional surface area and porosity, creating more adsorption sites for TcO4− and allowing for more efficient ion capture. This enhancement is particularly valuable for applications where high adsorption capacity is required for large ions such as TcO4− ions. (c) Facilitation of regeneration: the electrical conductivity and chemical inertness of carbon-based materials facilitate easier regeneration of the hybrid adsorbents. Carbon can help in desorbing TcO4− ions through simple methods like thermal treatment or electrochemical regeneration, making the material reusable and reducing the need for harsh chemicals.
(ii) Inorganic support materials for enhanced stability: integrating inorganic support materials, such as silica, alumina, or metal oxides, with MOFs can increase their chemical stability and thermal resistance, which is critical for applications involving TcO4− ion adsorption. The key advantages include: (a) prevention of MOF degradation: inorganic supports act as a protective matrix, stabilizing the MOF and preventing degradation of active sites (metal centers or organic linkers). This helps maintain the MOF's adsorption capacity over multiple cycles. (b) Improved chemical resistance: inorganic supports enhance the resistance to environmental factors, such as exposure to moisture, high temperatures, or fluctuating pH levels, which could otherwise degrade MOFs over time. For TcO4− ion adsorption, this means that hybrid materials are more stable under real-world conditions where stability is crucial. (c) Enhanced longevity: the incorporation of inorganic materials extends the MOF's lifetime by preventing structural collapse and leaching of active sites, allowing the hybrid material to undergo multiple adsorption–desorption cycles without significant loss of performance.
(iii) Polymer-MOF hybrid materials: the integration of polymers with MOFs enhances their mechanical flexibility and long-term usability. Polymers such as polystyrene, polyethylene glycol (PEG), polyvinyl alcohol (PVA), or conductive polymers can offer the following benefits: (a) enhanced flexibility and stability: polymers add flexibility to MOF structures, reducing the risk of mechanical failure during ion uptake or regeneration cycles. This is important when MOFs undergo swelling or shrinking due to changes in solvent or ionic concentration. (b) Improved reusability: the polymer matrix facilitates easier recovery and regeneration of the MOF material after ion adsorption. Polymers also help protect active sites by reducing direct exposure to solvents or reactive species, prolonging the lifespan of the MOF. (c) Controlled functionalization: polymers can be functionalized with specific groups that enhance the interaction between the MOF and TcO4− ions. For example, incorporating amine (–NH2) or carboxylate (–COOH) groups can enhance the affinity and selectivity of the hybrid material for TcO4−, improving both adsorption capacity and efficiency.
(iv) MOF-inorganic oxide composite materials: combining MOFs with metal oxides (such as titanium dioxide (TiO2), zinc oxide (ZnO), or iron oxide (Fe2O3)) results in hybrid materials that exhibit synergistic properties for enhanced TcO4− ion capture and stability. The key advantages of this hybridization include: (a) increased adsorption sites: metal oxides provide additional surface area and active sites for the adsorption of pertechnetate ions. For example, the hydroxyl groups on iron oxide or zinc oxide surfaces can interact with pertechnetate ions, enhancing adsorption. (b) Enhanced chemical and thermal stability: metal oxides are known for their stability at high temperatures and under extreme pH conditions. Integrating MOFs with metal oxides improves the long-term durability of the adsorbent, making it suitable for continuous use in dynamic environments. (c) Facilitated ion exchange: in some cases, metal oxides can promote ion exchange processes that facilitate the capture of TcO4− ions. These metal oxides also enhance the regeneration of the hybrid material through simple ion-exchange or thermal methods.
(v) Layered hybrid materials, where MOFs are combined with polymers or carbon-based materials, offer enhanced ion mobility and superior recyclability. These materials have several key benefits. (a) Increased ion diffusion: the layered structure allows for faster diffusion of TcO4− ions into the adsorbent, resulting in faster adsorption kinetics. Additionally, this structure enhances the overall adsorption capacity for large ions like TcO4−. (b) Improved structural integrity: the outer layers of polymer or carbon materials protect the MOFs from physical degradation, while maintaining accessibility to adsorption sites. This ensures that the material retains its effectiveness and reusability over extended use. (c) Long-term use: layered hybrids are less prone to the collapse or degradation that can occur in conventional MOF materials, thus extending their useable lifetime and increasing their cost-effectiveness over time.
(vi) MOF-inorganic–organic hybrid materials for enhanced selectivity and performance: combining inorganic materials (like metal oxides), organic linkers, and MOFs can create hybrid materials with superior selectivity for pertechnetate ions. The integration of functionalized polymers with inorganic supports offers: (a) tailored selectivity: the hybrid material can be functionalized to increase its affinity for TcO4−, improving its selectivity for pertechnetate ions over other potential contaminants. For example, incorporating specific amine or phosphonate functional groups can increase the material's affinity for TcO4−. (b) Enhanced durability: these materials offer excellent chemical stability and thermal resistance, making them suitable for long-term use in challenging environments, such as in the treatment of radioactive waste or environmental remediation applications. (c) Improved adsorption capacity: by optimizing the pore structure and enhancing the surface interaction with TcO4−, hybrid materials can achieve higher adsorption capacities compared to individual MOFs.
To sum up, the development of hybrid MOF-based materials that integrate carbon-based, polymer, and inorganic materials offers significant improvements in the recyclability, reusability, and long-term stability of adsorbents for TcO4− removal. These hybrids provide enhanced mechanical stability, adsorption capacity, and selectivity for pertechnetate ions, while also improving longevity and sustainability through simpler regeneration processes. The synergistic effects of hybridizing MOFs with other materials ensure that these adsorbents are better suited for continuous, long-term use in pertechnetate adsorption applications, making them a promising solution for environmental cleanup, radioactive waste management, and related fields.
Conclusion and future perspectives
In the relentless quest for efficient remedies against TcO4− contamination, MOFs have emerged as prominent contenders. Their remarkable tunable porosity architectures and precise control over surface chemistry offer tailor-made solutions for pollutant removal, marking a profound leap toward a sustainable future. Recent advancements in MOF-based TcO4− remediation underscore their potential to promote a sustainable future.The impressive adsorption capacities of MOFs can be attributed to the interactions between target ions and functional binding groups, as well as their highly ordered porous structures facilitating efficient diffusion. Nevertheless, notwithstanding these promising advancements, a myriad of challenges and unresolved issues persist, hindering the widespread adoption of MOFs in environmental remediation endeavors. One of the primary issues that MOFs face is their susceptibility to solution conditions, which can have a substantial influence on their overall performance. The challenges to overcome are exacerbated by the narrow pH operating ranges, sluggish adsorption kinetics, restricted water stability, and excessively expensive production costs. To surmount these obstacles and unlock the full potential of MOFs in environmental remediation, concerted research efforts are imperative. Overall, the recognition and capture of pertechnetate ions using MOFs as adsorbents represent a promising approach for addressing the challenges associated with TcO4− contamination, offering potential solutions for environmental remediation and nuclear waste management.
Future endeavors should prioritize the enhancement of MOFs' stability and the expansion of their pH tolerance, laying the groundwork for their utilization across a broader spectrum of environmental conditions. Moreover, comprehensive research is warranted to unravel the intricate adsorption behavior of MOFs under realistic conditions encountered in nuclear fuel cycle processes and waste management. This necessitates delving into their performance under extreme scenarios such as high acidity and intense radiation, which are pivotal for practical applications in radioactive wastewater decontamination.
In conclusion, addressing the outlined challenges through focused research endeavors holds the key to unlocking the widespread implementation of MOFs in radioactive waste management and environmental remediation efforts. By harnessing the inherent strengths of MOFs, namely, their high removal capacity and easy scalability in synthesis, we can pave the way for effective and sustainable solutions to radioactive waste management, thereby ushering in a cleaner, safer, and more sustainable future for generations to come.
Data availability
No new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge U. Dani, C. E., NRB, BARC, R. B. Bhatt, Dy C. E., NRB, BARC, D. B. Sathe, G. M., FF, NRB, BARC, Dr H. Pal, Homi Bhabha National Institute, Anushaktinagar, Mumbai 400 094, India, Chemistry Group, Bhabha Atomic Research Centre, Mumbai 400 085, India and Dr Biswajit Sadhu, Scientific Officer E, Bhabha Atomic Research Centre, Mumbai-400094, for constant encouragement. All the authors have given permission to the manuscript final version.References
- M. E.-S. Islam and S. Ma, Recent Development of Metal–Organic Frameworks for Water Purification, Langmuir, 2024, 40(10), 5060–5076, DOI:10.1021/acs.langmuir.3c03818.
- R. J. Drout, K. Otake, A. J. Howarth, T. Islamoglu, L. Zhu, C. Xiao, S. Wang and O. K. Farha, Efficient Capture of Perrhenate and Pertechnetate by a Mesoporous Zr Metal–Organic Framework and Examination of Anion Binding Motifs, Chem. Mater., 2018, 30(4), 1277–1284, DOI:10.1021/acs.chemmater.7b04619.
- J. G. Darab and P. A. Smith, Chemistry of technetium and rhenium species during low-level radioactive waste vitrification, Chem. Mater., 1996, 8(5), 1004–1021, DOI:10.1021/cm950418+.
- J. P. Icenhower, N. P. Qafoku, J. M. Zachara and W. J. Martin, The Biogeochemistry of Technetium: A Review of the Behavior of an Artificial Element in the Natural Environment, Am. J. Sci., 2010, 310(8), 721–752, DOI:10.2475/08.2010.02.
- N. M. Hassan, W. D. King, D. J. McCabe, L. L. Hamm and M. E. Johnson, Superlig® 639 Equilibrium Sorption Data For Technetium From Hanford Tank Waste Supernates, Solvent Extr. Ion Exch., 2002, 20(2), 211–226, DOI:10.1081/SEI-120003022.
- V. McKee, J. Nelson and R. M. Town, Caged oxoanions, Chem. Soc. Rev., 2003, 32(5), 309–325, 10.1039/B200672N.
- R. Amano, A. Ando, T. Hiraki, H. Mori, H. Matsuda and K. Hisada, Rapid uptake of technetium-99m-pertechnetate by several plants, Radioisotopes, 1990, 39(12), 585–586, DOI:10.3769/radioisotopes.39.12585.
- L. Zhu, C. Xiao, X. Dai, J. Li, D. Gui, D. Sheng, L. Chen, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Exceptional Perrhenate/Pertechnetate Uptake and Subsequent Immobilization by a Low-Dimensional Cationic Coordination Polymer: Overcoming the Hofmeister Bias Selectivity, Environ. Sci. Technol. Lett., 2017, 4(7), 316–322, DOI:10.1021/acs.estlett.7b00165.
- P. Samanta, P. Chandra, S. Dutta, A. V. Desai and S. K. Ghosh, Chemically Stable Ionic Viologen-Organic Network: an Efficient Scavenger of Toxic Oxo-Anions from Water, Chem. Sci., 2018, 9(40), 7874, 10.1039/C8SC02456A.
- M. J. Kang, S. W. Rhee and H. Moon, Sorption of MO4– (M=Tc, Re) on Mg/Al Layered Double Hydroxide by Anion Exchange, Radiochim, Acta, 1996, 75, 169–173, DOI:10.1524/ract.1996.75.3.169.
- M. J. Kang, K. S. Chun, S. W. Rhee and Y. Do, Comparison of Sorption Behavior of I– and TcO4– on Mg/Al Layered Double Hydroxide, Radiochim. Acta, 1999, 85, 57–63, DOI:10.1524/ract.1999.85.12.57.
- N. Daniels, C. Franzen, G. L. Murphy, K. Kvashnina, V. Petrov, N. Torapava, A. Bukaemskiy, P. Kowalski, H. Si, Y. Ji, A. Hölzer and C. Walther, Application of Layered Double Hydroxides for 99Tc Remediation, Appl. Clay Sci., 2019, 176, 1–10, DOI:10.1016/j.clay.2019.04.006.
- Y. Xiong, X. Cui, P. Zhang, Y. Wang, Z. Lou and W. Shan, Improving Re VII, Adsorption on Diisobutylamine-Functionalized Graphene Oxide, ACS Sustainable Chem. Eng., 2017, 5, 1010–1018, DOI:10.1021/acssuschemeng.6b02322.
- H. Zhang, F. Yang, C. Lu, C. Du, R. Bai, X. Zeng, Z. Zhao, C. Cai and J. Li, Effective Decontamination of 99TcO4–/ReO4– from Hanford Low-Activity Waste by Functionalized Graphene Oxide-Chitosan Sponges, Environ. Chem. Lett., 2020, 18, 1379–1388, DOI:10.1007/s10311-020-01005-w.
- K. Wang, Z. Yan, L. Fu, D. Li, L. Gong, Y. Wang and Y. Xiong, Gemini Ionic Liquid Modified Nacre-Like Reduced Graphene Oxide Click Membranes for ReO4–/TcO4– Removal, Sep. Purif. Technol., 2022, 302, 122073, DOI:10.1016/j.seppur.2022.122073.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341(6149), 1230444, DOI:10.1126/science.1230444.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Selective Gas Adsorption and Separation in Metal-Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1477–1504, 10.1039/b802426j.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Flexible Metal-Organic Frameworks, Chem. Soc. Rev., 2014, 43, 6062–6096, 10.1039/C4CS00101J.
- M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Carbon Capture and Conversion Using Metal-Organic Frameworks and MOF-Based Materials, Chem. Soc. Rev., 2019, 48, 2783–2828, 10.1039/C8CS00829A.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Chemical, Thermal and Mechanical Stabilities of Metal-Organic Frameworks, Nat. Rev. Mater., 2016, 1, 15018, DOI:10.1038/natrevmats.2015.18.
- K. Patra, S. A. Ansari and P. K. Mohapatra, Metal-organic frameworks as superior porous adsorbents for radionuclide sequestration: Current status and perspectives, J. Chromatogr. A, 2021, 1655, 462491, DOI:10.1016/j.chroma.2021.462491.
- K. Patra and A. Sengupta, Recent advances in functionalized porous adsorbents for radioactive waste water decontamination: Current status, research gap and future outlook, Mater. Today Sustainability, 2024, 25, 100703, DOI:10.1016/j.mtsust.2024.100703.
- K. Patra, A. Sengupta, V. K. Mittal and T. P. Valsala, Emerging functionalized magnetic nanoparticles: from synthesis to nuclear fuel cycle application: where do we stand after two decade?, Mater. Today Sustainability, 2023, 24, 100489, DOI:10.1016/j.mtsust.2023.100489.
- J. G. Darab and P. A. Smith, Chemistry of Technetium and Rhenium Species during Low-Level Radioactive Waste Vitrification, Chem. Mater., 1996, 8, 1004–1021, DOI:10.1021/cm950418+.
- R. Custelcean, Urea-functionalized crystalline capsules for recognition and separation of tetrahedral oxoanions, Chem. Commun., 2013, 49, 2173–2182, 10.1039/C2CC38252K.
- R. Custelcean and B. A. Moyer, Anion Separation with Metal–Organic Frameworks, Eur. J. Inorg. Chem., 2007, 2007(10), 1321–1340, DOI:10.1002/ejic.200700018.
- I. E. Burgeson, J. R. Deschane and D. L. Blanchard, Removal of Technetium from Hanford Tank Waste Supernates, Sep. Sci. Technol., 2005, 40, 201–223, DOI:10.1081/SS-200041916.
- W. R. Wilmarth, G. J. Lumetta, M. E. Johnson, M. R. Poirier, M. C. Thompson, P. C. Suggs and N. P. Machara, Review: Waste-Pretreatment Technologies for Remediation of Legacy Defense Nuclear Wastes, Solvent Extr. Ion Exch., 2011, 29, 1–48, DOI:10.1080/07366299.2011.539134.
- A. H. Bond, F. W. K. Chang, A. H. Thakkar, J. M. Williamson, M. J. Gula, J. T. Harvey, S. T. Griffin, R. D. Rogers and E. P. Horwitz, Study on Preferential Selectivity of Nuclear Grade Resin Indion-223 towards some Bivalent Ions, Ind. Eng. Chem. Res., 1999, 38, 1676–1682, DOI:10.56431/p-4b9z2i.
- J. Mor, A. Boda, S. K. Sharma, M. Ali and A. Sengupta, Understanding the sorption behavior of tri, tetra and hexavalent f-cations on metal–organic framework (MOF), ZIF-67: experimental and theoretical investigation, Sep. Sci. Technol., 2023, 58(15–16), 2806–2823, DOI:10.1080/01496395.2023.2255740.
- G. Salunkhe, A. Sengupta, A. Boda, R. Paz, N. K. Gupta, C. Leyva, R. S. Chauhan and S. M. Ali, Application of hybrid MOF composite in extraction of f-block elements: Experimental and computational investigation, Chemosphere, 2022, 287, 132232, DOI:10.1016/j.chemosphere.2021.132232.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Introduction to Metal–Organic Frameworks, Chem. Rev., 2012, 112, 673–674, DOI:10.1021/cr300014x.
- S. R. J. Oliver, Cationic inorganic materials for anionic pollutant trapping and catalysis, Chem. Soc. Rev., 2009, 38, 1868–1881, 10.1039/B710339P.
- B. F. Hoskins and R. Robson, Design and construction of a new class of scaffolding-like materials comprising infinite polymeric frameworks of 3D-linked molecular rods. A reappraisal of the zinc cyanide and cadmium cyanide structures and the synthesis and structure of the diamond-related frameworks [N(CH3)4][CuIZnII(CN)4] and CuI[4,4',4'',4'''-tetracyanotetraphenylmethane]BF4.xC6H5NO2, J. Am. Chem. Soc., 1990, 112, 1546–1554, DOI:10.1021/ja00160a038.
- O. M. Yaghi, H. L. Li and T. L. Groy, A Molecular Railroad with Large Pores: Synthesis and Structure of Ni(4,4‘-bpy)2.5(H2O)2(ClO4)2·1.5(4,4‘-bpy)·2H2O, Inorg. Chem., 1997, 36, 4292–4293, DOI:10.1021/ic970423a.
- D. Banerjee, W. Xu, Z. Nie, L. E. V. Johnson, C. Coghlan, M. L. Sushko, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan and P. K. Thallapally, Zirconium-based metal–organic framework for removal of perrhenate from water, Inorg. Chem., 2016, 55, 8241–8243, DOI:10.1021/acs.inorgchem.6b01004.
- D. Sheng, L. Zhu, C. Xu, C. Xiao, Y. Wang, Y. Wang, L. Chen, J. J. Chen, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Efficient and selective uptake of TcO4– by a cationic metal–organic framework material with open Ag+ sites, Environ. Sci. Technol., 2017, 51, 3471–3479, DOI:10.1021/acs.est.7b00339.
- C. P. Li, J. Y. Ai, H. Zhou, Q. Chen, Y. Yang, H. He and M. Du, Ultra-highly selective trapping of perrhenate/pertechnetate by a flexible cationic coordination framework, Chem. Commun., 2019, 55, 1841–1844, 10.1039/C8CC09364D.
- R. J. Drout, K. Otake, A. J. Howarth, T. Islamoglu, L. Zhu, C. Xiao, S. Wang and O. K. Farha, Efficient capture of perrhenate and pertechnetate by a mesoporous Zr metal–organic framework and examination of anion binding motifs, Chem. Mater., 2018, 30, 1277–1284, DOI:10.1021/acs.chemmater.7b04619.
- D. Sheng, L. Zhu, X. Dai, C. Xu, P. Li, C. Pearce, C. Xiao, J. Chen, R. Zhou, T. Duan, O. K. Farha, Z. Chai and S. Wang, Successful decontamination of 99 TcO4− in ground water at legacy nuclear sites by a cationic metal-organic framework with hydrophobic pockets, Angew. Chem., Int. Ed., 2019, 58, 4968–4972, DOI:10.1002/anie.201814640.
- H. Fei, M. R. Bresler and S. R. J. Oliver, A new paradigm for anion trapping in high capacity and selectivity: Crystal-to-crystal transformation of cationic materials, J. Am. Chem. Soc., 2011, 133(29), 11110–11113, DOI:10.1021/ja204577p.
- D. Banerjee, W. Xu, Z. Nie, L. E. V. Johnson, C. Coghlan, M. L. Sushko, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan and P. K. Thallapally, Zirconium-based metal–organic framework for removal of perrhenate from water, Inorg. Chem., 2016, 55, 8241–8243, DOI:10.1021/acs.inorgchem.6b01004.
- D. Sheng, Z. Lin, D. Xing, C. Xu, L. Peng, C. Pearce, C. Xiao, J. Chen, R. Zhou, T. Duan, O. K. Farha, Z. Chai and S. Wang, Successful Decontamination of 99TcO4- in Groundwater at Legacy Nuclear Sites by a Cationic Metal-Organic Framework with Hydrophobic Pockets, Angew. Chem., Int. Ed., 2019, 58, 4968–4972, DOI:10.1002/anie.201814640.
- D. Sheng, L. Zhu, X. Dai, C. Xu, P. Li, C. I. Pearce, C. Xiao, J. Chen, R. Zhou, T. Duan, O. K. Farha, Z. Chai and S. Wang, Three Mechanisms in One Material: Uranium Capture by a Polyoxometalate–Organic Framework through Combined Complexation, Chemical Reduction, and Photocatalytic Reduction, Angew. Chem., Int. Ed., 2019, 58, 4968–4972, DOI:10.1002/anie.201909718.
- S. Fajal, D. Ghosh, W. Mandala and S. K. Ghosh, Preferential Separation of Radioactive TcO4- Surrogate from a Mixture of Oxoanions by a Cationic MOF, Chem. Commun., 2024, 60, 1884–1887, 10.1039/D3CC05627A.
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Parts, K. W. Chapman, J. T. Hupp and O. K. Farha, High efficiency adsorption and removal of selenate and selenite from water using metal-organic frameworks, J. Am. Chem. Soc., 2015, 137, 7488–7494, DOI:10.1021/jacs.5b03904.
- J. Li, Y. Liu, X. Wang, G. Zhao, Y. Ai, B. Han, T. Wen, T. Hayat, A. Alsaedi and X. Wang, Experimental and theoretical study on selenate uptake to zirconium metal–organic frameworks: Effect of defects and ligands, Chem. Eng. J., 2017, 330, 1012–1021, DOI:10.1016/j.cej.2019.123325.
- S. Sharma, S. Let, A. V. Desai, S. Dutta, G. Karuppasamy, M. M. Shirolkar, R. Babarao and S. K. Ghosh, Rapid, selective capture of toxic oxo-anions of Se(iv), Se(vi) and As(v) from water by an ionic metal–organic framework (iMOF), J. Mater. Chem. A, 2021, 9, 6499–6507, 10.1039/D0TA04898D.
- S. Sharma, A. V. Desai, B. Joarder and S. K. Ghosh, A Water-Stable Ionic MOF for the Selective Capture of Toxic Oxoanions of SeVI and AsV and Crystallographic Insight into the Ion-Exchange Mechanism, Angew. Chem., Int. Ed., 2020, 59, 7788–7792, DOI:10.1002/anie.202000670.
- H. Ouyang, N. Chen, G. Chang, X. Zhao, Y. Sun, S. Chen, H. Zhang and D. Yang, Selective Capture of Toxic Selenite Anions by Bismuth-based Metal–Organic Frameworks, Angew. Chem., Int. Ed., 2018, 57, 13197–13201, DOI:10.1002/anie.201807891.
- N. Shen, Z. Yang, S. Liu, X. Dai, C. Xiao, K. T. Pashow, D. Li, C. Yang, J. Li, Y. Zhang, M. Zhang, R. Zhou, Z. Chai and S. Wang, 99 TcO4− removal from legacy defense nuclear waste by an alkaline-stable 2D cationic metal organic frame- work, Nat. Commun., 2020, 11, 5571, DOI:10.1038/s41467-020-19374-9.
- L. Cheng-Peng, H.-R. Li, J.-Y. Ai, J. Chen and M. Du, Optimizing Strategy for Enhancing the Stability and 99TcO4– Sequestration of Poly(ionic liquids)@MOFs Composites, ACS Cent. Sci., 2020, 6(12), 2354–2361, DOI:10.1021/acscentsci.0c01342.
- R. G. Pearson, Chemical hardness and density functional theory, J. Chem. Sci., 2005, 117, 369–377, DOI:10.1007/BF02708340.
- T. Zheng, et al., Overcoming the crystallization and designability issues in theultrastable zirconium phosphonate framework system, Nat. Commun., 2017, 8, 5369, DOI:10.1038/ncomms15369.
- T. F. Liu, et al., Topology guided design and syntheses of highly stable mesoporous porphyrinic zirconium metal-organic frameworks with high surface area, J. Am. Chem. Soc., 2015, 137, 413–419, DOI:10.1021/ja5111317.
- K. C. Wang, et al, A series of highly stable mesoporous metalloporphyrin Fe-MOFs, J. Am. Chem. Soc., 2014, 136, 13983–13986, DOI:10.1021/ja507269n.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, A water-stable porphyrin-based metal-organic framework active for visible-light photocatalysis, Angew. Chem., Int. Ed., 2012, 51, 7440–7444, DOI:10.1002/anie.201202471.
- K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie and J.-R. Li, Hong-Cai Zhou, Pyrazolate-based porphyrinic metal-organic framework with extraordinary base-resistance, J. Am. Chem. Soc., 2016, 138, 914–919, DOI:10.1021/jacs.5b10881.
- X. L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J. R. Li and H. C. Zhou, A Base-Resistant Metalloporphyrin Metal-Organic Framework for C-H Bond Halogenation, J. Am. Chem. Soc., 2017, 139(1), 211–217, DOI:10.1021/jacs.6b09463.
- V. Colombo, et al., High thermal and chemical stability in pyrazolate-bridged metal-organic frameworks with exposed metal sites, Chem. Sci., 2011, 2, 1311–1319, 10.1039/C1SC00136A.
- A. V. Desai, A. Roy, P. Samanta, B. Manna and S. K. Ghosh, Base-resistant ionic metal-organic framework as a porous ion-exchange sorbent, iScience, 2018, 3, 21–30, DOI:10.1016/j.isci.2018.04.004.
- X. Zhong, L. Wen, H. Wang, C. Xue and B. Hu, Aluminum-based metal-organic frameworks (CAU-1) highly efficient UO22+ and TcO4− ions immobilization from aqueous solution, J. Hazard. Mater., 2021, 407, 124729, DOI:10.1016/j.jhazmat.2020.124729.
- L. Mei, F. Li and J. Lan, et al., Anion-adaptive crystalline cationic material for 99TcO4− trapping, Nat. Commun., 2019, 10, 1532, DOI:10.1038/s41467-019-09504-3.
- J. Li, et al., 99TcO4− remediation by a cationic polymeric network, Nat. Commun., 2018, 9, 3007, DOI:10.1038/s41467-018-05380-5.
- J. Li, Y. Zhang, Y. Zhou, F. Fang and X. Li, Tailored metal-organic frameworks facilitate the simultaneously high-efficient sorption of UO22+ and ReO4− in water, Sci. Total Environ., 2021, 799, 149468, DOI:10.1016/j.scitotenv.2021.149468.
- S. Gu, Z. Yu, N. Li, Q. Zhang, H. Zhang, L. Zhang, L. Gong, R. Krishna and F. Luo, Structural flexibility in cationic metal–organic framework for boosting ReO4− capture, Chem. Eng. J., 2023, 466, 143139, DOI:10.1016/j.cej.2023.143139.
- G. Zhang, K. Tan, S. Xian, K. Xing, H. Sun, G. Hall, L. Li and J. Li, Ultrastable Zirconium-Based Cationic Metal-Organic Frameworks for Perrhenate Removal from Wastewater, Inorg. Chem., 2021, 60(16), 11730–11738, DOI:10.1021/acs.inorgchem.1c00512.
- Z. Lin, et al., Identifying the Recognition Site for Selective Trapping of 99TcO4− in a Hydrolytically Stable and Radiation Resistant Cationic Metal− Organic Framework, J. Am. Chem. Soc., 2017, 139(42), 14873–14876, DOI:10.1021/jacs.7b08632.
- Q. H. Hu, Y. Z. Shi and X. Gao, et al., An alkali-resistant metal–organic framework as halogen bond donor for efficient and selective removing of ReO4−/TcO4−, Environ. Sci. Pollut. Res., 2022, 29, 86815–86824, DOI:10.1007/s11356-022-21870-y.
- D. Li, N. B. Shustova, C. R. Martin, K. Taylor-Pashow, J. C. Seaman, D. I. Kaplan and J. W. Amoroso, Roman Chernikov, Anion-exchanged and quaternary ammonium functionalized MIL-101-Cr metal-organic framework (MOF) for ReO4−/TcO4− sequestration from groundwater, J. Environ. Radioact., 2020, 222, 106372, DOI:10.1016/j.jenvrad.2020.106372.
- H. Qing-Hua, X. Gao, S. Yu-Zhen, L. Ru-Ping, L. Zhang, S. Lin and Q. Jian-Ding, Tailor-Made Multiple Interpenetrated Metal–Organic Framework for Selective Detection and Adsorption of ReO4–, Anal. Chem., 2022, 94(48), 16864–16870, DOI:10.1021/acs.analchem.2c03983.
- M. Mei, Z. Hang, M. Yi-Ning, L. Hai-Ruo and J. Chen, Selective perrhenate/pertechnetate removal by a MOF-based molecular trap, Dalton Trans., 2022, 51, 4458–4465, 10.1039/D1DT04175D.
- C. W. Abney, K. M. L. Taylor-Pashow, S. R. Russell, Y. Chen, R. Samantaray, J. V. Lockard and W. Lin, Topotactic Transformations of Metal-Organic Frameworks to Highly Porous and Stable Inorganic Sorbents for Efficient Radionuclide Sequestration, Chem. Mater., 2014, 26, 5231–5243, DOI:10.1021/cm501894h.
- S. Dutta, P. Samanta, B. Joarder, S. Let, D. Mahato, R. Babarao and S. K. Ghosh, A Water-Stable Cationic Metal−Organic Framework with Hydrophobic Pore Surfaces as an Efficient Scavenger of Oxo-Anion Pollutants from Water, ACS Appl. Mater. Interfaces, 2020, 12, 41810–41818, DOI:10.1021/acsami.0c13563.
- Y. Li, Z. Yang and Y. Wang, et al., A mesoporous cationic thorium-organic framework that rapidly traps anionic persistent organic pollutants, Nat. Commun., 2017, 8, 1354, DOI:10.1038/s41467-017-01208-w.
- Q.-H. Hu, X. Gao, Y.-Z. Shi, R.-P. Liang, L. Zhang, S. Lin and J.-D. Qiu, Tailor-Made Multiple Interpenetrated Metal–Organic Framework for Selective Detection and Adsorption of ReO4–, Anal. Chem., 2022, 94, 16864–16870, DOI:10.1021/acs.analchem.2c03983.
- X. Zhao, X. Bu, T. Wu, S.-T. Zheng, L. Wang and P. Feng, Selective anion exchange with nanogated isoreticular positive metal-organic frameworks, Nat. Commun., 2013, 4, 23, DOI:10.1038/ncomms3344.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Metal-Organic Frameworks: Functional Luminescent and Photonic Materials for Sensing Applications, Chem. Soc. Rev., 2017, 46, 3242–3285, 10.1039/C6CS00930A.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal-Organic Framework Materials as Chemical Sensors, Chem. Rev., 2012, 112, 1105–1125, DOI:10.1021/cr200324t.
- C.-P. Li, H. Zhou, J. Chen, J.-J. Wang, M. Du and W. Zhou, A Highly Efficient Coordination Polymer for Selective Trapping and Sensing of Perrhenate/Pertechnetate, ACS Appl. Mater. Interfaces, 2020, 12, 15246–15254, DOI:10.1021/acsami.0c00775.
- S. Rapti, S. A. Diamantis, A. Dafnomili, A. Pournara, E. Skliri, G. S. Armatas, A. C. Tsipis, I. Spanopoulos, C. D. Malliakas, M. G. Kanatzidis, J. C. Plakatouras, F. Noli, T. Lazarides and M. J. Manos, Exceptional TcO4− sorption capacity and highly efficient ReO4− luminescence sensing by Zr4+ MOFs, J. Mater. Chem. A, 2018, 6, 20813–20821, 10.1039/C8TA07901C.
- S. Khan and S. K. Mandal, Luminescent 2D Pillared-Bilayer Metal-Organic Coordination Networks for Selective Sensing of ReO4- in Water, ACS Appl. Mater. Interfaces, 2021, 13, 45465–45474, DOI:10.1021/acsami.1c11606.
- H.-L. Jiang, T. A. Makal and H.-C. Zhou, Interpenetration Control in Metal-Organic Frameworks for Functional Applications, Coord. Chem. Rev., 2013, 257, 2232–2249, DOI:10.1016/j.ccr.2013.03.017.
- Y.-N. Gong, D.-C. Zhong and T.-B. Lu, Interpenetrating metal–organic frameworks, CrystEngComm, 2016, 18, 2596–2606, 10.1039/C6CE00371K.
- S. B. Choi, H. Furukawa, H. J. Nam, D.-Y. Jung, Y. H. Jhon, A. Walton, D. Book, M. O'Keeffe, O. M. Yaghi and J. Kim, Reversible Interpenetration in a Metal-Organic Framework Triggered by Ligand Removal and Addition, Angew. Chem., Int. Ed., 2012, 51(35), 791–8795, DOI:10.1002/anie.201202925.
- M. Gupta and J. J. Vittal, Control of interpenetration and structural transformations in the interpenetrated MOFs, Coord. Chem. Rev., 2021, 435, 213789, DOI:10.1016/j.ccr.2021.213789.
- Y. Wang, L. Cheng, K.-J. Wang, Z. Perry, W. Jia, R. Chen, Z.-L. Wang and J. Pang, Temperature-Controlled Degree of Interpenetration in a Single-Crystal-to-Single-Crystal Transformation within Two Co(II)-Triazole Frameworks, Inorg. Chem., 2019, 58, 18–21, DOI:10.1021/acs.inorgchem.8b01339.
- A. V. Desai, S. Sharma, S. Let and S. K. Ghosh, N-donor linker based metal-organic frameworks (MOFs): Advancement and prospects as functional materials, Coord. Chem. Rev., 2019, 395, 146–192, DOI:10.1016/j.ccr.2019.05.020.
- K. Kang, X. Dai, N. Shen, R. Xie, X. Zhang, L. Lei, S. Wang and C. Xiao, Unveiling the Uncommon Fluorescent Recognition Mechanism towards Pertechnetate Using a Cationic Metal-Organic Framework Bearing N-Heterocyclic AIE Molecules, Chem.–Eur. J., 2021, 27, 5632–5637, DOI:10.1002/chem.202005362.
- K. Kang, L. Li, M. Zhang, X. Zhang, L. Lei and C. Xiao, Constructing Cationic Metal−Organic Framework Materials Based on Pyrimidyl as a Functional Group for Perrhenate/Pertechnetate Sorption, Inorg. Chem., 2021, 60, 16420–16428, DOI:10.1021/acs.inorgchem.1c02257.
- K. Kang, L. Li, M. Zhang, X. Miao, L. Lei and C. Xiao, Two-Fold Interlocking Cationic Metal–Organic Framework Material with Exchangeable Chloride for Perrhenate/Pertechnetate Sorption, Inorg. Chem., 2022, 61, 11463–11470, DOI:10.1021/acs.inorgchem.2c01846.
- Q.-H. Hu, Y.-G. Wanga, X. Gaoa, Y.-Z. Shia, L. Sen, R.-P. Lianga and J.-D. Qiu, Halogen microregulation in metal-organic frameworks for enhanced adsorption performance of ReO4-/TcO4-, J. Hazard. Mater., 2023, 446, 130744, DOI:10.1016/j.jhazmat.2023.130744.
- A. Brown and P. D. Beer, Halogen bonding anion recognition, Chem. Commun., 2016, 52, 8645–8658, 10.1039/C6CC03638D.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi and G. Resnati, et al., The halogen bond, Chem. Rev., 2016, 116(4), 2478–2601, DOI:10.1021/acs.chemrev.5b00484.
- S. Scheiner, On the capability of metal–halogen groups to participate in halogen bonds, CrystEngComm, 2019, 21, 2875–2883, 10.1039/C9CE00496C.
- S. Zhou, X. Zhou, P. Zhao, Y. Huang, Y. Xu, X. Wu, S. Chen, C. Huang, Y. Jin, Y. Zhang and C. Xia, Selective removal of ReO4−/TcO4− by an imidazolyl-based polymer from 3 M HNO3 solution, Colloids Surf., A, 2024, 685, 133277, DOI:10.1016/j.colsurfa.2024.133277.
| This journal is © The Royal Society of Chemistry 2025 |