DOI:
10.1039/D4NR04725G
(Paper)
Nanoscale, 2025,
17, 2567-2576
Photogenerated charge carrier dynamics on Pt-loaded SrTiO3 nanoparticles studied via transient-absorption spectroscopy†
Received
12th November 2024
, Accepted 8th January 2025
First published on 20th January 2025
Abstract
Loading cocatalysts on semiconductor-based photocatalysts to create active reaction sites is a preferable method to enhance photocatalytic activity and a widely adopted strategy to achieve effective photocatalytic applications. Although theoretical calculations suggest that the broad density of states of noble metal cocatalysts, such as Pt, act as a recombination center, this has never been experimentally demonstrated. Herein, we employed pico–nano and nano–micro second transient absorption spectroscopy to investigate the often overlooked photogenerated holes, instead of the widely studied electrons on Pt- and Ni-loaded SrTiO3 to evaluate the effects of cocatalysts as a recombination center. It is demonstrated that Pt serves as the recombination center with no sacrificial agent; recombination can be suppressed by a hole scavenger, while recombination is not significant on Ni with localized density of states. It is also found that photo-generated holes in SrTiO3 tend to migrate to Pt within 400 ps, and photo-generated holes generated in the bulk gradually migrate to Pt cocatalysts in a micro-second regime.
1. Introduction
Photocatalysis has garnered significant attention as a green technology since it can utilize sunlight to convert ubiquitous molecules, such as water, carbon dioxide and nitrogen, into chemical energy.1–13 Despite extensive efforts, its efficiency still remains far from industrial demands, and its practical use has not been achieved yet. One approach to improving photocatalytic efficiency is loading “cocatalysts” on photocatalysts. Certain cocatalysts are known as rectifiers for efficient photocatalytic hydrogen evolution reactions (HERs) and oxygen evolution reactions (OERs). For example, Pt,14,15 Ni, NiO16–19 and CrOx/Rh20 are established cocatalysts for the HER, and IrO2,21,22 RuO2,21,23 CoPi (cobalt phosphate)24–27 and CoOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15,28,29 are frequently used as cocatalysts for the OER. There are two primary purposes of loading cocatalysts: (i) to reduce the overpotential required for the respective redox process and (ii) to inhibit the recombination of photogenerated charge carriers by securing and storing electrons or holes. While the former purpose has been verified using electrochemical measurements, there is limited experimental evidence to confirm the latter purpose with regard to carrier collection behavior.
15,28,29 are frequently used as cocatalysts for the OER. There are two primary purposes of loading cocatalysts: (i) to reduce the overpotential required for the respective redox process and (ii) to inhibit the recombination of photogenerated charge carriers by securing and storing electrons or holes. While the former purpose has been verified using electrochemical measurements, there is limited experimental evidence to confirm the latter purpose with regard to carrier collection behavior.
In the field of theoretical calculations, cocatalyst loading is predicted to act as a recombination center.30 When Pt is loaded on TiO2, the broad density of states of Pt overlaps with the conduction band minimum and valence band maximum of TiO2. Thus, it is expected that both photo-generated electrons and holes are captured by Pt cocatalysts. Consequently, the recombination is promoted. Furthermore, even though Pt is an excellent cocatalyst for hydrogen generation, the Pt cocatalyst is not suitable for complete water splitting owing to its backward reaction, in which the generated hydrogen and oxygen recombine and revert to water.31 Conversely, in the case of transition metals such as Ni and Co, localized 3d orbitals do not serve as a bridge between the conduction band and valence band.32,33 It is therefore expected to result in a relatively low recombination when such transition metals are loaded as cocatalysts. As described above, the cocatalyst plays a crucial but complicated role in the photocatalyst design, and it is essential to obtain experimental insights into how cocatalyst loading influences charge carrier dynamics.
Transient absorption spectroscopy is a very useful technique for measuring and evaluating such recombination processes in semiconductors and reactions proceeding on the surface of a photocatalyst.34–39 This method allows for the observation of non-emission carrier dynamics in the semiconductor, following photoexcitation. It has already been applied to investigate the carrier dynamics of Pt-TiO2,34 NiO-NaTaO3,40 Pt-LaTiO2N,15 CoPi-Fe2O3,41,42 Pt, Au-CdS,43,44 Ni-CdS,45 and MoS2-CdS.46 However, in many cases, the measurements are focused on the carriers that are anticipated to migrate, and the role of the cocatalyst as a recombination center has not been sufficiently discussed. For instance, in the case of Pt-TiO2 where electrons are highly expected to migrate to Pt cocatalysts, the migration behavior of the photogenerated holes is often overlooked.
In this study, we used transient absorption spectroscopy to observe the interaction between photo-excited holes and the broad density of states of Pt on SrTiO3, and compared these results with the situation using the Ni cocatalyst. By examining the carrier dynamics loading of Pt and Ni, the relationship between the cocatalyst type and recombination dynamics is discussed.
2. Experimental section
2.1 Photocatalyst preparation
SrTiO3 nanoparticles were synthesized through a polymerizable complex method.47 Specifically, Ti(OC4H9)4 (99.9%, Sigma-Aldrich) was dissolved in ethylene glycol, and the mixture was stirred under N2 atmosphere for 30 min. Afterwards, Sr(NO3)3 (99.9%, Sigma-Aldrich) and citric monohydrate (99.5%, Carl Roth) were added, and the stirring was continued until the solution became completely transparent. The reaction mixture was subsequently stirred for an additional 15 minutes to ensure full dissolution of the reagents, and then heated at 120 °C for 5 hours to promote polymerization. During heating, the solvent evaporated and the suspension transformed into a transparent brownish resin. This resin was further heated to 350 °C with a slow ramping temperature rate (1 °C min−1) and kept for 3 hours. The resulting cinders were ground to obtain fine nanoparticles, and further calcined at 750 °C for 6 hours.
2.2 Photo-deposition of the cocatalyst and photocatalytic reaction
Pt and Ni photo-deposition was carried out in 300 ml of 10 vol% methanol (MeOH) aqueous solution, and the evaluation of the photocatalytic hydrogen evolution was continued after the deposition. 50 mg SrTiO3 powder was placed in a Pyrex glass reactor, and subjected to ultrasonic treatment for 30 minutes. Then, H2PtCl6·6H2O or Ni(NO3)2 aqueous solution was added with vigorous stirring for the Pt and Ni photo-deposition, respectively. The photocatalytic activity evaluation was performed with a gas-closed circulation system, as shown in Fig. S1.† The Pyrex glass reactor and solution were degassed by linking to a gas-closed circulation system connected to a rotary pump. The solution was irradiated for 3 hours with the full arc of a 300 W Xe lamp for the photo-deposition and photocatalytic reaction. The produced H2 was analyzed by gas chromatography (GC) (GC-8A, Shimadzu, Japan; carrier gas, Ar) with a thermal conductivity detector (TCD), which was connected to the gas-closed circulation system.
2.3 Characterization
The sample was characterized by X-ray diffraction (XRD: D8 Advance; Bruker Corp, Germany) using Cu Kα radiation in the 2θ range of 10–70° with a step width of 0.01°. The diffused reflectance spectra of the sample were recorded on a UV-visible spectrometer (UV-2600; Shimadzu, Japan) with BaSO4 as the reference. The reflectance spectra were converted to absorption spectra by the Kubelka–Munk function. The photoluminescence (PL) spectra of the sample were recorded on a FP-8550 (JASCO, Japan) with the excitation wavelength of 355 nm. The microstructure observations were performed with transmission electron microscopy (TEM, JEM-ARM200F; JEOL Ltd, Japan) at an acceleration voltage of 60 kV. X-ray photoelectron spectroscopy (XPS) analysis was performed on a EnviroESCA (SPECS GmbH, Germany) equipped with Al Kα radiation, and the pass energy for all measurements was set to 20 eV. I–V measurements were performed by potentiostat (SP-300; Bio-Logic SAS, France) with the voltage range of −6 to 6 V.
Charge carrier dynamics were measured by transient absorption spectroscopy. The details of the set-up of the nano–micro48,49 and pico–nanosecond transient absorption are described in Fig. S2 and S3,† respectively. For the transient absorption experiments, the powder photocatalysts were packed in a quartz cuvette, which allowed for the introduction of reactant gases. Here, O2 gas and MeOH vapor were applied as electron and hole scavengers, respectively.34,35
3. Results and discussion
3.1 The characterization of Pt, Ni-loaded SrTiO3
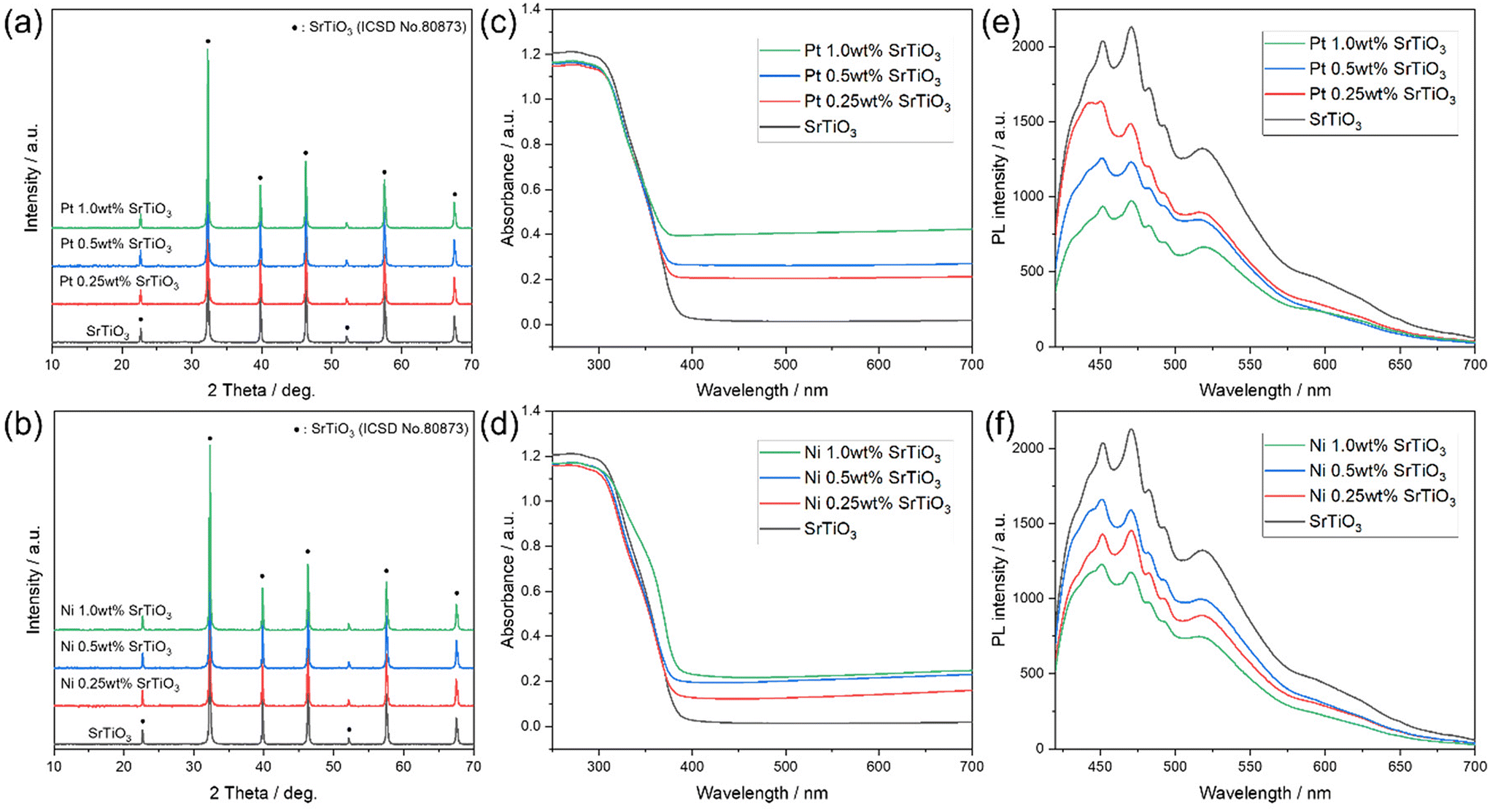
Fig. 1(a and b) shows the XRD patterns of the synthesized SrTiO3 and Pt and Ni-loaded SrTiO3. The synthesized SrTiO3 shows a perovskite structure with the lattice constant of a = 3.9233 Å, which is relatively consistent with the reported value of a = 3.911 Å.50 In addition, no other diffraction patterns were observed after the Pt and Ni cocatalyst is loaded, indicating that the Pt and Ni cocatalyst was loaded as relatively small particles. Fig. 1(c and d) shows the UV-Vis absorption spectra of (c) Pt and (d) Ni-loaded SrTiO3. SrTiO3 exhibits light absorption in the UV range, as reported in the previous literature.49Fig. 1(c) shows an increase in absorption in the 400–700 nm range with increasing loading amount of the Pt cocatalyst, which can be attributed to the absorption of Pt nanoparticles, as observed by TEM (Fig. S4†).51 In Fig. 1(d), the absorption of Ni-loaded SrTiO3 increased with increasing Ni loading amount, as in the case of Pt. A slight change in the shape of the shoulder of the absorption edge was observed when the Ni loading amount was 1.0 wt%, indicating that a part of the Ni cocatalyst was oxidized. Fig. 1(e and f) shows the PL spectra of Pt and Ni-loaded SrTiO3. The broad photoluminescence signal of SrTiO3 was derived from a radiative recombination between electrons located in numerous numbers of trapping states between the band gap and holes at the valence band. The PL signal intensity was weakened when the Pt or Ni cocatalysts were loaded. This is probably due to the non-radiative recombination pathway of the photoexcited electrons formed by cocatalyst loading. Fig. S5† shows the XPS spectra of the Pt and Ni 1.0 wt%-loaded SrTiO3. Fig. S5(a and b)† shows that the carbon species and –OH groups adsorbed on SrTiO3 were reduced by the loading process of the cocatalyst. This is considered to be due to the removal of the surface adsorbed species by the photocatalytic effect of SrTiO3 irradiated with light during the loading process. The chemical states of Ti and Sr shown in Fig. S5(c and d)† showed only limited changes due to the loading of the cocatalyst, indicating that the cocatalyst can be loaded without causing side reactions, such as alloying and or elemental interdiffusion. The chemical states of Pt and Ni shown in Fig. S5(e and f)† indicate that Pt was metallic, while Ni was partially oxidized. This partial oxidation of Ni is considered to correspond to the change in the shape of the absorption edge observed in the UV-Vis spectrum (Fig. 1(d)). Fig. S6† shows the I–V curves of SrTiO3 and Pt and Ni-loaded SrTiO3. The voltage–current response shows no linear response components even after the addition of the Pt or Ni cocatalysts, indicating that the junction between SrTiO3 and the cocatalyst is a Schottky junction.
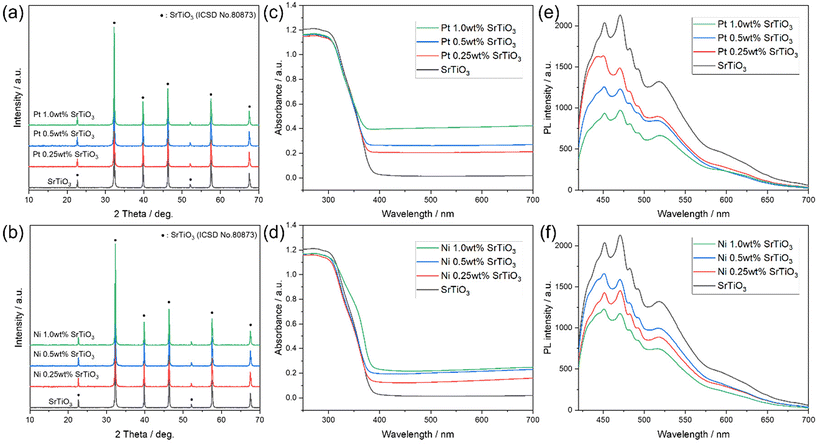 |
| | Fig. 1 XRD patterns of (a) Pt and (b) Ni-loaded SrTiO3. UV-vis absorption spectra of (c) Pt and (d) Ni-loaded SrTiO3. PL spectra of (e) Pt and (f) Ni-loaded SrTiO3 with an excitation wavelength of 355 nm. | |
3.2 The photocatalytic activity of the Pt, Ni-loaded SrTiO3
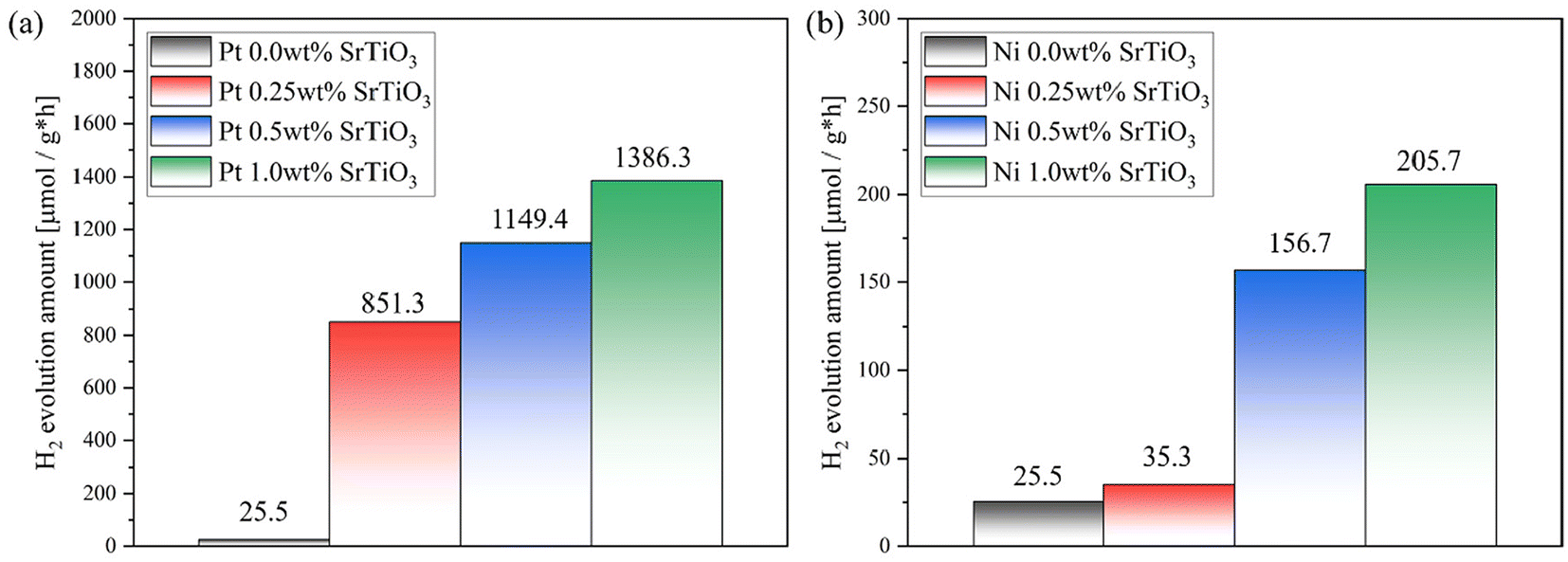
Fig. 2(a) shows the effect of Pt loading on the photocatalytic HER activities of the SrTiO3 nanoparticles. The results show that the photocatalytic activity was significantly enhanced from 25.5 μmol g−1 h−1 to 851.3, 1149.4, 1386.3 μmol g−1 h−1 with the increase of the Pt weight percent from 0.0 to 0.25, 0.5, and 1.0 wt%, respectively. This is reasonable as loading noble metals such as Pt is expected to create active sites for HER. However, the photocatalytic activity gets saturated with further increases in the Pt loading. Therefore, the loaded Pt contents affect the electron–hole recombination rate in SrTiO3 nanoparticles. Fig. 2(b) shows the photocatalytic HER activities with respect to the amount of Ni that is loaded. A similar trend was observed in the case of Ni loading up to 1.0 wt%, although the activity at 1.0 wt% was as low as 205 μmol g−1 h−1, approximately one-seventh of that achieved with Pt.
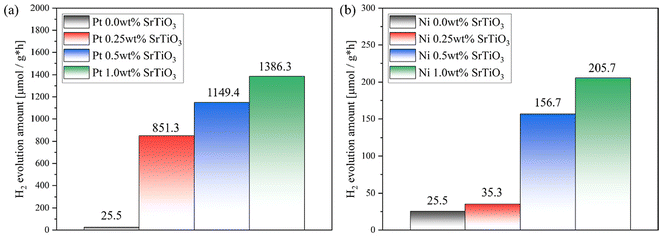 |
| | Fig. 2 Effect of the loaded platinum weight percent on the photocatalytic activity of (a) Pt- and (b) Ni-loaded SrTiO3 nanoparticles for photocatalytic hydrogen evolution. | |
3.3 Transient absorption of photogenerated charge carriers in SrTiO3
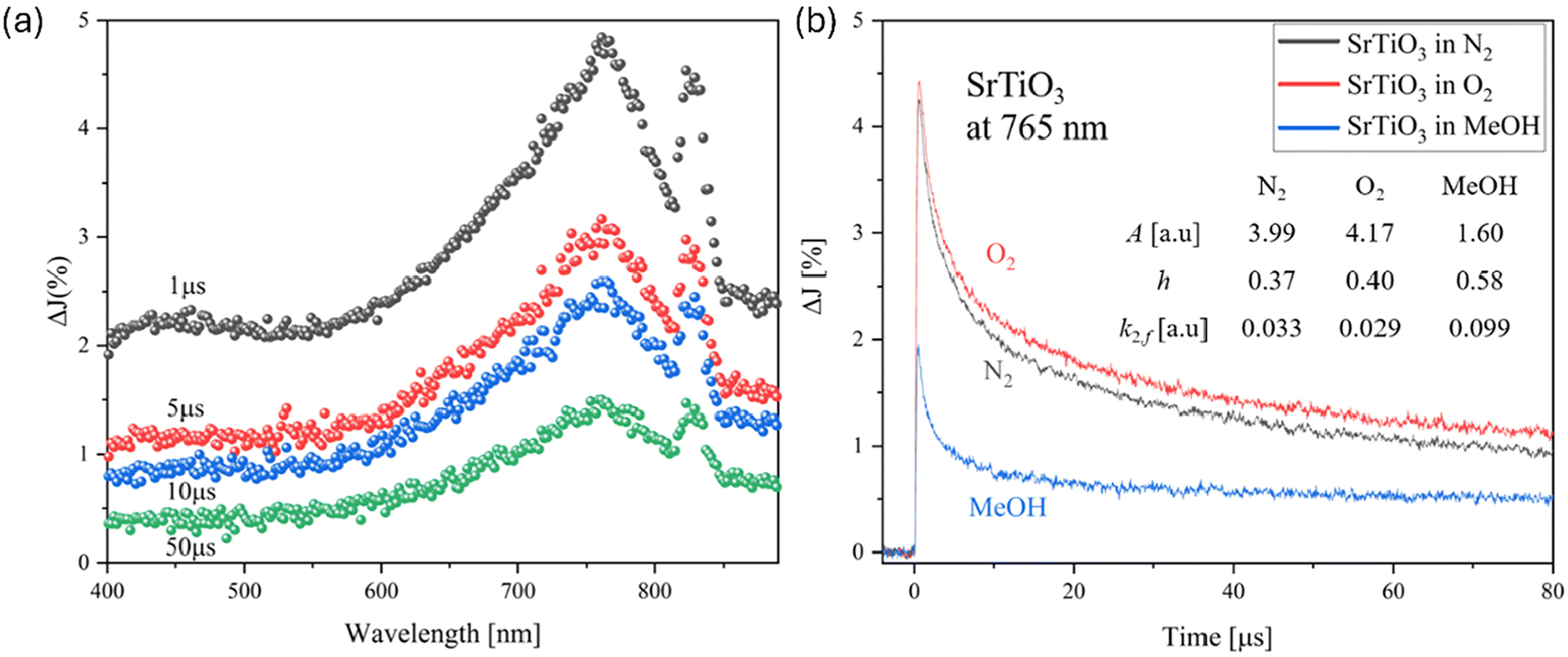
Fig. 3(a) shows the transient absorption spectrum after the irradiation with a 355 nm Nd:YAG laser over SrTiO3. The energy of the Nd:YAG laser is a suitable excitation wavelength to induce a band-to-band transition of SrTiO3. A broad absorption band is observed around 765 nm, and a relatively narrow absorption band is observed around 825 nm. To identify the observed carriers, decay measurements were taken at 765 nm with the presence of an electron scavenger (O2 gas) and a hole scavenger (MeOH vapor) (Fig. 3(b)). As a reference, a decay measurement was also performed in a N2 gas atmosphere, where no reactive species are present, representing a simple process of electron–hole recombination. In the measurement under O2 atmosphere, the decay began to slow down slightly after 1 μs. This is because the electrons that tend to recombine with holes are captured by O2.52
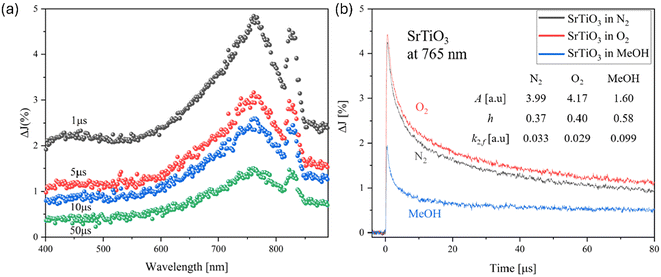 |
| | Fig. 3 (a) Transient absorption spectra of SrTiO3 particles irradiated by a UV (355 nm) pulsed laser under N2 gas. The pump energy was 1.3 mJ cm−2, and the repetition rate was 10 Hz. (b) Decay curves of transient absorption of the SrTiO3 at 765 nm. | |
Where (g) and (a) represent the gas phase and adsorbed phase, respectively. Interestingly, the decay was dramatically accelerated when measuring in MeOH. This is because the holes were collected by MeOH-derived adsorbates:
| | | CH3O−(a) + h+ → CH3O˙(a) | (2) |
From the above results, it can be concluded that the photoexcited carriers at 765 nm are transient absorption bands derived from holes. As shown in Fig. S7,† the decay of the signal was decelerated in O2 atmosphere and accelerated in MeOH vapor, similar to the signal seen at 765 nm, indicating a hole-derived transient absorption signal.
3.4 Transient absorption of photogenerated charge carriers over Pt, Ni-loaded SrTiO3
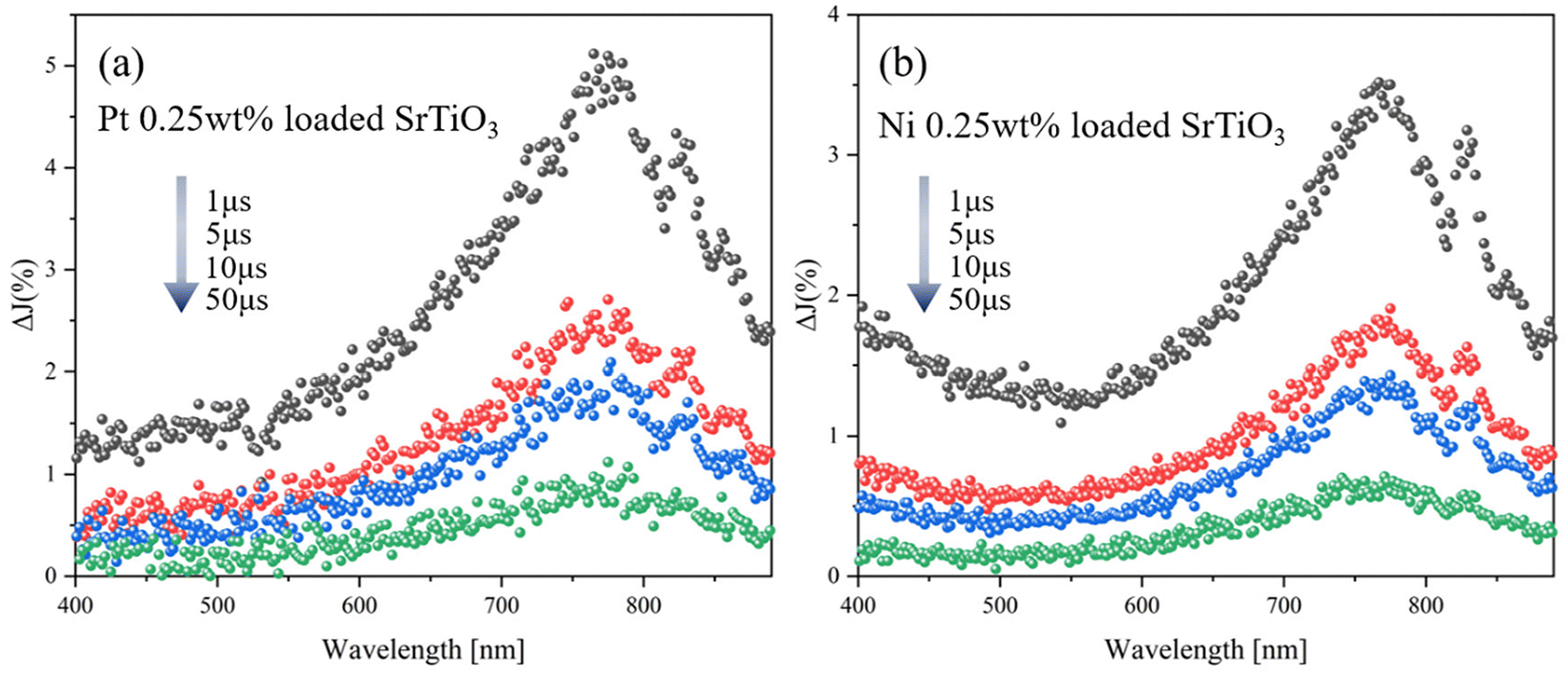
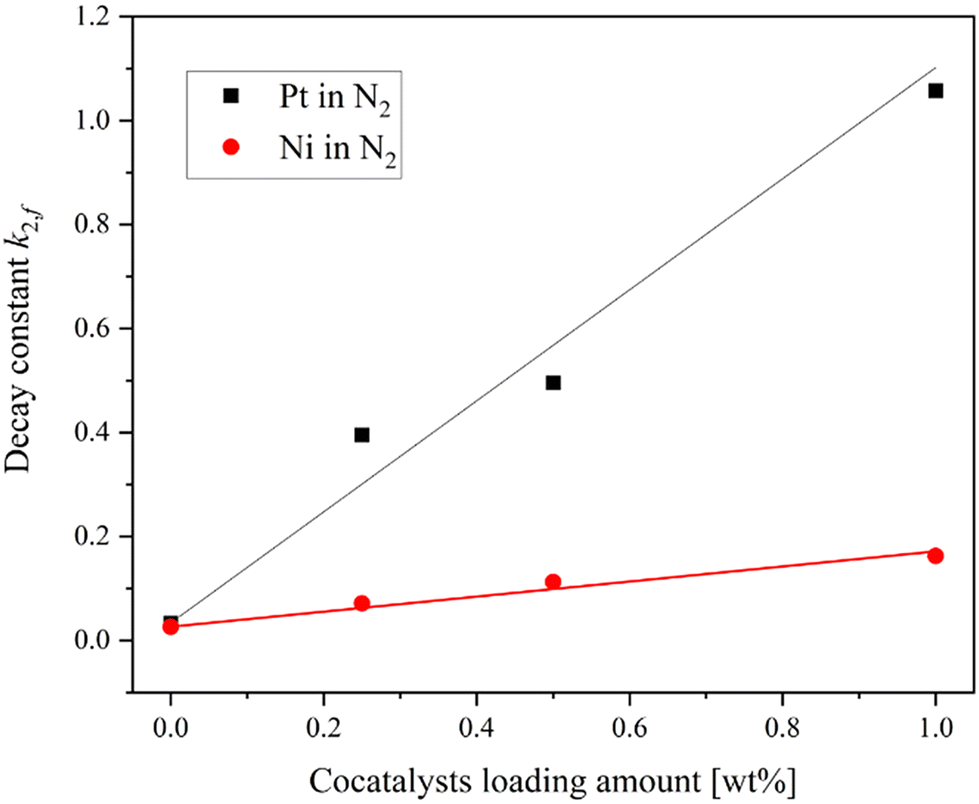
Fig. 4(a and b) shows the transient absorption spectra of SrTiO3 loaded with 0.25 wt% Pt and Ni, respectively. Both transient absorption spectra of Pt and Ni-loaded SrTiO3 show broad absorption peaks at around 765 nm and 825 nm, similar to those observed on the pure SrTiO3. This suggests that even after cocatalyst loading, the transient absorption signal reflects the transient feature of SrTiO3. To further investigate the interaction between the cocatalyst loading and the trapping behavior of photogenerated holes in SrTiO3, the relationship between the amount of cocatalyst loading and the decay constant in the inert N2 atmosphere was determined (Fig. 5). The transient absorption spectra of SrTiO3 loaded with 0.5 wt% and 1.0 wt% Pt or Ni can be found in Fig. S8.† The linear increase in k2,f, which indicates the rate of decay with respect to the amount of cocatalyst, indicates the transfer of holes to the cocatalyst. This phenomenon is also considered to be reasonable since holes can easily move through the Schottky junction between n-type semiconductors SrTiO3 and Pt or Ni cocatalysts, as shown in the I–V curves (Fig. S6†). The increase in the decay constant k2,f with Pt loading was much more pronounced than that with Ni, suggesting that the broad density of states of Pt act as a recombination center.
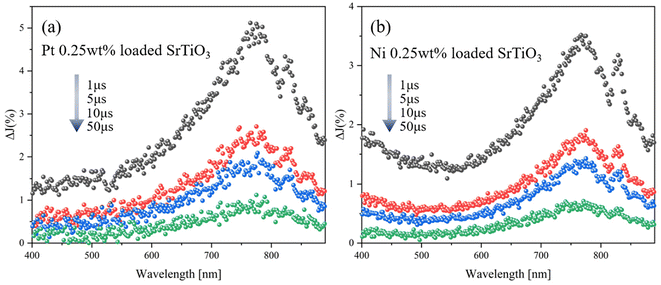 |
| | Fig. 4 (a) Transient absorption spectra of Pt 0.25 wt% SrTiO3 particles and (b) Ni 0.25 wt% SrTiO3 particles irradiated by a UV (355 nm) pulsed laser under N2 gas. The pump energy was 1.3 mJ cm−2, and the repetition rate was 10 Hz. | |
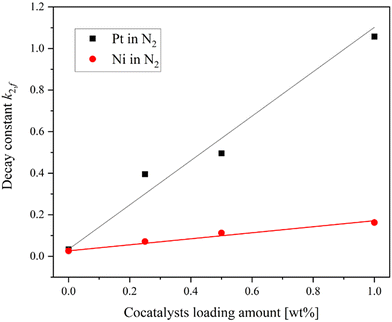 |
| | Fig. 5 The relationship between the loading amount of Pt and Ni cocatalysts and the decay constant k2,f. | |
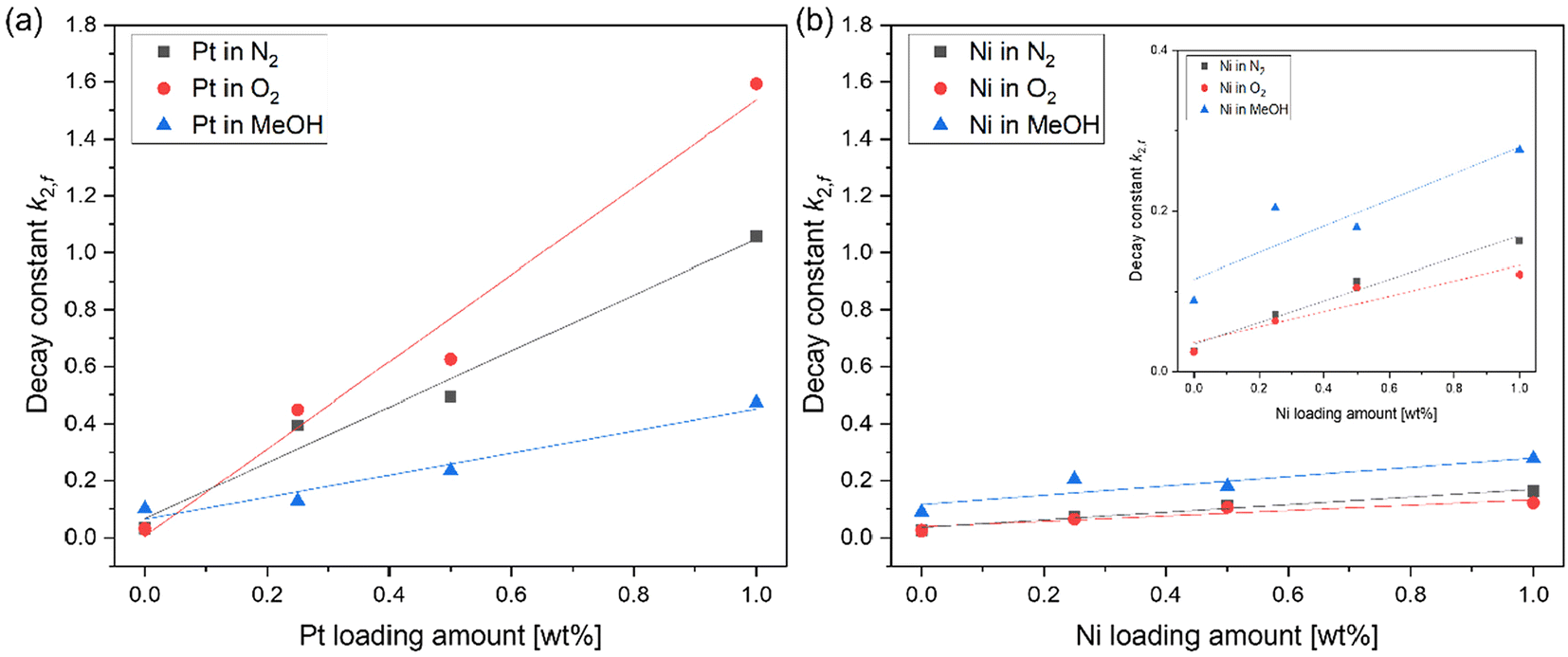
Fig. 6(a) exhibits the relationship between the amount of Pt loading and the decay constant of Pt-loaded SrTiO3 in different atmospheres. As described in Fig. 5, an increasing decay constant in N2 atmosphere with respect to the increase of the Pt amount has been observed. This increase in decay constant was further promoted in the presence of O2 gas and suppressed in MeOH vapor (Fig. 6(a)), suggesting that the adsorption of O2 and MeOH molecules influences the decay dynamics. In contrast, the decay constant of Ni-loaded SrTiO3, shown in Fig. 6(b), was less affected by the increase in the Ni amount and the various atmospheres.
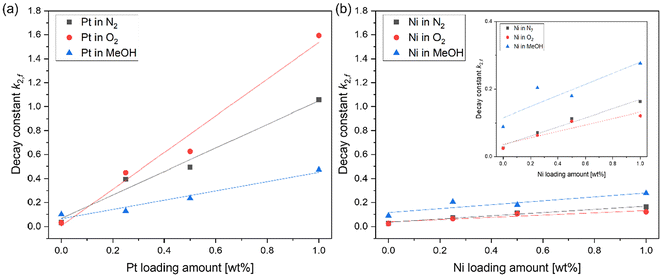 |
| | Fig. 6 (a) The relationship between the loading amount of Pt cocatalysts, (b) Ni cocatalyst and decay constant k2,f in different atmospheres. The inset figure shows the enlarged vertical axis of (b). | |
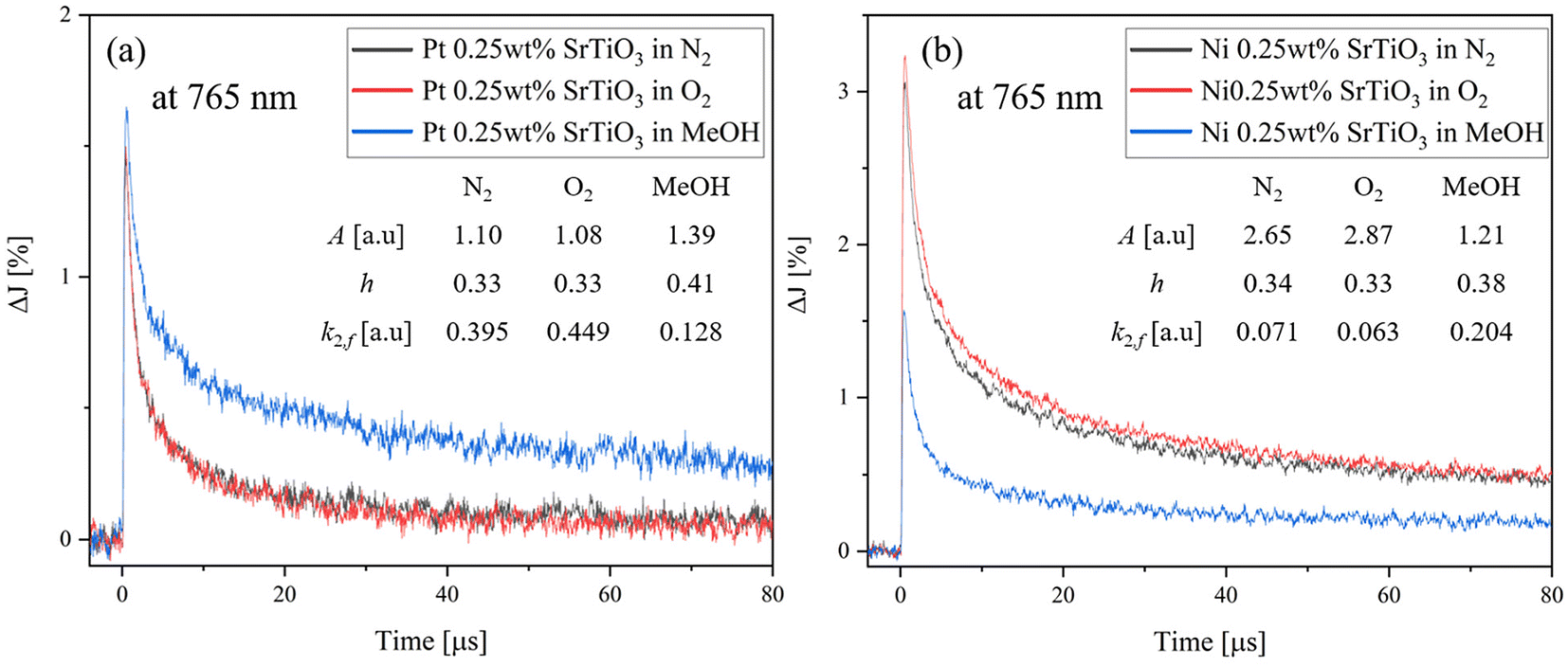
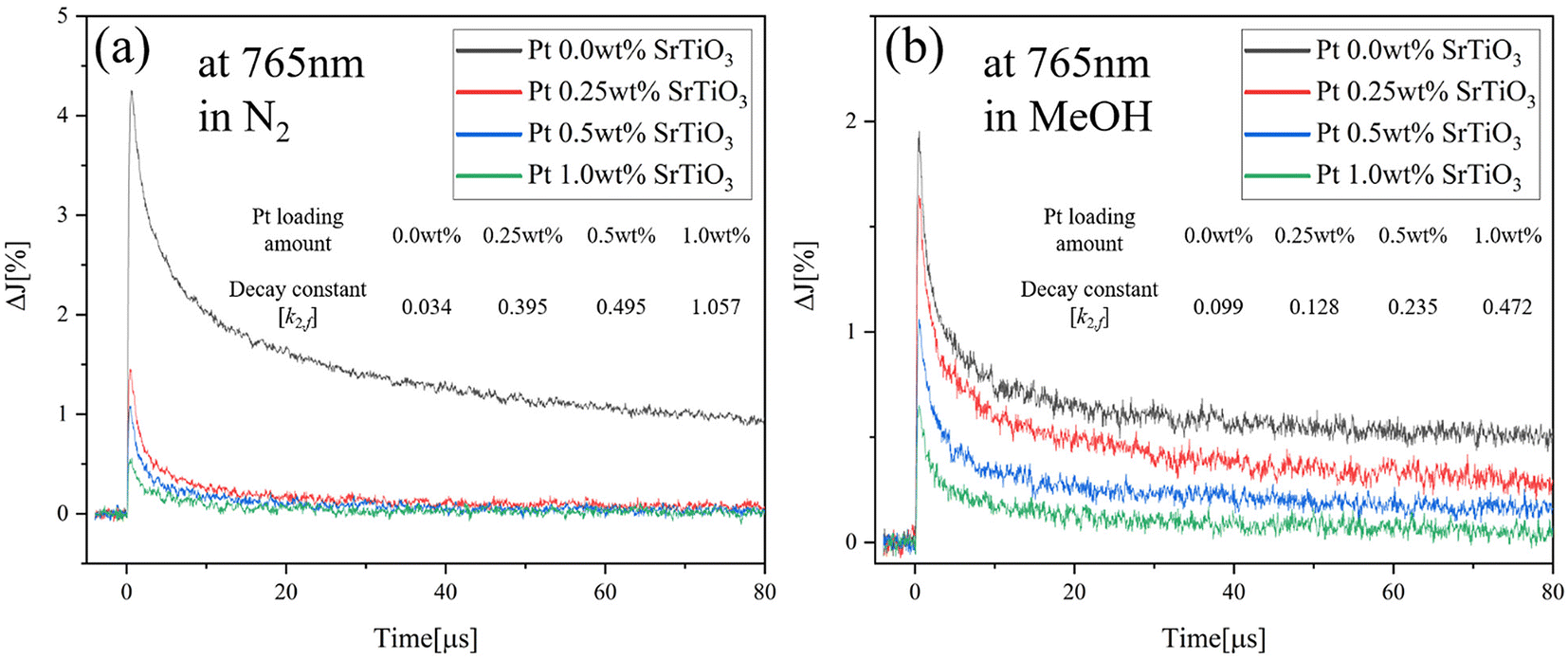
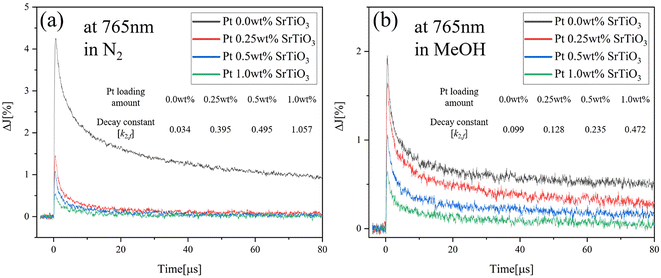
To further understand the effect of the adsorbed molecules on the Pt cocatalyst, the decay curves of Pt-loaded SrTiO3 at 765 nm in N2, O2 and MeOH atmospheres are compared, as shown in Fig. 7(a). As described in Fig. 6(a), the decay rate accelerates in the O2 atmosphere and decelerates in MeOH vapor, compared to that of N2 atmosphere. Unlike SrTiO3 (Fig. 3(b)), Pt loading causes the transient decay to be slowest in MeOH vapor. In the O2 atmosphere, the decay rate is slightly accelerated as the decay constant k2,f increases from 0.395 to 0.449, compared with that in N2 atmospheres. Moreover, Fig. 8 illustrates the decay curves of SrTiO3 with different loading amounts of Pt at 765 nm in N2 and MeOH atmospheres. In Fig. 8(a), the Pt loading dramatically decreases the signal intensity and increases the decay constant in a N2 atmosphere. Meanwhile, in a MeOH atmosphere (Fig. 8(b)), the decrease in signal intensity and increase in decay constant are not as significant as those observed in a N2 atmosphere. These limited changes in MeOH indicate that MeOH scavenges photogenerated holes in SrTiO3, thus mitigating the impact of Pt cocatalyst loading on the decay curves. Conversely, as seen in Fig. 7(b) and Fig. S9,† for Ni-loaded SrTiO3, the decay was decelerated in an O2 atmosphere and accelerated in a MeOH atmosphere compared with that in a N2 atmosphere, similar to that of SrTiO3, confirming that the Ni cocatalyst loading has a small effect on the carrier dynamics of SrTiO3 and does not act as a recombination center.
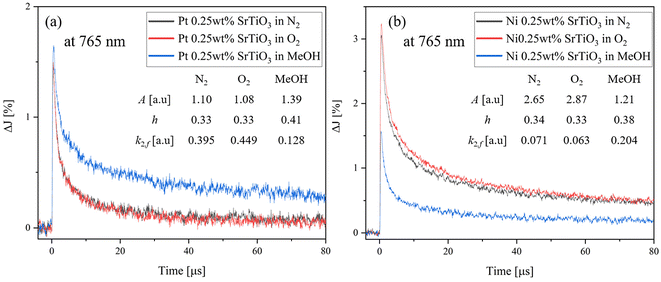 |
| | Fig. 7 Decay curves of the transient absorption of (a) Pt 0.25 wt%-loaded SrTiO3 and (b) Ni 0.25 wt%-loaded SrTiO3 in different atmospheres at 765 nm. | |
 |
| | Fig. 8 Decay curves of transient absorption of the Pt-loaded SrTiO3 in (a) N2 and (b) MeOH atmospheres at 765 nm. | |
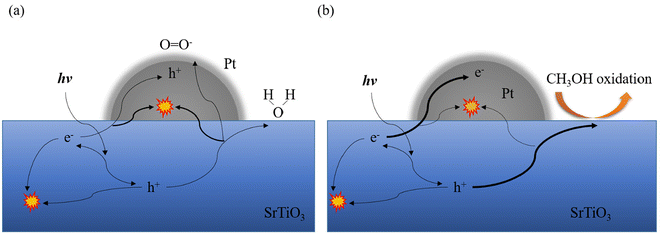
From the results discussed above, the dynamics shown in Fig. 9 can be considered as follows: in an O2 atmosphere, O2 adsorbed on Pt scavenges the electrons of the Pt cocatalyst to form charged oxygen species and produce holes.30,53 These charged oxygen species and the holes generated at the Pt cocatalyst are expected to trap the electron–hole pairs generated in SrTiO3, accelerating the consumption of charge carriers in the O2 atmosphere compared to the N2 atmosphere. In the case of MeOH vapor, MeOH molecules tend to scavenge the photogenerated holes in SrTiO3. Thus, there is no large effect observed over the carrier dynamics from loading different amounts of Pt cocatalyst. Therefore, the recombination at the Pt cocatalyst has been suppressed in the presence of hole sacrificial reagents.
 |
| | Fig. 9 Schematic representation of the photoexcited electron and hole path over Pt-loaded SrTiO3 in the presence of (a) O2 gas and (b) MeOH vapor. | |
3.5 Hole behavior observed by femtosecond transient absorption measurement
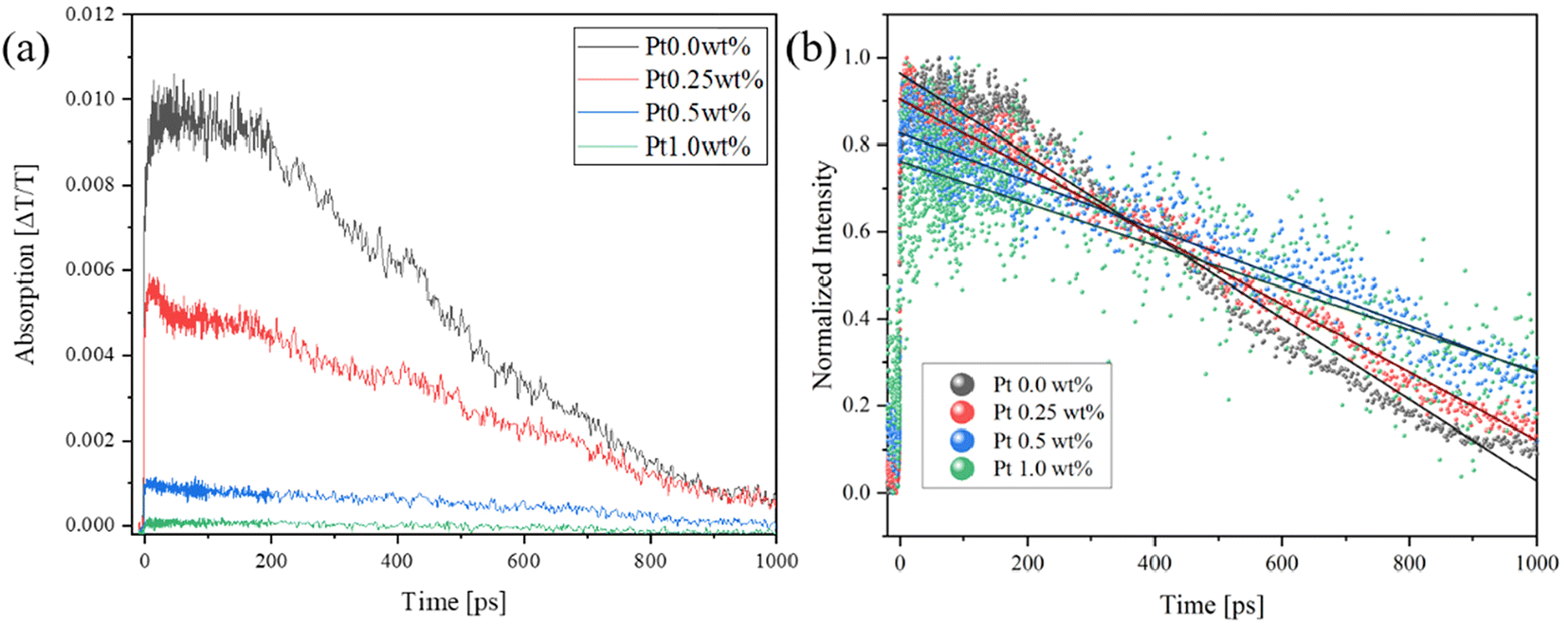
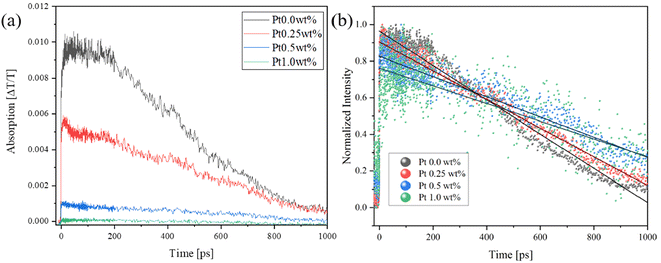
The photocatalytic reaction takes place in three steps, i.e., carrier generation, charge transfer, and surface reaction.52 In order to investigate the time scale on which the photo-generated holes transfer, we measured the transient absorption spectrum at 765 nm on the pico-second scale, as shown in Fig. 10(a). When Pt was loaded on SrTiO3, the sample itself absorbed light at 765 nm, and the signal intensity decreased with an increase in the Pt loading amount. After normalizing the spectra (Fig. 10(b)), it was observed that the photoexcitation showed a faster decay than pure SrTiO3 as the Pt loading increased up to about 400 ps. This is similar to the accelerated decay of LaTiO2N, which has the same perovskite structure as SrTiO3, in the range of 2–350 ps due to Pt loading.15 However, the lifetime was extended after 400 ps. This phenomenon indicates that photoexcited holes near the surface were effectively transferred to the Pt cocatalysts within 400 ps. Beyond this time frame, hole trapping behavior from the bulk did not occur within the pico–nano second time range of the transient absorption measurement. As described above, the holes were likely drawn and recombined in Pt cocatalysts in the micro-second range.
 |
| | Fig. 10 (a) Pico-second transient absorption spectra and (b) normalized spectra of Pt-loaded SrTiO3 nanoparticles irradiated by UV (355 nm) pulsed laser under ambient conditions at 765 nm. | |
4. Conclusion
SrTiO3 samples loaded with Pt and Ni cocatalysts by photodeposition were evaluated and compared using nano–micro and pico–micro second transient absorption spectroscopy. Focusing on the transient absorption signal derived from holes, rather than the normally studied electron signals, it was found that the decay constant, representing the speed of carrier decay, increased linearly with the loading amount of cocatalysts. When loading with the Pt cocatalyst, this increase in the decay constant was more significant than that with Ni cocatalyst loading, indicating that the broad density of state of Pt may act as a recombination center, as suggested by theoretical calculation. In MeOH vapor, this increasing recombination tendency was suppressed compared with that of a N2 atmosphere, indicating that the Pt cocatalyst did not act as a recombination center in the presence of hole sacrificial agents. As a contrast, for the transition metal Ni-loaded samples, the recombination was not accelerated. Therefore, this suggests that designing a cocatalyst with a localized density of states is desirable, and an appropriate hydrogen absorption/desorption feature is crucial for efficient overall water splitting. Furthermore, by measuring the charge dynamics in the picosecond-to-nanosecond range, it was observed that photo-generated holes near the surface were transferred to Pt cocatalysts within 400 ps following photoexcitation, while the holes generated in bulk migrated to Pt in a micro-second regime.
Data availability
The data supporting this article have been included within the article and its ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work received financial support from the World Premier International Research Center Initiative (WPI Initiative) on Materials Nanoarchitectonics (MANA), MEXT, Japan, and Photoexcitonix Project in Hokkaido University. The authors also thank Dr Jenny Schneider, Dr Fabian Sieland, Dr Anh-Thu Duong and Dr Yoshiki Wada for their fruitful discussion.
References
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst, Nature, 2001, 414(6864), 625–627, DOI:10.1038/414625a.
- H. Kato and A. Kudo, Visible-Light-Response and Photocatalytic Activities of TiO2 and SrTiO3 Photocatalysts Codoped with Antimony and Chromium, J. Phys. Chem. B, 2002, 106(19), 5029–5034, DOI:10.1021/jp0255482.
- J. W. Liu, G. Chen, Z. H. Li and Z. G. Zhang, Electronic structure and visible light photocatalysis water splitting property of chromium-doped SrTiO3, J. Solid State Chem., 2006, 179(12), 3704–3708, DOI:10.1016/j.jssc.2006.08.014.
- S. Ouyang, H. Tong, N. Umezawa, J. Cao, P. Li, Y. Bi, Y. Zhang and J. Ye, Surface-Alkalinization-Induced Enhancement of Photocatalytic H2 Evolution over SrTiO3-Based Photocatalysts, J. Am. Chem. Soc., 2012, 134(4), 1974–1977, DOI:10.1021/ja210610h.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Nano-photocatalytic Materials: Possibilities and Challenges, Adv. Mater., 2012, 24(2), 229–251, DOI:10.1002/adma.201102752.
- X. Meng, G. Zuo, P. Zong, H. Pang, J. Ren, X. Zeng, S. Liu, Y. Shen, W. Zhou and J. Ye, A rapidly room-temperature-synthesized Cd/ZnS:Cu nanocrystal photocatalyst for highly efficient solar-light-powered CO2 reduction, Appl. Catal., B, 2018, 237, 68–73, DOI:10.1016/j.apcatb.2018.05.066.
- P. Li, S. Ouyang, G. Xi, T. Kako and J. Ye, The Effects of Crystal Structure and Electronic Structure on Photocatalytic H2 Evolution and CO2 Reduction over Two Phases of Perovskite-Structured NaNbO3, J. Phys. Chem. C, 2012, 116(14), 7621–7628, DOI:10.1021/jp210106b.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Photocatalytic reduction of carbon dioxide by hydrous hydrazine over Au–Cu alloy nanoparticles supported on SrTiO3/TiO2 coaxial nanotube arrays, Angew. Chem., 2015, 127(3), 855–859 CrossRef.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye and Y. Zhou,
et al., A Room-Temperature Reactive-Template Route to Mesoporous ZnGa2O4 with Improved Photocatalytic Activity in Reduction of CO2, Angew. Chem., Int. Ed., 2010, 49(36), 6400–6404, DOI:10.1002/anie.201003270.
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Self-doped SrTiO3−δ photocatalyst with enhanced activity for artificial photosynthesis under visible light, Energy Environ. Sci., 2011, 4(10), 4211–4219, 10.1039/C1EE01594J.
- Z. Pei, H. Zhang, D. Luan and X. W. Lou, Electrocatalytic acidic oxygen evolution: From catalyst design to industrial applications, Matter, 2023, 6(12), 4128–4144, DOI:10.1016/j.matt.2023.11.007.
- B. Chang, H. Zhang, S. Sun and G. Zhang, Strategies to achieve effective nitrogen activation, Carbon Energy, 2024, 6(5), e491, DOI:10.1002/cey2.491.
- B. Chang, Z. Cao, Y. Ren, C. Chen, L. Cavallo, F. Raziq, S. Zuo, W. Zhou, Y. Han and H. Zhang, Electronic Perturbation of Isolated Fe Coordination Structure for Enhanced Nitrogen Fixation, ACS Nano, 2024, 18(1), 288–298, DOI:10.1021/acsnano.3c06212.
- A. Kudo and Y. Miseki, Heterogeneous photocatalyst materials for water splitting, Chem. Soc. Rev., 2009, 38(1), 253–278, 10.1039/B800489G.
- A. Yamakata, M. Kawaguchi, N. Nishimura, T. Minegishi, J. Kubota and K. Domen, Behavior and Energy States of Photogenerated Charge Carriers on Pt- or CoOx-Loaded LaTiO2N Photocatalysts: Time-Resolved Visible to Mid-Infrared Absorption Study, J. Phys. Chem. C, 2014, 118(41), 23897–23906, DOI:10.1021/jp508233z.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, Photocatalytic decomposition of water vapour on an NiO–SrTiO3 catalyst, J. Chem. Soc., Chem. Commun., 1980, 12, 543–544, 10.1039/C39800000543.
- K. Domen, S. Naito, T. Onishi and K. Tamaru, Photocatalytic decomposition of liquid water on a NiO-SrTiO3 catalyst, Chem. Phys. Lett., 1982, 92(4), 433–434, DOI:10.1016/0009-2614(82)83443-X.
- K. Domen, S. Naito, T. Onishi, K. Tamaru and M. Soma, Study of the photocatalytic decomposition of water vapor over a nickel(II) oxide-strontium titanate (SrTiO3) catalyst, J. Phys. Chem., 1982, 86(18), 3657–3661, DOI:10.1021/j100215a032.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, Photocatalytic decomposition of water into hydrogen and oxygen over nickel(II) oxide-strontium titanate (SrTiO3) powder. 1. Structure of the catalysts, J. Phys. Chem., 1986, 90(2), 292–295, DOI:10.1021/j100274a018.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Photocatalyst releasing hydrogen from water, Nature, 2006, 440, 295, DOI:10.1038/440295a.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, Metal oxides as heterogeneous catalysts for oxygen evolution under photochemical conditions, J. Chem. Soc., Faraday Trans. 1, 1988, 84(8), 2795–2806, 10.1039/F19888402795.
- B. H. Meekins and P. V. Kamat, Role of Water Oxidation Catalyst IrO2 in Shuttling Photogenerated Holes Across TiO2 Interface, J. Phys. Chem. Lett., 2011, 2(18), 2304–2310, DOI:10.1021/jz200852m.
- Y. Inoue, Photocatalytic water splitting by RuO2-loaded metal oxides and nitrides with d0- and d10-related electronic configurations, Energy Environ. Sci., 2009, 2(4), 364–386, 10.1039/B816677N.
- M. Barroso, C. A. Mesa, S. R. Pendlebury, A. J. Cowan, T. Hisatomi, K. Sivula, M. Grätzel, D. R. Klug and J. R. Durrant, Dynamics of photogenerated holes in surface modified α-Fe2O3 photoanodes for solar water splitting, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(39), 15640–15645, DOI:10.1073/pnas.1118326109.
- S. S. K. Ma, T. Hisatomi, K. Maeda, Y. Moriya and K. Domen, Enhanced Water Oxidation on Ta3N5 Photocatalysts by Modification with Alkaline Metal Salts, J. Am. Chem. Soc., 2012, 134(49), 19993–19996, DOI:10.1021/ja3095747.
- M. W. Kanan and D. G. Nocera, In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+, Science, 2008, 321(5892), 1072–1075, DOI:10.1126/science.1162018.
- J. A. Seabold and K.-S. Choi, Effect of a Cobalt-Based Oxygen Evolution Catalyst on the Stability and the Selectivity of Photo-Oxidation Reactions of a WO3 Photoanode, Chem. Mater., 2011, 23(5), 1105–1112, DOI:10.1021/cm1019469.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, Cobalt-Modified Porous Single-Crystalline LaTiO2N for Highly Efficient Water Oxidation under Visible Light, J. Am. Chem. Soc., 2012, 134(20), 8348–8351, DOI:10.1021/ja301726c.
- M. Higashi, K. Domen and R. Abe, Highly Stable Water Splitting on Oxynitride TaON Photoanode System under Visible Light Irradiation, J. Am. Chem. Soc., 2012, 134(16), 6968–6971, DOI:10.1021/ja302059g.
- C. L. Muhich, Y. Zhou, A. M. Holder, A. W. Weimer and C. B. Musgrave, Effect of Surface Deposited Pt on the Photoactivity of TiO2, J. Phys. Chem. C, 2012, 116(18), 10138–10149, DOI:10.1021/jp301862m.
- R. Abe, K. Sayama and H. Arakawa, Significant effect of iodide addition on water splitting into H2 and O2 over Pt-loaded TiO2 photocatalyst: suppression of backward reaction, Chem. Phys. Lett., 2003, 371(3), 360–364, DOI:10.1016/S0009-2614(03)00252-5.
- H. Peng, J. Li, S.-S. Li and J.-B. Xia, First-principles study of the electronic structures and magnetic properties of 3d transition metal-doped anatase TiO2, J. Phys.: Condens. Matter, 2008, 20(12), 125207, DOI:10.1088/0953-8984/20/12/125207.
- S. Tosoni, H.-Y. T. Chen and G. Pacchioni, A DFT study of Ni clusters deposition on titania and zirconia (101) surfaces, Surf. Sci., 2016, 646, 230–238, DOI:10.1016/j.susc.2015.04.004.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, Water- and Oxygen-Induced Decay Kinetics of Photogenerated Electrons in TiO2 and Pt/TiO2: A Time-Resolved Infrared Absorption Study, J. Phys. Chem. B, 2001, 105(30), 7258–7262, DOI:10.1021/jp010802w.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, Electron- and Hole-Capture Reactions on Pt/TiO2 Photocatalyst Exposed to Methanol Vapor Studied with Time-Resolved Infrared Absorption Spectroscopy, J. Phys. Chem. B, 2002, 106(35), 9122–9125, DOI:10.1021/jp025993x.
- A. Yamakata, J. J. M. Vequizo and H. Matsunaga, Distinctive Behavior of Photogenerated Electrons and Holes in Anatase and Rutile TiO2 Powders, J. Phys. Chem. C, 2015, 119(43), 24538–24545, DOI:10.1021/acs.jpcc.5b09236.
- A. Yamakata, J. J. M. Vequizo and M. Kawaguchi, Behavior and Energy State of Photogenerated Charge Carriers in Single-Crystalline and Polycrystalline Powder SrTiO3 Studied by Time-Resolved Absorption Spectroscopy in the Visible to Mid-Infrared Region, J. Phys. Chem. C, 2015, 119(4), 1880–1885, DOI:10.1021/jp510647b.
- A. J. Cowan, W. Leng, P. R. F. Barnes, D. R. Klug and J. R. Durrant, Charge carrier separation in nanostructured TiO2 photoelectrodes for water splitting, Phys. Chem. Chem. Phys., 2013, 15(22), 8772–8778, 10.1039/C3CP50318F.
- J. Tang, J. R. Durrant and D. R. Klug, Mechanism of Photocatalytic Water Splitting in TiO2. Reaction of Water with Photoholes, Importance of Charge Carrier Dynamics, and Evidence for Four-Hole Chemistry, J. Am. Chem. Soc., 2008, 130(42), 13885–13891, DOI:10.1021/ja8034637.
- A. Yamakata, T.-a. Ishibashi, H. Kato, A. Kudo and H. Onishi, Photodynamics of NaTaO3 Catalysts for Efficient Water Splitting, J. Phys. Chem. B, 2003, 107(51), 14383–14387, DOI:10.1021/jp036473k.
- S. R. Pendlebury, M. Barroso, A. J. Cowan, K. Sivula, J. Tang, M. Grätzel, D. Klug and J. R. Durrant, Dynamics of photogenerated holes in nanocrystalline α-Fe2O3 electrodes for water oxidation probed by transient absorption spectroscopy, Chem. Commun., 2011, 47(2), 716–718, 10.1039/C0CC03627G.
- M. Barroso, A. J. Cowan, S. R. Pendlebury, M. Grätzel, D. R. Klug and J. R. Durrant, The Role of Cobalt Phosphate in Enhancing the Photocatalytic Activity of α-Fe2O3 toward Water Oxidation, J. Am. Chem. Soc., 2011, 133(38), 14868–14871, DOI:10.1021/ja205325v.
- K. Wu, Z. Chen, H. Lv, H. Zhu, C. L. Hill and T. Lian, Hole Removal Rate Limits Photodriven H2 Generation Efficiency in CdS-Pt and CdSe/CdS-Pt Semiconductor Nanorod–Metal Tip Heterostructures, J. Am. Chem. Soc., 2014, 136(21), 7708–7716, DOI:10.1021/ja5023893.
- K. Wu, W. E. Rodríguez-Córdoba, Y. Yang and T. Lian, Plasmon-Induced Hot Electron Transfer from the Au Tip to CdS Rod in CdS-Au Nanoheterostructures, Nano Lett., 2013, 13(11), 5255–5263, DOI:10.1021/nl402730m.
- Y. Nakibli, Y. Mazal, Y. Dubi, M. Wächtler and L. Amirav, Size Matters: Cocatalyst Size Effect on Charge Transfer and Photocatalytic Activity, Nano Lett., 2018, 18(1), 357–364, DOI:10.1021/acs.nanolett.7b04210.
- Z. Yan, L. Du and D. L. Phillips, Multilayer, core–shell MoS2/CdS nanorods with very high photocatalytic activity for hydrogen production under visible-light excitation and investigation of the photocatalytic mechanism by femtosecond transient absorption spectroscopy, RSC Adv., 2017, 7(88), 55993–55999, 10.1039/C7RA12118K.
- S. Tonda, S. Kumar, O. Anjaneyulu and V. Shanker, Synthesis of Cr and La-codoped SrTiO3 nanoparticles for enhanced photocatalytic performance under sunlight irradiation, Phys. Chem. Chem. Phys., 2014, 16(43), 23819–23828, 10.1039/C4CP02963A.
- J. Schneider, K. Nikitin, M. Wark, D. W. Bahnemann and R. Marschall, Improved charge carrier separation in barium tantalate composites investigated by laser flash photolysis, Phys. Chem. Chem. Phys., 2016, 18(16), 10719–10726, 10.1039/C5CP07115A.
- F. Ichihara, F. Sieland, H. Pang, D. Philo, A.-T. Duong, K. Chang, T. Kako, D. W. Bahnemann and J. Ye, Photogenerated Charge Carriers Dynamics on La- and/or Cr-Doped SrTiO3 Nanoparticles Studied by Transient Absorption Spectroscopy, J. Phys. Chem. C, 2020, 124(2), 1292–1302, DOI:10.1021/acs.jpcc.9b09324.
- S. Keshri, L. Joshi and S. S. Rajput, Studies on La0.67Ca0.33MnO3−SrTiO3 composites using two-phase model, J. Alloys Compd., 2011, 509(19), 5796–5803, DOI:10.1016/j.jallcom.2011.02.122.
- K. K. R. Datta, M. Eswaramoorthy and C. N. R. Rao, Water-solubilized aminoclay–metal nanoparticle composites and their novel properties, J. Mater. Chem., 2007, 17(7), 613–615, 10.1039/B617198B.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Understanding TiO2 Photocatalysis: Mechanisms and Materials, Chem. Rev., 2014, 114(19), 9919–9986, DOI:10.1021/cr5001892.
- P. A. Pepin, J. D. Lee, C. B. Murray and J. M. Vohs, Thermal and Photocatalytic Reactions of Methanol and Acetaldehyde on Pt-Modified Brookite TiO2 Nanorods, ACS Catal., 2018, 8(12), 11834–11846, DOI:10.1021/acscatal.8b03081.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Hong
Pang
ab,
Hong
Pang