
Hyperconjugated linker design in giant dimeric donors enabled superior short-circuit current in organic solar cells†
Caixuan Wang‡
ab,
Mengying Wu‡ac,
Dan Deng *a,
Ruixiang Fangad,
Jianqi Zhanga,
Ruimin Zhou*c and
Zhixiang Wei
*ab
*a,
Ruixiang Fangad,
Jianqi Zhanga,
Ruimin Zhou*c and
Zhixiang Wei
*ab
aKey Laboratory of Nanosystem and Hierarchical Fabrication of Chinese Academy of Sciences, National Center for Nanoscience and Technology, Beijing, 100190, China. E-mail: dengd@nanoctr.cn; weizx@nanoctr.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cCollege of Chemistry, Zhengzhou University, Zhengzhou, 450001, China. E-mail: zhourm@zzu.edu.cn
dKey Laboratory of Flexible Electronics & Institute of Advanced Materials, Nanjing Tech University, Jiangsu 211816, China
First published on 7th June 2025
Abstract
Giant dimeric donors possess definite chemical structures and regulatable molecular skeletons and are expected to become alternative photovoltaic materials for polymer donors with batch differences. However, the design of giant dimeric donors is still at an early stage and needs to be further explored. Here, through creative semi-flexible and flexible linker design, we synthesized three interesting giant dimeric donors with relative monomer positions ranging from parallel to staircase to perpendicular in their optimized conformation. Unusually, the hyperconjugation effect in the semi-flexible linker stabilizes the perpendicular conformation, which results in the strongest homo-molecular interactions exhibiting non-planar molecular conformation. Combining calculations and multiple morphology characterization on dynamic and thermal packing, we systematically analyze the hyperconjugation effects, flexibility, and hetero-molecular interaction on the assembly. As a result, applying Y6 as an acceptor, the giant dimeric donor of BDT-Dimer3 with a semi-flexible non-planar linker achieved a satisfactory efficiency of 15.68% with a cutting-edge short-circuit current of 27.39 mA cm−2 and an improved photostability with a T80 of 630 hours. Our results provide hyperconjugated linker design for efficient and stable OSC devices with definite structures, as well as a deep understanding of the assembly in both pure and mixed systems.
 Zhixiang Wei | This is our second paper being published in Nanoscale Horizons, and we are proud to have sustained cooperation with such an excellent journal. We think the best way to express our gratitude is to continue to report our significant research advances, such as novel material synthesis, device technology and photophysical mechanism research, and flexible devices for organic solar cells. Congratulations on the 10th anniversary, and best wishes to the Nanoscale Horizons. |
New conceptsProposing a novel material design strategy and deeply demonstrating its effect on molecular interaction, assembly and device performances are crucial to develop highly efficient organic solar cells (OSCs). In this study, we proposed and analyzed a hyperconjugation strategy for constructing photovoltaic donor materials for the first time, and separately explored the molecular assembly behaviors during film-formation and annealing processes influenced by hyperconjugation. Based on the emerging dimeric donor materials, the hyperconjugation introduced in linker units imparted two unique properties: (1) enhanced homo-molecular interactions while stabilizing the non-planar conformation, thus weakening the crystallization driving force during film-formation but promoting it during the annealing process, which is quite different from traditional material systems and (2) optimized the distribution of surface electrostatic potential (ESP) to improve hetero-molecular interactions and compatibility. Hence, the device based on a molecule with a hyperconjugated linker achieves the most ordered packing with small domains, and ultimately the most efficient charge carrier management (FF × JSC) with a cutting-edge JSC of 27.39 mA cm−2 based on small molecule or dimeric donors. Our work provides guidance for simple and efficient molecular design using hyperconjugation effects. |
Introduction
Currently, polymers as donors obtain the highest power conversion efficiency (PCE) in organic solar cells (OSCs),1–4 but their chain disordered entanglement would cause initial burn-in loss5,6 and batch variations would largely enhance the cost of purification and device optimization technology, both of which are unfavorable for their commercial applications.7,8 On the other hand, though small-molecule donors (SMDs) eliminate the batch differences, their device parameters exhibit mutual constraints due to the conflicting crystallinity requirements, and their poor device stability cannot be separated from the high diffusion coefficient of small molecules.9–13 As promising photovoltaic materials, giant oligomeric donors compensate for the shortcomings of both small molecules and polymers, and additionally, the linker provides further optimization space relative to the small molecules. In our previous work, the design of giant dimeric donors14 enables all-oligomeric OSCs to achieve excellent device performance and photostability, confirming the feasibility and potential of dimeric donors.However, the design of giant dimeric donors is still in the early stage. Whether they are mixed with small-molecules or oligomeric acceptors, the formation of ideal morphology with simultaneous small domains and ordered packing that facilitate exciton dissociation and charge transport together with photostability is a primary challenge. Such a morphology is essentially decided by the homo/hetero-molecular interaction.15,16 Compared to SMDs, the extended molecular skeleton of giant dimeric donors with a smaller diffusion coefficient has the potential to fine-tune the morphology to weaken the constraint relationship between short-circuit current (JSC) and fill factor (FF), allowing better charge carrier management (JSC × FF) and higher efficiency;17,18 however, both interactions for dimeric donors are more difficult to regulate. For homo-molecular interaction, the decreased diffusion coefficient definitely impacts the growth of crystallinity19 (ordered aggregates); for hetero-molecular interaction, dimeric donors with double molecular weight (MW) reduce the mixed entropy, thus impairing the driving force for mixing.20 Combining our previous experiences in the design of small/dimeric donors, we attempt to regulate the molecular interaction by combining both monomer and linker modification,21 aiming at deeply investigating the unique assembly in emerging giant oligomeric materials.22–24
Herein, choosing an efficient benzodithiophene (BDT) based skeleton that is well mixed with the Y6 acceptor as a monomer,25 three BDT-Dimers with flexible or semi-flexible linkers were designed and synthesized, and their relative monomer positions were effectively regulated from parallel to staircase to perpendicular. Compared to BDT-Dimer1 and BDT-Dimer2 with flexible linkers and planar backbones, BDT-Dimer3 with a semi-flexible linker and a non-planar backbone possesses the following advantages: (1) the hyperconjugation effect resulted from a semi-flexible linker, promoting the most negative ESP distribution, and exhibiting the strongest compatibility with Y6 and highest hetero-molecular interaction and (2) the distorted molecular skeleton originated from the twisty linker, leading to weakened dynamic self-assembly but strengthened thermal-driven assembly. Therefore, BDT-Dimer3 and Y6 achieved the smallest domain size with the most ordered molecular packing, enabling excellent charge properties and photon utilization in optimized devices, resulting in a satisfied efficiency of 15.68% and a cutting-edge JSC of 27.39 mA cm−2 based on SMDs/dimeric donors. Additionally, the highest Tg of BDT-Dimer3/Y6 together with a small crystalline driving force makes its device obtain the best photostability with a T80 (time to maintain 80% of initial efficiency) of 630 hours. Our study provides an oligomeric molecular design strategy that utilising hyperconjugated linker, to promote satisfactory efficiency and stability of OSCs within well-defined molecular structure.
Results and discussion
Design and characterization of BDT-dimers
Based on our previous research, linked units significantly influence the assembly behaviours of dimeric donors.14 We choose the reported efficient BDT-based molecule as the monomer because of its good miscibility with Y6 and high cool crystalline temperature (TC), which would be beneficial to the efficiency and stability of devices,26,27 respectively. Applying flexible or semi-flexible linkers to tune the homo/hetero-molecular interactions, we designed and synthesized three BDT-Dimers (Fig. 1a). | ||
| Fig. 1 Molecular structures and DFT calculations. (a) Molecular structures and calculated optimized conformations. (b) NBO analyses for the hyperconjugation effect in the linker of BDT-Dimer3. Calculated ESP distribution for the linkers of (c) BDT-Dimer1, (d) BDT-Dimer2 and (e) BDT-Dimer3. | ||
Expectedly, linker modification effectively regulated the planarity and dominant conformation of molecular skeletons. Density functional theory (DFT) calculations show that the electron density of the frontier orbital energy levels in all three BDT-Dimers is distributed to one side of the monomer (Fig. S1, ESI†), while the two monomers of BDT-Dimer1 are arranged in a line, which are in a parallel staircase for BDT-Dimer2 and a perpendicular position for BDT-Dimer3. To uncover the essential reason behind the abnormal configuration in BDT-Dimer3, we performed DFT calculations in combination with natural bond orbital (NBO) analyses and found that all the C–H σ bonds in the linker overlapped with both the π* orbitals of the adjacent benzene ring group and rhodanine group (Fig. 1b and Fig. S2, ESI†), leading to a maximum hyperconjugation effect in its perpendicular conformation. Furthermore, the hyperconjugation effects also homogenize the electron cloud density as conjugation, leading to a much smaller average surface electrostatic potential (ESP) with a value of 0.52 kcal mol−1, which is 4 times smaller than that of flexible linkers in BDT-Dimer2. Due to the different electron-withdrawing ability between rhodanine and cyanoester, the linker of BDT-Dimer1 exhibits the largest ESP (Fig. 1c–e). Therefore, the hyperconjugation effect in the linker affects not only the optimized conformation but also the ESP distribution, and thus the homo/hetero-molecular interactions and assembly behaviors.
We first evaluated the homo-molecular interaction by their melting temperature (Tm) and melting enthalpy (ΔHm) tested by differential scanning calorimetry (DSC) (Fig. 2a and Fig. S3, ESI†). Unexpectedly, the most non-planar BDT-Dimer3 obtains the highest Tm (228.3 °C) and maximum ΔHm (22.8 J g−1), demonstrating its thermodynamically strongest homo-molecular interaction (Table S1, ESI†). However, BDT-Dimer1 with the most planar optimized conformation exhibits the lowest Tm of 194.3 °C with the smallest ΔHm of 15.7 J g−1. Furthermore, the driving force for crystals in the film (ΔGls =ΔHm × (Tm − TC)/Tm) was calculated, which was also the highest in BDT-Dimer3 (ΔGls = −2.51 J g−1, Table S1, ESI†). Consequently, for the three BDT-Dimers, the more planar the molecular backbone, the weaker the molecular interaction, the tendency of which is quite opposite to the common materials. This abnormal phenomenon should be due to the hyperconjugation effect originated from the semi-flexible linker in BDT-Dimer3,28 leading to stronger homo-molecular interaction than BDT-Dimer1 and BDT-Dimer2 with flexible linkers. The hyperconjugation effect in BDT-Dimer3 could be demonstrated by its more red-shifted absorption resulted from internal charge transfer (ICT) in dilute chloroform solution29,30 (Fig. 2b), which could be sabotaged by vibration reflected on the more obvious blue-shift absorption during heating (Fig. S4, ESI†).
 | ||
| Fig. 2 Molecular interactions and molecular packing of BDT-dimers. (a) Melting temperature and enthalpy. (b) Normalized solution absorption in dilute chloroform. (c) χ Calculated by DSC and the contact angle between BDT-dimers and Y6. (d) Normalized film absorption of BDT-dimers. CCL in (e) IP and (f) OOP directions before and after TA treatment. (g) Schematic illustrations of the packing process during film formation and the TA process for BDT-Dimer1 and BDT-Dimer3. | ||
Furthermore, the linkers’ ESP distribution is also reflected in the three BDT-Dimers (Fig. S5, ESI†). The result shows that BDT-Dimer3 achieves a decreased average ESP value of 1.36 kcal mol−1, which increases to 1.52 kcal mol−1 in BDT-Dimer2 and 1.87 kcal mol−1 in BDT-Dimer1, the tendency of which is the same as their linkers. The decreasing average ESP values from BDT-Dimer1 to BDT-Dimer3 sequentially enlarge their ESP difference to Y6 (a value of 5.06 kcal mol−1, Fig. S6, ESI†), thus gradually facilitating the enhancement of hetero-molecular interaction.31 This tendency is further proved using the Flory–Huggins parameter (χ) whether calculated from the contact angle or DSC tests32,33 (Fig. 2c and Fig. S3 and S7 and Table S2, ESI†). BDT-Dimer3 obtains the highest surface energy of 13.65 mN m−1 calculated from the contact angle and the lowest χ of 0.17 with Y6, while BDT-Dimer2 and BDT-Dimer1 obtain sequentially decreased surface energies with increasing χ, indicating decreasing compatibility. The tendency of compatibility was further supported by the DSC results, and the χ values from these results were 2.14, 1.71 and 1.46 for BDT-Dimer1/Y6, BDT-Dimer2/Y6 and BDT-Dimer3/Y6, respectively.
Summarily, the hyperconjugation resulted from the semi-flexible linker endows BDT-Dimer3 with the most non-planar skeleton but the strongest homo-molecular interaction and the best compatibility with Y6, resulting in the existence of contradiction properties.
Linkers’ effect on dynamic molecular packing and thermal packing
Molecular packing is a combination of kinetic and thermodynamic driving. For film formation, molecules are prone to relax in the solvent and easily adopt an optimized conformation with the lowest energy. Thus, the planarity of the optimized conformation would dynamically dominate the packing during film formation. This is further supported by the fitting absorbance ratio between π-stacking (I0–0) and the ICT (I0–1) in the film, which is decreased from 0.141 in BDT-Dimer1 to 0.096 in BDT-Dimer2 to 0.088 in BDT-Dimer3 (Fig. 2d and Fig. S8, ESI†). Grazing incident wide-angle X-ray scattering (GIWAXS) results of the three BDT-dimers further demonstrated that the planarity of the skeleton dominated the packing during film formation, inferring from the decreasing coherence crystalline length (CCL) of (010) peak from BDT-Dimer1 to BDT-Dimer3 (Fig. S9, ESI†). Thereinto, as the planarity decreased from BDT-Dimer1 to BDT-Dimer3, all three molecules presented a mixed face-on and edge-on packing, and the d-spacing increased from 3.72 to 3.83 Å to 3.85 Å, and the CCL decreased from 25.8 to 18.6 to 16.9 Å of π–π stacking in the in-plane (IP) direction. The poorer the planarity, the looser and poorer the stacking during film formation, which further confirms that the planarity dominated the kinetic film formation, and the twisted semi-flexible linker could decrease the molecular stacking during film formation (Fig. 2e and Table S3, ESI†).Thermal energy could improve molecular packing from the following two aspects: (1) provides additional energy to rotate to the conformation that facilitates molecular packing and (2) increases the diffusion rate and enhances the growth of aggregates or crystallinity. Unexpectedly, the packing tendency during annealing is totally different from that during film formation. After thermal annealing (TA), applying the IP direction as an example, BDT-Dimer3 with the most twisted molecular skeleton and the semi-flexible linker reaches the most ordered packing, which can be validated by the reduced d-spacing from 3.85 Å to 3.74 Å and significantly increased CCL from 16.9 Å to 26.6 Å during the TA process (Fig. 2e and Fig. S10 and Table S3, ESI†). However, TA has little effect on BDT-Dimer1 with the best planarity, which can be inferred from the almost constant d-spacing and CCL of π–π stacking. With an in-between skeleton planarity, TA makes an in-between impact on BDT-Dimer2 with an in-between packing. Noteworthily, the change tendency is also consistent with the out-of-plane (OOP) direction (Fig. 2f). These results demonstrate that the ΔGls in the film rather than the planarity dominated the molecular packing during TA. The driving force for the molecular packing of BDT-Dimer3 during TA is its strong homo-molecular interaction and ΔGls, which is higher than that of BDT-Dimer1 and BDT-Dimer2.
Consequently, the driving force for molecular packing in our system is well divided into two parts: one is kinetic-dominated by the planarity during film formation, while the other one is thermodynamic-dominated by the strength of ΔGls during TA. The unique packing process during film formation and the TA process of BDT-Dimer1 and BDT-Dimer3 is visualized as shown in Fig. 2g and Fig. S11 (ESI†). Our results demonstrate that for the low diffusion velocity of dimers, a twisted molecular backbone with proper molecular interaction resulted from the hyperconjugation effect appears to prevent dynamic packing during film formation but facilitate thermal-driven packing, which challenges the traditional understanding of aggregation and opens a new approach to well control the molecular assembly.
Pulsing miscibility on dynamic and thermal packing and charge properties
 | ||
| Fig. 3 Dynamic and thermal packing and charge properties in BDT-dimers/Y6 blends. In situ UV-visible-NIR spectra during film formation for (a) BDT-Dimer1/Y6, (b) BDT-Dimer2/Y6 and (c) BDT-Dimer3/Y6; 2-D GIWAXS for (d) BDT-Dimer1/Y6, (e) BDT-Dimer2/Y6 and (f) BDT-Dimer3/Y6 blends after film formation. (g) CCL of π–π stacking in the OOP direction and (h) mobilities in optimized blends. (i) TPC curves of optimized devices. | ||
Device performance and characterization
Based on BDT-Dimers and Y6, conventional devices with a structure of glass/ITO/EDOT:PSS/active layer/PNDIT-F3N/Ag were fabricated to investigate their photovoltaic properties. By adjusting the ratio of donor to acceptor (D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A), TA temperature and additive concentration (Table S5–S7, ESI†), the detailed device parameters based on optimized devices are summarized in Table 1, and the J–V curves and the corresponding external quantum efficiency (EQE) and statistical distribution of the optimized PCEs (≈10 devices) are shown in Fig. 4b–d, respectively. Thereinto, the BDT-Dimer3/Y6-based device obtained the best power conversion efficiency (PCE) of 15.68% with the maximum values of each device parameters, including a VOC of 0.800 V, an FF of 71.56% and a JSC of 27.39 mA cm−2. However, the device based on BDT-Dimer1/Y6 behaves in a completely opposite way and obtains the minimum values of each device parameter, especially an inferior PCE of 11.44% and a VOC of 0.758 V. The device based on BDT-Dimer2/Y6 achieved an in-between PCE of 14.4% with in-between device parameters.
:A), TA temperature and additive concentration (Table S5–S7, ESI†), the detailed device parameters based on optimized devices are summarized in Table 1, and the J–V curves and the corresponding external quantum efficiency (EQE) and statistical distribution of the optimized PCEs (≈10 devices) are shown in Fig. 4b–d, respectively. Thereinto, the BDT-Dimer3/Y6-based device obtained the best power conversion efficiency (PCE) of 15.68% with the maximum values of each device parameters, including a VOC of 0.800 V, an FF of 71.56% and a JSC of 27.39 mA cm−2. However, the device based on BDT-Dimer1/Y6 behaves in a completely opposite way and obtains the minimum values of each device parameter, especially an inferior PCE of 11.44% and a VOC of 0.758 V. The device based on BDT-Dimer2/Y6 achieved an in-between PCE of 14.4% with in-between device parameters.
| Active layer | VOC (V) | JSC (mA cm−2) | JSC,cal (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| BDT-Dimer1/Y6 | 0.758 (0.758 ± 0.007) | 23.13 (22.52 ± 0.56) | 22.56 | 65.21 (64.96 ± 1.00) | 11.44 (11.08 ± 0.17) |
| BDT-Dimer2/Y6 | 0.794 (0.802 ± 0.005) | 26.34 (25.63 ± 0.55) | 25.26 | 68.87 (67.68 ± 1.22) | 14.40 (14.00 ± 0.31) |
| BDT-Dimer3/Y6 | 0.800 (0.800 ± 0.002) | 27.39 (26.86 ± 0.50) | 26.19 | 71.56 (71.50 ± 0.97) | 15.68 (15.35 ± 0.15) |
 | ||
| Fig. 4 Device performances. (a) Chemical structure of Y6, (b) optimized J–V curves, (c) corresponding EQE curves and (d) statistical distribution of optimized PCE, (e) energy loss and (f) Veff–Jph curves for optimized devices. | ||
With a similar energy level of the highest occupied molecular orbital (HOMO, Fig. S18, ESI†), BDT-Dimer1/Y6-based devices obtained the lowest VOC, so the energy loss34 of three optimal devices was tested to investigate the essential reasons (Fig. 4e and Table S8 in the ESI†). As expected, the device based on BDT-Dimer1/Y6 exhibits the largest non-radiative energy loss (ΔE3), due to much reduced electroluminescence quantum efficiency (EQEEL) caused by significantly increased energy disorder (Fig. S19, ESI†), which results from the strongest and fastest aggregation of BDT-Dimer1 during the film-formation process.35 Therefore, due to the unfavorable molecular assembly, BDT-Dimer1/Y6 exhibits the worst device performance with completely attenuated device parameters.
The superior JSC and FF of BDT-Dimer3/Y6 are further confirmed by its most efficient charge transfer, photon utilization and less charge recombination. The strongest compatibility between BDT-Dimer3 and Y6 promotes electronic coupling in the blend film, as can be seen from the complete fluorescence quenching of Y6 (Fig. S20, ESI†), ensuring efficient charge transfer and exciton dissociation. The photon utilization is characterized by the exciton dissociation (Pdiss) and charge collection efficiency (Pcoll) (Fig. 4f) through the relationship between the photocurrent density (Jph) and the effective voltage (Veff). Thereinto, BDT-Dimer3/Y6 exhibits the highest Pdiss and Pcoll values of 98.45% and 80.64%, respectively, and BDT-Dimer2/Y6 (Pdiss = 95.96% and Pcoll = 64.03%) and BDT-Dimer1/Y6 (Pdiss = 94.35% and Pcoll = 58.89%) are in sequence. The significant difference in Pcoll may be attributed to well controlled blend morphology. Although with the best crystallinity, the optimal compatibility between BDT-Dimer3 and Y6 with ordered molecular assembly obtained the lowest root mean square roughness36 (RMS) of 0.96 nm measured by atomic force microscopy (AFM), followed by BDT-Dimer2/Y6 (RMS = 1.35 nm) and BDT-Dimer1/Y6 (RMS = 2.12 nm) (Fig. S21, ESI†). A smooth surface of an active layer facilitates charge transfer and balanced charge carrier mobilities in the interface,37,38 thereby enhancing the JSC and FF of the BDT-Dimer3/Y6 based device. The charge recombination was characterized by the α and n values, and the α is an indicator of bimolecular recombination deduced from the dependence of JSC on incident light intensity (Plight) (JSC ∝ (Plight)α), while the n is an indicator of trap-assisted recombination as inferred from the dependence of VOC on Plight (VOC ∝ nkT/qln(Plight)). Due to their similar α values (α = 0.98–0.99), BDT-Dimer based devices exhibit similar bimolecular recombination (Fig. S22, ESI†). However, the BDT-Dimer3/Y6 based device exhibits reduced trap-assisted recombination because its n value is equal to 1.00 compared to those of BDT-Dimer2/Y6 (n = 1.10) and BDT-Dimer1/Y6 (n = 0.97) based devices (Fig. S23, ESI†). In addition, the addition of DCBB further enhances the molecular assembly ability and achieves excellent charge carrier management (Fig. S24, ESI†). The highest Pdiss and Pcoll together with the effectively suppressed trap-assisted recombination well confirms the superior JSC and FF in the BDT-Dimer3/Y6 blend, and the essential reason is the formation of the smallest phase separation with the most ordered packing and smallest RMS, as previously analyzed in detail.
Device stability
We investigated the photostability of the devices based on the three systems together with their corresponding small-molecule systems and our previous G-Dimer-Ds14 (Fig. 5a and Fig. S25, ESI†). During continuous irradiation, BDT-Dimer3/Y6 exhibits the best photostability with a T80 of ca. 630 hours, followed by the BDT-Dimer2/Y6 based device with a T80 of ca. 400 hours and the BDT-Dimer1/Y6 based device with a T80 of ca. 300 hours. However, their corresponding SMDs of SM-CA-Reh and SM-Reh-based devices pairing with Y6 (the chemical structures, optimized J–V and EQE curves, optimization process and optimized device parameters are shown in Fig. S26 and Table S9 and S10 of the ESI†) performed attenuated PCE under the same conditions of light-soaking, which deteriorated to ca. 70% of the initial efficiency after 50 hours with sharp burn-in loss (Fig. 5a and Fig. S27, ESI†). Owing to the same linker as our previous design of G-Dimer-D2,6 the BDT-Dimer3/Y6 based device also exhibits further alleviated burn-in loss and the longest T80. That is to say, using the same small molecule acceptor of Y6, BDT-dimers especially the BDT-Dimer3 based devices exhibit the best photostability with the smallest burn-in loss.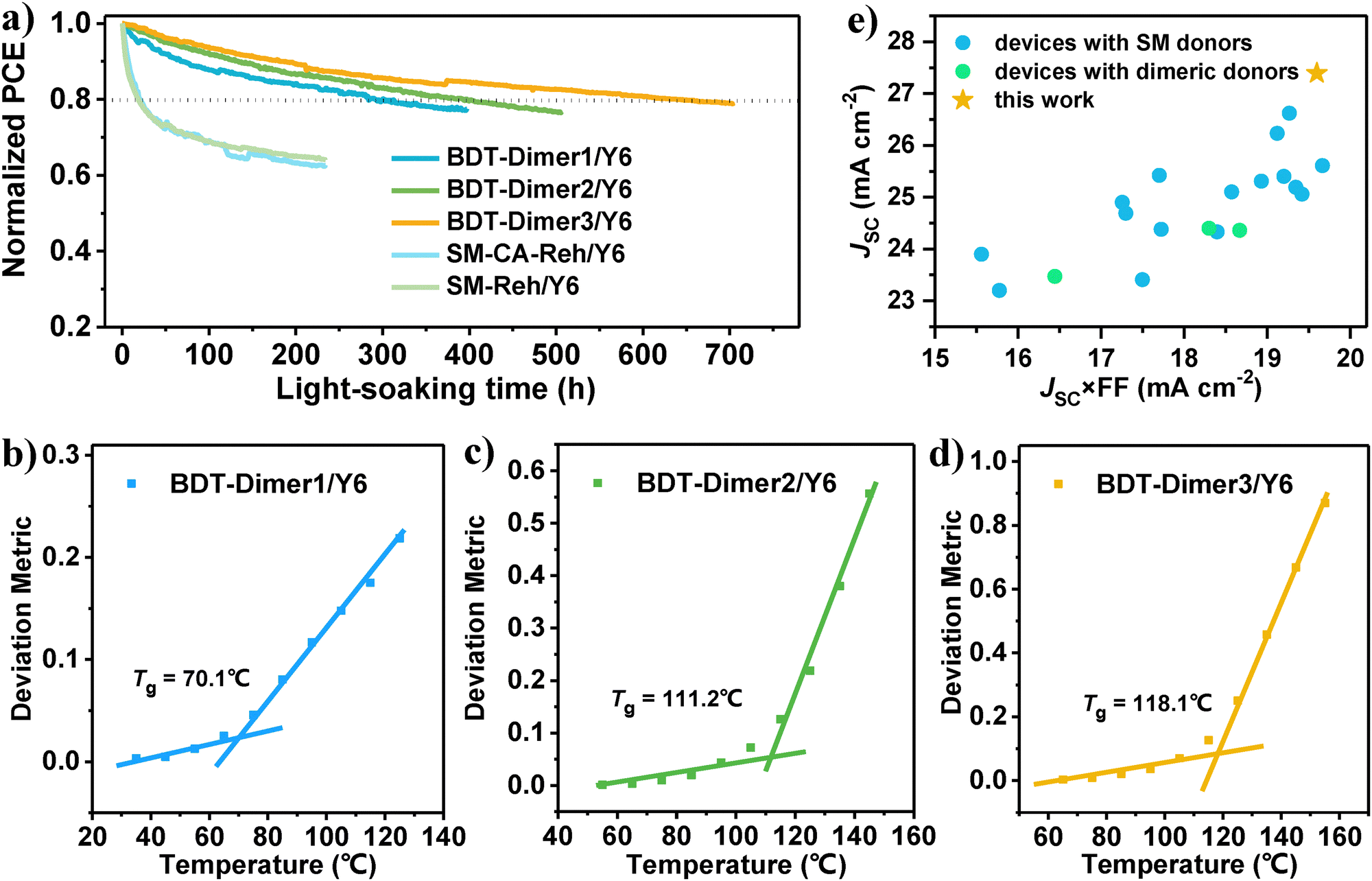 | ||
| Fig. 5 Device photostability. (a) Normalized PCE with light irradiation time for optimized devices of BDT-dimers and the corresponding small molecule with Y6. Blended Tg of (b) BDT-Dimer1/Y6, (c) BDT-Dimer2/Y6 and (d) BDT-Dimer3/Y6. (e) Charge carrier management (JSC × FF) and JSC based on SMDs or dimeric donors from reported literature studies and this work. | ||
As in our previous work, oligomeric dimeric donors would improve device stability from the following two aspects: (1) reduced crystallinity driving force and (2) increased blend glass transition temperature (Tg). For the former, the ΔGls value of the three BDT-dimers is −1.49, −1.40 and −2.51 J g−1 for BDT-Dimer1, BDT-Dimer2 and BDT-Dimer3, respectively. Whether compared to our previously reported G-Dimer-Ds (ΔGls = −8.86 J g−1 for G-Dimer-D1 and −5.72 J g−1 for G-Dimer-D2) or small molecules39 (i.e., ΔGls = −13.45 J g−1 for MPhS-C6 and −5.45 J g−1 for MPhS-C2), the ΔGls are quite smaller ones. Additionally, after adding the acceptor, the ΔGls would be further reduced depending on their hetero-molecular interaction. Generally, the stronger the hetero-molecular interaction, the smaller the blends’ ΔGls. For the BDT-Dimers, the addition of Y6 even leads to a negligible TC, indicating the reduced crystallinity in their blends. For the second aspect, we measured Tgs for both pure BDT-Dimers and their blends with Y6 by the temperature-dependent deviation metric of the absorption. The measured value is 63, 67 and 99 °C for BDT-Dimer1, BDT-Dimer2 and BDT-Dimer3 pure films, and 70, 111 and 118 °C for BDT-Dimer1/Y6, BDT-Dimer2/Y6 and BDT-Dimer3/Y6 blends, respectively (Fig. 5b–d and Fig. S28, ESI†). The higher the Tg, the superior the stability. Consequently, both the negligible driving force for crystallinity in blends and the highest Tg contributed to the longest T80 for the photostability in BDT-Dimer3/Y6 blends, thus ensuring its best morphological stability between optimized morphology and morphology after light soaking for 48 hours (Fig. S21 and S29, ESI†).
Discussions on oligomeric materials design
To further explore the design rules of dimeric donors, we first compared the BDT-Dimers with their corresponding SMDs of SM-CA-Reh and SM-Reh. Although the highest PCE is similar for the two kinds of OSCs, the JSC based on BDT-Dimer3/Y6 is much higher with a value of 27.39 mA cm−2, which is also currently the highest value for binary OSCs based on small-molecules or giant dimeric donors (Fig. 5e and Table S11, ESI†). Actually, we designed the linkers to different positions for the two monomers (Table S12, ESI†): for the semi-flexible linkers, we form both staircase and perpendicular molecular backbones; for the flexible linkers, we form parallel and staircase molecular backbones. Thereinto, the semi-flexible linker with the non-planar (perpendicular) skeleton has the following advantages: (1) appropriate ΔGls, which is smaller than the conjugate linker with the planar skeleton but larger than flexible ones, balancing both high efficiency and high stability and (2) preventing over stacking during film formation and facilitating thermal packing, leading to the morphology of the small domain size with ordered packing40 and thus obtaining simultaneously enhanced FF and JSC. Compared to the same linker as our previous design of G-Dimer-D2 (using G-Dimer-D2/Y6 as control), the advantage of BDT-Dimer3 is the smaller ΔGls and better compatibility with Y6; the former endows the further alleviated burn-in loss with longer T80 under light soaking, and the latter forms more interpenetrating networks with improved JSC and PCE. On the other hand, decreased ΔGls and inferior thermal-drive assembly not only caused a limitation on FF, restricting device performance pairing with oligomeric acceptors thus as DY (Table S13, ESI†), but also required a high annealing temperature (145 °C) of the optimized device, resulting in an increased non-radiative energy loss and a decreased VOC. Nevertheless, the efficiency of the dimeric donors still has significant room for improvement, and their further design principles can combine the advantages of BDT-Dimer3 and G-Dimer-D2 as follows: (1) deep HOMO energy levels, (2) improved hetero-molecular compatibility, and (3) proper thermal driving properties (moderate Tg). The first two facilitate obtaining high VOC and JSC, the last one is beneficial for promoting thermal-driven assembly and FF while maintaining ideal stability. Through effective chemical structure design of dimeric donors, using an unplanar semi-flexible linker with hyperconjugation to construct efficient and stable devices with a completely definite chemical structure is promising.Conclusions
In summary, we constructed three BDT-Dimers with flexible and semi-flexible linkers, leading to the monomer's relative position ranging from parallel to staircase to perpendicular. The dominant molecular conformation, linker properties and various hetero-molecular interactions with Y6 make unusual assembly behaviors in both pure dimeric donors and blends. Therefore, the most unplanar molecule of BDT-Dimer3 with the hyperconjugation effect exhibits the poorest stacking in the film formation process but the most ordered stacking after thermal packing. Combined with the strongest hetero-molecular interaction with Y6, BDT-Dimer3/Y6 based blends form a small domain size accompanied by ordered molecular assembly, thus facilitating the excellent charge properties and suppressed trap-assisted recombination, ultimately obtaining a satisfactory PCE of 15.68% with an outstanding JSC of 27.39 mA cm−2 and a neglectable burn-in loss under continuous irradiation. Our results provide a deep understanding of the molecular assembly in the pure materials and mixtures, especially for the emerging oligomeric materials, and also emphasize the design of the unplanar semi-flexible linker with the hyperconjugation effect in the preparation of devices with high efficiency and stability, and a completely definite structure.Author contributions
Caixuan Wang and Mengying Wu (first author): formal analysis, investigation, methodology, data curation, and writing – original draft. Ruixiang Fang: methodology. Jianqi Zhang: validation. Dan Deng, Ruimin Zhou, and Zhixiang Wei (corresponding author): conceptualization, funding acquisition, project administration, resources, supervision, validation, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support provided by the National Natural Science Foundation of China (52422308 and 22135001), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0770200), the Chinese Academy of Sciences Project for Young Scientists in Basic Research (No. YSBR-110), and the National Natural Science Foundation of China (52303258).References
- Y. Jiang, F. Liu and X. Zhu, Nat. Energy, 2024, 9, 930–931 CrossRef.
- C. Chen, L. Wang, W. Xia, K. Qiu, C. Guo, Z. Gan, J. Zhou, Y. Sun, D. Liu, W. Li and T. Wang, Nat. Commun., 2024, 15, 6865, DOI:10.1038/s41467-024-51359-w.
- Y. Sun, L. Wang, C. Guo, J. Xiao, C. Liu, C. Chen, W. Xia, Z. Gan, J. Cheng, J. Zhou, Z. Chen, J. Zhou, D. Liu, T. Wang and W. Li, J. Am. Chem. Soc., 2024, 146, 12011–12019 CrossRef CAS PubMed.
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS.
- Y. Cheng, B. Huang, Q. Mao, X. Huang, J. Liu, C. Zhou, W. Zhou, X. Ren, S. Kim, W. Kim, Z. Sun, F. Wu, C. Yang and L. Chen, Adv. Mater., 2024, 36, 2312938 CrossRef CAS PubMed.
- C. Wang, X. Ma, Y.-F. Shen, D. Deng, H. Zhang, T. Wang, J. Zhang, J. Li, R. Wang, L. Zhang, Q. Cheng, Z. Zhang, H. Zhou, C. Tian and Z. Wei, Joule, 2023, 7, 2386–2401 CrossRef CAS.
- J. Wu, G. Li, J. Fang, X. Guo, L. Zhu, B. Guo, Y. Wang, G. Zhang, L. Arunagiri, F. Liu, H. Yan, M. Zhang and Y. Li, Nat. Commun., 2020, 11, 4612 CrossRef CAS PubMed.
- X. Yuan, Y. Zhao, T. Zhan, J. Oh, J. Zhou, J. Li, X. Wang, Z. Wang, S. Pang, P. Cai, C. Yang, Z. He, Z. Xie, C. Duan, F. Huang and Y. Cao, Energy Environ. Sci., 2021, 14, 5530–5540 RSC.
- D. Hu, Q. Yang, H. Chen, F. Wobben, V. M. Le Corre, R. Singh, T. Liu, R. Ma, H. Tang, L. J. A. Koster, T. Duan, H. Yan, Z. Kan, Z. Xiao and S. Lu, Energy Environ. Sci., 2020, 13, 2134–2141 RSC.
- J. Qin, Z. Chen, P. Bi, Y. Yang, J. Zhang, Z. Huang, Z. Wei, C. An, H. Yao, X. Hao, T. Zhang, Y. Cui, L. Hong, C. Liu, Y. Zu, C. He and J. Hou, Energy Environ. Sci., 2021, 14, 5903–5910 RSC.
- Y. Gao, X. Yang, W. Wang, R. Sun, J. Cui, Y. Fu, K. Li, M. Zhang, C. Liu, H. Zhu, X. Lu and J. Min, Adv. Mater., 2023, 35, 2300531 CrossRef CAS PubMed.
- D. Hu, H. Tang, S. Karuthedath, Q. Chen, S. Chen, J. I. I. Khan, H. Liu, Q. Yang, J. Gorenflot, C. E. E. Petoukhoff, T. Duan, X. Lu, F. Laquai and S. Lu, Adv. Funct. Mater., 2023, 33, 2211873 CrossRef CAS.
- F. Xie, J. Fang, L. Zhang, D. Deng, Y. Chen, Z. Wei, F. Guo and C.-Q. Ma, ACS Appl. Mater. Interfaces, 2024, 16, 11767–11777 CrossRef CAS PubMed.
- C. Wang, X. Ma, D. Deng, H. Zhang, R. Sun, J. Zhang, L. Zhang, M. Wu, J. Min, Z.-G. Zhang and Z. Wei, Nat. Commun., 2024, 15, 8494 CrossRef CAS PubMed.
- Q. Zhu, J. Xue, L. Zhang, J. Wen, B. Lin, H. B. Naveed, Z. Bi, J. Xin, H. Zhao, C. Zhao, K. Zhou, S. Liu and W. Ma, Small, 2021, 17, 2007011 CrossRef CAS PubMed.
- C. Zhang, T. Heumueller, S. Leon, W. Gruber, K. Burlafinger, X. Tang, J. D. Perea, I. Wabra, A. Hirsch, T. Unruh, N. Li and C. J. Brabec, Energy Environ. Sci., 2019, 12, 1078–1087 RSC.
- L. Zhang, D. Deng, K. Lu and Z. Wei, Adv. Mater., 2024, 36, 2302915 CrossRef CAS PubMed.
- W. Gao, R. Ma, T. A. Dela Pena, C. Yan, H. Li, M. Li, J. Wu, P. Cheng, C. Zhong, Z. Wei, A. K. Y. Jen and G. Li, Nat. Commun., 2024, 15, 1946 CrossRef CAS PubMed.
- H. Lai, Y. Zhu, Y. Ouyang, X. Lai, M. Ou, Z. Deng, Y. Wang, D. Qiu, C. Zhang and F. He, Adv. Funct. Mater., 2024, 35, 2418106 CrossRef.
- A. D. de Zerio and C. Mueller, Adv. Energy Mater., 2018, 8, 1702741 CrossRef.
- M. Lv, Q. Wang, J. Zhang, Y. Wang, Z. G. Zhang, T. Wang, H. Zhang, K. Lu, Z. Wei and D. Deng, Adv. Mater., 2023, 36, 2310046 CrossRef PubMed.
- Y. Wei, J. Yu, L. Qin, H. Chen, X. Wu, Z. Wei, X. Zhang, Z. Xiao, L. Ding, F. Gao and H. Huang, Energy Environ. Sci., 2021, 14, 2314–2321 RSC.
- H. Hu, S. Liu, J. Xu, R. Ma, Z. Peng, T. A. D. Pena, Y. Cui, W. Liang, X. Zhou, S. Luo, H. Yu, M. Li, J. Wu, S. Chen, G. Li and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, 2400086 Search PubMed.
- L. Ma, H. Yao, J. Zhang, Z. Chen, J. Wang, J. Qiao, S. Wang, Z. Bi, Z. Li, X. Hao, Z. Wei, W. Ma and J. Hou, Chem, 2023, 9, 2518–2529 CAS.
- J. Ge, L. Hong, H. Ma, Q. Ye, Y. Chen, L. Xie, W. Song, D. Li, Z. Chen, K. Yu, J. Zhang, Z. Wei, F. Huang and Z. Ge, Adv. Mater., 2022, 34, 2202752 CrossRef CAS PubMed.
- J. Xin, X. Meng, X. Xu, Q. Zhu, H. B. Naveed and W. Ma, Matter, 2019, 1, 1316–1330 CrossRef.
- M. Ghasemi, H. Hu, Z. Peng, J. J. Rech, I. Angunawela, J. H. Carpenter, S. J. Stuard, A. Wadsworth, I. McCulloch, W. You and H. Ade, Joule, 2019, 3, 1328–1348 CrossRef CAS.
- Z. He, Y. Hong, G. Li, T. Shan, Y. Zhang and H. Zhong, Chin. J. Chem., 2024, 42, 725–730 CrossRef CAS.
- Q. Shen, C. He, B. Wu, Y. Lin, S. Chen, J. Gao, S. Li, Z. Ma, W. Ma, M. Shi, Y. Li and H. Chen, Chem. Eng. J., 2023, 471, 144472 CrossRef CAS.
- A. Y. Sosorev, M. K. Nuraliev, E. V. Feldman, D. R. Maslennikov, O. V. Borshchev, M. S. Skorotetcky, N. M. Surin, M. S. Kazantsev, S. A. Ponomarenko and D. Y. Paraschuk, Phys. Chem. Chem. Phys., 2019, 21, 11578–11588 RSC.
- C. Han, H. Gao, Y. Sun, Y. Kan, Z. Bi, W. Ma, Y. Zhang, J. A. Zapien, Y. Yang and K. Gao, J. Energy Chem., 2024, 93, 601–608 CrossRef CAS.
- Q. Ai, W. Zhou, L. Zhang, L. Huang, J. Yin, Z. Yu, S. Liu, W. Ma, J. Zeng and Y. Chen, J. Mater. Chem. C, 2017, 5, 10801–10812 RSC.
- L. Zhang, X. Zhu, D. Deng, Z. Wang, Z. Zhang, Y. Li, J. Zhang, K. Lv, L. Liu, X. Zhang, H. Zhou, H. Ade and Z. Wei, Adv. Mater., 2021, 34, 2106316 CrossRef PubMed.
- X.-K. Chen, D. Qian, Y. Wang, T. Kirchartz, W. Tress, H. Yao, J. Yuan, M. Huelsbeck, M. Zhang, Y. Zou, Y. Sun, Y. Li, J. Hou, O. Inganas, V. Coropceanu, J.-L. Bredas and F. Gao, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- X. Ma, C. Wang, D. Deng, H. Zhang, L. Zhang, J. Zhang, Y. Yang and Z. Wei, Small, 2023, 20, 2309042 CrossRef PubMed.
- H. Jiang, C. Han, Y. Li, F. Bi, N. Zheng, J. Han, W. Shen, S. Wen, C. Yang, R. Yang and X. Bao, Adv. Funct. Mater., 2020, 31, 2007088 CrossRef.
- R. K. Gupta, R. Garai, M. A. Afroz and P. K. Iyer, J. Mater. Chem. C, 2020, 8, 8191–8198 RSC.
- Y. Li, Z. Jia, Q. Zhang, Z. Wu, H. Qin, J. Yang, S. Wen, H. Y. Woo, W. Ma, R. Yang and J. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 33028–33038 CrossRef CAS PubMed.
- L. Zhang, R. Sun, Z. Zhang, J. Zhang, Q. Zhu, W. Ma, J. Min, Z. Wei and D. Deng, Adv. Mater., 2022, 34, 2207020 CrossRef CAS PubMed.
- J. Wang, C. Han, F. Bi, D. Huang, Y. Wu, Y. Li, S. Wen, L. Han, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2021, 14, 5968–5978 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00223k |
| ‡ Caixuan Wang and Mengying Wu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |