
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic probing of protein adsorption on protein-based nanoparticles in dependence of the particle surface charge†
Ben
Otange‡
a,
Tobias
Katenkamp‡
b,
Hendrik
Böhler
b,
Michael
Rütten
b,
Laurin
Lang
b,
Florian
Schulz
 a,
Wolfgang J.
Parak
*ac and
Tobias
Beck
*bc
a,
Wolfgang J.
Parak
*ac and
Tobias
Beck
*bc
aInstitute for Nanostructure and Solid State Physics, University of Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: wolfgang.parak@uni-hamburg.de
bInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: tobias.beck@uni-hamburg.de
cHamburg Centre for Ultrafast Imaging, University of Hamburg, Luruper Chaussee 149, Hamburg, Germany
First published on 3rd December 2024
Abstract
Understanding protein adsorption on the surface of nanoparticles (NPs) is crucial for determining their behavior in biological environments. Early research in this field faced challenges in producing high-quality NPs. Advancements in NP fabrication now allow for precise modifications of specific parameters, such as zeta potential. However, creating a series of NPs where only one parameter, such as surface charge, is independently varied remains challenging due to concurrent alterations in other properties. In this study, we address these challenges using the ferritin nanocage (Ftn) as a model system for NPs. By modifying only a few amino acids on the outer surface of Ftn, we produce NPs with highly defined properties, focusing solely on variations in surface charge. This approach enables us to generate a controlled series of protein-based nanocages, labeled with fluorophores inside the nanocage. We utilize fluorescent correlation spectroscopy (FCS) to investigate the adsorption of bovine serum albumin (BSA) on these NPs, analyzing the dependence of BSA binding on surface charge. This fundamental study enhances our understanding of the driving forces behind protein adsorption, contributing valuable insights into the design of NPs for biomedical applications.
Introduction
Successful pharmacodynamic application of nanoparticles (NPs) as a carrier system for pharmaceutically active substances requires a deeper exploration of NPs interaction with body components.1 One of the most extensively researched interactions with body components, which is highly relevant in understanding the fate of NPs in biological settings, is protein adsorption on their surface.2 The protein corona that is formed, for example, when NPs are injected into the blood stream, alters the NPs’ surface chemistry, size, and surface charge, which then co-determines their resulting physicochemical properties.3–6 In the biological environment, the adsorbed components on the NP surface affect the NP biodistribution, uptake, and general cellular fate.7,8 Therefore, it is crucial to understand the formation and driving forces of protein adsorption.As there are many parameters that regulate protein adsorption on the surface of NPs, e.g. size, surface charge, shape, colloidal stability, etc., it is hard to identify which of the many parameters has paramount importance.9–11 In general, there is a convolution of the effect due to the different parameters, and the parameters themselves are entangled.12 Therefore, for a fundamental understanding, nanoparticle libraries are helpful, in which only one physicochemical parameter is systematically varied, while the other parameters are preserved.12
In the initial stages of research that aimed at exploring the impact of physicochemical parameters of NPs on biological systems, it was difficult to fabricate high-quality NPs, involving limited colloidal stability, broad size distributions, etc. In order to investigate the influence of certain physicochemical parameters of NPs towards their interaction with biological environments, proteins were used as model particles. For example, in order to study charge dependent particle uptake by cells via endocytosis, peptide/protein families were used, such as native, anionized and cationized ferritins,13–15 anionized and cationized hemeundecapeptides,16 or native, aminated and succinylated human serum albumins,17 which have similar properties but differ in charge. This allowed for studies investigating the charge-dependent uptake of these proteins as a function of their charge characteristics. Likewise, such protein families were also used for quantifying protein adsorption to other particles for example native, aminated and succinylated human serum albumin,17 as their role on particle growth was investigated, for example avidin, streptavidin, and neutravidin, or aminated and succinylated bovine serum albumin, or aminated and succinylated catalase.18
Today, the fabrication of highly defined NPs is readily feasible,19 along with the modification of specific parameters, such as zeta potential, which is the electric potential on the outer surface of particles. However, when producing a series of surface coatings for such particles, typically also other parameters of the nanoparticles are altered simultaneously, together with the zeta potential. For example, colloidal stability of charge-stabilized nanoparticles depends on their charge, and in case the absolute surface charge of such NPs is reduced, also their colloidal stability goes down.20 It is then not possible to relate any change in the interaction of the NPs with their environment simply to changes in charge, as in fact it might be also related to the loss of colloidal stability (which in turn increased the effective size of the NPs). Consequently, it remains exceedingly challenging to create a series of similar NPs, in which only a single parameter is independently varied without affecting others.
In this study, we return to study protein adsorption on NPs in dependence of their surface charge.21,22 In order to make a NP series in which surface charge is varied, but other parameters, such as size, remain constant, we are using protein families, as described above, for the composition of the NPs. We use protein cages, namely ferritin (Ftn), as a model system for NPs: the outer surface can readily be modified, while fluorophores for tracking can be encapsulated into the cage, not interfering with the outside characteristics. By creating a series of protein-based NPs through the modification of only a few amino acids on the outer surface, NPs with very defined properties are produced. Due to their protein nature, the virtually sole parameter that is purposefully altered is the surface charge of the particles. Because the protein backbone remains largely intact, variations in other parameters are minimized. This methodology enables us to develop a controlled series of protein-based nanocages with different outer surface charges, labeled with fluorophores on the inside without changing the outer surface charge. Fluorescent correlation spectroscopy (FCS) can be employed to assess the size of proteins adsorbed to protein cage particles, thereby allowing for the determination of the binding affinity of these proteins in relation to the surface charge of the protein cage particles.23–25
In FCS, the signal comes from the fluorescent NPs (here the protein cages). Unbound excess proteins do not contribute to the signal and thus measurements can be performed in situ without the necessity to remove unbound proteins. A comparison of the results obtained with FCS for quantifying protein adsorption with results from other techniques is given in Hühn et al.26 For quantifying protein adsorption, bovine serum albumin (BSA), the most abundant blood protein, was used as model protein in this study.27 It shares 75.6% sequence homology with its human counterpart human serum albumin (HSA), making it an excellent subject for investigation. BSA consists of 583 amino acid residues and has a molecular weight of approximately 66 kDa.28,29 This study systematically examines the adsorption of BSA on protein cage particles, with a focus on the influence of surface charge on the adsorption.
Experimental section
Materials and reagents
Tris(hydoxymethyl)aminomethane buffer (Tris, 99.9%, Carl Roth), sodium chloride (NaCl, 99.5%, Carl Roth), 2-(N-morpholino)ethanesulfonic acid monohydrate (MES, 99%, Carl Roth), hydrochloric acid (HCl, 37%, Fisher Scientific), Rhodamin6G (Rho6G, ATTO-TEC), Rhodamine 6G (Lot. 80K1967, EC no. 213-584-9, Sigma), water free dimethyl sulfoxide (DMSO, 99.7%, Acros Organics), thioglycolic acid (TGA, 98%, Sigma-Aldrich), ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA, PanReac Applichem), tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl, Carl Roth), absolute ethanol (VWR chemicals). For production and purification of the Ftn variants, the following materials and reagents were used: calcium-competent E. coli BL21-gold (DE3) cells (Agilent), plasmid solutions (GenScript Biotech (Netherlands) B.V.), lysogeny broth medium (LB, Luria/Miller, Carl Roth), terrific broth medium (TB, Carl Roth), ampicillin sodium salt (PanReac Applichem), isopropyl β-D-1-thiogalactopyranoside (IPTG, 99%, Carl Roth), RNAse A (PanReac Applichem), bovine serum albumin (BSA, Cas number 9048-46-8 Sigma Aldrich).All chemicals were procured from commercial suppliers and were utilized without additional purification. All solutions were prepared using ultrapure water (Purelab Flex 2 system, resistivity of 18.2 MΩ cm) and analytical grade reagents were employed whenever feasible, unless stated otherwise.
Production and purification of different Ftn variants
The negatively charged variants Ftnneg-1C, HF-1C (wild-type ferritin) and Ftnneg-m8-1C as well as the positively charged variants Ftnpos-1C, Ftnpos-m4-1C and Ftnpos-A1-1C were produced and purified using a similar protocol as described before.34,42Fluorophore labelling
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and washed once with 15 mL of SEC buffer (50 mM Tris, pH 7.5, 1 M NaCl).
000 Da and washed once with 15 mL of SEC buffer (50 mM Tris, pH 7.5, 1 M NaCl).
Fluorescence correlation spectroscopy (FCS) measurements
The differential adsorption of bovine serum albumin (BSA) onto electrostatically differently charged protein nanocages loaded with the fluorescent dye Rhodamine 6G (Rho6G) was investigated using a confocal light scanning microscope (CLSM) and fluorescence correlation spectroscopy (FCS). First the fluorescent dye Rho6G with known diffusion coefficient was used to calibrate the confocal volume by measuring the diffusion time. Then, a solution of Rho6G, and subsequently the protein nanocages loaded with the fluorescent dye, were prepared in deionized water and introduced into the setup. A laser was focused on the sample, and the fluctuating fluorescence intensity of diffusing molecules was recorded. The measured values were used to determine the lateral and axial radii of the confocal volume. Additionally, the data were analyzed through software-assisted autocorrelation to determine the diffusion time and diffusion coefficient. For the actual measurement, the nanoparticles were mixed with varying concentrations of BSA and incubated to assess the effect of protein concentration on diffusion. The hydrodynamic radii were calculated from the diffusion coefficients using the Stokes–Einstein equation, considering the medium's viscosity.Results and discussion
As model system, we chose the protein cage Ftn, which is the major iron storage protein among all living organisms.30 This highly symmetrical and spherical protein cage has an outer diameter of 12 nm and the cavity has a diameter of 8 nm. It consists of 24 subunits with a molecular mass of around 500 kDa. The octahedral symmetry (432) of Ftn is based on the 4-fold, 3-fold and 2-fold symmetry axes present between the subunits.31–33 At the interfaces between subunits, pores are formed: through the 3-fold and 4-fold channels, small molecules and ions can be transported into the cavity.34Due to its high symmetry, a small change in the Ftn subunit through mutation leads to a large change of the surface charge, because the mutation is taking place in all 24 subunits. Therefore, a small number of mutations within one subunit of Ftn can induce significant alterations in the surface charge, profoundly impacting interactions with other biomolecules, such as other proteins. In this study, six different Ftn variants based on the human heavy chain wild type ferritin (HF) were examined with respect to their surface charge profiles (Fig. 1). Each variant was engineered to introduce specific mutations within the protein sequence, resulting in alterations in surface charge properties.
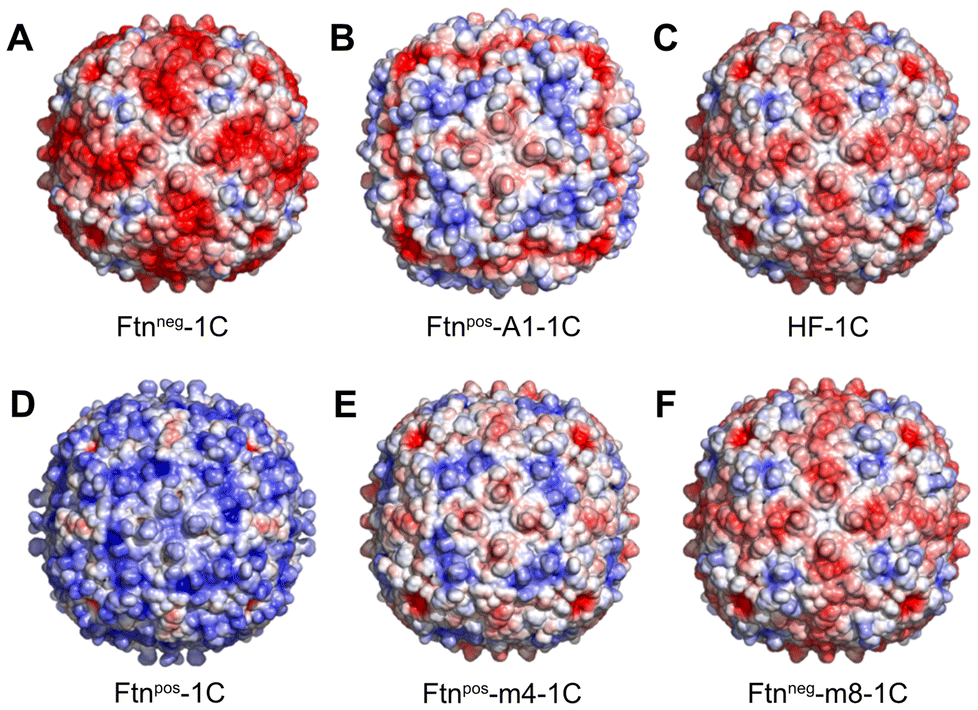 | ||
| Fig. 1 Ftn variants with different surface charges. The calculated electrostatic surface potential is shown (red: −5 kT e−1, blue: +5 kT e−1; see ESI methods†). Each variant contains a cysteine per subunit (1C). The variants are named as follows: A Ftnneg-1C, B Ftnpos-A1-1C, C HF-1C (human heavy chain wild type Ftn), D Ftnpos-1C, E Ftnpos-m4-1C and F Ftnneg-m8-1C. | ||
The calculated electrostatic potential, as depicted in Fig. 1, reveals a wide range of surface charge distributions among the Ftn variants.
Notably, the surface charge ranged from strongly negative in Ftnneg-1C to strongly positive in Ftnpos-1C (Fig. 1A and D). Additionally, the wild type (HF-1C; Fig. 1C) and its variant Ftnneg-m8-1C (Fig. 1F) exhibited negative surface charges, albeit slightly lower than Ftnneg-1C. Conversely, variants Ftnpos-m4-1C (Fig. 1E) and Ftnpos-A1-1C (Fig. 1B) displayed slightly lower positive surface charges compared to Ftnpos-1C. These variants exhibited distinct charge density distributions attributed to the specific mutations introduced.
Circular dichroism (CD) spectroscopy is a powerful tool for analyzing the secondary structure of proteins. Alterations in the secondary structure can be readily identified by variations in the spectral profile, as different secondary structure types produce distinct characteristic curves. In CD spectroscopy, all ferritin variants exhibited a characteristic spectrum indicative of a high α-helical content, as evidenced by pronounced minima at 210 nm and 222 nm (Fig. S4†). Moreover, negatively stained TEM images verified that all ferritin variants possess identical shape and size, with a consistent diameter of 12 nm, observed across all variants (Fig. S5 and S6†). These findings indicate that the introduced mutations neither affect the secondary structure nor alter the overall size and shape of the ferritin variants.
To enable NP detection, which is needed for the FCS measurements, without perturbing the outer surface charge or protein adsorption dynamics, we exploited the inner cavity of the Ftn cage. Here, we introduced one single amino acid cysteine per subunit as a chemical handle for further functionalization, replacing a lysine residue at position 53 (K53C) in the Ftn subunit. Subsequent mutations targeted the remaining native cysteine residues, replacing them with alanine, lysine, or glutamic acid residues, to avoid unwanted functionalization at these positions. The exact DNA and protein sequences for each variant are detailed in ESI Table S1,† along with a comprehensive list of the mutations carried out, based on the sequence of the human heavy chain wild type ferritin.
Several studies have investigated the adsorption of BSA onto various types of nanoparticles, including silicon dioxide, ZnO, CeO2, TiO2, polystyrene and Ag particles.35–38 In this study, protein adsorption onto the differently charged protein cages (Fig. 1) was examined using fluorescent correlation spectroscopy (FCS). By measuring the resulting hydrodynamic radius of the protein cages as exposed to different concentrations of BSA, the apparent dissociation coefficient Kd can be determined.23 Towards this goal, we functionalized the introduced cysteine residues on the inner surface with fluorophores, namely rhodamine 6G (Rho6G), through maleimide-cysteine coupling.39 We attached up to 24 fluorophore molecules per protein cage, enabling high fluorescent labeling of the inner cavity of the cages.
For the functionalization, as shown in Fig. 2, the protein cages were disassembled into individual subunits at pH 2 and maleimide functionalized fluorophores were added to the disassembled subunits.
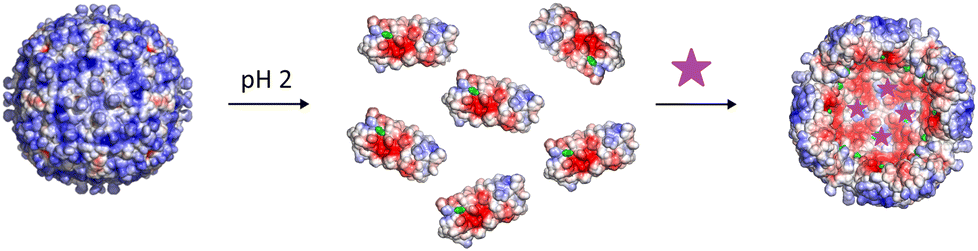 | ||
| Fig. 2 Schematic illustration of Ftnpos-1C fluorophore labelling. The ferritin cage is first disassembled at pH 2 into its subunits. Fluorophores (purple) are added to the disassembled Ftnpos-1C for protein functionalization (cysteine residues are outlined in green). After reassembly, the fluorophores are incorporated into the cavity of the protein cage. | ||
Via maleimide–thiol coupling, the fluorophores were covalently bound to the cysteine residues. After reassembly of the protein cage at neutral pH, the fluorophores were encapsulated and the inner cavity of the protein cage is labelled with fluorophores. The coupling of Rho6G with the subunits and the subsequent reassembly to the protein cage was initially confirmed by means of absorbance measurements during size exclusion chromatography. The absorption of Rho6G in the protein cage was detected at 507 and 536 nm (ESI Fig. S3†). The functionalization with fluorophores to the inner cavity of the protein cages was further confirmed by UV-Vis absorption and fluorescence measurements (ESI Fig. S7–S9†) and mass spectrometry (ESI Table S2†).
The negatively charged variants were characterized with electrospray ionization mass spectrometry (ESI-MS) and the positively charged variants with matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS). The mass of the subunit with coupled Rho6G could thus be detected (ESI Table S2†). The mass spectrometry data showed that most of the cysteine residues were coupled with Rho6G. The degree of labeling was also reproducible with the same absorption ratios from fluorophore to protein cage.
Interestingly, one variant, Ftnpos-m4-1C, exhibited challenges in fluorophore coupling, attributed to iron formation and low yield during protein production. An attempt was made to remove residual iron using thioglycolic acid (TGA) and ethylenediaminetetraacetic acid (EDTA), following a modified protocol by Moglia et al. (see ESI methods†).40 Fluorophore coupling worked to a certain extent.
However, MALDI-MS revealed that TGA had formed disulfide bridges to some of the cysteine residues. In order to achieve a higher fluorophore labeling, the Rho6G coupling reaction was repeated with tris(2-carboxyethyl)phosphine (TCEP) to cleave residual disulfide bonds.41 Based on absorption measurements and mass spectrometry results, there were less fluorophores per protein cage in this variant than in the others. A higher concentration of TCEP presumably led to the complete removal of the disulfide bridges between cysteine residues and TGA. Nevertheless, FCS measurements were also done with this protein variant.
After purification, positively and negatively charged cages are usually present in buffers with different salt concentrations. However, the FCS measurements were carried out in ultrapure water in order to achieve comparability of all protein variants labeled with fluorophores. pH measurements showed that a highly diluted protein Rho6G sample had a pH of 5.7.
Through FCS measurements with fluorophore-labeled protein cages, it was possible to measure and calculate the hydrodynamic radius of these cages (i.e. without having BSA added). All 6 different Ftn variants had similar hydrodynamic radii, confirming that variations in surface charge did not affect the size of the protein cages, as also shown by CD spectroscopy and TEM (see above and Fig. S4–S6†). By adding BSA to the NPs, their hydrodynamic radii increased due the adsorption of protein onto the surface of the protein cages, driven by their surface charge properties (Fig. 3). Remarkably, it was found that different surface charges of the protein cages led to varying affinities for BSA adsorption. Fitting of the curves led to the dissociation constant Kd, which describes the BSA concentration at which half saturation of the protein cage surface with BSA is achieved (Table 1).42
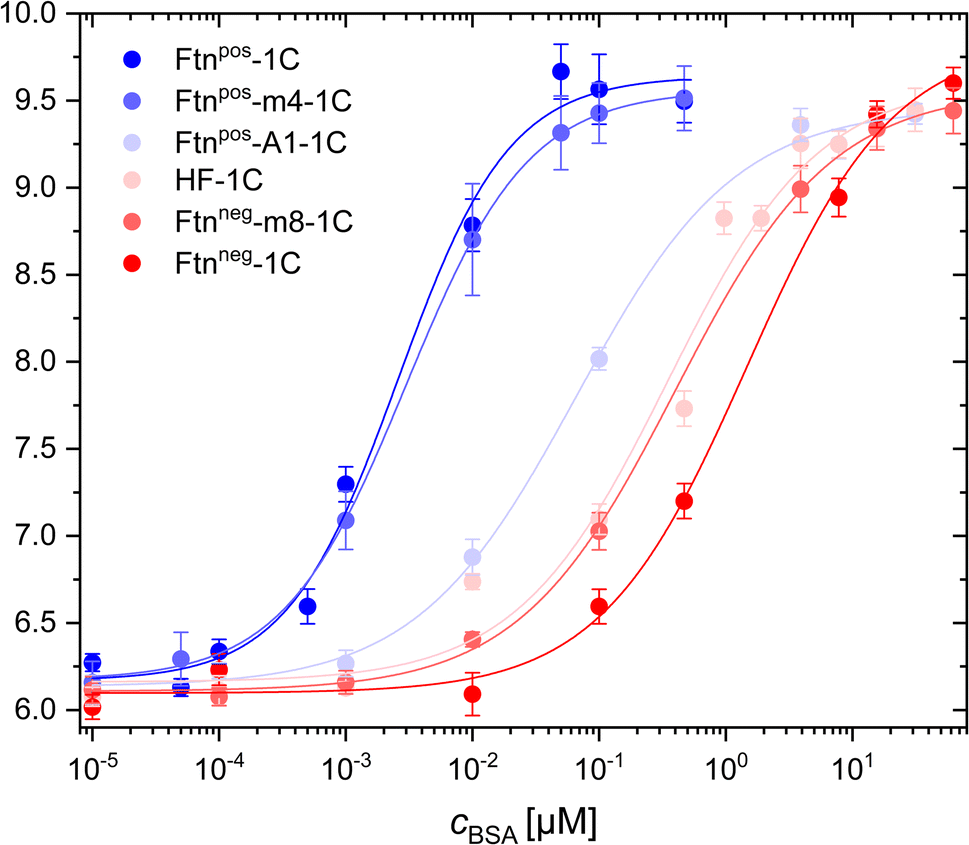 | ||
| Fig. 3 BSA adsorption on positively and negatively charged ferritin cages as measured with fluorescence correlation spectroscopy (FCS). The hydrodynamic radius rh of the ferritin cage/BSA conjugates is plotted versus the concentration cBSA of BSA that has been added to the ferritin cage solution. The hydrodynamic diameter at very low BSA concentrations (cBSA = 10−5 μM) corresponds to the hydrodynamic radius of the plain ferritin cages. The Kd values as extracted from these curves are listed in Table 1. | ||
| Protein variant | ζ [mV] | K d [μM] |
|---|---|---|
| Ftnpos-1C | 39.07 ± 0.52 | 0.00220 ± 0.00029 |
| Ftnpos-m4-1C | 27.54 ± 0.49 | 0.00313 ± 0.00051 |
| Ftnpos-A1-1C | 2.091 ± 0.053 | 0.0617 ± 0.0068 |
| HF-1C | −23.60 ± 1.1 | 0.303 ± 0.069 |
| Ftnneg-m8-1C | −23.26 ± 0.44 | 0.370 ± 0.084 |
| Ftnneg-1C | −37.78 ± 0.71 | 1.03 ± 0.25 |
In order to relate the BSA adsorption to the surface charge of the ferritin cages, in addition to the simulations shown in Fig. 1, also the zeta-potential ζ was measured in ultrapure water (pH 5.7), see Table 1.
The results revealed that the most positively charged protein cage (i.e. the one with the highest positive zeta-potential), Ftnpos-1C, exhibited the highest BSA adsorption, showing the largest hydrodynamic radius change at the lowest BSA concentrations (i.e. the smallest Kd value). The lowest BSA adsorption (i.e. the highest Kd value) was found for the most negatively charged protein cage Ftnneg-1C (i.e. the one with the most negative zeta-potential). Depending on the surface charge, the other variants fall between these two extremes. Interestingly, the variant Ftnpos-m4-1C showed an elution in ion exchange chromatography at higher salt concentration/conductivity (ESI Fig. S2†), and also has a higher zeta potential (ζ = 27.54 ± 0.49 mV) than the same variant Ftnpos-m4 without the cysteine modifications (ζ = 18.43 ± 0.060 mV).43 Therefore, based on the surface charge, Ftnpos-m4-1C is closer to the variant Ftnpos-1C (Table 1), which can also be seen in the BSA adsorption measurements (Fig. 3). We noted some experimental particularity with this sample Ftnpos-m4-1C, as there was also some lower loading of fluorophores. However, the correlation function of the FCS measurement does not depend on the fluorescence intensity of single NPs (ESI FCS measurements†).
At a pH value of 5.7, which was present in all highly diluted protein samples, BSA is negatively charged. This explains the strong adsorption to the positively charged protein cage Ftnpos-1C. The more negatively charged the protein variants are, the weaker the adsorption is with increasing BSA concentration. This is somehow in contrast with previous work with similar methodology (i.e. FCS), where negatively and positively charged nanoparticles had similar Kd values for the adsorption of HSA.44 The Kd value obtained for the negatively charged nanoparticles corresponds to the Kd value obtained here for the most negatively charged protein cage. However, in contrast, for the most positively charged protein cage in the present study, the Kd value is around 3 orders of magnitude lower (cf.Table 1). In a study by Hühn et al., FCS measurements were carried out in phosphate buffered saline (PBS), where the pH value was higher compared with the present study in water. As BSA/HSA change their charge in dependence of pH, this may be a reason for the differences. In addition, in the study by Hühn et al., the hydrodynamic diameter of the negatively and positively charged nanoparticles were not the same, and also their colloidal stability in different media was not the same. In other words, not only the surface charge was varied, but also other physicochemical parameters.44 This is why in the present study with the protein cages, a better library for investigating surface charge dependence than in the past is used.
In order to obtain a more quantitative analysis, for all Ftn variants the Kd values were plotted against the zeta potential ζ (Fig. 4). While we are not aware of a rational formula which would relate both entities, as guide to the eye the Kd(ζ) curves were fitted with different functions that were found to describe the experimental values. First, the power law function Kd(ζ) = a0/ζn (n = 1; a0 was the fit parameter) was used to fit the experimental values (Fig. 4A). This led to a reasonable fit. As on a linear scale the low Kd values are not represented well, we displayed Kd on a logarithmic scale: log(Kd(ζ)) = a1 + a2·ζ (a1 and a2 are fit parameters), see Fig. 4B.
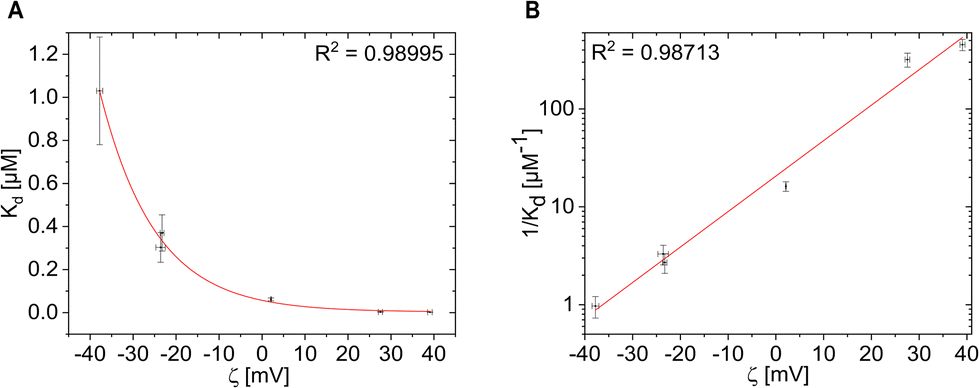 | ||
| Fig. 4 K d values plotted versus the zeta-potential ζ as obtained from Table 1. Data are displayed (A) in linear versus linear and (B) in logarithmic versus linear representation. Data were fitted with different functions and the quality of the fit is quantified by the coefficient of determination R2. | ||
The BSA affinity and the Kd value are overall in good agreement with the surface charge of the Ftn variants. As the zeta potential becomes more positive, the Kd value decreases (Fig. 4A) and the BSA affinity also increases (Fig. 4B). There is a large difference between the positively and negatively charged protein cages. The positively charged Ftn variants Ftnpos-1C (Fig. 1D) and Ftnpos-m4-1C (Fig. 1E) show a significantly higher BSA affinity than the negatively charged ones.
As shown in Fig. 4A, the more positive the surface charge, the lower the Kd value. For the variants Ftnpos-1C and Ftnpos-m4-1C, the Kd value approaches very small values close to 0, which shows a high affinity towards BSA. Such linear display on the other hand may be also missleading, as also for the small Kd values there is relation to the zeta potential, as can be seen in the logarithmic plot in Fig. 4B. For the negatively charged variants, the Kd value reach higher numbers, whereas Ftnneg-1C reaches a Kd value of around 1 μM, which is in the range of affinities reported for other negatively charged nanoparticles.45 A clear trend between the positively and negatively charged protein variants can be seen here.
There are many different ways to quantify protein adsorption to nanoparticles. In a recent review, several of such techniques and their interpretations are discussed.46 The main goal of the present work was to analyse the influence of charge on the adsorption of one model protein, BSA. For this, FCS is highly suited, as only one type of protein is involved. However, in general, FCS is not able to analyse the composition of the protein corona. Therefore, apart from the dependence of the Kd values from the surface charge, as detected in this work, also the composition of the protein corona could change in dependence of the surface charge, in case the NPs were incubated in mix of different proteins (such as present in blood). FCS cannot detect this composition, and other methods are required. A list of methods suitable for this are presented in a previous review.47
Conclusions and outlook
Through FCS analysis, the adsorption of BSA onto the outer surface of the protein cages was analysed, shedding light on the influence of surface charge on protein adsorption dynamics. This study highlights the utility of FCS in probing biomolecular interactions at the nanoscale level. The methodology allowed for precise quantitative measurements and characterization of protein adsorption processes, offering insights into biomolecular interactions essential for various biomedical and biotechnological applications. This study can be extended to other protein cage particles, such as Thermotoga maritima encapsulin, which possesses a larger diameter than ferritin and can also be labelled with fluorophores on the interior without altering the external surface.The results of this study are partly in disagreement with literature, including also our previous work.35 While the Kd values reported for the negatively charged protein cages are in general agreement with literature, in the present study there is much higher adsorption to the positively charged protein cages, which according to previous work was not expected to this degree.36 In general, in literature there is the trend that protein adsorption is increased for charged nanoparticle surfaces, regardless the sign of the charge.37,38 In the present work, however, protein adsorption increases the more positively charged the particle surfaces are, and here negatively charged surfaces show the lowest protein adsorption. This may come back to the entanglement of physicochemical parameters as already discussed above. In general, there are more negatively charged nanoparticles reported in literature than positively charged ones, and limited colloidal stability of some of the positively charged nanoparticles may be a reason for this. For analysing the direct influence of charge, therefore it is paramount that the positively charged nanoparticles have the same colloidal stability as the negatively charged ones (which not always is fully the case).35 In case nanoparticles are slightly agglomerated, differences in protein adsorption also might be due to the agglomeration, and not directly related to the charge.39 The protein cages are here a highly suitable system, as only few amino acids are exchanged and the similarity of the hydrodynamic radii of these particles (without having proteins added) shows that size is not affected by the charge variation. The “degree of manipulation” needed to vary charge is much lower than for other nanoparticles, in which the ligand structure has to be changed, highlighting again the suitability of the protein cages for systematic studies in which only one parameter is varied.
There is one additional difference to protein adsorption studies with other types of nanoparticles. In general, the more charged nanoparticles are, the higher their colloidal stability.40 For most charge-stabilized nanoparticles (e.g. ligand- or polymer-coated inorganic nanoparticles), therefore the absolute zeta potentials are mostly higher than in this study (30–40 mV).41 In the present study, charge was introduced by point mutations in the protein cages, and the charge-to-charge distance should be higher than in charged ligand- or polymer-coated nanoparticles. While having been investigated for decades now, protein corona formation at molecular level is still not completely quantified.
Author contributions
B. O. and T. K. carried out the experiments, supported by H. B., M. R., L. L. and F. S. W. J. P. and T. B. supervised the research and had the original idea for this work. B. O., T. K., W. J. P. and T. B. co-wrote the manuscript.Data availability
The data supporting this article including additional experimental data and details on the experimental procedures have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
BO was funded with a DAAD (Deutscher Akademischer Austauschdienst) PhD fellowship. This project was supported by the Cluster of Excellence ‘Advanced Imaging of Matter’ of the Deutsche Forschungsgemeinschaft (DFG) – EXC 2056 – project ID 390715994 (WJP, LL, TB) and the DFG graduate school ‘Nanohybrid’ (TK, HB, MR).References
- M. Haripriyaa and K. Suthindhiran, Future J. Pharm. Sci., 2023, 9, 113 CrossRef.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- B. Fadeel, N. Feliu, C. Vogt, A. M. Abdelmonem and W. J. Parak, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 111–129 CAS.
- I. Alberg, S. Kramer, M. Schinnerer, Q. Hu, C. Seidl, C. Leps, N. Drude, D. Mockel, C. Rijcken, T. Lammers, M. Diken, M. Maskos, S. Morsbach, K. Landfester, S. Tenzer, M. Barz and R. Zentel, Small, 2020, 16, e1907574 CrossRef.
- M. H. Akhter, H. Khalilullah, M. Gupta, M. A. Alfaleh, N. A. Alhakamy, Y. Riadi and S. Md, Biomedicines, 2021, 9, 1496 CrossRef CAS.
- M. Martinez-Negro, G. Gonzalez-Rubio, E. Aicart, K. Landfester, A. Guerrero-Martinez and E. Junquera, Adv. Colloid Interface Sci., 2021, 289, 102366 CrossRef CAS PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2015, 10, 221 CrossRef.
- G. Caracciolo, O. C. Farokhzad and M. Mahmoudi, Trends Biotechnol., 2017, 35, 257–264 CrossRef CAS.
- C. Ma, Y. Li, J. Li, L. Song, L. Chen, N. Zhao, X. Li, N. Chen, L. Long, J. Zhao, X. Hou, L. Ren and X. Yuan, J. Pharm. Anal., 2023, 13, 503–513 CrossRef.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernandez, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CAS.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- M. Xu, M. G. Soliman, X. Sun, B. Pelaz, N. Feliu, W. J. Parak and S. Liu, ACS Nano, 2018, 12, 10104–10113 CAS.
- L. Ghitescu and A. Fixman, J. Cell Biol., 1984, 99, 639–647 CAS.
- S. Mutsaers and J. Papadimitriou, J. Leukocyte Biol., 1988, 44, 17–20 CrossRef CAS PubMed.
- M. Williams, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6054–6058 CrossRef CAS.
- N. Ghinea and N. Simionescu, J. Cell Biol., 1985, 100, 606–612 CrossRef CAS PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS.
- I. Chakraborty, N. Feliu, S. Roy, K. Dawson and W. J. Parak, Bioconjugate Chem., 2018, 29, 1261–1265 CrossRef CAS PubMed.
- W. J. Parak, Science, 2011, 334, 1359–1360 CrossRef CAS PubMed.
- T. Pellegrino, S. Kudera, T. Liedl, A. Muñoz Javier, L. Manna and W. J. Parak, Small, 2005, 1, 48–63 CAS.
- A. Gessner, A. Lieske, B. R. Paulke and R. H. Müller, Eur. J. Pharm. Biopharm., 2002, 54, 165–170 CAS.
- M. P. Calatayud, B. Sanz, V. Raffa, C. Riggio, M. R. Ibarra and G. F. Goya, Biomaterials, 2014, 35, 6389–6399 CAS.
- P. Del Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 CAS.
- X. Jiang, S. Weise, M. Hafner, C. Röcker, F. Zhang, W. J. Parak and G. U. Nienhaus, J. R. Soc., Interface, 2010, 7, S5–S13 CAS.
- C. Röcker, M. Pötzl, F. Zhang, W. J. Parak and G. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577–580 CrossRef.
- J. Hühn, C. Fedeli, Q. Zhang, A. Masood, P. Del Pino, N. M. Khashab, E. Papini and W. J. Parak, Int. J. Biochem. Cell Biol., 2016, 75, 148–161 CrossRef PubMed.
- T. J. Peters, All About Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, 1995 Search PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- S. Ketrat, D. Japrung and P. Pongprayoon, J. Mol. Graphics Modell., 2020, 98, 107601 Search PubMed.
- G. Clegg, J. E. Fitton, P. M. Harrison and A. Treffry, Prog. Biophys. Mol. Biol., 1980, 36, 56–86 CAS.
- S. Chakraborti and P. Chakrabarti, Adv. Exp. Med. Biol., 2019, 1174, 313–329 CAS.
- P. D. Hempstead, S. J. Yewdall, A. R. Fernie, D. M. Lawson, P. J. Artymiuk, D. W. Rice, G. C. Ford and P. M. Harrison, J. Mol. Biol., 1997, 268, 424–448 CrossRef CAS.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CAS.
- T. Takahashi and S. Kuyucak, Biophys. J., 2003, 84, 2256–2263 CAS.
- B. E. Givens, N. D. Diklich, J. Fiegel and V. H. Grassian, Biointerphases, 2017, 12, 02D404 CrossRef PubMed.
- M. Bukackova and R. Marsalek, Biophys. Chem., 2020, 267, 106475 CrossRef CAS PubMed.
- G. Baier, C. Costa, A. Zeller, D. Baumann, C. Sayer, P. H. Araujo, V. Mailander, A. Musyanovych and K. Landfester, Macromol. Biosci., 2011, 11, 628–638 CrossRef CAS PubMed.
- A. Ravindran, A. Singh, A. M. Raichur, N. Chandrasekaran and A. Mukherjee, Colloids Surf., B, 2010, 76, 32–37 CAS.
- M. Budiarta, W. Xu, L. Schubert, M. Meledina, A. Meledin, D. Wöll, A. Pich and T. Beck, J. Colloid Interface Sci., 2021, 591, 451–462 CrossRef CAS PubMed.
- I. Moglia, M. Santiago, Á. Olivera-Nappa and M. Soler, J. Inorg. Biochem., 2018, 183, 184–190 CrossRef CAS.
- M. Budiarta, S. Roy, T. Katenkamp, N. Feliu and T. Beck, Small, 2023, 19, 2205606 CrossRef CAS.
- L. A. de Jong, D. R. Uges, J. P. Franke and R. Bischoff, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 829, 1–25 CrossRef CAS.
- L. Lang, H. Böhler, H. Wagler and T. Beck, Biomacromolecules, 2023, 25, 177–187 CrossRef PubMed.
- D. Hühn, K. Kantner, C. Geidel, S. Brandholt, I. De Cock, S. J. Soenen, P. Rivera_Gil, J.-M. Montenegro, K. Braeckmans, K. Müllen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253–3263 Search PubMed.
- J. Hühn, C. Fedeli, Q. Zhang, A. Masood, P. Del Pino, N. M. Khashab, E. Papini and W. J. Parak, Int. J. Biochem. Cell Biol., 2016, 75, 148–161 CrossRef PubMed.
- M. Barz, W. J. Parak and R. Zentel, Adv. Sci., 2024, 11, e2402935 Search PubMed.
- C. Carrillo-Carrion, M. Carril and W. J. Parak, Curr. Opin. Biotechnol., 2017, 46, 106–113 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details on experimental procedures and supporting tables and figures. See DOI: https://doi.org/10.1039/d4nr04069d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |