
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influential IPr: 25 years after its discovery
Vladislav A.
Voloshkin
 ,
Leandros P.
Zorba
and
Steven P.
Nolan
*
,
Leandros P.
Zorba
and
Steven P.
Nolan
*
Department of Chemistry and Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281, S-3, 9000 Ghent, Belgium. E-mail: Steven.Nolan@Ugent.be
First published on 9th January 2025
Abstract
N-Heterocyclic carbenes (NHCs) have emerged as a privileged ligand family in organometallic chemistry, widely recognized for their unique steric and electronic properties. Among them, the 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene (IPr) ligand has become a cornerstone of NHC chemistry for its remarkable versatility, stability, and broad use. Since its discovery by the Nolan group in 1999, IPr has played a pivotal role in advancing catalytic transformations and facilitating the utilization of NHC ligands in various domains. This article highlights major contributions where IPr has helped shape modern organometallic chemistry, with a focus on its influence in transition metal catalysis and ligand design. Twenty five years after its discovery, the IPr ligand continues to be a benchmark ligand, inspiring and driving innovation.
Introduction
N-Heterocyclic carbenes (NHCs) represent one of the most significant and extensively studied families of ligands in organometallic chemistry. Their robust σ-donating properties and ability to stabilize transition metals have made them indispensable in the chemistry of Late Transition Metals (LTM), where they have been widely employed as supporting ligands in a plethora of catalytic processes. Over the past few decades, the utility of NHCs has expanded far beyond traditional metal catalysis, finding applications in a range of fields such as in organocatalysis.1,2 Moreover, NHCs have been effectively incorporated into f-block metal chemistry,3 materials science,4 and surface chemistry,5 demonstrating their versatility and adaptability. These ligands have also been explored in biomedicinal research in the development of therapeutic agents,6 as well as in Main group chemistry,7 further illustrating their broad applicability across diverse areas of chemical research space.The rapid development and research into N-heterocyclic carbenes occurred shortly after Bertrand's isolation of the first stable λ3-phosphinocarbene (1989)8 and Arduengo in 1991, who isolated the first stable NHC – IAd (1,3-di(adamantyl)imidazole-2-ylidene).9 These breakthroughs built upon seminal work of Wanzlick and Öfele (dating back to 1968) who independently synthesized the first NHC complexes with mercury and chromium.10,11 The field saw further advances in 1995 with Herrmann's seminal report, showing the utility of NHC ligands in palladium-catalyzed Heck cross-coupling reactions.12 This reporting is a key moment where transition metal–NHC complexes were shown to have catalytic relevance.
The IPr ligand was first introduced by the Nolan group in 1999.13,14 The modified synthetic method is based on an important adaptation of the general protocol reported by Arduengo.15 This straightforward two-step process (Scheme 1) begins with the use of the readily available 1,3-diisopropylaniline to form a diimine, which, after simple collection as a solid via filtration, can be subjected to cyclization using paraformaldehyde. The imidazolium salt is formed with an accompanying counterion, which originates from the protic acid or TMSCl used. Following another filtration, the resulting bench-stable and easy-to-handle precursor can be immediately used either for the generation of the free carbene or for the direct synthesis of metal complexes. The simplicity of this method, which eliminates the need for rigorous purification steps, combined with the robustness and stability of the precursors, has further enhanced the appeal and widespread use of the IPr ligand in catalysis. Its practicality and stability have made it a highly attractive first choice for researchers in organometallic chemistry.
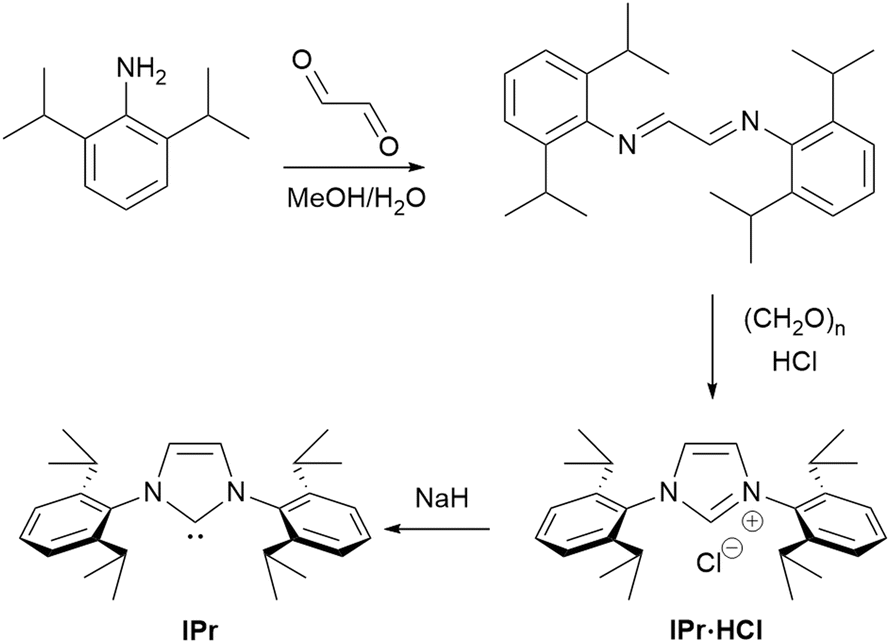 | ||
| Scheme 1 Synthesis of IPr·HCl and IPr. | ||
The synthetic protocol of the imidazolium salt (IPr·HCl) has proven quite robust and has been conducted in the Nolan labs on two continents and in 5 countries using high volume glass reactors and in a low-tech apparatus (Fig. 1). The detailed protocol for large scale synthesis of imidazolium salt and free carbene has been disclosed by the Nolan group.16 The free NHC IPr (1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) holds a unique and distinguished position among NHC ligands, despite not being the first NHC to be isolated or employed in catalysis. As it turns out, it is often the ligand of choice in any new catalyst screening involving an NHC. Its significance lies in its remarkable versatility, stability and broad applicability, making it one of the most widely used and influential ligands within the entire NHC family. The success of IPr can not only be attributed to its utility as a supporting ligand but also as inspiration in the development of related ligands. These evolutions in ligand design, oftentimes built around the IPr core, have permitted great advances in reactivity and catalysis.
 | ||
| Fig. 1 High- and low-tech reactors for the synthesis of IPr·HCl. | ||
Since its discovery, IPr has emerged as a cornerstone of NHC-based chemistry, featuring prominently in over 1800 scientific publications covering various domains of chemical science. However, the bibliographical metrics are somewhat misleading as the ligand and complexes are nowadays so commonly used that the primary literature goes often uncited. As one of the most readily accessible, its utilization as an ancillary ligand has often streamlined research efforts, causing a substantial increase in the exploration and application of NHCs in catalysis.
This account provides an overview of seminal contributions enabled using the IPr ligand, with particular focus on the pioneering advances that catalyzed the exploration of NHC as supporting ligands. While many examples of IPr–metal complexes have been reported, encompassing not only early and late transition metals17–20 but also Main group, and f-block elements,3,7 this contribution will focus on specific metals where IPr has had the greatest influence on their chemistry and catalytic applications. Additionally, we will examine the far-reaching influence of the IPr framework on ligand design and methodology development over the 25 years since its discovery, emphasizing its role in shaping modern organometallic chemistry.
Ruthenium chemistry and IPr
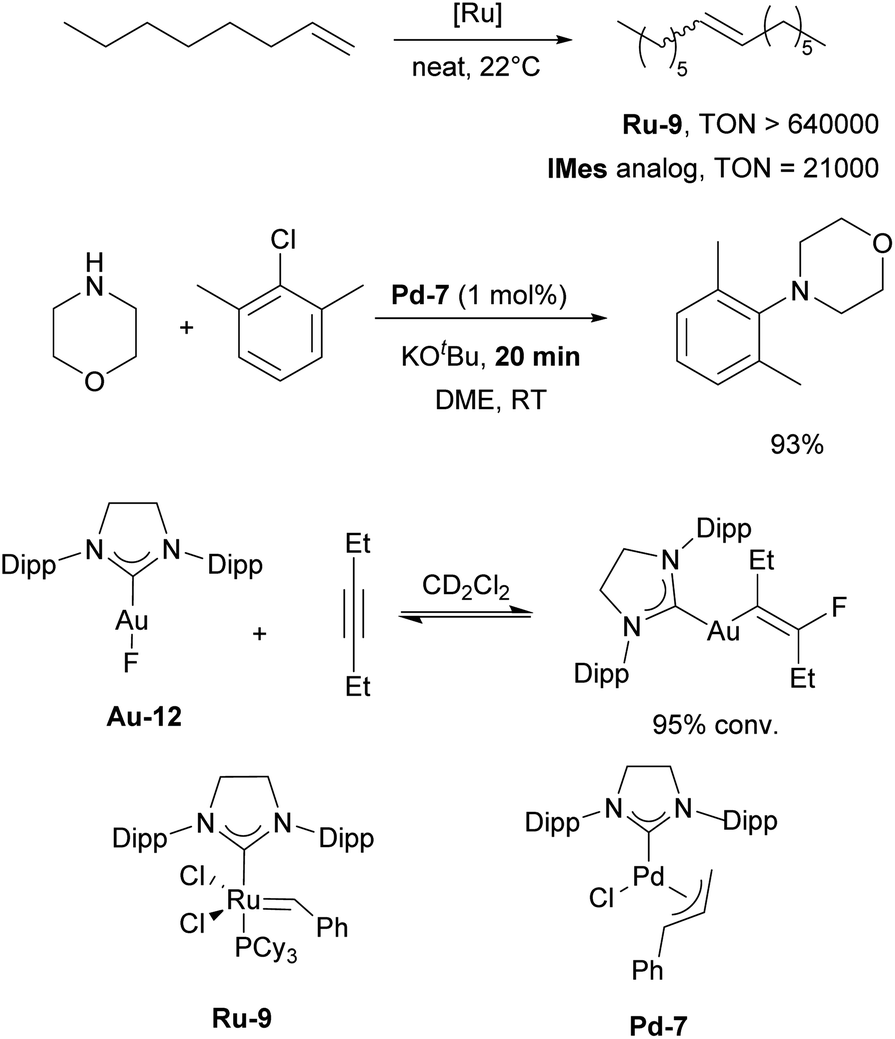
The first article employing IPr was devoted to the use of [Ru(p-cymene)(NHC)Cl2] complexes in ring-closing metathesis (RCM).21 Olefin metathesis plays a pivotal role in modern chemistry, as evidenced by the Nobel Prize in Chemistry in 2005 awarded for the development of this transformative method in organic synthesis. Interestingly, the research groups of Nolan,22 Grubbs,23 and Herrmann/Fürstner24 independently reported on mono-NHC-based ruthenium catalysts for olefin metathesis in the same year as when the IPr ligand was introduced. These catalysts, bearing IMes (1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene) and SIMes (1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene), mostly, are now known as second-generation Grubbs' catalysts.Catalytic activities of p-cymene IPr complex Ru-1 (Fig. 2) together with the IMes congener were tested in RCM employing diethyldiallylmalonate.21 Both IMes and IPr complexes showed high efficiency as catalyst precursors, with IMes being more active at 40 °C, achieving higher conversions than IPr under these conditions (Scheme 2a). However, at elevated temperatures (80 °C), both complexes exhibited similar catalytic activities, indicating that temperature played a key role in overcoming activation barriers in this transformation and using these two distinct NHCs. This study also highlighted the superior stability of these NHC ligands compared to phosphine-based systems, demonstrating their robustness at elevated temperatures.
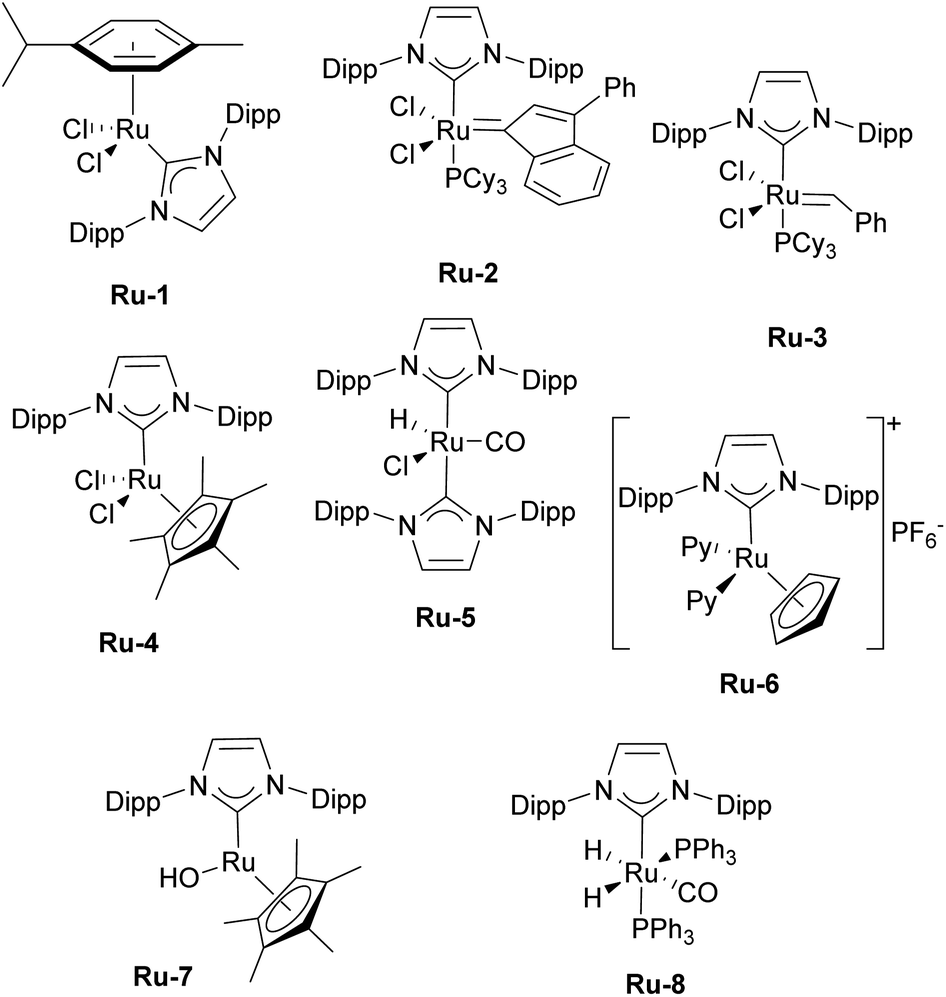 | ||
| Fig. 2 Selected examples of IPr–ruthenium complexes. Dipp = 2,5-diisopropylphenyl, Py = pyridine. | ||
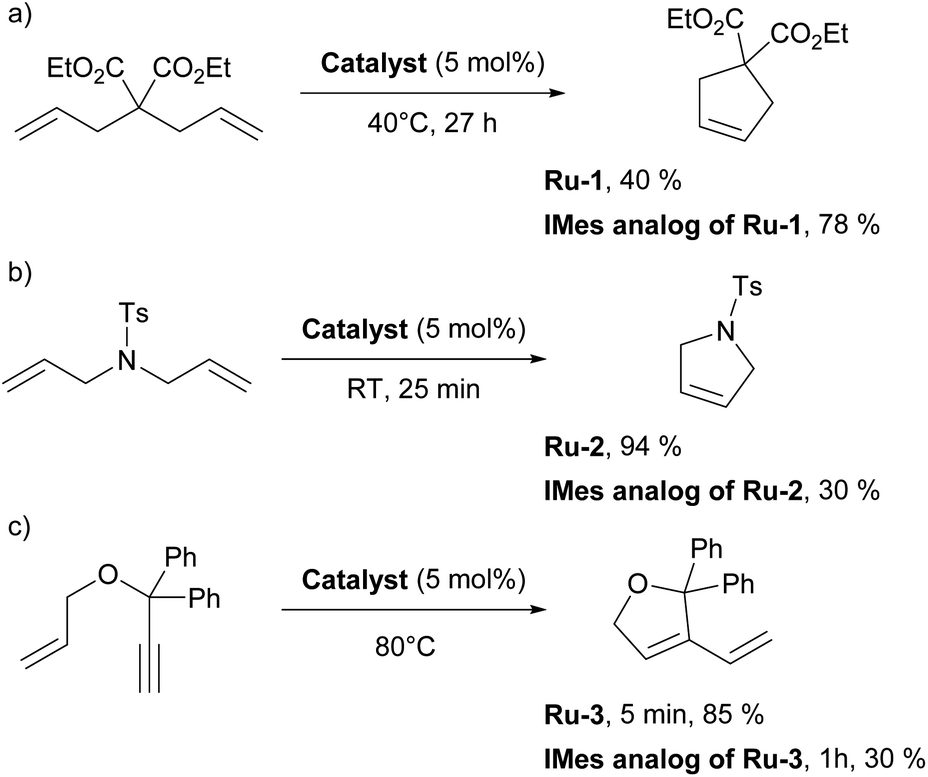 | ||
| Scheme 2 Examples of the use of Ru-1 (a), Ru-2 (b) and Ru-3 (c) in Ring Closing Metathesis (RCM). | ||
Subsequently, similar studies were performed with other precatalysts incorporating the alkylidene, 3-phenylindenylid-1-ene.25 Notably, while the IMes complex was more efficient in reactions involving diethyldiallylmalonate, the IPr complex Ru-2 outperformed IMes in reactions with diallyltosylamine (Scheme 2b). Stability tests further confirmed that both IMes and IPr complexes remain stable at 80 °C for over 10 days without any sign of decomposition, highlighting their robustness under prolonged thermal conditions. The advantageous role of the IPr ligand had yet to be fully demonstrated in olefin metathesis, but this was to come shortly (vide infra).
Subsequently, several comparative studies including IPr ruthenium complexes were reported. Fürstner showed that IPr–ruthenium complex Ru-3 (Fig. 2) in some cases outperformed IMes congeners and had as fast initiation rate for enyne metathesis as the state-of-the-art at a time, the SIMes analog (Scheme 2c).26 The nature of the increased catalytic activity between first and second generation ruthenium olefin metathesis catalysts was studied on the example of IPr, revealing that it slowed bimolecular carbene decomposition in the 14-electron ruthenium intermediate during catalysis,27 thus leading to significantly increased activity and thermal stability (because of the increased steric bulk of IPr) compared to first generation catalysts. In another study, [Ru(Cp*)(IPr)Cl] complex Ru-4 was studied to explain the difference in catalytic activity between saturated and unsaturated NHCs in ruthenium catalyzed olefin metathesis (Fig. 2), revealing that even slight differences in electronic and steric properties of the NHC could cause drastic differences in catalytic activity.28
Even though IMes and especially SIMes ruthenium complexes became go-to catalysts in olefin metathesis, those highly cited important studies, featuring the IPr ligand, contributed to the increased interest and subsequent improved understanding and popularity of the NHC-containing ruthenium metathesis catalysts. The Ru–IPr based systems proved to be excellent olefin metathesis systems but are less frequently employed, we feel, because the SIMes-based system had, by then, taken a strong hold on the market.
Although highly impactful, the IPr–ruthenium chemistry is not only limited to olefin metathesis. Another field where IPr–ruthenium complexes were implemented is in hydrogenation reactions. One of the first examples of the use of IPr–Ru complex in hydrogenation was reported by the Whittlesey group29 who showed that among a series of [Ru(NHC)(L)(CO)(H)Cl] complexes, the most efficient catalyst for the hydrogenation of acetophenone (Scheme 3) was the IPr-based Ru-5 (Fig. 2). Later on, the Nikonov group reported efficient transfer hydrogenation of nitriles and olefins, catalyzed by half-sandwich ruthenium complexes incorporating IPr as ancillary ligand, with best results obtained using Ru-6 (Fig. 2 and Scheme 3).30 The developed protocol allowed for the hydrogenation of nitriles, activated heterocycles such as quinoline, and various olefins. It was shown that these catalysts perform better than phosphine-based cousins.
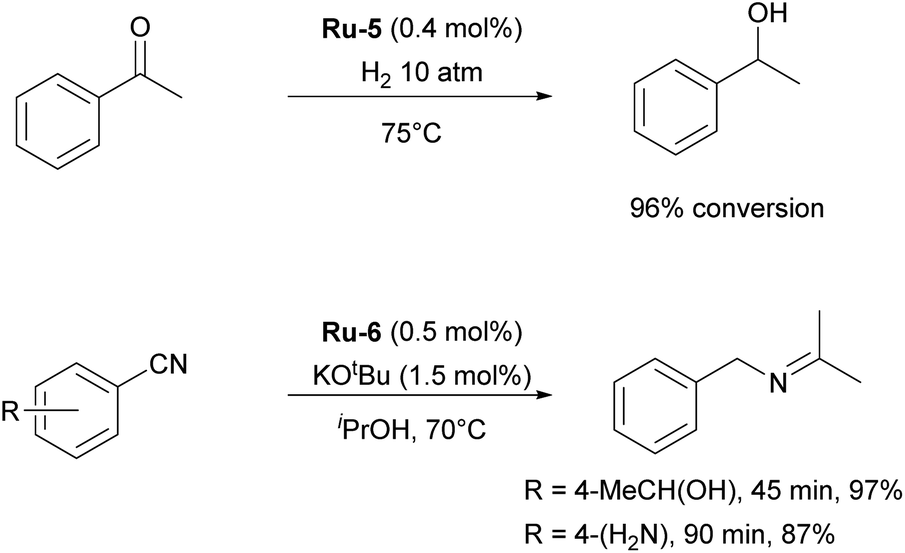 | ||
| Scheme 3 Examples of the use of Ru-5 and Ru-6 in hydrogenation reactions. | ||
Another interesting example of the use of IPr as a ligand is its role as a stabilizing ligand for the isolation of reactive species. It has been shown that heterobimetallic catalyst (IMes)AgRuCp(CO)2 is a highly efficient and selective catalyst for E-selective semi-hydrogenation of alkynes.31 However, attempts to isolate intermediates with IMes as ancillary ligand in order to study the mechanism failed. Therefore, authors turned to IPr analogs of those intermediates and managed their isolation and proposed a mechanism for the transformation. This is also a recurring theme encountered in Transition Metal (TM) IPr chemistry: the improved crystallinity/stability of the IPr-derived complexes. NHC–ruthenium complexes have also been employed in the racemization of chiral alcohols. Although ICy-based complex proved to be more efficient than the IPr counterpart, in the presence of strong base,32 ruthenium hydroxide complex Ru-7 (Fig. 2) bearing IPr could catalyze this transformation for secondary alcohols without base (Scheme 4).33
 | ||
| Scheme 4 Examples of the use of Ru-7 and Ru-8 in racemization of alcohols and hydrodefluorination, respectively. | ||
The Whittlesey group has reported on the catalytic hydrodefluorination of aromatic fluorocarbons using [Ru(NHC)(PPh3)2(CO)H2] complexes. During their studies, the IPr-based catalyst Ru-8 (Fig. 2) showed the highest catalytic activity in hydrodefluorination of pentafluorobenzene in the presence of Et3SiH (Scheme 4).34 The follow-up mechanistic study of this reaction revealed the effects behind the unusual selectivity of this process which turned out to be dependent on the nature and properties of the NHC ligand.35
In summary, the IPr ligand has played a pivotal role in advancing ruthenium chemistry, particularly in the fields of olefin metathesis and hydrogenation. These contributions have not only improved reaction efficiency and selectivity, but also broadened the scope of ruthenium-catalyzed transformations, solidifying the importance of IPr in established areas of ruthenium catalysis.
Nickel catalysis and IPr
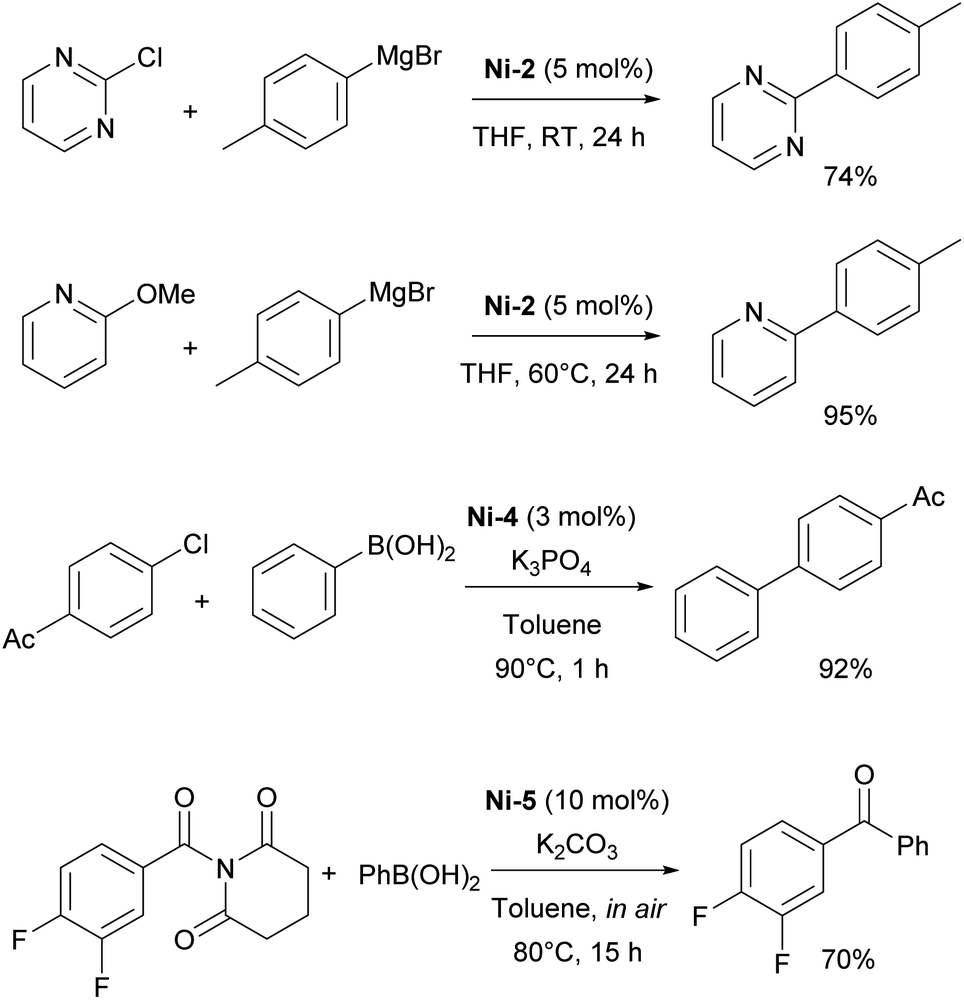
The first report on the IPr–nickel complexes dates back to 2001, when Hermann used IPr2Ni(0) complex Ni-1 (Fig. 3) for room temperature Kumada–Tamao–Corriu coupling between aryl fluorides and aryl Grignard reagents.36 Interestingly, in situ generated catalyst from [Ni(acac)2] and imidazolium salt proved to be slightly more efficient than well-defined complex (Scheme 5a). This was attributed to the fact that mono-ligated IPr–Ni(0) was the active species in this transformation and a species bearing two such units would necessitate a ligand displacement to form the active species. Simultaneously, the Fort group reported the use of the [Ni(acac)2]/IPr·HCl catalytic system for amination of aryl chlorides (Scheme 5b).37 Louie et al. also reported on the use of in situ generated IPr–Ni catalyst for the [2 + 2 + 2] cycloaddition of CO2 and diynes (Scheme 5c).38 A year later, Dible and Sigman reported the isolation of the first mono-ligated Ni(II) complex Ni-2 with IPr as ancillary ligand (Fig. 3).39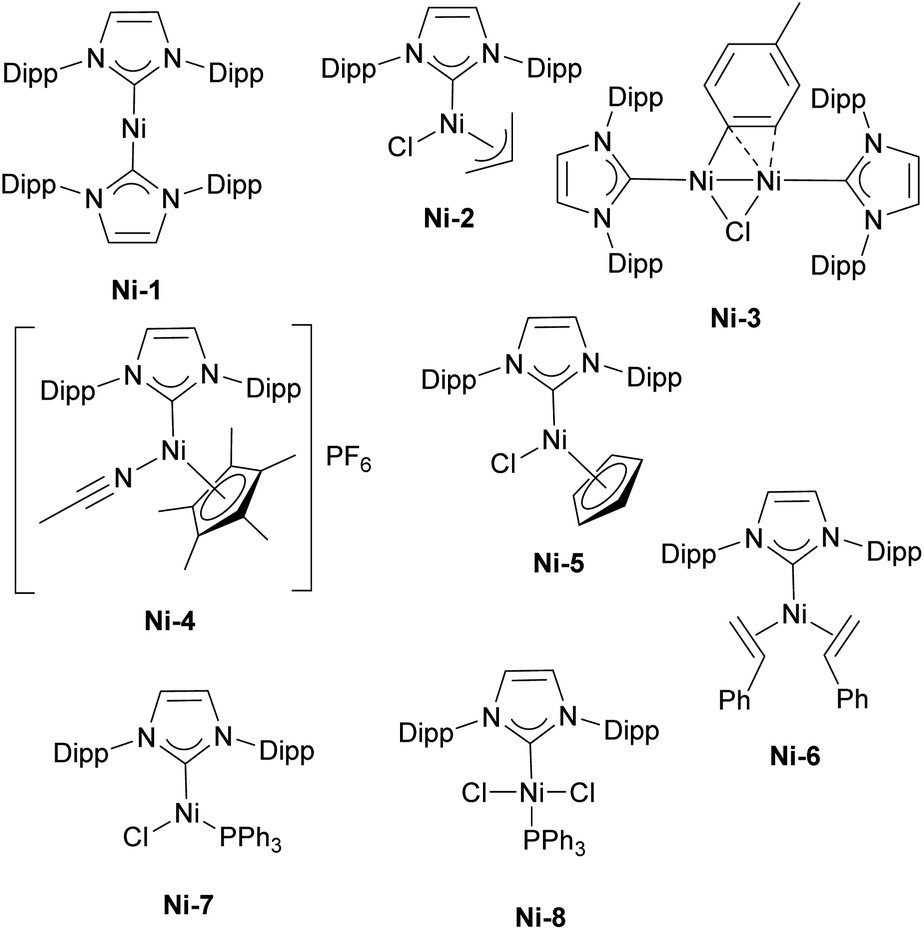 | ||
| Fig. 3 Selected examples of the IPr–nickel complexes used in catalysis. | ||
 | ||
| Scheme 5 Early examples of the use of IPr ligand in Ni catalysis. | ||
Another highly influential study, not only for Ni–NHC chemistry, but one affecting the broader field of NHCs, was conducted by Nolan in the elucidation and quantification of the steric and electronic properties of NHCs. The synthesis and characterisation via IR spectroscopy of [Ni(CO)3(NHC)] complexes were used to measure the CO stretching frequencies leading to determination of Tolman electronic parameter (TEP).40 At the same time, the results from diffraction studies on single crystals were used to calculate the percent buried volume (%Vbur) of the NHC ligands, providing valuable insights into their steric properties.41 This work became the basis for further studies and development of steric descriptors for NHCs which are widely used in explaining the catalytic activity of NHC metal complexes.42–46
These pioneering studies have permitted significant advances in the development of NHC–Ni chemistry. Unlike in ruthenium chemistry, where IMes-based catalysts dominate, IPr remains one of the most widely used monodentate NHC ligands in nickel chemistry and catalysis. The incorporation of NHC ligands not only enabled milder reaction conditions and enhanced selectivity but also facilitated new catalytic transformations. Among the most explored reactions in this area are cross-couplings—particularly Suzuki–Miyaura, Kumada–Tamao–Corriu, and Buchwald–Hartwig reactions—along with α-arylation, C–H functionalization, reductive couplings, cycloadditions, and hydrosilylation, although this short listing is by no means exhaustive.
Building on the Herrmann work, several subsequent studies explored Kumada–Tamao–Corriu (KTC) couplings with IPr–Ni complexes, with two particularly influential examples being the use of Ni-2 and the dinuclear Ni(I)–IPr complex, Ni-3 (Fig. 3). In the first study, the authors successfully performed KTC coupling reactions with both heteroaromatic chlorides and anisoles under relatively mild conditions.47 The second study provided crucial insights into the catalytic cycle, specifically highlighting the involvement of dinuclear monovalent nickel complexes in the reaction mechanism. These findings have greatly improved the understanding of Ni-catalyzed cross-coupling reactions and the role of IPr in enhancing their efficiency.48
Regarding the Suzuki–Miyaura coupling, polydentate NHC nickel complexes have dominated the field. However, it should be mentioned that in a comparative study of half-sandwich NHC–nickel(II) complexes, the IPr complex Ni-4 (Fig. 3) showed the best reaction rate in the Suzuki–Miyaura coupling (Scheme 6).49 Another Ni–IPr complex Ni-5 (Fig. 3) recently showed high catalytic activity in Suzuki–Miyaura coupling of amides under aerobic conditions.50
 | ||
| Scheme 6 IPr–nickel complexes in KTC and Suzuki–Miyaura couplings. | ||
Following Fort's work, several studies have appeared on the use of Ni–NHC catalytic systems for amination reactions. In 2005, Schneider introduced the in situ generation of the Ni-1 complex for efficient amination of aryl chlorides, demonstrating that IPr provided superior reactivity compared to other ligands.51
The Nicasio group utilized the previously mentioned allyl complex, Ni-2, for the amination of (hetero)aryl chlorides under mild conditions, showcasing, for the first time, that this transformation could be achieved at room temperature with a nickel pre-catalyst (the IMes-coordinated congener of Ni-2 exhibited very poor reactivity).52 Building on this work, the same group developed another pre-catalyst, [Ni(IPr)(styrene)2] (Ni-6, Fig. 3), which was successfully applied to the amination of aryl tosylates,53 and later to the N-arylation of indoles and carbazoles (Scheme 7).54 Another important report featuring IPr was devoted to the use of Y-shaped monovalent nickel complex Ni-7 (Fig. 3), which enabled the amination of arylhalides with diphenylamine at room temperature, an unprecedented result, at the time.55 One of the latest examples where IPr ligand was used to stabilize active intermediates in nickel catalysis is the mechanistic study of the Ni(I)–Ni(III) catalytic cycle in the Buchwald–Hartwig amination.56
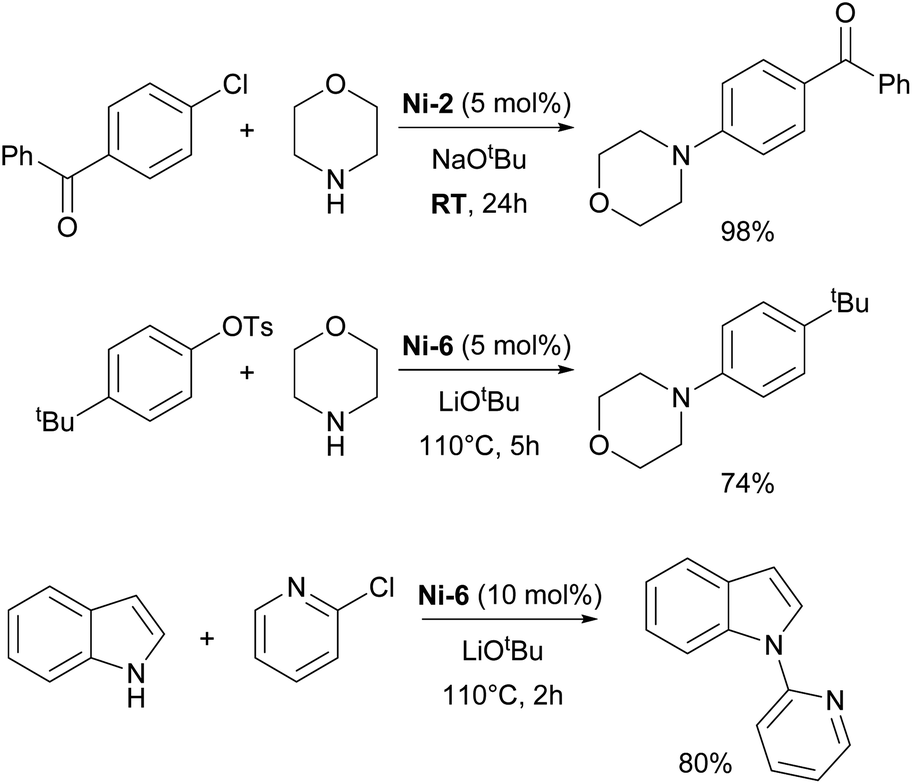 | ||
| Scheme 7 IPr–nickel complexes in amination reactions. | ||
In 2014, Hartwig and co-workers reported the hydroarylation of olefins with trifluoromethyl-substituted arenes using the [Ni(IPr)2] Ni-1 complex. This transformation displayed unprecedented selectivity for the linear anti-Markovnikov product.57
The α-arylation transformation is another reaction where IPr–Ni complexes have been successfully employed. Matsubara et al. utilized a mixed NHC/PR3 nickel complex containing IPr (Ni-8, Fig. 3) for the arylation of propiophenones with various aryl halides, although this required a relatively high catalyst loading of 10 mol% at an elevated temperature of 110 °C (Scheme 8).58 Several years later, the Ritleng group reported the use of the half-sandwich complex Ni-5, which permitted to reduce the catalyst loading to 3 mol% and to broaden the reaction scope (the IMes-congener of Ni-5 was not very efficient, leading to a 25% yield, using 5 mol% catalyst loading, whereas Ni-5 afforded a 78% yield with 3 mol% catalyst loading).59 Further advances were made by increasing the steric bulk of the IPr core structure with phenyl substituents, leading to improved efficiency in the α-arylation reaction.60 This ground-breaking work laid the foundation for a series of studies in which either IPr or modified IPr-based ligands were employed to further enhance selectivity and expand the reaction scope to include very challenging substrates.61–63
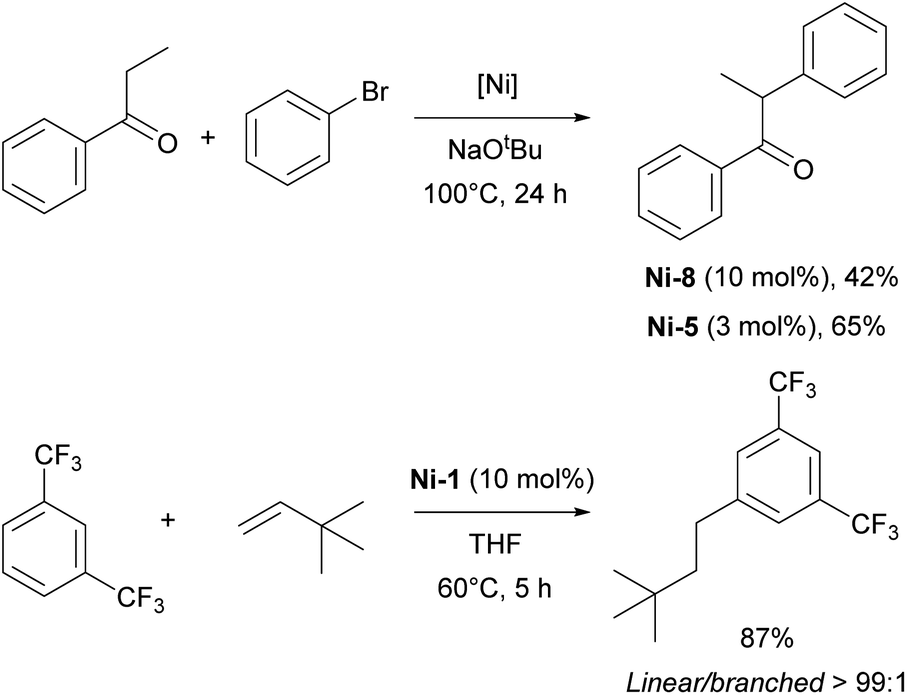 | ||
| Scheme 8 IPr–nickel complexes in α-arylation and hydroarylation reactions. | ||
Additionally, it is important to highlight other key contributions to NHC–nickel chemistry, where the IPr ligand has played a pioneering role in facilitating the development of novel methods and reactions. These include (but are not limited to) the use of IPr–Ni complexes in hydrosilylation,64,65 C–H functionalization polycondensation,66 dual photoredox catalysis67 and metalloradical catalysis.68,69
Copper catalysis and IPr
The history of NHC–copper chemistry began in 1993 when Arduengo reported the synthesis of the first homoleptic NHC complexes of copper.70 Since then, numerous reports featuring NHC–copper complexes in organometallic chemistry and their deployment in catalysis have been published. A significant milestone came with the work of Buchwald and co-workers, who synthesized the first well-defined IPr-containing copper complex, Cu-1 (Fig. 4), which played a pivotal role in advancing this field. This complex proved to be highly efficient in the conjugate reduction of α,β-unsaturated carbonyl compounds, setting a new standard for catalytic performance at the time (Scheme 9a).71 The reaction mechanism was proposed to involve the formation of an NHC–copper hydride intermediate, generated through the reaction of in situ formed [Cu(NHC)(OtBu)] and polymethylhexasilazane (PMHS), a hypothesis later confirmed by stoichiometric experiments conducted by Sadighi.72 NHC ligands played a pivotal role in the use of copper complexes in catalysis in general, with IPr being often the first NHC ligand successfully employed in the development of a new transformation or improvement of existing ones.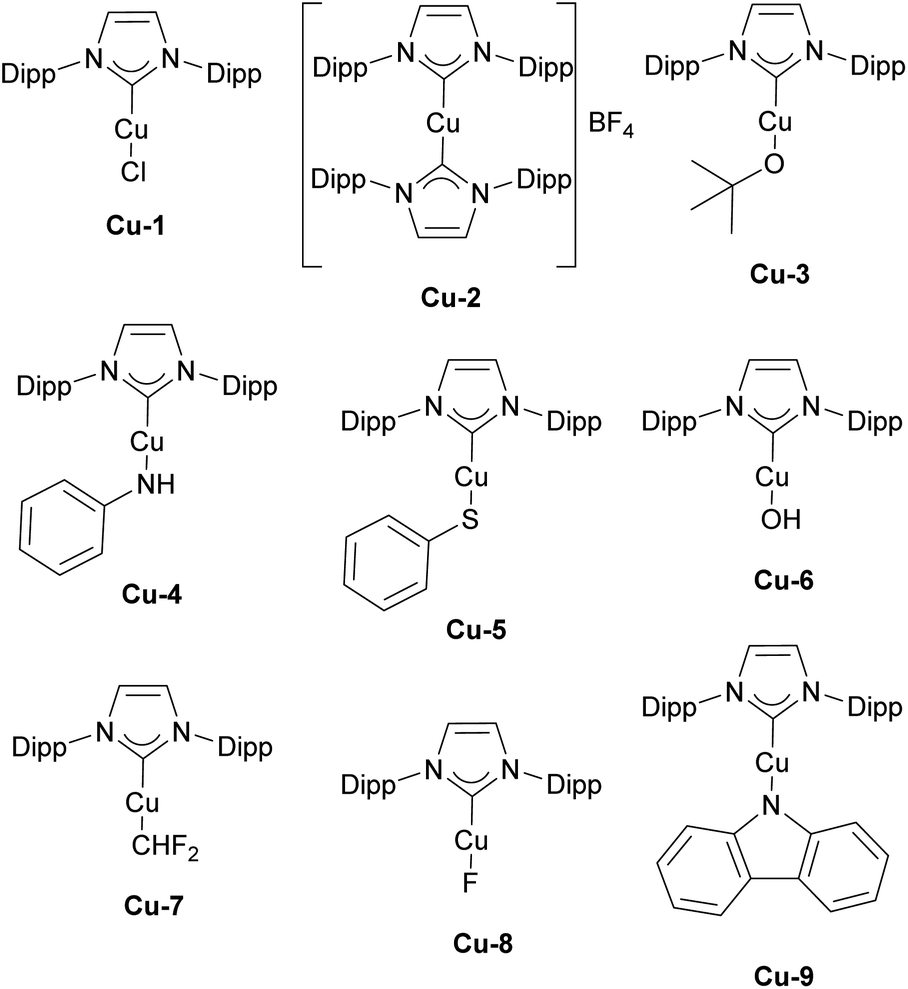 | ||
| Fig. 4 A selection of IPr–copper complexes used in catalysis. | ||
 | ||
| Scheme 9 Use of Cu-1 (a and b) and Cu-2 (c) in conjugate reduction and hydrosilylation of ketones. | ||
In the same year as Buchwald reported the synthesis of the first IPr–copper complex, Nolan and co-workers utilized this catalytic system in the hydrosilylation of ketones at room temperature (Scheme 9b).73 In the follow-up study, the Nolan group introduced cationic complex Cu-2 (Fig. 4) to mediate the same transformation, which proved to be slightly more efficient and allowed for an expanded reaction scope (Scheme 9c).74 A slightly different reaction mechanism was proposed for the cationic complex, where dissociation of one of the IPr ligands occurs prior to formation of the tert-butoxide intermediate.75
Other important contributions opening the way for various catalytic transformations mediated by NHC–copper complexes came from Sadighi and Petersen on insertions into Cu–alkyl bond. The insertion of CO2 into Cu–CH3 under mild conditions to form copper acetate was established in the first report.76 This reactivity, along with Sadighi's earlier findings on the interaction of copper alkoxide species with B–B bonds,77 paved the way for Hou's important development of the carboxylation of organoboronic esters, catalyzed by [Cu(IPr)Cl] (Scheme 10).78 The Hou research group also reported on the hydrosilylation of carbon dioxide using [Cu(IPr)(OtBu)] (Cu-3, Fig. 4) as the catalyst, further expanding the scope of copper-mediated transformations to CO2 activation/conversion (99% vs. 74% yield for the hydrosilylation of carbon dioxide, between Cu-3 and the corresponding IMes-coordinated congener, at 100 °C).79
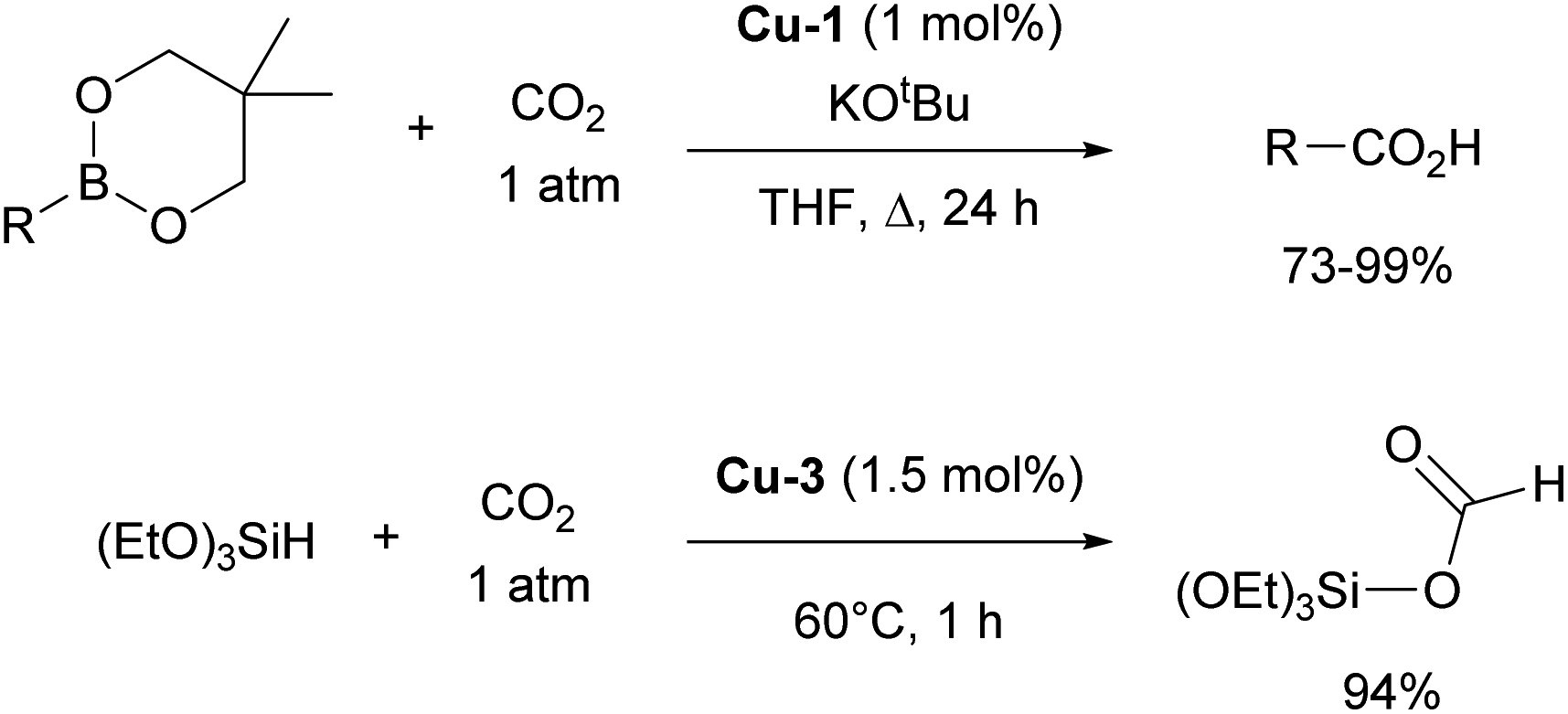 | ||
| Scheme 10 IPr–Cu complexes in CO2 insertion chemistry. | ||
Further studies revealed that the [Cu(IPr)(CH3)] complex reacts with various compounds containing acidic N–H, C–H, and O–H bonds, leading to the formation of [Cu(IPr)X] species and the release of methane.80,81 These [Cu(IPr)X] complexes were subsequently employed as catalysts for anti-Markovnikov additions of N–H and O–H bonds to olefins (Scheme 11a).82 The same strategy was extended to the hydrothiolation of olefins using [Cu(IPr)(SR)] catalysts (Scheme 11b).83
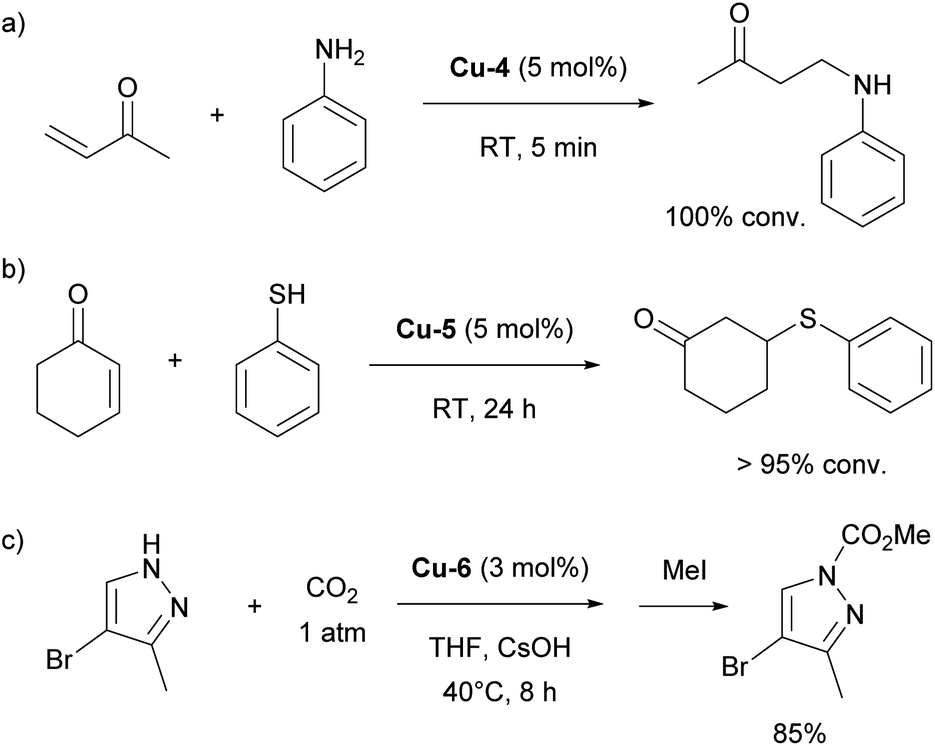 | ||
| Scheme 11 Use of IPr–copper in hydroamination, hydrothiolation and carboxylation. | ||
In 2010, the Nolan group introduced a new IPr–copper hydroxide complex, Cu-6 (Fig. 4), which possessed the ability to activate various C–H bonds under mild conditions.84 This property allowed for the in situ formation of [Cu(IPr)R] species containing Cu–C bonds, which were later used successfully in the catalytic carboxylation of C–H and N–H bonds (Cu-6 exhibited higher reactivity than the SIPr-, IMes- and SIMes-coordinated congeners, 69% vs. 28% vs. 16% vs. 23%, using 2-methyl-1H-imidazole as reagent and 1 mol% catalyst loading, respectively), utilizing CO2 as a reagent (Scheme 11c).85
Recently, numerous reports have focused on the use of NHC–copper complexes, particularly IPr–Cu complexes, in carbonylative and borylative transformations. The Mankad group has reported on hydrocarbonylative couplings of alkynes with alkyl halides, where the [Cu(IPr)Cl] complex proved to be the most efficient for coupling alkyl iodides with both terminal and internal alkynes (Scheme 12).86,87 Following these reports, a carbonylative C–C coupling of arylboronic esters with alkyl halides for the synthesis of ketones was developed, using a dual Mn/Cu catalytic system. This method is compatible with various arylboronic ester nucleophiles and works efficiently with both primary and secondary alkyl iodide electrophiles (Scheme 12).88 Another important contribution utilizing this chemistry was devoted to carbonylative silylation of alkyl halides, providing access to valuable acylsilane synthons.89
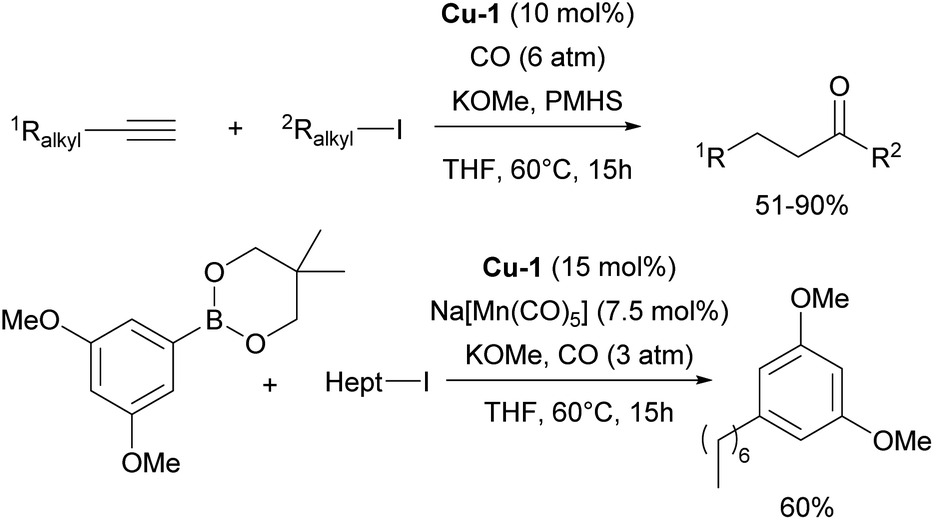 | ||
| Scheme 12 Use of IPr–copper complexes in carbonylative transformations. | ||
In 2015, Miura and Hirano reported a ligand-controlled regiodivergent Cu-catalyzed aminoboration of unactivated terminal alkenes. Interestingly, while using [Cu(Xantphos)Cl], boration occurs at the terminal position, however, the use of [Cu(IPr)Br] provides access to borated product at internal positions with very good regioselectivity (Scheme 13a).90 This methodology was later extended to include regiodivergent arylboration of the dienes using Cu/Pd catalytic system, where [Cu(IPr)Cl] played a key role (Scheme 13b).91
 | ||
| Scheme 13 Examples of use of IPr–Cu complexes in borylation chemistry. | ||
The Procter group reported on the borylative cross-coupling of allenes and imines using the [Cu(IPr)Cl]/KOtBu catalytic system.92 In this case, boration occurred selectively at the internal position of the allene, yielding branched α,β-substituted-γ-boryl homoallylic amines (Scheme 13c). This approach was further applied to the synthesis of quaternary α-amino esters (Scheme 13d).93
In addition, it is important to highlight that the IPr ligand allowed for the isolation and characterization of the first copper difluoromethyl complex Cu-7 (Fig. 4).94 This important intermediate had never been isolated previously, despite copper-based catalytic systems being among the oldest and most widely used for the introduction of the CHF2 group. Following stoichiometric experiments, a catalytic version of this transformation was developed using [Cu(IPr)Cl] as pre-catalyst and (TMS)CHF2 as the difluoromethyl source (Scheme 14).94
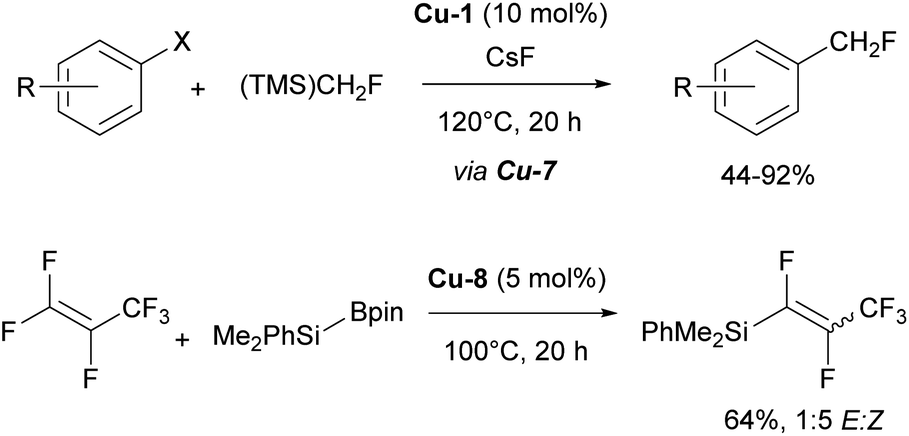 | ||
| Scheme 14 The use of IPr–Cu complexes in fluorination and defluorination chemistry. | ||
Another noteworthy contribution in fluorine chemistry, featuring the IPr–Cu complex Cu-8 (Fig. 4), is the defluorosilylation of fluoroalkene feedstocks providing access to fluorinated silanes.95
In closing this section, the synthesis of copper carbene–metal–amido (CMA) complexes, such as Cu-9 (Fig. 4), which feature carbazole as an ancillary ligand is also worth mentioning. These complexes exhibited promising dual-emission properties and ultralong room-temperature phosphorescence (RTP), which hold great potential for the development of advanced RTP materials.96
Gold catalysis and IPr
The report describing the synthesis and characterization of the first IPr gold complex dates to 2005.97 The [Au(IPr)Cl] complex Au-1 (Fig. 5) was prepared by mixing free carbene with the [Au(SMe2)Cl] precursor, in a simple ligand displacement reaction. These [Au(NHC)Cl] complexes have quickly become widely used as pre-catalysts in gold-catalyzed transformations. Although much more cost-efficient and sustainable synthetic methods were developed to access NHC gold complexes,98 this important report represents the onset of an avalanche of publications devoted to the use of these in various catalytic transformations. Notably, since the initial report, more than 800 publications have featured the IPr–gold core structure, establishing gold organometallic chemistry as a leading platform for the application of IPr as an ancillary ligand. While it is beyond the scope of this contribution to cover all these studies, we will focus on key milestones that have reinforced the central role of IPr in gold catalysis.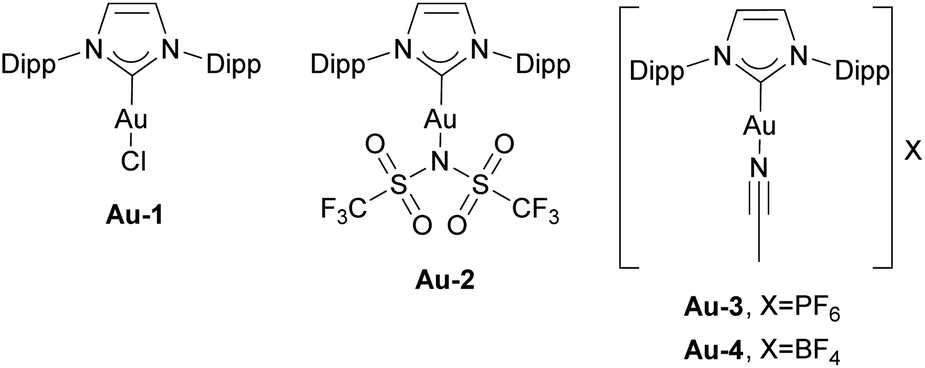 | ||
| Fig. 5 Examples of the IPr gold complexes. | ||
Upon gaining access to this new class of pre-catalysts, researchers naturally began exploring their potential in transformations previously mediated by phosphine–gold and NHC–copper complexes. In many instances, these investigations resulted in the development of milder reaction conditions, attributed to the unique steric and electronic properties of the NHC ligand, which enhance both the stability and reactivity of the active LAu+ species. Interestingly, the use of NHC–gold complexes has sometimes led to the discovery of entirely new catalytic reactions or significant alterations in reactivity compared to previously known systems. Notably, in most of these studies, IPr–gold complexes were the primary candidates tested and employed, underscoring the critical role of IPr in advancing gold-catalyzed transformations.
One particularly interesting example of a new catalytic reaction developed using gold NHC complexes, rather than the more traditional copper NHC complexes, is related to carbene-transfer reactions. Nolan and co-workers demonstrated that [Au(IPr)Cl] not only catalyzed the expected cyclopropanation of olefins but also facilitated the formal insertion of carbenes into N–H and O–H bonds. Remarkably, the gold catalyst enabled unprecedented carbene insertion into the aromatic C–H bonds of styrene, benzene and toluene (Scheme 15a).99,100 Among the many studies highlighting the enhanced reactivity of NHC–gold complexes, several early seminal reports stand out. Widenhoefer and co-workers reported that using [Au(IPr)Cl] for the intramolecular hydroamination of N-alkenylacetamides allowed the reaction to proceed at lower temperatures than those required with phosphine-based analogues (Scheme 15b)101 Similarly, the Toste group observed higher catalytic activity with IPr–gold complexes in the oxidative rearrangement of enynes compared to PPh3-ligated gold complex (Scheme 15c).102
 | ||
| Scheme 15 Early examples of the use of IPr–gold complexes exhibiting superior reactivity in comparison with phosphine–gold analogues. | ||
The Zhang group utilized [Au(IPr)NTf2] complex Au-2 (Fig. 5) in the generation of α-oxo gold carbenoids and their insertion into R–CO bonds (Scheme 15d).103 Again, while an IPr complex facilitated this transformation, the related PPh3 analogue failed to do so.
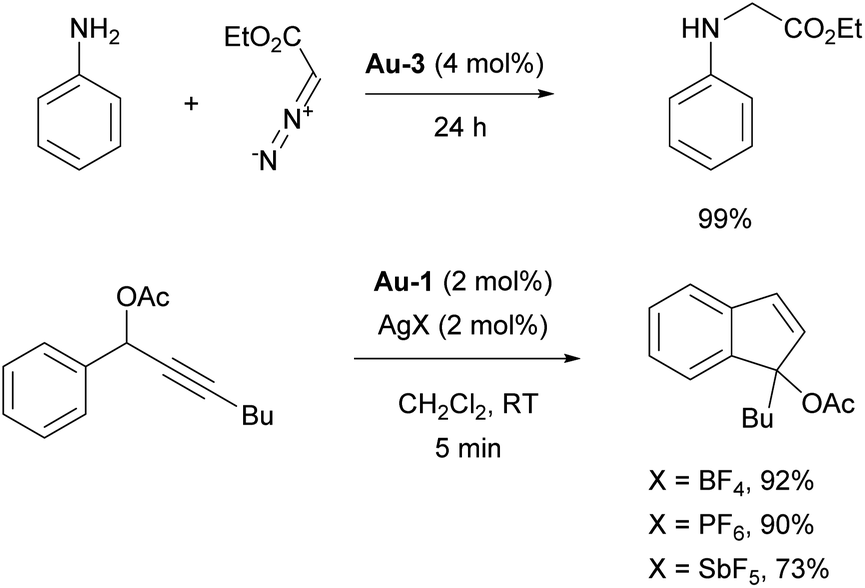
In 2006 the Nolan group reported the isolation of well-defined cationic gold complex Au-3 (Fig. 5), allowed to perform catalysis without activating AgX or NaBAr4 salts (Scheme 16).104
 | ||
| Scheme 16 The use of Au-3 in carbene insertion chemistry, and effect of counterion on hydroarylation. | ||
The catalytic activity of Au-3 (Fig. 5) was tested in carbene insertion of EDA and revealed the significant effect of the counterion on the reactivity. The same conclusion was drawn while using [Au(IPr)Cl]/AgX catalytic system for novel tandem [3,3]-rearrangement-intramolecular hydroarylation to access indenes (Scheme 16; note that the SIMes-coordinated congener afforded a moderate yield, in contrast to Au-1/AgX).105 The silver-free route, under Au-6/HBF4·Et2O (Scheme 18), supported by theoretical calculations is also efficient, where pre-catalyst activation is mediated by the HBF4·Et2O that acts as Brønsted acid.106 In 2007, Ricard and Gagosz reported the synthesis of already mentioned IPr gold catalyst Au-2 with weakly coordinating NTf2 moiety.107 Although simultaneously reported by Zhang,103 the synthesis and characterization of the complex was reported only in the former. These complexes demonstrated enhanced stability, particularly under ambient conditions, and proved highly active in a variety of gold-catalyzed transformations without the need of activating agent.107 Based on these and related work, numerous reports dealing with the effect of the counterion in gold–NHC catalysis have appeared recently, revealing new aspects of the mechanisms of these transformations. Most of these reports once again utilized IPr–gold complexes as the “gold standard” for this chemistry.108–110
The remarkable ability of gold to activate π-bonds has long been recognized as one of its most distinctive features, accounting for the predominant focus on π-bond activation in gold-catalyzed reactions as seen above. Another important contribution in this area is the work of Echavarren in comprehensive studies of the gold-catalyzed intermolecular addition of carbon nucleophiles to 1,5- and 1,6-enynes.111 From this report, it became clear that the reaction pathway could be directed by the auxiliary ligand, leading either to cyclopropane or carbene-site selectivity. The presence of NHC ligands, such as IPr, leads to carbene-site selectivity in most cases (Scheme 17).
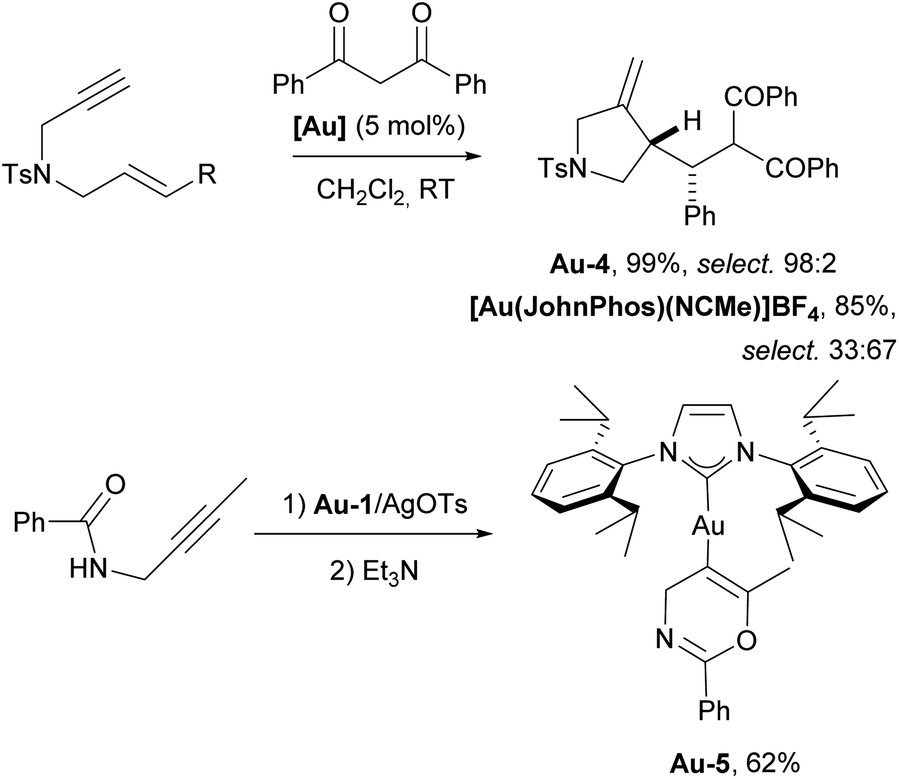 | ||
| Scheme 17 The use of IPr–gold complex for intermolecular addition of carbon nucleophiles to 1,6-enyne and isolation of the first gold–vinyl complex Au-5. | ||
The Hashmi group successfully isolated the first vinylgold complex Au-5 (Scheme 17) using N-propargylcarboxamides as substrates and IPr as an auxiliary ligand on gold. This achievement provided direct evidence for the existence of these species in gold-catalyzed reactions with alkynes, which had been proposed as key intermediates but had never been isolated prior to this work.112 Here again we have an example of the use of IPr to isolate reactive species.
The Nolan group reported on the use of [Au(IPr)Cl]/AgSbF6 catalytic system for both formation of conjugated enols and enals, and acid-free alkyne hydration at ppm level catalyst loadings (Scheme 18, top).113 In both cases, the presence of H2O in the reaction mixture proved to be an important factor, allowing catalytic reaction to occur. Based on experimental and computational data, the reaction was hypothesized to proceed through the intermediate formation of [Au(IPr)OH] species. Soon afterwards, complex Au-6 was isolated using a simple procedure of mixing [Au(IPr)Cl] with a hydroxide source. The obtained complex was found to be air- and moisture-stable, although highly basic and under the right conditions, very reactive.114 The reactivity of this “golden synthon” was illustrated by reactions with various acids and compounds containing acidic C–H and O–H bonds, performed in air and at ambient temperature. Among several products accessible using this synthon, the gold hydride [Au(IPr)(H)] Au-7, gold aryl and alkynyl complexes Au-8 and Au-9 (Scheme 18) could be easily accessed. Furthermore, this development allowed for the use of gold hydroxide as a precatalyst for various transformations, using a Brønsted acid as activator to generate the LAu+ active species.115 These silver-free protocols are of significant importance, as silver was found to not just act as an innocent activator but an actor for several transformations.116 One of the most significant uses of the gold hydroxide is in the carboxylation of C–H bonds directly with CO2.117
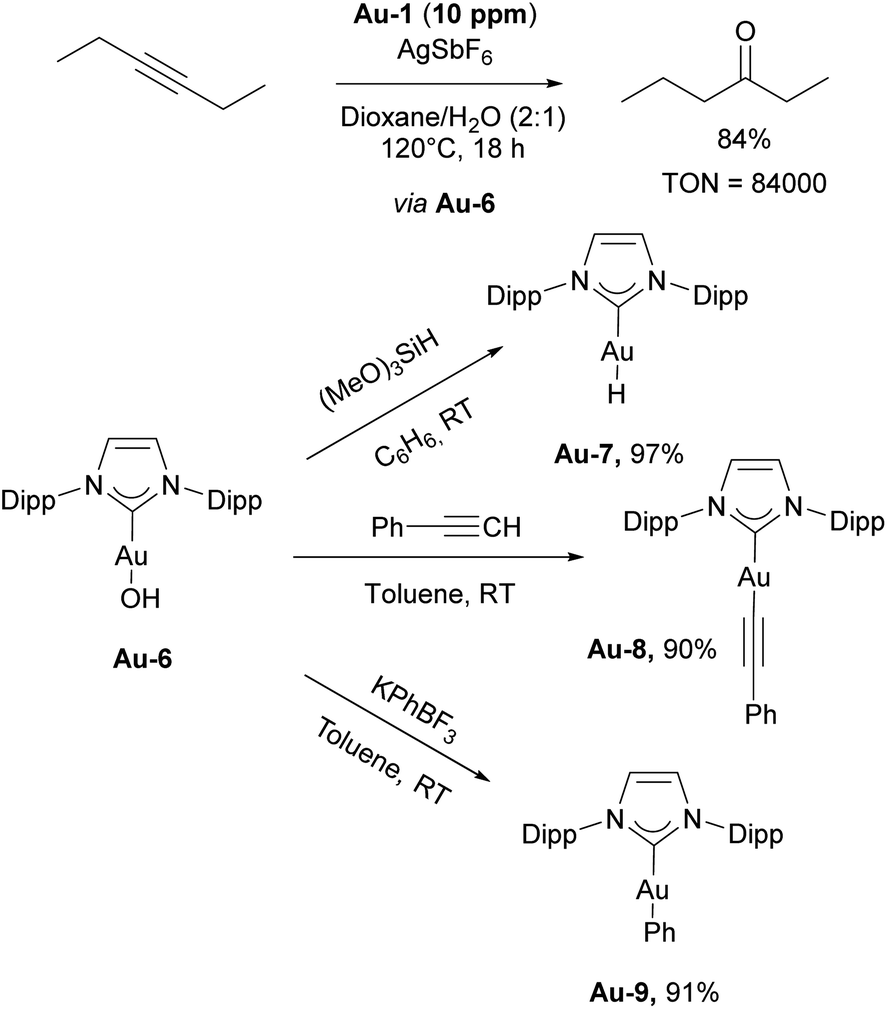 | ||
| Scheme 18 Gold-catalyzed hydration of alkynes and reactivity of Au-5 hydroxide. | ||
In the report describing silver-free protocols, the authors noted the formation of unreported species in the reaction mixture, which upon further investigation turned out to be a dinuclear species, [{Au(IPr)}2(μ-OH)]+.115 The isolated complex [{Au(IPr)}2(μ-OH)][BF4] Au-10 (Scheme 19) demonstrated excellent catalytic performance in the hydration of nitriles118 and hydrophenoxylation of alkynes,119,120 attributed to a “dual activation”121 mechanism. This dual activation arises from the equilibrium between dinuclear complex and [Au(IPr)OH]/[Au(IPr)(BF4)] pair, where the former functions as a Brønsted base and the latter operates as a Lewis acid (Scheme 19). This catalyst was later successfully employed in the hydroalkoxylation and hydrocarboxylation of alkynes.122,123
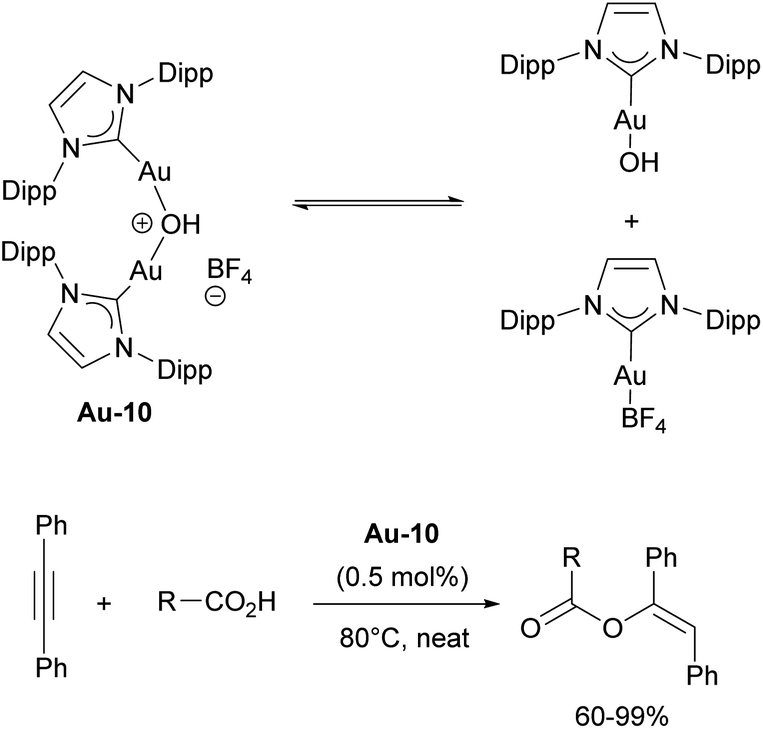 | ||
| Scheme 19 Equilibrium of Au-10 complex and its use in carboxylation of alkynes. | ||
Just prior to reports on dinuclear gold catalysts, the Hashmi group explored various gold-catalyzed arylative cyclizations of 1,x-diynes. This series of reports was of major importance in gold catalysis, as it shed light on and provided evidence for various important intermediates in gold-catalyzed reactions involving activation of π-bonds, confirming the dual catalytic role of gold.
In most cases, the IPr ligand was used as ancillary ligand, allowing for the detection or/and isolation of important intermediates. In 2012, Hashmi showed that formation of Au–C σ-bond plays an important role in the activation of triple bonds involving further π-binding of the second bond atom.124 In the same report, the authors managed to isolate the first NHC-containing gem-diaurated aryl species Au-11 and provided the rationale for its role in the catalytic cycle (Scheme 20). The follow-up report provided further compelling evidence for the dual activation role of gold and the involvement of gold vinylidene intermediates in the formation of dibenzopentalenes from arene-1,2-diynes.125 Finally, those σ,π-alkyne gold acetylide complexes were isolated and fully characterized, including X-ray diffraction on single crystals, which provided insights in their structure (Scheme 20). Those complexes were probed as on-cycle catalysts for hydroarylating aromatization and proved efficient without additives.126 In contrast to these findings, the Echavarren group successfully isolated several σ,π-digold alkyne complexes bearing the IPr ligand. Following mechanistic investigations, including both experimental and theoretical studies, they concluded that these complexes represent “dead ends” in gold catalysis involving enynes.127
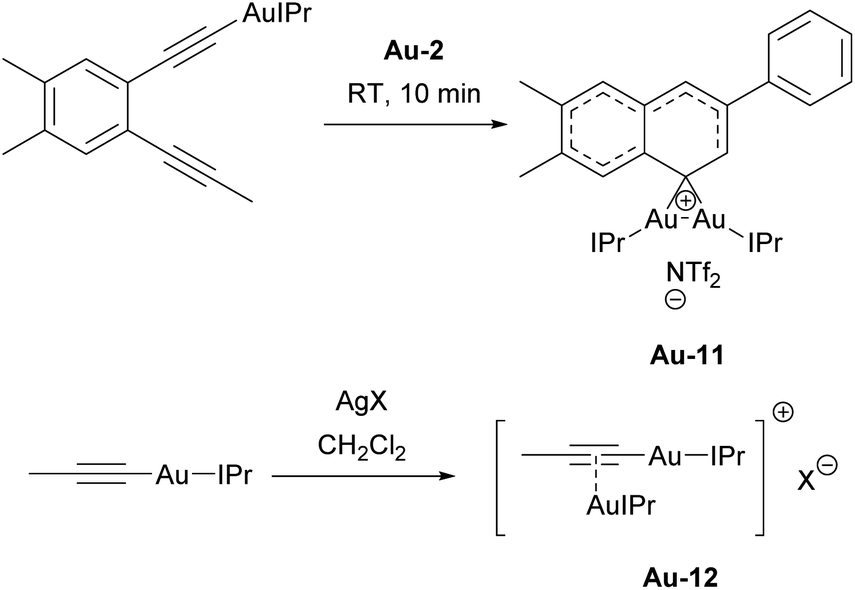 | ||
| Scheme 20 Isolation of important intermediates Au-11 and Au-12. | ||
With ongoing advances in gold catalysis, IPr has emerged as the preferred ligand for NHC–gold catalytic systems, reinforcing its position as one of the most widely employed ligands in this field. Recent applications of IPr include a variety of transformations including the domino128 and cascade cyclization,129 cycloaddition,130 and annulation131 reactions, as well as for hydrofluorination132 and hydration133 of triple bonds. The versatility displayed by IPr extends even to more unconventional areas of gold chemistry, including the encapsulation134 of gold complexes and their use in energy transfer photocatalysis.135,136
Palladium catalysis and IPr
Catalytic studies involving IPr in palladium catalysis began shortly after the discovery of the ligand itself, in 1999. The initial reports focused on the amination of aryl halides13 and the Kumada–Tamao–Corriu coupling,14 both utilizing in situ generated catalysts from Pd2(dba)3 and imidazolium salts as precursors. This approach was later extended to Hiyama and Stille couplings, using Pd(OAc)2 as the palladium source (Scheme 21).137,138 After the development of such efficient protocols for those transformations, the next significant development was the design of Pd/NHC well-defined complexes.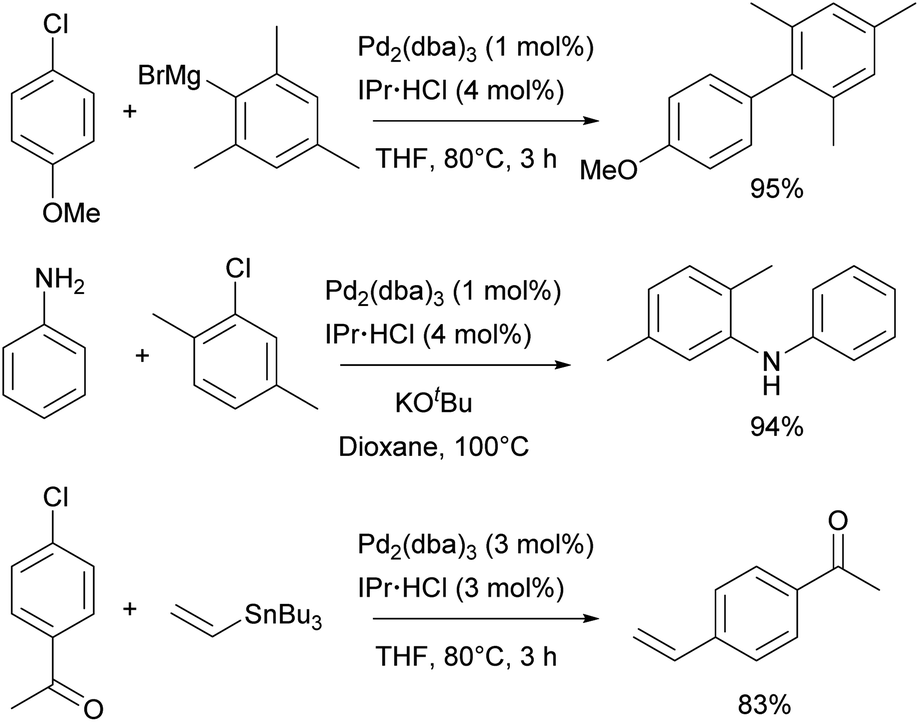 | ||
| Scheme 21 Some early examples of IPr ligand use in palladium chemistry. | ||
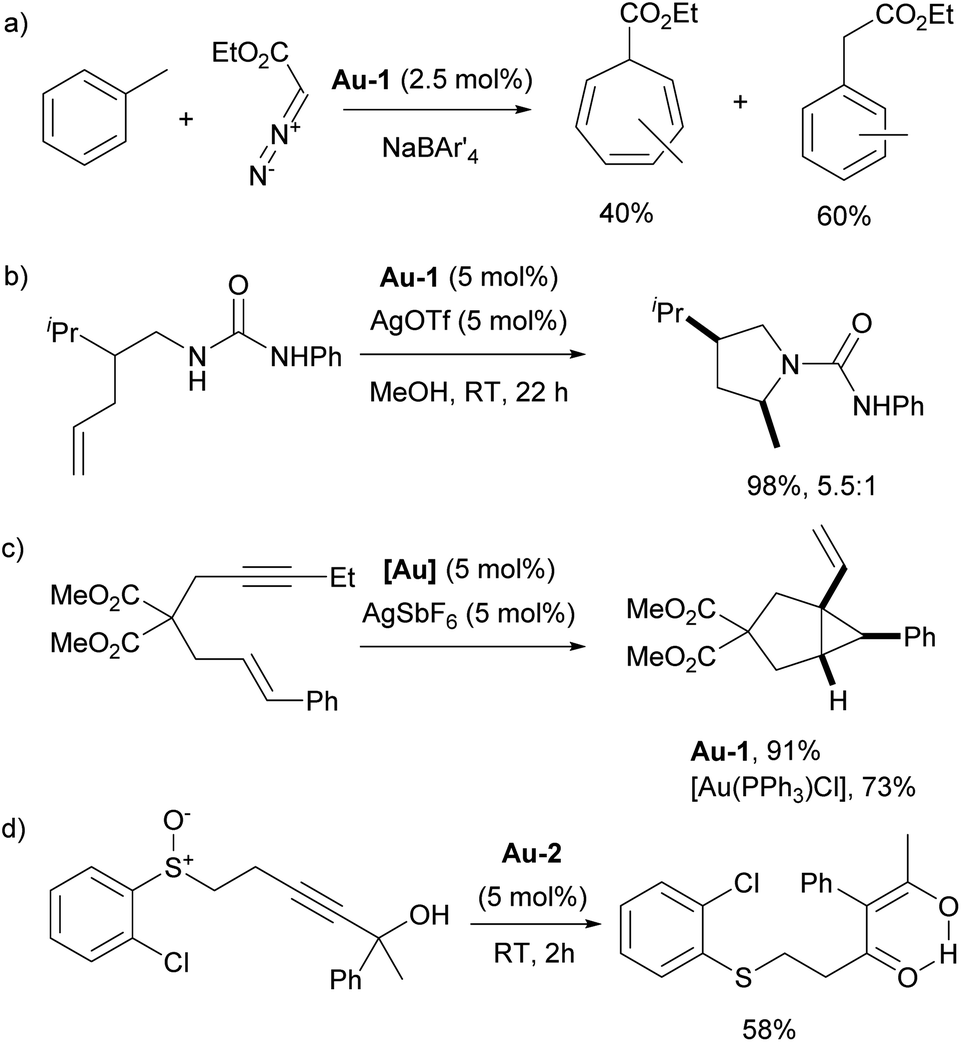
In 2002, the Nolan group introduced two families of the well-defined Pd–NHC precatalysts – NHC palladium μ-chloro-bridged dimers [Pd(NHC)Cl2]2139 and NHC palladium allyl complexes [Pd(NHC)(allyl)Cl].140 Subsequently, both of these families played a substantial role in further advancing the area of NHC–palladium catalysis.
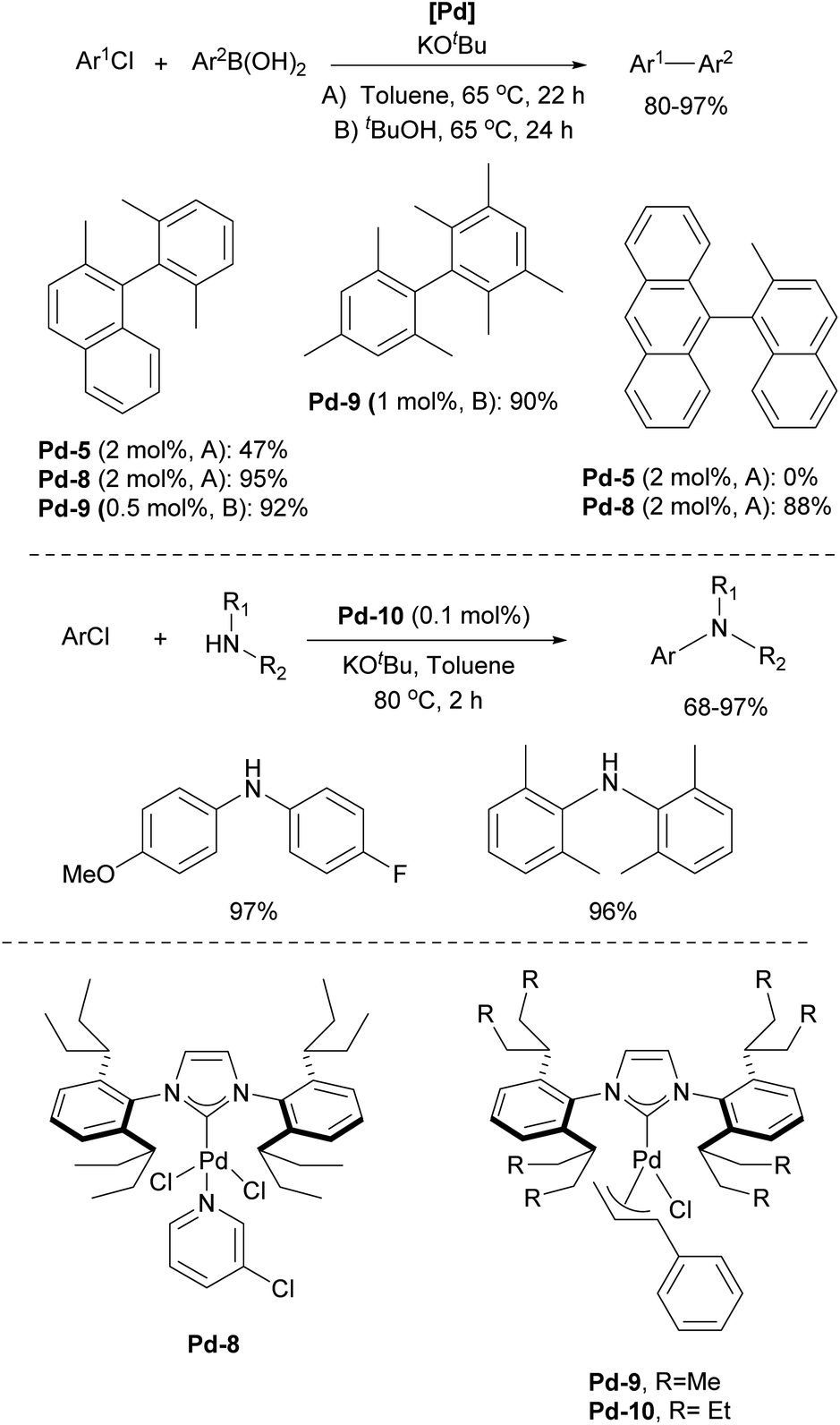
In particular, [Pd(IPr)Cl2]2 (Pd-1, Fig. 6) showed remarkable catalytic activity in various aryl halide aminations, including very challenging versions and operating under surprisingly mild reaction conditions, i.e. without the need for anhydrous and anaerobic conditions (Scheme 22a).139 Subsequently, this catalyst was employed in room-temperature Suzuki–Miyaura couplings,141,142 and a recent comprehensive study on its activation has been published.143 This study included a direct comparison with various other precatalysts developed later and revealed that this previously “overlooked” family of pre-catalysts remains among the most active. Consequently, the authors suggested that these pre-catalysts should be considered as a preferred option in catalytic applications.
 | ||
| Fig. 6 Selected examples of IPr–palladium complexes used in catalysis. | ||
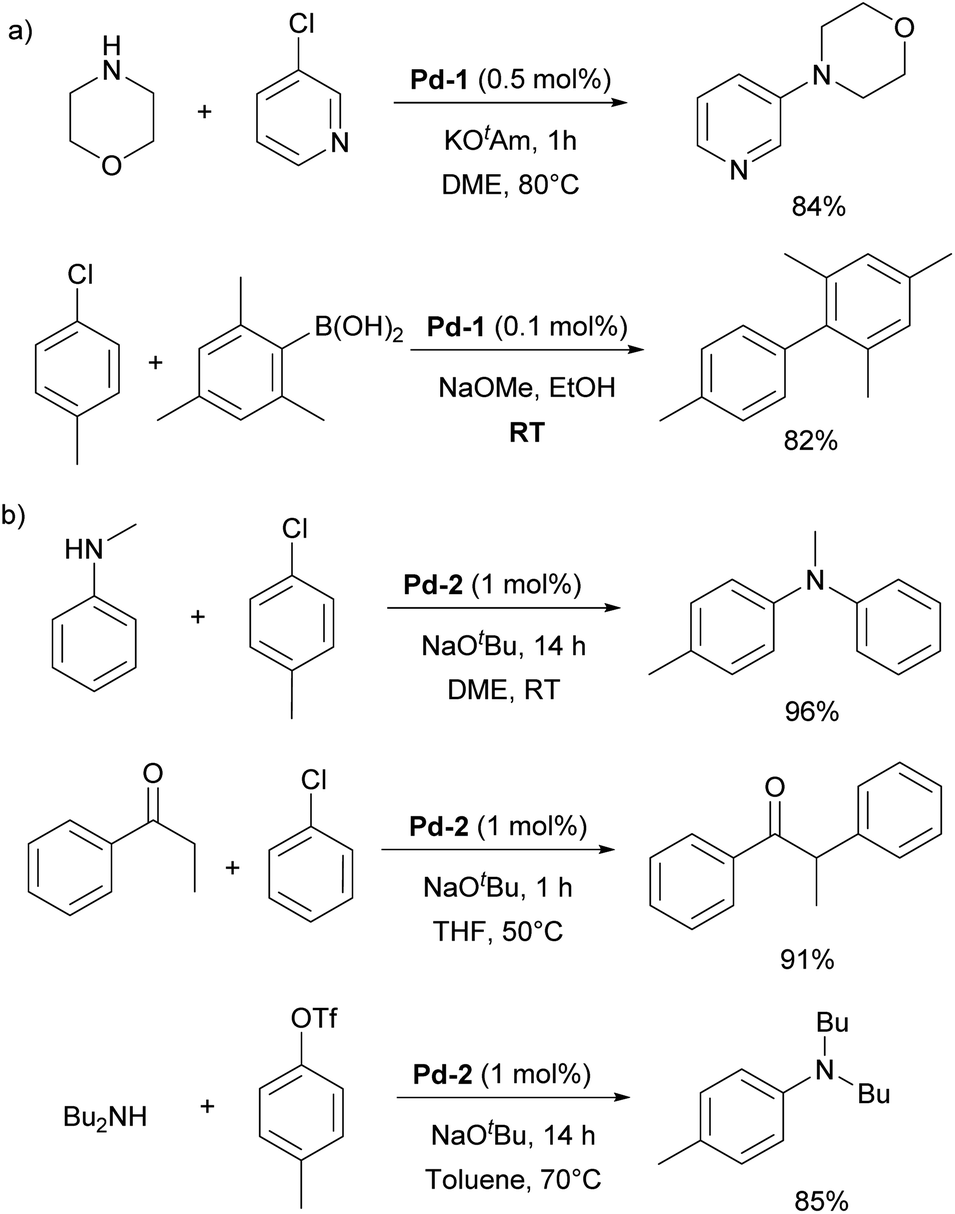 | ||
| Scheme 22 Utilization of Pd-1 and Pd-2 in cross-coupling reactions. | ||
[Pd(NHC)(allyl)Cl] complexes, being air- and moisture-stable compounds, were successfully tested in the arylation of ketones, Suzuki–Miyaura and Buchwald–Hartwig cross-coupling reactions.140,144,145 Later on, a more detailed study on these reactions was reported revealing a very high efficiency of [Pd(IPr)(allyl)Cl] (Pd-2, Fig. 6) (Scheme 22b).146
Since then, considerable amount of research has focused on optimizing the structure of NHC–palladium precatalysts to develop more readily activated and efficient complexes. Numerous families of precatalysts have been synthesized and evaluated, with varying degrees of success in terms of catalytic efficiency. However, it is noteworthy that precatalysts containing IPr as an ancillary ligand are almost always among the first to be synthesized and tested, serving as a benchmark in comparative studies.
One of such families was NHC palladacycles, which combined the stability of the metallacycle scaffold with strong σ-donating properties of the NHC ligands. In a series of reports IPr congener Pd-3 (Fig. 6) showed high catalytic activity in various room temperature cross-coupling reactions, including Suzuki–Miyaura coupling with hindered (di)ortho-substituted aryl chlorides (Scheme 23).147,148 A more comprehensive study on Suzuki–Miyaura, α-ketone arylation and dehalogenation reactions provided efficient protocols for the use of these precatalysts, with IPr complex Pd-3 being the most efficient.149 The Morandi group recently used the same precatalyst for C–S and C–P bonds metathesis by reversible arylation.150
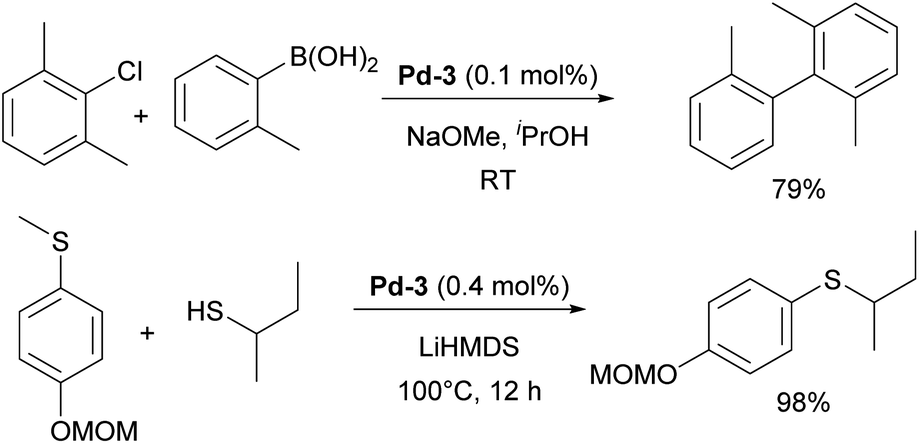 | ||
| Scheme 23 The use of Pd-3 pre-catalyst in catalysis. | ||
During the same period, the Sigman group worked on the palladium-catalyzed aerobic oxidation of alcohols. After successfully using the aforementioned [Pd(IPr)Cl2]2 dimer (Pd-1) for oxidative kinetic resolution of secondary alcohols151 and based on mechanistic insights, they introduced IPr–palladium–acetato complex (Pd-4, Fig. 6) for aerobic oxidation of alcohols (Scheme 24).152 The acetate ligand plays a key role in facilitating intramolecular deprotonation, which enhances catalytic efficiency. Further in-depth mechanistic investigations and screenings153 led to significant advances in this field.154 The introduction of the tosylato congener [Pd(IPr)(OTs)2] obtained in situ from Pd-1 and AgOTf, permitted the use of tert-butyl hydroperoxide (TBHP) as the oxidant instead of copper additives in the Wacker oxidation of styrenes, leading to high conversion rates and selectivity for acetophenone as a substrate.155 This precatalyst was also employed in the oxidative 1,1-diarylation of terminal olefins156 and oxidative Heck coupling.157
 | ||
| Scheme 24 Use of IPr acetato and tosylato palladium complexes in catalysis. | ||
In 2006, two of the most successful and widely utilized families of NHC–palladium precatalysts were introduced. In both cases, IPr-ligated precatalysts were synthesized and evaluated in a variety of reactions to demonstrate their robustness, versatility and efficiency. The Organ group pioneered the use of 3-chloropyridine as a “throw-away” ligand, resulting in the development of PEPPSI precatalysts, which stands for Pyridine-Enhanced Precatalyst Preparation, Stabilization, and Initiation.158 These precatalysts can be produced on a large scale under ambient conditions and are highly bench-stable, allowing for long-term storage. Initially, these catalysts were prepared using a non-environmentally friendly procedure with 3-chloropyridine as the solvent. However, a more atom-economical and environmentally friendly method has recently been developed.159 The well-defined IPr–Pd–PEPPSI complex Pd-5 (Fig. 6) was first tested in Negishi reactions for C(sp3)–C(sp3) coupling, as well as in Suzuki–Miyaura couplings with boronic acids and trifluoroborates. Notably, the well-defined system exhibited a much faster initial reaction rate and significantly higher turnover numbers (TON) compared to in situ generated catalysts. This complex alone appears in more than 360 publications since its discovery, including reports featuring its implementation in Buchwald–Hartwig,160 Negishi161 and Kumada–Tamao–Corriu162 cross-couplings (Scheme 25). The non-chlorinated version of Pd-5 has recently been used in a mechanistic study devoted to R–NHC coupling under catalytic conditions,163 which was shown to dictate whether the so-called NHC-connected or disconnected mode of catalysis is at play in particular transformations.164
 | ||
| Scheme 25 Representative examples of the IPr–Pd–PEPPSI complex (Pd-5) employed in diverse cross-coupling reactions. | ||
The Nolan group developed a modified version of [Pd(NHC)(allyl)Cl] complexes (let us call these, the second generation Pd-allyl family) by altering the substituents on the allyl moiety.165 It was found that the incorporation of a phenyl group at the terminal position of the allyl ligand facilitated faster activation of the precatalysts, leading to enhanced catalytic activity across a broad range of transformations. Note that this lesson learned in Pd–NHC catalyst development has been adapted to Pd-Buchwald phosphine by Colacot and co-workers at Johnson Matthey.166
The resulting NHC Pd–cinnamyl (and crotyl and prenyl) complexes, particularly [Pd(IPr)(cinnamyl)Cl] (Pd-6, Fig. 6), exhibited exceptional efficiency, enabling Suzuki–Miyaura couplings with challenging substrates at very low catalyst loadings and at room temperature. In some cases, reactions were successfully conducted with as little as 50 ppm of catalyst, achieving 93% yield within 3 hours, demonstrating the remarkable efficiency of the developed catalytic system (Scheme 26). The Pd-6 catalyst demonstrated exceptional reactivity in Suzuki–Miyaura couplings with non-traditional electrophiles, including aryl esters and amides, as reported by the Szostak group.167,168 In both instances, the developed protocol enabled the efficient synthesis of diaryl ketones with a broad substrate scope and high yields, utilizing a commercially available palladium precatalyst. Studying the effect of the substitution on the backbone of the N-heterocyclic ring of Pd-6 in the Suzuki–Miyaura coupling experimentally and through DFT calculations provided crucial insights into the activation pathway of precatalysts and reactivity of complexes.169 Additionally, this catalyst was employed in the first general method for the transamidation of secondary carboxamides, significantly expanding the scope of amide N–C bond cross-coupling reactions.170,171 In this arena, it should be clearly stated that Pd–phosphine complexes have been found lacking in terms of catalytic effectiveness and that Pd–NHC systems rule the day in this area.
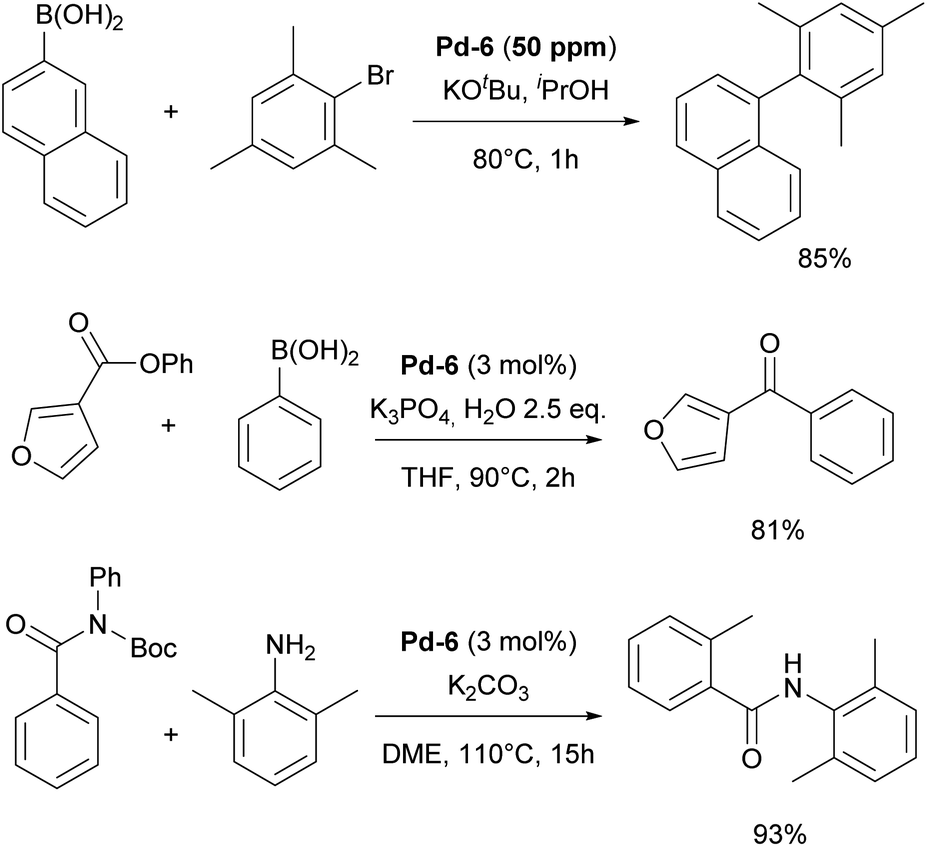 | ||
| Scheme 26 Representative examples of the IPr–palladium cinnamyl complex (Pd-6) employed in various cross-coupling reactions. | ||
The pursuit of more efficient, robust, universal, and sustainable NHC–palladium precatalysts remains an ongoing area of research, leading to the development and evaluation of numerous new pre-catalysts, such as the [Pd(NHC)(acac)Cl]172 and [Pd(NHC)(η3-1-tBu-indenyl)Cl] systems.173 Notably, the IPr ligand continues to serve as a benchmark for comparison within these novel structures, underscoring its pivotal role in NHC palladium catalysis. However, following the success of highly efficient NHC–Pd–cinnamyl and NHC–Pd–PEPPSI pre-catalysts, researchers have increasingly focused on modifying the NHC ligand itself to further enhance catalytic efficiency and selectivity, especially for more challenging substrates. This shift has led to the development of various NHC ligands inspired by IPr, which will be discussed in the following section, providing insights into the design, evolution, and applications of these new NHC ligands in catalysis.
The far-reaching influence of the IPr structure
As could be seen from the previous sections, the role of IPr as an indispensable ligand in the vast array of NHCs is evident. The use of the ligand consistently enhances stability, catalytic activity, and selectivity in various catalytic processes featuring a variety of transition metals, making it a privileged construct in the design of efficient catalytic systems. The versatility of IPr in facilitating a wide range of transformations, from cross-couplings to hydroaminations and metathesis, underscores its adaptability to different metals and reaction conditions. As we look to the impact (past and future) of IPr on NHC ligand design, the following section will offer an overview of the evolution of IPr-inspired ligands, discussing the emerging modifications that aim to move the boundaries of catalytic performance forward and meet the demands for increasingly efficient and sustainable transformations.174Over the course of the previous two decades, substantial efforts have been made by several research groups to exploit the unique properties that are inherent to the IPr architecture, to design and synthesize new families of NHC ligands, with the goal of exploring their potential in catalysis. The closest analogue to IPr is its saturated counterpart, (1,3-bis(2,6-diisopropylphenyl))imidazolidin-2-ylidene (SIPr). Arduengo reported the synthesis of the free SIPr in the same year as when IPr ligand was introduced,175 so it cannot be said that the development of SIPr was directly inspired by IPr. However, the IPr-inspired modifications, we feel, has led to the design of four major NHC architectures: IPr* (1,3-bis[2,6-bis(diphenylmethyl)-4-methylphenyl] imidazole-2-ylidene, IPr** (1,3-bis(2,6-di(p-tertbutylphenyl)methyl-4-methylphenyl)imidazole-2-ylidene, IPr# (1,3-bis-(2,4,6-tribenzhydrylphenyl)-1H-imidazole-2-ylidene), and all members of the ITent (Tent = tentacular) family (Fig. 7).
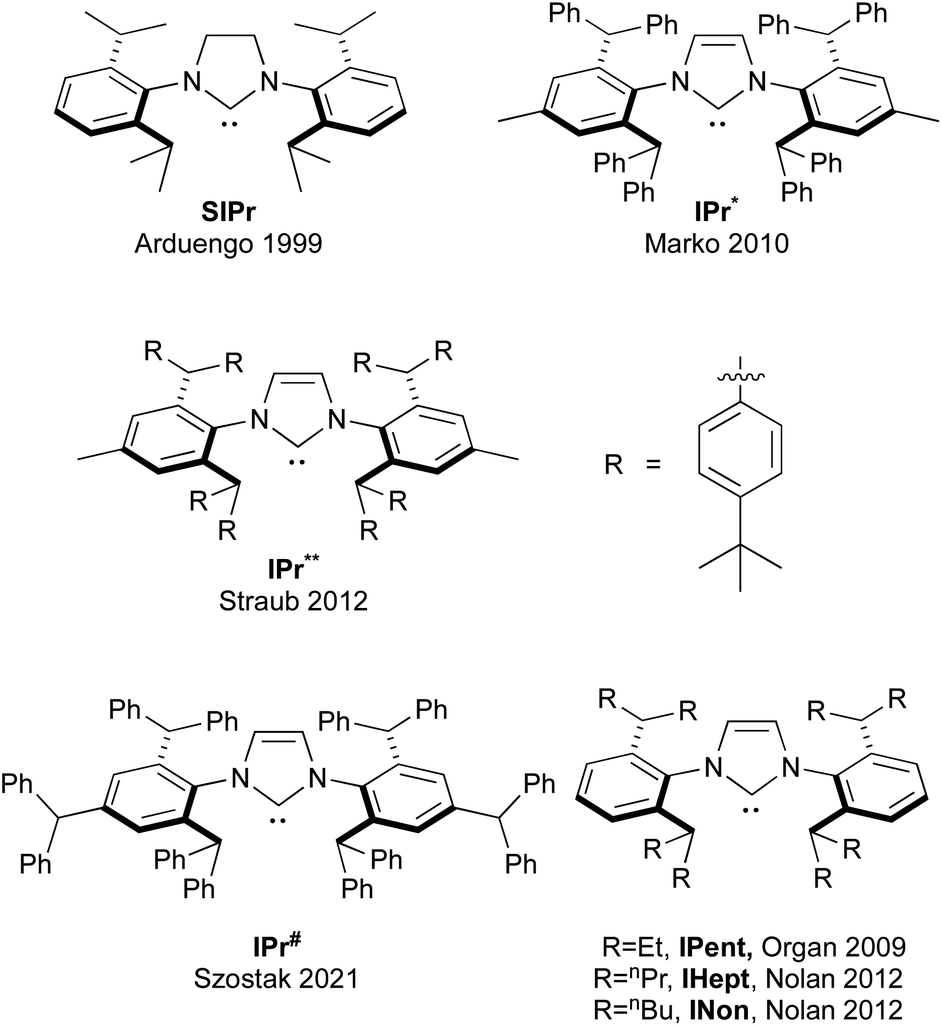 | ||
| Fig. 7 SIPr and other NHC ligands inspired and developed based on the IPr structure. | ||
In the following section, we will highlight selected examples of applications of these IPr-inspired NHCs in catalysis. The IPr and SIPr, being structurally similar, have often been examined side-by-side in many of the aforementioned-reactions. In most cases, IPr demonstrated somewhat better catalytic activity, stability, and/or selectivity. However, there are several notable instances where SIPr outperformed its counterpart. Saturated NHCs occupy a unique position among ligands in ruthenium metathesis, as even slight variations in the electronic and steric properties of NHC ligands can significantly impact reactivity.28 Shortly after the development of SIPr, it was shown that complexes containing SIPr exhibited substantially higher catalytic activity, with 6- and 20-fold increases in TON and TOF, respectively, for the metathesis of 1-octene, compared to the (S)IMes catalyst, which was the standard at the time (Scheme 27).176 Similarly, SIPr-based complexes have been found to be highly active in acyclic diene metathesis polymerization (ADMET).177 In palladium chemistry, SIPr-ligated complex Pd-7 allowed N-aryl amination with aryl chlorides at room temperature in minutes.178 [Au(SIPr)F] complex was used to detect and isolate β-(fluorovinyl)gold complexes for the first time, showing the reversible nature of their formation (Scheme 27). This observation resulted in the first gold-catalyzed hydrofluorination of alkynes under mild conditions.179 Interestingly, SIPr was also a ligand of choice in one of the widely known studies by the Fokin group, where the mechanism of copper-catalyzed azide–alkyne cycloaddition (CuAAC) was explored and the ligand used to stabilize dinuclear copper intermediates.180
 | ||
| Scheme 27 Examples of the use of SIPr in catalysis. | ||
One of the most influential classes of NHCs inspired by the IPr scaffold is the ITent (tentacular NHCs) family, which includes IPent (1,3-bis(2,6-di(3-pentyl)phenyl)imidazole-2-ylidene), IHept (1,3-bis(2,6-di(4-heptyl)phenyl)imidazole-2-ylidene), and INon (1,3-bis(2,6-di(5-nonyl)phenyl)imidazole-2-ylidene). Although IPent was first described by the Organ group,181 its synthesis was not reported in the literature until the Nolan group disclosed its detailed synthetic protocol.182 The IPent PEPPSI complex Pd-8 demonstrated exceptional activity in Suzuki–Miyaura cross-coupling reactions, particularly in the synthesis of challenging tetra-ortho-substituted biaryls, catalytically outperforming its IPr counterpart (Scheme 28).181 Additionally, the IPent cinnamyl complex Pd-9 was also found to be highly active for the same transformation, while the IHept congener Pd-10 exhibited the highest reactivity in Buchwald–Hartwig amination (Scheme 28).182 However, in both case studies, the increased steric bulk of INon did not result in higher catalytic activity, offering valuable insights into the limitations of the “bulky yet flexible” concept in NHC design.
 | ||
| Scheme 28 Use of ITent family ligands in palladium chemistry. | ||
Subsequently, the Organ group discovered that introducing halide atoms on the backbone (C4 and C5 positions) of the NHC ligands in the ITent family, could enhance catalytic activity. In particular, Pd–PEPPSI–IPentClPd-11 was found to be an excellent precatalyst for selective monoarylation of primary amines with aryl halides, affording monoarylation products in 49–99% yields (Scheme 29).183 The IHeptCl analog of Pd-12 was also utilized in the selective coupling of secondary alkyl zinc reagents and heteroaryl bromides, accelerating the reductive elimination, which leads to minimal amount of rearranged products caused by migratory insertion.184
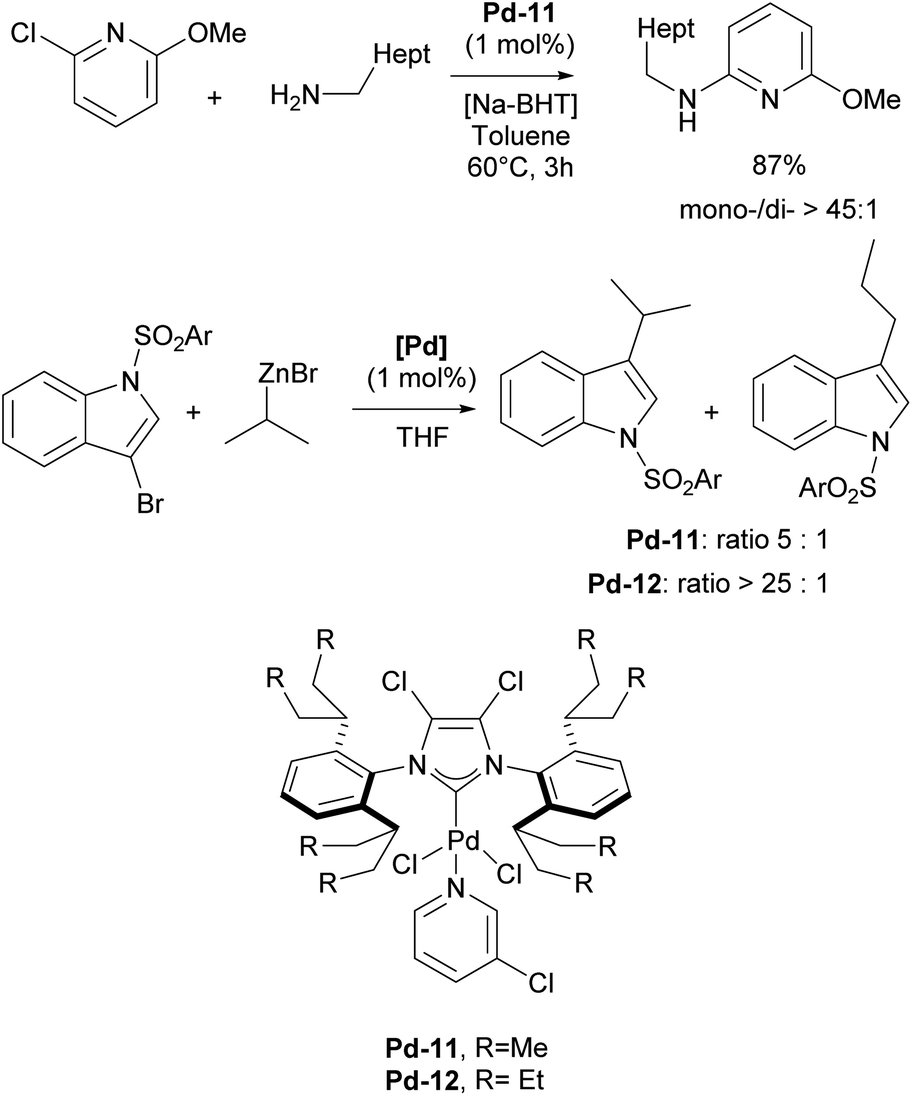 | ||
| Scheme 29 Use of backbone-chlorinated IPent and IHept ligands in catalysis. | ||
The higher reactivity of the Pd–PEPPSI–IHeptCl precatalyst, in contrast to Pd–PEPPSI–IPentCl, was attributed to the increased bulkiness of IHeptCl, which substantially affects the competitive β-hydride elimination when secondary alkyl zinc reagents are employed (Scheme 29).
The synthesis of the IPr* imidazolium salt was first reported by Markó and coworkers, following a modified route akin to that of the IPr imidazolium salt.185 The process involves dialkylation of p-toluidine with benzhydrol in the presence of concentrated HCl and ZnCl2 to afford the corresponding aniline, which, after treatment with glyoxal, yields the diimine intermediate. Finally, the IPr*·HCl salt is obtained by refluxing the diimine and paraformaldehyde in chloroform in the presence of ZnCl2. IPr* is significantly more sterically bulky than IPr, which leads to notable differences in its structural and catalytic properties. A representative example is the carboxylation of organoboronates, catalyzed by the [Ni(IPr*)(η3-allyl)Cl] complex.186 Nolan and coworkers demonstrated that nickel precatalyst Ni-9 affords the corresponding carboxylic acid in quantitative yield, whereas the [Ni(IPr)(η3-allyl)Cl] complex (Ni-2) yielded only 7% of the product (Scheme 30). The superior performance of the bulkier IPr*-coordinated nickel complex Ni-9, compared to Ni-2, may be attributed to the stabilization of reactive intermediates or the acceleration of a key step (reductive elimination, most likely) along the reaction pathway. Although DFT studies provided valuable mechanistic insights, they were insufficient to fully explain the necessity of the sterically bulky NHC for achieving high catalytic activity.187
 | ||
| Scheme 30 Higher activity of IPr*–Ni precatalyst in comparison with IPr–Ni congener. | ||
Further increasing the steric bulk of IPr* by introducing an OMe group at the para-position was found to have a positive effect on Ni-catalyzed C–N coupling between morpholine and 1-chloro-4-methylbenzene.188 It was postulated that the enhanced steric bulk of IPr*OMe stabilized the [Ni0(NHC)] active species, preventing their rapid decomposition. However, this increased bulkiness compromised the ligand's flexibility, limiting the incorporation of sterically hindered aryl halides near the metal center, which consequently could not be efficiently converted into the corresponding products (Scheme 31).
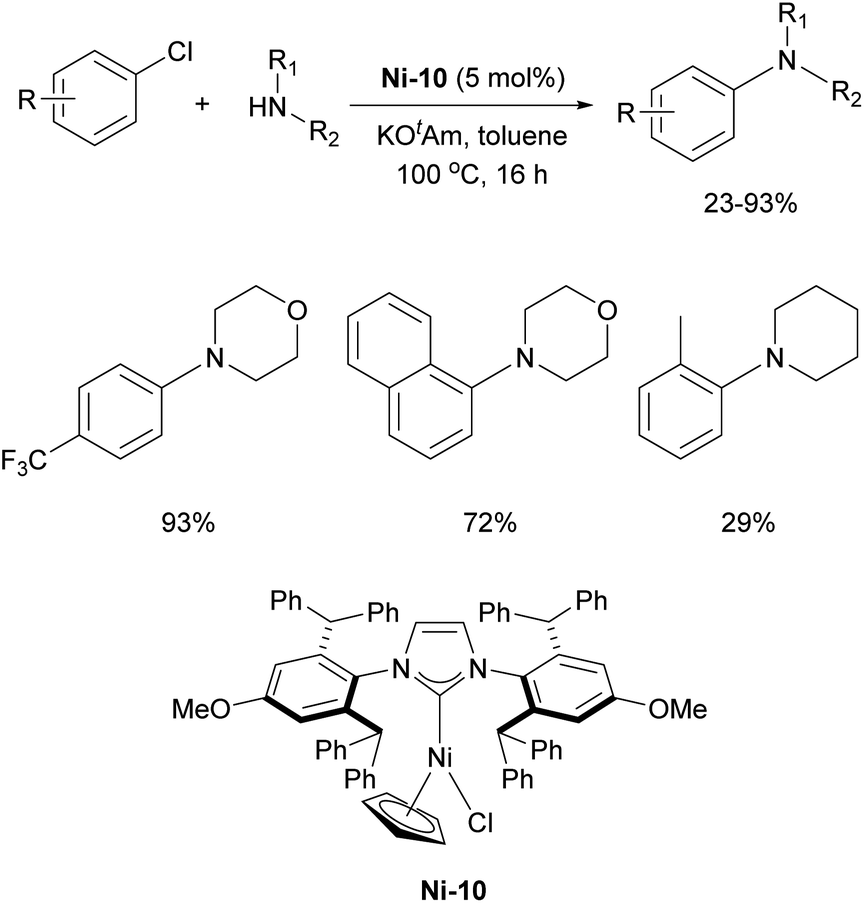 | ||
| Scheme 31 Ni-10 precatalyst in Buchwald–Hartwig amination. | ||
One of the most notable examples of the use of the IPr* architecture in catalysis is related to palladium chemistry. Shortly after the introduction of IPr*, the Nolan group synthesized the [Pd(IPr*)(cin)Cl] complex, analogous to the successful [Pd(IPr)(cin)Cl] and investigated its catalytic performance in Suzuki–Miyaura cross-coupling reactions, particularly with challenging tetra-ortho-substituted biaryls. Remarkably, this palladium precatalyst, featuring the sterically bulky yet flexible IPr* ligand, performed efficiently with sterically hindered substrates, achieving 67–99% yields at room temperature.189 Noteworthy examples include the reaction between mesityl chloride and 2,6-dimethylbenzene boronic acid, 1-chloro-2-methoxynaphthalene and mesityl boronic acid, as well as 4-bromo-1,3,5-trimethyl-1H-pyrazole and 2,6-dimethylbenzene boronic acid (Scheme 32).
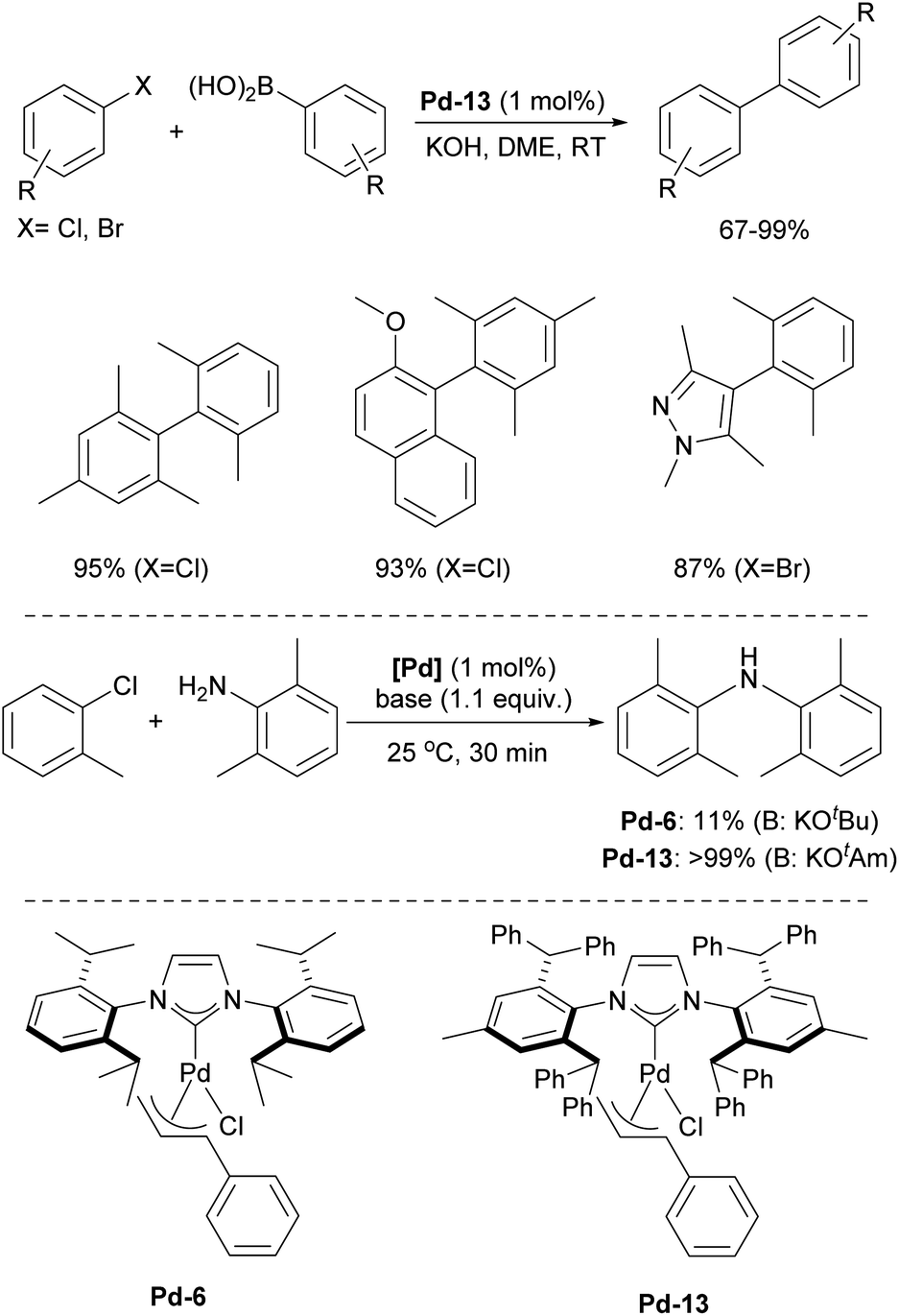 | ||
| Scheme 32 Performance of Pd-8 in Suzuki–Miyaura and Buchwald–Hartwig cross-coupling reactions. | ||
Given the remarkable reactivity of Pd-13 (Scheme 32) in Suzuki–Miyaura coupling, it was soon tested in Buchwald–Hartwig amination. Similarly, Pd-13 demonstrated high catalytic activity, even at very low catalyst loadings (0.05 mol%), affording a broad scope of products.190 Deactivated and heteroaryl chlorides were reactive under these conditions, and the typically challenging dibutylamine also underwent successful coupling, although a slightly higher loading of Pd-13 (0.1 mol%) was required to achieve a 58% yield. This is noteworthy compared to other studies that required higher catalyst loadings.191Pd-13 was also evaluated in solvent-free Buchwald–Hartwig cross-coupling reactions of unactivated aryl chlorides with secondary and tertiary amines, yielding the corresponding products in up to >99%.192 When comparing the reactivity of precatalysts Pd-13 and its IPr analogue, Pd-6, the former achieved >99% conversion to the coupling product, while Pd-6 provided only 11% conversion (Scheme 32).
Once again, it is evident that the increased steric bulkiness of IPr* could permit to overcome some of the catalytic limitations of the IPr ligand. Subsequently, Pd-13 was also employed in the S-arylation of unactivated arylsulfoxides, where it again outperformed Pd-6. In the reaction between methylphenylsulfoxide and p-bromotoluene, Pd-13 afforded 85% conversion, compared to just 9% with Pd-6 as the precatalyst.193
Straub and coworkers reported an even bulkier version of IPr*, namely, IPr**, which is decorated by eight tert-butyl groups at para-positions of phenyl substituents on the wingtips (Fig. 7). Despite a substantial effort needed to synthesize this ligand and its relatively poor catalytic activity in palladium catalysis, IPr** is uniquely useful catalyst due to its increased bulkiness.194 It provides excellent stabilization to the metal center, which was confirmed by stabilizing a gold–arene complex, originating from the C–B cleavage of one of the aryl groups of BArF24 and gold–silver intermediate allegedly formed during activation of gold complexes with silver salts.195,196
Additionally, IPr** gold complexes have shown promising catalytic activity in several gold-catalyzed transformations including phenol synthesis and hydration of alkynes (Scheme 33).197 The benefits of further extending the steric properties along this direction appeared to have reached its optimum peak, until recent reports inspired by the invaluable applications of IPr and IPr* in catalysis, where Szostak and coworkers have developed a new ligand, IPr#, which was synthesized using a procedure similar to that of IPr*.198,199 Structural analysis of [Au(NHC)Cl] complexes demonstrated that IPr# is bulkier than IPr*, but slightly less sterically hindered than IPr**. This ligand has been successfully applied in various palladium-catalyzed cross-coupling reactions, using the cinnamyl precatalyst Pd-14 (Scheme 34). It proved efficient in Suzuki–Miyaura, Buchwald–Hartwig, and transamidation reactions (Scheme 34).198 Additionally, the Ghosh group utilized IPr# to synthesize silver and copper chloride and bromide complexes, which were successfully employed in A3 (amine–aldehyde–alkyne) coupling reactions.200 The chase here continues and the original design of the IPr appears to still hold promise for further improvements in catalytic performances.
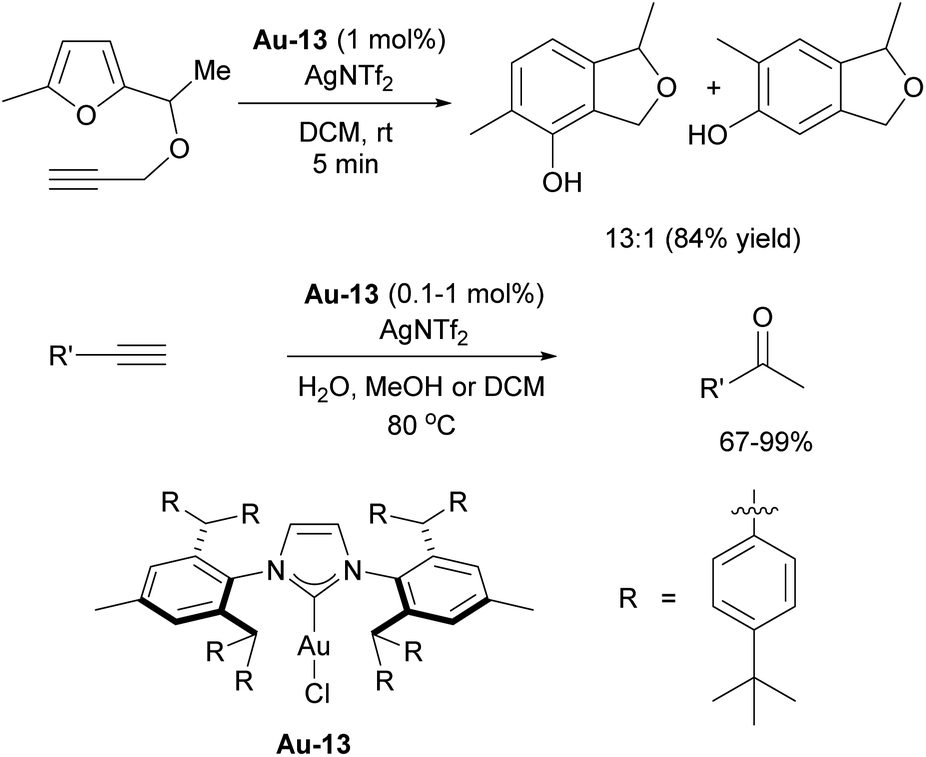 | ||
| Scheme 33 Catalytic activity of Au-13 in phenol synthesis. | ||
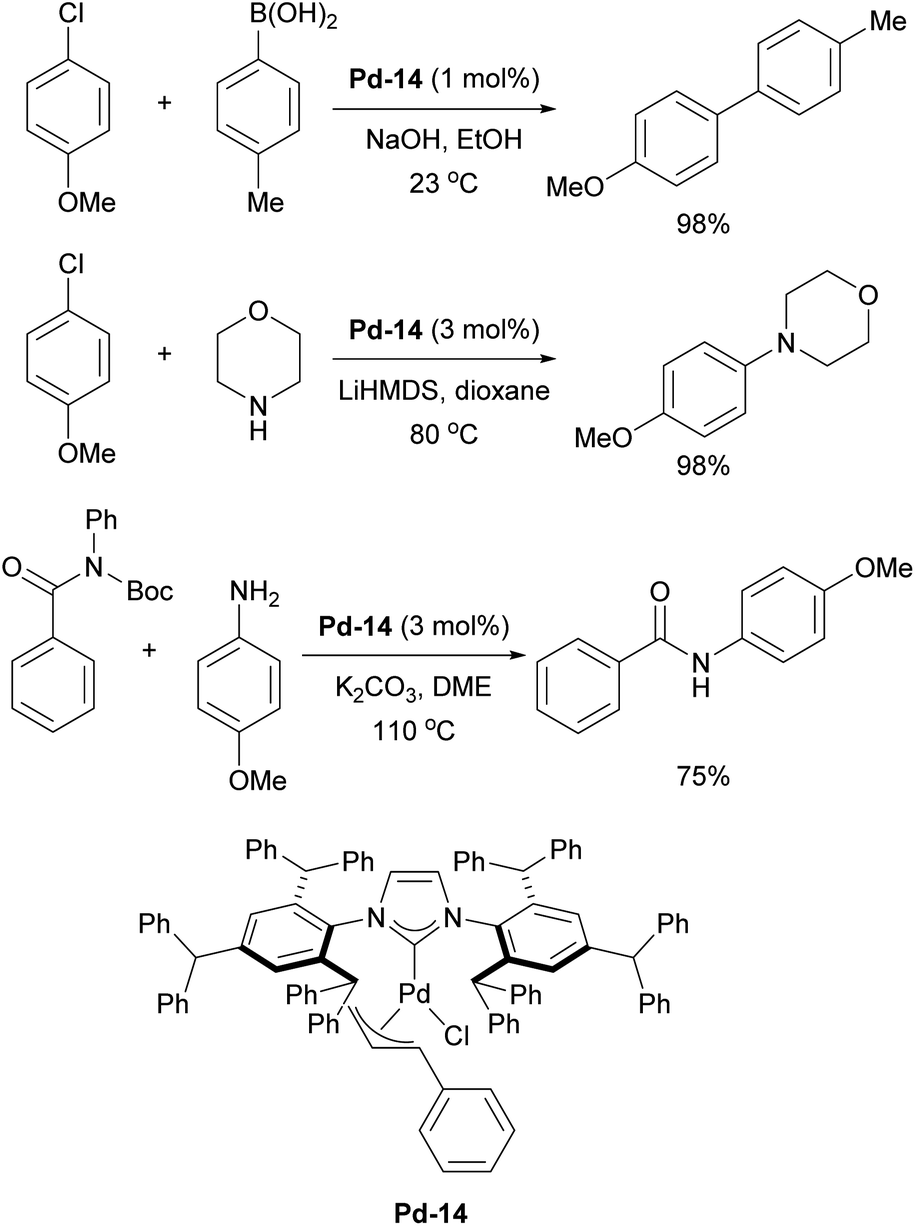 | ||
| Scheme 34 Examples of use of IPr# in palladium-catalyzed cross-coupling reactions. | ||
Conclusions
The IPr ligand has been shown as a transformative element in the field of N-heterocyclic carbene (NHC) chemistry, profoundly influencing the development of organometallic catalysis across a wide range of metals and reactions. Its unique combination of steric bulk and electronic properties has not only led to enhanced stability and reactivity of metal complexes but has also enabled catalytic transformations that were previously challenging or inaccessible. Over the past 25 years, IPr has been instrumental in advancing both the fundamental understanding of NHC ligands and their practical applications in catalysis, especially in the chemistry of transition metals.Moreover, the versatility and robustness of IPr have inspired the development of new NHC ligands tailored to specific catalytic needs, broadening the scope of NHC-based chemistry. As research continues to evolve, the IPr framework remains a cornerstone in the design of next-generation catalysts, shaping future advances in catalysis and materials science. Its lasting impact on the field underscores its significance as a privileged/benchmark ligand, ensuring its continued relevance in modern chemistry and still holds surprises in the evolution and understanding of modern catalyst design.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
VAV, LPZ and SPN wrote the manuscript and have given approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The FWO is gratefully acknowledged for support of this work as are collaborators who have greatly contributed to the development of the area and whose names can be found in the references.Notes and references
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef PubMed
.
-
N-Heterocyclic Carbenes in Organocatalysis, ed. A. T. Biju, Wiley-VCH, Weinheim, 2019 Search PubMed
- P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef PubMed
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef PubMed
- M. Koy, P. Bellotti, M. Das and F. Glorius, Nat. Catal., 2021, 4, 352–363 CrossRef CAS
-
I. Ott, in Advances in Inorganic Chemistry, ed. P. J. Sadler and R. van Eldik, Academic Press, 2020, vol. 75, pp. 121–148 Search PubMed
- A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS
- A. J. I. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS
- H.-W. Wanzlick and H.-J. Schönherr, Angew Chem. Int. Ed. Engl., 1968, 7, 141–142 CrossRef CAS
- K. Öfele, J. Organomet. Chem., 1968, 12, P42–P43 CrossRef
- W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew Chem. Int. Ed. Engl., 1995, 34, 2371–2374 CrossRef CAS
- J. Huang, G. Grasa and S. P. Nolan, Org. Lett., 1999, 1, 1307–1309 CrossRef CAS
- J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 9889–9890 CrossRef CAS
-
A. J. Arduengo III, United States, US Pat., 5077414, 1991 Search PubMed
- X. Bantreil and S. P. Nolan, Nat. Protoc., 2011, 6, 69–77 CrossRef CAS PubMed
- C. Lorber and L. Vendier, Dalton Trans., 2009, 6972–6984 RSC
- C. Romain, S. Bellemin-Laponnaz and S. Dagorne, Coord. Chem. Rev., 2020, 422, 213411 CrossRef CAS
-
L. Cerquera-Montealegre, D. Gallego and E. A. Baquero, in Advances in Organometallic Chemistry, Elsevier, 2024, vol. 81, pp. 181–270 Search PubMed
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef
- L. Jafarpour, J. Huang, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 3760–3763 CrossRef
- J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef
- M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef
- L. Ackermann, A. Fürstner, T. Weskamp, F. J. Kohl and W. A. Herrmann, Tetrahedron Lett., 1999, 40, 4787–4790 CrossRef
- L. Jafarpour, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5416–5419 CrossRef
- A. Fürstner, L. Ackermann, B. Gabor, R. Goddard, C. W. Lehmann, R. Mynott, F. Stelzer and O. R. Thiel, Chem.–Eur. J., 2001, 7, 3236–3253 CrossRef
- L. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49–54 CrossRef
- A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef
- V. L. Chantler, S. L. Chatwin, R. F. R. Jazzar, M. F. Mahon, O. Saker and M. K. Whittlesey, Dalton Trans., 2008, 2603–2614 RSC
- V. H. Mai and G. I. Nikonov, Organometallics, 2016, 35, 943–949 CrossRef
- M. K. Karunananda and N. P. Mankad, Organometallics, 2017, 36, 220–227 CrossRef
- J. Bosson and S. P. Nolan, J. Org. Chem., 2010, 75, 2039–2043 CrossRef PubMed
- P. Nun, G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2011, 30, 6347–6350 CrossRef
- S. P. Reade, M. F. Mahon and M. K. Whittlesey, J. Am. Chem. Soc., 2009, 131, 1847–1861 CrossRef
- D. McKay, I. M. Riddlestone, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, ACS Catal., 2015, 5, 776–787 CrossRef
- V. P. W. Böhm, C. W. K. Gstöttmayr, T. Weskamp and W. A. Herrmann, Angew. Chem., Int. Ed., 2001, 40, 3387–3389 CrossRef
- B. Gradel, E. Brenner, R. Schneider and Y. Fort, Tetrahedron Lett., 2001, 42, 5689–5692 CrossRef
- J. Louie, J. E. Gibby, M. V. Farnworth and T. N. Tekavec, J. Am. Chem. Soc., 2002, 124, 15188–15189 CrossRef PubMed
- B. R. Dible and M. S. Sigman, J. Am. Chem. Soc., 2003, 125, 872–873 CrossRef
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef
- R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef
- R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC
- F. Vermersch, L. Oliveira, J. Hunter, M. Soleilhavoup, R. Jazzar and G. Bertrand, J. Org. Chem., 2022, 87, 3511–3518 CrossRef
- M. J. Iglesias, A. Prieto and M. C. Nicasio, Org. Lett., 2012, 14, 4318–4321 CrossRef PubMed
- K. Matsubara, H. Yamamoto, S. Miyazaki, T. Inatomi, K. Nonaka, Y. Koga, Y. Yamada, L. F. Veiros and K. Kirchner, Organometallics, 2017, 36, 255–265 CrossRef
- V. Ritleng, A. M. Oertel and M. J. Chetcuti, Dalton Trans., 2010, 39, 8153–8160 RSC
- J. Buchspies, M. M. Rahman and M. Szostak, Catalysts, 2020, 10, 372 CrossRef
- S. Kuhl, Y. Fort and R. Schneider, J. Organomet. Chem., 2005, 690, 6169–6177 CrossRef
- M. J. Iglesias, A. Prieto and M. C. Nicasio, Adv. Synth. Catal., 2010, 352, 1949–1954 CrossRef
- M. J. Iglesias, J. F. Blandez, M. R. Fructos, A. Prieto, E. Álvarez, T. R. Belderrain and M. C. Nicasio, Organometallics, 2012, 31, 6312–6316 CrossRef
- S. G. Rull, J. F. Blandez, M. R. Fructos, T. R. Belderrain and M. C. Nicasio, Adv. Synth. Catal., 2015, 357, 907–911 CrossRef
- S. Nagao, T. Matsumoto, Y. Koga and K. Matsubara, Chem. Lett., 2011, 40, 1036–1038 CrossRef
- T. Inatomi, Y. Fukahori, Y. Yamada, R. Ishikawa, S. Kanegawa, Y. Koga and K. Matsubara, Catal. Sci. Technol., 2019, 9, 1784–1793 RSC
- J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098–13101 CrossRef PubMed
- K. Matsubara, K. Ueno, Y. Koga and K. Hara, J. Org. Chem., 2007, 72, 5069–5076 CrossRef PubMed
- M. Henrion, M. J. Chetcuti and V. Ritleng, Chem. Commun., 2014, 50, 4624–4627 RSC
- J. A. Fernández-Salas, E. Marelli, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2015, 21, 3906–3909 CrossRef PubMed
- Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215–12218 CrossRef CAS
- J. Diesel, A. M. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489–4493 CrossRef CAS
- N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nat. Chem., 2020, 12, 276–283 CrossRef CAS PubMed
- M. R. Chaulagain, G. M. Mahandru and J. Montgomery, Tetrahedron, 2006, 62, 7560–7566 CrossRef CAS
- Z. D. Miller, W. Li, T. R. Belderrain and J. Montgomery, J. Am. Chem. Soc., 2013, 135, 15282–15285 CrossRef CAS
- S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2011, 133, 9700–9703 CrossRef CAS PubMed
- S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531–9534 CrossRef CAS PubMed
- A. Kapat, T. Sperger, S. Guven and F. Schoenebeck, Science, 2019, 363, 391–396 CrossRef CAS PubMed
- M. Mendel, T. M. Karl, J. Hamm, S. J. Kaldas, T. Sperger, B. Mondal and F. Schoenebeck, Nature, 2024, 631, 80–86 CrossRef CAS PubMed
- A. J. Arduengo, H. V. R. Dias, J. C. Calabrese and F. Davidson, Organometallics, 1993, 12, 3405–3409 CrossRef CAS
- V. Jurkauskas, J. P. Sadighi and S. L. Buchwald, Org. Lett., 2003, 5, 2417–2420 CrossRef CAS
- N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef CAS
- H. Kaur, F. K. Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157–1160 CrossRef CAS
- S. Díez-González, N. M. Scott and S. P. Nolan, Organometallics, 2006, 25, 2355–2358 CrossRef
- S. Díez-González, E. D. Stevens, N. M. Scott, J. L. Petersen and S. P. Nolan, Chem.–Eur. J., 2008, 14, 158–168 CrossRef
- N. P. Mankad, T. G. Gray, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 1191–1193 CrossRef
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef
- T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792–5795 CrossRef PubMed
- L. Zhang, J. Cheng and Z. Hou, Chem. Commun., 2013, 49, 4782–4784 RSC
- L. A. Goj, E. D. Blue, C. Munro-Leighton, T. B. Gunnoe and J. L. Petersen, Inorg. Chem., 2005, 44, 8647–8649 CrossRef PubMed
- L. A. Goj, E. D. Blue, S. A. Delp, T. B. Gunnoe, T. R. Cundari, A. W. Pierpont, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2006, 45, 9032–9045 CrossRef PubMed
- C. Munro-Leighton, E. D. Blue and T. B. Gunnoe, J. Am. Chem. Soc., 2006, 128, 1446–1447 CrossRef PubMed
- S. A. Delp, C. Munro-Leighton, L. A. Goj, M. A. Ramírez, T. B. Gunnoe, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2007, 46, 2365–2367 CrossRef PubMed
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966–3972 CrossRef
- I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef PubMed
- L.-J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2017, 139, 10200–10203 CrossRef CAS PubMed
- L.-J. Cheng, S. M. Islam and N. P. Mankad, J. Am. Chem. Soc., 2018, 140, 1159–1164 Search PubMed
- D. R. Pye, L.-J. Cheng and N. P. Mankad, Chem. Sci., 2017, 8, 4750–4755 Search PubMed
- L.-J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2020, 142, 80–84 CrossRef CAS PubMed
- R. Sakae, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 6460–6463 CrossRef CAS PubMed
- S. R. Sardini and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 9823–9826 CrossRef CAS
- J. Rae, K. Yeung, J. J. W. McDouall and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 1102–1107 CrossRef CAS
- K. Yeung, F. J. T. Talbot, G. P. Howell, A. P. Pulis and D. J. Procter, ACS Catal., 2019, 9, 1655–1661 CrossRef CAS
- J. R. Bour, S. K. Kariofillis and M. S. Sanford, Organometallics, 2017, 36, 1220–1223 CrossRef CAS
- H. Sakaguchi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2018, 57, 328–332 CrossRef CAS PubMed
- J. Li, L. Wang, Z. Zhao, X. Li, X. Yu, P. Huo, Q. Jin, Z. Liu, Z. Bian and C. Huang, Angew. Chem., Int. Ed., 2020, 59, 8210–8217 CrossRef CAS
- P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC
- M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS PubMed
- M. R. Fructos, P. de Frémont, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2006, 25, 2237–2241 CrossRef CAS
- C. F. Bender and R. A. Widenhoefer, Org. Lett., 2006, 8, 5303–5305 CrossRef
- C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef PubMed
- G. Li and L. Zhang, Angew. Chem., Int. Ed., 2007, 46, 5156–5159 CrossRef PubMed
- P. de Frémont, E. D. Stevens, M. R. Fructos, M. M. Díaz-Requejo, P. J. Pérez and S. P. Nolan, Chem. Commun., 2006, 2045–2047 RSC
- N. Marion, S. Díez-González, P. de Frémont, A. R. Noble and S. P. Nolan, Angew. Chem., Int. Ed., 2006, 45, 3647–3650 CrossRef PubMed
- P. Nun, S. Gaillard, A. Poater, L. Cavallo and S. P. Nolan, Org. Biomol. Chem., 2010, 9, 101–104 RSC
- L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef
- M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638–1652 CrossRef
- D. Zuccaccia, A. Del Zotto and W. Baratta, Coord. Chem. Rev., 2019, 396, 103–116 CrossRef
- Z. Lu, G. B. Hammond and B. Xu, Acc. Chem. Res., 2019, 52, 1275–1288 CrossRef PubMed
- C. H. M. Amijs, V. López-Carrillo, M. Raducan, P. Pérez-Galán, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef PubMed
- A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247–8249 CrossRef
- N. Marion, R. S. Ramón and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448–449 CrossRef CAS
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC
- S. Gaillard, J. Bosson, R. S. Ramón, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2010, 16, 13729–13740 CrossRef CAS
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed
- I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858–8859 CrossRef CAS PubMed
- R. S. Ramón, S. Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2011, 17, 1238–1246 CrossRef
- Y. Oonishi, A. Gómez-Suárez, A. R. Martin and S. P. Nolan, Angew. Chem., Int. Ed., 2013, 52, 9767–9771 Search PubMed
- A. Gómez-Suárez, Y. Oonishi, A. R. Martin, S. V. C. Vummaleti, D. J. Nelson, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, S. P. Nolan and A. Poater, Chem.–Eur. J., 2016, 22, 1125–1132 CrossRef PubMed
- I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902–1907 CrossRef CAS
- R. M. P. Veenboer, S. Dupuy and S. P. Nolan, ACS Catal., 2015, 5, 1330–1334 CrossRef CAS PubMed
- S. Dupuy, D. Gasperini and S. P. Nolan, ACS Catal., 2015, 5, 6918–6921 CrossRef CAS
- A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS
- A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555–562 CrossRef CAS
- A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem.–Eur. J., 2013, 19, 1058–1065 CrossRef PubMed
- S. Ferrer and A. M. Echavarren, Organometallics, 2018, 37, 781–786 CrossRef
- Y. He, Z. Li, K. Robeyns, L. Van Meervelt and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2018, 57, 272–276 CrossRef PubMed
- Z. Li, L. Song, L. Van Meervelt, G. Tian and E. V. Van der Eycken, ACS Catal., 2018, 8, 6388–6393 CrossRef
- Y.-B. Bai, Z. Luo, Y. Wang, J.-M. Gao and L. Zhang, J. Am. Chem. Soc., 2018, 140, 5860–5865 CrossRef PubMed
- P. D. Jadhav, X. Lu and R.-S. Liu, ACS Catal., 2018, 8, 9697–9701 CrossRef
- R. Gauthier, N. V. Tzouras, Z. Zhang, S. Bédard, M. Saab, L. Falivene, K. Van Hecke, L. Cavallo, S. P. Nolan and J.-F. Paquin, Chem.–Eur. J., 2022, 28, e202103886 CrossRef
- M. Gatto, A. Del Zotto, J. Segato and D. Zuccaccia, Organometallics, 2018, 37, 4685–4691 CrossRef
- M. Jin, R. Ando, M. J. Jellen, M. A. Garcia-Garibay and H. Ito, J. Am. Chem. Soc., 2021, 143, 1144–1153 CrossRef PubMed
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Beliš, K. V. Hecke, C. S. J. Cazin and S. P. Nolan, Chem. Sci., 2022, 13, 6852–6857 RSC
- V. A. Voloshkin, M. Villa, E. A. Martynova, M. Beliš, K. V. Hecke, P. Ceroni and S. P. Nolan, Chem. Sci., 2024, 15, 4571–4580 RSC
- H. M. Lee and S. P. Nolan, Org. Lett., 2000, 2, 2053–2055 Search PubMed
- G. A. Grasa and S. P. Nolan, Org. Lett., 2001, 3, 119–122 CrossRef CAS
- M. S. Viciu, R. M. Kissling, E. D. Stevens and S. P. Nolan, Org. Lett., 2002, 4, 2229–2231 CrossRef CAS
- M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2002, 4, 4053–4056 CrossRef PubMed
- O. Diebolt, P. Braunstein, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2008, 3190–3192 RSC
- S. Yang, T. Zhou, A. Poater, L. Cavallo, S. P. Nolan and M. Szostak, Catal. Sci. Technol., 2021, 11, 3189–3197 RSC
- T. Zhou, S. Ma, F. Nahra, A. M. C. Obled, A. Poater, L. Cavallo, C. S. J. Cazin, S. P. Nolan and M. Szostak, iScience, 2020, 23, 101377 CrossRef
- M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470–5472 CrossRef
- G. M. Meconi, S. V. C. Vummaleti, J. A. Luque-Urrutia, P. Belanzoni, S. P. Nolan, H. Jacobsen, L. Cavallo, M. Solà and A. Poater, Organometallics, 2017, 36, 2088–2095 CrossRef
- O. Navarro, H. Kaur, P. Mahjoor and S. P. Nolan, J. Org. Chem., 2004, 69, 3173–3180 CrossRef PubMed
- O. Navarro, R. A. Kelly and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 16194–16195 CrossRef
- M. S. Viciu, R. A. Kelly, E. D. Stevens, F. Naud, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479–1482 CrossRef PubMed
- O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly and S. P. Nolan, J. Org. Chem., 2006, 71, 685–692 CrossRef PubMed
- Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef PubMed
- D. R. Jensen and M. S. Sigman, Org. Lett., 2003, 5, 63–65 CrossRef PubMed
- D. R. Jensen, M. J. Schultz, J. A. Mueller and M. S. Sigman, Angew. Chem., Int. Ed., 2003, 42, 3810–3813 CrossRef PubMed
- J. A. Mueller, C. P. Goller and M. S. Sigman, J. Am. Chem. Soc., 2004, 126, 9724–9734 CrossRef
- M. J. Schultz, S. S. Hamilton, D. R. Jensen and M. S. Sigman, J. Org. Chem., 2005, 70, 3343–3352 CrossRef
- C. N. Cornell and M. S. Sigman, J. Am. Chem. Soc., 2005, 127, 2796–2797 CrossRef
- E. W. Werner, K. B. Urkalan and M. S. Sigman, Org. Lett., 2010, 12, 2848–2851 CrossRef PubMed
- E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 13981–13983 CrossRef CAS
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743–4748 CrossRef
- S. G. Guillet, V. A. Voloshkin, M. Saab, M. Beliš, K. V. Hecke, F. Nahra and S. P. Nolan, Chem. Commun., 2020, 56, 5953–5956 RSC
- M. G. Organ, M. Abdel-Hadi, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien, M. Sayah and C. Valente, Chem.–Eur. J., 2008, 14, 2443–2452 CrossRef
- M. G. Organ, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien and C. Valente, Chem.–Eur. J., 2006, 12, 4749–4755 CrossRef PubMed
- M. G. Organ, M. Abdel-Hadi, S. Avola, N. Hadei, J. Nasielski, C. J. O'Brien and C. Valente, Chem.–Eur. J., 2007, 13, 150–157 CrossRef PubMed
- E. D. Patil, J. V. Burykina, D. B. Eremin, D. A. Boiko, K. E. Shepelenko, V. V. Ilyushenkova, V. M. Chernyshev and V. P. Ananikov, Inorg. Chem., 2024, 63, 2967–2976 Search PubMed
- V. M. Chernyshev, E. A. Denisova, D. B. Eremin and V. P. Ananikov, Chem. Sci., 2020, 11, 6957–6977 RSC
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef
- C. C. C. Johansson Seechurn, S. L. Parisel and T. J. Colacot, J. Org. Chem., 2011, 76, 7918–7932 CrossRef
- T. Ben Halima, W. Zhang, I. Yalaoui, X. Hong, Y.-F. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311–1318 CrossRef
- P. Lei, G. Meng and M. Szostak, ACS Catal., 2017, 7, 1960–1965 CrossRef
- F. Izquierdo, C. Zinser, Y. Minenkov, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, F. Nahra, C. S. J. Cazin and S. P. Nolan, ChemCatChem, 2018, 10, 601–611 CrossRef
- G. Meng, P. Lei and M. Szostak, Org. Lett., 2017, 19, 2158–2161 CrossRef PubMed
- G. Li, T. Zhou, A. Poater, L. Cavallo, S. P. Nolan and M. Szostak, Catal. Sci. Technol., 2020, 10, 710–716 RSC
- N. Marion, E. C. Ecarnot, O. Navarro, D. Amoroso, A. Bell and S. P. Nolan, J. Org. Chem., 2006, 71, 3816–3821 CrossRef PubMed
- P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari, D. P. Hruszkewycz, H. P. Shah and M. T. Tudge, ACS Catal., 2015, 5, 3680–3688 CrossRef CAS
- F. Nahra, D. J. Nelson and S. P. Nolan, Trends Chem., 2020, 2, 1096–1113 CrossRef CAS
- A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef CAS
- M. B. Dinger and J. C. Mol, Adv. Synth. Catal., 2002, 344, 671–677 CrossRef CAS
- F. C. Courchay, J. C. Sworen and K. B. Wagener, Macromolecules, 2003, 36, 8231–8239 CrossRef CAS
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS
- J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef CAS PubMed
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed
- M. G. Organ, S. Çalimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CrossRef CAS
- S. Meiries, G. Le Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2013, 19, 17358–17368 CrossRef CAS
- S. Sharif, R. P. Rucker, N. Chandrasoma, D. Mitchell, M. J. Rodriguez, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2015, 54, 9507–9511 CrossRef CAS PubMed
- B. Atwater, N. Chandrasoma, D. Mitchell, M. J. Rodriguez, M. Pompeo, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2015, 54, 9502–9506 CrossRef CAS PubMed
- G. Berthon-Gelloz, M. A. Siegler, A. L. Spek, B. Tinant, J. N. H. Reek and I. E. Markó, Dalton Trans., 2010, 39, 1444–1446 RSC
- Y. Makida, E. Marelli, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 8010–8013 RSC
- M. Delarmelina, E. Marelli, J. W. d. M. Carneiro, S. P. Nolan and M. Bühl, Chem.–Eur. J., 2017, 23, 14954–14961 CrossRef CAS PubMed
- A. R. Martin, Y. Makida, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 6265–6270 CrossRef
- A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem.–Eur. J., 2012, 18, 4517–4521 CrossRef PubMed
- A. Chartoire, X. Frogneux and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 1897–1901 CrossRef
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef PubMed
- A. Chartoire, A. Boreux, A. R. Martin and S. P. Nolan, RSC Adv., 2013, 3, 3840–3843 RSC
- F. Izquierdo, A. Chartoire and S. P. Nolan, ACS Catal., 2013, 3, 2190–2193 CrossRef
- S. G. Weber, C. Loos, F. Rominger and B. F. Straub, Arkivoc, 2012, 2012, 226–242 Search PubMed
- S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, Chem. Commun., 2012, 48, 11325–11327 RSC
- S. G. Weber, F. Rominger and B. F. Straub, Eur. J. Inorg. Chem., 2012, 2012, 2863–2867 CrossRef
- S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, ChemCatChem, 2013, 5, 2330–2335 CrossRef
- Q. Zhao, G. Meng, G. Li, C. Flach, R. Mendelsohn, R. Lalancette, R. Szostak and M. Szostak, Chem. Sci., 2021, 12, 10583–10589 RSC
- G. Utecht-Jarzyńska, S. Jarzyński, M. M. Rahman, G. Meng, R. Lalancette, R. Szostak and M. Szostak, Organometallics, 2024, 43, 2305–2313 Search PubMed
- R. Manne, S. Sharma, S. Dey, S. K. Patil, N. Nehra, H. Rawool, G. Rajaraman and P. Ghosh, J. Mol. Struct., 2024, 1316, 139022 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |