
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConnecting the complexity of stereoselective synthesis to the evolution of predictive tools
Jiajing
Li
 and
Jolene P.
Reid
*
and
Jolene P.
Reid
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: jreid@chem.ubc.ca
First published on 23rd January 2025
Abstract
Synthetic methods have seemingly progressed to an extent where there is an apparent and increasing need for predictive models to navigate the vast chemical space. Methods for anticipating and optimizing reaction outcomes have evolved from simple qualitative pictures generated from chemical intuition to complex models constructed from quantitative methods like quantum chemistry and machine learning. These toolsets are rooted in physical organic chemistry where fundamental principles of chemical reactivity and molecular interactions guide their development and application. Here, we detail how the evolution of these methods is a successful outcome and a powerful response to the diverse synthetic challenges confronted and the innovative selectivity concepts introduced. In this review, we perform a periodization of organic chemistry focusing on strategies that have been applied to guide the synthesis of chiral organic molecules.
 Jiajing Li | Jiajing Li was born in Guangxi, China. He received a BSc degree (2020) in Chemistry from Zhejiang University. After spending a year as a research assistant in Zhejiang University, he joined the group of Prof. Jolene Reid at the University of British Columbia. His current research focuses on application of machine learning in organocatalysis. |
 Jolene P. Reid | Jolene Reid was born in Ballymena, County Antrim, Northern Ireland. She received a MSci from Queens University Belfast and a PhD from the University of Cambridge working with Professor Jonathan Goodman. She was a Marie Curie research fellow in Professor Matthew Sigman's group at the University of Utah. In 2020, she joined the faculty of the University of British Columbia as an assistant professor. Here, her research program combines computational and experimental chemistry techniques for the development of selective organic reactions and catalysts. |
Introduction
Organic chemistry has been and continues to be categorized by unique eras of discovery, each marked by transformative technologies enabling the preparation of new molecular structures. Indeed, as reactions evolved from foundational to complexity generating synthetic strategies, new methods for their design and study emerged. Perhaps, it can be stated that organic synthesis has persistently shaped the broad landscape of physical organic chemistry, an area that has traditionally been focused on understanding reaction outcomes. Typically, mechanistic tools are deployed after the empirical identification of an effective transformation or catalyst structure rather than in predictive fashion. However, this strategic decision is beginning to change as a consequence of exceptional advances in computational power and methods that now allow modelling of complex interactions accurately. Moreover, machine learning (ML) and artificial intelligence (AI) have emerged as powerful tools for predicting and understanding the role of catalysts/substrates and act as a useful complement to the traditional techniques. Given the significant uptake of data science in organic synthesis we thought in the context of this review, that it would be pragmatic to demonstrate in parallel the evolution of the techniques used to generate organic molecules in addition to ways for rationalizing stereochemical outcomes and making predictions. Through this historical survey, we aim to explore whether the integration of data science is considered an advantageous choice—enhancing research capabilities—or if it has become an essential element for addressing the complexities and challenges inherent in modern organic chemistry.This review focuses on the area of stereoselective synthesis, a field that has witnessed significant advancements in physical organic tool sets, from meticulously designed experiments to computational modelling. Progress in this area has been propelled by several factors that serve to exemplify the complexities and challenges of modern organic chemistry. One key factor is the role of transition states (TS) in imparting enantioselectivity, which relies heavily on core non-covalent interactions (NCIs) that define their geometries and energies. These NCIs are energetically weak and highly sensitive to the molecular characteristics of every reaction component, including the catalyst, substrates, reagents, and solvent. Consequently, stereoselective synthesis exemplifies an area where even minor structural variations in any component can significantly and often unpredictably impact the observed enantioselectivity. By examining the contributions of physical organic chemistry to this field, we aim to elucidate its role in refining mechanistic hypotheses and improving reaction outcomes. As this review demonstrates, there is a clear gradual progression from simple molecular projections to the application of quantum chemistry, and ultimately to the use of large-data ML approaches.
As our interest is in connecting the physical organic tools to reaction complexity, this article will primarily detail the stereochemical models that have been commonly used and/or rigorously studied. Given space limitations, we cannot cover all subfields of stereoselective synthesis. Instead, we will focus on selected reagent types, including catalysts and their corresponding reactions. To reflect the evolving toolsets of physical organic chemistry and the growing complexity of reactions, we will, where possible, highlight examples from the same reaction type within a given field to maintain continuity and coherence in the discussion. We decided to organize this review by types of techniques, providing a structured examination of the various methods used in the study of stereoselective synthesis and the progression of strategies for synthesizing chiral molecules.
Periodization of selective organic synthesis
There are several ways to generate new stereogenic centers in asymmetric synthesis. Fundamentally, creating new stereogenic centers with a specific absolute configuration relies on the transfer of chirality from one or more of the reaction components. In this context, naturally occurring chiral compounds, known as the “chiral pool”, have been valuable resources of the initial chirality. These compounds can be used directly, in derivative forms, or in chiral resolutions to produce other enantioenriched compounds, which can engage in downstream chiral transfer events. The strategy for constructing new stereogenic centers with control over absolute configuration is divided into three major approaches: substrate control, chiral stoichiometric reagent control, and catalyst control (asymmetric catalysis). Substrate control utilizes the inherent chirality of the starting material, with chiral auxiliaries being the stereotype, to achieve asymmetric induction. This strategy is particularly effective when at least one stereogenic center is already present, and additional stereogenic centers are needed. In these cases, a chiral moiety is pre-installed on the substrate, with the option of removal at a later stage. Substrate control has proved to be widely applicable in asymmetric synthesis, especially for cyclic scaffolds that benefit from their more rigid conformations. Chiral stoichiometric reagent control involves deploying a stoichiometric amount of a chiral reagent to direct stereoselectivity, thereby rendering inherent chirality on the substrate unnecessary. Classical examples include asymmetric allylations of achiral aldehydes mediated by chiral allylmetal reagents, such as those involving boron, tin, or silicon, as well as sparteine-mediated asymmetric deprotonation. In addition, asymmetric catalysis has emerged as another elegant and powerful tool for stereoselective organic synthesis. In contrast to stoichiometric chiral reagent control, a smaller amount, usually 20% or less, is required. Asymmetric catalysts often operate by providing kinetic discrimination between enantiomers through the formation of diastereomeric TS structures.Indeed, a question this organizational scheme provokes is, why are reaction outcomes involving chiral auxiliaries or stoichiometric reagents more straightforward to rationalize and predict? Are there specific types of reactions or substrate classes where certain physical organic tools, such as qualitative models and quantum calculations, are particularly advantageous or limited? Additionally, how do these tools compare in effectiveness and predictability? Herein, we begin to probe these questions by systematically comparing the performance of various physical organic tools and linking them to the predictability of different chiral transfer strategies. Our goal is to identify the strengths and limitations of each approach and provide a clearer understanding of how these tools can be leveraged to improve reaction outcomes and mechanistic insights.
Simple molecular projections
The mechanisms and stereochemical models of fundamental reactions have been developed based on experimental evidence, especially through meticulously designed control experiments. While bonding and molecular interactions ultimately arise from the quantum mechanical behavior of electrons, in an overly simplistic sense, all stereoselectivity can be attributed to steric and electronic effects, serving as a practical framework for understanding structural influences on reaction outcomes. With the help of chemists' intuition and expertise in steric and electronic effects, these stereochemical models were initially represented as simple qualitative pictures with molecular projections. While many of these models have proven robust across various reactions, adaptations to established rules have often been necessary when applying these models to include new classes of substrates or reaction conditions. Here, we aim to highlight representative qualitative pictures and experimental studies that have served to enhance our understanding of reaction mechanisms across the three major strategies for controlling enantioselectivity. A list of representative stereochemical models in the area of stereoselective transformations of carbonyl compounds are summarized in Table 1.| Name of models/reactions | Substrate type | Use case |
|---|---|---|
| Cram's rule1 | α-Chiral carbonyls | Empirical model in predicting nucleophilic additions to chiral aldehydes/ketones |
| Felkin–Anh model2,3 | α-Chiral carbonyls | |
| Cram's chelation rule4 | α-Chiral carbonyls | Empirical model in predicting nucleophilic additions to chiral aldehydes/ketones with a chelating metal ion |
| Zimmerman–Traxler model5 | Enolates and carbonyls | Empirical model in predicting aldol and aldol-type reactions of boron and titanium enolates, etc., which proceed through six-membered-ring TS |
| Mukaiyama-aldol reaction6 | Silyl enol ethers and carbonyls | Empirical model in predicting Mukaiyama-aldol reactions that proceed through acyclic TS |
| Cram–Reetz model7,8 | β-Alkoxy aldehyde | Empirical model in predicting 1,3-stereocontrol for carbonyls without chelation |
| Evans model9 | β-Alkoxy aldehyde | |
| Reetz chelate model10 | β-Alkoxy aldehyde | Empirical model in predicting 1,3-stereocontrol for carbonyls with chelation |
In the early stages of asymmetric synthesis, nucleophilic addition to carbonyl compounds was one of the most representative reaction classes. The prochiral face of a carbonyl group can be distinguished by steric hindrance or beneficial non-covalent interactions, which enable effective stereocontrol. In 1952, Cram et al. proposed one of the earliest stereochemical models for asymmetric synthesis, which could be easily sketched by hand, to describe the asymmetric induction at carbonyl compounds based on studies of diastereoselective nucleophilic addition to aldehydes and ketones with α-stereogenic centers (Fig. 1A).1 Logically, this model prioritizes the minimization of steric contacts in the TS that would lead to the major product. The steric hindrance of the carbonyl increases when the oxygen coordinates with a metal ion in the reaction, thereby dictating the conformation of the α-stereogenic center. The bulkiest substituent RL is placed at the anti-position to the carbonyl group and adopts an eclipsed conformation with the substituent R at the other side of the carbonyl group. The less bulky substituents RM and RS is positioned adjacent to the carbonyl group. To minimize the steric repulsion, the nucleophile preferentially approaches from the less hindered side, giving the favored product. Cram's rule was well supplemented by the Felkin–Anh model,2,3 which suggested a staggered conformation with RL perpendicular to the carbonyl group. Similarly, the nucleophile approaches from the less hindered side (Fig. 1B). Together, Cram's rule and the Felkin–Anh model provide a robust framework for understanding the stereochemical outcomes of such reactions. Modifications to these models are required when additional interactions, such as hydrogen bonding or chelation with external Lewis acids,4,11 alter the predicted stereochemistry from the aforementioned traditional expectations (Fig. 1C).
 | ||
| Fig. 1 Illustrative model of (A) Cram's rule; (B) Felkin–Anh model; (C) Cram's chelating model (RL = large substituent, RM = medium substituent, RS = small substituent, depending on steric size; Nu = nucleophile, M = external Lewis acid). | ||
Another well-established stereochemical rationale is the Zimmerman–Traxler model (Fig. 2A), which is based on a pseudo-six-membered ring TS designed to resemble the ground state conformation of a cyclohexane ring. Initially proposed by Zimmerman and Traxler5 to explain the stereochemistry of diastereoselective Ivanov and Reformatsky reactions, the pseudo-six-membered ring typically involves coordination with a metal ion and two reactants. The substituents on the enolate and the aldehyde adopt the favored conformation to minimize steric repulsions in the TS, thus determining the relative configuration of the major product. The configuration of the enolate determines the position of the methyl substituent on the chair conformation and forces the large R1 substituent on the axial position, while the large substituent R2 on the aldehyde prefers to occupy the equatorial position in order to avoid 1,3-diaxial interactions with R1. Such a highly organized TS results in the syn-diastereoisomer being the major product.
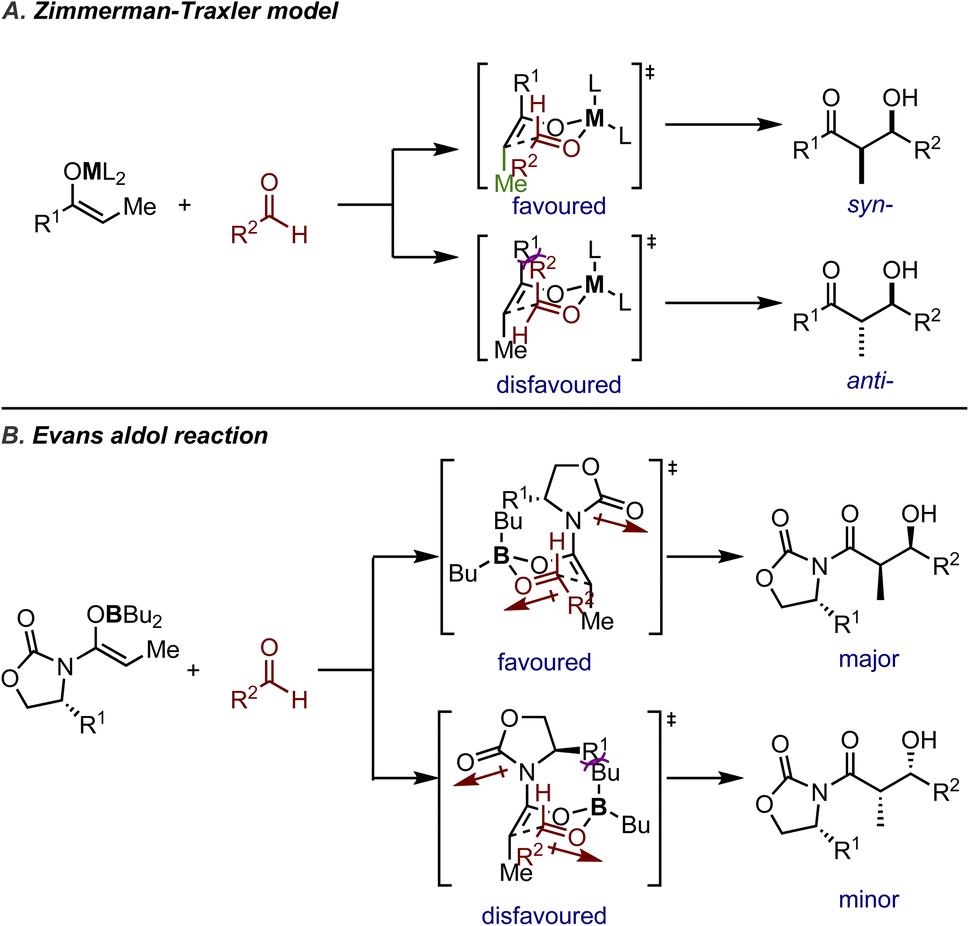 | ||
| Fig. 2 A) Zimmerman–Traxler model (determination of relative configuration); (B) an illustrative example for an Evans aldol reaction (absolute configuration controlled by the chiral auxiliary). | ||
The Zimmerman–Traxler model not only demonstrated robustness in the reactions studied in the seminal work, but has also influenced future reaction design and mechanistic rationale. Over time, it has proven particularly useful in explaining boron-mediated asymmetric reactions, such as Evans' auxiliaries-mediated asymmetric aldol reactions12–15 (or Evans' aldol) and boron-mediated allylation of aldehydes. Taking the boron-mediated Evans aldol reaction as an illustrative example, the boron reagents will coordinate with both the enolates and the carbonyl group of the approaching electrophile. The diastereoselectivity is rationalized in the same way as shown in Fig. 2A. Upon examining the TSs leading to two enantiomers, one TS is favored due to the auxiliary substituent pointing towards free space to minimize 1,3-diaxial interactions (Fig. 2B). It should be noted that the carbonyl group on the auxiliary has to rotate away and adopt the conformation where the dipoles of the enolate oxygen and the carbonyl group are opposed, instead of rotating freely to further avoid 1,3-diaxial interactions.
In the process of proposing stereochemical models, chemists often rely on designing control experiments when the key factors for steric control are less straightforward. In the area of chiral stoichiometric reagent control, (−)-sparteine, a naturally occurring chiral diamine, has proven to be one of the most useful chiral ligands for lithium, especially in asymmetric α-deprotonation reactions.16 An illustrative example is the asymmetric lithiation of N-Boc pyrrolidine (Fig. 3A). The rigid multicyclic scaffold of sparteine, which consists of four rings, provides a chiral environment that is highly effective for stereocontrol by restricting the conformational freedom of reactant structures. Recent studies, however, indicate that the selectivity outcomes are not solely due to this effect. Specifically, certain portions of the ligand profoundly contribute to the energy differences between the TSs leading to the competing products. Part of these mechanistic efforts has been motivated by the need to identify simpler structures or substitutes for use in these reactions, including seeking a surrogate for the commercially unavailable (+)-sparteine. Simply put, understanding the role of each ring would enable the identification of portions that could be replaced or simplified. Indeed, control experiments have revealed the A-wing as the crucial element for achieving high enantioselectivity. In 2002, O'Brien et al. demonstrated that the D-ring had a minimal impact on enantioselectivity in asymmetric lithiation through their efforts towards a (+)-sparteine surrogate.17 This conclusion was further supported one year later by Kozlowski et al., who synthesized a simplified version of sparteine containing only the B- and C-rings (Fig. 3B).18 Their comparative studies revealed that the absence of the A-ring led to significantly reduced enantioselectivity, thus emphasizing the crucial role of this structural feature in controlling the stereoselectivity. Indeed, such a strategy has become a standard experimental test when investigation of a specific moiety in the ligand is desired.
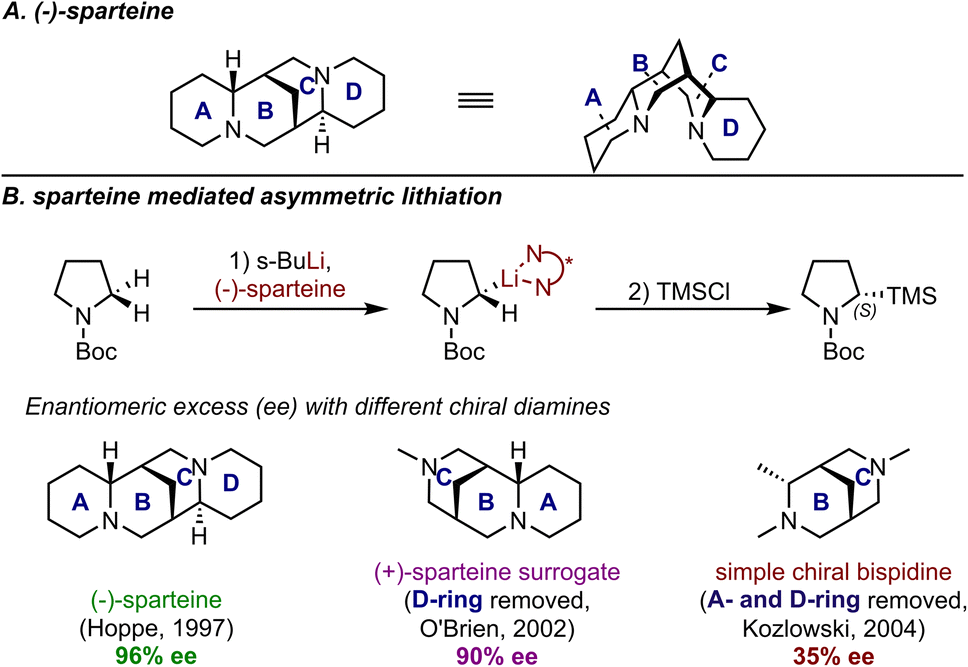 | ||
| Fig. 3 (A) 3D structure of (−)-sparteine; (B) sparteine-mediated asymmetric lithiation and results with other diamines as comparisons. | ||
In the early development of asymmetric catalysis, considerable achievements were made using metal catalysts with chiral ligands to impart stereocontrol. These metal catalysts either simply coordinate with substrates as chiral Lewis acids, or engage in organometallic reactions. The most common strategy in ligand design is to incorporate large groups at particular positions to exert influence at the TS for the enantio-determining step, typically through steric repulsion. C2-symmetric chiral ligands, particularly chiral diphosphine ligands in transition metal catalysis, emerged as early structures capable of achieving high enantioselectivities.19 To illustrate these interactions in a clear and qualitative manner, a communicative visualization of the spatial arrangement of the ligand around the metal ion is essential. Quadrant diagrams were developed for this purpose, where the environment around the metal center is divided into four portions.20 Shaded quadrants represent areas blocked by substituents, while the less hindered quadrants are left unfilled. In the picture that leads to the major product, larger substituents on the substrate tend to fill the open areas and smaller substituents occupy the blocked areas. Overall, by imagining how portions of the ligand structure occupy specific pockets of space within the molecular environment, the quadrant model offers a rational framework for understanding the experimental outcomes and predicting the stereoselectivity.
An illustrative example of asymmetric hydrogenation with Ru/DIPAMP is shown in Fig. 4. In this case, the phenyl groups on the phosphine ligand block the top left and the bottom right quadrants, while the top right and bottom left quadrants remain relatively unhindered. For alkene substrates approaching the metal center, two possible modes of activation leading to the opposite configurations of the product are drawn out. One of them is obviously disfavored because it forces larger substituents into the blocked quadrants, resulting in steric repulsion between the substrate and the catalyst. Through such simple diagrams, the steric environment is shown in a straightforward way, and the stereochemistry of the reaction can be well explained.
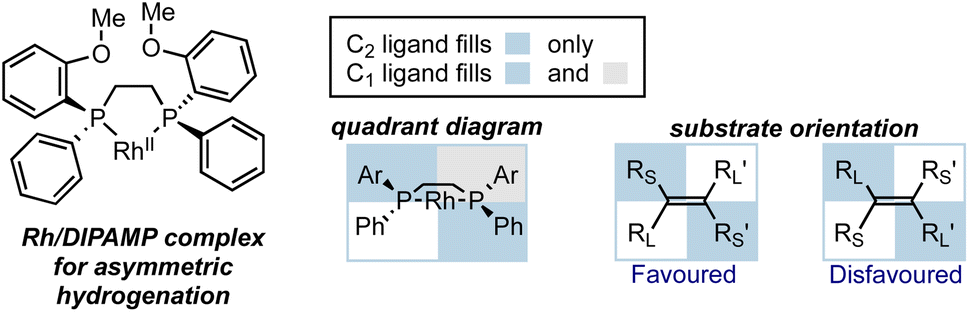 | ||
| Fig. 4 The quadrant diagram for Ru/DIPAMP catalyst in asymmetric hydrogenation and the design of a non-symmetric ligand based on quadrant diagram. | ||
In addition to describing the stereocontrol and predicting products, quadrant diagrams also serve as a template for ligand modification. For example, Hoge et al. designed a diphosphine ligand with three hindered quadrants by replacing a methyl group with a bulkier tert-butyl group, thereby blocking the third quadrant and leaving only one quadrant open (Fig. 4).21 The non-symmetric ligand was proven also successful in Rh-catalyzed asymmetric hydrogenation of dehydroamino acids, giving similar levels of enantioselectivity to the C2-symmetric ligand.22,23 Such rational design was among the earliest attempts at the design and synthesis of non-symmetric diphosphine ligands.
When asymmetric reactions involve similar molecules with consistent structural features, such as identical functional groups or comparable steric environments, predicting outcomes becomes more straightforward. In such cases, simple qualitative models obtained from empirical trends and chemical intuition provide reliable guides for understanding and anticipating the behavior of these transformations.
Quantum chemistry
While applying qualitative models has enabled the use of simple pictures for rationalizing and predicting reaction outcomes, a challenge with this approach is the necessary oversimplification of complex interactions and mechanisms. This limits the depth of insight obtainable in more complex scenarios, making it difficult to meet specific synthetic demands in certain cases, such as the reversal of the sense of stereoinduction and improving the performance of challenging substrates. As mentioned above, simple models are effective when one or a few interactions determine stereoselective outcomes. However, chemical reactions can be highly complex, especially with large catalyst structures, and are dependent on numerous factors including subtle attractive non-covalent contacts. Simple qualitative models often fall short of revealing critical interactions between components, especially when multiple activation modes are involved in asymmetric catalysis. Therefore, it is essential to move beyond basic qualitative models and incorporate quantitative assessments of stereoselectivity.The challenge of modelling reactions influenced by complex noncovalent interactions has been investigated throughout the history of asymmetric reaction development. With the increasing reliance on quantum calculations in many fields, ab initio methods have significantly expanded the types of structures that can be modeled accurately. From molecular mechanics to quantum chemical methods, the ability of computational chemistry tools to study organic reactions has progressively improved. Computational chemistry techniques now offer capabilities ranging from predicting molecular structures and energies for comparison with experimental data to modelling and visualizing molecular orbitals, charge distributions and molecular interactions.24–35
For a kinetically controlled reaction, which is the most common scenario under mild conditions, the most straightforward approach towards understanding the selectivity outcome is to calculate the TS structures leading to the competing products, based on a mechanism supported by experimental evidence. Within this framework, at a given temperature, the selectivity is determined by the ratio of the competing reaction rates (Fig. 5). By correlating the difference in activation free energy barriers (ΔΔG‡) with selectivity outcomes—such as diastereomeric ratio (d.r.), enantiomeric ratio (e.r.), or enantiomeric excess (ee)—the computationally predicted mechanism can be validated by comparing the predicted stereoselectivity, both in direction and magnitude, with experimental observations. While the commonly used ee formula shown in Fig. 5 provides a convenient approximation for relating ΔΔG‡ to the observed selectivity, it is important to note that this formula is empirically derived and simplifies the relationship between ΔΔG‡ and the equilibrium constant (K).36
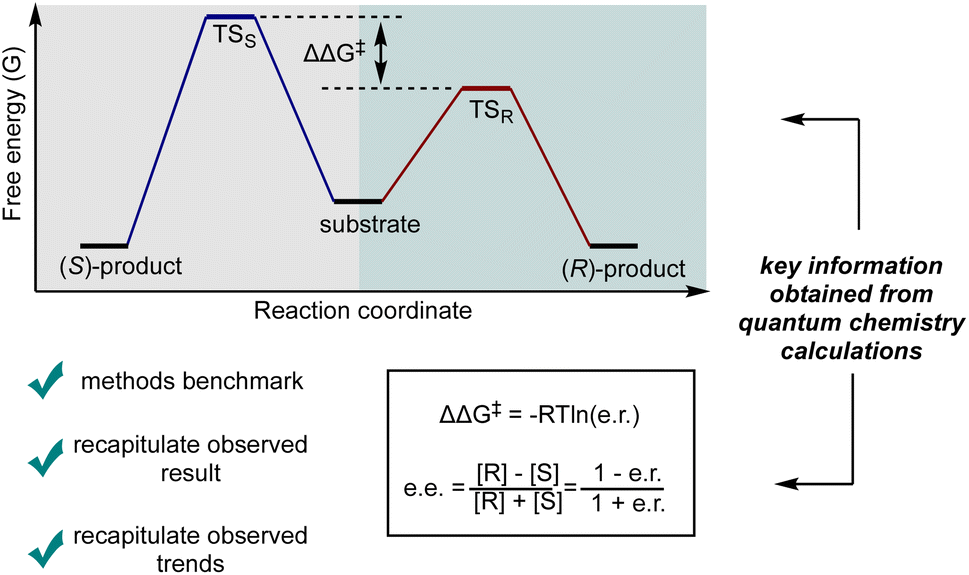 | ||
| Fig. 5 Illustration of the relationship between ΔΔG‡ and enantioselectivity (e.r. and ee). [R] and [S] represent the concentration of R- and S-product after the reaction, respectively. | ||
If multiple activation modes are possible or several stereogenic centers are formed in a single chemical step, it becomes necessary to locate TS structures for many possibilities. Furthermore, examining the TS structures can reveal key features such as beneficial non-covalent interactions, energetically repulsive contacts and notable distortion of some reaction components.
Pioneering work from Houk et al. demonstrated the quality of this approach and enabled the recapitulation of mechanistic insights into well-studied reactions. An illustrative example reported in the late 1980s demonstrated the use of ab initio calculations and molecular mechanics for TS modelling. Results from calculations and supplementary experiments quantitatively support previous mechanistic proposals based on the Felkin–Anh model (Fig. 6).37 The authors investigated the additions of hydride and Grignards to ketones. By calculating the TS structures leading to different diastereomers and comparing the calculated e.r. with the experimental values, the authors found excellent agreements for the Felkin–Anh models. Although the qualitative model only predicts the major product, transition state calculations provided quantitative support on stereoselectivities.
 | ||
| Fig. 6 Selected results of calculated and experimental results on hydride addition to (A) acyclic ketones and (B) cyclic ketones. Results generated by the MM2 force field. | ||
An important consideration in the progression of computational models is the availability of accurate methods. Compared to molecular mechanics, the development of quantum chemical methods has significantly improved the reliability of the investigations of organic systems. By far the most successful approach has been the utilization of density functional theory (DFT), as the relatively good accuracy at low computational cost has facilitated the use of this method in studying a wide range of chemical processes. The use of DFT optimizations with modest double-zeta valence polarized basis sets, such as B3LYP/6-31G*,38–43 has historically been a cost-effective approach for studying organic transformations. The comparison of chemically related structures, as in stereoselective reactions, often benefits from the cancellation of systematic errors, leading to quantitative agreements with experiments in some cases. However, it is crucial to recognize the inherent limitations of these early methods, particularly in the absence of dispersion corrections or sufficient polarization functions. Although the cost-effective methods such as B3LYP/6-31G* were very useful in the 2000's, they are not routinely used today as they have been replaced by more accurate methods which will be discussed below.
In the area of asymmetric organocatalysis, secondary amine-catalyzed aldol and aldol-type reactions are important examples that demonstrate how quantum chemical calculations can enhance our understanding of reaction mechanisms, give a quantitative account of the selectivity, and guide new catalyst design. Proline-catalyzed intramolecular aldol reactions were reported in the 1970's,44,45 while the intermolecular version emerged in 2000.46 Although experimental studies provided significant mechanistic insights, they failed to convincingly explain the origin of enantioselectivity.
In 2003, List and Houk et al. applied quantum chemical calculations to investigate the mechanism of the intermolecular proline-catalyzed aldol reaction47 (Fig. 7A). The mechanism for this intramolecular aldol reaction can be rationalized by a Zimmerman–Traxler-type TS. Several variants of the reaction including acyclic and cyclic enamines and a few aldehydes were investigated. Ultimately, their results corroborated the presence of hydrogen bonding in the TSs and highlighted that steric repulsion between reactants governed the relative energies of different transition states. By calculating and comparing transition states leading to four different diastereomeric products, they proposed a set of nine-membered ring TSs organized by hydrogen bonding (Fig. 7B), and the activation energy barrier aligned well with experimental results. This mechanistic model, now known as the Houk–List model, serves as a state-of-the-art example of how quantum chemical calculations can probe asymmetric catalytic reactions.
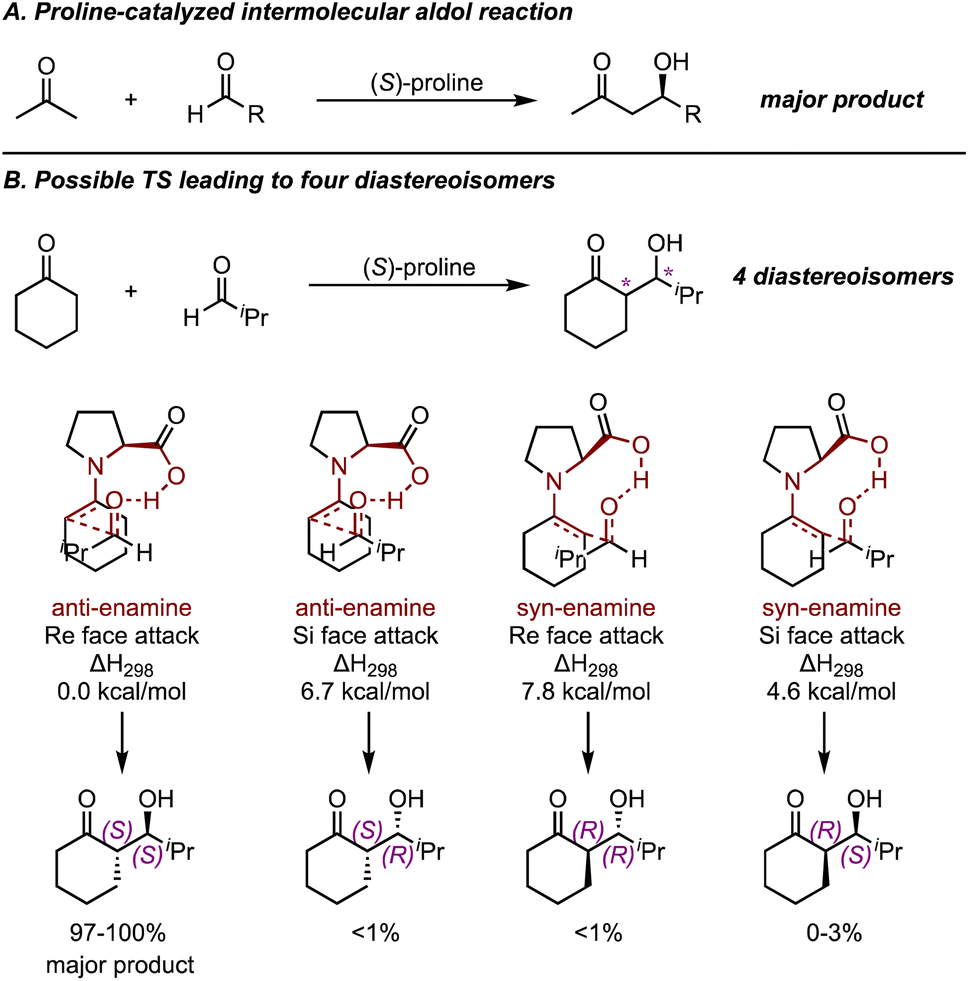 | ||
| Fig. 7 (A) Proline-catalyzed intermolecular aldol reactions. (B) Possible TSs leading to four diastereoisomers for a proline-catalyzed reaction of cyclohexanone and isobutyraldehyde. Calculated at B3LYP/6-31G* level and under 298 K.47 | ||
Quantum chemical calculations provide quantitative explanations of the stereochemical outcomes for many reactions. However, it is often useful to summarize these complex insights into simple qualitative models, which can complement and enhance the understanding gained from calculations. When supported by quantum chemical data, these stereochemical models become more robust, and can be generalized to other reactions, enabling predictions about the major product stereoisomer formed without the need for additional calculations.
In this context, the nine-membered-ring TS developed to explain the stereochemistry of the proline-catalyzed aldol reaction was successfully extended to the Mannich reaction enabled by the same catalyst structure.48–50 While the Mannich reaction yields the syn-product as the major diastereomer, and the Houk–List model predicts the anti-product for the aldol reaction, the recognition of prochiral faces was rationalized in the same way (Fig. 8A).51 By considering the difference in steric profiles between the imine electrophile in the Mannich reaction and the carbonyl electrophile in the aldol reaction, the model could be adjusted accordingly. Specifically, because the nitrogen in the imine contains a larger substituent, the substrate undergoes a 180-degree rotation to position this bulkier group away from the catalyst (Fig. 8A). Recognizing the importance of accessing anti-Mannich products, coupled with insights from TS analysis, motivates the modification of the catalyst structure to alter the stereochemistry of this particular reaction. This proved possible in 2006, when Houk and Barbas developed a modified proline catalyst to realize a preference for the s-cis-enamine predicted by TS modelling (Fig. 8B).52 The calculations suggested that the modified proline would lead to a highly selective outcome for the anti-Mannich, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr and ∼98% ee. Subsequent validation experiments demonstrated that the product was obtained in 94:6 dr and >99% ee showing excellent agreement with the calculations.
:5 dr and ∼98% ee. Subsequent validation experiments demonstrated that the product was obtained in 94:6 dr and >99% ee showing excellent agreement with the calculations.
 | ||
| Fig. 8 Proline-catalyzed syn-Mannich reaction and the computational design of a new catalyst for anti-Mannich reaction. (A) Proline-catalyzed syn-Mannich reaction, TS for major product (calculated at B3LYP/6-31G* level) and designed catalyst TS for anti-Mannich reaction (calculated at HF/6-31G* level); (B) experimental validation. PMP = p-methoxyphenyl. | ||
As demonstrated, small modifications to the catalyst or substrate structure can lead to significant and often unexpected changes in selectivity, making the generalization of asymmetric catalytic reactions challenging. At the core of these changes are subtle NCIs that drive selectivity. However, such sensitivities, often viewed as limitations in reaction development, can be harnessed to explore opportunities for stereodivergent strategies. Given the unique NCIs established between organocatalysts and various reactants, it is not surprising that employing these systems has emerged as a promising approach for accessing different products. Examples illustrating this concept can be found in the field of chiral phosphoric acid (CPA) catalysis, where many effective catalysts feature a chiral pocket defined by the 1,1′-binaphthol (BINOL) backbone and constrained by large 3,3′ groups.53 Since the seminal work by Akiyama54 and Terada55 in 2004, CPA catalysis has experienced significant growth in methodology development and mechanistic studies. While several research groups have contributed to understanding how these catalysts function, Goodman56 and Himo57 were among the first. Indeed, Goodman et al. conducted in-depth computational studies on a CPA-catalyzed transfer hydrogenation of imines using Hantzsch esters.56 Their work identified possible activation modes, initially through a truncated CPA catalyst system, revealing that the bifunctional mechanism, characterized by hydrogen bonding between the catalyst and the two substrates, was the most plausible, as it exhibited the lowest activation free energy barrier (Fig. 9). Further investigations with full catalyst systems and work by Himo et al.57 also supported this conclusion.
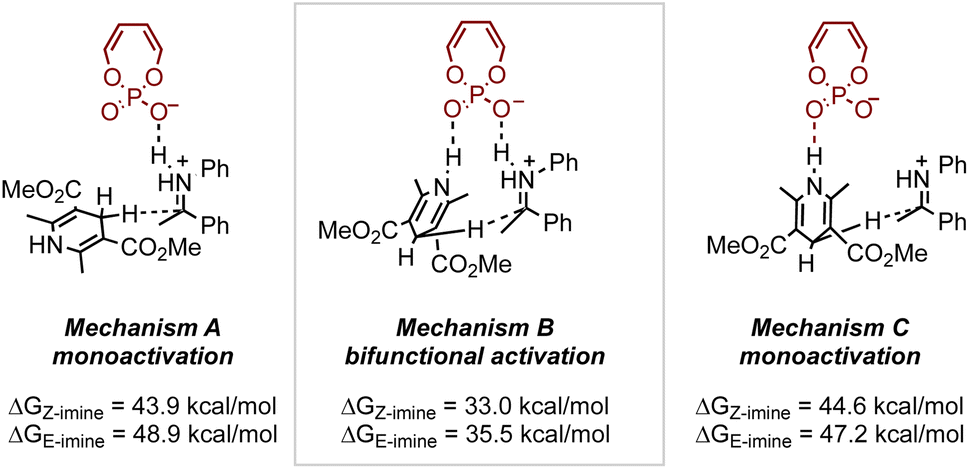 | ||
| Fig. 9 Investigation of different activation modes on a CPA-catalyzed transfer hydrogenation with a truncated phosphoric acid catalyst. Results calculated at PCM(toluene)-B3LYP/6-311++G**//B3LYP/6-31+G* level. | ||
Based on these mechanisms, simple qualitative models can be employed to quickly predict the stereochemistry of CPA-catalyzed reactions. Two commonly-used projections for this purpose are the quadrant projection, developed by Himo and Terada,57,58 and the Goodman projection (Fig. 10A).56 In the quadrant projection, the catalyst is aligned so that the POOH moiety is vertical, while the naphthol oxygens lie along the horizontal axis. Here, the BINOL backbone and 3,3′-groups occupy two of the four quadrants. The substrate's lowest energy orientation typically minimizes steric clashes by placing larger groups in the unoccupied quadrants. Alternatively, the Goodman projection views the catalyst with the POOH moiety extending above and below the plane, and the 3,3′-groups positioned on either side. The steric demands of various parts of the reactants influence how they fit into the chiral cavity, determining which pathway is favored. The preferred pathway is identified by considering the size of the substituents on the nitrogen and carbon atoms of the imine electrophile and the positioning of the nucleophilic site relative to the hydrogen bond that secures it within the catalyst's active site (Fig. 10B). Goodman's model has been shown to be highly general, successfully explaining the outcomes of a wide range of reactions,59 including the transfer hydrogenation of enamides,60 Friedel–Crafts reactions of indoles,61 and Mannich reactions with enamines.62 Mechanisms for many of these reactions were supported through TS calculations on full CPA catalysts, with detailed comparisons of different types of mechanisms and configuration of imines. More recently, these models have been updated to incorporate structural descriptions of catalysts, allowing for the explanation of new experimental observations. Notably, they account for phenomena such as the enantioreversal in stereoselectivity observed with 2,4,6-(iPr)3C6H2 (TRIP) and SiPh3 (TIPSY).63,64
 | ||
| Fig. 10 (A) Two alternative views of the BINOL–CPA catalysts. (B) Goodman's model on mechanisms of CPA-catalyzed imine additions. | ||
An important consideration in the use of DFT methods is the reliability of the density functional and basis set for modelling complex interactions accurately. Since its introduction in 1993, B3LYP has become one of the mainstream functionals in DFT calculations due to its versatility in organic systems.38–40 However, the shortcomings of B3LYP in effectively describing dispersion effects have been well-documented.65 As a result, critical binding energies are often underestimated in many reactions. To address this, new functionals have been continuously developed. For example, meta-GGA functionals developed by Truhlar66–69 have been parameterized to improve the accuracy of medium-range correlation, including some dispersion effects. Another approach is implementing dispersion correction on available density functionals—a method popularized by Grimme.70–73 The existing functionals can be augmented with an additional energy term to account for dispersion. Such dispersion corrections not only revitalized old functionals like B3LYP (in the form of B3LYP-D3(BJ)71), but also enabled higher accuracy on top of the functionals containing medium-range dispersion effects by introducing long-range dispersion corrections, leading to the applications of functionals like M06-2X-D3 in studying dispersion-dominated systems. Indeed, while ab initio methods such as CCSD(T)74,75 with large basis sets have long been able to accurately model attractive noncovalent interactions, recent developments in DFT-based methods have made it more feasible to model NCIs in larger systems.
Since some selectivity models were derived from calculations performed before the introduction of dispersion-inclusive functionals, re-evaluating these reactions has refined our mechanistic understanding of selectivity, giving the same results with higher accuracy or providing new insights into the mechanisms. For example, the Houk–List model was revisited with the introduction of new computational techniques, revealing greater accuracy and deeper insights into the non-covalent interactions governing transition states.76 Another example is the re-visit of iminium intermediates derived from MacMillan imidazolidinones.77 Previous calculations without considering dispersion interactions by Houk et al. suggested the conformation of the benzyl group placed on top of the π-system,78 while the study combining X-ray structures and calculations including dispersion corrections suggested that the conformer where the benzyl group is on top of the heterocycles has only ∼2 kcal mol−1 energy differences from the previous one across various calculation methods. These results indicated the necessity of accounting for dispersion corrections when studying such systems (Fig. 11A). More recent examples include investigations into the stereochemical models of the Corey–Bakshi–Shibata (CBS) reduction by Schreiner et al. In re-examining the TS of CBS reduction, they purposely aimed to interrogate attractive non-covalent interactions by employing dispersion corrections in their computational method.79 These efforts led to the proposal of a chair-type hydride transfer TS, offering an alternative to the previously-studied boat-like TS model,80–84 and providing more reasonable activation free energy barriers that aligned better with experimental results. Unlike the previous stereochemical model in which the origin of enantioselectivity was attributed to steric repulsion in the TS structures, the attractive London dispersion interactions between the substrate and the catalyst were found to also determine the enantioselectivity (Fig. 11B).
 | ||
| Fig. 11 Incorporation of dispersion correction provides new mechanistic insights on organocatalyzed reactions: (A) possible conformers of iminium intermediates of MacMillan imidazolidinone catalysis (energy calculated at various theoretical methods); (B) attractive dispersion interactions instead of steric repulsion determine the enantioselectivity of CBS reductions (previously-studied TS calculated at B3LYP/6-31+G(d,p) in ref. 83; proposed alternative TS calculated at B3LYP-D3(BJ)/6-311+G(d,p)-SMD(THF)//B3LYP-D3(BJ)/6-311G(d,p) in ref. 79). | ||
Overall, the sharp downtick in reliance on empirical pictures coincided with a notable increase in the use of DFT, a reflection of the increased accessibility to methods capable of modelling geometries and energies accurately. Concurrently, as reaction complexity increased, intricate molecular interactions and subtle steric effects emerged as pivotal factors in determining stereochemical outcomes. In such cases, traditional chemical intuition may have proven insufficient in elucidating the underlying mechanisms driving stereoselectivity, necessitating the utilization of complex computational approaches for comprehensive understanding and prediction.
Correlations
TS calculations are generally not scalable and often limited to a few substrate–catalyst combinations, which restricts their effectiveness in analyzing structure–selectivity relationships and in benchmarking computational methods against a broader set of experimental results. While computational approaches are significant in studying TS features under optimal reaction conditions, experimentally determined substituent effects on rate or selectivity are crucial for gaining insights into critical NCIs in reactions of interest. In this context, developing models that accurately reflect the relationship between reaction outcome and chemical structure is essential. At the core of these modelling efforts are featurization – the process of representing complex chemical structures with simple numerical descriptors – and regression analysis, which estimates the relationship between a dependent variable (the response) and independent variables (the features). Linear regression, and more recently multivariate linear regression (MLR), is particularly noteworthy for its simplicity, interpretability, and robustness, as well as its resistance to overfitting. Consequently, linear free energy relationships (LFERs) offer a well-established and powerful method to correlate reactivity with chemical structure. Traditionally, this approach has been applied to reactivity, but by recognizing that ΔΔG‡ = −RTln(e.r.), this method can be extended to selectivity data. Depending on the factors influencing the experimental outcome, one or multiple descriptors may be employed, categorizing the regression techniques into univariate or MLR. Although LFERs were originally developed for mechanistic insight, once a quantitative structure–selectivity relationship (QSSR)85 is established, chemists can effectively predict enantioselectivities in underexplored chemical spaces. As discussed in this section of the review, concepts like LFER, MLR, QSSR, and statistical modelling exhibit significant overlap both chronologically and in content. Specifically, LFER and MLR often facilitate the development of a QSSR, while statistical modelling serves as a broad term encompassing all these related concepts. These models have been deployed to correlate the selectivity of various reactions, including those facilitated by the catalysts and ligand types discussed above. In many cases, enantioselectivity is the primary target for correlation, as it has historically been challenging to optimize. However, these tools have also been applied to predict other selectivity outcomes, such as diastereoselectivity, site-selectivity, and regioselectivity. In the first example of multivariate statistical modelling approach in predicting selectivity outcomes reported by Norrby et al. in 1997, chiral ligands and achiral ligands were combined in the same dataset of Pd-catalyzed allylation reactions. Enantioselectivities were predicted for datapoints with chiral ligands, and regioselectivities were predicted for datapoints with achiral ligands.86
The earliest examples of LFERs to probe stereoselective reaction outcomes predominantly relied on the use of experimentally derived parameters, such as Hammett descriptors, which assess the electronic effects of substituents (Fig. 12A). As quantum chemical methods have improved, the use of computationally derived descriptors emerged as an alternative to experimental parameters to describe important molecular features. The major benefit to this analysis is that reaction features generally only require computationally inexpensive ground-state (GS) calculations, thus allowing for the evaluation of a much larger set of structures compared to traditional transition state analysis. We acknowledge that a more complete understanding of reaction rates requires consideration of transition-state structures and energies,87,88 but we believe that ground-state descriptors serve as a valuable starting point for predictive models, especially when computational resources are limited.
 | ||
| Fig. 12 Early examples on building linear correlations between enantioselectivity and reaction parameters. | ||
The development and evolution of various descriptors have significantly expanded the capabilities and scope of model development, enabling LFER analysis to be applied to more complex reactions and encompass broader reaction classes. For example, Sigman et al. have highlighted the limitations of traditional experimentally derived descriptors, such as Hammett, Charton, and A-values, in accurately reflecting underlying mechanistic phenomena.89 As a result, more comprehensive descriptors, like IR vibrations and NMR chemical shifts, were introduced to capture a broader range of mechanistically relevant features.90 Presently, common statistical modelling approaches typically leverage a wide array of structural and molecular descriptors, gathered from techniques such as DFT, Quantitative Structure–Activity Relationships (QSAR),91,92 and Molecular Mechanics (MM).93,94 The choice of descriptors often depends on the specific structures and processes being examined. Featurization methods typically aim to capture steric effects (e.g., Sterimol parameters, cone angles) or electronic properties (e.g., Hammett parameters,95 Natural Bond Orbital96–100 (NBO) charge) of reaction partners, catalysts, and solvent. The introduction of these advanced descriptors has been pivotal in transforming the field from developing simple correlations that explain only a subset of reaction outputs to addressing larger, more complex datasets generated during the reaction development process. While these techniques are often described as mechanism-agnostic – meaning they are not inherently dependent on mechanistic details – it is still valuable to have a solid understanding of the underlying mechanisms to fully harness their potential. Importantly, when leveraging equations to describe reactions based on one or several features, these features must influence the transformations in a similar manner and to a comparable extent, allowing for their representation using simple, singular equations with high goodness of fits (i.e. R2 values). However, to accommodate a broader range of reactions, which may exhibit different sensitivities to the molecular features commonly captured in larger datasets, non-linear machine learning algorithms have been applied. These algorithms enable the modelling of more complex relationships and this topic will be explored further in the next section.
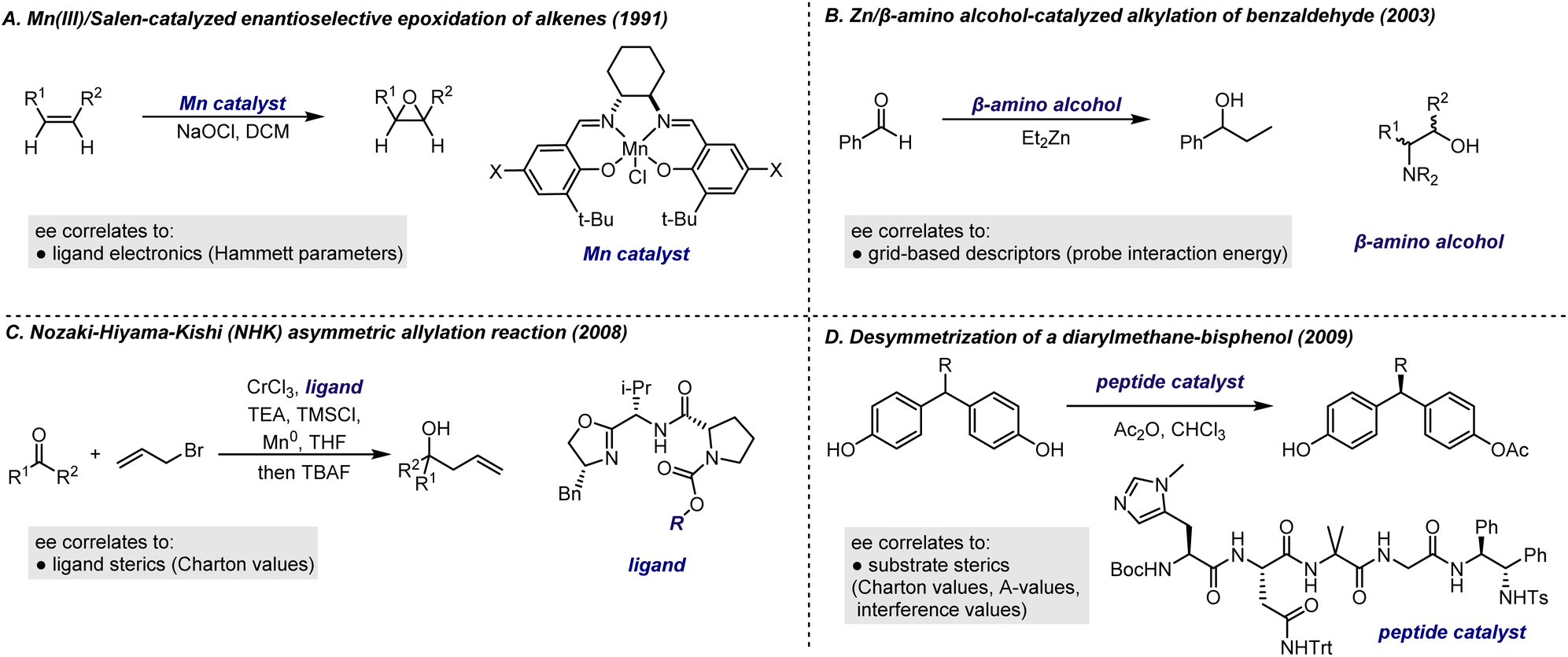
An oft-cited example of applying LFERs to the study of asymmetric catalysis is the Mn(III)/Salen-catalyzed enantioselective epoxidation of alkenes reported by Jacobsen et al. in 1991101 (Fig. 12A). Notably, electron-donating substituents on the Salen ligand led to higher enantioselectivities for alkenes prompting the authors to use LFER to investigate how these substituents influenced enantioselectivity. A linear correlation between enantioselectivity and the Hammett parameter, σpara, was obtained for a few alkene substrates. These observations also offered insights into the reaction mechanism. Based on substituent effects and other mechanistic experiments, the authors attributed the influence of substituents on enantioselectivity to their effect on the positioning of relevant transition states along the reaction coordinate. Electron-donating groups were found to stabilize transition states that are more product-like, while electron-withdrawing groups stabilize transition states that are more reactant-like.102 This hypothesis was confirmed through density functional theory (DFT) calculations, which demonstrated that substituents affect the strength of the Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the intermediate, thereby influencing the distance between the alkene and oxygen in the transition states.103 This successful outcome highlights the effectiveness of Hammett analysis when the chemical space under investigation is small and the structural perturbation affects a single transition state feature.
O bond in the intermediate, thereby influencing the distance between the alkene and oxygen in the transition states.103 This successful outcome highlights the effectiveness of Hammett analysis when the chemical space under investigation is small and the structural perturbation affects a single transition state feature.
An outstanding limitation of early-stage experimentally derived descriptors, such as Hammett, is the narrow scope of molecules that they can describe. To obtain a descriptor value for a new substituent, relevant experiments must be performed, which can be challenging due to material availability and measurement accuracy. In the early 2000's, when experimental descriptors were still predominantly employed, a few efforts focused on addressing this limitation by assembling QSSR models using computationally-derived parameters.
