
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereochemical expression of Bi 6s2 lone pairs mediates fluoride-ion (De)insertion in tunnel-structured Bi2PdO4 and Bi1.6Pb0.4PtO4†
George
Agbeworvi
 a,
Anindya
Pakhira
a,
Shruti
Hariyani
a,
Wasif
Zaheer
a,
Alice
Giem
a,
Jaime R.
Ayala
a,
John D.
Ponis
a,
Saul
Perez-Beltran
b,
Cherno
Jaye
c,
Conan
Weiland
c,
Daniel A.
Fischer
c,
Hassan S.
Bazzi
d,
Mohammed
Al-Hashimi
d and
Sarbajit
Banerjee
*aef
a,
Anindya
Pakhira
a,
Shruti
Hariyani
a,
Wasif
Zaheer
a,
Alice
Giem
a,
Jaime R.
Ayala
a,
John D.
Ponis
a,
Saul
Perez-Beltran
b,
Cherno
Jaye
c,
Conan
Weiland
c,
Daniel A.
Fischer
c,
Hassan S.
Bazzi
d,
Mohammed
Al-Hashimi
d and
Sarbajit
Banerjee
*aef
aDepartment of Chemistry and Department of Material Science and Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: banerjee@chem.tamu.edu; Web: https://twitter.com/SarbajitBanerj
bDepartment of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA
cMaterial Measurement Laboratory, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA
dCollege of Science and Engineering, Hamad Bin Khalifa University, P.O. Box: 34110, Doha, Qatar
eLaboratory for Battery Science, Paul Scherrer Institute, Forschungsstrasse 111 CH-5232, Villigen PSI, Switzerland
fLaboratory for Inorganic Chemistry, Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 2, CH-8093 Zürich, Switzerland
First published on 18th February 2025
Abstract
Fluoride-ion batteries are a promising alternative to lithium-ion batteries by dint of the greater crustal abundance of fluorine and the potential to alleviate the need for metal electrodeposition. However, conventional metal fluoride cathodes typically rely on conversion-type reactions that require propagation of a reaction—diffusion front, thereby limiting cycling performance and rate capability. In contrast, the topochemical insertion of fluoride-ions in periodic solids remains a relatively unexplored approach. Here, we explore the mechanisms of fluoridation of Bi2PdO4 and Bi1.6Pb0.4PtO4 insertion hosts that possess capacious tunnels that can accommodate fluoride-ions with a particular emphasis on elucidating the role of stereochemical expression of bismuth 6s2 lone pairs in mediating anion diffusion. We reveal that the topochemical solution-phase insertion and deinsertion of fluoride-ions at room temperature is mediated by redox reactions at platinum and palladium centers but involves multi-center synergies between d- and p-block atoms across the one-dimensional (1D) tunnel structure. While Pt and Pd centers mediate redox reactions, the stereochemically active lone pair electrons of Bi3+ play a pivotal role in facilitating reversible fluoride-ion diffusion. Consequently, Bi1.6Pb0.4PtO4 and Bi2PdO4 can be reversibly fluoridated with full recovery of the crystal lattice and with minimal alteration of the unit cell volume. The results reveal a key principle that the stereochemical activity of p-block electron lone pairs can be harnessed to modulate anion–lattice interactions and mediate facile anion diffusion.
Introduction
Lithium-ion batteries are the current paradigm for electrochemical energy storage across a diversity of formats; however, their widespread adoption in electromobility and grid-scale energy storage is currently stymied by materials criticality and supply chain constraints,1,2 which often give rise to vexatious trade-offs between performance and cost efficiency. Recent efforts have increasingly focused on enhancing technological diversity as a means of alleviating material criticality concerns.3,4 Anion batteries represent an orthogonal construct to Li-ion batteries by utilizing anions, commonly fluoride- and chloride-ions, as charge carriers.5 Such battery concepts rival Li-ion batteries in terms of theoretical energy densities and hold promise for improved safety by entirely alleviating the need for metal electrodeposition.5–7 Much of the current palette of fluoride-ion anodes leverage conversion reactions to store fluoride-ions. This poses inevitable challenges of large volume changes and sluggish progression of the reaction—diffusion front, which are manifested as poor rate performance and cycle life.6,8–11 In contrast, the insertion chemistry of fluoride-ions in periodic solids to form mixed-anion (oxyfluorides) periodic solids remains poorly developed. As such, fully integrated fluoride-ion insertion batteries remain to be realized despite promising steps toward this goal, which is a reflection of fundamental challenges in achieving reversible topochemical fluoride-ion diffusion in redox-active insertion hosts.12–14While topochemical cation insertion is well studied as a proxy for electrochemical coupled electron—ion transport processes,15 anion insertion reactions in periodic inorganic crystals are much less explored.16,17 Beyond elucidation of fundamental mechanisms relevant to energy storage, topochemical anion insertion holds promise for accessing a rich palette of mixed-anion periodic solids,18,19 especially far-from-equilibrium structures with anions precisely positioned in specific interstitial or interlayer sites that can be challenging to access from direct synthesis.20–22 Here, we have adopted a solution-phase topochemical fluoridation approach as a proxy for electrochemical fluoridation using XeF2 as a fluoridating agent, C4H9Li as a defluoridating agent and Bi1.6Pb0.4PtO4 and Bi2PdO4 hosts as model systems to understand mechanisms of charge compensation and the modulation of electronic structure changes during reversible fluoride-ion insertion.23 We apply a broad range of X-ray scattering as well as X-ray absorption and emission spectroscopy tools to our model systems to probe the mechanistic basis of reversible fluoride-ion insertion and diffusion, as well as to unravel key design principles, an approach that would have been rather challenging to implement in fully assembled fluoride-ion batteries.
Similar to Li-ion batteries, fluoride-ion insertion requires hosts to have a high density of available interstitial sites and accessible redox centers capable of accommodating relatively large F-ions (contrast ionic radii of 1.32 Å for fluoride-ions as compared to 0.79 Å for Li-ions)24 with relatively modest structural distortions. However, a crucial distinction is that fluoride-ion insertion reactions must induce oxidation rather than the reduction of the redox active sites upon ion insertion. Recent studies have indicated the synergistic role of redox reactions at d-block transition metal redox sites and electron repulsion at polarizable p-block sites in mediating reversible fluoride-ion diffusion.25,26 Notably, reversible fluoride-ion insertion has been achieved in schafarzikite-type FeSb2O4 at room temperature. Kageyama and co-workers recently demonstrated intriguing low-temperature fluoride-ion insertion in the 1D tunnels of Bi2PdO4.27 We explore here the mechanisms underlying fluoride-ion insertion in Bi2PdO4 and Bi1.6Pb0.4PtO4 compounds with a particular emphasis on elucidating the role of stereochemical expression of bismuth 6s2 lone pairs in mediating anion diffusion.
p-block cations with stereochemically active electron lone pairs manifest lattice anharmonicity and distinctive structural distortions in periodic solids because of the expression of the stereochemical activity of filled 4/5/6s2 electron lone pairs.28–31 In both schafarzikite-type FeSb2O4 and Bi2PdO4/Bi1.6Pb0.4PtO4 structures, the lone-pair repulsions of p-block cations underpin the opening of sizable one-dimensional tunnels, which can be used to accommodate fluoride-ions. As per the revised lone pair model, stereochemical expression of 4/5/6s2 lone pairs is predicated on their effective hybridization with anion p states (mediated by cation 4/5/6p states), which gives rise to filled slightly anti-bonding states at the valence band maximum.28 Here, we examine the electronic structure consequences of reversible room-temperature solution-phase (de)insertion of fluoride-ions in tunnel-structured insertion hosts, Bi1.6Pb0.4PtO4 and Bi2PdO4. Using X-ray scattering and spectroscopy probes, we map structural distortions induced by anion insertion to electronic structure modulations. Next, we use energy-variant hard X-ray photoemission spectroscopy (HAXPES) and first-principles density functional theory (DFT) calculations along with crystal orbital Hamiltonian population (COHP) analyses to examine how the stereochemical expression of Bi 6s2 lone pairs and their interactions with inserted fluoride ions mediates the strength of Pd/Pt–F interactions and enables reversible fluoride-ion insertion in Bi1.6Pb0.4PtO4 and Bi2PdO4 periodic solids.
Results and discussion
Structure evolution upon topochemical fluoridation and defluoridation
Bi2PdO4 and Bi1.6Pb0.4PtO4 have been prepared from precursor powders of stoichiometric oxides by ball milling and annealing in a fused silica ampoule as per:32| Bi2O3 (s) + PdO (s) → Bi2PdO4 (s) | (1) |
An attempt to synthesize stoichiometric Bi2PtO4 did not succeed; instead, partial substitution of Bi3+ by Pb2+ is needed to stabilize the Pt variant as a Bi–Pb solid-solution as per:32,33
| 4Bi2O3 (s) + 2PbO (s) + 5 Pt (s) + 3O2 (g) → 5Bi1.6Pb0.4PtO4 (s) | (2) |
Mixed valence platinum is required to stabilize the structure, which can formally be denoted as Bi1.6Pb0.4PtII0.8PtIV0.2O4. Fig. 1 illustrates the structures of Bi1.6Pb0.4PtO4 and Bi2PdO4 as inferred from Rietveld refinements (Fig. S2C and D†) to powder X-ray diffraction (XRD) patterns shown in Fig. 2A, S2A and S2B† respectively. Bi1.6Pb0.4PtO4 and Bi2PdO4 crystallize in the tetragonal space group, P4/ncc; Tables S1 and S2† list the unit cell parameters and cell volume. The compounds are isostructural with Bi2CuO4 and comprise edge-sharing Pt/PdO4 square-planar units bonded to corner-sharing BiO6 octahedra that are asymmetrically distorted as a result of Bi3+ 6s2 stereoactive electron lone pairs. Electron lone-pair repulsions between Bi sites open sizable 1D tunnels along the [001] and [110] directions of the crystal structure (Fig. 1A). These capacious tunnels (see also structure representations in Fig. S1†) are sufficiently large to serve as diffusion channels for F-ions between interstitial sites bounded by Bi centers.12,25,26
 | ||
| Fig. 1 Reversible topochemical fluoridation of Bi 1.6 Pb 0.4 PtO 4 and Bi 2 PdO 4 . (A) Reaction scheme for topochemical fluoride-ion insertion in Bi1.6Pb0.4PtO4 and Bi2PdO4. (B) Expanded view of fluoridated Bi1.6Pb0.4O4 and Bi2PdO4 along the c-axis indicating F-ion positions along the 1D tunnel defined by Bi lone-pair repulsions; and (C) a view down the crystallographic axis, showing chains of edge-sharing square planar PtO4/PdO4 units sharing two oxygens with BiO6 octahedra that are asymmetrically distorted owing to the stereochemically expression of Bi3+ 6s2 electrons. The PtO4/PdO4 and BiO6 structural motifs separate the redox site from the F-ion binding site. Bi adopts a capped trigonal prismatic electron geometry with one vertex occupied by an electron lone pair (see Fig. S1†). Two long Bi–O (2.803 Å) bonds are weakly coordinated, and as such, have been removed to highlight the 1D tunnels with interstitial sites that accommodate F-ions. | ||
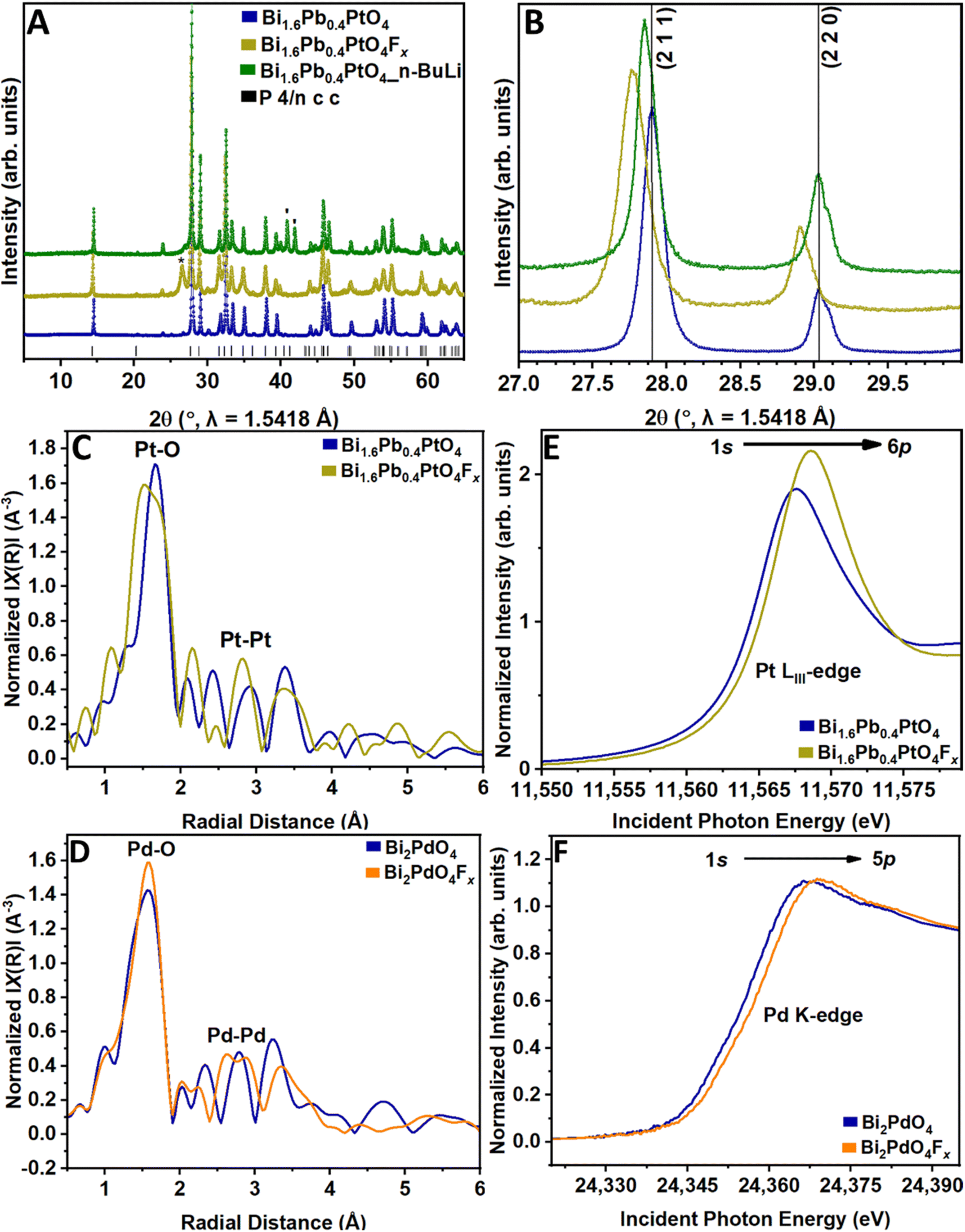 | ||
| Fig. 2 Structure evolution of Bi 1.6 Pb 0.4 PtO 4 and Bi 2 PdO 4 upon topochemical fluoride-ion insertion. (A) Comparison of powder XRD patterns collected for Bi1.6Pb0.4PtO4, Bi1.6Pb0.4PtO4Fx obtained by treatment with XeF2 in acetonitrile, and Bi1.6Pb0.4PtO4 recovered after treatment with n-butyllithium. The reflections observed around 2θ = 26° (denoted with *) for Bi1.6Pb0.4PtO4Fx were similar to that observed by Kageyama et al. and can be ascribed to interphasic products (e.g. PtF2, PtF4, BiF3, BiOF).14,27 The reflection at 2θ = 43° (denoted with ′) for Bi1.6Pb0.4PtO4_n-BuLi is further likely an interphasic lithiated species.26,34 (B) Expanded view showing evolution of the (211) and (220) reflections across the F-ion insertion and deinsertion reactions. Normalized k3-weighted Fourier-transformed R-space data extracted from (C) Pt LIII-edge EXAFS spectra for Bi1.6Pb0.4PtO4 and Bi1.6Pb0.4PtO4Fx; and (D) Pd K-edge EXAFS spectra for Bi2PdO4, and Bi2PdO4Fx. Comparison of normalized (E) Pt LIII-edge XANES spectra for Bi1.6Pb0.4PtO4 and Bi1.6Pb0.4PtO4Fx; and (F) Pd K-edge XANES spectra for Bi2PdO4 and Bi2PdO4Fx. | ||
Topochemical fluoride-ion insertion has been performed by reacting the as-prepared materials with XeF2 in acetonitrile at room temperature, as also described by Kageyama, as per:27
| 2Bi2PdO4 (s) + xXeF2 (acetonitrile) → 2Bi2PdO4Fx (s) + xXe (g) | (3) |
| 2Bi1.6Pb0.4PtO4 (s) + xXeF2 (acetonitrile) → 2Bi1.6Pb0.4PtO4Fx (s) + xXe (g) | (4) |
Topochemical fluoridation serves here as a proxy for electrochemical fluoridation, which has recently been achieved for CsMnFeF6 and ReO3 using tetra-n-butylammonium fluoride (TBAF) liquid electrolyte dissolved in THF at room temperature.11,35 In this study, by examining the mechanisms of reversible topochemical insertion of F-ions into the quasi-1D tunnels of Bi1.6Pb0.4PtO4 and Bi2PdO4, we have attempted to identify the interplay between atomistic and electronic structure required to mediate fluoride-ion insertion with a view towards designing generalizable design principles. Solution-phase topochemical fluoridation provides a means to explore the mechanistic basis for F-ion insertion and diffusion within periodic inorganic solids without having to identify a suitable electrolyte compatible with the positive and negative electrodes or assembling a complete electrochemical cell. Defluoridation has been performed by reaction with n-butyllithium as per25,26 and the nominally defluoridated products are denoted with the modifier (Bi2PdO4_n-BuLi and Bi1.6Pb0.4PtO4_n-BuLi):
| 2Bi2PdO4Fx (s) + 2xC4H9Li (hexane) → 2Bi2PdO4 (s) + 2xLiF (s) + xC8H18 (l) | (5) |
| 2Bi1.6Pb0.4PtO4Fx (s) + 2xC4H9Li (hexane) → 2Bi1.6Pb0.4PtO4 (s) + 2xLiF (s) + xC8H18 (l) | (6) |
Rietveld refinement of powder XRD data shown in Fig. 2A and S3A† for fluoridated samples infers a nominal stoichiometry (x) of 0.5 ± 0.02 in Bi1.6Pb0.4PtO4Fx and Bi2PdO4Fx, with notable disorder observed in F-ion sites arrayed along the 1D tunnel (Tables S3 and S4†). The tetragonal (P4/ncc) structure is preserved, albeit with some modification. The (211), (220), (411), and (420) reflections of Bi1.6Pb0.4PtO4 are shifted to lower 2θ values upon F-ion insertion (Fig. 2B and S3BD† shows comparable data for Bi2PdO4 before and after fluoridation), indicating an anisotropic expansion of the 1D tunnel. This expansion brings about an outwards flexion of PtO4 and PdO4 units concomitantly with the inward buckling of BiO6 octahedra, as depicted in Fig. 1. Rietveld refinements to powder XRD data for fluoridated samples are shown in Fig. S4; Tables S3 and S4† show that the a and b lattice parameters are increased upon fluoridation, whereas the c parameter is decreased. Beyond the reflections corresponding to topochemical fluoridation, some additional reflections are also observed corresponding to interphasic products. Analogous to topochemical cation reactions, the formation of corrosion products competes with insertion reactions at high local chemical potentials.14,36
The unit cell volume of Bi1.6Pb0.4PtO4, decreases from 435.01(3) Å3 to 434.84(5) Å3, a change of 0.17 Å3 (0.04%), whereas in Bi2PdO4, the volume decreases from 439.12(2) Å3 to 439.082(19) Å3, a change of 0.038 Å3 (0.009%). Overall, fluoride-ion insertion induces a modest (<1%) lattice contraction, which is characteristic of oxidative insertion.25,26 These minimal volume changes upon fluoride-ion insertion reflect the structural resilience afforded by the stereochemical expression and polarizability of 6s2 lone pairs on Bi and Pb centers,37 which introduce localized lattice distortions that accommodate fluoride ions ameliorating the need for large structural distortions or phase transformations. In this regard, while Bi and Pb both express stereochemical activity proportionate to the strength of 6s–2p hybridization with O 2p states,38 as quantified by ΔEs–p.28 Since Pb 6s atomic states (−11.9 eV) closer to O 2p states as compared to Bi 6s (−14.4 eV), the introduction of Pb induces a greater lone pair distortion.39 An empirical estimate of the extent of lone pair distortion can also be derived from the fractional contribution of O 2p to lowest metal ns valence band states (vide infra).39 As such, Pb–F interactions are stronger than Bi–F interactions. The stronger Pb–F hybridization induces relatively greater lattice distortions and contributes to the larger volume contraction observed in Bi1.6Pb0.4PtO4 as compared to Bi2PdO4.
Corroboration of F-ion reactivity with Bi1.6Pb0.4PtO4 and Bi2PdO4 is derived from energy dispersive spectroscopy (EDS) mapping (Fig. S5 and S6†), which shows a homogeneous spatial distribution of F-atoms co-localized with Bi and Pt/Pd in the fluoridated materials. Upon reaction with n-butyllithium as per eqn (5) and (6), Fig. 2A, B and S3A–D† illustrate restoration of the starting crystal structure of Bi1.6Pb0.4PtO4 and Bi2PdO4, respectively (see also Tables S6 and S7†). These results demonstrate the facile reversibility of the solution-phase F-ion insertion reaction.
Extended X-ray absorption fine structure (EXAFS) spectroscopy data collected at Pt L3-and Pd K-edges for Bi1.6Pb0.4PtO4 and Bi2PdO4, respectively, have further been used to obtain a local structure perspective of structural modifications induced by fluoridation. The EXAFS data has been fitted starting from initial structural models derived from Rietveld refinements of the powder XRD patterns. Fourier-transformed R-space EXAFS data for Bi1.6Pb0.4PtO4 and Bi2PdO4 with and without fluoridation are presented in Fig. 2C and D, respectively. Two prominent features at Pt L3-and Pd K-edges are assigned to Pt–O/Pd–O (ca. 2.0 Å) and Pt–Pt/Pd–Pd (ca. 3.0 Å) scattering paths. The fitting results in Fig. S7† indicate that upon fluoridation, the Pt–O bond correlations are reduced from 2.000 Å to 1.986 Å; Pd–O bond distances are similarly decreased from 2.010 Å to 1.991 Å. The reduced bond lengths are consistent with oxidation of the platinum and palladium centers. Models of PtO4 and PdO4 square-planar coordination environments shown in Fig. S7E and F† have been constructed based on the Pt–O and Pd–O distances derived from EXAFS fits in Fig. S7 and Table S5.† Further evidence for topochemical insertion of F-ions derives from F K-edge XANES spectra of Bi1.6Pb0.4PtO4Fx and Bi2PdO4Fx shown in Fig. S8A and B,† respectively, which are overlapped with Bi NIII-edge (Bi 4d3/2 → Bi 6p) resonances. The local structure results thus show that fluoride-ion insertion in Bi1.6Pb0.4PtO4 and Bi2PdO4 causes reversible, anisotropic structural changes, expanding the 1D tunnels and contracting the a and b lattices as a result of shortened Pt–O and Pd–O bonds. Topochemical anion insertion oxidizes Pt and Pd centers whilst maintaining tetragonal symmetry, which is fully reversible upon defluoridation.
Electronic structure implications of topochemical fluoridation and defluoridation
X-ray absorption and emission spectroscopies have been employed to investigate the electronic structure changes in Bi1.6Pb0.4PtO4 and Bi2PdO4 upon F-ion insertion. Pt L3 (Pt 1s → Pt 6p) and Pd K-edge (Pd 1s → Pd 5p) XANES spectra in Fig. 2E and F, respectively, reveal a shift in the primary white-line absorption features to higher energies by ca. 2 eV upon fluoride-ion insertion, which is indicative of the oxidation of nominally divalent Pt and Pd centers.40 This observation further squares with the observed shrinkage of Pd–O and Pt–O bonds in the EXAFS measurements. Soft X-ray Pt NIII-edge (Pt 4d3/2 → Pt 6p), Pd MIII-edge (3d3/2 → Pd 5p), and O K-edge (1s → 2p) XANES spectra for Bi1.6Pb0.4PtO4/Bi1.6Pb0.4PtO4Fx and Bi2PdO4/Bi2PdO4Fx are shown in Fig. S8C and D.†41–43 Consistent with the redox activity of the transition-metal centers, Pt and Pd NIII/MIII-edge features are shifted to higher energy upon topochemical F-ion insertion.Further corroboration of transition-metal redox accompanying topochemical fluoride-ion insertion in these systems is derived from core-level hard X-ray photoemission spectroscopy (HAXPES) measurements (Fig. 3 and S9†). The high incident photon energy of HAXPES (as compared to surface-sensitive laboratory X-ray photoemission spectroscopy) enhances the kinetic energy of the emitted electron, and consequently, its inelastic mean free path,14 enabling interrogation of bulk (and not just surface) electronic structure.44,45Fig. 3A and B plot high-resolution core-level Pt 4f and Pd 3d spectra, respectively. Both features are shifted to higher binding energy upon fluoride-ion insertion but restored upon removal of fluoride-ions by treatment with n-BuLi. These results attest to the oxidation of the transition metal centers upon fluoridation and reduction upon fluoride-ion de-insertion. In contrast, high-resolution Bi 4d5/2 core-level HAXPES spectra in Fig. 3C and D show features centered at ca. 440.2 eV corresponding to trivalent Bi centers in Bi1.6Pb0.4PtO4 and Bi2PdO4, which remain essentially unmodified by fluoride-ion insertion and de-insertion. Fig. S9A and B† show core-level F 1s HAXPES spectra obtained for Bi1.6Pb0.4PtO4Fx and Bi2PdO4Fx, further corroborating bulk fluoridation, with the fluorine position for both compounds at 685 eV. This binding energy is typical of ionic fluoride species in metal oxide matrices.46 These results attest to the transition metal centers, Pt and Pd, mediating redox reactivity upon fluoride-ion insertion, whereas the p-block cation, Bi, remains nominally redox innocent.
 | ||
| Fig. 3 HAXPES and magnetic measurements to probe evolution of electronic structure upon topochemical fluoridation. High-resolution core-level HAXPES plots at 2 keV incident energy for (A) Pt 4f, (B) Pd 3d, and Bi 4d core excitations collected for (C) Bi1.6Pb0.4PtO4, Bi1.6Pb0.4PtO4Fx, and Bi1.6Pb0.4PtO4 recovered after treatment with n-butyllithium and (D) Bi2PdO4, Bi2PdO4Fx, and Bi2PdO4 recovered after treatment with n-butyllithium. FC magnetic susceptibility as a function of temperature for (E) Bi1.6Pb0.4PtO4/Bi1.6Pb0.4PtO4Fx and (F) Bi2PdO4/Bi2PdO4Fx under an applied field of 0.1T. | ||
Further corroboration of redox insertion is derived from magnetic susceptibility measurements. Fig. 3E and F show the temperature-dependent magnetic susceptibilities of Bi1.6Pb0.4PtO4/Bi1.6Pb0.4PtO4Fx and Bi2PdO4/Bi2PdO4Fx, during field cooling (FC), and reveal that F-ion insertion and subsequent Pt2+ and Pd2+ oxidation bring about a noteworthy alteration in magnetic behavior. Both Bi1.6Pb0.4PtO4 and Bi2PdO4 exhibit diamagnetic behavior, which is in stark contrast to the paramagnetic behavior of Bi1.6Pb0.4PtO4Fx and Bi2PdO4Fx. This pronounced alteration of magnetic structure upon F-ion insertion is consistent with the change from d8 (M2+) to nominally d7 (M3+) electronic structure. The extracted effective magnetic moment on the basis of the CW fit in the temperature range between 100 K and 350 K is increased from μeff = 0 of (Bi1.6Pb0.4PtO4 and Bi2PdO4) to 1.30 μB (Bi1.6Pb0.4PtO4Fx) and 1.25 μB (Bi2PdO4Fx), which is consistent with a change in magnetic spins from S = 0 to almost S = ½ upon F-ion insertion.
Stereochemical expression of Bi 6s2 lone pairs
Energy-variant valence band (VB) HAXPES measurements have been performed on Bi1.6Pb0.4PtO4 and Bi2PdO4 before and after reaction with XeF2 to examine the role of electronic structure modulations in mediating fluoride-ion insertion and to elucidate fundamental design principles associated with specific anion–cation interactions. Photoionization cross-sections in photoemission spectroscopy are generally suppressed as a function of incident photon energy; however, the steepness of the decrease is greater for subshells with higher orbital angular momentum. As such, HAXPES spectra obtained at higher incident photon energies more prominently manifest orbital contributions from subshells with lower angular momentum values (e.g., s and p orbitals). In the context of periodic solids, energy-variant HAXPES serves as an excellent probe to selectively spotlight filled 4/5/6s2-derived states of p-block cations at the valence band edge.28,38,44,47Fig. 4 and S10† display VB HAXPES spectra for Bi1.6Pb0.4PtO4 and Bi2PdO4 acquired at two distinct incident photon energies, 2.0 keV and 5.0 keV, before and after F-ion insertion. HAXPES features have been assigned based on COHP calculations shown in Fig. 4E, F, S10C and D† and DFT-calculated atom-projected density of states plotted in Fig. S11.† The VB of Bi1.6Pb0.4PtO4 and Bi2PdO4 comprises O 2p states hybridized with Pt 5d, Pd 4d, and Bi 6s; additional contributions from F 2p states appear after F-ion insertion. Notably, the relative intensity of the features at ca. 14 eV for Bi1.6Pb0.4PtO4 and ca. 12 eV for Bi2PdO4 increases with higher incident photon energy (Fig. 4A and B), suggesting pronounced 6s2 contributions derived from bonding and antibonding Bi–O interactions.25,28,38 As per the revised lone pair model,28 Bi 6s–O 2p bonding states occur deep in the valence band (at ca. 14 eV and ca. 12 eV as shown in Fig. 4F and S10D†), whereas Bi 6s, 6p–O 2p antibonding states are positioned at the top of the VB (at ca. 4.0 eV and 3.9 eV), just below Pt 5d–O 2p and Pd 4d–O 2p AB states, respectively. The latter anti-bonding states manifest the stereochemical expression of Bi 6s2 lone pairs.28,38
 | ||
| Fig. 4 Evolution of the electronic structure of Bi 1.6 Pb 0.4 PtO 4 and Bi 2 PdO 4 upon F-ion insertion, examined using valence-band HAXPES and COHP analysis. Overlay of valence-band HAXPES spectra collected at incident photon energies of 2.0 keV and 5.0 keV for (A) Bi1.6Pb0.4PtO4 (Pb 6s2 states also contribute to bonding (B) and anti-bonding (AB) interactions) and (B) Bi2PdO4. Differentials in spectral weight as a function of incident photon energy are shaded in light blue. High-resolution HAXPES data collected for Bi1.6Pb0.4PtO4 and Bi1.6Pb0.4PtO4Fx at incident photon energy of (C) 2 keV and (D) 5 keV. All valence-band HAXPES spectra have been normalized to internal core level Bi 5d5/2 peak at a binding energy of 24 eV near the valence band. COHP analysis of (E) Pt–O and (F) Bi–O interactions. | ||
Fig. S10A and B† contrast analogous spectra acquired at 2 and 5 keV for Bi2PdO4 and Bi2PdO4Fx. At an incident photon energy of 2 keV, where considerable spectral weight derives from transition metal d-states, VB HAXPES spectra show a notable decrease in the intensity of the states at the VB maximum upon fluoride-ion insertion. The loss of intensity at the valence band maximum corresponds to emptying of filled Pd/Pt 4d/5d states at the valence band edge upon oxidative F-ion insertion. Bonding Pt 5d–O 2p and Pd 4d–O 2p are concomitantly shifted to higher binding energies, which further corroborates oxidation of Pt and Pd centers, respectively. COHP analyses of Pt–O and Pd–O interactions in Fig. 4E and S10C† illustrate that fluoride-ion insertion significantly reduces Pt 5d–O 2p and Pd 4d–O 2p anti-bonding interactions at the valence band maximum, while introducing new anti-bonding interactions at the bottom of the conduction band in the case of Pt 5d–O 2p.
Conversely, VB HAXPES spectra acquired at an incident photon energy of 5 keV, which feature enhanced contributions from Bi 6s2 states show the opposite trend upon fluoride-ion insertion.28,47,48 The occupied electronic states at the valence band edge are higher for the F-ion-inserted compounds as compared to the pristine Bi1.6Pb0.4PtO4 and Bi2PdO4 insertion hosts (Fig. 4F and S10D†). With the loss of anti-bonding Pt/Pd–O 2p states, the lone-pair-derived anti-bonding states constitute the valence band edge. Furthermore, new Bi–F-derived bonding and anti-bonding interactions appear with the latter contributing considerable spectral weight at the valence band maximum. Fig. S12† shows that Bi–F anti-bonding interactions emerge at higher binding energies as compared to Bi–O interactions.
In contrast to La-ions in LaSrMnO4 and La2CoO4, which primarily play a structural role, stereochemically active Bi3+ centers play an important role in facilitating F-ion (de)insertion. Energy-variant HAXPES spectra in Fig. 4 provide insights into the modification of hybrid Bi 6s, Bi 6p–O 2p states upon fluoride-ion insertion. Upon fluoride-ion insertion, the Bi 6s2 lone pairs are further hybridized with F 2p states near the VBM in the mixed anion compound as evident in Fig. 4D and S10B† (which have been assigned with the help of COHP analyses in Fig. S12A and S12B†). F 2p states are nominally lower in energy as compared to O 2p states because of the higher electronegativity of fluorine as compared to oxygen. As such, given the lower ΔEs–p values, these states are well positioned to hybridize with filled Bi 6s2 states through dative covalent interactions.49 The electron lone pairs on fluoride-ions back-donate electron density to the Bi centers.50 COHP analysis of Bi–O interactions in Bi1.6Pb0.4PtO4Fx (Fig. 4F and S12A†)and Bi2PdO4Fx (Fig. S10D and 12SB†), reveals these newly occupied Bi 6s, 6p–O 2p lone pair states at the VBM, alongside the Bi 6s, 6p–F 2p lone pair states. Considering that Bi 6s, 6p–O 2p lone pair states have a strong antibonding character at the VBM, addition of electron density upon fluoride-ion insertion results in the decrease of the Bi–O bond order, which is evident in the elongation of Bi–O bonds (Tables S8 and S9†). Notably, the Pb 6s2 lone pairs exhibit stronger lone pair distortions as compared to Bi 6s2 for both oxide and fluoride ion interactions, as can be quantified by the fractional contribution of O 2p states to the lowest valence band metal ns state.28,38,39 Comparing PbO and Bi2O3, Walsh and co-workers have determined values of ca. 0.4 and 0.2, respectively, attesting to the much stronger anion hybridization of Pb 6s2 states. This differential contributes to the larger lattice contraction observed for Bi1.6Pb0.4PtO4Fx as compared to Bi2PdO4Fx.
To further examine the changes in electronic and atomistic structure resulting from F-ion insertion, we have calculated charge density differences (CDD) and electron localization function (ELF) plots for Bi1.6Pb0.4PtO4 and Bi2PdO4 before and after fluoride-ion insertion. Fig. 5A, B, S13A and B† illustrate the ELFs for Bi1.6Pb0.4PtO4 and Bi2PdO4 with and without inserted fluoride-ions. The stereochemical expression of Bi 6s2 lone pairs in Bi1.6Pb0.4PtO4 and Bi2PdO4 define the 1D open tunnels (Fig. 5A and S13A†). After insertion of a fluoride-ion, repulsions between Bi–O and Bi–F bonding pairs induce significant polarization of Bi 6s2 electron density in the vicinity of the fluoride-ion (Fig. 5B and S13B†). Additionally, Fig. 5C, D, S13C and D† present a 2D cross-section of the 3D ELF map shown in Fig. 5A, B, S13A and B† highlighting the local changes in Bi 6s2 electron density upon fluoride-ion bonding.
 | ||
| Fig. 5 Charge distribution and molecular orbital perspective of topochemical fluoride-ion insertion. Electron localization function (ELF) map of (A) Bi1.6Pb0.4PtO4, and (B) Bi1.6Pb0.4PtO4Fx. A 2-D cross-section of the 3-D ELF map shown in (C) A and (D) B. (E) Charge density difference (CDD) of Bi1.6Pb0.4PtO4 with a fluoride ion inserted. (F) Charge redistribution around the oxidized Pt atoms. (G) Molecular orbital sketch showing the relative position of Bi 6s, 6p–O 2p, and Bi 6s, 6p–F 2p lone pair states relative to the Pt 5d–O 2p AB states. | ||
CDD induced as a result of F-ion insertion have been plotted in Fig. 5E and F, S13E and F† as per
| Δρ(r) = ρBi2MO4F0.25 (r) − ρBi2MO4(r) − ρF(r) | (7) |
Fig. 5G sketches a simplified molecular orbital diagram to summarize the electronic structure modulation of Bi1.6Pb0.4PtO4 and Bi2PdO4 upon fluoride-ion insertion based on electronic structure calculations in conjunction with HAXPES, XANES, and COHP analysis. Anti-bonding Pt 5d–O 2p and Pd 4d–O 2p states are the highest occupied states in Bi1.6Pb0.4PtO4 and Bi2PdO4 respectively, as also observed in the DOS plots in Fig. S11A and S11C.† These states are positioned in close proximity of but higher in energy as compared to Bi 6s2-derived lone-pair states. Fluoride-ion insertion results in oxidation of Pt and Pd, which brings about loss of electron density from the Pt 5d–O 2p and Pd 4d–O 2p AB states at the valence band edge. Oxidation of the transition metal centers is observed as contraction of Pt–O and Pd–O bonds in powder XRD and EXAFS measurements. Filled Bi–F AB states emerge at the VBM as F-ions form dative covalent bonds with Bi atoms lining the edges of the 1D tunnel. The BiO6LP polyhedra is transformed to a distorted BiO6FLP polyhedra, with considerable polarization of the Bi 6s2 lone pair density.23,51 In essence, the polarization of Bi lone pairs stabilizes the inserted F-ions within the 1D tunnel of Bi1.6Pb0.4PtO4 and Bi2PdO4 and mediates the redox reaction at the Pt and Pd centers (Fig. 4, S10 and S12†). The repulsions between Bi 6s2 and F 2p lone pairs yield a relatively weak Bi–F bond that can be formed and broken at room temperature. As such, the stereochemical expression of the Bi 6s2 lone pair is pivotal to enabling bulk diffusion and homogenous F-ion insertion as observed in the EDS data.
Conclusions
In summary, we evidence reversible room-temperature topochemical insertion and bulk diffusion of fluoride-ions in tetragonal Bi1.6Pb0.4PtO4 and Bi2PdO4. Solution-phase fluoridation using XeF2 yields a nominal fluoride-ion stoichiometry of x ≈ 0.5 in Bi1.6Pb0.4PtO4Fx and Bi2PdO4Fx; fluoride-ions can be extracted by reaction with n-butyllithium at room-temperature. Powder XRD measurements reveal that the inserted F-ions reside in interstitial sites within one-dimensional tunnels defined by electrostatic repulsions between stereochemically active 6s2 electron lone pairs of four bismuth centers. Fluoride-ion insertion is mediated by oxidation of Pt and Pd centers and brings about a modest contraction of the unit cell volume (<1% change), a significant improvement over conventional conversion electrodes, suggesting potential viability as an insertion electrode for fluoride-ion batteries.Based on X-ray scattering and spectroscopy characterization of modulations of local atomistic and electronic structure upon fluoride-ion insertion, several fundamental design principles can be delineated. While Pt and Pd centers mediate redox reactions, the stereochemically active lone pair electrons of Bi3+ play a critical role in facilitating reversible fluoride-ion diffusion. Although Bi centers are not formally oxidized, Bi 6s2 (and Pb 6s2) electron lone pairs interact with F 2p states, resulting in the emergence of new states in the valence band. The inserted fluoride-ions polarize the Bi 6s2 lone pairs and donate electron density through covalent dative interactions, thereby weakening Bi–O bonds. The repulsions between Bi 6s2 and F 2p lone pairs in BiO6FLP polyhedra gives rise to a relatively weak Bi–F bond, which can be formed and broken at room temperature resulting in facile F-ion diffusion along 1D tunnels. Decoupling the transition metal (Pt, Pd) redox-active center from the fluoride-ion coordination site (Bi) enables the uniform distribution of the structural distortions between the Pt/PdO4 and BiO4 structural units within the lattice, which is critical to the small volume change and topochemical nature of anion insertion. As such, insertion hosts combining transition metal redox centers with p-block cations expressing stereochemical activity represent a promising design motif for fluoride-ion insertion batteries. Future work will explore electrochemical fluoridation in full cell configurations and implement operando cells for X-ray spectroscopy and scattering investigations under electrochemical fluoridation from solid or liquid electrolytes.
Data availability
All experimental procedures, computational sections, and associated data are included in the article and ESI.†Author contributions
S. B. directed the research. G. A. and S. B. conceptualized and designed the experiments. G. A. conducted and analyzed the data from powder X-ray diffraction. G. A. performed Rietveld analysis of the powder X-ray diffraction data. A. P., W. Z., and S. P. performed and analyzed the DFT calculations. G. A., S. H., A. G., and, J. D. P. conducted and analyzed the data from HAXPES, EXAFS, and XANES. C. W., C. J., and D. A. F. helped with setting up HAXPES and XANES experiments and data collection. S. G. A. conducted the magnetic measurements and analyzed the data. J. R. A. conducted and analyzed the data from EDS. G. A. and S. B. contributed to writing the paper. H. S. B., M. A., G. A., and S. B. revised the paper. All authors discussed the results and contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge support from Welch Foundation under award A-1978-20190330. Use of the Texas A&M Microscopy and Imaging Center is acknowledged. Part of the research described in this paper was performed at National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Basic Energy Science. We acknowledge support from the Qatar Research, Development and Innovation Council, Qatar National Research Fund under ARG01-0522-230270.References
- P. Xu, D. H. S. Tan and Z. Chen, Emerging Trends in Sustainable Battery Chemistries, Trends Chem., 2021, 3(8), 620–630, DOI:10.1016/j.trechm.2021.04.007.
- A. Zeng, W. Chen, K. D. Rasmussen, X. Zhu, M. Lundhaug, D. B. Müller, J. Tan, J. K. Keiding, L. Liu, T. Dai, A. Wang and G. Liu, Battery Technology and Recycling Alone Will Not Save the Electric Mobility Transition from Future Cobalt Shortages, Nat. Commun., 2022, 13, 1341, DOI:10.1038/s41467-022-29022-z.
- J. L. Andrews and S. Banerjee, It's Not Over until the Big Ion Dances: Potassium Gets Its Groove On, Joule, 2018, 2(11), 2194–2197, DOI:10.1016/j.joule.2018.11.003.
- J. W. Choi and D. Aurbach, Promise and Reality of Post-Lithium-Ion Batteries with High Energy Densities, Nat. Rev. Mater., 2016, 1(4), 16013, DOI:10.1038/natrevmats.2016.13.
- A. W. Xiao, G. Galatolo and M. Pasta, The Case for Fluoride-Ion Batteries, Joule, 2021, 5(11), 2823–2844, DOI:10.1016/j.joule.2021.09.016.
- M. A. Nowroozi, I. Mohammad, P. Molaiyan, K. Wissel, A. R. Munnangi and O. Clemens, Fluoride Ion Batteries - Past, Present, and Future, J. Mater. Chem. A, 2021, 9(10), 5980–6012, 10.1039/d0ta11656d.
- C. Rongeat, M. Anji Reddy, T. Diemant, R. J. Behm and M. Fichtner, Development of New Anode Composite Materials for Fluoride Ion Batteries, J. Mater. Chem. A, 2014, 2(48), 20861–20872, 10.1039/c4ta02840f.
- D. T. Thieu, M. H. Fawey, H. Bhatia, T. Diemant, V. S. K. Chakravadhanula, R. J. Behm, C. Kübel and M. Fichtner, CuF2 as Reversible Cathode for Fluoride Ion Batteries, Adv. Funct. Mater., 2017, 27(31), 1–11, DOI:10.1002/adfm.201701051.
- T. Yoshinari, D. Zhang, K. Yamamoto, Y. Kitaguchi, A. Ochi, K. Nakanishi, H. Miki, S. Nakanishi, H. Iba, T. Uchiyama, T. Watanabe, T. Matsunaga, K. Amezawa and Y. Uchimoto, Kinetic Analysis and Alloy Designs for Metal/Metal Fluorides toward High Rate Capability for All-Solid-State Fluoride-Ion Batteries, J. Mater. Chem. A, 2021, 9(11), 7018–7024, 10.1039/d0ta12055c.
- S. T. Hartman and R. Mishra, Layered Electrides as Fluoride Intercalation Anodes, J. Mater. Chem. A, 2020, 8(46), 24469–24476, 10.1039/d0ta06162j.
- J. L. Andrews, E. T. McClure, K. K. Jew, M. B. Preefer, A. Irshad, M. J. Lertola, D. D. Robertson, C. Z. Salamat, M. J. Brady, L. F. J. Piper, S. H. Tolbert, J. Nelson Weker, B. F. Chmelka, B. S. Dunn, S. R. Narayan, W. C. West and B. C. Melot, Room-Temperature Electrochemical Fluoride (De)Insertion into CsMnFeF6, ACS Energy Lett., 2022, 7(7), 2340–2348, DOI:10.1021/acsenergylett.2c01324.
- B. P. De Laune, G. J. Rees, J. F. Marco, H. Y. Hah, C. E. Johnson, J. A. Johnson, F. J. Berry, J. V. Hanna and C. Greaves, Topotactic Fluorine Insertion into the Channels of FeSb2O4-Related Materials, Inorg. Chem., 2017, 56(16), 10078–10089, DOI:10.1021/acs.inorgchem.7b01613.
- M. A. Nowroozi, K. Wissel, J. Rohrer, A. R. Munnangi and O. Clemens, LaSrMnO4: Reversible Electrochemical Intercalation of Fluoride Ions in the Context of Fluoride Ion Batteries, Chem. Mater., 2017, 29(8), 3441–3453, DOI:10.1021/acs.chemmater.6b05075.
- A. R. Giem, J. R. Ayala, C. Jingxiang, C. Weiland, C. Jaye, D. Fischer and S. Banerjee, Interphase Formation Versus Fluoride-Ion Insertion in Tunnel-Structured Transition Metal Antimonites, Chem. Commun., 2024, 60(98), 14589–14592, 10.1039/D4CC04331F.
- A. Sood, A. D. Poletayev, D. A. Cogswell, P. M. Csernica, J. T. Mefford, D. Fraggedakis, M. F. Toney, A. M. Lindenberg, M. Z. Bazant and W. C. Chueh, Electrochemical Ion Insertion from the Atomic to the Device Scale, Nat. Rev. Mater., 2021, 6(9), 847–867, DOI:10.1038/s41578-021-00314-y.
- M. L. Aubrey and J. R. Long, A Dual-Ion Battery Cathode via Oxidative Insertion of Anions in a Metal-Organic Framework, J. Am. Chem. Soc., 2015, 137(42), 13594–13602, DOI:10.1021/jacs.5b08022.
- N. Shpigel, A. Chakraborty, F. Malchik, G. Bergman, A. Nimkar, B. Gavriel, M. Turgeman, C. N. Hong, M. R. Lukatskaya, M. D. Levi, Y. Gogotsi, D. T. Major and D. Aurbach, Can Anions Be Inserted into MXene?, J. Am. Chem. Soc., 2021, 143(32), 12552–12559, DOI:10.1021/jacs.1c03840.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Expanding Frontiers in Materials Chemistry and Physics with Multiple Anions, Nat. Commun., 2018, 9, 1–15, DOI:10.1038/s41467-018-02838-4.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Heteroanionic Materials by Design: Progress Toward Targeted Properties, Adv. Mater., 2019, 31(19), 1–26, DOI:10.1002/adma.201805295.
- M. Udayakantha, P. Schofield, G. R. Waetzig and S. Banerjee, A Full Palette: Crystal Chemistry, Polymorphism, Synthetic Strategies, and Functional Applications of Lanthanide Oxyhalides, J. Solid State Chem., 2019, 270, 569–592, DOI:10.1016/j.jssc.2018.12.017.
- M. Udayakantha, J. V. Handy, R. D. Davidson, J. Kaur, G. Villalpando, L. Zuin, S. Chakraborty and S. Banerjee, Halide Replacement with Complete Preservation of Crystal Lattice in Mixed-Anion Lanthanide Oxyhalides, Angew. Chem. Int. Ed., 2021, 60(28), 15582–15589, DOI:10.1002/anie.202104231.
- C. H. Wood and R. E. Schaak, Topochemical Anionic Subunit Insertion Reaction for Constructing Nanoparticles of Layered Oxychalcogenide Intergrowth Structures, J. Am. Chem. Soc., 2023, 145(34), 18711–18715, DOI:10.1021/jacs.3c05200.
- J. He, S. Hao, Y. Xia, S. S. Naghavi, V. Ozoliņš and C. Wolverton, Bi2PdO4: A Promising Thermoelectric Oxide with High Power Factor and Low Lattice Thermal Conductivity, Chem. Mater., 2017, 29(6), 2529–2534, DOI:10.1021/acs.chemmater.6b04230.
- P. F. Lang and B. C. Smith, Ionic Radii for Group 1 and Group 2 Halide, Hydride, Fluoride, Oxide, Sulfide, Selenide and Telluride Crystals, Dalton Trans., 2010, 39(33), 7786–7791, 10.1039/c0dt00401d.
- W. Zaheer, G. Agbeworvi, S. Perez-Beltran, J. L. Andrews, Y. Aierken, C. Weiland, C. Jaye, Y. S. Yu, D. A. Shapiro, S. C. Fakra, D. A. Fischer, J. Guo, D. Prendergast and S. Banerjee, Lessons Learned from FeSb2O4 on Stereoactive Lone Pairs as a Design Principle for Anion Insertion, Cell Rep. Phys. Sci., 2021, 2(10), 100592, DOI:10.1016/J.XCRP.2021.100592.
- W. Zaheer, J. L. Andrews, A. Parija, F. P. Hyler, C. Jaye, C. Weiland, Y. S. Yu, D. A. Shapiro, D. A. Fischer, J. Guo, J. M. Velázquez and S. Banerjee, Reversible Room-Temperature Fluoride-Ion Insertion in a Tunnel-Structured Transition Metal Oxide Host, ACS Energy Lett., 2020, 5(8), 2520–2526, DOI:10.1021/acsenergylett.0c01328.
- F. Takeiri, T. Yajima, S. Hosokawa, Y. Matsushita and H. Kageyama, Topochemical Anion Insertion into One-Dimensional Bi Channels in Bi2PdO4, J. Solid State Chem., 2020, 286, 121273, DOI:10.1016/j.jssc.2020.121273.
- A. Walsh, D. J. Payne, R. G. Egdell and G. W. Watson, Chemical Society Reviews Stereochemistry of Post-Transition Metal Oxides: Revision of the Classical Lone Pair Model Stereochemistry of Post-Transition Metal Oxides: Revision of the Classical Lone Pair Model, Chem. Soc. Rev., 2011, 40(9), 4455–4463, 10.1039/c1cs15098g.
- J. V. Handy, W. Zaheer, A. R. M. Rothfuss, C. R. McGranahan, G. Agbeworvi, J. L. Andrews, K. E. García-Pedraza, J. D. Ponis, J. R. Ayala, Y. Ding, D. F. Watson and S. Banerjee, Lone but Not Alone: Precise Positioning of Lone Pairs for the Design of Photocatalytic Architectures, Chem. Mater., 2022, 34(4), 1439–1458, DOI:10.1021/acs.chemmater.1c03762.
- G. Laurita and R. Seshadri, Chemistry, Structure, and Function of Lone Pairs in Extended Solids, Acc. Chem. Res., 2022, 55(7), 1004–1014, DOI:10.1021/ACS.ACCOUNTS.1C00741.
- D. Sarkar, A. Bhui, I. Maria, M. Dutta and K. Biswas, Hidden Structures: A Driving Factor to Achieve Low Thermal Conductivity and High Thermoelectric Performance, Chem. Soc. Rev., 2024, 53, 6013–6628, 10.1039/d4cs00038b.
- N. Bettahar, P. Conflant, J. C. Boivin, F. Abraham and D. Thomas, Electrical Conductivity of (Bi,Pb)2MO4 (M = Pd,Pt) Linear Chain Compounds, J. Phys. Chem. Solids, 1985, 46(3), 297–299, DOI:10.1016/0022-3697(85)90169-6.
- N. Bettahar, Z. Derriche, F. Abraham and P. Conflant, Etude Par Diffraction Des Rayons x et Spectroscopie Des Photoelectrons de l’environnement et Des Degres d’oxydation Du Metal Dans Les Oxydes: Pb2PtO4, PbPt2O4 ET Bi1.6Pb0.4PtO4, Ann. Chim.: Sci. Mater., 2000, 25(4), 297–306, DOI:10.1016/S0151-9107(00)80006-9.
- K. Wissel, T. Vogel, S. Dasgupta, A. D. Fortes, P. R. Slater and O. Clemens, Topochemical Fluorination of n = 2 Ruddlesden-Popper Type Sr3Ti2O7 to Sr3Ti2O5F4 and Its Reductive Defluorination, Inorg. Chem., 2020, 59(2), 1153–1163, DOI:10.1021/acs.inorgchem.9b02783.
- N. H. Bashian, M. Zuba, A. Irshad, S. M. Becwar, J. Vinckeviciute, W. Rahim, K. J. Griffith, E. T. McClure, J. K. Papp, B. D. McCloskey, D. O. Scanlon, B. F. Chmelka, A. Van Der Ven, S. R. Narayan, L. F. J. Piper and B. C. Melot, Electrochemical Oxidative Fluorination of an Oxide Perovskite, Chem. Mater., 2021, 33(14), 5757–5768, DOI:10.1021/acs.chemmater.1c01594.
- J. Cabana, B. J. Kwon and L. Hu, Mechanisms of Degradation and Strategies for the Stabilization of Cathode-Electrolyte Interfaces in Li-Ion Batteries, Acc. Chem. Res., 2018, 51(2), 299–308, DOI:10.1021/acs.accounts.7b00482.
- T. Takami, N. Yasufuku, M. Ivonina, T. Tada, K. Tani, C. Pattanathummasid and K. Mori, Fluoride-Ion Conduction by Synergic Rotation of the Anion Sublattice for Tl4.5SnF8.5 Analogues, Chem. Mater., 2024, 36(17), 8488–8495, DOI:10.1021/acs.chemmater.4c01626.
- S. A. Razek, M. R. Popeil, L. Wangoh, J. Rana, N. Suwandaratne, J. L. Andrews, D. F. Watson, S. Banerjee and L. F. J. Piper, Designing Catalysts for Water Splitting Based on Electronic Structure Considerations, Electron. Struct., 2020, 2(2), 023001, DOI:10.1088/2516-1075/ab7d86.
- D. J. Payne, R. G. Egdell, A. Walsh, G. W. Watson, J. Guo, P. A. Glans, T. Learmonth and K. E. Smith, Electronic Origins of Structural Distortions in Post-Transition Metal Oxides: Experimental and Theoretical Evidence for a Revision of the Lone Pair Model, Phys. Rev. Lett., 2006, 96(15), 1–4, DOI:10.1103/PhysRevLett.96.157403.
- S. J. Kim, S. Lemaux, G. Demazeau, J. Y. Kim and J. H. Choy, X-Ray Absorption Spectroscopic Study on LaPdO3, J. Mater. Chem., 2002, 12(4), 995–1000, 10.1039/b106795h.
- A. Smekhova, D. Schmitz, N. V. Izarova, M. Stuckart, S. F. Shams, K. Siemensmeyer, F. M. F. de Groot, P. Kögerler and C. Schmitz-Antoniak, Intramolecular Crossover from Unconventional Diamagnetism to Paramagnetism of Palladium Ions Probed by Soft X-Ray Magnetic Circular Dichroism, Commun. Chem., 2020, 3(1), 1–10, DOI:10.1038/s42004-020-0327-9.
- V. Srot, M. Watanabe, C. Scheu, P. A. Van Aken, U. Salzberger, B. Luerßen, J. Janek and M. Rühle, Characterization of Chemical Composition and Electronic Structure of Pt/YSZ Interfaces by Analytical Transmission Electron Microscopy, Solid State Ionics, 2010, 181(35–36), 1616–1622, DOI:10.1016/j.ssi.2010.08.026.
- F. Frati, M. O. J. Y. Hunault and F. M. F. De Groot, Oxygen K-Edge X-Ray Absorption Spectra, Chem. Rev., 2020, 120(9), 4056–4110, DOI:10.1021/acs.chemrev.9b00439.
- C. Kalha, N. K. Fernando, P. Bhatt, O. L. Fredrik, R. Lindblad, S. Siol, L. P. H. Jeurgens, C. Cancellieri, K. Rossnagel, K. Medjanik, M. Simon and A. X. Gray, Hard X-Ray Photoelectron Spectroscopy: A Snapshot of the State-of-the-Art in 2020, J. Phys.:Condens. Matter, 2021, 33, 233001 CrossRef CAS PubMed.
- K. Artyushkova, S. R. Leadley and A. G. Shard, Introduction to Reproducible Laboratory Hard X-Ray Photoelectron Spectroscopy, J. Vac. Sci. Technol., A, 2024, 42, 052801, DOI:10.1116/6.0003740.
- J. R. Ebner, D. L. McFadden and R. A. Walton, The X-Ray Photoelectron Spectra of Inorganic Molecules. XI1. Fluorine 1s Binding Energies in Metal Fluorides, J. Solid State Chem., 1976, 17(4), 447–449, DOI:10.1016/S0022-4596(76)80016-3.
- A. Regoutz, M. Mascheck, T. Wiell, S. K. Eriksson, C. Liljenberg, K. Tetzner, B. A. D. Williamson, D. O. Scanlon and P. Palmgren, A Novel Laboratory-Based Hard X-Ray Photoelectron Spectroscopy System, Rev. Sci. Instrum., 2018, 89, 073105, DOI:10.1063/1.5039829.
- U. Dang, W. Zaheer, W. Zhou, A. Kandel, M. Orr, R. W. Schwenz, G. Laurita, S. Banerjee and R. T. MacAluso, Lattice Anharmonicity of Stereochemically Active Lone Pairs Controls Thermochromic Band Gap Reduction of PbVO3Cl, Chem. Mater., 2020, 32(17), 7404–7412, DOI:10.1021/acs.chemmater.0c02342.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Tetrel-Bonding Interaction: Rediscovered Supramolecular Force?, Angew. Chem. Int. Ed., 2013, 52(47), 12317–12321, DOI:10.1002/anie.201306501.
- B. L. Murphy and F. P. Gabbaï, Tunable Pnictogen Bonding at the Service of Hydroxide Transport across Phospholipid Bilayers, J. Am. Chem. Soc., 2024, 146(11), 7146–7151, DOI:10.1021/jacs.4c00202.
- M. Retuerto, A. Muñoz, M. J. Martínez-Lope, M. Garcia-Hernandez, G. André, K. Krezhov and J. A. Alonso, Influence of the Bi3+ Electron Lone Pair in the Evolution of the Crystal and Magnetic Structure of La1-xBixMn2O5 Oxides, J. Phys. Condens. Matter, 2013, 25, 216002, DOI:10.1088/0953-8984/25/21/216002.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08111k |
| This journal is © The Royal Society of Chemistry 2025 |