
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of Co3Te4–Fe3C for efficient overall water-splitting in an alkaline medium
M.
Abdul
 *ac,
Miao
Zhang
a,
Tianjun
Ma
a,
Nouf H.
Alotaibi
b,
Saikh
Mohammad
b and
Yin-Sheng
Luo
a
*ac,
Miao
Zhang
a,
Tianjun
Ma
a,
Nouf H.
Alotaibi
b,
Saikh
Mohammad
b and
Yin-Sheng
Luo
a
aSchool of Electronics and Communication Engineering, Quanzhou University of Information Engineering, Quanzhou, Fujian, China. E-mail: mabdul@mail.ustc.edu.cn
bDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
cResearch Institute of Electronic Science and Technology of UESTC, Chengdu, China
First published on 21st December 2024
Abstract
The large amounts of attention directed towards the commercialization of renewable energy systems have motivated extensive research to develop non-precious-metal-based catalysts for promoting the electrochemical production of H2 and O2 from water. Here, we report promising technology, i.e., electrochemical water splitting for OER and HER. This work used a simple hydrothermal method to synthesize a novel Co3Te4–Fe3C nanocomposite directly on a stainless-steel substrate. Various physical techniques like XRD, FESEM/EDX, and XPS have been used to characterize the good composite growth and confirm the correlation between the structural features. It has been shown that the composite's morphology consists of interconnected particles, each uniformly coated with a thin layer of carbon. This structure then forms a porous network with defects, which helps stabilize the material and improve its charge conductivity. XPS analysis shows that combining Fe3C with Co3Te4 adjusts the atomic structure of both metals. This interaction creates redox sites (Fe3+/Fe2+ and Co3+/Co2+) at the Co₃Te₄–Fe₃C interface, which are crucial for activating redox reactions and enhancing electrochemical performance. The results also confirm the presence of multiple synergistic active sites, which contribute to improved catalytic activity. The optimized chemical composition and conductive structure result in enhanced electrocatalytic activity of Co3Te4–Fe3C towards electron transportation between the material interface and medium. It is found that the Co3Te4–Fe3C catalyst exhibits robust OER/HER activity with reduced overpotential values of 235/210 mV@10 mA cm−2 and Tafel slopes of 62/45 mV dec−1 in an alkaline solution. For overall water-splitting, cell voltages of 1.44, 1.88, and 2.0 V at current densities of 10, 50, and 100 mA cm−2 were achieved with a stability of 102 h. The electrochemically active surface area of the composite is 1125 cm2, indicating that a large surface area offered numerous reactive sites for electron transfer in the promotion of the electrochemical activity. The enhancement in catalytic performance was also checked using chronoamperometry analysis, reflecting long-term stability. Our results provide a novel idea for designing a composite of carbide with chalcogenide with robust catalytic mechanisms, which is useful for various applications in environmental and energy conversion fields.
1. Introduction
Increased exploitation of fossil fuels for energy production has caused a serious environmental crisis and climate change, motivating the scientific community to urgently increase research into clean and renewable alternatives for the future.1 For example, a sustainable hydrogen economy is an important concept to utilize green energy fuels for society's demands. Hydrogen, with high gravimetric energy density, no carbon footprint, and an abundant supply, is an attractive energy carrier that can generate chemical energy when needed.2 Thus, we need an effective technology such as water electrolysis, which is a favorable solution for H2 and O2 evolution on a large scale, because of its high efficiency, environmental friendliness, selectivity, and ability to convert renewable energy with an affordable cell setup.3 The electrocatalysis of water consists of OER at the anode for O2 gas generation and HER at the cathode for H2 gas generation; both are heterogeneous processes at the catalyst surface with KOH solution.4 On the other hand, water electrolysis is a difficult process and suffers from high overpotentials caused by sluggish electrode kinetics of the reactions. From kinetics studies, the hydrogen evolution reaction involves a double electron transfer process to generate hydrogen and hydroxyl ions from splitting water molecules. In contrast, OER is kinetically slow due to the complex 4e− transfer mechanism with a high overpotential, and the need for O–H bond breaking, thus there is a tendency to form oxygen–oxygen bonds and water again.5,6 Additionally, a high voltage of more than 1.23 V is generally needed for the OER, and HER usually operates from 0 V and achieves a benchmark current density to initiate the processes.7 Notably, greater overpotentials are required to reduce the major energy barriers for the actual reaction and bring high overall efficiency under many active electrocatalytic materials.8 Thus, ongoing research is based on developing efficient catalysts with durability, optimum efficiency, low cost, and good fabrication/design, which have always exhibited reduced overpotentials and small Tafel slopes in water splitting systems and must be utilized for widespread industrial applications. Various noble metals like Pt-based materials have shown the best performance for water reduction, whereas IrO2 and RuO2 have the most potential in electrocatalysts for water oxidation.9,10 However, these precious materials have limitations, such as unfavorable cost, tricky storage, poor stability, short supply, and poor electroactivity, impeding their commercial use.11,12 Therefore, the continued search to replace noble metal electrocatalysts with cost-effective non-noble metal-based electrocatalysts must be concerned with significant features of abundance, stability, low overpotentials, and high activities. Furthermore, the effectiveness of the electrocatalyst in catalytic progress is also based on the atomic structure, surface morphology, and composition.13,14 Accordingly, transition metals (e.g., Fe, Co, and Ni, etc.) show outstanding characteristics of smaller d orbitals, particular structural properties, catalytic activity, and intrinsic semiconducting behavior, as well as metallic characteristics, numerous active sites, and adjustable electronic behavior with good mechanical and chemical stability.15 Therefore, they have attracted lots of significant interest for solving catalytic problems.16–18 Specifically, transition metals, including oxides, hydroxides, phosphides, nitrides, tellurides, sulfides, selenides, and carbides, have been reported to explore superior catalysts towards OER/HER activity.3,7,19–24 Among these, transition metal carbides (TMCs), including iron ones, are sufficient for electrocatalytic activity in electrocatalysts. Fe3C, with its unique structure and cost-effective advantages, shows that carbon atoms are induced in the voids of densely packed metallic lattices.25–27 In addition, introduced interactions between Fe–Fe and covalent Fe–C endow Fe3C with prominent mechanical strength, excellent stability, and corrosion resistance, to which the interfacial electron transfer in electrocatalysis can be attributed to.28–32 Extensive work has been reported on different compositions of iron carbide, such as Fe/Fe3C,33 Fe3C@NG,27 and Fe3C@N-CNT,34 in the electrochemical field. In contrast, the electronic conductance of Fe3C-based electrocatalysts is poor and shows insufficient reactive sites. Therefore, iron carbide materials must be optimized further for electrocatalytic oxygen/hydrogen evolution. Different effective modification methods, such as the synthesis of nanostructures, interfacial engineering, composite formation, and elemental alloying/doping, are widely used in electrochemistry because they can be used to adjust the electronic structure.35 Notably, interface engineering by adopting a composite strategy has proved a feasible way to enhance the bifunctional activity of transition-based catalysts, as the adaptive coupling effects optimize the rate of dissociation/proton activity for HER and OER, thus providing electron modulation with transition metal active sites.36,37 Therefore, designing heterogeneous interfaces of a constructed Fe3C compound with Co3Te4 has resulted in large quantities of interfacial defects that serve as beneficial sites for reactant/intermediate adsorption and lead to the promotion of electrocatalytic activity. However, iron carbide's significance can never be ignored because embedded carbon develops a porous structure, and porous materials result in the adsorption of reactant intermediates.For example, cobalt chalcogenides such as Co3Te4 have been shown to have the highest rate of electrochemically catalysis for the OER and HER due to their versatile structures and covalencies.38 Compared with S and Se chalcogenides with electronegativities of 2.55, participating element Te has attracted the most attention due to its smaller electronegativity of 2.1, atomic dimensions, and even strong metallic connection characteristics with multiple oxidation states (−2, +2, +4, +6), which are currently essential in improving OER/HER reaction.39–42 Besides the good electronic conductivity and electronic properties of Co3Te4, the low-spin Co(II) coordination (t2g6,eg1) may increase the Te band efficiency, which opens up new pathways to facilitate the charge transportation mode at the electrolyte/electrocatalyst interphase.43–45 Owing to these excellent features, researchers have introduced cobalt telluride-based catalysts, including CoTe2/TM,46 CoP–CoTe2,47 CoTeNR/NF,48 and S–CoTe/CC49 for electrochemical application. In this paper, we synthesize and fabricate a porous Co3Te4–Fe3C material with interconnectivity by choosing an effective hydrothermal approach. The coordination of transition metal species contributed to exposing more active sites and larger specific surface areas, thus improving the electrocatalytic activity.
Furthermore, the redox ability of iron (Fe2+/Fe3+) and cobalt (Co2+/Co3+) during electronic transition can reduce oxidation/reduction potentials with facile electron transfer, thus making the electrocatalyst more energy efficient. Moreover, this bifunctional electrocatalyst results in a synergistic effect of the active structure due to the typical coexistence of intrinsically active Fe/Co sites and carbon-encapsulated metallic nanoparticles. The crystal structure, morphology, composition, and electronic interaction of the synthesized samples were analyzed through XRD, SEM/EDX, and XPS. According to the above capabilities, Co3Te4–Fe3C exhibits a better performance with an overpotential of 227 mV (10 mA cm−2) and a Tafel slope of 68.4 mV dec−1 for OER, and similarly, a lower overpotential of 211 mV for HER. All electrochemical studies are carried out using LSV, EIS, ECSA, and chronoamperometry tests. ECSA leads to a higher capacitance of 45.0 mF cm−2 and an increased specific surface area of 1125 cm2, which is favorable for water electrolysis. Additionally, electrocatalysts provide long-term stability of 60/90 h for OER/HER reactions in an alkaline medium. In summary, it is found that the novel couple Fe3C with Co3Te4 reduces kinetic energy barriers and achieves superiority in fundamental and practical insights.
2. Experimental
2.1. Reagents
Cobalt(II) acetate [Co(CH3COO)2·4H2O], potassium hydroxide (KOH, 98%), Nafion solution (5 wt%), N,N-dimethylformamide (DMF), ferric nitrate [Fe(NO3)39H2O], and ethanol (99.9%) were used. All chemicals were purchased from Sigma-Aldrich. Deionized water was used for all experiments.2.2. Synthesis of Co3Te4
Co3Te4 was prepared using a facile hydrothermal method. In a typical procedure, 0.2 mM of cobalt acetate and 0.2 mM of tellurium powder were immersed in 10 mL of homogenous aqueous electrolyte containing 6 M KOH solution. At the same time, 5 mL of hydrazine monohydrate was added to a precursor suspension under magnetic stirring for 3 h to obtain a good mixture. Then, the resulting solution was transferred to a Teflon-lined autoclave and hydrothermally treated at 180 °C in an oven for 5 h. After natural cooling, the obtained product was washed repeatedly with ethanol and DI water and dried at 60 °C. Then, the obtained material was precisely annealed in a tube furnace at 70 °C under an Ar atmosphere. The as-synthesized product was labeled Co3Te4 and saved for characterization.2.3. Synthesis of Fe3C and Co3Te4–Fe3C
To fabricate the Fe-based MOF precursor, 7 mL of cyclohexane, 7 mL of deionized water, and 140 mmol of 1,3,5-benzene tricarboxylic acid (BTC) were dissolved in 200 mmol of ferric nitrate, gently stirring for 30 min. Then 1 mL of 2 M NaOH was added dropwise into the above mixture, which was further stirred to start the reaction. Subsequently, the mixed solution was transferred to an 80 mL sealed autoclave reactor and maintained at 370 °C for 48 h for a complete reaction. Then, the autoclave reactor was allowed to cool naturally, and the fabricated sample, denoted as Fe-based MOF, was filtered, washed with DI water and ethanol multiple times, and then dried at 333 K in a drying oven overnight. The obtained powder was then carbonized at 673 K under an N2 carrier gas at a flow rate of 5 K min−1, and the final product (Fe3C) was ground. The Co3Te4–Fe3C composite was prepared in parallel under the same conditions mentioned above, but 0.5 g of already synthesized Co3Te4 was added to the above mixture before thermal treatment.2.4. Electrochemical measurements
All electrochemical investigations used an AutoLab PGSTAT-204 electrochemical workstation at ambient temperature. A Teflon-covered Pyrex glass cell is employed with a standard three-electrode electrochemical setup to investigate all electrochemical parameters. A basic medium (1.00 M KOH solution) is adopted with a pH of 13.8. The electrochemical cell is cleaned from surface impurities by boiling in a mixture of H2SO4 and HNO3, then inserted into ultrapure water acetone and dried in an oven at 80 °C for 30–40 min. Before measurement, the electrolyte is deaerated with pure N2 for at least 30 min. Co3Te4–Fe3C/SS, Co3Te4/SS, and Fe3C/SS were prepared as working electrodes for OER/HER analyses.Platinized platinum wire and Ag/AgCl electrodes functioned as counter and reference electrodes. To make catalyst ink using synthesized powder including Co3Te4–Fe3C, Co3Te4, and Fe3C, the testing process is as follows: about 5 mg of the hydrothermally synthesized compound is dispersed in the solution of de-ionized water and Nafion, then the mixture is subjected to ultrasonication for 30 min to obtain catalytic ink. Then, the ink is pipetted onto a cleaned stainless-steel substrate, and the as-prepared electrodes are dried naturally at room temperature. The potential vs. Ag/AgCl cited in this research is changed into the RHE scale via the following formula:50
| ERHE = EAg/AgCl + 0.059(pH) + EOAg/AgCl | (1) |
All potentials and voltages of polarization curves are acquired to correct with iR-compensation:51
| Ecompensated = Emeasured − iRu | (2) |
Some extra potential is required to achieve a complete electrochemical reaction; the overpotential is expressed in terms of η. For water-splitting reactions, the following expression is used to obtain the overpotential value:52
| η(V) = ERHE − 1.23 | (3) |
Electrochemical properties are evaluated through LSV, EIS, ECSAs, and chronoamperometry (i–t) to study the electrochemical performance of OER/HER. Linear sweep voltammetry (LSV) graphs are recorded at a scan rate of 5 mV s−1 using an appropriate potential window to obtain polarization curves. Tafel slopes were utilized to determine the kinetics of reactions and gain insight into the reaction mechanism of catalysts for OER and HER. The Tafel equation is applied to the linear part of the polarization curve. Tafel plots are obtained with the log of the current density vs. overpotential (η) plot according to the following relationship:53
η = α + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj logj | (4) |
The transfer of electrons and the effect of the solution resistance across the electrode–electrolyte interphase are measured using a Nyquist plot, which is analyzed from EIS by applying a bias of 5 mV within a frequency range from 100 kHz to 0.01 Hz in KOH media. The electrochemically active surface area (ECSA) of the as-prepared working electrodes is computed using Cdl. It is estimated using CV in a non-faradaic window from 0.8 to 1.5 V vs. RHE under different scan rates from 10 to 70 mV s−1 for OER/HER. The Cdl is obtained by plotting the difference between the anodic and cathodic currents (ja–jc) against potential vs. different scan rates, in which the half-slope of the obtained straight line is used to determine the Cdl value. To calculate the ECSA, the double-layer capacitance is divided by the specific capacitance of the flat electrode, which is 0.04 mF cm−2 using the following equation:54
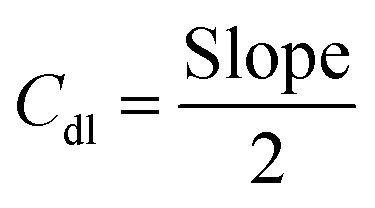 | (5) |
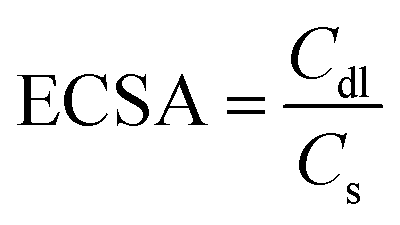 | (6) |
Long-term stability evaluation for the electrocatalysts is performed using a chronoamperometry (i–t) test with fixed potential in KOH aqueous solution for an extended period.
2.5. Characterization
The structural purity of the synthesized material is analyzed using a Bruker D8 XRD. The external morphology and elemental mapping along with the composition and distribution are obtained on a scanning electron microscope coupled with energy-dispersive X-ray (EDX) spectroscopy. An X-ray photoelectron spectrometer (XPS, ESCALab 50i, λ = 1486.7 eV) equipped with an Al X-ray source is employed to test the surface elemental states and binding energies of the elements in the samples. The electrochemical properties of the samples are evaluated utilizing LSV, EIS, ECSA, and CA tests, using the Potentiostat instrument of Metrohm AUTOLAB (PGSTAT-204). Electrochemical tests are performed under ambient temperature.3. Results and discussion
3.1. X-ray diffraction (XRD)
X-ray diffraction explains the phase composition and crystalline nature of as-synthesized products. The XRD patterns of pure Fe3C, Co3Te4, and the Co3Te4–Fe3C composite are shown in Fig. 1. The XRD pattern of Co3Te4 clearly shows diffraction peaks for planes of (021), (030), (004), (212), and (035) (JCPDS No. 00-044-1057). Besides, the diffraction planes of the Fe3C crystal system corresponding to (110), (111), (112), (300), and (113) are indexed and closely match with standard JCPDS Card No-00-006-0670. The Co3Te4–Fe3C composite system is attributed to forming mixed phases (Fe3C and Co3Te4) with strong and sharp peak intensity. This indicates strong contact between Fe3C and Co3Te4 to improve the electrochemically active area towards good electrochemical activity and stability of OER and HER reactions. No prominent peaks related to impurities were found, implying the crystalline nature of materials.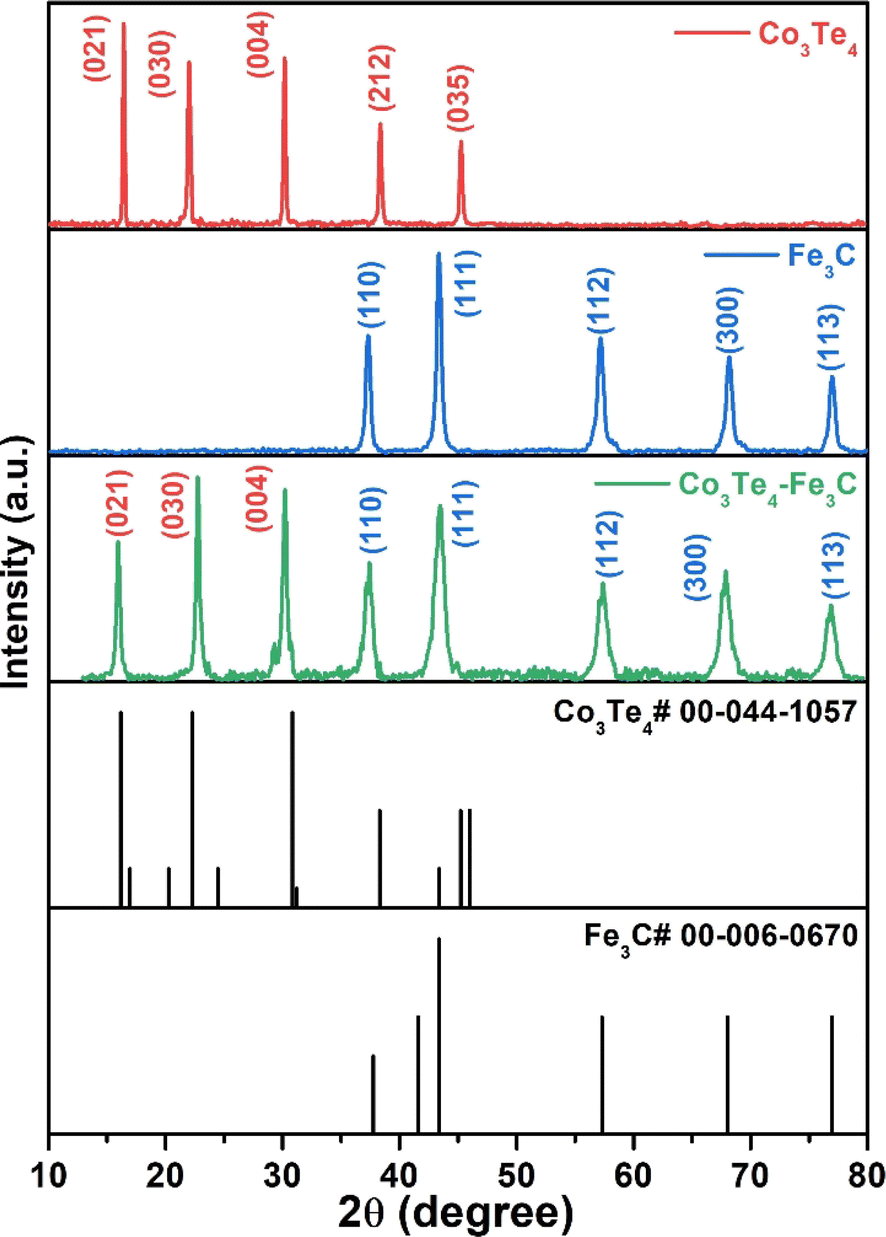 | ||
| Fig. 1 XRD patterns of as-synthesized Co3Te4, Fe3C, and Co3Te4–Fe3C catalysts. | ||
3.2. X-ray photoelectron spectroscopy (XPS)
The XPS technique provides information on the chemical makeup and bonding states of the samples. The survey pattern of Co3Te4–Fe3C in Fig. 2(a) shows the existence of distinct peaks corresponding to Fe (2p), C (1s), Co (2p), and Te (3d) at their corresponding binding energies. Interestingly, no evidence of other elements on the catalyst surface proves the formation of a high-purity composite material. The orbital spectra of Co (2p) are shown in Fig. 2(b), showing that Co 2p was split into two prominent peaks centered at 780.9 eV and 796.5 eV, related to Co3+ 2p3/2 and Co3+ 2p1/2, respectively.55 In addition, the oxidation state of Co3+ (2p3/2) is deconvoluted into 779.9 and 783.3 eV, while deconvolution at 800.2 eV corresponded to the Co2+ (2p1/2) species in cobalt telluride.56,57 The presence of mixed oxidation states of Co2+/Co3+ gives information about more Co atoms in the catalyst. Moreover, shake-up satellite peaks at 786.5 eV (Co2+ 2p3/2) and 802.3 eV (Co2+ 2p1/2) suggest the bivalent formation of cobalt.58,59 This observation indicates Co's electron donor ability to decrease the required energy for CoOOH formation, further successfully showing the surface reconstruction phenomena to enhance catalyst activity.60–62 In Fig. 2(c), the Te 3d core region of the sample is deconvoluted into metallic Te 3d3/2 and Te 3d5/2 peaks at BEs of 585.95 and 575.535 eV, ascribed to the Te–Co bond, increased due to superficial oxidation of the Te+4 and Te−2 oxidation states of Te in the Co3Te4–Fe3C network.63–66 As presented in Fig. 2(d), the Fe 2p region shows two signals at 711.28 and 725.19 eV arising from Fe 2p3/2 and Fe 2p1/2 with a +3 state of Fe in Fe3C. Moreover, the apparent positive shifts of the binding energies at 711.28 (2p3/2) and 724.29 (2p1/2) eV are related to Fe2+ while the peaks at 712.98 (2p3/2), 714.58 (2p3/2), 725.79 (2p1/2) and 727.89 (2p1/2) eV correspond well with Fe3+.45,67,68 In addition, the respective satellite signals are found at approximately 718.49, 721.19, and 733.196 eV, further supporting mixed oxidation states of Fe2+/Fe3+ in Co3Te4–Fe3C.69,70 This indicates that Fe exists in the forms Fe2+ and Fe3+ in the Co3Te4–Fe3C, which is consistent with the XRD result. Besides, the dominant peaks of the C 1s spectrum in Fig. 2(e) are at binding energies of 284.8 and 286.0 eV and are related to M–C (M = Fe, Co, Te) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2, respectively.71,72 Furthermore, the BE peak at 284.8 eV is assigned to the metal–carbon bonding, suggesting that iron atoms are coordinated with the carbon in iron carbide, thus promoting the successful formation of the Fe3C phase,73 and the peaks at 288 and 289 eV are assigned to CO due to environmental oxygen. The modulation in the C 1s spectra analysis results by iron, therefore, demonstrate that optimized carbon defects have been generated in Co3Te4–Fe3C; thus, defect sites with low oxygen coordination led to the reduction of the overpotential and result in high efficiency for adsorption and reduction of the intermediates (H*, H2O*, OH*, O*, and OOH*) for OER/HER reaction. Moreover, the changed oxidation states of Co and Fe are mainly due to the difference in electronegativity of Co and Fe. The series of CoFe hydroxides increases the intrinsic conductivity of the Co3Te4–Fe3C to enhance the electrocatalytic performance.74,75 All the above results prove the phenomenon of interfacial charge transfer efficiency due to the conductivity of the coupled surface, meanwhile making the catalyst active by reducing electron density on CoOOH, while FeOOH accepts electrons. Thus the distribution of charge densities leads to abundant active sites, resulting in faster H2 and O2 evolution for the Co3Te4–Fe3C catalyst.
C sp2, respectively.71,72 Furthermore, the BE peak at 284.8 eV is assigned to the metal–carbon bonding, suggesting that iron atoms are coordinated with the carbon in iron carbide, thus promoting the successful formation of the Fe3C phase,73 and the peaks at 288 and 289 eV are assigned to CO due to environmental oxygen. The modulation in the C 1s spectra analysis results by iron, therefore, demonstrate that optimized carbon defects have been generated in Co3Te4–Fe3C; thus, defect sites with low oxygen coordination led to the reduction of the overpotential and result in high efficiency for adsorption and reduction of the intermediates (H*, H2O*, OH*, O*, and OOH*) for OER/HER reaction. Moreover, the changed oxidation states of Co and Fe are mainly due to the difference in electronegativity of Co and Fe. The series of CoFe hydroxides increases the intrinsic conductivity of the Co3Te4–Fe3C to enhance the electrocatalytic performance.74,75 All the above results prove the phenomenon of interfacial charge transfer efficiency due to the conductivity of the coupled surface, meanwhile making the catalyst active by reducing electron density on CoOOH, while FeOOH accepts electrons. Thus the distribution of charge densities leads to abundant active sites, resulting in faster H2 and O2 evolution for the Co3Te4–Fe3C catalyst.
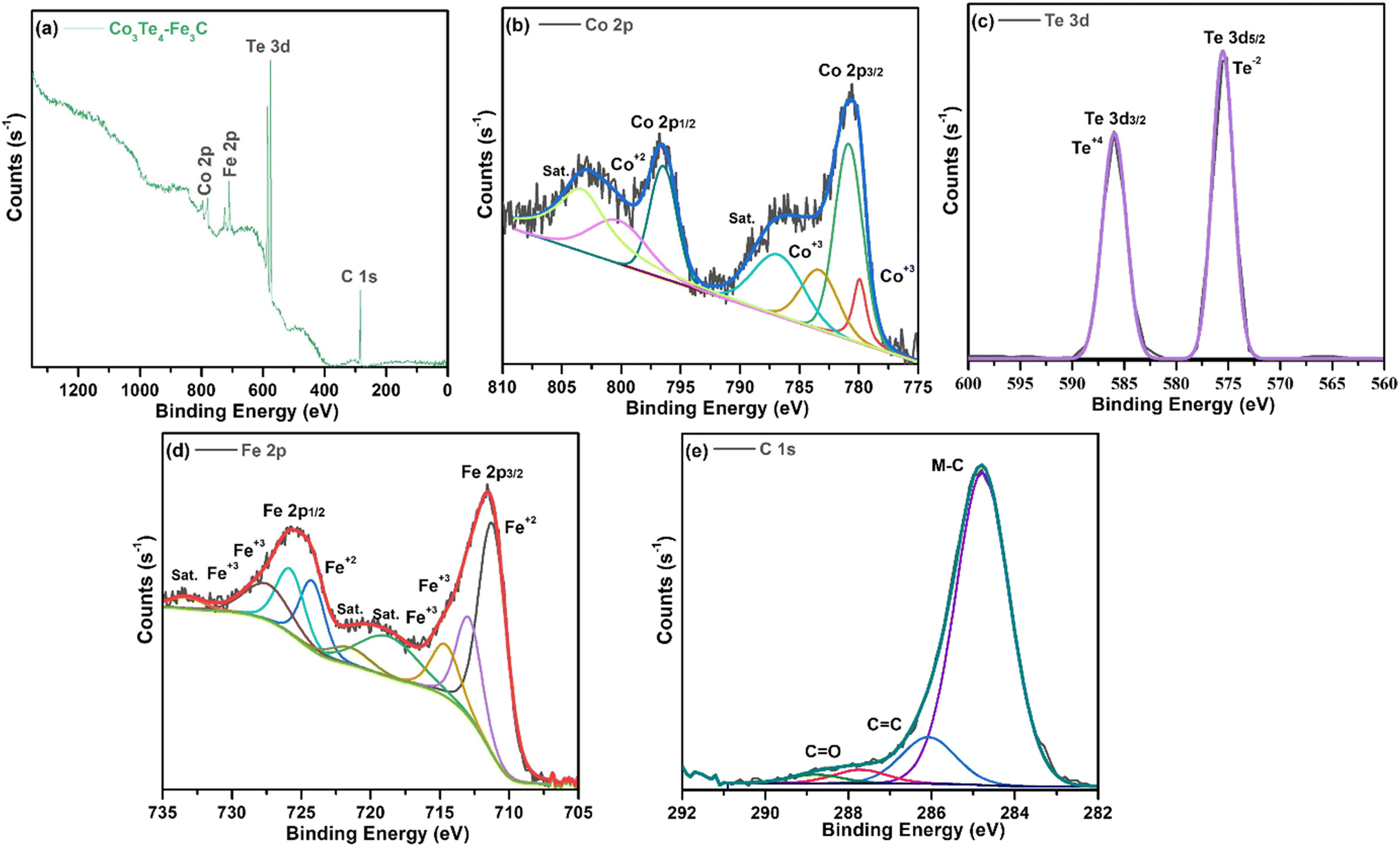 | ||
| Fig. 2 High resolution XPS spectrum of Co3Te4–Fe3C: (a) survey spectrum, and deconvoluted curves of (b) Co 2p, (c) Te 3d, (d) Fe 2p and (e) C 1s. | ||
3.3. SEM and EDX analysis
The size and surface morphology of the prepared electrocatalysts are investigated with SEM. Fig. 3 shows the SEM image of Co3Te4–Fe3C, where information on the smooth surface of the nanoparticles encapsulated into the carbide structure is confirmed. The synthesized Fe3C is composed of several large particles with encapsulation of the doped phase of carbon; the carbon feature effectively leads to a nanosheet-like morphology of the Fe3C,76,77 which is manifested by the abundant dark Fe3C nanoparticles on the catalyst surface as presented in Fig. 3. A literature survey shows that Co3Te4 has irregularly shaped nanoparticles without apparent aggregation, indicating the successful yield of cobalt telluride. During composite formation, when Fe3C is closely connected with Co3Te4 prepared using the hydrothermal synthesis procedure, as displayed in Fig. 3, a change in the arrangement of nanocrystals with agglomeration and a tight interface between Fe3C and Co3Te4 is observed, which corresponds to the generation of dislocations in the lattice and provides exposure of sufficient active sites. Meanwhile, a significant transformation in morphology, i.e., abundant defects, caused the formation of the porous structure, which exhibited a convenient charge/ionic diffusion pathway on the Co3Te4–Fe3C surface. The mean width of the composite is equivalent to 133 ± 0.4 nm.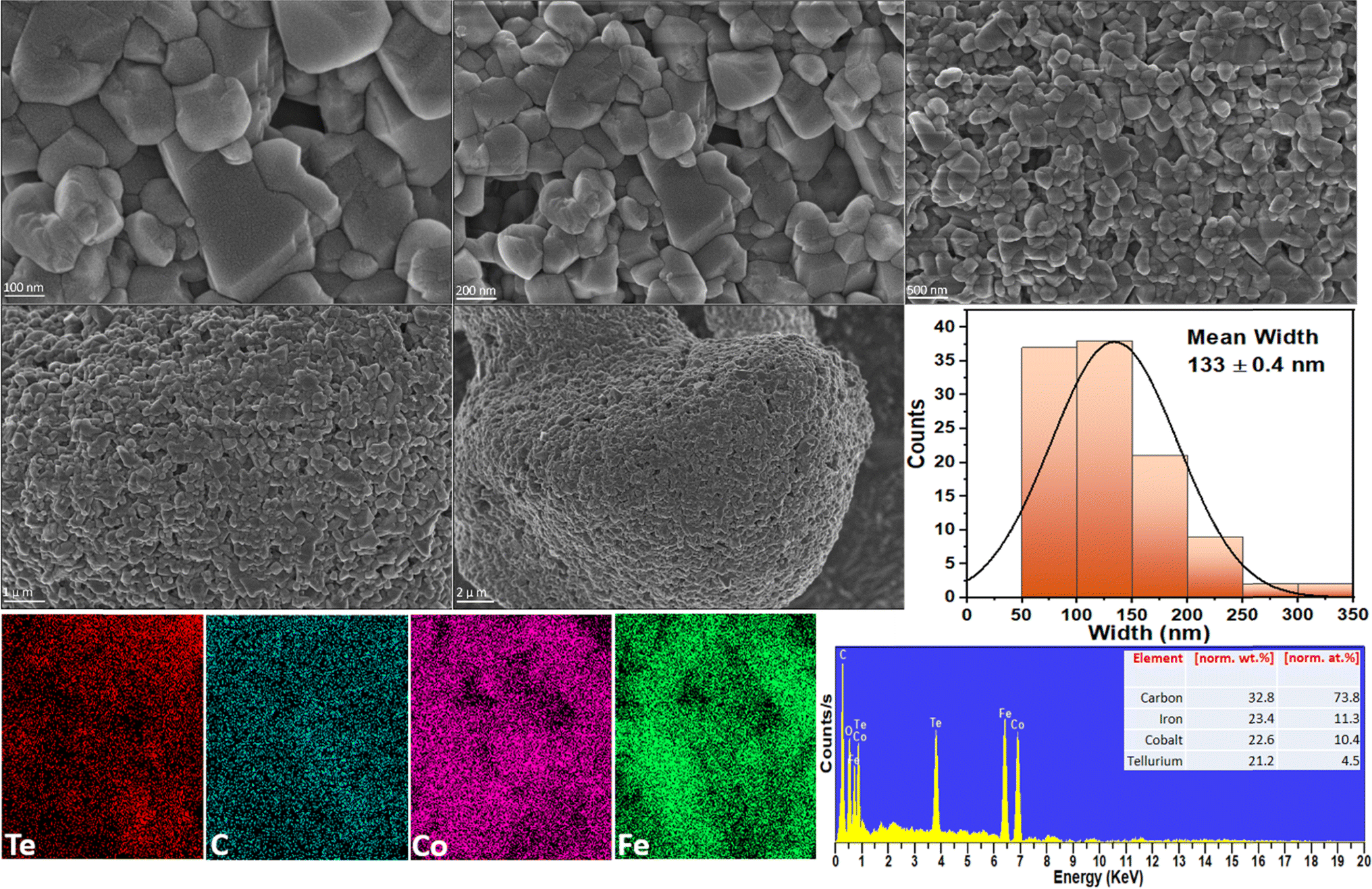 | ||
| Fig. 3 High and low magnification FE-SEM analysis, and the corresponding widths, and EDX spectrum/mapping of the Co3Te4–Fe3C composite. | ||
Moreover, the uniform distribution of Fe, Co, Te, and C elements in the catalyst composition without adding impurities is shown in Fig. 3 by EDX spectra along with the atomic (at%) and weight (wt%) percentages. Fig. 3 also shows EDX spectra and line scans for Co3Te4–Fe3C, revealing the uniform distribution of carbon and telluride, whereas iron and cobalt are scattered in the nanoparticles. From the elemental line scan analysis, homogeneity features in Co3Te4–Fe3C are verified, suggesting the random mixing of Fe, Co, Te, and C elements in their respective atomic ratios, ultimately leading to confirmed interaction between the metal particles. Except for pure Fe3C and Co3Te4, the trend of the Co3Te4–Fe3C nanostructure suggests that the interconnected particles and carbide support in the catalyst form a flexible, conducting network with an enhanced active surface area providing advantageous interfacial charge transfer and structural stability during electrochemical (OER/HER) operation.
3.4. Electrochemical activity
A study on the electrocatalytic performance of the as-synthesized Fe3C, Co3Te4, and Co3Te4–Fe3C is conducted to research their oxidation/reduction activity. All the products are deposited on stainless steel (SS) substrates as electrocatalysts using 1 M KOH aqueous electrolyte. Control of electrochemical parameters such as aqueous electrolyte, scan rate, charge transfer, high current density, small overpotential, adsorption/desorption phenomena, diffusion, stability, and electrochemical reaction kinetics are necessary to optimize electrochemical water splitting. A standard current density of 10 mA cm−2 shows efficient activity, which can be achieved by considering 10% solar-to-fuel conversion efficiency when comparing electrochemical activities.First, the OER performance of all the samples (Fe3C, Co3Te4, and Co3Te4–Fe3C) were investigated by LSV at a scan rate of 5 mV s−1 in 1.0 mol alkaline solution using three-electrode configurations. According to the LSV profile, as shown in Fig. 4(a), all samples with an onset potential of 1.41 V vs. RHE demonstrate satisfactory OER activity, whereas Co3Te4–Fe3C exhibits excellent activity within the potential window (1.0–2.2 V vs. RHE). The onset potential is the minimal starting potential to initiate OER, presented at a small current density. The LSV pattern of Co3Te4–Fe3C followed a sharp catalytic wave, which indicates that the peroxidative peak feature can be assigned to the oxidation of iron (Fe3+ to Fe2+) and cobalt (Co3+ to Co2+) for O2 generation on the electrode surface. However, an effective catalyst with overpotential to afford a current density of 10 mA cm−2 is required to start OER catalysis. For this, LSV displayed the best OER activity for Co3Te4–Fe3C with a heterostructure at an overpotential of 227 mV to provide 10 mA cm−2. This overpotential is much lower than pristine electrocatalysts like Fe3C (288 mV) and Co3Te4 (317 mV). A reduced overpotential for a heterostructured catalyst is also confirmed by a sharp and steep pre-oxidation peak, which involves the activation process of intermediate hydroxo and peroxide production on the catalyst surface to speed up the sluggish OER. In addition, coupling between Fe3C and Co3Te4 led to a highly defective porous nature (confirmed via SEM), making it easier for Fe and Co atoms to interact, so effective conductivity is achieved. The most extensive electrocatalytic surface area with many exposed active sites thus promoted electron transportation at the interphase during catalysis. Note that the modified electronic structure of Co3Te4–Fe3C favorably initiates water oxidation at a lower overpotential relative to pure combinations like metal/non-metal, so carbon and telluride contact increases the degree of covalency around the transition metal. Besides, the better oxidation performance of Co3Te4–Fe3C can also be credited to the lower electronegativity and strength of metal carbide/metal chalcogenides during composite formation. A decrease of the catalytic activity for Fe3C and Co3Te4, as shown in the LSV, reveals the formation of single phases with limited active sites. Fig. 4(b) and Table 1 show a comparison of the overpotential of Co3Te4–Fe3C and pristine Fe3C and Co3Te4 electrocatalysts at the desired 10 mA cm−2.
 | ||
| Fig. 4 OER electrocatalysis: (a) LSV curves, (b) bar chart of the overpotential at 10 mA cm−2 current density, (c) Tafel plots, (d) EIS plots, (e) comparison of Rp and Rs, and (f) stability (i–t) curve of Co3Te4, Fe3C and Co3Te4–Fe3C. | ||
| Catalyst | Electrolyte | Overpotential (η) mV@10 mA cm−2 | Tafel slope mV dec−1 | Stability (h) | Ref. |
|---|---|---|---|---|---|
| CuO/Co3O4 | 1 M KOH | 227 | — | — | 78 |
| CuO/Fe–Co3O4 | 1 M KOH | 232 | — | — | 79 |
| Cu(OH)2@NiFe | 1 M KOH | 283 | 88 | 10 | 80 |
| Fe–Co3O4/CNT | 1 M KOH | 300 | 62 | 25 | 81 |
| NiFe2O4@Co3O4 | 1 M KOH | 251 | 36 | 50 | 82 |
| Co3O4/Co9S8 | 1 M KOH | 281 | 37 | 12 | 83 |
| Fe3C–Mo2C | 1 M KOH | 274 | 36.18 | 25 | 84 |
| Fe3C/NF | 1 M KOH | 262 | 49 | 100 | 85 |
| Co5Fe5–C | 1 M KOH | 245 | 58.2 | 20 | 86 |
| FeCoP/C | 1 M KOH | 282 | 53 | — | 87 |
| FeMn-MOF | 1 M KOH | 290 | 87.02 | 12 | 88 |
| V–Co/CoO@C | 1 M KOH | 320 | 143 | — | 89 |
| Fe-doped NiO | 1 M KOH | 274 | 79.1 | 10 | 90 |
| MnFeCoNi | 1 M KOH | 302 | 83.7 | 20 | 91 |
| Sm2O3/Fe2O3 | 1 M KOH | 272 | 75 | 200 s | 92 |
| CoTe | 1 M KOH | 290 | 92 | 24 | 93 |
| Co 3 Te 4 –Fe 3 C | 1 M KOH | 227 | 68.4 | 59 | TW |
The Tafel slope is another crucial factor in providing information about the mechanism and kinetic behavior of the OER, as it is associated with the RDS and ion transport mechanisms. It is discovered that the linear part of the polarization curve of each sample is consistent with the Tafel equation. Thus, Tafel slopes are determined via Tafel plots (η vs. logj). The lower the Tafel slope is, the faster the electron transfer kinetic process toward oxygen evolution reaction. As shown in Fig. 4(c), the fluctuations of Tafel slopes are due to differences in catalyst reactive sites as the Co3Te4–Fe3C catalyst showed a reduced Tafel slope value of 68.4 mV dec−1, which is superior those of to Fe3C and Co3Te4 with Tafel slopes of 85.5 and 129 mV dec−1, respectively. Generally, a Tafel slope of 70 mV dec−1 involves the 4e− transfer with one electron in each step, and Tafel slopes near 80 and 120 mV dec−1 are identical to the RDS with 3e− and 2e− transfer. Such a low Tafel slope value indicates possible synergistic effects of Fe3C with Co3Te4 accompanied by introduced defect sites, and it confirms that the participation of carbon and tellurium enhances the catalytic efficiency. Mainly, a low Tafel result is beneficial for the proton-coupled electron transfer (PCET) mechanism, which involves the formation of oxo-intermediates to accelerate the acceleration of OER in an alkaline medium.
For the lowest Tafel slope, a proposed OER electrocatalytic mechanism at the anode electrode as proton-coupled electron transfer in an alkaline solution includes a sequence of intermediates (OH*, O*, and OOH*) as follows:94
| M + OH− → M − OHads + e− | (7) |
| M − OHads + OH− → M − O + H2O + e− | (8) |
| M − O + OH− → M − HOO + e− | (9) |
| M − HOO + OH− → M + O2↑ + H2O + e− | (10) |
The water oxidation reaction involves attachment of the metal surface's anionic group (OH−). Coupling of Fe3C with Co3Te4 leads to alteration in the electronic structure through the oxidation of Fe3+ to Fe2+ and Co3+ to Co2+, giving rise to the adsorption of metal hydroxide and oxide intermediates on the surface of the electrocatalyst. Electron transfer is ascribed to the peroxide formation (M–O) based on the many exposed active sites. This peroxide further interacts with 1e− to generate hydroperoxide (M–OOH), leading to the evolution of the oxygen molecule from attachment to another OH− under another electron transfer, thus leaving the catalyst with no structural modifications. The initial step is the adsorption of the (O, OH) species, whereas the OOH* formation (step iii) is the rate-determining step.
Additionally, the Co3Te4–Fe3C structure has a porous, defective nature that helps adsorb O and OH species, promoting the formation of O–O bonds. The bond is more easily formed at the catalyst surface. The synergistic effects of the carbon (C) and tellurium (Te) nanomaterials improve electrical conductivity, which in turn enhances oxygen adsorption. This is beneficial for the oxygen evolution reaction (OER).
The facile electrochemical kinetics of the reactions on the electrocatalyst surface during OER are estimated by using electrochemical impedance spectroscopy (EIS). The associated Nyquist plots through EIS measurements for Fe3C, Co3Te4, and Co3Te4–Fe3C electrocatalysts at a specific potential are shown in Fig. 4(d), and the Rs and Rp values are summarized in Fig. 4(e) in bar chart form. The Nyquist plot indicates how fast electrons participate over the catalyst surface during the OER process. There are two semicircles in the EIS curves, and a high-frequency region on the left side is always a perfect half-circle representing Rp at the interface. In contrast, the low-frequency region on the right side is always an inclined line, meaning uncompensated solution resistance (Rs) in the electrolyte. In addition, by fitting the raw data of the EIS measurements, a simplified Randles circuit with quantitative charge transfer resistance, solution resistance, and a constant phase element is provided in the inset of Fig. 4(d). It is well known that small values of Rp give rise to a fast electron transport rate between the catalyst and electrolyte, with quicker return. As shown in Fig. 4(e), coordination of Co3Te4 with the Fe3C skeleton results in the smallest Rp (1.2 Ω) in comparison with Fe3C (Rp, 1.55 Ω) and Co3Te4 (Rp, 1.8 Ω), which is consistent with the increasing Tafel slopes of the corresponding pure samples. The smallest values of Rp and Rs indicate facile catalytic processes occurring by excellent electronic transfer on the catalytic surface, while the increased CPE parameter indicates the highest conductivity of the composite catalyst. This may also result from the defective porous structure and many active sites associated with redox couples. Thus, EIS measurements also indicate a high surface area that facilitates OER activity when there is a small applied overpotential and Tafel slope.
The long-term stability of the modified electrode in the OER reaction is studied using chronoamperometry under alkaline electrolyte conditions. As shown in Fig. 4(f), the Co3Te4–Fe3C heterostructure showed notable durability without evident changes even after 60 h of CA measurement with a constant current density of 10 mA cm−2, demonstrating the superior stability in the alkaline electrolyte. However, a slight drop in the current density may be caused by the electrolyte infiltrating the electrode, indicating the excellent adsorption and diffusion of the porous material with the KOH solution. The stability experiment revealed that no oxygen bubbles immediately leave the electrode surface; this signifies electrolyte transfer to the exposed active sites, corresponding to a reduced potential.
After successfully testing the Co3Te4–Fe3C electrocatalyst towards oxygen evolution reaction, the electrocatalytic activity of different cathodes towards HER activity is investigated in a 1 M alkaline medium using 3 electrode configurations.
Fig. 5(a) shows LSV with a scanning rate of 5 mV s−1 at a potential window of −1.0 to 0.0 (vs. RHE) for Fe3C, Co3Te4, and Co3Te4–Fe3C. A 10 mA cm−2 current density is standard for comparing the over potential toward HER. As shown, the polarization curve of Co3Te4–Fe3C slopes very sharply downward, corresponding to a very large hydrogen adsorption/desorption peak, and mainly has a larger particular area, which confirms plenty of active sites for hydrogen gas storage ability of the heterostructured catalysts. As observed, Co3Te4–Fe3C exhibited a reduced overpotential of 211 mV compared to Fe3C (267 mV) and Co3Te4 (327 mV) at a current density of 10 mA cm−2. The high catalytic activity is related to the large surface area and the influence of C and Te in coordinating Fe–Co-based compounds, suggesting a more favorable electronic structure for electrocatalytic proton reduction. Thus, it is apparent that a rapid increase in cathodic current provides more electron transfer for superior hydrogen evolution performance than unconnected species. Fig. 5(b) and Table 2 show the comparison of the overpotential to provide 10 mA cm−2 current density for cathodic electrodes of Fe3C, Co3Te4, and Co3Te4–Fe3C.
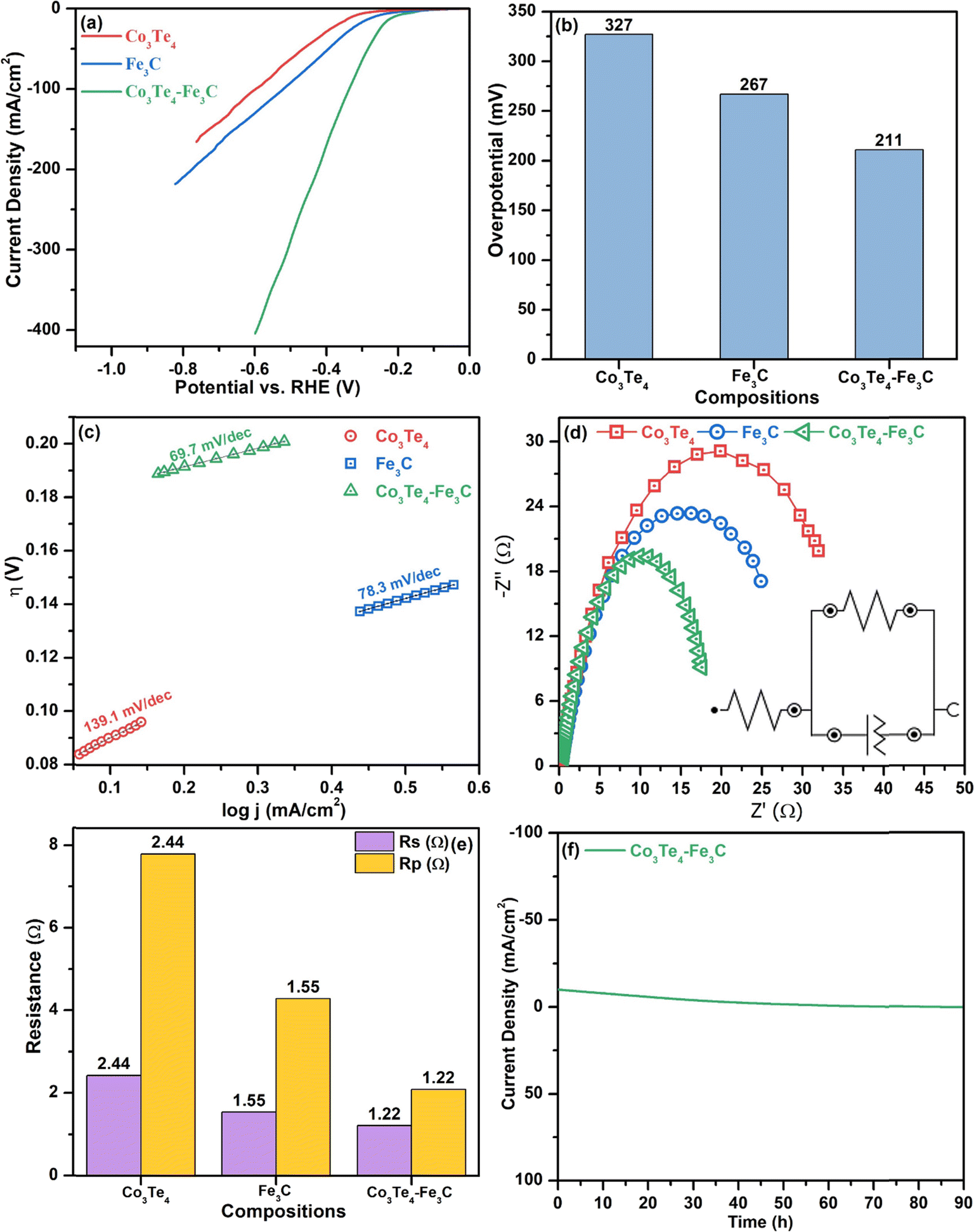 | ||
| Fig. 5 HER electrocatalysis: (a) LSV curves, (b) bar chart of overpotential at 10 mA cm−2 current density, (c) Tafel plots, (d) EIS plots, (e) comparison of Rp and Rs, and (f) stability (i–t) curve of Co3Te4, Fe3C, and Co3Te4–Fe3C. | ||
| Catalyst | Electrolyte | Overpotential (η) mV@10 mA cm−2 | Tafel slope mV dec−1 | Stability (h) | Ref. |
|---|---|---|---|---|---|
| NiFeOF | 1 M KOH | 253 | 96 | 18 | 95 |
| CuCoP/NC | 1 M KOH | 122 | 220 | 50 | 96 |
| Co3O4/Ppy/MWCNT | 1 M KOH | 490 | 110 | 3600 s | 97 |
| Fe@C | 0.5 M H2SO4 | 520 | 94 | — | 98 |
| Mo-NC@CoFe | 1 M KOH | 280 | 110 | 16 | 99 |
| Co2B/CoSe2 | 1 M KOH | 300 | 76 | 30 | 100 |
| NFC@CNSs | 1 M KOH | 213 | 115.1 | 42 | 101 |
| Fe@C-SN/50 | 0.5 M H2SO4 | 358 | 123 | — | 98 |
| CoTe/CoTe2 | 0.5 M H2SO4 | 230 | 57.1 | 20 | 102 |
| MoC–Cu | 1 M KOH | 233 | 73 | — | 103 |
| Co 3 Te 4 –Fe 3 C | 1 M KOH | 211 | 69.7 | 90 | TW |
Tafel analysis is commonly used to determine the reaction kinetics of HER through the derived Tafel slope. Fig. 5(c) shows the Tafel plots (log. J vs. E) of Fe3C, Co3Te4, and Co3Te4–Fe3C catalysts. A proposal for the mechanism of the hydrogen evolution reaction is provided either with a Volmer–Heyrovsky or Volmer–Tafel relationship. It can be shown that the cathodic Tafel slopes in the linear portions are 139.1, 78.3, and 69.7 mV dec−1 for Co3Te4, Fe3C and Co3Te4–Fe3C, as the Tafel slope of the heterostructured catalyst is relatively low in the 40–120 mV dec−1 range, indicating that the HER process outperforms the Volmer–Heyrovsky mechanism with fast kinetics.
Generally, the HER mechanism consists of proton discharge for hydrogen adsorption on the catalyst surface through three possible reaction steps in primary media:56,104
| M + H2O + e− → M − Hads + OH− (Volmer) | (11) |
| M − Hads + H2O + e− → M + H2 + OH− (Heyrovsky) | (12) |
| M − Hads + M − Hads → 2M + H2↑ (Tafel) | (13) |
The synergy between Fe3C and Co3Te4 results in fast kinetics for the discharge of protons from the catalyst surface. Notably, a rapid rate of discharging protons enhances the recombination rate of hydrogen bubbles, thus resulting in the stronger evolution of hydrogen gas. These results suggest the following. The substitution of the nanomaterial connecting to the transition-based catalyst in the 40–120 mV dec−1 range demonstrates that the Volmer–Heyrovsky (adsorption–desorption) pathway takes place when anionic doping favors a suitable electronic structure for the adsorption of the HER intermediate for efficient HER performance.
Additionally, the EIS technique is an indicator that explains the Rp and conductivity at the interfaces under alkaline conditions. Using EIS, the Nyquist plots for Fe3C, Co3Te4, and Co3Te4–Fe3C are shown in Fig. 5(d). A smaller radius of the semicircle demonstrates a smaller value of Rp, which indicates the tendency for stronger kinetics in cathodic electrochemical reactions. The corresponding electrical fitted circuit model consisting of solution resistance (Rs), polarization resistance (Rp), and a constant phase element (CPE) is presented in Fig. 5(d). Impressively, Fe3C taken together with Co3Te4 exhibited the lowest Rp of 1.22 Ω compared with the other samples (Fe3C: 1.55 Ω and Co3Te4: 2.44 Ω). In contrast, the CPE parameter of Co3Te4–Fe3C is much higher than that of Fe3C and Co3Te4, which indicates the effective surface area available for cathodic reaction. The lowest Rp indicates superior conductivity, which might be related to the defective porosity providing active points in the structure (analyzed using SEM) and intimate interfaces between the redox electrolyte and Co3Te4–Fe3C (consistent with XPS), and thus the designated structure results in a faster rate of the Faradaic process. Fig. 5(e) shows the values of Rp/Rs through a bar chart diagram.
The electrochemical stability of the proposed material is investigated to determine its importance to HER. Fig. 5(f) shows the i–t stability test for long-term durability with continuous H2 bubbles escaping from the electrode surface for 90 h, with the current density of 10 mA cm−2 maintained with a slight current attenuation. Therefore, the porous Co3Te4–Fe3C is a potential practical catalyst on both the cathode and anode associated with efficient activity and the heterostructure's low-cost, simple, and suitable composition.
To investigate the intrinsic catalytic behavior (exposure of active sites) towards OER and HER, the ECSA of the electrocatalysts is determined by evaluating the Cdl, which directly relates to the ECSA. First, to derive Cdl according to eqn (6), CV curves of all catalysts are determined in non-faradic areas in the 0.8–1.5 V (vs. RHE) window by scanning with various scan rates at 10–70 mV s−1. The CVs of Fe3C, Co3Te4, and Co3Te4–Fe3C in 1.0 M KOH are shown in Fig. 6(a–c). Plotting the scan rates against different current densities at the anode and cathode allows the calculation of Cdl, equivalent to half of the fitted line slopes, as shown in Fig. 6(d). The calculated Cdl of Co3Te4–Fe3C (45.0 mF cm−2) is much larger than those of Fe3C (29.8 mF cm−2) and Co3Te4 (5.6 mF cm−2), implying that a large exposed surface area is created by the hybridization of Fe3C with Co3Te4. Meanwhile, a low Cdl of Fe3C and Co3Te4 indicated poor catalytic activity, consistent with the result of LSV measurement and Tafel analysis, as explained below. Thus, using the specific capacitance of 0.04 mF cm−2 from the literature, the ECSA value of the Co3Te4–Fe3C (1125 cm2) is higher than for Fe3C (745 cm2) and Co3Te4 (140 cm2). On the strength of the above results, the higher ECSA value originated from an enlarged electrochemically active region and a more exposed reactive site achieved by an enlarged defective porous structure. This data confirmed that the more active region of Co3Te4–Fe3C exhibits considerable adsorption/desorption energy for the OER/HER intermediates and enhanced charged transport characteristics, showing superior catalytic activity. Moreover, more accessible active centres of the Co3Te4–Fe3C catalyst are attributed to the unique electronic structure composed of both Co3+ and Fe3+, found from XPS results.
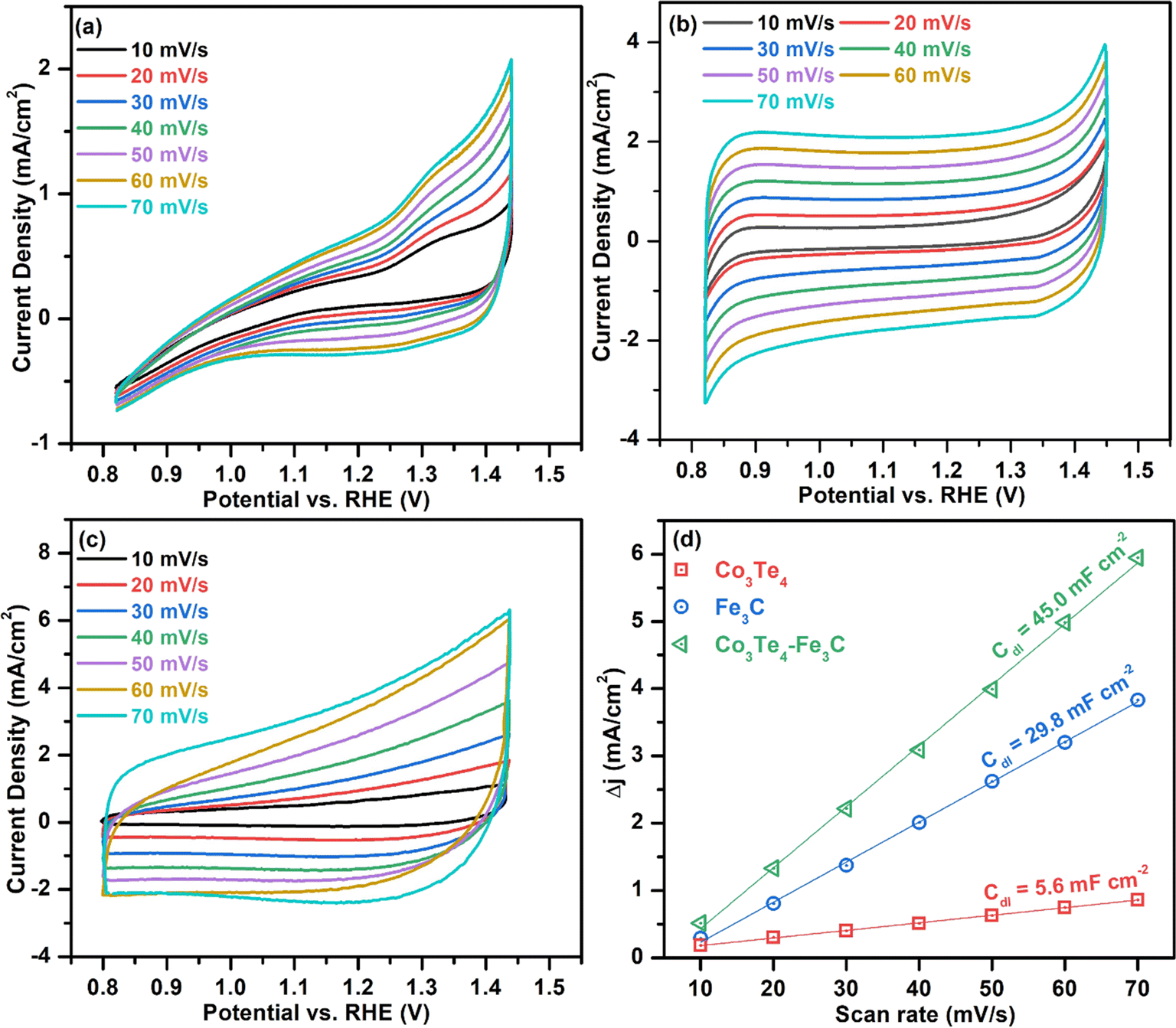 | ||
| Fig. 6 Cyclic voltammograms without the faradaic zone at 10–70 mV s−1 for (a) Co3Te4, (b) Fe3C, and (c) Co3Te4–Fe3C, and (d) comparison of Cdl plots for ECSA evaluation. | ||
The overall water-splitting performance was also investigated by taking Co3Te4–Fe3C as a cathode and anode (Co3Te4–Fe3C (+)//Co3Te4–Fe3C (−)) in 1.0 M KOH. Fig. 7(a) shows cell voltages of 1.44, 1.88, and 2.0 V at current densities of 10, 50, and 100 mA cm−2. H2 and O2 bubbles were generated at the surfaces of both electrodes, indicating the presence of HER and OER at the same time under the conversion of low-voltage electricity. Fig. 7(b) shows the chronoamperometry test in an alkaline medium, exhibiting superior stability of the Co3Te4–Fe3C catalyst for a period of 102 h, and there is no significant loss of current density or overpotential. The XRD pattern of Co3Te4–Fe3C after the water splitting process (Fig. 7(c)) revealed good crystallinity similar to the initial pattern, but a small decrease in peak intensities proved the structural durability of the bifunctional electrocatalyst.
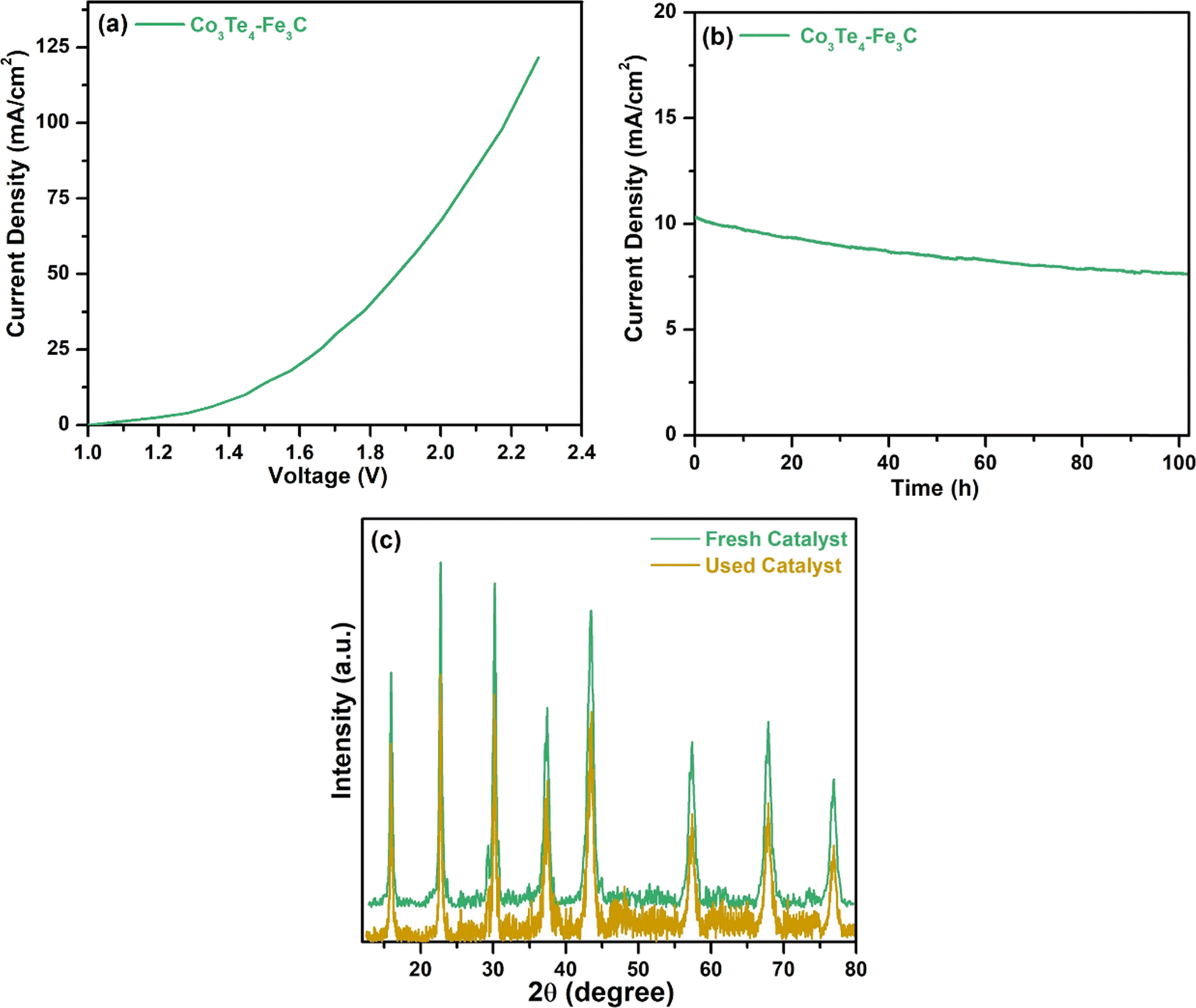 | ||
| Fig. 7 (a) LSV polarization curve for overall water splitting, (b) chronoamperometric curve of Co3Te4–Fe3C using an overall two electrode set-up, and (c) XRD pattern of the used catalyst after overall water-splitting. | ||
Based on the above characterization results of Co3Te4–Fe3C related to structural, compositional, and electrochemical analyses, the following reasons are responsible for its excellent activity towards OER and HER. The synergistic effect between Fe3C and Co3Te4 can lead to the cooperation of active sites. It is well known that the incorporation of Te element with Co ions results in a large covalent nature and small electronegativity of the anionic network around the transition metal centre; thanks to carbonaceous doping in Fe ions, this could provide a porous structure and structural defects, which is advantageous to provide well-defined heterointerfaces between the Fe3C and Co3Te4 heterostructure. This network with outstanding metallic/non-metallic characteristics results in a large surface area and good electrical conductivity, providing more electron transfer access to promote electrocatalytic performance. The high oxidation states of iron (Fe3+/Fe2+) and cobalt (Co3+/Co2+) help to break the barriers during O–O formation. Moreover, H–H bond formation on the composite material's surface increases the electron transfer rate. The coating of the Co3Te4–Fe3C on the SS and the close interconnection of this defective structure facilitate better contact to maintain good structural stability. The randomly oriented bonds, as confirmed by XPS, increase the activation of reactants and result in higher catalytic activity.
Moreover, the interaction between Fe3C and Co3Te4 is also revealed by SEM measurements, which showed direct nucleation and growth of a disordered structure in the functional groups on carbon, showing a large increase in OER/HER activity. The unique morphology of the hybrid is attributed to the ingenious synthetic strategy using a facile hydrothermal route for an abundance of active sites. The advantages mentioned above for the Co3Te4–Fe3C composite provide it with excellent electrocatalytic performance as it possesses long-term stability, lower overpotential/Tafel slope rate, and charge transfer resistance.
4. Conclusion
In summary, we report a hydrothermally produced Co3Te4–Fe3C material supported by SS as a 3D integrated electrocatalyst, which performs exceptionally well towards HER and OER. Different techniques like XRD and EDX show structural and compositional advantages with no phase impurities. Profiting from a defective porous structure analyzed using SEM, Co3Te4–Fe3C shows the merits of high surface area and intrinsic conductivity, thus enabling a drastically enhanced catalytic activity. The phase transformation of iron (Fe3+/Fe2+) and cobalt (Co3+/Co2+) offers more catalytically active sites with promising features of rapid charge transfer and energetically favorable intermediates; these effects are analyzed using the XPS technique. Compared with two pure catalysts, the outstanding electrochemical activity of Co3Te4–Fe3C is evidenced by requiring overpotentials of only η10 227 mV for OER and η10 211 mV for HER with minimal Tafel slopes (68.4 and 69.7 mV dec−1) in 1.00 M KOH solution. For overall water-splitting, cell voltages of 1.44, 1.88, and 2.0 V at current densities of 10, 50, and 100 mA cm−2 were achieved with a stability of 102 h. Furthermore, the catalyst also exhibits good stability continuously over a long time period with a constant current, which promotes the catalyst's charge/ion transfer efficiency and durability, as shown using chronoamperometry. We believe this highly interconnected novel heterojunction generated through electrochemical interfacial engineering could offer a cost-effective route toward generating green hydrogen and oxygen fuel for future study.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was funded by the Researchers Supporting Project Number (RSPD2024R551) at King Saud University, Riyadh, Saudi Arabia.References
- V. S. Kumbhar, H. Lee, J. Lee and K. Lee, Carbon Resour. Convers., 2019, 2, 242–255 CrossRef CAS.
- J. Zhang, Q. Zhang and X. Feng, Adv. Mater., 2019, 31, 1–19 Search PubMed.
- K. S. Bhat and H. S. Nagaraja, Recent Trends and Insights in Nickel Chalcogenide Nanostructures for Water-Splitting Reactions, Mater. Res. Innov., 2021 DOI:10.1080/14328917.2019.1703523.
- Y. Chu, D. Wang, X. Shan, C. Liu, W. Wang, N. Mitsuzaki and Z. Chen, Int. J. Hydrogen Energy, 2022, 47, 38983–39000 CrossRef CAS.
- X. Li, L. Zhao, J. Yu, X. Liu, X. Zhang, H. Liu and W. Zhou, Nanomicro Lett., 2020, 12, 131 CAS.
- P. J. McHugh, A. D. Stergiou and M. D. Symes, Adv. Energy Mater., 2020, 10, 1–21 Search PubMed.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- C. Zhang, H. Nan, H. Tian and W. Zheng, J. Alloys Compd., 2020, 838, 155685 CrossRef CAS.
- R. Zahid, M. R. Abdul Karim, M. Waqas Khan and M. A. Marwat, Electrocatalytic Water Splitting: A Review under the Shade of Metal-Organic Frameworks, Int. J. Hydrogen Energy, 2024, 958–982, DOI:10.1016/j.ijhydene.2023.12.225.
- Z. Chen, W. Wei and B. J. Ni, Curr. Opin. Green Sustainable Chem., 2021, 27, 100398 CrossRef CAS.
- B. A. Yusuf, W. Yaseen, M. Xie, R. S. Zayyan, A. I. Muhammad, R. Nankya, J. Xie and Y. Xu, Recent Advances in Understanding and Design of Efficient Hydrogen Evolution Electrocatalysts for Water Splitting: A Comprehensive Review, Adv. Colloid Interface Sci., 2023 DOI:10.1016/j.cis.2022.102811.
- X. Huang, L. Chang, H. Zhao and Z. Cai, Mater. Des., 2022, 224, 111408 CrossRef.
- Y. Liu, Y. Guo, Y. Liu, Z. Wei, K. Wang and Z. Shi, A Mini Review on Transition Metal Chalcogenides for Electrocatalytic Water Splitting: Bridging Material Design and Practical Application, Energy Fuels, 2023, 2608–2630, DOI:10.1021/acs.energyfuels.2c03833.
- Y. X. Yu, J. L. Xu, L. W. Zhang, Y. C. Ma and J. M. Luo, Int. J. Hydrogen Energy, 2024, 72, 209–219 CrossRef CAS.
- T. Han, C. Zhang and J. Luo, Langmuir, 2018, 34, 11281–11291 CrossRef CAS PubMed.
- M. R. Ashwin Kishore, K. Larsson and P. Ravindran, ACS Omega, 2020, 5, 23762–23768 CrossRef CAS PubMed.
- M. Rezaei, A. Nezamzadeh-Ejhieh and A. R. Massah, Energy Fuels, 2024, 38, 7637–7664 CrossRef CAS.
- J. Wang, X. Yue, Y. Yang, S. Sirisomboonchai, P. Wang, X. Ma, A. Abudula and G. Guan, J. Alloys Compd., 2020, 819, 153346 CrossRef CAS.
- S. Gupta, M. K. Patel, A. Miotello and N. Patel, Metal Boride-Based Catalysts for Electrochemical Water-Splitting: A Review, Adv. Funct. Mater., 2020 DOI:10.1002/adfm.201906481.
- M. M. Najafpour, S. Madadkhani and M. Tavahodi, Int. J. Hydrogen Energy, 2017, 42, 60–67 CrossRef CAS.
- M. Ali, E. Pervaiz, U. Sikandar and Y. Khan, A Review on the Recent Developments in Zirconium and Carbon-Based Catalysts for Photoelectrochemical Water-Splitting, Int. J. Hydrogen Energy, 2021, 18257–18283, DOI:10.1016/j.ijhydene.2021.02.202.
- Y. Peng, K. Jiang, W. Hill, Z. Lu, H. Yao and H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 3971–3977 CrossRef CAS PubMed.
- B. Ren, D. Li, Q. Jin, H. Cui and C. Wang, Chemelectrochem, 2019, 6, 413–420 CrossRef CAS.
- P. M. Bodhankar, P. B. Sarawade, P. Kumar, A. Vinu, A. P. Kulkarni, C. D. Lokhande and D. S. Dhawale, Nanostructured Metal Phosphide Based Catalysts for Electrochemical Water Splitting: A Review, Small, 2022 DOI:10.1002/smll.202107572.
- Y. Zhang, W. J. Jiang, L. Guo, X. Zhang, J. S. Hu, Z. Wei and L. J. Wan, ACS Appl. Mater. Interfaces, 2015, 7, 11508–11515 CrossRef CAS PubMed.
- J. Zhang, J. Chen, Y. Jiang, F. Zhou, G. Wang and R. Wang, Appl. Surf. Sci., 2016, 389, 157–164 CrossRef CAS.
- H. Jiang, Y. Yao, Y. Zhu, Y. Liu, Y. Su, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 21511–21520 CrossRef CAS PubMed.
- M. Hu, X. Li, J. Xiong, L. Zeng, Y. Huang, Y. Wu, G. Cao and W. Li, Nano-Fe3C@PGC as a Novel Low-Cost Anode Electrocatalyst for Superior Performance Microbial Fuel Cells, Biosens. Bioelectron., 2019, 142 DOI:10.1016/j.bios.2019.111594.
- M. M. Mohideen, A. V. Radhamani, S. Ramakrishna, Y. Wei and Y. Liu, Recent Insights on Iron Based Nanostructured Electrocatalyst and Current Status of Proton Exchange Membrane Fuel Cell for Sustainable Transport, J. Energy Chem., 2022, 466–489, DOI:10.1016/j.jechem.2022.01.035.
- J. Gautam, T. D. Thanh, K. Maiti, N. H. Kim and J. H. Lee, Carbon, 2018, 137, 358–367 CrossRef CAS.
- Y. Zhang, Y. Zhang, B. Tian, H. Li, Z. Zeng and D. Ho, D-Band Center Optimization of Iron Carbide via Cr Substitution for Enhanced Alkaline Hydrogen Evolution, Mater. Today Energy, 2022, 29 DOI:10.1016/j.mtener.2022.101133.
- M. Shkir, M. Arif, A. Singh, I. S. Yahia, H. Algarni and S. AlFaify, Mater. Sci. Semicond. Process., 2019, 100, 106–112 CrossRef CAS.
- A. Jaiswal, R. Kumar and R. Prakash, Energy Fuel., 2021, 35, 16046–16053 CrossRef CAS.
- W. Yu, J. Yu, Y. Wang, X. Li, Y. Wang, H. Yuan, X. Zhang, H. Liu and W. Zhou, Appl. Catal., B, 2022, 121291, DOI:10.1016/j.apcatb.2022.121291.
- A. Ali, F. Liang, H. Feng, M. Tang, S. Jalil Shah, F. Ahmad, X. Ji, P. Kang Shen and J. Zhu, Mater. Sci. Energy Technol., 2023, 6, 301–309 CAS.
- R. Yoo, K. Min, H. Kim, D. Lim and S. H. Baeck, Appl. Surf. Sci., 2022, 152405, DOI:10.1016/j.apsusc.2021.152405.
- K. S. Bhat, H. C. Barshilia and H. S. Nagaraja, Int. J. Hydrogen Energy, 2017, 42, 24645–24655 CrossRef CAS.
- D. A. Alshammari, Y. M. Riyad, S. Aman, N. Ahmad, H. M. Tahir Farid and Z. M. El-Bahy, J. Electroanal. Chem., 2023, 117701, DOI:10.1016/j.jelechem.2023.117701.
- L. Nisar, M. Sadaqat, A. Hassan, N. U. A. Babar, A. Shah, M. Najam-Ul-Haq, M. N. Ashiq, M. F. Ehsan and K. S. Joya, Fuel, 2020, 118666, DOI:10.1016/j.fuel.2020.118666.
- K. C. Majhi, P. Karfa, S. De and R. Madhuri, in IOP Conference Series: Materials Science and Engineering, IOP Publishing Ltd, 2019, vol. 577 Search PubMed.
- Q. Gao, C. Q. Huang, Y. M. Ju, M. R. Gao, J. W. Liu, D. An, C. H. Cui, Y. R. Zheng, W. X. Li and S. H. Yu, Angew. Chem. Int. Ed., 2017, 56, 7769–7773 CrossRef CAS PubMed.
- A. Padmanaban, N. Padmanathan, T. Dhanasekaran, R. Manigandan, S. Srinandhini, P. Sivaprakash, S. Arumugam and V. Narayanan, J. Electroanal. Chem., 2020, 114658, DOI:10.1016/j.jelechem.2020.114658.
- H. Wang, Y. Wang, L. Tan, L. Fang, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, Appl. Catal., B, 2019, 244, 568–575 CrossRef CAS.
- A. Sivanantham, S. Hyun, M. Son and S. Shanmugam, Electrochim. Acta, 2019, 312, 234–241 CrossRef CAS.
- W. Li, J. Chen, Y. Zhang, W. Gong, M. Sun, Y. Wang, X. Wang, H. Rao, J. Ye and Z. Lu, Int. J. Hydrogen Energy, 2021, 46, 39912–39920 CrossRef CAS.
- K. S. Joya, M. A. Ehsan, N. U. A. Babar, M. Sohail and Z. H. Yamani, J. Mater. Chem. A, 2019, 7, 9137–9144 RSC.
- I. Amorim, J. Xu, N. Zhang, Z. Yu, A. Araújo, F. Bento and L. Liu, Chem. Eng. J., 2021, 130454, DOI:10.1016/j.cej.2021.130454.
- L. Yang, H. Xu, H. Liu, D. Cheng and D. Cao, Small Methods, 2019, 1900113, DOI:10.1002/smtd.201900113.
- L. Yang, H. Qin, Z. Dong, T. Wang, G. Wang and L. Jiao, Small, 2021, 2102027, DOI:10.1002/smll.202102027.
- W. Yaseen, S. Meng, W. Li, M. Xie, M. Rafiq, B. Adegbemiga Yusuf, S. A. Shah, I. Khan, J. Xie and Y. Xu, Int. J. Hydrogen Energy, 2024, 51, 565–577 CrossRef CAS.
- Z. Yang, H. Tan, Y. Qi, S. Ma, J. Bai, L. Liu and D. Xiong, Supplementary Information Preparation and characterization of nanostructured Fe-doped CoTe2 electrocatalysts for the oxygen evolution reaction, 2023.
- Y. Deng, S. Yun, J. Dang, Y. Zhang, C. Dang, Y. Wang, Z. Liu and Z. Gao, J. Colloid Interface Sci., 2022, 624, 650–669 CrossRef CAS PubMed.
- Q. U. Khan, M. W. Ishaq, N. Begum, K. Khan, U. Khan, A. T. Khan, M. A. U. Din, M. Idrees and L. Zhu, Nanoscale CuTe Electrocatalyst Immobilized at Conductor Surface for Remarkable Hydrogen Evolution Reaction, Int. J. Hydrogen Energy, 2021, 46(36), 18729–18739, DOI:10.1016/j.ijhydene.2021.03.031.
- J. Xing, X. Wang, Y. Zhang and X. Fu, ChemistrySelect, 2022, e202203656, DOI:10.1002/slct.202203656.
- X. Cao, J. E. Medvedeva and M. Nath, ACS Appl. Energy Mater., 2020, 3, 3092–3103 CrossRef CAS.
- G. Wang, C. Hua, W. Chen, H. Fan, P. Feng and Y. Zhu, Electrochim. Acta, 2023, 142133, DOI:10.1016/j.electacta.2023.142133.
- D. Khalafallah, W. Huang, M. Zhi and Z. Hong, Energy Environ. Mater., 2024, e12528, DOI:10.1002/eem2.12528.
- M. Liu, X. Lu, C. Guo, Z. Wang, Y. Li, Y. Lin, Y. Zhou, S. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 36146–36153 CrossRef CAS PubMed.
- Y. Wang, H. Li, Q. Yao, R. Li, Z. Guo, H. Chen, K. Qu and R. Li, Int. J. Hydrogen Energy, 2021, 46, 6513–6521 CrossRef CAS.
- T. Kshetri, T. Ibomcha Singh, Y. Sun Lee, D. Devi Khumujam, N. Hoon Kim and J. Hee Lee, Metal organic framework-derived cobalt telluride-carbon porous structured composites for high-performance supercapacitor, Composites, Part B, 2021, 211, 108624 CrossRef CAS.
- S. Yang, G. D. Park and Y. C. Kang, Appl. Surf. Sci., 2020, 147140, DOI:10.1016/j.apsusc.2020.147140.
- N. Bhuvanendran, M. G. Choi, D. Kim and S. Y. Lee, J. Alloys Compd., 2023, 168079, DOI:10.1016/j.jallcom.2022.168079.
- E. K. Kim, H. T. Bui, N. K. Shrestha, C. Y. Shin, S. A. Patil, S. Khadtare, C. Bathula, Y. Y. Noh and S. H. Han, Electrochim. Acta, 2018, 260, 365–371 CrossRef CAS.
- L. Ji, Z. Wang, H. Wang, X. Shi, A. M. Asiri and X. Sun, ACS Sustain. Chem. Eng., 2018, 6, 4481–4485 CrossRef CAS.
- Y. Qi, J. Wu, J. Xu, H. Gao, Z. Du, B. Liu, L. Liu and D. Xiong, Inorg. Chem. Front., 2020, 7, 2523–2532 RSC.
- P. Guo, S. Cao, Y. Wang, X. Lu, Y. Zhang, X. Xin, X. Chi, X. Yu, I. Tojiboyev, H. Salari, A. J. Sobrido, M. Titirici and X. Li, Appl. Catal., B, 2022, 121355, DOI:10.1016/j.apcatb.2022.121355.
- W. Li, X. Yu, L. Luo, Y. Du and Q. Wu, J. Alloys Compd., 2022, 165369, DOI:10.1016/j.jallcom.2022.165369.
- Z. Wang, M. Liu, J. Du, Y. Lin, S. Wei, X. Lu and J. Zhang, Electrochim. Acta, 2018, 264, 244–250 CrossRef CAS.
- W. Li, Y. Deng, L. Luo, Y. Du, X. Cheng and Q. Wu, J. Colloid Interface Sci., 2023, 639, 416–423 CrossRef CAS PubMed.
- X. Ji, Y. Lin, J. Zeng, Z. Ren, Z. Lin, Y. Mu, Y. Qiu and J. Yu, Nat. Commun., 2021, 1380, DOI:10.1038/s41467-021-21742-y.
- S. Yang, J. Yu, C. Yao, S. Li and A. Xie, J. Alloys Compd., 2022, 163874, DOI:10.1016/j.jallcom.2022.163874.
- W. Fang, J. Wang, Y. Hu, X. Cui, R. Zhu, Y. Zhang, C. Yue, J. Dang, W. Cui, H. Zhao and Z. Li, Electrochim. Acta, 2021, 137384, DOI:10.1016/j.electacta.2020.137384.
- M. Chen, J. Liu, W. Zhou, J. Lin and Z. Shen, Sci. Rep., 2015, 10389, DOI:10.1038/srep10389.
- R. Li, Y. Li, P. Yang, P. Ren, D. Wang, X. Lu, R. Xu, Y. Li, J. Xue, J. Zhang, M. An, J. Ma, B. Wang, H. Liu and S. Dou, Appl. Catal., B, 2022, 121834, DOI:10.1016/j.apcatb.2022.121834.
- P. Babar, A. Lokhande, H. H. Shin, B. Pawar, M. G. Gang, S. Pawar and J. H. Kim, Small, 2018, 1702568, DOI:10.1002/smll.201702568.
- Z. Cao, M. Qin, C. Zuo, Y. Gu and B. Jia, J. Colloid Interface Sci., 2017, 491, 55–63 CrossRef CAS PubMed.
- T. Gao, S. Yu, Y. Chen, X. Li, X. Tang, S. Wu, B. He, H. Lan, S. Li, Q. Yue and D. Xiao, J. Colloid Interface Sci., 2023, 642, 120–128 CrossRef CAS PubMed.
- X. Li, G. Guan, X. Du, J. Cao, X. Hao, X. Ma, A. D. Jagadale and A. Abudula, Chem. Commun., 2015, 51, 15012–15014 RSC.
- X. Li, C. Li, A. Yoshida, X. Hao, Z. Zuo, Z. Wang, A. Abudula and G. Guan, J. Mater. Chem. A, 2017, 5, 21740–21749 RSC.
- X. Ma, X. Li, A. D. Jagadale, X. Hao, A. Abudula and G. Guan, Int. J. Hydrogen Energy, 2016, 41, 14553–14561 CrossRef CAS.
- H. Begum and S. Jeon, Int. J. Hydrogen Energy, 2018, 43, 5522–5529 CrossRef CAS.
- J. Zhu, Q. Zhang, P. Zhao, L. Chen, S. Yang, Q. Yan and H. Li, J. Alloys Compd., 2022, 165705, DOI:10.1016/j.jallcom.2022.165705.
- N. A. Khan, I. Ahmad, N. Rashid, M. N. Zafar, F. K. Shehzad, Z. ullah, A. Ul-Hamid, M. F. Nazar, M. Junaid, M. Faheem, S. S. Shafqat, U. Jabeen and A. Dahshan, Int. J. Hydrogen Energy, 2022, 47, 30970–30980 CrossRef CAS.
- D. Yu, M. Ye, S. Han, Y. Ma, F. Hu, L. Li and S. Peng, ChemSusChem, 2020, 13, 5280–5287 CrossRef CAS PubMed.
- S. Zhu, J. Lei, L. Zhang and L. Lu, Int. J. Hydrogen Energy, 2019, 44, 16507–16515 CrossRef CAS.
- Y. Yan, Y. Han, F. Wang, Y. Hu, Q. Shi, G. Diao and M. Chen, J. Alloys Compd., 2022, 163126, DOI:10.1016/j.jallcom.2021.163126.
- R. Zhao, B. Ni, L. Wu, P. Sun and T. Chen, Colloids Surf., A, 2022, 128118, DOI:10.1016/j.colsurfa.2021.128118.
- H. Guan, N. Wang, X. Feng, S. Bian, W. Li and Y. Chen, Colloids Surf., A, 2021, 126596, DOI:10.1016/j.colsurfa.2021.126596.
- H. Huang, Y. Li, W. Li, S. Chen, C. Wang, M. Cui and T. Ma, Inorg. Chem. Commun., 2019, 103, 1–5 CrossRef CAS.
- Y. Dong, J. Yang, Y. Liu, Y. Wang, Z. Dong, M. Cui, M. Li, X. Yuan, X. Zhang and X. Dai, Dalton Trans., 2020, 49, 6355–6362 RSC.
- W. Dai, T. Lu and Y. Pan, J. Power Sources, 2019, 430, 104–111 CrossRef CAS.
- A. G. Abid, S. Manzoor, M. Usman, T. Munawar, M. U. Nisa, F. Iqbal, M. N. Ashiq, M. Najam-Ul-Haq, A. Shah and M. Imran, Energy Fuel., 2021, 35, 17820–17832 CrossRef CAS.
- M. Nath, U. De Silva, H. Singh, M. Perkins, W. P. R. Liyanage, S. Umapathi, S. Chakravarty and J. Masud, ACS Appl. Energy Mater., 2021, 4, 8158–8174 CrossRef CAS.
- F. Gao, J. He, H. Wang, J. Lin, R. Chen, K. Yi, F. Huang, Z. Lin and M. Wang, Te-Mediated Electro-Driven Oxygen Evolution Reaction, Nano Res. Energy, 2022 DOI:10.26599/NRE.2022.9120029.
- A. Kumar and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2017, 9, 41906–41915 CrossRef CAS PubMed.
- J. Song, C. Zhu, B. Z. Xu, S. Fu, M. H. Engelhard, R. Ye, D. Du, S. P. Beckman and Y. Lin, Bimetallic Cobalt-Based Phosphide Zeolitic Imidazolate Framework: CoPx Phase-Dependent Electrical Conductivity and Hydrogen Atom Adsorption Energy for Efficient Overall Water Splitting, Adv. Energy Mater., 2017, 7(2), 1601555, DOI:10.1002/aenm.201601555.
- S. S. Jayaseelan, N. Bhuvanendran, Q. Xu and H. Su, Int. J. Hydrogen Energy, 2020, 45, 4587–4595 CrossRef CAS.
- A. Jaiswal, R. Kumar and R. Prakash, Energy Fuel., 2021, 35, 16046–16053 CrossRef CAS.
- S. Wang, G. Bendt, S. Saddeler and S. Schulz, Energy Technol., 2019, 1801121, DOI:10.1002/ente.201801121.
- Y. Guo, Z. Yao, C. Shang and E. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 39312–39317 CrossRef CAS PubMed.
- W. Yaseen, N. Ullah, M. Xie, B. A. Yusuf, Y. Xu, C. Tong and J. Xie, Surface. Interfac., 2021, 101361, DOI:10.1016/j.surfin.2021.101361.
- K. Wang, Z. Ye, C. Liu, D. Xi, C. Zhou, Z. Shi, H. Xia, G. Liu and G. Qiao, ACS Appl. Mater. Interfaces, 2016, 8, 2910–2916 CrossRef CAS PubMed.
- W.-C. Chen, S.-C. Wang, S. Brahma, J.-L. Huang, D. R. Sahu and Y.-M. Shen, J. Alloys Compd. Commun., 2024, 3, 100027 Search PubMed.
- R. Khan, M. T. Mehran, M. M. Baig, B. Sarfraz, S. R. Naqvi, M. B. Muhammad, M. Z. Khan and A. H. Khoja, Fuel, 2021, 119174, DOI:10.1016/j.fuel.2020.119174.
| This journal is © The Royal Society of Chemistry 2025 |