A representative example is the QSSR model for Zn/β-amino alcohol-catalyzed alkylation of benzaldehyde, reported by Kozlowski et al. (Fig. 12B).87 In this study, grid-based descriptors known as comparative molecular field analysis (CoMFA) – commonly used in biochemistry and medicinal chemistry to probe interactions between small molecules and large biological molecules104,105 – were applied to describe the steric and electronic effect of the catalysts. The TS structure for each catalyst was located and then aligned within a uniform 3D grid space, where a probe (a carbon 2s electron in this study) was assigned to each grid point. The interaction energy between the probe and the molecule, calculated at each grid point, effectively captured the molecular shape and charge distribution in the 3D space. These interaction energies were then utilized as descriptors to construct a model that showed a good correlation and predictive capability for a dataset of 22 amino alcohol ligands. In a contemporary study by Lipkowitz et al., a similar approach based on CoMFA was used to analyze the Cu-catalyzed asymmetric Diels–Alder reaction. In addition to establishing a predictive model, insights into key features of catalysts for higher enantioselectivities were also provided.106
Because the TS structure often resembles the reaction components, it is also possible to collect parameters from the ground states. This insight, combined with advances in quantum chemical calculations, has provided an accurate and reproducible method for obtaining molecular structures and deriving features from calculated geometries. However, the adoption of computational techniques was initially slow, likely due to their perception as specialized knowledge, and their widespread use did not gain traction until a decade later. Today, with the support of specialized software and automated scripts, the process of extracting these parameters has become more streamlined.
Many stereoselective reactions are highly sensitive to steric effects, making it essential to establish a robust base of steric descriptors in order to effectively apply LFER for probing these reactions. The challenge of modelling reactions affected by steric factors has been a longstanding focus in the development and application of LFERs. Several steric descriptors, including Taft descriptors,107 Charton values,108–111 A-values,112,113 and interference values,114 have been created or repurposed to quantify these effects. Successful application of these descriptors includes early studies by Sigman and co-workers, which demonstrated that the enantioselectivity of various reactions, such as the Nozaki–Hiyama–Kishi (NHK) asymmetric allylation reaction115 and the desymmetrization of a diarylmethane-bisphenol,116 can be correlated with the Charton values of a single substituent on the catalyst or substrate (Fig. 12C and D). In the latter study,116 it was discovered that correlating three distinct parameters – Charton values, A-values, and interference values – revealed that Charon values were effective for substrates with steric bulk near the chiral center, while their effectiveness diminished with more distal steric bulk. In these contexts, A-values and interference values provided better correlations, despite the limited availability of data. This highlights a modern approach to feature extraction, where all possible parameters are collected to relate ee and differentiate between structures. The model then performs feature selection, leading to optimal mathematical relationships.
In some cases, the descriptors may not accurately capture the changes in structure and enantioselectivity, indicating that additional descriptors might be necessary to achieve strong correlations. This can result in complex correlations that are difficult to interpret. Alternatively, incorporating a parameter that more comprehensively describes these changes can help. To do this effectively, it is crucial to recognize the limitations of the current descriptor set, which will inform the selection of alternative descriptors or the design of entirely new ones.
For example, A-values were defined as the free energy differences between the axial and equatorial conformers of a mono-substituted cyclohexane. The assumption was that the conformational preferences were solely dependent on steric repulsion, and London dispersion interactions were not considered. However, in the case of large substituents, dispersion interactions should be carefully considered, as they will undoubtedly contribute to the observed free energy difference. This resulted in diminished predictive power when they were used to build correlations for larger datasets and more complicated substituents. Therefore, A-values cannot be treated as purely steric descriptors and need to be further refined.117 Moreover, Charton values assume that substituents can be treated as spheres, which simplifies their behavior by accounting for rapid rotation around their axes. However, in kinetically-controlled reactions, which are common in asymmetric catalysis, only one or a few conformers of a substituent are relevant. In such cases, the descriptors derived under the assumption of spherical, rapidly rotating substituents become less realistic, especially for anisotropic, non-symmetrical substituents in specific confirmations. As part of their study on the NHK allylation, Sigman et al. observed breaks in linearity when several non-symmetrical substituents were introduced. They extrapolated from a linear model developed with substituents of smaller Charton values, proposing that larger Charton values might yield higher enantioselectivity. However, new catalysts with larger substituents resulted in lower enantioselectivities than the optimal t-Bu substituted catalyst. Interestingly, this new set of data points formed a separate linear correlation among themselves, effectively splitting the linear relationship into two groups. The break in linearity was initially explained as a change of the activation mode, though several years later, imperfections in the descriptors were also considered as a possible cause.
To address this issue, Sterimol parameters, first developed by Verloop et al. in the 1970's for describing the steric effects of substituents, were introduced.118,119 Deriving Sterimol parameters for substituents requires establishing a reference point by defining an axis that passes through the atoms linking the substituent and the substrate. Three subsets of Sterimol parameters can be derived: the length (L), which represents the maximum extension of the substituent parallel to the axis; and the minimum (B1) and maximum (B5) widths, which indicate the expansion of the substituent perpendicular to the axis. Some early efforts were made using Sterimol parameters to establish linear correlations. In 2004, Andersson et al. correlated the enantioselectivities with the Sterimol parameters B1 of the alkyl substituents on the substrates when studying Ru-catalyzed asymmetric transfer hydrogenation of ketones, thereby revealing the impact of steric hindrance of substituents on the enantioselectivities.120 In Sigman's study on NHK allylation, a good linear correlation can be established incorporating all substituents when Sterimol parameters were used instead of Charton values.89 These data analysis tools were also found to be well-suited to study the interdependence between substrate and catalyst structure. One of the first investigations in this context focused on the prediction of both substrate and catalyst performance in the enantioselective propargylation of dialkyl ketones using Sterimol parameters (Fig. 13).121
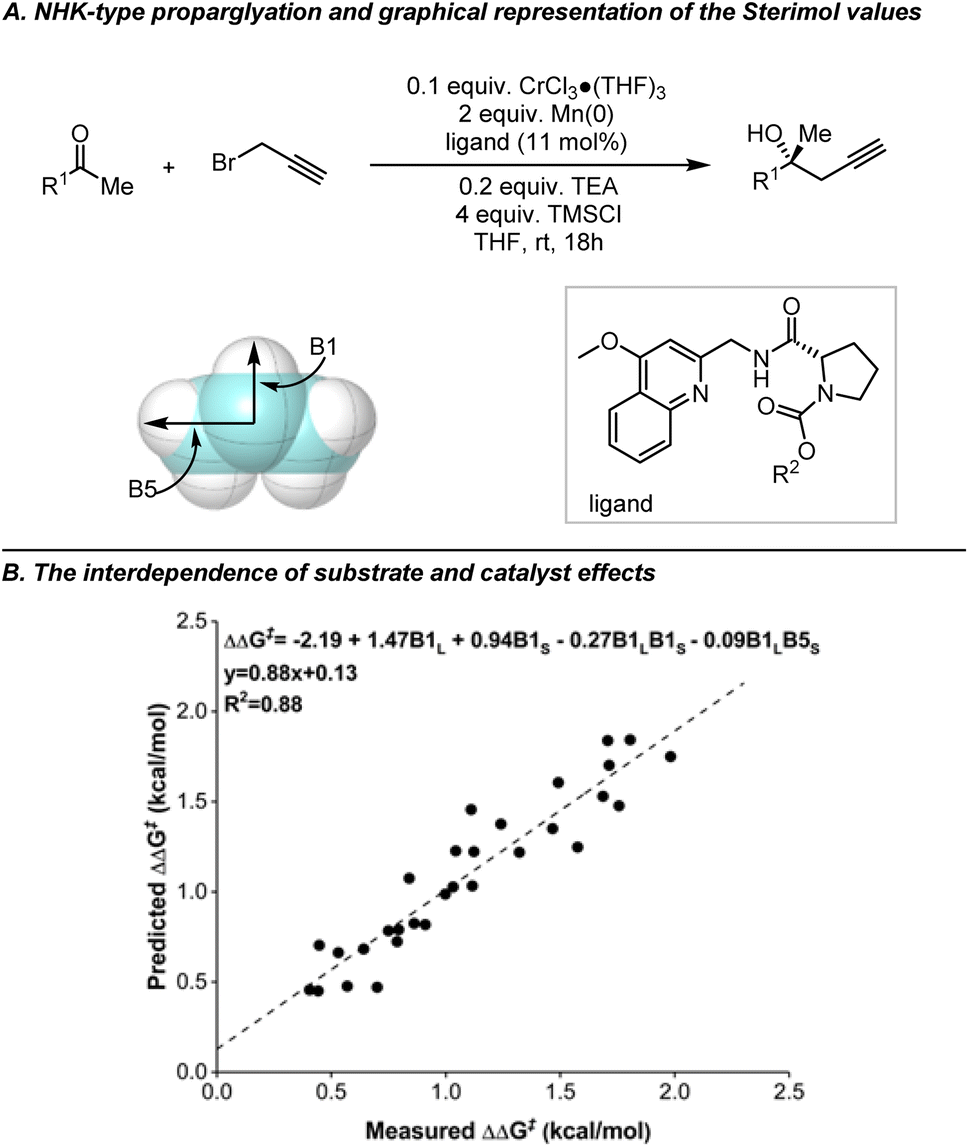 | ||
| Fig. 13 Exploring the interdependence of substrate and catalyst effects in NHK-type propargylation reactions through Sterimol parameters B1 and B5. The graphical representation shows Sterimol values using an isopropyl group as an example, oriented along the primary bond axis. The two width measurements, B1 (minimum width) and B5 (maximum width), are taken perpendicular to this axis. Adapted with permission from ref. 121. Copyright 2013, American Chemical Society. | ||
In principle, given the molecular structure and the defined axis for the substituents, the Sterimol parameters can be readily calculated. For common and simple substituents, these parameters can often be extracted from the literature. Modern approaches to obtaining Sterimol parameters rely on computed structures to maintain consistency within a statistical model. In many cases, the molecular structure used is the ground state; however, the ground state may undergo conformational changes to adopt the transition state geometry. Calculating from the ground state provides a standardized reference point, facilitating comparisons between molecules. While analyzing a single conformer can streamline the acquisition of parameters for analysis and prediction, flexible molecules may require consideration of averaged, maximum, or minimum parameter values, as previously highlighted by various groups.122 Paton et al. merged the Sterimol parameters with the Boltzmann weighting of different conformers to develop the weighted Sterimol (wSterimol) parameters.123 Today, Sterimol parameters are widely employed as tools in physical organic chemistry, particularly in the field of asymmetric catalysis. However, Sterimol parameters should not be considered universal. In the case where Sterimol parameters fail to accurately describe the steric effects within the targeted reaction systems, chemists can still define new sets of steric descriptors to improve the performance of the correlation.124
Likewise, descriptors focused on electronic effects have advanced rapidly due to both an increased fundamental understanding of reaction mechanisms and greater access to routine quantum chemistry calculations. In more recent MLR studies, steric parameters are often required alongside one or more electronic descriptors, which are typically represented by NBO charges, the IR stretching frequency of covalent bonds, orbital energies, or NMR chemical shifts.90
These tools have demonstrated the ability to streamline reaction optimization by narrowing the parameter space that needs physical testing to the regions with predicted high performance. While several impressive studies highlight the potential of this approach,125–135 practical limitations arise due to built-in constraints that restrict its applicability to specific reaction types and starting materials. Generally, these models only predict outcomes that closely resemble the training set. Accordingly, this approach requires developing a new correlation for each rection type, which can be both inefficient and resource-intensive. Such limitations can be a highly valuable direction for future work. By explicitly defining the limits of parameterization, chemists can unveil the factors leading to deviation or failure of a predictive model. This would offer critical insights into the reliability and applicability of the model.136,137
Recalling from the previous section that qualitative predictions about stereoselectivity for genuinely new reactions can be made, it is also possible to extend this capability to quantitative predictions using these tools. This was first demonstrated by Reid and Sigman, who showed that statistical models can be generated for entire reaction classes in which multiple reaction components (substrates, catalyst, solvent, temperature, and so on) are varied.138 This approach contrasts with the traditional MLR analysis described above, which typically limits studies to systematically modifying a catalyst or substrate structure (Fig. 14).
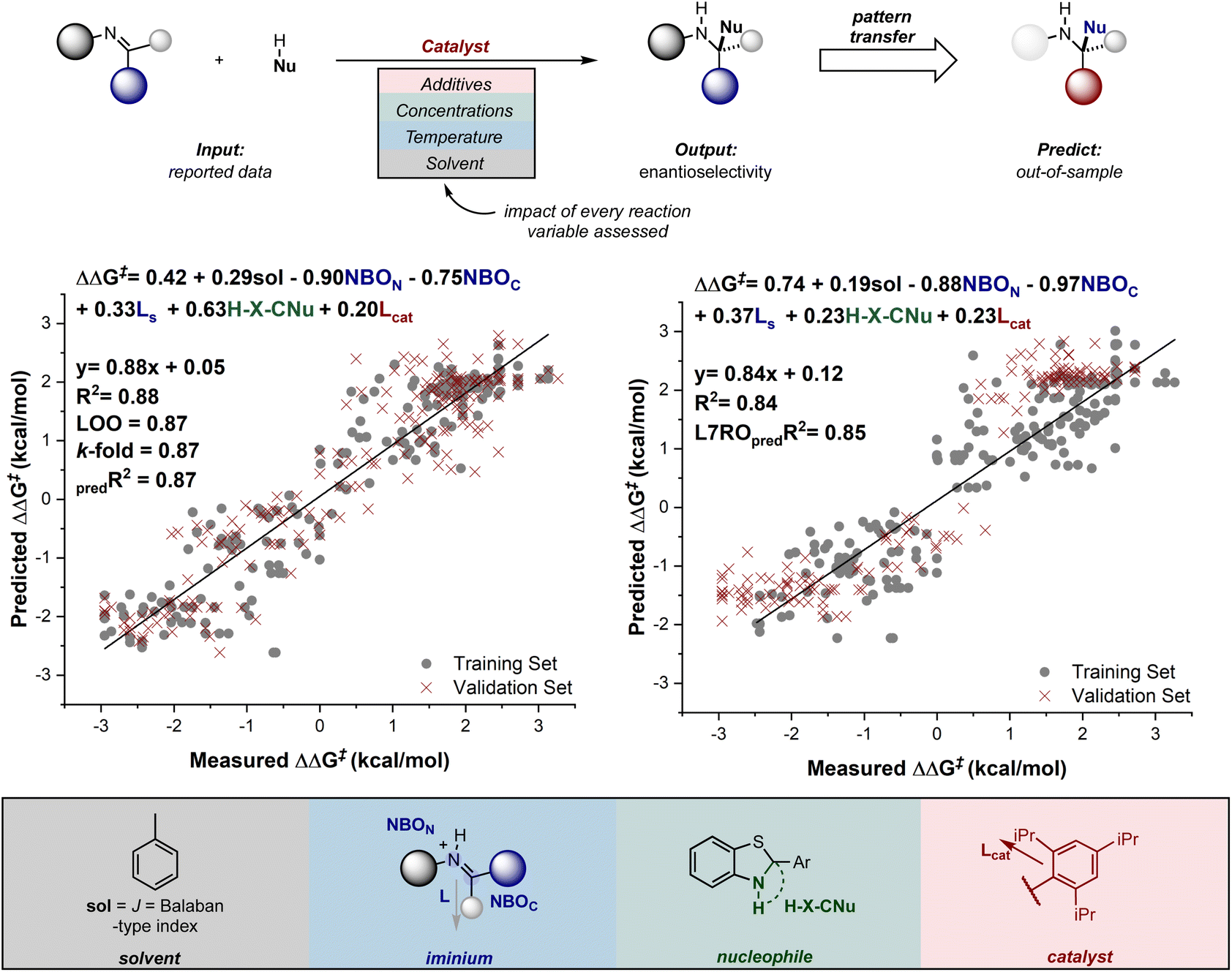 | ||
| Fig. 14 Comprehensive MLR models for CPA-catalyzed nucleophilic addition to imines. | ||
The models developed by Reid and Sigman revealed the general interactions that influence asymmetric induction and enabled the quantitative transfer of this information to new reaction components. More specifically, by curating a dataset of 367 reactions from the literature, with a wide range of enantioselectivity data (spanning a free energy range of about 6 kcal mol−1), they were able to explore both enantiomeric amine products, a result of active E and Z configurations in the reactions. Using this comprehensive dataset, correlations were developed between the experimentally determined enantioselectivity and molecular descriptors collected from DFT-optimized geometries. These descriptors captured the structural features of each imine, nucleophile, catalyst, and solvent. A comparison of the terms in the correlations derived from E and Z data revealed that most interactions are similar and driven by repulsive catalyst–substrate contacts. However, a clear difference in the models is the result of nucleophile steric effects which become the dominant selectivity discriminant in Z pathways. As a final step in the workflow, the ability of these models to transfer mechanistic principles to entirely different structural motifs not included in the training set was evaluated. Excitingly, the models predicted each result accurately, even in situations where multiple components are varied.
The Reid group has demonstrated that this methodology, which focuses on leveraging small, well-understood portions of chemical space to predict the behavior of much larger, unexplored regions, is broadly applicable.139 By combining a fundamental understanding of reaction mechanisms and structure–function relationships, insights can be generalized to predict the behavior of other molecules, even if those specific structures have never been characterized.140,141 The effectiveness of this approach has been demonstrated across increasingly complicated reactions, including multi-catalysis142 and reaction application to substrates that lead to complex molecule formation.143
Clearly, the complexity and functions of correlation models have evolved alongside the development and application of new molecular descriptors. Initially derived from experimental data, these descriptors have now expanded to include calculated values, particularly for designer systems (i.e., those applicable to one or a few systems). Advances in descriptor sets have enabled the prediction of increasingly complex and diverse reactions, allowing multiple phenomena (e.g., steric and electronic features) to be captured within a single descriptor or through several descriptors to accurately describe structural effects. This progress has demonstrated tremendous potential and has been well-received in addressing various practical research demands including the optimization of reaction conditions and catalyst structures.85,125,133,144–146
Non-linear machine learning
While MLR is a valuable tool for identifying linear relationships between features and outcomes, it is inherently limited in its ability to capture the complex, non-linear relationships often characteristic of asymmetric catalysis. Additionally, substrate–catalyst matching—where a specific catalyst performs well with certain starting materials but poorly with others, even structurally similar ones—further complicates the use of MLR, as it requires interaction terms that increase model complexity and can obscure interpretability (Fig. 13).147,148Non-linear ML methods offer promising alternatives to address these challenges.149–151 These algorithms are capable of modelling intricate, non-linear relationships, allowing for more nuanced predictions by capturing complex interactions between catalysts and substrates. However, to fully leverage non-linear algorithms, large, high-quality datasets (i.e., datasets that include sufficient chemical diversity and distribution of points) are typically required, as smaller datasets increase the risk of overfitting. Additionally, ML models may lack transparency, making it challenging to interpret the specific features or interactions driving predictions. Despite these limitations, non-linear ML methods represent a significant advancement toward building predictive models that can accommodate the complexities of asymmetric catalysis.
Some early efforts were made in 2000's using non-linear ML to predict enantioselectivity values and absolute configurations of major products given a certain reaction class.152,153 However, the resurgence of interest in applying non-linear ML in asymmetric catalysis has been in the past few years. A landmark contribution in this area was reported by Denmark et al. in 2019, who developed average steric occupancy (ASO) descriptors—an extension of the CoMFA descriptors previously utilized by Kozlowski and Lipkowitz et al. and described above—and applied them with ML to predict highly selective CPA catalysts for thiol additions to imines (Fig. 15A).154 Essentially, this grid-based descriptor is obtained by modelling the presence or absence of an atom at various grid points, and averaging these values for all low-energy conformers of the catalyst. With a dataset of 2150 CPA-catalyzed thiol additions to N-acylimines, they found support vector regression and deep feed-forward neural networks to be the most effective algorithms for predicting enantioselectivity. Notably, the deep feed-forward neural network model could accurately predict reactions achieving >80% ee, even when the training set included only reactions with <80% ee. This demonstrated the extrapolative capacity of ML algorithms, highlighting their potential to assist in reaction optimization, even when datasets primarily contain low to moderate enantioselectivities. Due to its size (over 1000 reactions) and high quality, this dataset has since become a benchmark for evaluating new descriptors, as discussed below. The Sunoj and Doyle groups have explored various ML approaches to predict selectivity and reactivity in complex catalytic systems, and their efforts represent other important early contributions to ML-based reaction prediction.155,156
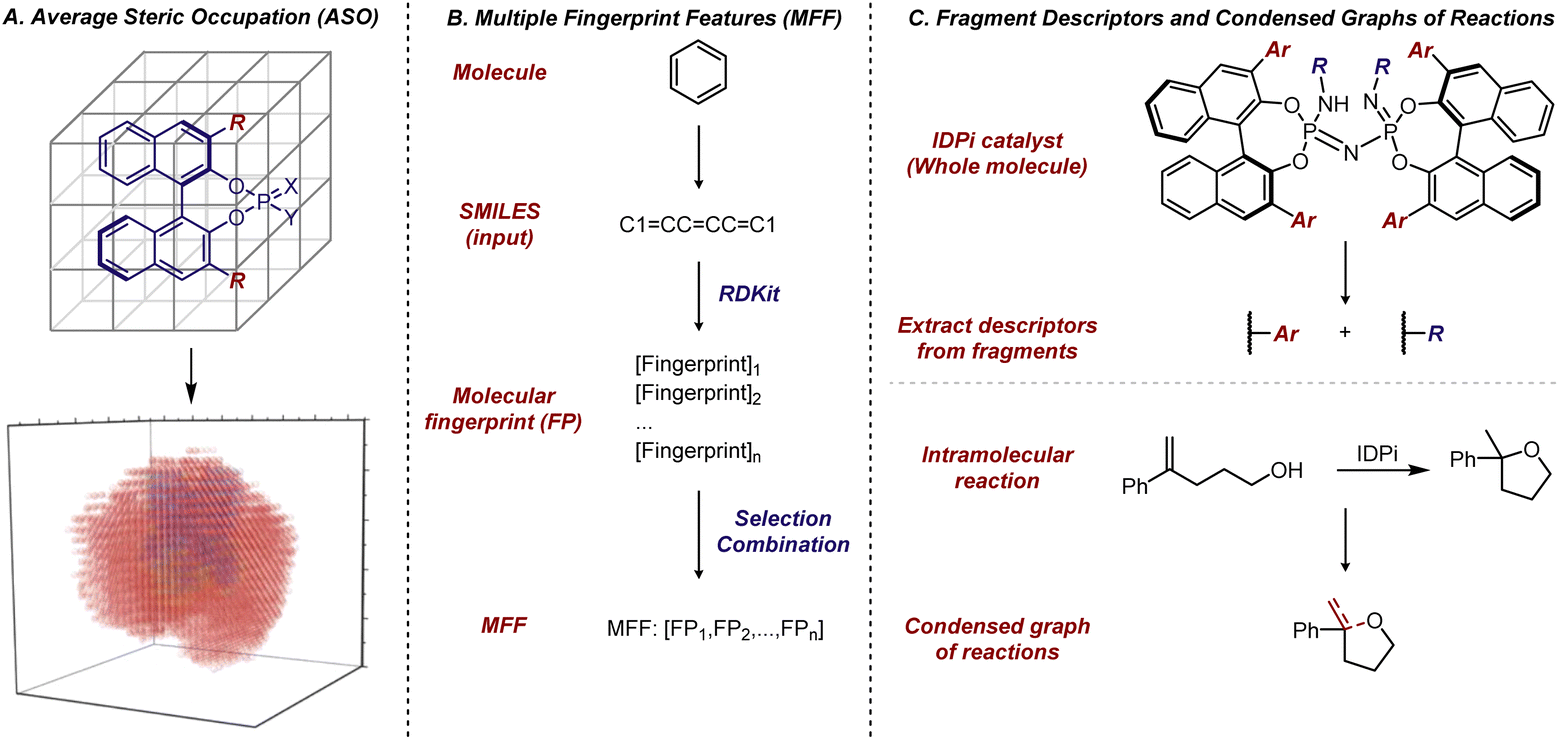 | ||
| Fig. 15 (A) Average steric occupation (ASO) for description of CPA catalyst. Adapted with permission from ref. 154. Copyright 2019, The American Association for the Advancement of Science. (B) Multiple fingerprint features as molecular descriptors. (C) Fragment descriptors for IDPi catalysts and condensed graph of reactions (CGR) for intramolecular reactions. | ||
ML models typically offer better fits to training sets than MLR, as demonstrated by the Reid group's efforts in revisiting the datasets of CPA-catalyzed nucleophilic addition to imines using the XGBoost algorithm.157 While the statistics improve in this context, interpretation can be challenging due to the often ‘black-box’ nature of ML models. Interestingly, this group leveraged the interpolative ability of the ML model to construct virtual datasets, allowing them to derive catalyst generality values, a new metric for measuring broad catalyst success, without bias.
As described throughout, MLR typically involves selecting simple, interpretable descriptors that linearly relate to the outcome, such as electronic properties, steric effects, and thermodynamic properties. In contrast, non-linear ML can accommodate a wider variety of descriptors, including those that may not have simple, direct relationships with the outcome and can interact in more complex, non-linear ways. It also supports richer, more abstract representations like molecular fingerprints, graph-based features, or embeddings, which do not necessarily require the 3D structures of molecules. Due to the various encoding approaches, these descriptors may be presented in formats that are more accessible to computers, but less interpretable and straightforward for chemists.
In 2020, Glorius et al. developed a type of molecular representation based on multiple fingerprint features (MFF).158 This platform is based on the assumption that a molecule's reactivity can be directly derived from its structure and relies solely on SMILES, a string notation for molecules, as input. For each molecule, an array of 24 diversely configured fingerprints, which describe structure-based molecular properties in binary strings, was generated using the open-source Python package RDKit. Subsequently, these MFF representations of molecules were used to solve various problems relevant to small organic molecules. Notably, the authors revisited the dataset of CPA-catalyzed thiol additions to N-acylimines reported by Denmark et al.,154 and the models trained with molecular fingerprints demonstrated training set fits comparable to those of the original study, where the model was trained with more complicated descriptors. From a predictive perspective, the approach using molecular fingerprints significantly simplified the process of obtaining molecular descriptors, as these MFF descriptors can be generated error-free within seconds (Fig. 15B).
In addition to generating molecular fingerprints for whole molecules, especially in the case of smaller organic compounds, there are also examples of applying fragment descriptors for substrates and catalysts. In 2023, List et al. introduced fragment descriptors for BINOL-derived imidodiphosphorimidate (IDPi) catalysts (Fig. 15C). Since all IDPi catalysts share the same backbone and phosphorimidate moiety, but have different 3,3′-substituents and perfluoro-substituents on the nitrogen, only these substituents were used to represent the distinct features of the IDPi catalysts.159 The Circular Substructure (CircuS) descriptors, derived from the ISIDA (In Silico Design and Data Analysis) platform,160 were employed for the catalysts. This approach was applied to study an IDPi-catalyzed intramolecular cyclization reaction that forms substituted cyclic ethers. Fragment descriptors for the substrates were obtained through Condensed Graphs of Reaction (CGR),161,162 a function of the ISIDA platform that allows the combination of reactants and products into a single pseudo-molecule with dynamic bonds. While the CGR approach has been utilized in modelling practices in other fields, its application in asymmetric catalysis had not been reported prior to this study. The combination of CircuS descriptors for the catalysts, CGR for the substrates, and common descriptors for other reaction parameters facilitated model construction. This integration demonstrated applicability in predicting selective IDPi catalysts and effective substrates. Furthermore, retraining models for the CPA-catalyzed thiol addition to N-acylimines using fragment descriptors for the 3,3′-substituents of BINOL–CPAs also exhibited promising performance.
Most of the parameters examined are designed to differentiate one molecule from another while also identifying relevant features that link structural changes to enantioselectivity. The implication of these parameter types is that if similar molecules, as defined by a descriptor, exhibit comparable levels of enantioselectivity, that descriptor is considered significant. However, this approach can lead to local chemical neighborhoods—defined by structural similarity—performing similarly in many instances. To complement these existing parameter sets, the Reid group implemented a descriptor set that incorporates information about the performance and characteristics of neighboring molecules into their model (Fig. 16A).163 The focus of this effort was to build a multi-reaction model for predicting IDPis reaction outcomes across various reactions, including Mukaiyama aldol,164,165 Michael additions,166 Diels-Alder,167–171 Nazarov,172 Prins,173 and Hosomi–Sakurai reactions.174
 | ||
| Fig. 16 (A) Features and descriptors derived from chemical space networks (CSN) enable better predictions for IDPi-catalyzed reactions. (B) “Key intermediate” graph as a new representation of reaction intermediates for graphical neural networks (GNN). | ||
To construct local neighborhoods of molecules, the authors employed chemical space networks (CSN), a method that builds a network of nodes corresponding to molecules, with edges typically representing some form of similarity index. Networks naturally illustrate chemical spaces by depicting how molecules are structured and interrelated without needing to establish a coordinate system or reduce dimensionality. Similarity metrics, such as Tanimoto and maximum common substructure (MCS), are derived from molecular structures (such as those represented by SMILES strings), thereby circumventing challenges associated with high-dimensional data. Since molecules tend to share similarities, a threshold is employed to prevent a fully connected network, making the network properties somewhat reliant on this threshold. This approach allows for the collection of experimental and local structural information that reflects the average historical performance of all neighbors of a given molecule, as well as the maximum and minimum average values among its neighbors. These features supplement the traditional descriptors and reduce prediction error.
Another innovative featurization approach was recently introduced by Schreiner et al., who developed a ‘key-intermediate graph’ to investigate the enantioselectivity of CBS reduction using graphical neural networks (GNN) (Fig. 16B).175 The use of the “key-intermediate graph” resulted in slightly higher accuracy, indicated by a lower root-mean-square error (RMSE), compared to using separate graphs of starting materials and catalyst structures for model construction and prediction. By leveraging a dataset of only ∼100 reactions, the authors were able to increase the enantioselectivity for the CBS reduction of 2-butanone to 80% ee.
ML models provide new opportunities for employing innovative featurization techniques, enabling more accurate predictions of enantioselectivity outcomes. These advancements have addressed critical synthetic challenges, such as the development of a catalyst for the steric differentiation of nearly equal-sized groups, as demonstrated in a previous case study by Schreiner.175 One limitation is the requirement for larger datasets, which can make these approaches less accessible to bench chemists and more complex, potentially rendering the models less intuitive. However, new innovations in explainable AI are expected to facilitate the transformation of predictions into straightforward, executable experimental directions.
Conclusions
Reaction outcomes involving chiral auxiliaries or stoichiometric reagents are generally more straightforward to rationalize and predict, as these systems often proceed through well-defined intermediates and transition states, allowing for clear mechanistic insights. In contrast, catalytic systems introduce dynamic interactions and competing pathways, adding complexity to predictions. Certain physical organic tools, such as qualitative models and quantum calculations, are particularly advantageous for specific reactions and substrate classes. Qualitative models excel in cases where one or two steric or electronic effects dominate, providing quick insights, though they may struggle with complex systems involving multiple competing factors. Quantum chemical calculations, on the other hand, offer detailed and precise predictions, particularly for smaller or rigid molecules with well-defined transition states; however, they can be computationally expensive and less effective for larger, flexible systems.Correlation-based methods rely on structurally diverse, modestly sized datasets and can be challenging to apply if suitable descriptors that accurately capture the interactions are not identified. Therefore, much focus has been directed toward well-known reactions and catalysts with established molecular representations and extensive, available datasets. By strategically leveraging qualitative models for rapid insights and quantum calculations for refining predictions, chemists can improve reaction outcomes and gain deeper mechanistic understanding, optimizing reaction conditions and catalyst designs in a more targeted manner. The main uses for different tools discussed in this review, as applied to stereoselective synthesis are summarized in Table 2. However, it is reasonable to acknowledge that others may have different perspectives on their applications.
| Main uses | Other uses | |
|---|---|---|
| Qualitative pictures | Rationalizing stereochemical outcomes | Estimating selectivity trends |
| Predicting configurations of major products | ||
| Quantum chemistry | Modelling TS structures and molecular interactions | Predicting the selectivity trends |
| Investigating reaction mechanisms | Optimizing reaction outcomes (i.e., mechanism-guided catalyst modification) | |
| Linear correlations | Providing mechanistic insights | Predicting selectivity values (ΔΔG‡) |
| Non-linear ML | Predicting selectivity values (ΔΔG‡) | Providing mechanistic insights |
An integrative approach will enhance both the predictability and scope of chiral transfer strategies. The next steps in this field will likely focus on integrating ML with innovative experimental techniques, improving the interpretability of ML models, applying diverse algorithms, including generative modelling, and fostering interdisciplinary collaboration. Together, these initiatives will pave the way for more accurate predictions, efficient catalyst designs, and transformative breakthroughs in synthetic chemistry. Ultimately, we believe that the complexity of physical organic tools for investigating organic chemistry will advance alongside developments in synthetic methods.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
J. L. and J. P. R. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support to J. P. R. was provided by the University of British Columbia, the Natural Sciences and Engineering Research Council of Canada (NSERC) and the CFI John R. Evans Leaders Fund. J. L. thanks the University of British Columbia for support through four-year fellowship (4YF).References
- D. J. Cram and F. A. A. Elhafez, J. Am. Chem. Soc., 1952, 74, 5828–5835 CrossRef CAS.
- M. Chérest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 9, 2199–2204 CrossRef.
- N. T. Anh, O. Eisenstein, J. M. Lefour and M. E. Tran Huu Dau, J. Am. Chem. Soc., 1973, 95, 6146–6147 CrossRef.
- M. T. Reetz, M. Hüllmann and T. Seitz, Angew Chem. Int. Ed. Engl., 1987, 26, 477–479 CrossRef.
- H. E. Zimmerman and M. D. Traxler, J. Am. Chem. Soc., 1957, 79, 1920–1923 CrossRef CAS.
- C. H. Heathcock, K. T. Hug and L. A. Flippin, Tetrahedron Lett., 1984, 25, 5973–5976 CrossRef CAS.
- T. J. Leitereg and D. J. Cram, J. Am. Chem. Soc., 1968, 90, 4011–4018 CrossRef CAS.
- M. T. Reetz, K. Kesseler and A. Jung, Tetrahedron Lett., 1984, 25, 729–732 CrossRef CAS.
- D. A. Evans, J. L. Duffy and M. J. Dart, Tetrahedron Lett., 1994, 35, 8537–8540 CrossRef CAS.
- M. T. Reetz and A. Jung, J. Am. Chem. Soc., 1983, 105, 4833–4835 CrossRef CAS.
- M. T. Reetz, Acc. Chem. Res., 1993, 26, 462–468 CrossRef CAS.
- D. A. Evans and T. R. Taber, Tetrahedron Lett., 1980, 21, 4675–4678 CrossRef CAS.
- D. A. Evans, J. V. Nelson, E. Vogel and T. R. Taber, J. Am. Chem. Soc., 1981, 103, 3099–3111 CrossRef CAS.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- D. A. Evans and L. R. McGee, J. Am. Chem. Soc., 1981, 103, 2876–2878 CrossRef CAS.
- D. Hoppe and T. Hense, Angew Chem. Int. Ed. Engl., 1997, 36, 2282–2316 CrossRef CAS.
- M. J. Dearden, C. R. Firkin, J.-P. R. Hermet and P. O'Brien, J. Am. Chem. Soc., 2002, 124, 11870–11871 CrossRef CAS.
- P.-W. Phuan, J. C. Ianni and M. C. Kozlowski, J. Am. Chem. Soc., 2004, 126, 15473–15479 CrossRef CAS.
- T. Imamoto, Chem. Rev., 2024, 124, 8657–8739 CrossRef CAS PubMed.
- K. E. Koenig, M. J. Sabacky, G. L. Bachman, W. C. Christopfel, H. D. Bamstorff, R. B. Friedman, W. S. Knowles, B. R. Stults, B. D. Vineyard and D. J. Weinkauff, Ann. N. Y. Acad. Sci., 1980, 333, 16–22 CrossRef CAS.
- G. Hoge, H.-P. Wu, W. S. Kissel, D. A. Pflum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966–5967 CrossRef CAS.
- T. Imamoto, J. Watanabe, Y. Wada, H. Masuda, H. Yamada, H. Tsuruta, S. Matsukawa and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1635–1636 CrossRef CAS.
- H.-P. Wu and G. Hoge, Org. Lett., 2004, 6, 3645–3647 CrossRef CAS PubMed.
- K. N. Houk and P. H.-Y. Cheong, Nature, 2008, 455, 309–313 CrossRef CAS PubMed.
- P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm and K. N. Houk, Chem. Rev., 2011, 111, 5042–5137 CrossRef CAS.
- E. H. Krenske and K. N. Houk, Acc. Chem. Res., 2013, 46, 979–989 CrossRef CAS PubMed.
- G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706–1725 CrossRef CAS.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- Q. Peng, F. Duarte and R. S. Paton, Chem. Soc. Rev., 2016, 45, 6093–6107 RSC.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- S. Ahn, M. Hong, M. Sundararajan, D. H. Ess and M.-H. Baik, Chem. Rev., 2019, 119, 6509–6560 CrossRef CAS PubMed.
- P. Nakliang, S. Yoon and S. Choi, Org. Chem. Front., 2021, 8, 5165–5181 RSC.
- Y. P. Chin, N. W. See, I. D. Jenkins and E. H. Krenske, Org. Biomol. Chem., 2022, 20, 2028–2042 RSC.
- N. Melnyk, I. Iribarren, E. Mates-Torres and C. Trujillo, Chem.–Eur. J., 2022, 28, e202201570 CrossRef CAS.
- H. Hayashi, S. Maeda and T. Mita, Chem. Sci., 2023, 14, 11601–11616 RSC.
- M. Ruth, T. Gensch and P. R. Schreiner, Angew. Chem., Int. Ed., 2024, 63, e202410308 CrossRef CAS.
- Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 1987, 109, 908–910 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- U. Eder, G. Sauer and R. Wiechert, Angew Chem. Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS PubMed.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS.
- B. List, P. Pojarliev, W. T. Biller and H. J. Martin, J. Am. Chem. Soc., 2002, 124, 827–833 CrossRef CAS.
- J. W. Yang, M. Stadler and B. List, Angew. Chem., Int. Ed., 2007, 46, 609–611 CrossRef CAS.
- S. Bahmanyar and K. N. Houk, Org. Lett., 2003, 5, 1249–1251 CrossRef CAS PubMed.
- S. Mitsumori, H. Zhang, P. Ha-Yeon Cheong, K. N. Houk, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2006, 128, 1040–1041 CrossRef CAS.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741–8747 CrossRef PubMed.
- T. Marcelli, P. Hammar and F. Himo, Chem.–Eur. J., 2008, 14, 8562–8571 CrossRef CAS.
- I. D. Gridnev, M. Kouchi, K. Sorimachi and M. Terada, Tetrahedron Lett., 2007, 48, 497–500 CrossRef CAS.
- L. Simón and J. M. Goodman, J. Org. Chem., 2011, 76, 1775–1788 CrossRef PubMed.
- G. Li and J. C. Antilla, Org. Lett., 2009, 11, 1075–1078 CrossRef CAS.
- Y.-X. Jia, J. Zhong, S.-F. Zhu, C.-M. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 5565–5567 CrossRef CAS PubMed.
- M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2006, 45, 2254–2257 CrossRef CAS PubMed.
- Q. Kang, Z.-A. Zhao and S.-L. You, Org. Lett., 2008, 10, 2031–2034 CrossRef CAS PubMed.
- J. P. Reid and J. M. Goodman, J. Am. Chem. Soc., 2016, 138, 7910–7917 CrossRef CAS.
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Valero, R. Costa, I. de, P. R. Moreira, D. G. Truhlar and F. Illas, J. Chem. Phys., 2008, 128, 114103 CrossRef.
- D. Jacquemin, E. A. Perpète, I. Ciofini, C. Adamo, R. Valero, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2071–2085 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- S. Ehrlich, J. Moellmann and S. Grimme, Acc. Chem. Res., 2013, 46, 916–926 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS PubMed.
- G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- A. Armstrong, R. A. Boto, P. Dingwall, J. Contreras-García, M. J. Harvey, N. J. Mason and H. S. Rzepa, Chem. Sci., 2014, 5, 2057–2071 RSC.
- D. Seebach, U. Grošelj, W. B. Schweizer, S. Grimme and C. Mück-Lichtenfeld, Helv. Chim. Acta, 2010, 93, 1–16 CrossRef CAS.
- R. Gordillo, J. Carter and K. N. Houk, Adv. Synth. Catal., 2004, 346, 1175–1185 CrossRef CAS.
- C. Eschmann, L. Song and P. R. Schreiner, Angew. Chem., Int. Ed., 2021, 60, 4823–4832 CrossRef CAS.
- E. J. Corey and C. J. Helal, Tetrahedron Lett., 1995, 36, 9153–9156 CrossRef CAS.
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986–2012 CrossRef CAS PubMed.
- M. P. Meyer, Org. Lett., 2009, 11, 4338–4341 CrossRef CAS.
- H. Zhu, D. J. O'Leary and M. P. Meyer, Angew. Chem., Int. Ed., 2012, 51, 11890–11893 CrossRef CAS.
- Z. Lachtar, A. Khorief Nacereddine and A. Djerourou, Struct. Chem., 2020, 31, 253–261 CrossRef CAS.
- A. F. Zahrt, S. V. Athavale and S. E. Denmark, Chem. Rev., 2020, 120, 1620–1689 CrossRef CAS PubMed.
- J. D. Oslob, B. Åkermark, P. Helquist and P.-O. Norrby, Organometallics, 1997, 16, 3015–3021 CrossRef CAS.
- M. C. Kozlowski, S. L. Dixon, M. Panda and G. Lauri, J. Am. Chem. Soc., 2003, 125, 6614–6615 CrossRef CAS PubMed.
- L.-C. Xu, J. Frey, X. Hou, S.-Q. Zhang, Y.-Y. Li, J. C. A. Oliveira, S.-W. Li, L. Ackermann and X. Hong, Nat. Synth., 2023, 2, 321–330 CrossRef CAS.
- K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366–374 CrossRef CAS PubMed.
- A. Milo, E. N. Bess and M. S. Sigman, Nature, 2014, 507, 210–214 CrossRef CAS.
- C. Hansch, P. P. Maloney, T. Fujita and R. M. Muir, Nature, 1962, 194, 178–180 CrossRef CAS.
- A. Cherkasov, E. N. Muratov, D. Fourches, A. Varnek, I. I. Baskin, M. Cronin, J. Dearden, P. Gramatica, Y. C. Martin, R. Todeschini, V. Consonni, V. E. Kuz’min, R. Cramer, R. Benigni, C. Yang, J. Rathman, L. Terfloth, J. Gasteiger, A. Richard and A. Tropsha, J. Med. Chem., 2014, 57, 4977–5010 CrossRef CAS.
- U. Burkert and N. L. Allinger, Molecular Mechanics, American Chemical Society, 1982 Search PubMed.
- N. L. Allinger, in Theoretical and Computational Models for Organic Chemistry, ed. S. J. Formosinho, I. G. Csizmadia and L. G. Arnaut, Springer Netherlands, Dordrecht, 1991, pp. 125–135 Search PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 610–627 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop and F. Weinhold, J. Comput. Chem., 1998, 19, 628–646 CrossRef CAS.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- E. N. Jacobsen, W. Zhang and M. L. Guler, J. Am. Chem. Soc., 1991, 113, 6703–6704 CrossRef CAS.
- M. Palucki, N. S. Finney, P. J. Pospisil, M. L. Güler, T. Ishida and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 948–954 CrossRef CAS.
- L. Cavallo and H. Jacobsen, J. Org. Chem., 2003, 68, 6202–6207 CrossRef CAS PubMed.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- R. D. Cramer, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS PubMed.
- K. B. Lipkowitz and M. Pradhan, J. Org. Chem., 2003, 68, 4648–4656 CrossRef CAS PubMed.
- R. W. Jr. Taft, J. Am. Chem. Soc., 1952, 74, 3120–3128 CrossRef CAS.
- M. Charton, J. Am. Chem. Soc., 1975, 97, 1552–1556 CrossRef CAS.
- M. Charton, J. Am. Chem. Soc., 1975, 97, 3691–3693 CrossRef CAS.
- M. Charton, J. Am. Chem. Soc., 1975, 97, 3694–3697 CrossRef CAS.
- M. Charton, J. Org. Chem., 1976, 41, 2217–2220 CrossRef CAS.
- S. Winstein and N. J. Holness, J. Am. Chem. Soc., 1955, 77, 5562–5578 CrossRef CAS.
- F. R. Jensen, C. H. Bushweller and B. H. Beck, J. Am. Chem. Soc., 1969, 91, 344–351 CrossRef CAS.
- G. Bott, L. D. Field and S. Sternhell, J. Am. Chem. Soc., 1980, 102, 5618–5626 CrossRef CAS.
- J. J. Miller and M. S. Sigman, Angew. Chem., Int. Ed., 2008, 47, 771–774 CrossRef CAS PubMed.
- J. L. Gustafson, M. S. Sigman and S. J. Miller, Org. Lett., 2010, 12, 2794–2797 CrossRef CAS PubMed.
- E. Solel, M. Ruth and P. R. Schreiner, J. Am. Chem. Soc., 2021, 143, 20837–20848 CrossRef CAS PubMed.
- A. Verloop, W. Hoogenstraaten and J. Tipker, in Drug Design, ed. E. J. Ariëns, Academic Press, Amsterdam, 1976, vol. 11, pp. 165–207 Search PubMed.
- A. Verloop, in Pesticide Chemistry: Human Welfare and Environment, ed. P. Doyle and T. Fujita, Pergamon, 1983, pp. 339–344 Search PubMed.
- P. Brandt, P. Roth and P. G. Andersson, J. Org. Chem., 2004, 69, 4885–4890 CrossRef CAS.
- K. C. Harper, S. C. Vilardi and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 2482–2485 CrossRef CAS.
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610–617 CrossRef CAS.
- A. V. Brethomé, S. P. Fletcher and R. S. Paton, ACS Catal., 2019, 9, 2313–2323 CrossRef.
- B. Yang, A. J. Schaefer, B. L. Small, J. A. Leseberg, S. M. Bischof, M. S. Webster-Gardiner and D. H. Ess, Chem. Sci., 2024, 15, 18355–18363 RSC.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef CAS PubMed.
- T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296–1310 CrossRef CAS PubMed.
- R. Ardkhean, M. Mortimore, R. S. Paton and S. P. Fletcher, Chem. Sci., 2018, 9, 2628–2632 RSC.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, J. Am. Chem. Soc., 2019, 141, 19178–19185 CrossRef CAS.
- V. Dhayalan, S. C. Gadekar, Z. Alassad and A. Milo, Nat. Chem., 2019, 11, 543–551 CrossRef CAS PubMed.
- J. Miró, T. Gensch, M. Ellwart, S.-J. Han, H.-H. Lin, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 6390–6399 CrossRef.
- J. M. J. M. Ravasco and J. A. S. Coelho, J. Am. Chem. Soc., 2020, 142, 4235–4241 CrossRef CAS PubMed.
- M. Escudero-Casao, G. Licini and M. Orlandi, J. Am. Chem. Soc., 2021, 143, 3289–3294 CrossRef CAS.
- J. M. Crawford, C. Kingston, F. D. Toste and M. S. Sigman, Acc. Chem. Res., 2021, 54, 3136–3148 CrossRef CAS.
- J. J. Dotson, L. van Dijk, J. C. Timmerman, S. Grosslight, R. C. Walroth, F. Gosselin, K. Püntener, K. A. Mack and M. S. Sigman, J. Am. Chem. Soc., 2023, 145, 110–121 CrossRef CAS.
- J. P. Liles, C. Rouget-Virbel, J. L. H. Wahlman, R. Rahimoff, J. M. Crawford, A. Medlin, V. S. O'Connor, J. Li, V. A. Roytman, F. D. Toste and M. S. Sigman, Chem, 2023, 9, 1518–1537 CAS.
- J. Paul Janet, C. Duan, T. Yang, A. Nandy and H. J. Kulik, Chem. Sci., 2019, 10, 7913–7922 RSC.
- M. Yu, Y.-N. Zhou, Q. Wang and F. Yan, Digital Discovery, 2024, 3, 1058–1067 RSC.
- J. P. Reid and M. S. Sigman, Nature, 2019, 571, 343–348 CrossRef CAS.
- J. P. Reid, I. O. Betinol and Y. Kuang, Chem. Commun., 2023, 59, 10711–10721 RSC.
- A. Shoja and J. P. Reid, J. Am. Chem. Soc., 2021, 143, 7209–7215 CrossRef CAS PubMed.
- A. Shoja, J. Zhai and J. P. Reid, ACS Catal., 2021, 11, 11897–11905 CrossRef CAS.
- Y. Kuang, J. Lai and J. P. Reid, Chem. Sci., 2023, 14, 1885–1895 RSC.
- I. O. Betinol, Y. Kuang and J. P. Reid, Org. Lett., 2022, 24, 1429–1433 CrossRef CAS PubMed.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398–2412 RSC.
- J. P. Reid and M. S. Sigman, Nat. Rev. Chem, 2018, 2, 290–305 CrossRef CAS.
- D. M. Lustosa and A. Milo, ACS Catal., 2022, 12, 7886–7906 CrossRef CAS.
- K. C. Harper and M. S. Sigman, Science, 2011, 333, 1875–1878 CrossRef CAS PubMed.
- K. C. Harper and M. S. Sigman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2179–2183 CrossRef CAS.
- J. C. A. Oliveira, J. Frey, S.-Q. Zhang, L.-C. Xu, X. Li, S.-W. Li, X. Hong and L. Ackermann, Trends Chem., 2022, 4, 863–885 CrossRef CAS.
- W. L. Williams, L. Zeng, T. Gensch, M. S. Sigman, A. G. Doyle and E. V. Anslyn, ACS Cent. Sci., 2021, 7, 1622–1637 CrossRef CAS.
- S.-Q. Zhang, L.-C. Xu, S.-W. Li, J. C. A. Oliveira, X. Li, L. Ackermann and X. Hong, Chem.–Eur. J., 2023, 29, e202202834 CrossRef CAS PubMed.
- J. Aires-de-Sousa and J. Gasteiger, J. Chem. Inf. Comput. Sci., 2001, 41, 369–375 CrossRef CAS PubMed.
- J. Aires-de-Sousa and J. Gasteiger, J. Comb. Chem., 2005, 7, 298–301 CrossRef CAS.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- S. Singh, M. Pareek, A. Changotra, S. Banerjee, B. Bhaskararao, P. Balamurugan and R. B. Sunoj, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1339–1345 CrossRef CAS.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS.
- I. O. Betinol, J. Lai, S. Thakur and J. P. Reid, J. Am. Chem. Soc., 2023, 145, 12870–12883 CrossRef CAS.
- F. Sandfort, F. Strieth-Kalthoff, M. Kühnemund, C. Beecks and F. Glorius, Chem, 2020, 6, 1379–1390 CAS.
- N. Tsuji, P. Sidorov, C. Zhu, Y. Nagata, T. Gimadiev, A. Varnek and B. List, Angew. Chem., Int. Ed., 2023, 62, e202218659 CrossRef CAS.
- A. Varnek, D. Fourches, F. Hoonakker and V. P. Solov’ev, J. Comput.-Aided Mol. Des., 2005, 19, 693–703 CrossRef CAS PubMed.
- F. Hoonakker, N. Lachiche, A. Varnek and A. Wagner, Int. J. Artif. Intell., 2011, 20, 253–270 Search PubMed.
- R. I. Nugmanov, R. N. Mukhametgaleev, T. Akhmetshin, T. R. Gimadiev, V. A. Afonina, T. I. Madzhidov and A. Varnek, J. Chem. Inf. Model., 2019, 59, 2516–2521 CrossRef CAS PubMed.
- M. Sanocki, H. C. Russell, J. Handjaya and J. P. Reid, ACS Catal., 2024, 16849–16860 CrossRef CAS.
- H. Y. Bae, D. Höfler, P. S. J. Kaib, P. Kasaplar, C. K. De, A. Döhring, S. Lee, K. Kaupmees, I. Leito and B. List, Nat. Chem., 2018, 10, 888–894 CrossRef CAS.
- L. Schreyer, P. S. J. Kaib, V. N. Wakchaure, C. Obradors, R. Properzi, S. Lee and B. List, Science, 2018, 362, 216–219 CrossRef CAS.
- T. Gatzenmeier, P. S. J. Kaib, J. B. Lingnau, R. Goddard and B. List, Angew. Chem., Int. Ed., 2018, 57, 2464–2468 CrossRef CAS.
- L. Liu, H. Kim, Y. Xie, C. Farès, P. S. J. Kaib, R. Goddard and B. List, J. Am. Chem. Soc., 2017, 139, 13656–13659 CrossRef CAS.
- T. Gatzenmeier, M. Turberg, D. Yepes, Y. Xie, F. Neese, G. Bistoni and B. List, J. Am. Chem. Soc., 2018, 140, 12671–12676 CrossRef CAS.
- H. Kim, G. Gerosa, J. Aronow, P. Kasaplar, J. Ouyang, J. B. Lingnau, P. Guerry, C. Farès and B. List, Nat. Commun., 2019, 10, 770 CrossRef PubMed.
- S. Ghosh, S. Das, C. K. De, D. Yepes, F. Neese, G. Bistoni, M. Leutzsch and B. List, Angew. Chem., Int. Ed., 2020, 59, 12347–12351 CrossRef CAS PubMed.
- S. Ghosh, J. E. Erchinger, R. Maji and B. List, J. Am. Chem. Soc., 2022, 144, 6703–6708 CrossRef CAS PubMed.
- J. Ouyang, J. L. Kennemur, C. K. De, C. Farès and B. List, J. Am. Chem. Soc., 2019, 141, 3414–3418 CrossRef CAS.
- Y. Xie, G.-J. Cheng, S. Lee, P. S. J. Kaib, W. Thiel and B. List, J. Am. Chem. Soc., 2016, 138, 14538–14541 CrossRef CAS.
- P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200–13203 CrossRef CAS.
- O. Pereira, M. Ruth, D. Gerbig, R. C. Wende and P. R. Schreiner, J. Am. Chem. Soc., 2024, 146, 14576–14586 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |