
PVP pre-intercalation engineering combined with the V4+/V5+ dual-valence modulation strategy for energy storage in aqueous zinc-ion batteries†
Wenhui Mia,
Bosi Yina,
Haixi Gua,
Hanyu Wena,
Zhibiao Wanga,
Hui Libc,
Ziqian Yuana,
Siwen Zhang*a and
Tianyi Ma
 *bc
*bc
aInstitute of Clean Energy Chemistry, Key Laboratory for Green Synthesis and Preparative Chemistry of Advanced Material, College of Chemistry, Liaoning University, Shenyang 110036, P. R. China
bCentre for Atomaterials and Nanomanufacturing (CAN), School of Science, RMIT University, Melbourne, VIC 3000, Australia
cARC Industrial Transformation Research Hub for Intelligent Energy Efficiency in Future Protected Cropping (E2Crop), Melbourne, VIC 3000, Australia
First published on 15th May 2025
Abstract
Aqueous zinc-ion batteries (AZIBs) have become a potential energy storage technology due to their inherent safety, environmental compatibility, and cost-effectiveness. Vanadate compounds have demonstrated considerable potential for AZIB applications among various cathode materials. However, their practical implementation is significantly constrained by intrinsic limitations, including sluggish ion diffusion kinetics, structural instability, and vanadium framework collapse during cycling. To address these challenges, we developed a novel strategy involving polyvinylpyrrolidone (PVP) pre-intercalation into CaV6O16·3H2O (CaVO), resulting in a phase transformation to Ca0.24V2O5·H2O (PVP–CaVO). The embedded PVP acts as a “pillar” between the interlayer spaces, stabilizing the structural stability and thereby enhancing cycling performance. Incorporating PVP introduces additional functional advantages through its amide groups, which possess strong polar characteristics. These groups serve as hydrogen bond acceptors, with nitrogen and oxygen atoms acting as coordination sites. This unique configuration facilitates chemical bond rearrangement and promotes partial reduction of vanadium from higher oxidation states (V5+) to lower ones (V4+), establishing a V4+/V5+ hybrid valence system. Such electronic structure modification not only enables multi-step redox reactions but also alleviates the strong polarization effect of Zn2+ ions. Benefiting from these synergistic effects, the PVP–CaVO cathode demonstrates remarkable electrochemical performance in AZIBs, delivering a specific capacity of 323 mA h g−1 at 0.5 A g−1 and maintaining a specific capacity of 169 mA h g−1 at 10 A g−1, coupled with excellent cycling stability. Comprehensive ex situ characterization studies further elucidated the energy storage processes, verifying a reversible Zn2+/H+ co-insertion mechanism. This innovative approach of structural and phase engineering through PVP intercalation provides a valuable approach for optimizing vanadate-based materials.
 Tianyi Ma | I am honoured to contribute to the 10th anniversary collection of Nanoscale Horizons. My connection with the Horizons journals dates back to 2015, when I was invited to join the inaugural Materials Horizons Community Board, a role I proudly held until 2020. In 2021, I was recognised as a Materials Horizons Emerging Investigator, and I published my first paper in the journal in 2019. Over the years, I’ve remained actively involved with the Horizons community, including organising a themed issue for Nanoscale Horizons in 2019. I truly value the opportunity to share my group's latest research through such a respected platform. Congratulations to Nanoscale Horizons on this important milestone—I look forward to many more years of collaboration. |
New conceptsCurrently, aqueous zinc-ion batteries (AZIBs) with the advantages of low cost and environmental friendliness are gradually emerging as highly promising alternatives in the field of next-generation electrochemical energy storage. However, the poor stability of cathode materials and the presence of strong electrostatic interactions pose significant obstacles to the practical application of ZIBs. Therefore, the development of high-performance cathode materials with stable structures is crucial for the advancement of ZIBs. This work reports a dual-synergistic strategy that successfully addresses the issues of poor multi-step and multi-electron reactivity as well as inferior cycling stability. Based on these strategies, phase transformation occurs in CaVO after polyvinylpyrrolidone (PVP) intercalation, resulting in PVP–CaVO, where the high-valence state of V is partially reduced to a low-valence state, forming hybrid valences of V4+/V5+. Meanwhile, PVP acts as a pillar. During this process, PVP not only stabilizes the material structure between vanadium oxide layers to enhance cycling stability but also facilitates the multi-step and multi-electron reactions of vanadium through the formation of hybrid valences of V4+/V5+. The dual-synergistic strategy proposed in this study is expected to promote the further development of ZIB energy storage systems. |
1. Introduction
In recent years, lithium-ion batteries (LIBs) have dominated the rechargeable battery market, functioning as the foundation for diverse applications, ranging from portable electronic gadgets to electric vehicles.1,2 However, the widespread adoption of LIBs has revealed critical limitations, including lithium resource scarcity, risks linked to flammable organic electrolytes, and environmental hazards. These challenges have prompted the exploration of alternative energy storage solutions to meet the increasing requirements of the energy storage market. Among the emerging candidates, ZIBs have garnered significant interest for their innate benefits, such as affordability, enhanced safety, and environmental sustainability, positioning them as a prospective contender for future electrochemical energy storage systems.3–6 A key advantage of ZIBs lies in their aqueous electrolytes, which demonstrate a remarkable ionic conductivity reaching 1 S cm−1, surpassing that of conventional non-aqueous electrolytes (typically 1–10 mS cm−1). This superior ionic conductivity translates into improved rate capability, enhanced cycling stability, and reduced resistance polarization, making ZIBs a viable alternative to traditional LIBs for high-performance energy storage applications.7,8 While substantial efforts have been devoted to developing modification strategies for zinc anodes, the broad application of ZIBs remains constrained by the lack of a perfect cathode material capable of concurrently providing high capacity and maintaining enduring cycling stability. The inherent characteristics of Zn2+, particularly its large hydrated ion radius and pronounced polarization effect, pose significant challenges to cathode materials in aqueous ZIBs, resulting in severe structural degradation, limited cycle life, reduced specific capacity, and compromised rate performance.9 Consequently, the design and development of advanced cathode materials that can effectively enhance Zn2+ storage efficiency and accelerate battery kinetics have become crucial for propelling the progress of zinc-ion battery technology.To date, extensive research has been conducted on a diverse array of cathode materials for ZIBs, including manganese oxides, vanadium oxides, vanadates, Prussian blue analogues, and conductive organic polymers, among others.10–16 The electrochemical behavior of these materials in ZIBs is mainly determined by their crystal structure, which directly influences the Zn2+ insertion/extraction storage mechanism.17–19 Among various structural configurations, layered cathode materials exhibit distinct advantages due to their large and tunable interlayer distances, which not only enhance the reversibility of ion insertion/extraction processes but also significantly reduce ion diffusion resistance. In particular, vanadium-based oxides and vanadates have garnered substantial attention owing to their open layered frameworks and the presence of multiple elemental valence states, which contribute to their high capacity.20 Notably, vanadate CaV6O16·3H2O stands out as a prospective cathode material for future energy storage systems, as the incorporation of Ca2+ and structural water (H2O) within its framework effectively stabilizes and supports the interlayer spacing, addressing a critical challenge in ZIB cathode design. Despite their promising characteristics, vanadium-based cathode materials still face significant challenges, including inevitable vanadium dissolution and the strong electrostatic repulsion of Zn2+, which collectively result in poor structural reversibility and sluggish reaction kinetics. These limitations manifest as slow zinc-ion diffusion and suboptimal electrochemical performance, hindering their practical applications.21,22 To overcome these obstacles, researchers have developed a range of advanced engineering strategies, with the pre-insertion approach emerging as a particularly effective method for enhancing cathode performance and, consequently, improving the overall battery efficiency.23,24 The pre-inserted guest species interact with the host material's atoms or ionic carriers through chemical bonding, thereby modifying the material's structure and significantly enhancing the reaction kinetics and ion diffusion rates. Among the various pre-insertion species, interstitial ions—including metal ions (Li+, K+, Mg2+, and Ca2+),25–28 non-metal ions such as NH4+, H2O molecules29 and organic molecules (e.g., dimethyl sulfoxide, PEDOT, and PANI)30–32—play a crucial role in stabilizing the material's framework and acting as structural “pillars” during ion insertion/extraction processes. For instance, the incorporation of organic guest molecules like polyaniline (PANI) and poly(3,4-ethylenedioxythiophene) (PEDOT) has been demonstrated to significantly expand the distance between layers in these materials, addressing a major limitation in their performance. For example, the insertion of polyaniline (PANI) as an organic molecule into MnO2 results in an approximate interlayer distance of 10 Å, enabling the composite to deliver an impressive discharge specific capacity of 280 mA h g−1 at 0.2 A g−1 while maintaining stable performance over 300 cycles. Similar enhancements have been successfully demonstrated in V2O5 systems.33,34 Furthermore, the incorporation of poly(3,4-ethylenedioxythiophene) (PEDOT) into NH4V3O8 has been demonstrated to enhance the layer distance from 7.8 Å to 10.8 Å, achieving exceptional cycling stability with over 5000 cycles at 10 A g−1.31 Recent advancements have revealed that the strategic introduction of trace elements or organic molecules can induce phase transitions, effectively modulating the crystalline growth behavior of materials. This approach enables precise control over particle sizes and interlayer spacing, thereby enhancing energy storage capabilities. Among these modifiers, polyvinylpyrrolidone (PVP), an organic polymer featuring a lactam ring with strong metal ion coordination ability, has emerged as a versatile surface morphology modifier. PVP has demonstrated its efficacy in expanding the interlayer distances of diverse materials, such as VS4 and MoS2.35,36 Therefore, layered vanadates with hybrid valence and pillared by the intercalation of organic molecules may emerge as promising cathode materials for zinc-ion batteries (ZIBs) with superior performance.
In this research, we put forward an innovative dual synergistic engineering approach that combines PVP pre-intercalation with V4+/V5+ hybrid valence to optimize cathode performance. The PVP pre-intercalation induces a phase transition in the precursor, resulting in the formation of PVP–CaVO. Owing to the PVP's ability to pillar the interlayer spacing of PVP–CaVO, the resulting pillar structure stabilizes the fragile layered structure and enhances the diffusion dynamics, thereby facilitating Zn2+ diffusion and increasing ion conduction rates. More importantly, the amide groups in PVP molecules, due to their pronounced polar characteristics, readily participate in hydrogen bonding, with nitrogen (N) and oxygen (O) atoms serving as coordination centers. This unique property enables the dynamic breakage and reformation of chemical bonds, thereby modifying the coordination environment within the material's lattice. The introduction of V4+/V5+ hybrid valence states creates additional active regions for Zn2+ storage, promoting vanadium's multi-step redox reactions during the processes of Zn2+ intercalation and de-intercalation. This synergistic effect not only improves electrochemical performance but also mitigates Zn2+ polarization on the cathode surface. Electrochemical assessments showcase the excellent performance of the PVP–CaVO cathode material, delivering a remarkable charge/discharge capacity of 323 mA h g−1 at 0.5 A g−1. Even at 10 A g−1, the battery maintains a specific discharge capacity of 169 mA h g−1, with an impressive retention of nearly 80% of capacity after 2700 cycles. To elucidate the energy storage mechanism, comprehensive characterization techniques including ex situ XRD, ex situ XPS, and electrochemical analyses were applied, confirming the reversible co-insertion mechanism of Zn2+/H+ ions in the battery system.
2. Results and discussion
Fig. 1 depicts the synthesis process of two nanorod-structured materials in a schematic manner, CaVO and PVP–CaVO, along with the corresponding structural transformations. Both target materials were synthesized via a simple, single-step hydrothermal process, with the main difference lying in the addition of a tiny amount of the PVP precursor in the hydrothermal reaction at 180 °C for PVP–CaVO synthesis.37 While the morphological characteristics of the two products remain largely unchanged before and after PVP incorporation, as evidenced in Fig. S1 (ESI†), a notable color difference is observed in the resulting powders: CaVO appears as a red powder, whereas PVP–CaVO exhibits a green coloration. This color variation suggests that the partial intercalation of PVP molecules into the interlayer spacing of CaVO may have altered the lattice atoms' coordination scenario, leading to the partial reduction of vanadium (V) elements. This hypothesis will be further substantiated through subsequent comprehensive characterization studies. | ||
| Fig. 1 Preparation process of CaVO and PVP–CaVO. | ||
To elucidate the structural differences between PVP–CaVO and CaVO, we performed analysis of their XRD patterns. Fig. 2a illustrates that all diffraction peaks of CaVO are accurately indexed to the monoclinic phase (PDF#33-0317 for CaV6O16·3H2O).37 However, the introduction of PVP induces significant alterations in the diffraction peak positions, confirming the successful synthesis of the new phase-transformed product, PVP–CaVO. The monoclinic phase of PVP–CaVO exhibits excellent agreement with the standard PDF#88-0579 for Ca0.24V2O5·H2O. This structural transformation can be attributed to the strong polarity of amide groups within PVP molecules, which readily participate in hydrogen bonding. The coordination atoms in these groups facilitate the breaking and reformation of chemical bonds, thereby modifying the atomic coordination environment within the material's lattice. Consequently, PVP intercalation induces an internal restructuring of the crystal architecture, transforming CaV6O16·3H2O, which originally consisted of [VO6] octahedra and [VO5] square pyramids, into a new Ca0.24V2O5·H2O phase consisting solely of [VO6] octahedra. These XRD results provide conclusive evidence that PVP incorporation triggers a phase transition in the material.38 It is noteworthy that vanadium (V) in aqueous solution forms various vanadates depending on the pH value, which significantly influences the final product formation. When the pH exceeds 7.0, green needle-like crystals of CaV2O6 (JCPDS 73-0971) coexist with CaVO.39 Given the presence of Ca(OH)2 during the synthesis process, the solution pH remains above 7.0, resulting in characteristic diffraction peaks at 18.23° and 19.52° in the XRD pattern of CaVO, corresponding to the (200) and (−201) planes of CaV2O6. A similar phenomenon is observed in PVP–CaVO. Additionally, PVP–CaVO exhibits a minor impurity peak at approximately 10° in its diffraction pattern, which may be attributed to a small fraction of PVP that failed to intercalate into the interlayer spacing of the precursor CaVO during the reaction stage. This observation is particularly interesting as the (002) crystal plane of CaVO demonstrates its strongest diffraction peak at 10.79°, which serves as a characteristic feature of CaVO. The structural characteristics of CaVO and PVP–CaVO are characterized through the utilization of FT-IR spectroscopy, as presented in Fig. 2b. The peaks of absorption detected within the 3000–3700 cm−1 range and at approximately 1605 cm−1 are respectively ascribed to the stretching and bending vibrations of H2O molecules.40 To demonstrate the successful embedding of PVP in PVP–CaVO, the stretching and bending vibrations of C–H and C–N bonds were identified at distinct peaks located approximately at 1410 cm−1 and 1090 cm−1, respectively, which can be directly associated with the functional groups in the molecular structure of PVP. However, these characteristic peaks are exclusively present in PVP–CaVO but absent in CaVO, thereby confirming the successful incorporation of PVP into the CaVO matrix. To further investigate the presence of crystalline water molecules, thermogravimetric analysis (TGA) was conducted. The TGA curves of CaVO and PVP–CaVO (Fig. 2c) reveal distinct weight loss patterns. Specifically, CaVO exhibits a weight loss of 15.10% between 30 °C and 450 °C, while PVP–CaVO shows a reduced weight loss of 9.26% within the same temperature range. This significant difference in weight loss not only confirms the presence of water molecules in both materials but also indicates that PVP–CaVO contains a lower content of crystalline water compared to CaVO. The reduction in the crystalline water content can be attributed to the insertion of PVP molecules into the interstitial sites within the lattice structure, which displaces some of the water molecules. This observation provides additional evidence for the successful intercalation of PVP into the precursor CaVO material, which is in line with the crystalline water content predicted by the molecular formulas of CaVO and PVP–CaVO.40 We performed full XPS measurements of the spectra, as depicted in Fig. 2d, which elucidate information about the valence states of the constituent elements in the sample. Both CaVO and PVP–CaVO samples showed the coexistence of the elements Ca, V, and O. Notably, the presence of PVP in the PVP–CaVO composite is evidenced by the significant increase in the signal intensities of the C 1s and N 1s spectral components, providing further concrete evidence for PVP embedding. The high-resolution C 1s spectrum (Fig. 2e) reveals three distinct peaks at 284.7, 286.5, and 288.6 eV, which are respectively assigned to C–C, C–N, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.41 Furthermore, the O 1s spectrum of PVP–CaVO (Fig. S2, ESI†) displays peaks at 532.7 eV and 530.1 eV, which correspond to H2O and O2− species present in both materials. Additionally, the presence of a characteristic peak at 532.1 eV in PVP–CaVO is attributed to the CO bond.42,43 The N 1s spectrum of PVP–CaVO displays a prominent peak at 400.1 eV, corresponding to the nitrogen atom in the five-membered heterocyclic ring of PVP, thereby providing additional confirmation of PVP incorporation.44 To investigate the valence state variations of vanadium, high-resolution and fitted spectra were analyzed in the range of 514–528 eV. For the CaVO precursor, the V 2p1/2 and V 2p3/2 peaks are respectively fitted at 524.7 eV and 517.3 eV, indicating the exclusive presence of V5+ species. In contrast, the V 2p spectrum of PVP–CaVO demonstrates the coexistence of both V5+ (V 2p3/2 at 517.4 eV and V 2p1/2 at 524.8 eV) and V4+ (V 2p3/2 at 516.1 eV and V 2p1/2 at 523.3 eV) states, as shown in Fig. 2f.45 This observation confirms the successful construction of V4+/V5+ hybrid-valent PVP–CaVO. Collectively, the XPS analysis provides conclusive evidence for the presence of V4+/V5+ hybrid valence and the successful pre-intercalation of PVP pillars in PVP–CaVO.
O bonds.41 Furthermore, the O 1s spectrum of PVP–CaVO (Fig. S2, ESI†) displays peaks at 532.7 eV and 530.1 eV, which correspond to H2O and O2− species present in both materials. Additionally, the presence of a characteristic peak at 532.1 eV in PVP–CaVO is attributed to the CO bond.42,43 The N 1s spectrum of PVP–CaVO displays a prominent peak at 400.1 eV, corresponding to the nitrogen atom in the five-membered heterocyclic ring of PVP, thereby providing additional confirmation of PVP incorporation.44 To investigate the valence state variations of vanadium, high-resolution and fitted spectra were analyzed in the range of 514–528 eV. For the CaVO precursor, the V 2p1/2 and V 2p3/2 peaks are respectively fitted at 524.7 eV and 517.3 eV, indicating the exclusive presence of V5+ species. In contrast, the V 2p spectrum of PVP–CaVO demonstrates the coexistence of both V5+ (V 2p3/2 at 517.4 eV and V 2p1/2 at 524.8 eV) and V4+ (V 2p3/2 at 516.1 eV and V 2p1/2 at 523.3 eV) states, as shown in Fig. 2f.45 This observation confirms the successful construction of V4+/V5+ hybrid-valent PVP–CaVO. Collectively, the XPS analysis provides conclusive evidence for the presence of V4+/V5+ hybrid valence and the successful pre-intercalation of PVP pillars in PVP–CaVO.
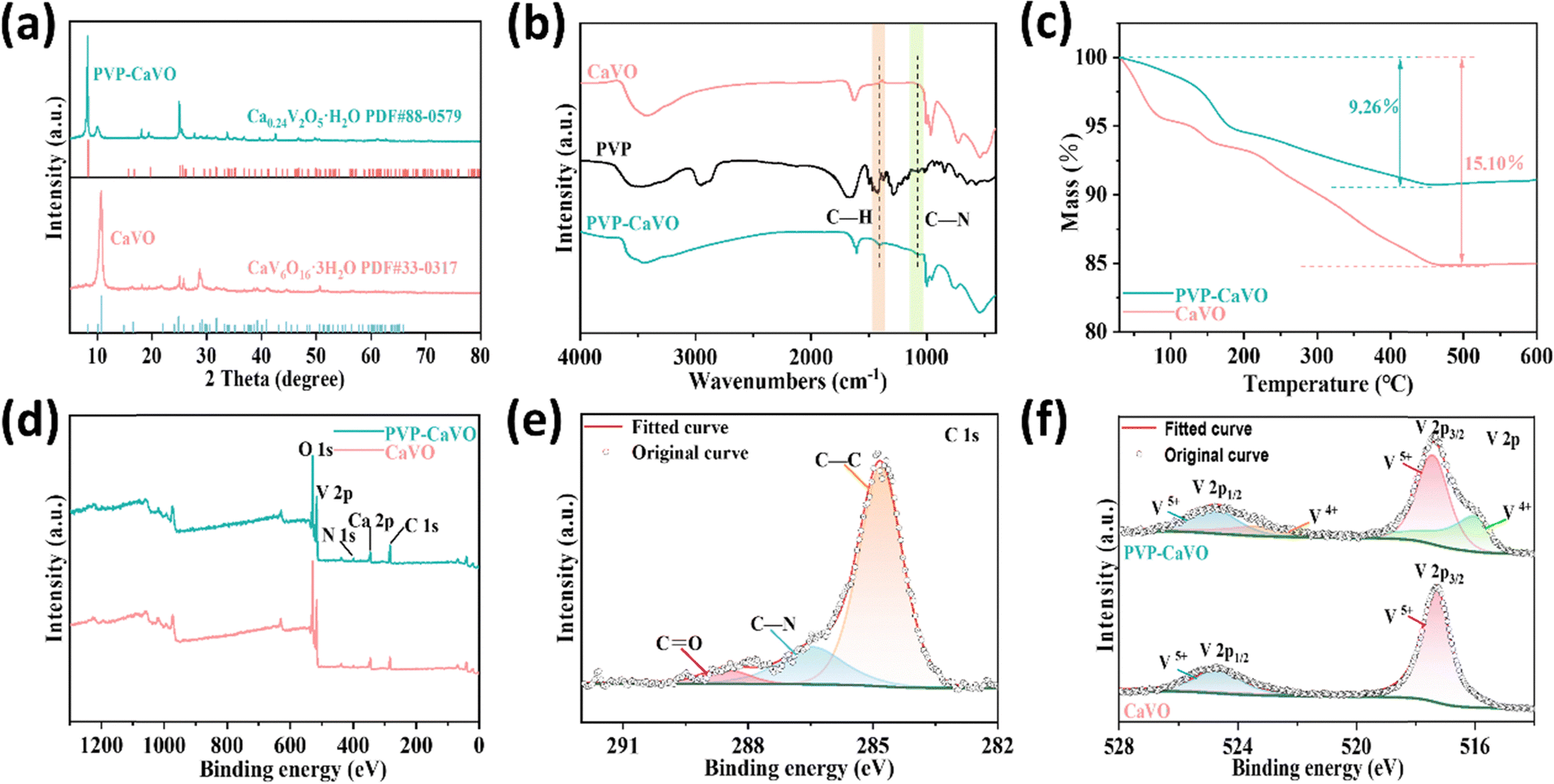 | ||
| Fig. 2 (a) XRD patterns of CaVO and PVP–CaVO. (b) FT-IR plots of CaVO and PVP–CaVO. (c) TGA curves of CaVO and PVP–CaVO. (d) The full XPS survey spectra of CaVO and PVP–CaVO. (e) C 1s XPS spectra of PVP–CaVO. (f) V 2p XPS spectra of CaVO and PVP–CaVO. | ||
Fig. 3a and Fig. S3 (ESI†) show the SEM images of PVP–CaVO and pristine CaVO, respectively, revealing a uniform one-dimensional nanorod morphology with widths of several hundred nanometers. TEM analysis (Fig. 3b) confirms the PVP–CaVO width (200–300 nm). At the same time, it can be observed that the morphology of the two materials before and after the process has not changed significantly, indicating that the insertion of PVP still retains the nanorod structure. SEM-EDS analysis (Fig. 3c and Fig. S5, ESI†) demonstrates the consistent distribution of C, N, Ca, V, and O in PVP–CaVO, contrasting with only Ca, V, and O in pristine CaVO. Furthermore, SEM-EDS (Fig. S6, ESI†) reveals a Ca/V atomic ratio change from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11.8 in CaVO to 1:6.4 in PVP–CaVO, indicating PVP intercalation into the lattice spacing, replacing V positions and altering the elemental composition. The HR-TEM (as depicted in Fig. 3d–f) results indicate that the lattice fringes in PVP–CaVO possess d-spacings of 1.68 Å and 1.74 Å, which correspond to the (−222) and (−221) crystal planes, further indicating the successful synthesis of the PVP–CaVO material and the transformation from the precursor CaVO to PVP–CaVO. Meanwhile, Fig. S7 (ESI†) demonstrates the d-spacing for CaVO lattice fringes of 1.57 Å, corresponding to the (406) crystal plane in CaVO, indicating the successful synthesis of the CaVO material. The selected area electron diffraction (SAED) pattern, composed of diffuse diffraction rings presented in Fig. 3g, points to a monoclinic crystal structure corresponding to PVP–CaVO (PDF#88-0579), demonstrating that PVP–CaVO possesses excellent crystallinity, and this finding aligns with the HR-TEM results.38
:11.8 in CaVO to 1:6.4 in PVP–CaVO, indicating PVP intercalation into the lattice spacing, replacing V positions and altering the elemental composition. The HR-TEM (as depicted in Fig. 3d–f) results indicate that the lattice fringes in PVP–CaVO possess d-spacings of 1.68 Å and 1.74 Å, which correspond to the (−222) and (−221) crystal planes, further indicating the successful synthesis of the PVP–CaVO material and the transformation from the precursor CaVO to PVP–CaVO. Meanwhile, Fig. S7 (ESI†) demonstrates the d-spacing for CaVO lattice fringes of 1.57 Å, corresponding to the (406) crystal plane in CaVO, indicating the successful synthesis of the CaVO material. The selected area electron diffraction (SAED) pattern, composed of diffuse diffraction rings presented in Fig. 3g, points to a monoclinic crystal structure corresponding to PVP–CaVO (PDF#88-0579), demonstrating that PVP–CaVO possesses excellent crystallinity, and this finding aligns with the HR-TEM results.38
 | ||
| Fig. 3 (a) SEM image of PVP–CaVO. (b) TEM image of PVP–CaVO. (c) SEM-EDS elemental mapping of PVP–CaVO. (d)–(f) HR-TEM images of PVP–CaVO. (g) SAED pattern of PVP–CaVO. | ||
To elucidate the synergistic effect of the embedding of trace amounts of PVP being able to support the layer spacing of PVP–CaVO, which in turn yields the synergistic effect of having a stable lamellar structure as well as the formation of a V4+/V5+ hybridized valence state creating additional active sites for Zn2+ storage on the storage performance of PVP–CaVO for zinc ions, a series of electrochemical tests were performed within a voltage window of 0 to 1.8 V. Fig. 4a presents the CV profiles of PVP–CaVO and pristine CaVO at a scan rate of 0.8 mV s−1. Both materials exhibit two distinct redox pairs (relative to Zn2+/Zn), with PVP–CaVO demonstrating a slight negative shift in redox potentials. This shift is attributed to the presence of V4+ in PVP–CaVO, corroborating the formation of hybrid V4+/V5+ valence states, which aligns with the XPS V 2p spectra presented in Fig. 2f.41 To further assess the electrochemical reversibility, the initial three cycles of the CV profiles were examined for both PVP–CaVO and CaVO and were recorded at a scan rate of 0.2 mV s−1 (Fig. S8, ESI†). The CV curves for both materials exhibit an excellent overlap across the three cycles, indicating high electrochemical reversibility. Noteworthily, the CV profiles of both PVP–CaVO and CaVO display the same two major redox pairs (relative to Zn2+/Zn) at 0.2 mV s−1, with PVP–CaVO showing a slight shift compared to CaVO, further confirming the hybrid V4+/V5+ valence states. The consecutive redox peaks observed in these electrodes suggest a multi-step Zn2+ insertion/extraction process accompanied by vanadium valence changes. Importantly, the redox peaks of PVP–CaVO are closer together and exhibit broader reaction platforms compared to those of CaVO, indicating reduced polarization in PVP–CaVO with hybrid V4+/V5+ valence states.46 This reduced polarization is a direct consequence of the enhanced electronic and ionic conductivities facilitated by the PVP intercalation and the hybrid valence states, which collectively contribute to the superior electrochemical performance of PVP–CaVO. Furthermore, the CV curve of PVP–CaVO exhibits a significantly larger integrated peak area compared to that of CaVO at an identical scan rate, demonstrating its superior charge storage capability and higher specific capacity. Fig. 4b presents the GCD curves of both materials at 0.5 A g−1. The sloping discharge plateaus observed in both PVP–CaVO and CaVO electrodes are the characteristic of Zn2+ insertion behavior. Notably, PVP–CaVO delivers a substantially higher initial specific capacity than CaVO under identical testing conditions. The excellent reversibility of PVP–CaVO is further corroborated by the close correspondence between the charge–discharge plateau voltages in its GCD curves (Fig. S9a, ESI†) and the redox peak positions in the CV curves. The detailed analysis of the rate performance reveals that PVP–CaVO achieves discharge specific capacities of 323, 284, 230, 207, 197, and 169 mA h g−1 at progressively increasing current densities. In contrast, CaVO exhibits significantly lower capacities of 255, 226, 172, 147, 134, and 120 mA h g−1 under the identical conditions (Fig. S9b, ESI†). The more pronounced IR drop observed in CaVO, primarily attributed to zinc anode passivation, further highlights its inferior reversibility compared to PVP–CaVO. This performance enhancement in PVP–CaVO can be directly attributed to the successful PVP incorporation and the improved redox reversibility enabled by the multi-valent V4+/V5+ system. Additional evidence supporting this conclusion is furnished by the first four GCD cycles of both materials at 0.5 A g−1 (Fig. S10, ESI†), where PVP–CaVO demonstrates a significantly higher capacity (≈323 mA h g−1) compared to CaVO (≈255 mA h g−1). To explore further into the electrochemical kinetics and interfacial characteristics of the materials, we conducted electrochemical impedance spectroscopy (EIS) and its equivalent circuit diagram analyses on both PVP–CaVO and CaVO, as illustrated in Fig. 4c. The semicircle is attributed to the charge transfer resistance (Rct) and the diagonal line is attributed to Zn2+ diffusion into the electrode; a comparative analysis reveals that PVP–CaVO exhibits a significantly smaller semicircular diameter in the high-frequency range, as opposed to CaVO, suggesting enhanced ion diffusion kinetics and reduced charge transfer resistance. Quantitative fitting results demonstrate that the Rct values for PVP–CaVO and CaVO are 113.3 Ω and 326.9 Ω, respectively, confirming the superior electrochemical activity, faster ion diffusion capability, and higher conductivity of PVP–CaVO. The rate performance of PVP–CaVO and CaVO, as illustrated in Fig. 4d, further corroborates these findings. PVP–CaVO delivers a significantly higher average discharge specific capacity over diverse current densities (0.5 to 10 A g−1) compared to CaVO, underscoring its superior charge storage capability. Moreover, the enduring cycling stability of PVP–CaVO, evaluated at 10 A g−1 (Fig. 4e), demonstrates exceptional performance, retaining nearly 80% of its original capacity following 2700 cycles. In stark comparison, CaVO exhibits a significantly lower capacity retention of merely 66% following 2100 cycles, with a final discharge specific capacity of 120 mA h g−1. The remarkable electrochemical performance of PVP–CaVO can be attributed to two key factors: (1) the structural stabilization and enhanced ion diffusion pathways provided by the intercalated organic compound, and (2) the improved redox kinetics enabled by the hybrid V4+/V5+ valence states. The combined synergistic effects are conducive to the material's impressive reversible capacity, outstanding rate performance, and exceptional cycling stability, making PVP–CaVO a greatly promising candidate for the cathode material in advanced zinc-ion batteries. Fig. 4f summarizes a plot illustrating the correlation between specific capacity and current density. The outstanding performance of this work surpasses that of many other previously reported aqueous zinc-ion battery cathode materials, including D-MoS2-O, Na5V12O32, HNaV6O16·4H2O,  , PA-VOP, H2V3O8, and Na2V6O16·1.63H2O. Table S1 (ESI†) provides the relevant references and detailed data. Additionally, as shown in Fig. S11 (ESI†), the PVP–CaVO battery exhibits an energy density of 239 W h kg−1 at a power density of 370 W kg−1 with high energy density, and it sustains a high energy density of 116 W h kg−1 even when the power density increases to 6.4 kW kg−1, outperforming CaVO (184 W h kg−1 at 360 W kg−1 and 73 W h kg−1 at 6.2 kW kg−1).
, PA-VOP, H2V3O8, and Na2V6O16·1.63H2O. Table S1 (ESI†) provides the relevant references and detailed data. Additionally, as shown in Fig. S11 (ESI†), the PVP–CaVO battery exhibits an energy density of 239 W h kg−1 at a power density of 370 W kg−1 with high energy density, and it sustains a high energy density of 116 W h kg−1 even when the power density increases to 6.4 kW kg−1, outperforming CaVO (184 W h kg−1 at 360 W kg−1 and 73 W h kg−1 at 6.2 kW kg−1).
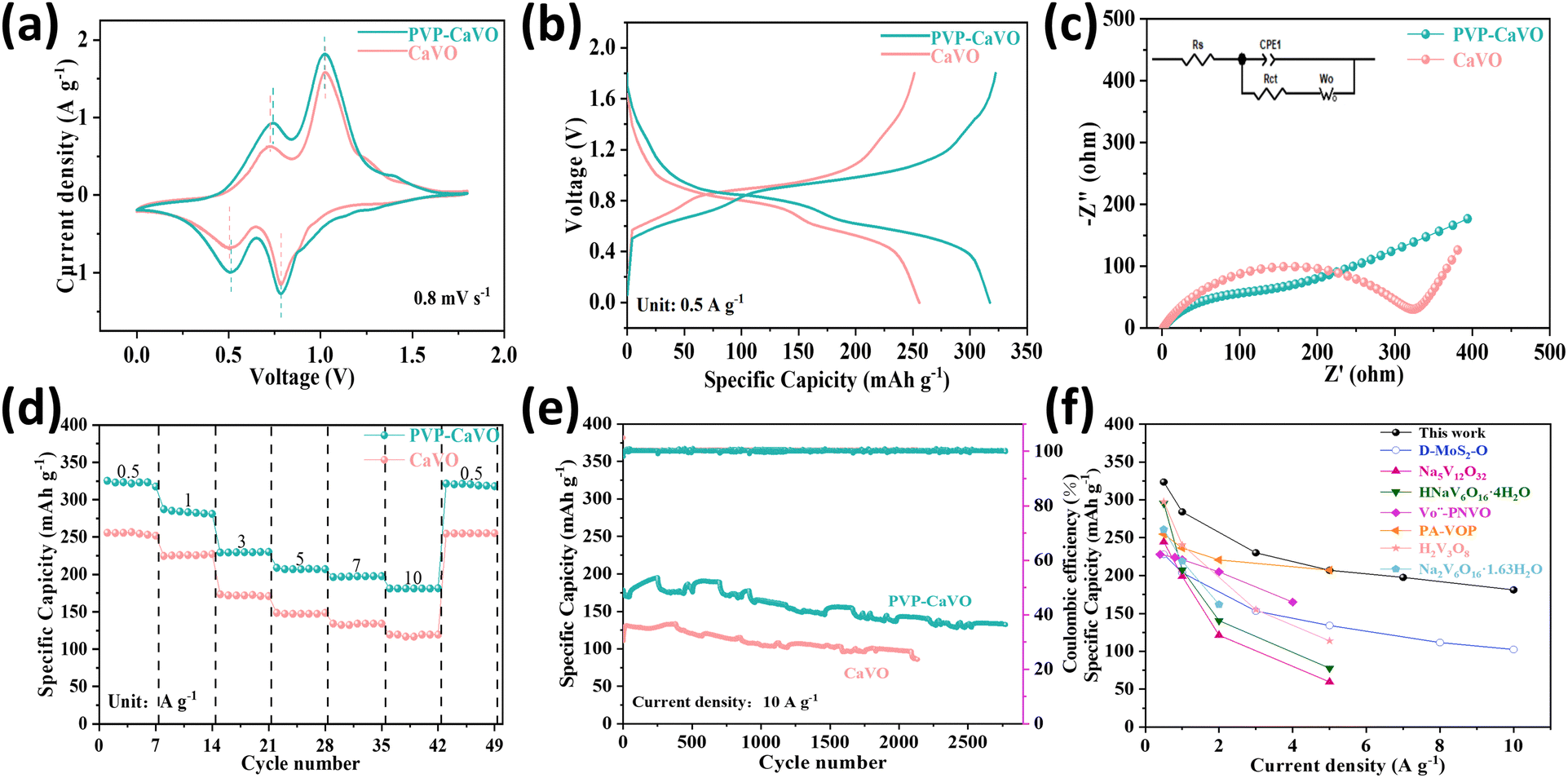 | ||
| Fig. 4 (a) CV curves of CaVO and PVP–CaVO at 0.8 mV s−1. (b) GCD curves of CaVO and PVP–CaVO at 0.5 A g−1. (c) Comparison of the Nyquist plots of CaVO and PVP–CaVO before cycling. (d) Rate performance comparison of CaVO and PVP–CaVO. (e) Cycling performance comparison of CaVO and PVP–CaVO. (f) Comparison of the discharge specific capacity of PVP–CaVO as a cathode with other reported cathodes used in aqueous zinc-ion batteries. | ||
To explain the excellent electrochemical performance exhibited by the PVP–CaVO electrode, we conducted multi-scan cyclic voltammetry (CV) measurements to investigate the kinetics of the electrochemical reaction. Fig. 5a and b shows the CV curves of the PVP–CaVO cathode within the scan rate range of 0.2 to 1.0 mV s−1. Even at varying scan rates, the CV curves generally retain a similar shape, with the reduction peaks and oxidation peaks shifting towards the low-voltage and high-voltage regions, respectively. According to previous literature reports, the scan rate (v) and peak current (i) satisfy a power-law relationship, as shown in the following equation:47
| i = avb | (1) |
|
log(i) = blog(v) + log(a)
| (2) |
 | ||
| Fig. 5 (a) CV profiles of PVP–CaVO. (b) Correlation between log(i) and log(v) corresponding to the redox peaks of the PVP–CaVO electrode. (c) Capacitance contribution at different scan rates. (d) Capacitive contribution at 0.8 mV s−1 of PVP–CaVO. (e) GITT curves of CaVO and PVP–CaVO. (f) Zn2+ diffusion coefficient (DZn2+) of CaVO and PVP–CaVO. | ||
In the equation, a and b are the variable parameters; for pseudocapacitive reactions, b = 1.0. A b-value closer to 1 indicates a stronger Faraday pseudocapacitive control. The PVP–CaVO electrode's peaks 1, 2, 3, and 4, respectively, exhibit b-values of 0.98, 0.81, 0.87, and 0.75. However, as shown in Fig. S12 and S13 (ESI†), the CaVO electrode's peaks 1, 2, 3, and 4 show b-values of 0.89, 0.65, 0.57, and 0.63, respectively. It is evident that the PVP–CaVO electrode exhibits a stronger pseudocapacitive control.48 Consequently, the capacity of the PVP–CaVO electrode is not only related to the ion intercalation and deintercalation processes but also contributes to pseudocapacitance, indicating that partial intercalation pseudocapacitance enhances the capacity of PVP–CaVO, thereby enabling rapid Zn2+ diffusion. To determine the exact capacitive contribution at different scan rates and distinguish between the surface capacitive effect (k1v) and the diffusion-controlled process (k2v1/2) of the current, as shown in the following equation:49
| i = k1v + k2v1/2 | (3) |
| i/v1/2 = k1v1/2 + k2 | (4) |
The capacitive contribution is illustrated in Fig. 5c and d, and we calculated the pseudocapacitive contributions of PVP–CaVO at different scanning rates in the range of 0.1–0.8 mV s−1. The results showed that the pseudocapacitive contributions of PVP–CaVO at different scanning rates were 62.16%, 66.06%, 74.72%, 78.33%, and 81.72%, respectively, suggesting that the scan rate is correlated with an increase in the capacitive contribution. In contrast, the corresponding values of CaVO were 39.88%, 50.66%, 58.13%, 62.60%, and 65.97% (Fig. S14 and S15, ESI†). Therefore, it is evident that the main contribution to the total capacity, crucial for achieving high-rate electrochemical performance, is capacitance. To further demonstrate the advantages of the PVP–CaVO cathode, the diffusion coefficient of zinc ions can be measured using GITT and calculated according to the following equation:50
 | (5) |
DZn2+ during discharge for PVP–CaVO is −10.28 to −9.83 cm2 s−1, and for CaVO, the range of lgDZn2+ is −10.67 to −9.89 cm2 s−1. This indicates that the Zn2+ diffusion coefficient in PVP–CaVO is greater than that in CaVO and the Zn2+ diffusion transfer rate of PVP–CaVO is faster, together with a smaller energy barrier. Obviously, the DZn2+ of PVP–CaVO is larger than that of CaVO (Fig. 5f), indicating that Zn2+ intercalation and deintercalation are faster in PVP–CaVO. This suggests that the successful doping of PVP leads to increased interlayer spacing, and the presence of multiple valence states V4+/V5+ greatly promotes ion diffusion rates, resulting in an increased D value for Zn2+. The results show that PVP–CaVO has lower charge transfer resistance and excellent Zn2+ diffusion rates, which reflect the improved rate performance and increased storage capacity of the PVP–CaVO battery. It exhibits advantages such as multiple active sites caused by multiple valence states and high kinetics due to intercalation.
To elucidate the transformation in the structure and the mechanism for Zn2+ storage within the PVP–CaVO material during electrochemical cycling, we performed a comprehensive suite of ex situ characterization studies under various discharge/charge conditions corresponding to reversible redox reactions. Ex situ XRD analysis was applied to monitor the phase transitions and structural changes in PVP–CaVO throughout the charge and discharge processes (Fig. 6a–c). During discharge to 0.3 V, a distinct (001) diffraction peak manifested around 8.0° (Fig. 6b), which subsequently exhibited a reversible shift during the charging process. Detailed examination of the (001) peak evolution (Fig. 6c) revealed a systematic shift toward lower angles during discharge, followed by a gradual return to higher angles during charging, ultimately recovering to its initial position. This reversible peak shift can be attributed to the dynamic variation in the V4+/V5+ ratio and the formation/release of interlayer V–V bonds, which modulate the interlayer spacing during Zn2+ insertion/extraction.41 The complete recovery of the (001) peak position demonstrates the exceptional structural reversibility of the PVP–CaVO electrode during electrochemical cycling. Concurrently, the appearance of novel diffraction peaks at 12.29°, 30.10°, and 36.46° indicates the formation of a secondary phase, Zn3(OH)2V2O7·H2O (PDF#50-0570, denoted as ZnVO). This phase formation results from the complex interplay between Zn2+ and oxygen atoms, leading to structural rearrangement within the vanadium oxide layers. The electrostatic forces acting between Zn2+ and the vanadium oxide structural arrangement cause a concomitant reduction in lattice spacing. Similar structural transformations are documented in other vanadium-based cathode materials, where the interaction between electrolyte water molecules and vanadium oxide layers enhances their binding energy, thereby promoting ZnVO formation on the electrode surface.51 Additionally, the presence of weaker diffraction peaks near 33.0° suggests the generation of additional by-products, Znx(OH)y(CF3SO3)2x−y·nH2O (BZS), which originates from the simultaneous insertion/extraction of H+ during cycling.52,53 The formation of these secondary phases can be rationalized as follows: ZnVO results from the increased interlayer binding energy induced by Zn2+ insertion, while BZS formation is driven by the co-insertion of H+, causing the generation of OH−, CF3SO32−, Zn2+, and crystalline water.50 These observations provide compelling evidence pointing to the greatly reversible Zn2+ storage mechanism and the synergistic co-insertion of Zn2+ and H+ in the PVP–CaVO host material.
 | ||
| Fig. 6 (a) Discharge/charge curves of PVP–CaVO in AZIBs. (b) Ex situ XRD patterns. (c) Enlargements of patterns. (d) XPS spectra of Zn 2p. (e) XPS spectra of V 2p. (f) Schematic illustration of the Zn2+ storage mechanism of PVP–CaVO. | ||
The ex situ XPS measurements at various charge/discharge states are conducted. Fig. S16a (ESI†) presents the survey XPS spectra of PVP–CaVO in three distinct states, revealing characteristic peaks corresponding to V 2p, Zn 2p, O 1s, and C 1s, along with notable variations in peak intensities across different electrochemical states. The Zn 2p spectrum (Fig. 6d) demonstrates the dynamic evolution of Zn2+ during cycling. In the pristine stage, no detectable Zn 2p indication is observed, confirming the absence of Zn2+ in the initial material. Upon discharge to 0 V, distinct Zn 2p3/2 peaks at 1022.4 eV and Zn 2p1/2 peaks at 1045.5 eV emerge, revealing the successful insertion of Zn2+. During the subsequent charging process, the Zn 2p peak intensity gradually diminishes, reflecting the extraction of Zn2+. Notably, a slight shift in the Zn 2p peak position observed at the fully charged state suggests the presence of residual Zn2+ on the cathode surface, which can be ascribed to incomplete extraction and the generation of ZnVO by-products. The V 2p spectrum (Fig. 6e) reveals the reversible redox behavior of vanadium during cycling. Compared to the pristine state, discharge to 0 V induces the incomplete diminution of V5+ to V4+, accompanied by the emergence of a new peak at 515.4 eV, corresponding to the further reduction of V4+ to V3+ during Zn2+ intercalation.54 Upon charging to 1.8 V, the V3+ peak completely disappears, and the V5+/V4+ peaks return to their original states, illustrating the excellent reversibility of vanadium redox reactions, consistent with the Zn 2p XPS results. The O 1s spectrum (Fig. S16b, ESI†) exhibits three distinctive characteristic peaks, 530.1 eV for O2−, 531.6 eV for OH−, and 532.7 eV for H2O.27 The increased intensity ratio of H2O and OH− peaks during discharge provides additional evidence for the co-insertion of Zn2+/H+ ions and the subsequent generation of Zn3(OH)2V2O7·2H2O.55 On the basis of the complete ex situ XRD and XPS analyses, we propose the Zn2+ storage mechanism in PVP–CaVO, as illustrated in Fig. 6f. The material demonstrates excellent structural stability and reversible redox behavior during Zn2+ insertion/extraction. As shown in Fig. S17 (ESI†), two series-connected button cells can effectively power and sustain high-speed operation of a small fan, while three series-connected cells successfully illuminate an LED light requiring 3.8 V. These demonstrations not only validate the high power output capability of PVP–CaVO//Zn batteries but also highlight their promising prospects for practical utilization in energy storage technologies, paving the way for their widespread implementation in various electronic systems.
3. Conclusions
In conclusion, we have accomplished the successful development of PVP preintercalated Ca0.24V2O5·H2O (PVP–CaVO) nanorod composite materials through innovative engineering strategies that combine organic compounds as “pillar” and V4+/V5+ hybrid valence state modulation. The structural and compositional advantages of PVP–CaVO, including its stable layered structures and additional tetravalent vanadium ions generated through coordination environment modification, enable multi-step, multi-electron redox reactions of vanadium, resulting in exceptional electrochemical performance. Meanwhile, our comprehensive experimental analyses demonstrate that PVP preintercalation holds a pivotal position in regulating the ionic reaction kinetics, effectively mitigating severe structural collapse of vanadium and reducing the strong polarization effect of Zn2+ ions. The optimized PVP–CaVO cathode delivers an impressive specific capacity of 169 mA h g−1 at 10 A g−1, demonstrating high-rate capability and cycling stability. The reaction mechanism was investigated through detailed ex situ XRD and XPS investigations, and we have elucidated the Zn2+/H+ dual-insertion mechanism of the PVP–CaVO cathode, accompanied by reversible structural and morphological transformations during cycling. These findings offer new perspectives regarding the electrochemical behavior of vanadate-based cathode materials.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52071171 and 52202248), the Key Research Project of Department of Education of Liaoning Province (LJKZZ20220015), the Chunhui Program of the Ministry of Education of the People's Republic of China (202201135), the Liaoning University Youth Research Fund project (LDZDJB2302), the Fundamental Research Funds for Public Universities in Liaoning (LJKLJ202425), the Natural Science Foundation of Liaoning Province (2024-MS-007), the Natural Science Foundation of Liaoning Province (2024-MSLH-184), the Natural Science Foundation of Yingkou City (YKSCJH2024-025), the Fundamental Research Funds for Public Universities in Liaoning (LJ232410140033), and the Advanced Plan Project of Liaoning Province (2023JH2/1060014). T. M. acknowledged the Australian Research Council (ARC) through the future fellowship (FT210100298), the Discovery Project (DP220100603), the Linkage Project (LP210200504, LP220100088, and LP230200897) and the Industrial Transformation Research Hub (IH240100009) schemes, the Australian Government through the Cooperative Research Centres Projects (CRCPXIII000077), the Australian Renewable Energy Agency (ARENA) as part of ARENA's Transformative Research Accelerating Commercialisation Program (TM021), and European Commission's Australia-Spain Network for Innovation and Research Excellence (AuSpire).Notes and references
- C. Zhuang, S. Zhang, Z. Yu, J. Yang, Y. Sun, H. Wen, H. Wen, H. Li, B. Yin and T. Ma, Adv. Funct. Mater., 2025, 15, 2419351 Search PubMed.
- Q. Liu, Y. Ji, X. Yin, J. Li, Y. Liu, X. Hu and Z. Wen, Energy Storage Mater., 2022, 46, 384–393 Search PubMed.
- R. Ge, Y. Yang, H. Li, S. Zhang, Y. Hu, Y. Shi, J. Li, H. Fan, B. Yin and T. Ma, J. Power Sources, 2025, 640, 236691 CrossRef CAS.
- Y. Zhu, G. Liang, X. Cui, X. Liu, H. Zhong, C. Zhi and Y. Yang, Energy Environ. Sci., 2024, 17, 369–385 Search PubMed.
- Y. Tian, Y. An, C. Liu, S. Xiong, J. Feng and Y. Qian, Energy Storage Mater., 2021, 41, 343–353 CrossRef.
- Y. An, Y. Tian, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2021, 15, 11828–11842 CrossRef CAS PubMed.
- Z. Yuan, B. Yin, W. Mi, M. Liu and S. Zhang, Polymers, 2025, 17, 1003 CrossRef CAS PubMed.
- Z. Li and A. Robertson, Battery Energy, 2023, 2, 20220029 CrossRef CAS.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 Search PubMed.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- L. Yan, B. Liu, J. Hao, Y. Han, C. Zhu, F. Liu, X. Zou, Y. Zhou and B. Xiang, J. Energy Chem., 2023, 82, 88–102 Search PubMed.
- F. Meng, Y. Ren, B. Ping, J. Huang, P. Li, X. Chen, N. Wang, H. Li, L. Zhang, S. Zhang, Y. Hu, Z. Yu, B. Yin and T. Ma, Adv. Mater., 2024, 45, 2408475 CrossRef PubMed.
- M. Liao, J. Wang, L. Ye, H. Sun, Y. Wen, C. Wang, X. Sun, B. Wang and H. Peng, Angew. Chem., Int. Ed., 2019, 59, 2273–2278 CrossRef PubMed.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- T. Gupta, A. Kim, S. Phadke, S. Biswas, T. Luong, B. Hertzberg, M. Chamoun, K. Evans-Lutterodt and D. Steingart, J. Power Sources, 2016, 305, 22–29 Search PubMed.
- K. Nam, S. Park, R. Reis, V. Dravid, H. Kim, C. Mirkin and J. Stoddart, Nat. Commun., 2019, 10, 4948 CrossRef PubMed.
- A. Liu, F. Wu, Y. Zhang, J. Zhou, Y. Zhou and M. Xie, Small, 2022, 18, 2201011 CrossRef CAS PubMed.
- X. Zhao, L. Mao, Q. Cheng, F. Liao, G. Yang, X. Lu and L. Chen, Energy Storage Mater., 2021, 38, 397–437 CrossRef.
- N. Bensalah and Y. Luna, Energy Technol., 2021, 9, 2100011 CrossRef CAS.
- F. Wan and Z. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS PubMed.
- A. Konarov, N. Voronina, J. Jo, Z. Bakenov, Y. Sun and S. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- H. Tang, Z. Peng, L. Wu, F. Xiong, C. Pei, Q. An and L. Mai, Electrochem. Energy Rev., 2018, 1, 169–199 CrossRef CAS.
- Y. Zhao, C. Han, J. Yang, J. Su, X. Xu, S. Li, L. Xu, R. Fang, H. Jiang, X. Zou, B. Song, L. Mai and Q. Zhang, Nano Lett., 2015, 15, 2180–2185 Search PubMed.
- M. Clites and E. Pomerantseva, Energy Storage Mater., 2018, 11, 30–37 CrossRef.
- Y. Hu, S. Zhang, Y. Ren, R. Ge, Y. Shi, X. Feng, H. Li, B. Jia, B. Yin and T. Ma, ChemElectroChem, 2024, 11, e202400504 CrossRef CAS.
- G. Zhang, T. Wu, H. Zhou, H. Jin, K. Liu, Y. Luo, H. Jiang, K. Huang, L. Huang and J. Zhou, ACS Energy Lett., 2021, 6, 2111–2120 CrossRef CAS.
- Y. Shi, B. Yin, Y. Sun, R. Ge, Y. Hu, J. Li, H. Li, S. Zhang and T. Ma, Chem. Commun., 2024, 60, 8597–8600 RSC.
- K. Zhu, T. Wu and K. Huang, ACS Nano, 2019, 13, 14447–14458 CrossRef CAS PubMed.
- L. Zhang, J. Hu, B. Zhang, J. Liu, H. Wan, L. Miao and J. Jiang, J. Mater. Chem. A, 2021, 9, 7631–7639 Search PubMed.
- F. Wan, Z. Hao, S. Wang, Y. Ni, J. Zhu, Z. Tie, S. Bi, Z. Niu and J. Chen, Adv. Mater., 2021, 33, 2102701 CrossRef CAS PubMed.
- D. Bin, W. Huo, Y. Yuan, J. Huang, Y. Liu, Y. Zhang, F. Dong, Y. Wang and Y. Xia, Chem, 2020, 6, 968–984 CAS.
- W. Li, C. Han, Q. Gu, S. Chou, J. Wang, H. Liu and S. Dou, Adv. Energy Mater., 2020, 10, 2001852 CrossRef CAS.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- S. Liu, H. Zhu, B. Zhang, G. Li, H. Zhu, Y. Ren, H. Geng, Y. Yang, Q. Liu and C. C. Li, Adv. Mater., 2020, 32, 2001113 Search PubMed.
- C. Wu, G. Zhao, S. Gong, N. Zhang and K. Sun, J. Mater. Chem. A, 2019, 7, 4426–4430 RSC.
- S. Ding, X. Dai, Z. Li, C. Wang, A. Meng, L. Wang, G. Li, J. Huang and S. Li, Energy Storage Mater., 2022, 47, 211–222 CrossRef.
- J. Wang, J. Wang, Y. Jiang, F. Xiong, S. Tan, F. Qiao, J. Chen, Q. An and L. Mai, Adv. Funct. Mater., 2022, 32, 2113030 CrossRef CAS.
- H. Huang, X. Xia, J. Yun, C. Huang, D. Li, B. Chen, Z. Yang and W. Zhang, Energy Storage Mater., 2022, 52, 473–484 CrossRef.
- L. Kong, M. Shao, Q. Xie, J. Liu and Y. Qian, J. Cryst. Growth, 2004, 260, 435–439 Search PubMed.
- S. Niragatti, S. Madhukar, J. Kim and K. Yoo, J. Power Sources, 2024, 613, 234852 Search PubMed.
- Y. Wang, M. Zhao, G. Gao, C. Zheng, D. He, C. Wang and G. Diao, Small Methods, 2023, 7, 2300606 CrossRef CAS PubMed.
- Y. Tong, X. Li, S. Su, J. Li, J. Fang, B. Liang, J. Hou and M. Luo, J. Colloid Interface Sci., 2022, 606, 645–653 CrossRef CAS PubMed.
- M. Sathiya, A. Prakash, K. Ramesha, J. Tarascon and A. Shukla, J. Am. Chem. Soc., 2011, 133, 16291–16299 Search PubMed.
- L. Lin, S. Tian, L. Fang, L. Chang, W. Liu and Q. Wang, J. Energy Storage, 2023, 72, 108356 Search PubMed.
- Y. Liu, Y. Zou, M. Guo, Z. Hui and L. Zhao, Chem. Eng. J., 2022, 433, 133528 CrossRef CAS.
- F. Liu, Z. Chen, G. Fang, Z. Wang, Y. Cai, B. Tang, J. Zhou and S. Liang, Nano-Micro Lett., 2019, 11, 25 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. Lowe, J. Kim, P. Taberna, S. Tolbert, H. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- B. Wu, C. Lu, F. Ye, L. Zhang, L. Jiang, Q. Liu, H. Dong, Z. Sun and L. Hu, Adv. Funct. Mater., 2021, 31, 2106816 CrossRef CAS.
- B. Liu, X. Shi, X. Lang, L. Gu, Z. Wen, M. Zhao and Q. Jiang, Nat. Commun., 2018, 9, 1375 Search PubMed.
- T. Zhou, X. Du and G. Gao, J. Energy Chem., 2024, 95, 9–19 Search PubMed.
- Y. Yang, Y. Tang, S. Liang, Z. Wu, G. Fang, X. Cao, C. Wang, T. Lin, A. Pan and J. Zhou, Nano Energy, 2019, 61, 617–625 CrossRef CAS.
- X. Liang, Y. Yang, W. Di, S. Chen, R. Zhang, J. Hu, D. Lin and Y. Huo, Chem. Eng. J., 2024, 495, 153255 CrossRef CAS.
- T. Wang, S. Li, X. Weng, L. Gao, Y. Yan, N. Zhang, X. Qu, L. Jiao and Y. Liu, Adv. Energy Mater., 2023, 13, 2204358 CrossRef CAS.
- Y. Du, X. Wang, Y. Zhang, H. Zhang, J. Man, K. Liu and J. Sun, Chem. Eng. J., 2022, 434, 134642 Search PubMed.
- Q. Wu, X. Li, H. Fan, J. Cao, X. Liu, M. Wei and L. Yang, J. Alloys Compd., 2023, 941, 168936 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/D5NH00236B |
| This journal is © The Royal Society of Chemistry 2025 |