
Engineering tough, energy-dissipating soft materials via sacrificial chemical bonds
Parbhat
Kumar†
 ,
Abhishek
Roy†
and
Benjamin E.
Partridge
*
,
Abhishek
Roy†
and
Benjamin E.
Partridge
*
Department of Chemistry, University of Rochester, Rochester, NY 14627-0216, USA. E-mail: benjamin.partridge@rochester.edu
First published on 9th June 2025
Abstract
Impact-resistant materials such as Kevlar provide impact protection but lack flexibility and processability due to their highly crystalline, rigid structures. In contrast, the natural world is replete with examples of soft materials that can resist applied load. For example, sea cucumbers and other echinoderms modulate the stiffness of their outer skin in response to predators. This dynamic change in mechanical properties is regulated by transient, non-covalent interactions between the collagenic protein fibers that comprise the dermis. Upon the application of force, these transient interactions break, thereby acting as sacrificial bonds and providing a means of energy dissipation without damaging the protein fibers. This mechanism has been mimicked in synthetic materials, typically based on covalent polymers augmented with non-covalent bonding motifs. In this review, we survey the current state-of-the-art in the field of energy-dissipating soft materials. Specifically, we present recent (post-2017) highlights from the primary literature, organized by the chemical nature of the weak sacrificial bonds to outline the chemical toolkit available for programming material properties. We conclude by highlighting some opportunities to advance the development of soft yet tough energy-dissipating materials that harness the sacrificial bond.
 Parbhat Kumar Parbhat Kumar | Dr Benjamin E. Partridge (left) is an Assistant Professor of Chemistry at the University of Rochester (UR), where he is also an affiliated faculty member of the Materials Science Program. His research group leverages supramolecular chemistry to design novel materials that mimic and manipulate natural systems. Prior to joining the UR faculty in 2022, Dr Partridge completed postdoctoral training as an International Institute for Nanotechnology Postdoctoral Fellow at Northwestern University and earned his PhD in Chemistry as a Howard Hughes Medical Institute International Student Research Fellow at the University of Pennsylvania. Parbhat Kumar (center) and Abhishek Roy (right) are PhD candidates in the Partridge group. Close friends for many years, they both earned integrated BS-MS degrees in Chemistry (with a specialization in Organic Chemistry) from the Indian Institute of Science Education and Research (IISER) in Mohali and joined the Chemistry PhD program at UR in the 2022–23 academic year. Parbhat's research focuses on the design of bifacial nucleobases as building blocks for energy-dissipating hierarchical fibrous materials. Abhishek's research focuses on the development of novel photoswitchable nucleobase derivatives to engineer dissipative pathways into protein assembly. |
1. Introduction
The ability of natural materials to withstand mechanical force without damage is crucial for the survival of living organisms; consider the thousands of impacts endured by a human foot during a day's walking or by a woodpecker's skull during a day's drilling.1–3 The resilience of these materials is even more remarkable when one considers that strength (force withstood by a material) and toughness (resistance to fracture) are generally considered to be mutually exclusive in canonical materials,4,5 especially soft materials.6,7 Consequently, nature has evolved various approaches to prevent irreversible (plastic) material damage under applied load, including architectural hierarchy, porosity, and the use of composite structures that utilize small amounts of soft materials to absorb and dissipate energy (Fig. 1).3,5 For example, the extraordinary toughness of abalone shells and nacre—the iridescent internal layer of mollusc shells known also as mother of pearl—derives from their 5% biopolymer content rather than their major aragonite component.8 This biopolymer comprises a mixture of proteins such as lustrin, which reversibly folds and unfolds in response to force, and polysaccharides such as chitin, which engages in polyvalent hydrogen-bonding. Similarly, collagen molecules endow bone with its hallmark toughness by forming thin, glue-like filaments that bridge mineral components (Fig. 1, bottom right).9,10 Individual collagen fibers and fibrils within the filament can slip against each other due to the breakage and subsequent re-formation of sacrificial non-covalent interactions that bind these filaments together. | ||
| Fig. 1 Biological strategies to achieve impact resistance in natural materials. Top left to bottom right: architectural hierarchy in bamboo's culm wall (SEM, scale bar: 100 μm; adapted from ref. 11, copyright 2010 Budapest University of Technology and Economics); porosity in a turtle's shell (X-ray CT, scale bar: 2 mm; reprinted from ref. 12 with permission from Elsevier, copyright 2009); composite materials such as nacre in seawater snails (SEM, scale bar: 10 μm; reprinted from ref. 13 with permission from Elsevier, copyright 2008), and collagen-based sacrificial bonds [white arrows] within fractured bone (AFM, scale bar: 200 nm; adapted from ref. 14 with permission from Springer Nature, copyright 2005). | ||
The soft components of these composite materials harness sacrificial bonds to absorb and dissipate energy.15 Sacrificial bonds are comparatively weak bonds within a polymer chain or network that, under applied force, break before the (typically covalent) bonds within the strong polymer network.16,17 Under mechanical stress, rupture of these weak bonds dissipates energy while protecting the integrity of the strong matrix.6 If the fission and formation of these sacrificial bonds is reversible, their presence can enhance a material's toughness under multiple cycles of loading and unloading. In this way, natural biomaterials harness structural failure, through the rupture of deliberately weak sacrificial bonds, to tolerate mechanical force.1
Due to the attractiveness of strong yet tough soft materials, there has been substantial research effort expended towards incorporating sacrificial mechanisms into synthetic materials. These efforts can broadly be categorized based on whether weak sacrificial bonds are incorporated as a separate material component or as part of the primary material matrix (Fig. 2).18 In the former strategy, materials containing two interpenetrating polymer networks rely on the preferential rupture of the weaker sacrificial network to dissipate energy while maintaining the integrity of the stronger interpenetrated matrix (Fig. 2a).18,19 Such materials, known as double network materials,20 are not discussed further in this review due to an excellent recent discussion by Gong and coworkers.21 Instead, this review focuses on the latter strategy, whereby a single polymer network is responsible both for the material's strength and toughness, aided by weak sacrificial interactions that, at rest, hold together strong polymer chains, but rupture preferentially under mechanical stress (Fig. 2b and c). This approach has also garnered substantial research interest, as excellently summarized by Hu and coworkers in 2017.17
 | ||
| Fig. 2 Strategies to incorporate sacrificial bonds into synthetic materials. (a) In double network materials, application of force breaks a weaker sacrificial network (pink), thereby protecting the stronger structural network (blue); these materials were reviewed recently elsewhere.21 (b, c) In single network materials, applying force breaks the weak, sacrificial bonds that exist between interacting motifs (green) on the structural network (blue); such materials are the focus of this review. Intrinsic sacrificial bonds (b) are those resulting from direct interaction between polymer chains, whereas extrinsic sacrificial bonds (c) rely on small molecule mediators (yellow) to define intra-chain interactions. | ||
Our aim in this review is to present recent (post-2017) highlights from the primary literature, organized by the chemical nature of the weak sacrificial bonds. This organizational scheme was motivated by our belief that the key challenges in the field have evolved from preparing energy-dissipating materials to designing specific properties into those materials. Reviews by others have focused on mesoscale design strategies, including the nature of the polymer backbone itself.22–26 Instead, we offer herein a chemist's perspective on the design of sacrificial interactions and thus hope to provide the reader with a better sense of the chemical toolkit available for programming material properties (Fig. 3). We begin by defining some key terms used to describe materials properties and then introduce recent advances in the theoretical treatment of sacrificial bond-containing materials. The bulk of the review, describing recent experimental progress, follows. We conclude by highlighting some outstanding challenges and proposing potential research avenues to continue progress towards soft materials with extraordinary strength and toughness enabled by the sacrificial bond.
 | ||
| Fig. 3 Chemical toolkit described in this review. In all structures, R denotes an arbitrary group and differs from structure to structure. | ||
2. Terminology for mechanical properties
In this section, we briefly define the terms used in this review to describe the mechanical properties of materials. Many rheological properties focus on how materials respond to external applied forces; the measure of amount of force applied to a given area of material is denoted stress. Tensile stress is the most reported type of stress, measured by stretching a material along the axis of measurement. This stretching of the material is a form of deformation; the extent of deformation (i.e., the ratio of a material dimension under deformation vs. at rest) is defined as strain. Thus, strain is a direct measure of physical change in a material and may be associated with a change in length (axial strain) or change in volume (volumetric strain). Experimentally, the rate at which a material is forcibly deformed (i.e., the strain rate) plays a substantial role in the material's stress response. At small strains, the relationship between force applied (stress) and material deformation (strain) is linear and elastic (that is, reversible); in this case, the stress–strain relationship adheres to Hooke's law and the ratio of tensile stress over axial strain is described by the Young's (elastic) modulus. This constant indicates the extent of deformation under a given applied stress and is characteristic of each material, ranging from ∼103 Pa for gels to ∼109 Pa for metals. The limit of proportionality denotes the maximum stress at which Hooke's law applies; beyond this limit, stress is no longer directly proportional to strain.The resistance of a material to deformation may be characterized by different terms depending on the mode of failure. Tensile strength is defined as the maximum stress that a material can withstand when pulled before permanent deformations occur, such as irreversible shape changes or material fission. Some materials tend to crack or fracture when stress is applied; fracture stress is the stress at which a material fails due to fractures, and the amount of energy that leads to crack propagation is known as fracture energy.
Viscoelastic materials behave differently depending on the applied strain rate; this time-dependent response to stress leads to different regimes in which the material exhibits more solid-like or liquid-like behavior at different strain rates. Viscoelastic materials with the ability to stretch and return to their original shape without damage have high stretchability, also termed extensibility. Typically, this ability is limited: the maximum elongation (or elongtion-at-break) measures the maximum (axial) strain before permanent fission occurs. Materials subjected to constant stress over time might experience a gradual, time-dependent deformation, called creep elongation.
Energy dissipation is commonly quantified using cyclic loading–unloading experiments, whereby materials are subjected to an increasing external load which is gradually removed (unloaded). Stress–strain plots of the loading and unloading phases differ: the loading cycle represents the strain energy required to generate a given stress within the material under measurement, while the unloading cycle represents the residual elastic energy of the material at that stress. This residual elastic energy is typically lower due to the energy dissipation via heat or fission of sacrificial bonds; consequently, the stress–strain plot of a loading–unloading cycle exhibits a hysteresis loop whose area denotes the total energy dissipated by the material. After a waiting period (relaxation time), this loading–unloading cycle is repeated and compared to the initial cycle. If a material does not return to its original length after removal of the external load, the material has deformed and its recovered length is called the residual length.
For viscoelastic materials, rheological measurements also quantify two key measures of a material's response to external stress: the storage and loss moduli. The storage modulus (G′) represents a material's ability to store elastic energy, while the loss modulus (G′′) represents its ability to dissipate energy as heat. The ratio of G′′ to G′, known as tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ, quantifies a material's ability to absorb and dissipate energy: values of tanδ less than 1 denote a more elastic material, while values greater than 1 denote a predominance of viscous behavior and a more plastic material.
δ, quantifies a material's ability to absorb and dissipate energy: values of tanδ less than 1 denote a more elastic material, while values greater than 1 denote a predominance of viscous behavior and a more plastic material.
3. Theoretical studies
Much interest in sacrificial bond-containing materials stems from their non-linear mechanical properties. However, this non-linearity complicates efforts to faithfully describe such materials using canonical polymer physics descriptions such as the worm-like chain (WLC) model, which assumes that the motion of each segment in a semiflexible polymer chain is correlated to some extent with its neighboring segments.1 In particular, theoretical treatments of energy-dissipating materials must consider the topology of the strong polymer network before and after load is applied, as well as the rate dependence of sacrificial bond rupture and formation. Thus, the faithful modeling of energy-dissipating materials requires consideration of both thermodynamic and kinetic factors.Kroy and coworkers have approached this problem by considering the inelasticity intrinsic to biomechanics.1 Their initial model proposed a ‘glassy’ WLC model that accounts for the reduced dynamics of a polymer under confinement in a network,27 and was subsequently updated to include transient crosslinks, in a revised inelastic glassy WLC (iGWLC) model (Fig. 4).28,29 The presence of transient crosslinks, akin to sacrificial bonds, are parametrized with a single time-dependent variable that represents the fraction of such bonds that are ‘closed’. Despite the simplicity of this treatment, the authors are able to reproduce non-linear properties including hysteresis and strain-rate dependent mechanical response.1
 | ||
| Fig. 4 Kroy and coworkers’ inelastic glassy wormlike chain (iGWLC) model reproduces the non-linear mechanical response of biopolymers. At low rates where power-law rheology is exhibited (top panel, log scales), the model predicts entropic stiffening of the polymer backbone at low amplitudes (left panel, linear scale). This stiffening is superseded at higher amplitudes by exponential bond softening. At high rates and high amplitudes, rapid stiffening is followed by fluidization. Reprinted from ref. 29. Copyright 2012, Wolff et al., licensed under CC BY. | ||
Incorporating thermodynamic details of specific chemical bonds was achieved by Elbanna and coworkers, who developed a polymer network model (based on the WLC) supplemented with rate-dependent damage behavior.30 In their model, sacrificial bonds are described using a transition state theory (TST) approach that considers each bond to have two stable states (associated or dissociated), separated by a transition state with an accompanying activation energy (Ea). This Ea term contains a contribution that accounts for the force-dependent difference in nuclear coordinate between a given bond and the transition state. A term is also incorporated to account for the rate of sacrificial bond-breaking. Using this model, the authors found that increasing the concentration of sacrificial crosslinkers does not lead to a monotonic increase in the peak force withstood by the polymer network, but instead a maximum was reached due to the prevalence of short chain lengths between adjacent crosslinks. The authors also used their model to probe the relationship between the velocity at which tensile force is applied to a material and peak force withstood by that material. However, the re-formation of sacrificial bonds was not investigated by the authors in that report.30 A similar, damage-induced, energy dissipation mechanism was incorporated into a recent model by Sun, Yin, and Liew that sought to correlate microscale structural damage with macroscale mechanical properties in double-network materials.31
The topic of breaking and re-forming sacrificial bonds has been modelled through various approaches. Suhail, Banerjee, and Rajesh developed a course-grained kinetic model to describe energy dissipation in fibrils of collagen.32 Their model not only accounts for the reformation of sacrificial bonds, but also considers how hidden length—segments of a polymer chain released upon rupture of an intra-chain sacrificial bond—affects energy dissipation under cyclic loading. The authors compare their kinetic model against experimental data and find that several aspects of the stress–strain response, including cycle-dependent energy dissipation (known also as moving hysteresis loops), are well reproduced.
An alternative approach by Liu and coworkers employed molecular dynamics (MD) simulations to investigate the breaking and re-forming of specific chemical hydrogen-bonding motifs in polymer networks based on poly(isoprene) (Fig. 5).33,34 Polymer backbones were investigated with different extents of modification with a hydrogen bond-capable motif, either 3-amino-1,2,4-triazole33 or a tetraalanine peptide.34 Fully atomistic MD studies allowed the authors to observe the extent of sacrificial bond clustering in these materials and reproduced the findings of Elbanna and coworkers.30 Specifically, desirable mechanical properties initially increase with increasing sacrificial bond content before reaching a maximum and decrease as the number of sacrificial bond motifs continues to increase. Atomistic MD studies were also employed by Xie et al. in their recent development of a multiscale model to describe the fracture mechanics of self-healing, energy-dissipating composite materials such as nacre and bone.35 MD simulations on the nanoscale were coupled with shear-lag and crack-bridging models at the meso- and macroscales, respectively, to produce an integrated framework for predicting materials properties based on structural parameters.
 | ||
| Fig. 5 (a) Molecular dynamics (MD) simulations of poly(isoprene) with different monomer ratios (α) of a hydrogen bond (HB) competent motif. Red and blue spheres denote oxygen and nitrogen atoms, respectively, of the HB motif. (b and c) Variation of (b) storage modulus, G′, and (c) loss modulus, G′′, as a function of frequency (ω). Reprinted from ref. 33 with permission from Elsevier, copyright 2023. | ||
One emerging area for experimental investigation is the incorporation of multiple different types of sacrificial bond within the same material (as will be discussed later). Such materials are also being explored at the vanguard of current theoretical efforts. In 2022, Yu et al. reported a course-grained MD treatment of networks of carbon nanotube fibers crosslinked with two distinct sets of sacrificial bonds, one much weaker than the other.36 The authors explore several features of their simulated material, including conductivity and Young's modulus, as a function of both overall crosslinking density and the ratio of strong and weak crosslinks. Weak crosslinks are observed to enhance the toughness of the doubly-crosslinked networks, while stronger crosslinks contribute little to material toughness but underlie material strength. Analysis of the spatial distribution of broken crosslinks after stress suggests that heterogeneous crosslinking leads to stress concentrations within the material, resulting in synergistic rupture and a marked decrease in overall resistance to deformation. These results emphasize the importance of the relative interaction strengths and spatial distribution of multiple sacrificial bonds within a single material.
4. Hydrogen bonding
Hydrogen bonds (HBs) are particularly attractive candidates for sacrificial bonds, due to their tunable binding strength, directional interactions, and readily reversible binding modality.37 Biological materials, such as silk and muscles, often utilize HBs to crosslink polypeptides to form a primary structural network.38 Following a similar strategy, several synthetic materials have been reported in the past two decades that exhibit excellent energy dissipating and mechanical properties.39–41 The most commonly employed approach has been to modify polymer chains with prosthetic HB-capable motifs (for example, ureidopyrimidinones and amides) to define sacrificial interactions between polymers.42–45 Additionally, some studies have leveraged intrinsic HB acceptors and donors in the chemical structure of the strong polymer network (for example, poly(vinyl alcohol) and poly(ether-thiourea)) to augment the prosthetic hydrogen-bonding motifs.4.1. Ureidopyrimidinones
Inspired by DNA base-pairing, Meijer and coworkers designed the ureidopyrimidinone (UPy) motif (Fig. 6), a self-complementary quadruple H-bond unit that is attractive for materials design due to its strong binding affinity and facile synthetic accessibility.46 In the solution state, UPy exists in an equilibrium between two isomers, 2-ureido-4[1H]-pyrimidinone and 2-ureido-4[1H]-pyrimidinol, due to keto–enol tautomerization (Fig. 6, top). The pyrimidinone derivative has an AADD (A = acceptor, D = donor) H-bonding pattern and the other derivative has a DADA H-bonding pattern. Both conformers are interconvertible and the ratio of the two tautomers in a specific environment depends on the nature of the substituents at the R1 and R2 positions (Fig. 6). The presence of n-alkyl chains at the R1 and R2 positions favors the AADD tautomer, while in the presence of any electron-withdrawing group, the DADA tautomer predominates. Both tautomers are self-complementary, that is, a given tautomer can associate with itself while forming four HBs. Due to this quadruple hydrogen bond, UPy exhibits high dimerization constants (Ka > 105 M−1 for the DADA tautomer and 5.7 × 107 M−1 for the AADD tautomer).47 | ||
| Fig. 6 UPy exists as two tautomers at equilibrium (top). Each tautomer can respectively dimerize due to self-complementary quadruple hydrogen bonding to form homodimers (bottom). Reprinted from ref. 47 with permission from Elsevier, copyright 2023. | ||
Li and coworkers employed the UPy motif to design bioinspired, HB-crosslinked healable polymers with remarkable energy dissipating properties.44 Poly(propylene glycol) (PPG) or poly(tetramethylene ether glycol) (PTMEG) chains (soft segment) were combined with cyclohexylmethylmethane-4,4′-diisocyanate (HMDI) or isophorone diisocyanate (IPDI) (hard segment) as well as UPy units (Fig. 7). The reported polymer has several units, such as UPy, urea, and urethane, capable of forming hierarchical H-bonding networks that lead to hard–soft nanophase segregation. Regulating these nanophases allowed the mechanical and energy dissipating properties of the bulk material to be modified. The samples prepared with PTMEG and HMDI showed elongations of up to 1960% at their breakage point. To investigate the energy dissipation properties, cyclic tensile tests were conducted. Samples showed hysteresis loops as well as energy dissipation at low and high strain levels. At low strain levels, energy dissipation occurs via the breakage of weak HBs in urethane while at high strain levels, energy is further dissipated via the breakage of strong HBs between urea and UPy. In addition to energy dissipation and self-healing, the material showed an exceptionally high toughness of 345 MJ m−3, which compares favorably to synthetic materials such as Kevlar (33 MJ m−3) and natural materials such as spider silk (50–500 MJ m−3).48
 | ||
| Fig. 7 Copolymers of hard and soft segments generate hierarchical H-bonding domains that are responsible for self-healing and energy dissipative properties. Figure adapted from ref. 44. Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Biobased elastomers are attractive materials due to their low modulus, high elasticity, and excellent recovery upon removal of external load,49 but their mechanical weakness and limited molecular structures reduce the scope of their potential applications.50,51 To overcome this challenge, Guo and coworkers employed a sacrificial bond strategy to conserve material integrity.52 Elastomers were synthesized by dissolving various ratios of UPy-based methacrylate (UPyMA) and biobased polyester elastomer (BPE) in DMF (Fig. 8a). To confirm the network structure and UPy dimerization, variable temperature (30 to 150 °C) FTIR tests were conducted. The elastomers with higher proportions of UPyMA showed better shape memory behavior (restoration within 30 s), suggesting that increased HB density increases the energy dissipation and shape recovery of the materials. Stress–strain plots of polymers with 0% and 30% UPyMA showed that the addition of UPy increased the Young's modulus from 0.6 to 3.5 MPa and tensile strength from 0.9 to 5.2 MPa (at 100% strain). Moreover, cyclic loading–unloading tests indicated that the amount of dissipated energy increased with increasing UPyMA content (almost 20-fold from 0% to 30% UPyMA, Fig. 8b).
 | ||
| Fig. 8 (a) Schematic representation of BPE/UPyMA-x polymeric networks, where x denotes the relative proportion of UPyMA, with UPy quadruple HBs. (b) Energy dissipation (hysteresis loop area) for BPE/UPyMA-x, x = 0 to 30. Reprinted from ref. 52 with permission from Elsevier, copyright 2019. | ||
Impact-hardening polymers (IHPs) are a class of energy-absorbing materials with loading rate-dependent viscoelasticity, commonly applied as impact protectors.53 IHPs exhibit an increase in viscosity with increasing shear stress and can be categorized by their physical form under no stress as either shear-thickening fluids (STFs) or shear-stiffening gels (SSGs). STFs, known also as non-Newtonian fluids, have limited applications due to their fluid nature and inability to hold their shape; therefore, SSGs have been more explored for applications.54 In 2021, Yan and coworkers designed an impact-protective SSG based on a supramolecular polymeric material (SPM) comprising the triblock copolymer poly(propylene glycol)-b-poly(ethylene glycol)-b-poly(propylene glycol) (PPG-PEG-PPG) and UPy motifs.55 Loading–unloading cycles showed an increase in hysteresis with increasing compression rates, representing a higher degree of energy dissipation. Hammer drop tests were performed to mimic destructive impact in the real world and the SPM slowed down the hammer to rest. To demonstrate the protective properties of the SPM as a casing, an egg placed between two SPM cups and hammered several times remained intact (Fig. 9).
 | ||
| Fig. 9 Hammer test on an egg encapsulated between two SPM cups. Energy dissipation by the SPM cups protected the structural integrity of the egg. Adapted with permission from ref. 55. Copyright 2020 American Chemical Society. | ||
The role of multiple sacrificial bonds in IHPs was recently investigated by Zhang and coworkers.54 The authors prepared a single IHP containing H-bonding UPy motifs, reversible boron–oxygen linkages, and Cu2+-coordinating imidazoline ligands, as multiple energy-dissipating pathways (Fig. 10a). To investigate the mechanism of energy dissipation, loading–unloading cycles at different strain rates were performed. Samples showed greater hysteresis at higher strain rates, indicating higher energy-dissipating capabilities. The authors proposed that at low strain rates, boron–oxygen bonds break, increasing the mobility of polymer chains within the network. At higher strain rates, chain motion becomes too fast for boron–oxygen bonds to break, causing chain entanglement, followed by the dissociation of HBs and metal coordination (Fig. 10b). Further increasing the strain rate breaks the covalent bonds of the polymer backbone and leads to irreversible microcracks. To confirm that the sacrificial bonds break reversibly, the authors spliced the sample in two halves (30 × 10 × 3 mm) and subsequently kept them in close contact for 10 s at room temperature. These combined segments were able to withstand 400 g of external load. Hence, the authors concluded that the reported IHP showed effective impact-absorbing capabilities due to reversible dynamic bonds. This work highlights the ability of multiple non-covalent interactions to act in synergy to achieve impact resistance. Moreover, it provides a rare example where the relative kinetics of strain rate and bond breaking lead to selectivity over which type of sacrificial bond is broken. Such control is desirable for envisaged applications including soft robotics and personal protective equipment.
 | ||
| Fig. 10 (a) Molecular structure of the reported impact hardening polymer (IHP) with trisubstituted triisocyanate (TTI) junctions, multiple HBs, and metal-coordination sacrificial bonds. (b) Illustration of energy dissipation mechanism. Reprinted from ref. 54 with permission from Elsevier, copyright 2023. | ||
In 2017, Ikkala and coworkers reported a similar strategy to enhance the mechanical and energy-dissipating properties of an acrylate-based polymer, but utilized three H-bonding motifs with different association strengths rather than three different bonding interactions.56 Poly(2-hydroxyethyl methacrylate) (pHEMA) is known to have a brittle structure and poor mechanical properties despite having hydroxyl groups as HB acceptors and donors. The resultant HBs alone are not strong enough to provide toughness or dissipate substantial amounts of energy. To increase the HB density, UPy motifs were attached to a fraction of chains and the resulting copolymer, p(HEMA-co-UPyMA), displayed an array of single and quadruple HBs as interchain links (Fig. 11). The strong UPy-mediated HBs slowed crack propagation and increased yield strength and Young's modulus, by 65% and 69%, respectively, indicating that these links act as sacrificial bonds to dissipate the crack energy. To further enhance the mechanical properties of the copolymer, carbon nanotubes (CNTs) were incorporated into the material. CNTs exhibit extraordinary thermal, electrical, and mechanical properties, and there have been several reports on harnessing these properties in the bulk.57,58 However, materials with CNTs incorporated non-covalently into soft materials had previously shown poor mechanical properties due to highly heterogeneous dispersion of CNTs in the polymer matrix.59,60 To further increase these properties of p(HEMA-co-UPyMA), oxidized multiwall carbon nanotubes (MWCNTs) were used as another crosslinker to strengthen the material. The surface of the oxidized CNTs presented UPy, –OH, and –COOH groups to facilitate interactions with the polymer chains (Fig. 11). The resulting material showed increases in Young's modulus, tensile strength, and yield strength of 321%, 51%, and 100%, respectively. This report demonstrates that the incorporation of sacrificial bonds in nanocomposites can synergistically increase solid-state toughness and defect tolerance. This strategy of combining hierarchical HBs with CNTs and harnessing the properties of both materials mirrors natural approaches to composite materials with ultra-strong toughness and energy-dissipating properties.3 Jiang and coworkers also demonstrated the utility of UPy motifs as one of several sacrificial HB motifs in a polyacrylate hydrogel.61 Notably, the authors found that incorporating UPy at a concentration of just 0.8 mol% was sufficient to increase the Young's modulus from 74 ± 10 kPa to 1252 ± 108 kPa, surpassing the mechanical properties of human tissues such as nasal cartilage (250–440 kPa)62 and skin (200–250 kPa).63
 | ||
| Fig. 11 Different levels of hierarchical HBs in the supramolecular network. The first level of HBs is between the –OH groups of pHEMA chains (weak interactions). The second level of HBs is between the UPy motifs (strong interactions) and the last level is between p(HEMA-co-UPyMA) and OxMWCNT-g-UPy. Adapted with permission from ref. 56. Copyright 2017 American Chemical Society. | ||
Hierarchical H-bonding was also explored by Sun and coworkers, who used carboxylic acids and UPy to crosslink polyurethane (PU) chains, supplementing the carbamate-based H-bonding inherent to the PU backbone.64 To compare the relative contributions of each HB motif to the polymer's mechanical properties, three elastomers were prepared with varying molar ratios of UPy and DPA (3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid), where DPA acts as a chain extender and provides a carboxylate H-bonding motif. The authors propose that weak carbamate–carbamate and carboxyl–carboxyl HBs dissociate first and contribute to the toughness of the elastomer, while the UPy–UPy dimer provides high strength (Fig. 12a). The sample with the lowest UPy content (10 mol% UPy) exhibited low tensile strengths, presumably due to an insufficient number of strong HBs, while the sample with the highest UPy content (30 mol% UPy) showed an increased Young's modulus but had poor stretchability, attributed to an insufficient number of weak HBs. However, a sample with a UPy:DPA ratio of 20:80 exhibited a good balance of enhanced toughness and mechanical strength. This elastomer was characterized by cyclic loading–unloading tests at 400% strain. The first cycle showed a hysteresis loop with an area of 5.29 MJ m−3 that was significantly reduced in an immediate second cycle (no relaxation time) with 44.8% residual strain (Fig. 12b). However, with increasing relaxation time, the residual strain increased and was indistinguishable from the first cycle after a relaxation time of 90 min (Fig. 12c). This study demonstrates how mechanical properties can be tuned by tailoring the relative amounts of different sacrificial bonds as well as their relative interaction strengths.
 | ||
| Fig. 12 (a) Structural representation of PU-UPyx-DPAy polymer network and sacrificial HBs in the network. (b) Two successive loading-unloading cycles of PU-UPy0.2DPA0.8. (c) Cyclic loading–unloading test with different relaxation times. Adapted with permission from ref. 64. Copyright 2023 American Chemical Society. | ||
UPy motifs have also been explored as sacrificial bonds in elastomers, whose elastic properties make them excellent materials for applications in tires, sealants, protective coatings, and wearable electronics.65,66 Elastomers can tolerate large deformations and stress due to their elasticity, but they accumulate internal microcracks that expand over time and result in functional failure and material degradation. To address this problem, Cai and coworkers combined PU with UPy and imidazole motifs using thiolactone chemistry (Fig. 13).67 In brief, a thiolactone derivative, α-isocyanato-γ-thiolactone, was functionalized with pendent UPy and imidazole groups, and incorporated into PU chains via typical synthesis strategies using PTMEG and HMDI. Subsequent treatment with Zn(CF3SO3)2 to form the metal–ligand bonds between Zn2+ and imidazole (Im), followed by molding and drying, afforded the polyurethane-based elastomer, PU-Im-UPy-Zn. A control sample prepared via a similar strategy, PU-Im-BA-Zn, replaced UPy with butyl acrylate to eliminate the H-bonding network. Rheological studies showed that the introduction of Zn2+ ions increased the storage modulus (G′) two-fold compared to metal-free PU-Im-UPy and four-fold compared to the butyl acrylate control, demonstrating the synergistic cross-linking of UPy units and Zn2+ coordination with imidazole ligands. At higher strain rates, PU-Im-UPy and PU-Im-BA-Zn showed enhanced tensile strength but poor extensibility. In contrast, PU-Im-UPy-Zn showed enhanced toughness (62.1 MJ m−3, 136% vs. control), tensile strength (9.1 MPa, 198% vs. control), and extensibility, due to the rapid rearrangement of dynamic HBs and metal–ligand coordination bonds. This work highlights the ability of dynamic, rapidly reformed sacrificial bonds to bolster the mechanical properties of elastomers.
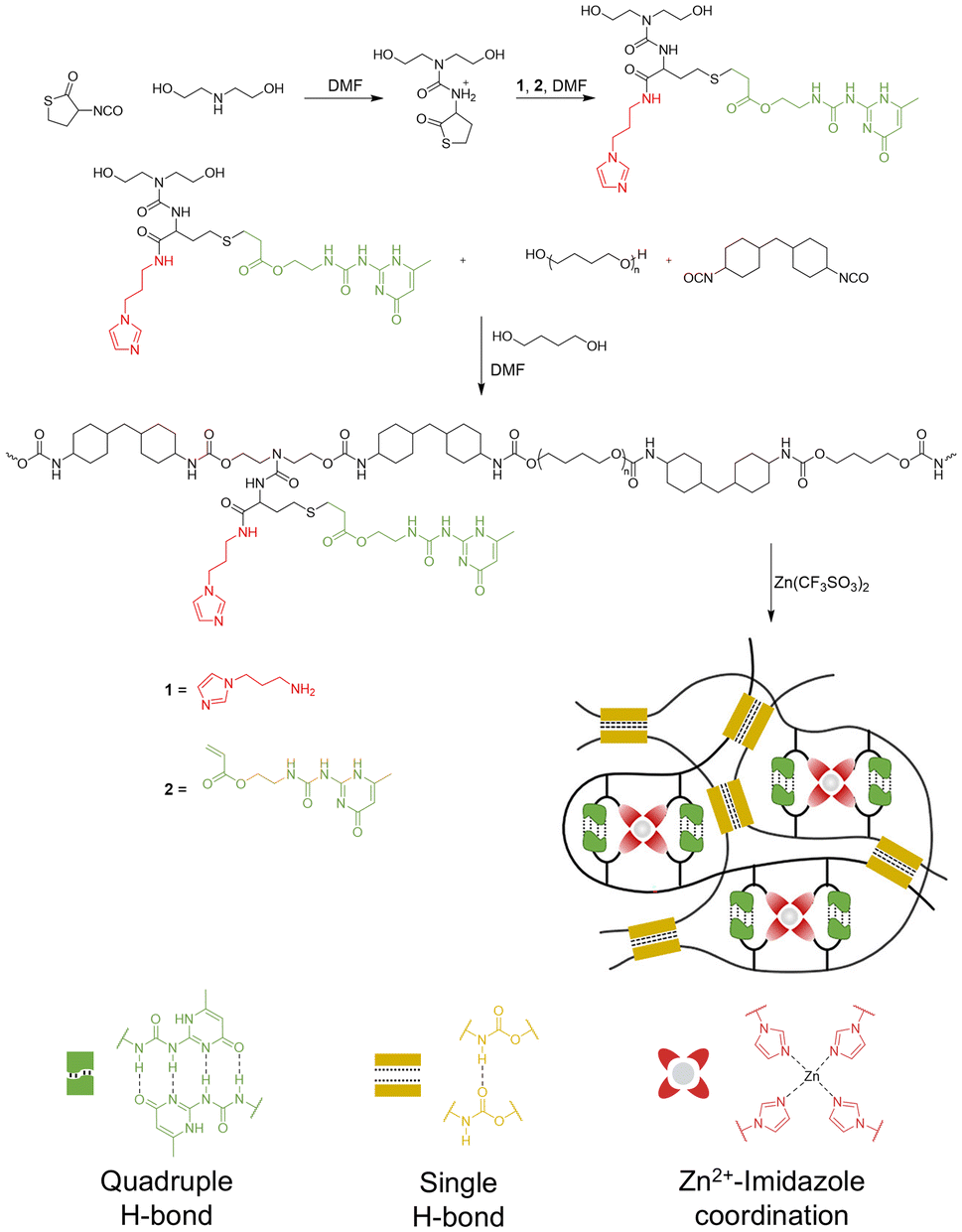 | ||
| Fig. 13 Synthetic route for PU-Im-UPy-Zn and sacrificial bonds involved in the network. Adapted with permission from ref. 67. Copyright 2023 American Chemical Society. | ||
A 2019 report by Guo and co-workers showed that incorporation of UPy in poly(urea) elastomers improved their strength, stiffness, and toughness due to the formation of quadruple HBs.68 Samples with UPy showed threefold higher toughness than the pristine sample. The dimerization of UPy has also been used to improve the toughness of polymer networks comprising poly(ether imide)69 and other weak polymers.70,71
4.2. Amides
In biological systems, amides serve as the primary chemical motifs for HB formation: for example, proteins have hierarchical structures scaffolded by NH–carbonyl, NH–water, and carbonyl–water H-bonding interactions. These interactions provide structural stability to the proteins and ensure proper functioning.72 Accordingly, amides have been explored extensively as H-bonding motifs in synthetic polymeric materials because of their biocompatibility, straightforward synthesis, and potential to enhance mechanical and energy-dissipating properties, as discussed in an excellent recent review by Li et al.73In 2015, Guan and co-workers combined amide functionality with a covalent self-healing polymeric network and observed enhancements in mechanical and energy-dissipating properties.45 Specifically, secondary amide groups were attached to a cyclooctene-based polymer backbone which was cross-linked and appended with Grubbs’ second-generation ruthenium catalyst (G2), to give a final material (amide-containing cyclooctene network, ACON) with both covalent and non-covalent cross-links. The HB cross-links broke under stress and provided toughness to the material, while the G2 catalyst mediated dynamic olefin metathesis at the fracture interface in the case of mechanical damage to the covalent network (Fig. 14a). A control material in which the amide hydrogen was replaced with a methyl group (blocked amide-containing cyclooctene network, BACON) exhibited decreased tensile toughness (7-fold) and strain-at-break (6-fold), confirming the importance of the amide-mediated H-bonding. The reversible nature of amide sacrificial bonds was demonstrated via cyclic tensile tests, in which BACON showed an extensibility of 167% while ACON showed up to 300%. At first, 10 immediate cycles of stress loading and unloading were performed. The first cycle showed large energy dissipation, but this decreased by 80% in the second cycle and decreased further over subsequent cycles. An 11th cycle was performed after 30 min of relaxation time in ambient conditions, and ACON exhibited 90% of the energy dissipation observed in the first cycle (Fig. 14b and c). This study validated amides as an effective H-bonding motif to improve the energy-dissipating properties of synthetic polymer networks, demonstrated by a battery of mechanical testing studies.
 | ||
| Fig. 14 (a) Schematic illustration of reversible sacrificial HB breaking under external force. (b) Hysteresis loops for 11 loading–unloading cycles of ACON at 300% strain. The first ten cycles were done immediately and the last was performed after 30 min of relaxation time. (c) Energy dissipation corresponding to each cycle. Adapted with permission from ref. 45. Copyright 2015 American Chemical Society. | ||
Hydrogels are particularly attractive in applications like tissue engineering due to their high modulus, softness yet toughness, resistance to mechanical shear, and biocompatibiity.74,75 Early work by Sheiko and coworkers reported that hydrogels formed by copolymerizing N,N-dimethylacrylamide and methacrylic acid dissipate large amounts of energy (1.7 MJ m−3) and exhibit a near-complete recovery of dissipative capacity after a relaxation time of 60 min.76 Later work by Malmström and coworkers achieved tunable energy dissipation in synthetic hydrogels.77 Three different semi-interpenetrating double networks were made by photopolymerizing two different monomers (acrylic acid and tannic acid) in cross-linked poly(acrylamide) (PAAm) hydrogels (Fig. 15a and b). The two different monomers had unique H-bonding capabilities when copolymerized with the PAAm network (Fig. 15c). Poly(acrylic acid) (PAA) adopts a charged state at physiological pH and forms complexes (via H-bonding) with PAAm. Poly(tannic acid) (PTA) is capable of UV polymerization, has multiple HB sites, and forms shorter, more highly branched chains compared to PAA. In comparison to unmodified PAAm, in which limited H-bonding is expected, both PAAm-PTA and PAAm-PAA showed increased energy dissipation as reflected by improved mechanical properties: PAAm-PTA showed a 19-fold increase in tensile strength and a 55-fold increase in toughness, while the PAAm-PAA hybrid exhibited 4.3- and 3.6-fold improvements, respectively. DLS studies indicated that PAAm-PTA formed larger aggregates than PAAm-PAA, which might account for the increase in toughness. The reported hydrogels have similar mechanical properties to soft tissues and comparable relaxation times to skin. Such materials could be used to explore hydrogel applications in cellular mechanotransduction studies.
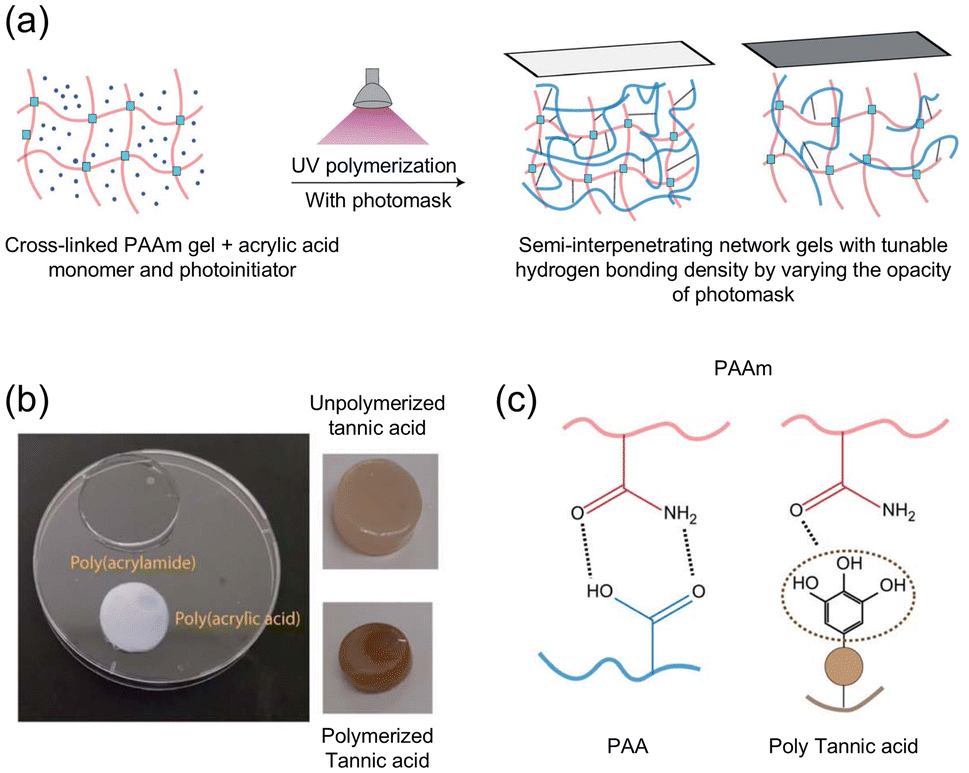 | ||
| Fig. 15 (a) Photopolymerization of monomers (acrylic acid, acrylamide or tannic acid) in a cross-linked PAAm network using UV light. The HB density can be easily tuned by varying the intensity of UV light with different photomasks. (b) Macroscale samples of double network hydrogels. (c) H-bonding interactions between PAAm and poly(acrylic acid) or poly(tannic acid). Figure adapted from ref. 77 with permission from the Royal Society of Chemistry, copyright 2021. | ||
As noted earlier, polyurethanes are desirable materials whose toughness is limited due to their low inherent H-bonding strength. In contrast to Cai and coworkers’ study of UPy-functionalized polyurethanes (section 4.1),67 Liu and coworkers employed an alternative approach that utilized H-bonding chain extenders made from the dipeptide N-acryloyl glycinamide (NAGA) (Fig. 16).78 These extenders were reacted with copolymers of PTMEG and either hexamethylene diisocyanate (HDI) or IPDI to generate PUHD-NAGA and PUIP-NAGA, respectively. The aim was to supplement the main chain amide HBs with the amide HBs of the NAGA extenders to make a mechanically stiff, tear-resistant, self-healing material. By varying the molar ratio of NAGA units to PTMEG segments, different mechanical properties could be optimized. For example, PUIP-NAGA with a NAGA:PTMEG ratio of 2.1 showed the highest energy dissipation (2.52 MJ m−3 at 200% strain), while a thin-film sample of PUIP-NAGA with a NAGA:PTMEG ratio of 2.6 could hold up to 4 kg of weight with minimal deformation.
 | ||
| Fig. 16 Synthetic schemes for PUHD-NAGA and PUIP-NAGA PU elastomers. H-bonding in the polymer network comprises two different types of HB: strong HBs between NAGA motifs and weak HBs between amides in the polyurethane backbone. Figure adapted from ref. 78. Copyright 2020 Wiley-VCH GmbH. | ||
To achieve high toughness and tensile strength, one of the most common strategies is the crosslinking of polymeric chains, but this can cause other issues like poor reprocessibility and difficulties in degradation, which create environmental issues. Modulating the number of amide HBs was also recently explored by Shi and co-workers in the context of curable epoxy materials based on polyurethanes.79 The authors synthesized epoxy resins with different H-bonding capabilities by curing an epoxy-terminated monomer (IDV-EP) with three different bisamines (Fig. 17a): diamino-diphenylmethane (DDM), which introduces the fewest HBs; 2-(4-aminophenyl)-1H-benzimidazol-5-amine (APBIA), which introduces an imidazole moiety; and 4,4-diaminobenzanilide (DABA), which introduces an amide motif. All three samples surpass a commercially available resin (diglycidyl ether-bisphenol A, DGEBA) in terms of mechanical properties. Notably, IDV-EP-DABA showed a tensile strength of 90 MPa and toughness of 9.73 MJ m−3, which are 1.8 and 6.7 times higher than DGEBA/DDM (Fig. 17b). Moreover, the reported epoxy materials showed complete degradation when exposed to a mixture of 0.1 M HCl and THF for 24 h at room temperature, opening a potential route to reprocessing.
 | ||
| Fig. 17 (a) Three different curing agents used for IDV-EP. (b) Sacrificial HBs between alcohol, amide, urea, and imine functionalities in IDV-EP polymers. Reprinted from ref. 79 with permission from Elsevier, copyright 2024. | ||
Ureas offer many of the same benefits of amides for sacrificial bond formation due to their HB-accepting carbonyl oxygen atom and the presence of two HB-donating N–H bonds, compared to the single HB donor in a traditional amide. Consequently, ureas and related structures have been utilized extensively in tough materials. Zhang and co-workers recently reported a poly(urethane-urea) based elastomer with HB-rich macrocyclic 2D polyamide (2DPA).80 2DPA was synthesized from melamine and 1,3,5-benzenetricarbonyl trichloride and was subsequently integrated into a soft matrix of poly(urethane-urea) (Fig. 18a and b). Varying the proportion of 2DPA (1, 2, 3, or 4 wt%) gave elastomers with different numbers of sacrificial HB cross-links between the urea and urethane functional groups of poly(urethane-urea) and the amide groups of 2DPA. The pristine polymer network was soft and weak with a tensile strength of 1.1 MPa, toughness of 5.8 MJ m−3, and 641% elongation at break. All samples with 2DPA outperformed the pristine sample; the sample with 3 wt% 2DPA (PITAT@2DPA3) showed the highest tensile strength (54.6 MPa) and toughness (116.7 MJ m−3) with an excellent elongation at break (705%). Additionally, the reported elastomer can lift a mass of 10 kg, 40000 times its own weight. In cyclic tensile tests, the sample with 3 wt% 2DPA showed increased energy dissipation with increased strain (strain increased from 50% to 400%, Fig. 18c). When the specimen was subjected to five consecutive cycles at a constant strain of 200%, all five cycles showed energy dissipation (denoted by the hysteresis loop area) with substantial loss after the first cycle but minimal loss thereafter (Fig. 18d). A recovery time of 3 h was sufficient to restore energy dissipation almost completely, to 95.6% of its original value (Fig. 18e).
 | ||
| Fig. 18 (a) Chemical structures of poly(urethane-urea) made from polytetrahydrofuran (PTMG), isophorone diisocyanate (IPDI), toluene-2,4-diisocyanate (TDI), and 2-(tert-butylamino)ethyl methacrylate (TBEMA). Different H-bonding sites are represented by different colors. (b) Chemical structure of 2D polyamide (2DPA). (c–e) Cyclic tensile test results for PITAT@2DPA3 with varying (c) strain, (d) cycle number at a constant strain of 200%, and (e) recovery time. Figure adapted from ref. 80. Copyright 2024 Wiley-VCH GmbH. (f) Chemical structure of poly(urethane-urea) with acylsemicarbazide unit (PU-ASC). Adapted with permission from ref. 81. Copyright 2022 American Chemical Society. (g) Chemical structure of PU with imidazolidinyl urea (IU), hexamethylene diisocyanate (HDI), and poly(caprolactone) (PCL). (h) H-bonding cross-links due to the presence of IU. Figure adapted from ref. 82 with permission from the Chinese Chemical Society (CCS), Institute of Chemistry of Chinese Academy of Sciences (IC), and the Royal Society of Chemistry, copyright 2021. | ||
Sun and coworkers made a similar poly(urethane-urea)-based elastomer containing poly(caprolactone) (PCL) segments (Fig. 18f).81 The reported elastomer has multiple cross-linking HBs between urethane and urea-containing acylsemicarbazide (ASC) groups, achieving a tensile strength of 72.6 MPa and 1440% of elongation at break with extremely high toughness of 375 MJ m−3. Wang and coworkers used a similar strategy with IPDI and PCL to synthesize an elastomer with HBs between the hard and soft segments.83 Their supramolecular poly(urethane-urea) showed a tensile strength of 64 MPa and 2160% of elongation at break along with robust energy dissipation properties.
The urea moiety has been derivatized to further expand the palette of amide-like HB motifs for sacrificial bonds. Zhang and co-workers employed imidazolidinyl urea (IU) as an HB motif in poly(urethane)s based on PCL and hexamethylene diisocyanate (Fig. 18g and h).82 The resulting polymer exhibited a toughness of 168.2 MJ m−3. Cyclic tensile strength results showed that HBs in IU dissipate energy during stretching. The mechanical properties could be further enhanced by adding cellulose nanocrystals as additional HB partners.84 Similarly high densities of HBs were achieved in thermoplastic PU elastomers by Cai and co-workers.85 Like typical urea, thioureas have also shown promise as sacrificial bond motifs due to their ability to engage in H-bonding between amide-like N–H bonds and thiocarbonyl sulfur atoms. Additionally, thioureas can form dynamic covalent bonds due to the reversible nature of the thiourea linkage. Kim and coworkers leveraged both of these binding modes in a biomimetic polymeric network using thioureas as a key sacrificial motif.42 The H-bonding capabilities of thiourea enhanced the polymer's mechanical properties (maximum strain = 360%, toughness = 71 MJ m−3) while its dynamic covalent character improved malleability and processibility.
4.3. Alcohols
Unlike ureidopyrimidinones and amides, in which oxygen atoms act solely as HB acceptors, alcohols include a polar O–H bond that serves as an HB donor. This labile hydrogen atom, in addition to the HB acceptor ability of the oxygen atom, makes alcohols privileged motifs capable of engaging in multiple HBs simultaneously. This ability is leveraged throughout natural materials, from the molecular recognition of sugars in the glycocalyx86 and the structural integrity of polysaccharides such as cellulose,87 to the strong adhesive properties of polyphenols such as catechols even in challenging aqueous environments.88 Unsurprisingly, materials scientists have sought to leverage the O–H group as a motif for sacrificial bonds in synthetic systems too.In 2020, Chen and coworkers reported hydrogels based on poly(vinyl alcohol) (PVA) and cellulose nanocrystals (CNC) with tannic acid (TA), a large polyphenol, as a H-bonding crosslinker (Fig. 19).89 Cyclic tensile tests showed the expected increase in the area of the hysteresis loop (i.e., dissipated energy) in the presence of HB-mediated crosslinking between TA and PVA/CNCs. Notably, a sample with 15 wt% of TA and 20 wt% of CNC (both with respect to PVA) exhibited increased amounts of energy dissipation with increasing strain levels (0.96 MJ m−3 at 100% strain and 11.89 MJ m−3 at 600% strain). When this sample was subjected to multiple loading–unloading cycles (at 200% strain) with no relaxation time, a significant decrease in the hysteresis area was observed, suggesting that the H-bonds and PVA crystalline domains were broken. Re-testing after 180 min of relaxation time caused the hysteresis loop to recover to almost 60% of its original size.
 | ||
| Fig. 19 (a) Fabrication process of TA–PVA–CNC hydrogels. (b) Sacrificial HBs in the hydrogel network. Figure adapted from ref. 89 with permission from the Royal Society of Chemistry, copyright 2020. | ||
In 2021, Duan and coworkers reported a strategy to make tough hydrogels using chitin and polyphenols.90 Their approach employed two separate non-covalent networks: a strong and rigid non-covalent network to achieve high mechanical performance and a weak non-covalent network for efficient energy dissipation. Chitin acted as the strong network because of the extensive H-bonding, hydrophobic, and ionic interactions between its polymer chains, while the weak and reversible interactions between polyphenols defined a second, weaker network. Of several polyphenols screened for hydrogelation with chitin (Fig. 20a), TA showed the highest compressive strength (8 times higher than that of chitin alone, Fig. 20b). The authors attribute this strength to the large number of weak chitin–TA interactions acting as sacrificial bonds under stress loading.
 | ||
| Fig. 20 (a) Different polyphenols used to make chitin–polyphenol. (b) Comparison of compressive strength of chitin and chitin–polyphenol hydrogels (CH: chitin, PG: pyrogallic acid, GA: gallic acid, TA: tannic acid, PCA: protocatechuic acid, and QC: quercetin). Figure adapted from ref. 90 with permission from the Royal Society of Chemistry, copyright 2021. | ||
In 2022, Zhou and coworkers reported an unusual strategy to build sacrificial bonds by incorporating a molecular solvent into a double network-based hydrogel (Fig. 21).91 The double-network structure, termed PAAN, comprised interpenetrated polyaniline and hydrophobic-associating poly(acrylic acid) (HAPAA). Extrinsic sacrificial bonds were introduced by incorporating glycerol into the PAAN gel using a solvent exchange method. The glycerol content in the resultant organohydrogels was varied by immersing PAAN samples in glycerol for different lengths of time. Increasing the immersion time enhanced the mechanical properties of the organohydrogels, which showed a fracture energy ∼4 times higher than that of human skin (∼3.6 kJ m−2) and ∼14 times that of cartilage (∼1 kJ m−2).92,93
 | ||
| Fig. 21 Chemical structure and preparation scheme for the PAAN/Gly organohydrogels with different types of sacrificial bonds involved in the hydrogel. Adapted with permission from ref. 91. Copyright 2022 American Chemical Society. | ||
Several recent reports have also examined alcohols as HB motifs for sacrificial bonds. Zhang and coworkers showed that molecules with multiple –OH groups, such as tannic acid, can act as H-bonding bridging agents between the –OH groups of natural bamboo fiber and the carbonyl groups of the synthetic polymer poly(butylene succinate-co-butylene adipate) (PBSA).94 The resulting biocomposite showed a 22% increase in tensile strength compared to the composite without tannic acid. Karak and coworkers reported a poly(ester-amide-urethane) network with different ratios of IPDI, leading to excellent elongation-at-break values (641% to 1876%) and tensile strengths (1125 to 1896 MJ m−3).95 Additionally, Leng and coworkers explored the role of H-bonding on the mechanical properties of cross-linked epoxy resins.96
5. Metal coordination
Metal–ligand interactions are formed when a Lewis base (ligand) donates electrons to a Lewis acidic (metal) center. Interestingly, the dynamic properties of resultant metal complexes can range anywhere between covalent and supramolecular systems depending on the specific metals and ligands used: in some cases, a ligand formally donates a pair of electrons to the metal center, making the resultant bond stronger than strictly97 non-covalent interactions such as H-bonding. Furthermore, multidentate ligands introduce the potential for multiple interactions between ligands and the same metal center, in both intramolecular and intermolecular fashions, providing additional options for tuning the overall strength of their interactions. Indeed, mussels’ unique ability to attach themselves to solid surfaces and avoid being swept off by intertidal waves is attributed to the polydentate coordination between iron and DOPA molecules in mussel cuticles.98 Moreover, metal–ligand systems often possess fast bond exchange kinetics that makes them more versatile than similarly strong covalent systems.16 These properties, coupled with the vast chemical space of metal–ligand systems, have spurred substantial exploration of metal coordination chemistry in the context of self-healing polymers,99–104 and make such bonds attractive candidates to serve as programmable sacrificial bonds.Inspired by the adhesive mechanism of mussels (vide supra),98,105 several groups have explored iron–catechol interactions as sacrificial bonds. Waite and coworkers examined the role of pH on iron–catechol interactions in PEG-based materials.106 Upon deprotonation of the acidic catechol protons, the resulting catecholate motifs formed bidentate coordinate bonds to the iron center that could then serve as sacrificial bonds to enhance the polymer's mechanical properties (Fig. 22a). The authors noted that a significant challenge in this process was that the alkaline pH necessary for bis- and tris-complex formation between Fe3+ ions and catechol made the metal ions insoluble, but this was overcome by slowly adjusting the system's pH. Under acidic conditions, Fe3+ ions were primarily coordinated by a single catechol motif (Fig. 22b, green). Increasing the pH favored additional catechol coordination, and at pH 12, complete conversion to the tris-coordinated complex was achieved, similar to that observed in mussel cuticles (Fig. 22b, red). The authors compared the mechanical properties of this modified polymer with a control polymer without catechol motifs or metal–ligand interactions. Although both samples showed similar hardness (comparable G′ values) at low strain, the iron–catechol-modified gel dissipated 10-fold more energy (G′′) compared to the control gel. This study validates the energy dissipating potential of metal–ligand bonds. Using similar coordination chemistry, Holten-Andersen and coworkers reported a strategy to decouple spatial and temporal hierarchy in a material.107 By establishing different metal-based coordination networks in the same polymeric matrix, the authors were able to design the hierarchy of sacrificial bond rupture. This allowed the authors to program the viscoelastic properties of the bulk polymeric material.
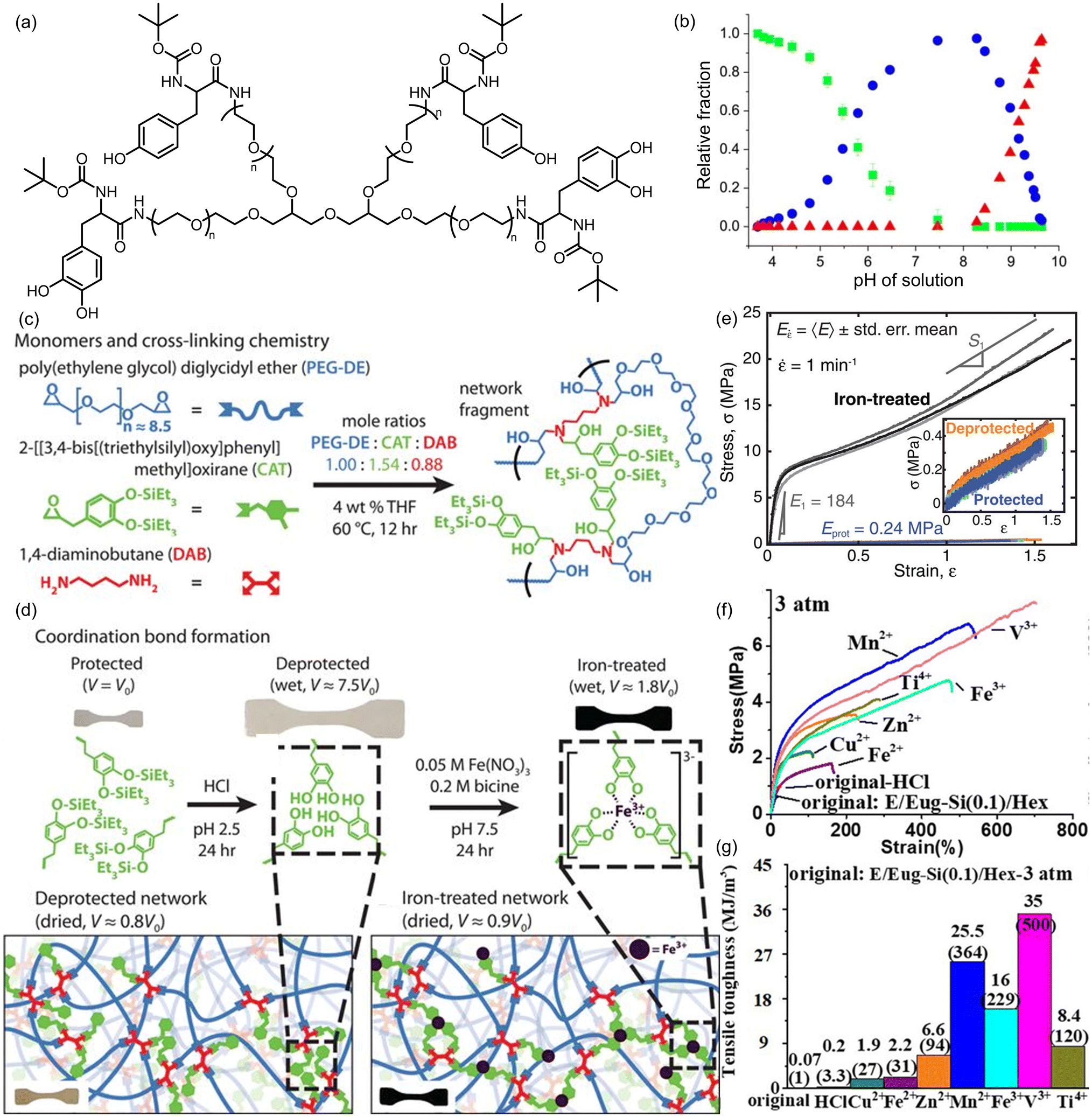 | ||
| Fig. 22 Metal–catechol coordination interactions as sacrificial bonds to improve mechanical properties. (a) DOPA-modified PEG units containing catechols for iron coordination. (b) Relative fractions of mono- (green), di- (blue) and tris- (red) coordinated Fe3+ in solution of PEG-dopa4 at different pH values. Figures adapted from ref. 106. (c) Synthesis of catechol-containing PEG polymers. (d) Scheme showing preparation of dry, iron-containing catechol-PEG networks. (e) Uniaxial tensile tests demonstrating the impact of metal coordination on mechanical properties of catechol–PEG polymers. Figures adapted from ref. 108. Reprinted with permission from AAAS. (f) Stress–strain curves showing different mechanical properties of eugenol-based polymers. (g) Comparing toughness of different polymers with different metals or HCl in eugenol-based polymers. Figures adapted from ref. 109. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Valentine and co-workers have noted that, in soft, wet systems such as hydrogels, high stiffness and high maximum elongation strain are often orthogonal and cannot be achieved simultaneously.108 To circumvent this problem, they designed a dry polymeric system in which sacrificial metal-coordination networks yield a material with both high stiffness and extensibility. The covalent polymer network was formed by copolymerizing a short-chain bisepoxide (poly(ethylene glycol) diglycidyl ether, PEG-DE), a monoepoxide carrying a silyl-protected catechol, and a diamine crosslinker (Fig. 22c). Subsequent deprotection of the silyl groups to reveal the catechol motifs, followed by chelation of Fe3+ ions and drying, produced the crosslinked metal-coordination network (Fig. 22d). To quantify the enhancement of mechanical properties upon the introduction of iron-catechol coordination bonds, uniaxial tensile tests were used to compare silyl-protected polymers (no Fe3+–OH interactions) with deprotected polymers (with free catechols capable of metal coordination): the latter were shown to exhibit a 770-fold increase in elastic modulus, a 58-fold increase in tensile strength, a 76-fold improvement in yield stress and a 92-fold increase in tensile toughness (Fig. 22e). These results provide a seminal example of metal–ligand coordination being successfully deployed in dry polymer materials.
The coordination of metal cations beyond Fe3+ was employed by Na and coworkers in their study of eugenol-based polyolefins.109 These polymers were prepared via copolymerization of a silyl-protected version of eugenol, a low cost and renewable allylic alkene, and various linear alkenes, followed by acid-catalyzed hydrolysis of the silyl ether protecting group to enable metal coordination. This increased coordination of Fe3+–catechol sacrificial bonds led not only to an 8-fold increase in tensile strength and 28-fold increase in strain-at-break, but also a remarkable 229-fold increase in tensile toughness when compared to the silyl-protected polymer with no metal-coordination bonds. The authors also found that increasing the chain length of the alkene component from ethene to 1-hexene led to increased flexibility and hence better sacrificial capacity of the iron–catechol coordination. The authors also investigated the effect of replacing iron with other metals. Mn2+ and V3+ were found to show even better property enhancement compared to Fe3+, with V3+ showing a 500-fold increase in tensile toughness compared to the coordination-free silyl-protected network (Fig. 22f and g).
Recent work by Chen and coworkers has further explored how the identity of the metal component in a metal–ligand sacrificial bond can be changed to tune the mechanical properties of polymers (Fig. 23).110 Specifically, polyesters containing pendent terpyridine (TPY; shown in blue) groups, which serve as high-affinity tridentate ligands (Fig. 23a), were doped with a series of divalent transition metals (Fe2+, Co2+, Ni2+, Cu2+, Zn2+) (Fig. 23b). Interestingly, the identity of the metal cation did not affect the modulus, strength, and toughness of the resultant materials equivalently. The authors found that the maximum enhancement in Young's modulus, observed for the Fe2+–polymer (3.3-fold compared to the undoped polymer), was consistent with the coordination strength of Fe2+ being the highest of the metals investigated. However, the enhancement in maximum elongation was higher for Ni2+ compared to Fe2+ (Fig. 23b), which the authors attribute to the inability of the strong Fe2+–TPY coordination to break before other interactions are (irreversibly) disrupted.
 | ||
| Fig. 23 Effect of different metal cations on sacrificial bond strength. (a) Structure of polyester that incorporates pendent tridentate terpyridine ligands (shown in blue). (b) Schematic depiction of the sacrificial nature of the metal coordination network and the relative effect of different metals on modulus, tensile strength and toughness. Adapted with permission from ref. 110. Copyright 2024 American Chemical Society. | ||
Metal-coordination sacrificial bonds have also been deployed to improve the mechanical properties of rubbers.111–113 Zhang and coworkers used the coordination of iron clusters by free hydroxy groups to reinforce rubber.114 Hydroxy groups were introduced into solution-polymerized styrene–butadiene rubber (SSBR) via thiol–ene click chemistry, after which the material was soaked in a FeCl3/THF solution to incorporate the metal ions. TEM studies on model compounds suggest that Fe3+ ions form clusters of iron-rich segments instead of a homogenous solution. Hence, the resultant material had sulfur crosslinks, hydroxy-based H-bonding and hydroxy-based coordination with Fe3+ ions in the iron cluster (Fig. 24a). The presence of Fe3+ ions led to a marked increase in mechanical properties, with an almost 7-fold increase in tensile strength and 9-fold increase in maximum elongation strain compared to unadulterated SSBR (Fig. 24b). Furthermore, metal coordination enhanced the extent of energy dissipation, as demonstrated by cyclic tensile loading experiments. While the unadulterated polymer and metal-free hydroxy-containing polymer show only 12% and 21% relaxation of the initial stress, respectively, Fe3+-doped polymers showed 71% relaxation of the initial stress. These enhancements were achieved with only 4.2 wt% iron, whereas achieving this strength in SBR modified by other methods would require a much higher content of fillers.115 Moreover, control experiments without metal ions showed limited improvement in tensile strength and modulus compared to unadulterated SBR, suggesting that the hydroxy-based H-bonding alone does not have a drastic effect on mechanical properties, thereby validating the efficacy of metal coordination as a sacrificial bond strategy in rubbers.
 | ||
| Fig. 24 Modification of rubbers using metal–ligand coordination interactions as sacrificial bonds. (a) Scheme showing the formation of cluster Fe3+–OH coordination bonds (red) and HBs (magenta) in solution-polymerized styrene–butadiene rubber (SSBR). (b) A stress–strain graph shows higher yield stress in the presence of metal coordination in iron cluster-based SSBR. Reprinted from ref. 114 with permission from Elsevier, copyright 2020. (c) Formation of SBR with dangling benzimidazole ligands (SBR-BET). (d) Scheme showing sacrificial nature of the Zn2+-imidazole coordination network under loading and unloading conditions. (e) Change in Young's modulus and tensile toughness under different concentrations of Zn2+ ions in SBR-BET. (f) Cyclic tensile loading tests show higher dissipation of energy in the presence of the Zn2+-imidazole coordination network in SBR-BET. Figure adapted from ref. 116 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Liu and coworkers reported Zn2+–imidazole coordination as an alternative sacrificial network in SBR.116 Their study utilized SBR crosslinked with sacrificial boronic ester-based dynamic covalent bonds to impart vitrimer-like properties to the rubber, rendering it melt-processible. However, vitrimers usually lack mechanical toughness, owing to their ability to melt at higher temperatures. To address this problem, 2-(2-benzimidazolyl)ethanethiol was incorporated during the thiol–ene click reaction to functionalize the SBR vitrimer with pendent benzimidazole motifs (Fig. 24c). Upon the introduction of Zn2+ ions, the benzimidazole motifs serve as ligands to form a sacrificial coordinate network (Fig. 24d), which not only provides added mechanical toughness to the mechanically weak vitrimer but also imparts creep resistance: the authors report a maximum 4.6-fold increase in tensile toughness and a 5.4-fold increase in Young's modulus, compared to the Zn2+-free polymer, without compromising the maximum elongation of the material (Fig. 24e). Cyclic tensile loading–unloading experiments showed an increase in hysteresis loop area, characteristic of greater energy dissipation, in the Zn2+-modified polymer versus the unmodified polymer (Fig. 24f). Other groups have employed similar strategies, using pyridine as an alternative ligand in modified SBR117 and Zn2+–carboxylate interactions to reinforce isoprene rubber.118 Notably, Weder and coworkers achieved an 81-fold improvement in toughness over neat SBR using pendent 2,6-bis(1′-methylbenzimidazolyl)-pyridine groups to coordinate Zn2+ ions.119 Recently, Shangguan and coworkers have demonstrated the mechanical toughening of SBR vulcanizates by incorporating sacrificial Zn2+–pyridine coordination linkages.120
As noted earlier, hydrogels are another class of materials with many desirable properties, such as shape retention and ease of formation, that often have weak mechanical properties, in this case due to their high content of liquid water. Employing metal-coordination sacrificial bonds in hydrogels requires additional considerations due to the ability of water to bind competitively to metal ions, typically necessitating multidentate or cooperative ligands to successfully make mechanically tough hydrogels.121–124 Recently, Li and coworkers have reported a polyacrylamide hydrogel with isotropically-distributed deacetylated chitin nanowhiskers (ChNWs).125 The monomers within these ChNWs are comprised of pyranose rings decorated with free amine and hydroxyl groups that can form a H-bonding network or, upon introduction of Zn2+ ions, form a multidentate metal coordination network (Fig. 25a). The metal-doped material can be stretched up to 13 times its original length, and can also be bent, twisted, and even knotted to give different complex shapes (Fig. 25b). Furthermore, increasing the concentration of the ChNWs and Zn2+ ions led to increasing tensile toughness, compared to unadulterated polyacrylamide gels or chitin-modified gels without Zn2+ (Fig. 25c). These results validate that the energy dissipation behavior can be attributed primarily to the rupture of the sacrificial metal coordination network. The authors also noted that, at higher stress, the relative viscous character of the metal-coordinated material was higher than the unadulterated polyacrylamide gels, due to the more fluid-like behavior caused by sacrificial bond breaking.
 | ||
| Fig. 25 Mechanical properties of hydrogels enhanced by metal coordination sacrificial bonds. (a) Scheme showing polyacrylamide gels with chitin nanowhiskers and Zn2+ ions (PAAm-ChNW-Zn2+). (b) Different shapes of PAAm-ChNW-Zn2+ achieved by twisting and bending. (c) Tensile and compressive stress versus strain curves of PAAm-ChNW-Zn2+ gels show that addition of Zn2+ increases both tensile strength and toughness. Reprinted from ref. 125 with permission from Elsevier, copyright 2020. (d) Scheme showing the formation of a PAA matrix with cellulose nanofibrils and Fe3+ ions (PAA-CNF-Fe3+). (e) Uniaxial tensile tests reveal enhancement of mechanical properties upon addition of CNF and Fe3+ ions. (f) Tensile toughness varies with [Fe3+] in PAA-CNF-Fe3+. (g) PAA-CNF-Fe3+ gels can be (from top) stretched, knotted, twisted and compressed. Adapted with permission from ref. 126. Copyright 2017 American Chemical Society. | ||
Shao and coworkers enhanced the mechanical properties of a PAA-based hydrogel using modified cellulose nanofibrils (CNFs).126 Prior to their incorporation into the gel, the CNFs were oxidized using TEMPO, which converted the 5′-hydroxyl group on each pyranose ring to a carboxylate group. This carboxylate group, together with the residual acid groups from the PAA backbone, served as ligands when Fe3+ ions were introduced, forming a sacrificial coordination network (Fig. 25d). Unlike pristine PAA gels, which showed low tensile strength (0.035 MPa) and low maximum elongation (320%), PAA-Fe3+ gels with no CNFs show enhanced tensile strength (20-fold increase) and elongation ratio (800%). This was further improved by the introduction of CNFs: PAA-CNF-Fe3+ showed a tensile strength of 1.37 MPa (∼40-fold improvement over pristine PAA) and an elongation ratio of 1803% (Fig. 25e). This proved that the enhancement in toughness of the material could be primarily attributed to the synergistic dual sacrificial metal coordination network formed between the metal ion and both PAA and CNF matrices. Increasing [Fe3+] led to an increase in tensile toughness, likely due to a greater number of sacrificial metal coordination interactions. However, the authors note that material toughness and maximum elongation reach a maximum at 1 mol% Fe3+ (Fig. 25f), allowing the material to be knotted, twisted and stretched (Fig. 25g).
Hussain and coworkers used a similar strategy to make mechanically tough hydrogels by incorporating the natural polymer glycogen into a synthetic polymeric matrix composed of PVA and PAA.127 Adding Fe3+ ions established a coordination network in which the hydroxyl groups of PVA and glycogen and the carboxylic acid motifs of PAA acted as ligands. Varying the concentrations of glycogen and PVA allowed the tensile strength, maximum elongation ratio, and tensile toughness of the material to be tuned, with a maximum toughness of 0.65 MJ m−3. Recently Zhou and coworkers have developed a dual network hydrogel that incorporates HB interactions in addition to ionic interactions between EDTA ligands and amine groups on the amino-functionalized polysiloxane backbone.128 Tuning the relative amount of each motif enabled precise control over the hydrogel's tensile strength (0.07 to 1.46 MPa) and tensile toughness (0.06 to 2.17 MJ m−3).
Silicon-based elastomers are becoming increasing important due to their applications in electronic materials. However, there is often a trade-off between the heat-dissipating characteristics and mechanical properties of these materials. To address this drawback, metal-coordination bonds have also been employed as sacrificial bonds to improve mechanical toughness of silicon-based elastomers.129–132 Bao and coworkers innovated this approach by positioning weakly- and strongly-coordinating binding sites next to each other in a single multidentate ligand incorporated within a polydimethylsiloxane (PDMS) elastomer.133 To achieve this, 2,6-pyridinedicarboxamide (pdca) was selected as a tridentate Fe3+-binding ligand, in which the pyridine N atom coordinates most strongly and the O and N atoms of the amide form weaker, secondary coordination bonds (Fig. 26a). Under stress, the authors hypothesized that the weaker coordination bonds would break to dissipate energy, but the metal ions would remain immobilized due to the stronger, Fe–Npyridyl bonds, enabling rapid reformation of the weak coordination linkages, when the stress is removed. This design strategy proved successful: the pdca-modified PDMS achieved a maximum elongation of up to 10000% (Fig. 26b). This result compares favorably to unadulterated PDMS, which has a maximum elongation of up to 1100%, and PDMS strengthened using other supramolecular approaches, which produced a maximum elongation of 2000%.134 The authors attributed the enhanced mechanical properties to the ability of intrachain linkages to form, leading to the release of chain length upon fission of the weak metal-coordination bonds. Zhang and coworkers also used a similar strategy to strengthen PDMS in which Fe-pdca complexes act as weak coordination bonds which break to dissipate energy under stress, and stronger interactions between Fe3+ ions and carboxylate groups on N-acetyl-L-cysteine (Fe-NAC) held the metal ions in place.135 This also led to enhanced tensile toughness of 0.866 MJ m−3, ∼3-fold higher than controls without either ligand (Fig. 26c).
 | ||
| Fig. 26 Modification of silicone-based elastomers using metal–ligand interactions as sacrificial bonds. (a) Scheme showing reversible formation and breaking of coordination bonds with various strengths in pdca-doped PDMS-based elastomers under application of force. (b) PDMS-pdca-Fe3+ can be stretched up to 10000%. Figure adapted from ref. 133 with permission from Springer Nature, copyright 2016. (c) Stress–strain graph shows that Fe/NAC-pdca-PDMS has considerably higher toughness than coordination-free controls. Figure adapted from ref. 135 with permission from the Royal Society of Chemistry, copyright 2020. | ||
6. Electrostatic interactions
Electrostatic interactions arise from the coulombic forces of attraction between species that have formal opposite charge. Though such attraction contributes in part to the strength of metal–ligand coordination bonds (see previous section), electrostatic interactions are far more universal and can be observed in a wide range of structural motifs. The strength of these interactions depends solely on the formal charge of interacting species and the distance between them. Consequently, these interactions have unique characteristics distinct from H-bonding and metal coordination, such as their non-directional nature and the ability to form multiple interactions with the same charged center.Wang and coworkers recently reported an SPM based on an ion-dipole network generated by photopolymerizing 2,2,3,4,4,4-hexafluorobutyl acrylate (HFBA) monomers and introducing lithium bis(trifluoromethane)sulfonamide.136 Li+ ions interacted with the polar carbonyl bond on the acrylate species to crosslink the polymer chains (Fig. 27a). The addition of these ion–dipole interactions led to increased tensile strength and tensile toughness, with maxima of 1.32 MPa and 6.67 MJ m−3, respectively, at 1.5 M Li+, which are 14.8- and 4.8-fold higher than the corresponding values for pure pHFBA (Fig. 27b). Cyclic loading–unloading experiments showed excellent energy dissipation (Fig. 27c). The authors also noted that the high density of ionic crosslinks restricts the dynamic motion of the polymeric segments, decreasing the entropic gain upon heating and maintaining these dynamic interactions at elevated temperatures. These materials have showed excellent stretchability of over 600%, even after being cut and allowed to self-heal (Fig. 27d).
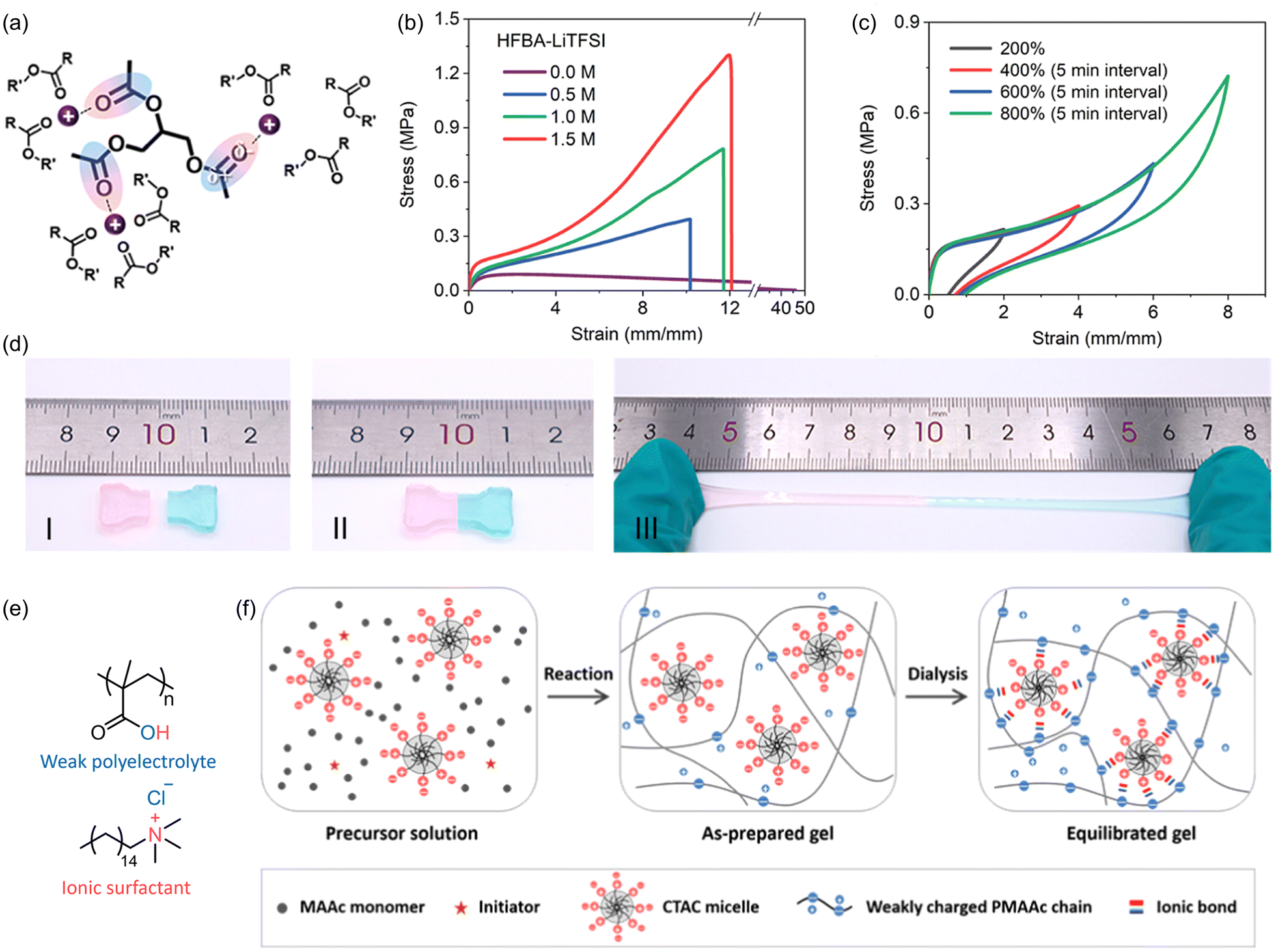 | ||
| Fig. 27 Ion-dipole electrostatic interactions in polymers and hydrogels. (a) Schematic representation of ion–dipole interactions between Li+ ions and carbonyl motifs. (b) Effect of increasing [Li+] on the mechanical properties of pHFBA hybrid polymers. (c) Cyclic tensile loading experiments on pHFBA hybrid polymers reveal large hysteresis loops under different applied strains. (d) pHFBA polymeric systems show excellent stretchability even after self-healing. Adapted with permission from ref. 136. Copyright 2023 American Chemical Society. (e) Structure of polyelectrolyte and zwitterionic surfactant in methacrylic acid-based plastic-like supramolecular hydrogels. (f) Scheme showing preparation of methacrylate-based hydrogels incorporating polyelectrolyte surfactant complexes (PESCs). Adapted with permission from ref. 137. Copyright 2021 American Chemical Society. | ||
Wu and coworkers have reported plastic-like supramolecular hydrogels formed by the polymerization of methacrylic acid in the presence of hexadecyltrimethylammonium chloride (CTAC) (Fig. 27e).137,138 Hydration of the polymeric matrix induces strong electrostatic interactions between the weak polyelectrolyte and zwitterionic surfactant. These polyelectrolyte-surfactant complexes (PESCs) serve as crosslinks between polymer chains and, in conjunction with the hydrophobic effect induced by the surfactants, act as dynamic sacrificial interactions that enhance the hydrogel's mechanical properties (Fig. 27f). For example, while the pure polyacrylamide gel is too weak to perform any tensile tests, increasing [CTAC] to 10 mol% leads to a tensile strength of 900 kPa. Interestingly, the authors also report that the extent of ionization of the polyelectrolyte can be affected by temperature, pH, and salt concentration providing additional handles with which to tune the mechanical properties of these materials.
Ionic liquid-based polymers form an important class of polymeric gels as they have highly tunable ionic conductivity, leading to wide applications in electronics and robotics.139 However, their practical applications demand high stretchability and robustness which are usually difficult to achieve in gel-like materials. To circumvent this problem, Zhang and coworkers developed a fluorinated polymeric matrix using HFBA and oligoethylene glycol monomers (OEGA) with various amounts of an embedded ionic liquid (IL), 1-butyl-3-methylimidazolium-bis((trifluoromethylsulfonyl)imide) ([BMIM][TFSI], Fig. 28a).140 Sacrificial ion–dipole crosslinks between the imidazolium cations of the ionogel and the carbonyl groups of the acrylate component led to very high tensile strength and stretchability, reaching values of 251 kPa and 6000%, respectively, for the polymer containing 12.5 mol% IL. Furthermore, this material maintained its excellent stretchability even after being cut and allowed to self-heal (Fig. 28b). Other groups have explored the effect of incorporating H-bonding alongside ion–dipole interactions on the mechanical toughness and stretchability of ionogels.141,142
 | ||
| Fig. 28 Electrostatic interactions in ionogels lead to enhanced mechanical properties. (a) Schematic depiction of ion–dipole interactions inside a polyethylene glycol-based matrix with embedded ionic liquid. (b) This material can be cut and self-healed and retains its high stretchability (scale in cm). Adapted with permission from ref. 140. Copyright 2021 American Chemical Society. | ||
7. The hydrophobic effect
The hydrophobic effect describes the tendency of hydrophobic components to associate in aqueous solutions to minimize unfavorable enthalpic interactions with water of solvation and maximize the entropy of bulk water. This effect is a significant contributor to the formation and stability of tertiary and quaternary structures of large biomacromolecules such as proteins. In synthetic materials, the hydrophobic effect has been explored thoroughly in the context of water-soluble polymers, where hydrophilic components impart solubility whereas hydrophobic components induce polymer chain assembly.143 Subsequent studies have explored the disruption of hydrophobic domains as a mechanism to dissipate energy,144 due to their non-directional nature, which allows hydrophobic groups to re-associate quickly after force-induced dissipation. Moreover, the hydrophobic effect is compatible with a broad range of functional groups, spurring their investigation in materials alongside other non-covalent interactions to optimize mechanical properties.137,145Traditional methods to synthesize hydrogels that incorporate hydrophobic domains have been challenging due to the insolubility of the hydrophobic components in aqueous media.146 These solubility issues not only frustrate materials synthesis but can also lead to a loss of structural control at the macroscopic and microscopic scales. Candau and coworkers overcame this synthetic bottleneck using a simple, robust synthesis based on a micellar copolymerization technique.144 In this, the hydrophobic monomer is solubilized using micelles (formed from surfactants such as sodium dodecyl sulfate, SDS) in an aqueous environment.144 Then, via a free radical mechanism, micellar hydrophobic comonomer and solubilized hydrophilic comonomer copolymerize to form the hydrogel. Due to the high local concentration of hydrophobes within the micelles, the resultant material contains blocks of randomly distributed hydrophobic components.147 This method has been widely employed to incorporate hydrophobic associations as sacrificial motifs in materials.
Okay and coworkers were among the first to demonstrate that increasing hydrophobic associations, by incorporation of hydrophobic segments in a synthetic hydrogel via a copolymerization technique, led to drastically improved mechanical properties of the resultant material.148 Hydrogels were constructed from acrylamide monomers and N,N′-methylenebis(acrylamide), which served as an interchain crosslinker. Modified acrylamides containing alkyl chains including N-butyl, N-hexyl and N-octyl were also incorporated, to act as the hydrophobic segment. While pure PAAm gels were too weak to conduct any mechanical experiments, increasing the amount of hydrophobic N-alkylacrylamide components led to increasing toughness. Indeed, PAAm hydrogels modified with 10 mol% of N-octylacrylamide achieved 313% elongation at a stress of 71 kPa (Fig. 29a). Furthermore, these gels could also withstand knotting and extreme deformation. The authors also addressed the role of the micellar polymerization technique in generating favorable materials properties. Comparative studies showed that the creation of blocks of hydrophobic segments via the micellar copolymerization technique using SDS yielded enhanced toughness over randomly distributed hydrophobic units. However, the authors hypothesized that the presence of the SDS surfactant in the polymeric matrix might affect energy dissipation by increasing the solubility of the hydrophobic components. In a subsequent study,149 the authors found that the removal of surfactant led to the loss of the dynamic mechanical properties associated with reversible hydrophobic effects (Fig. 29b). This is because SDS solubilizes the hydrophobic blocks, leading to enhanced stability of the hydrophobic groups, but the removal of the surfactant forces the hydrophobic groups to form much stronger associations to avoid any interaction with the aqueous environment. Hence the presence of surfactants produces weaker gels with higher G′′ values but leads to better dynamic properties. For example, hydrogels with an elongation ratio of almost 1700% in the presence of SDS achieved only a 700% elongation ratio in the absence of SDS (Fig. 29b).
 | ||
| Fig. 29 Hydrophobic dynamic interactions. (a) Stress–strain curves of hydrogels containing 10 mol% of various alkyl chains such as N-octyl (8), N-hexyl (6) and N,N-dihexyl (6/6). Reprinted from ref. 148 with permission from Elsevier, copyright 2009. (b) Without SDS, higher tensile strength is obtained but with lower elongation ratio. Adapted with permission from ref. 149. Copyright 2012 American Chemical Society. (c) Scheme showing the formation of hydrophobic interactions in water without surfactants by using natural silk fibroin and sodium alginate. (d) Strain-at-break values are much higher in the presence of hydrophobic monomers. (e) Shear rheology experiments show higher G′ and G′′ values in the presence of the hydrophobic component (C18M). Reprinted with permission from ref. 150. Copyright 2020 American Chemical Society. | ||
However, the dependence of this category of hydrogels on surfactants, which are often cytotoxic, renders them unsuitable for applications in biological systems. Hence, Meng and coworkers recently targeted the formation of hydrogels via hydrophobic associations in the absence of any synthetic surfactants.150 Alginate and stearyl methacrylate (C18M) were selected as the hydrophilic and hydrophobic comonomers, respectively. Instead of using a surfactant, amphiphilic regenerated silk fibroin (RSF) allowed micelle formation by solubilizing the hydrophobic C18M via RSF's conserved hydrophobic sequences. Additionally, Ca2+ ions were added to induce hydrophilic ionic interactions between the metal cation and the negatively charged carboxylate ion on the alginate polymer chain (Fig. 29c). This hybrid material exhibited increasing toughness as the concentration of the hydrophobic component (C18M) was increased. Compared to an alginate-RSF gel without C18M, the stress at 50% strain increased more than 200% when [C18M] was increased to 30 mM. Additionally, the yield strength of the material was significantly increased when C18M was incorporated (Fig. 29d). Moreover, strain sweep measurements revealed that the intersection of G′ and G′′ occurred at 109% strain for the hybrid material ([C18M] = 30 mM), which was much higher than for pure alginate gels (1.59%) or alginate-RSF gels without C18M (10%) (Fig. 29e). These results clearly indicated that the enhancement in mechanical properties was primarily due to hydrophobic associations acting as sacrificial bonds and not the ionic interactions present in the material. Importantly, this study highlights a path forward to leverage the hydrophobic effect in biocompatible energy-dissipating materials.
8. Host–guest complexation
While previous sections have focused on individual types of non-covalent interaction, the complexation of a suitable guest by a macrocyclic host is driven by multiple effects that may include electrostatic interactions, H-bonding, the hydrophobic effect, and others.151 Unlike these individual interaction modes, (macrocyclic) host–guest interactions can result in a precise co-location of two components (i.e., host and guest) in 3-D space, often with a preferred relative orientation. Furthermore, both the host and guest can be independently tailored to tune the thermodynamic and kinetic favorability of association. Consequently, host–guest interactions have been explored extensively for the generation of supramolecular polymer networks, in which both polymer backbones and the crosslinks between them are scaffolded by non-covalent interactions.151 Furthermore, the supramolecular nature of these interactions has also been exploited as sacrificial bonds which has led to enhanced mechanical properties of the bulk polymer.Among supramolecular hosts, cyclodextrins (CDs) have garnered particular interest due to their water solubility, synthetic accessibility, and their interior cavity that is capable of binding various hydrophobic guests including azobenzenes and adamantanes.152 Yang and coworkers leveraged the selective host–guest recognition of adamantane by β-cyclodextrin (β-CD) to modify the mechanical properties of PAAm hydrogels.153 To synthesize these materials, a host–guest macromer (HGM) was prepared from an adamantane-functionalized hyaluronic acid and mono-acrylated β-CD, and subsequently copolymerized with acrylamide using a methylene bisacrylamide (MBA) cross-linker to synthesize MBA-HGM polymers (Fig. 30a). Control samples without HGM, crosslinked solely using different concentrations of MBA, were also prepared (denoted MBA-L and MBA-H for relatively low and high concentrations, respectively). Dynamic oscillatory experiments established that increasing the concentration of MBA in the HGM-free polymers increased the storage modulus G′ but did not change the loss modulus G′′, indicating that the material became stronger but also more brittle. Conversely, the MBA-HGM polymer exhibited an increase in both G′ and G′′, suggesting that the addition of host–guest sacrificial bonds endowed the material with overall higher tensile toughness. Uniaxial tensile tests showed that, compared to MBA-H, MBA-HGM had much higher stretchability (750% vs. 300%) and higher fracture stress (an additional 100% compared to controls, Fig. 30b). The ability of MBA-HGM to dissipate energy was confirmed through cyclic loading–unloading studies, which showed that MBA-HGM polymers dissipated 22.8% of the loading energy, compared to negligible energy dissipation by the control materials, as evidenced by negligible area inside the hysteresis loop.
 | ||
| Fig. 30 (a) Scheme showing the preparation of MBA-HGM polyacrylamide gels. (b) Uniaxial tensile tests showing superior performance of MBA-HGM polyacrylamide gels showing much higher overall toughness. Figure adapted from ref. 153. (c) Compression tests comparing host guest-based silk fibroin (HG-SF) and unadulterated silk fibroin (SF). Reprinted with permission from ref. 154. Copyright 2020 American Chemical Society. (d) Scheme showing energy dissipation mechanism as well as ultra-stretchability of slide-ring (SR) gels. (e) Tensile tests showing the superior performance of SR-gels (SR-x, where x denotes the volume fraction of PEG) compared to tetra-PEG (tetra-x) control gels. Figure adapted from ref. 155. Reprinted with permission from AAAS. | ||
Bai and coworkers used a similar strategy to enhance the mechanical properties of silk fibroin (SF) based hydrogels.154 To chemically modify the SF polymers, carboxylic acid functional groups were introduced to serine and tyrosine residues by oxidation and diazo linkages, respectively. These modified SFs were then coupled with either cholesterol or β-CD to give Chol-SF and CD-SF respectively. Mixing these two polymers allowed the hydrophobic cholesterol moieties to be encapsulated by β-CD, forming hydrogels (HG-SF). Tensile tests revealed a dramatic increase in both fracture stress (3.16 MPa vs. 0.1 MPa) and maximum elongation (65% vs. 31%) compared to a SF-only control. The combined effect of these changes resulted in a 65-fold enhancement in the tensile toughness of HG-SF vs. its counterpart without sacrificial host–guest interactions. This was also demonstrated by a compression test (Fig. 30c), in which the control polymer disintegrated while HG-SF recovered its shape.
Ito and coworkers used an innovative host–guest strategy that utilizes poly(rotaxane)s to form slide-ring (SR) hydrogels (Fig. 30d).155 Specifically, PEG chains were first incubated with hydroxy-propyl-α-cyclodextrin (HP-α-CD), to form rotaxane-like assemblies in which PEG interpenetrated the CDs. Subsequent cross-linking of the CDs with divinyl sulfone led to hydrogels in which the CDs retained their mobility along the PEG chains. Under tensile stress, CD molecules were hypothesized to slide toward each other, leading to extended lengths of PEG chains between increasingly congested cross-linking points. Under large stress, these PEG regions were expected to become highly ordered and form a close packed structure via strain-induced crystallization, leading to further increased mechanical toughness. SR hydrogels with different volume fractions (x) of PEG (denoted SR-x) were compared to control polymers containing tetra-arm PEG as an immobile cross-linker. Uniaxial tensile tests demonstrated that SR-gels had much higher tensile toughness than their slide-free counterparts, with SR-0.38 showing a 40-fold improvement over the best performing tetra-PEG variant. The authors also noted that the toughness of the gels is reduced by using PEG chains of lower molecular weight, perhaps due to the smaller sliding distance of the cross-links as well as more defects in the PEG network. The ability of polymer chains to interpenetrate CDs was also observed by Takashima and coworkers, who unexpectedly observed favorable toughness in copolymers of ethyl acrylate and a γ-CD methylacrylamide monomer (PAcγCDAAmMe).156 These copolymers exhibited a comparable fracture energy to nitrile rubber but with much higher Young's modulus.
Cucurbit[n]urils (CB[n]) are another classic barrel-like host used for molecular recognition.157 Scherman and coworkers recently employed CB[8] to improve the mechanical properties of PAAm gels.158 PAAm gels were prepared by copolymerizing 1-benzyl-3-vinylimidazolium ions with hydrophilic acrylamide monomers using MBA as crosslinker. Subsequent addition of CB[8] led to complexation-driven crosslinking of the gel network, in which the CB[8] motifs formed reversible 1:2 host–guest complexes with the pendent benzylimidazolyl chains on the polymer backbone (Fig. 31a). This hydrogel exhibited phenomenal mechanical properties, with a tensile toughness (750 MJ m−3) comparable to cartilage, and was able to support 500 times its own weight without material failure. Control gels with either no CB[8] motifs or with only smaller CB[7] motifs, incapable of binding two benzylimidazolyl groups simultaneously, led to the loss of mechanical strength, demonstrating the essential and specific nature of the host–guest linkages. Uniaxial tensile tests showed that the CB[8]-containing material was highly stretchable, achieving a maximum elongation of 2400%. The authors also note that, above a strain rate of 100 mm min−1, there was a significant decrease in the overall toughness of the material. They hypothesize that this could arise from the inability of the dynamic host–guest components to effectively rearrange on small timescales, which could lead to decreased energy dissipation (Fig. 31b). Other work from the same group has focused on exploring the effect of changing the size of the guest motif.159 The Scherman group has also explored CB[n]-based single network hydrogels without the presence of covalent cross-linking160 as well as hybrid double-network hydrogels with both CB[n]-based and DNA-mediated cross-linking.161
 | ||
| Fig. 31 (a) Scheme showing cucurbit[8]uril-based gels and their energy dissipation mechanism under loading and unloading (blue rod is guest; cylinder is host). (b) Tensile tests at different strain rates show lowered toughness at higher strain rates. Figure adapted from ref. 158. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Crown ethers have also found widespread utility as supramolecular hosts for sacrificial host–guest interactions. Yan and coworkers exploited the molecular recognition of a dialkylammonium salt by benzo-21-crown-7 (B21C7) molecules to prepare supramolecular polymeric networks (SPNs).162 Specifically, a covalent polymer network was synthesized by ring-opening metathesis polymerization (ROMP) of B21C7-functionalized norbornene monomers. This was then mixed with different molar ratios of a dialkylammonium crosslinker (DAAS) to form different SPNs (SPN-x, where the molar ratio of dialkylammonium salt to B21C7 is x:10; Fig. 32a). The charged ammonium species interacted with the crown ether oxygens to form a reversible non-covalent interaction which could rupture to dissipate energy under mechanical stress and reform when the stress had been removed. Tensile tests established that increasing the amount of the dialkylammonium crosslinker increased both the Young's modulus and the tensile toughness of the hybrid gels. SPN-5 showed the best mechanical properties, with maximum toughness (22 MJ m−3), fracture stress (5.1 MPa) and elongation-at-break (648%) (Fig. 32b and c). Subsequent work from the same group compared SPNs to covalent polymeric networks (CPNs) and dual polymer networks (DPNs) that have both supramolecular as well as covalent linkages (Fig. 32d).163 Tensile tests on the three materials revealed that SPN and DPN exhibited 10.3- and 9.1-fold improvements, respectively, of tensile toughness over CPN, further emphasizing the importance of sacrificial non-covalent host–guest interactions. Furthermore, DPNs also exhibited almost a 100% increase in tensile strength over SPNs, showing that a dual network system comprising both covalent and supramolecular networks can exhibit both high strength and toughness (Fig. 32e).
 | ||
| Fig. 32 (a) Synthesis of supramolecular networks (SPNs) composed of B21C7-norbornene-based hosts and dialkylammonium guests. (b) Uniaxial tensile tests of SPN-x (molar ratio of guest:host = x:10) shows increasing toughness and yield strength with increasing guest concentration. (c) Comparative studies show increasing Young's modulus and tensile toughness with an increasing concentration of host–guest interactions. Figure adapted from ref. 162. (d) Dually cross-linked polymers with both supramolecular and covalent cross-links. Three polymers (SPN, CPN and DPN) were constructed with different compositions of supramolecular and covalent networks. (e) Tensile tests comparing the mechanical performance of SPN, CPN and DPN show DPN having higher overall toughness. Figure adapted from ref. 163. Copyright 2023 Wiley-VCH GmbH. (f) PPS1 can be extended to a length of more than 1800% without fracturing. (g) Scheme showing the design of zipper gels with monomers PS1 and PS2 as well as controls R1, R2, A1 and A2. (h) Mechanical performance of PPS1 gels with different concentrations of PS1. Figure adapted from ref. 164. Copyright 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Host–guest supramolecular networks have also been used in conjunction with other types of sacrificial bond to further improve energy dissipation. Qu and coworkers combined the host–guest complexation of a dibenzylammonium motif by a dibenzo-crown ether with amide-mediated H-bonding to make mechanically robust polymers that undergo zipper-like ring sliding.164 Central to their design was the incorporation of pseudorotaxane monomers (PS1), comprising a dibenzo-24-crown-8 motif and a dibenzylammonium-based axle, into PU networks that are inherently decorated with extensive HBs. When tensile force is applied across the resultant material (PPS1), the host–guest interaction breaks, leading to ring sliding of the benzo-crown ether along the PU backbone, disrupting the H-bonding between linear PU chains (Fig. 32g). This phenomenon, termed ‘molecular zippering’, dissipates a large amount of energy. Tensile tests showed that PPS1 could be stretched by 1890% without fracture (Fig. 32f) and had higher tensile strength and fracture energy than pristine PU (8.5- and 45-fold improvements, respectively). Upon decreasing the crosslinking density (PPS2), the tensile strength, stretchability and toughness all fall significantly, further demonstrating the profound effect of the host–guest interactions. The authors also showed that a low concentration of PS1 (0.5 mol%) was optimal for mechanical properties and noted that this value was one of the lowest compared to other ring-sliding crosslinkers. Increasing the concentration of PS1 beyond this value led to decreased tensile toughness (Fig. 32h). This work, together with the PEG-CD slide-ring gels by Ito and coworkers (vide supra),155 highlights the potential for the mechanical bond165 to act as a distinct sacrificial bond motif.166
9. Covalent bonds
A key feature of a sacrificial bond is its ability to re-form after mechanical force or applied load is removed. For these reasons, non-covalent interactions, as discussed in the previous sections, are natural choices for sacrificial bonds due to their inherent reversibility. Covalent bonds, in comparison, initially seem unsuitable for reversible energy dissipation. However, two major classes of covalently-bonded motifs that can spontaneously recover after fission are known: dynamic covalent bonds167 and mechanophores.168,1699.1. Dynamic covalent bonds
As opposed to traditional, stable covalent bonds, dynamic covalent bonds readily break and subsequently reform.170 A recent review of the rich chemistry of dynamic covalent bonds167 emphasizes the sheer number of different reaction modes that lead to dynamic bonding, including Diels–Alder cyclization,171 transetherification,172 transamination,173 photodimerization,174 boronic acid exchange,175 transcarbamoylation,176 and imine metathesis,177 among others. Consequently, the kinetics of bond cleavage and formation can be finely tuned to achieve desirable bond lifetimes, leading to widespread study of dynamic covalent bonds in polymeric materials and even deployment in industrial processes.178 However, most polymer research that utilizes dynamic covalent chemistry has been geared towards the preparation of self-healing materials, which are often flexible and have poor strength and toughness.171The mechanism by which dynamic covalent bonds break and reform to achieve reversible molecular network rearrangements (MNR) can be classified as either concerted or stepwise. In a concerted mechanism, bond breaking and bond formation occur simultaneously (Fig. 33a), such that the number of covalent bonds remains constant at all times. In contrast, stepwise molecular rearrangement consists of distinct bond cleavage and bond formation steps (Fig. 33b). If the bond formation happens before the bond cleavage step, the total number of covalent bonds temporarily increases and the process is termed an associative stepwise MNR. Conversely, if the bond cleavage precedes the bond forming step, dissociative stepwise MNR occurs, with a momentary decrease in the total number of bonds.179 Given that energy dissipation requires that there is a reduction in the extent of bonding, i.e. bonds are broken, in response to external force, dynamic covalent bonds that exchange via a dissociative stepwise mechanism are desirable as sacrificial bonds.
 | ||
| Fig. 33 Mechanism of bond exchange in dynamic covalent bonds. (a) Concerted and (b) Stepwise molecular network rearrangements (MNRs). Stepwise MNRs can proceed via associative and dissociative pathways. Figure adapted from ref. 179 with permission from the Royal Society of Chemistry, copyright 2019. | ||
In 2018, Lv and coworkers reported one of the first examples of using dynamic covalent bonds as sacrificial bonds for energy dissipation.180 Amine-terminated PDMS was copolymerized with 1,3,5-triformylbenzene to generate a polymer network crosslinked with strong yet dynamic imine bonds (Fig. 34a). To introduce weaker dynamic covalent bonds, 2-aminophenyl disulfide (ADPS) was incorporated as an additional crosslinker during polymerization. Crucially, the disulfide linkages in ADPS were hypothesized to serve as sacrificial dynamic bonds which would cleave to dissipate energy. The authors reported that increasing the ADPS concentration enhanced the material's stretchability, up to a maximum elongation of 4300%. A control sample in which ADPS was replaced with an alternative crosslinker that did not contain a disulfide gave a maximum elongation of only 580%, confirming that the dynamic aromatic disulfide linkages act as sacrificial bonds. Reducing the rate of elongation allowed the material to be stretched further but resulted in a reduction in yield strength, as the slower elongation rate provides more time for the dynamic covalent bonds to reorient and reorganize (Fig. 34b). Cyclic tensile loading experiments produced large hysteresis loops characteristic of energy dissipation; however, extended recovery times (48 h) were required to fully recover the material's dissipative capacity (Fig. 34c). Wang and coworkers used a similar strategy to design a PDMS-based material that utilized dynamic imine and boroxine bonds to dissipate energy.181 Boron–oxygen linkages have also been used to modulate the mechanical properties of alginate-based materials.182
 | ||
| Fig. 34 Siloxane-based elastomers containing dynamic covalent bonds. (a) Schematic representation of the cross-linked PDMS polymer containing imine and aromatic disulfide dynamic bonds. (b) With decreasing rate of elongation, the yield strength decreases and the maximum elongation increases. (c) Cyclic tensile tests reveal that a recovery time of 48 h is necessary to reproduce the same extent of energy dissipation. Figure adapted from ref. 180. Copyright 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (d) Schematic of PDETAS elastomer incorporating imine and cyclic aminal dynamic covalent bonds. Reprinted from ref. 183 with permission from Elsevier, copyright 2023. (e) PDMS elastomers showing highest mechanical properties when dynamic covalent linkages are present. (f) Schematic representation and (g) chemical structure of PDMS polymer incorporating three sacrificial bond motifs: H-bonding between urea motifs, disulfide bonds, and Diels–Alder-type dynamic covalent linkages. Figure adapted from ref. 184. Copyright 2024 Wiley Periodicals LLC. | ||
Deng and coworkers leveraged the different amine sites in diethylenetriamine-functionalized polysiloxanes (PDETAS) to prepare two types of sacrificial dynamic covalent bond in a single polymer network.183 Reaction of the crosslinker 1,4-diformylbenzene (DFB) with the primary amines on the PDETAS side-groups formed imine linkages whereas reaction with the secondary amines formed aminal linkages via a ring-closing mechanism (Fig. 34d). The imine and the aminal bonds not only underwent dynamic exchange with the free amines but could also metathesize (exchange with) each other. This highly dynamic behavior of the system led to efficient energy dissipation, leading to a tensile strength of ∼25 MPa, much higher than previously reported silicone materials. Increasing the concentration of DFB initially increased the tensile strengths due to an increase in the extent of crosslinking, but excess DFB saturated the amine sites of the polymer network, favoring single-point attachment and therefore reducing the extent of crosslinking.
As with other types of sacrificial bonds, dynamic covalent bonds can be employed synergistically with other types of sacrificial bond. Wang and coworkers polymerized amine-terminated PDMS with IPDI to generate urea motifs capable of H-bonding (Fig. 34e–g).184 Unreacted isocyanates were subsequently reacted with ADPS, to introduce a disulfide linkage, and 2,5-furandimethanol, with acts as a functional handle for Diels–Alder reactions with maleimide moieties. Hence the resultant material, denoted PDMS-IPDI-SS-F-M, incorporated three sacrificial networks: one based on H-bonding and two based on dynamic covalent bonds (Fig. 34e). Compared to PDMS-IPDI polymers without further functionalization (i.e., polymers that could engage only in H-bonding), PDMS-IPDI-SS-F-M hybrids exhibited enhanced strength without compromising the maximum elongation of the material, suggestive of energy dissipation enabled by the sacrificial nature of the dynamic covalent bonds (Fig. 34f). Moreover, eliminating either of the types of dynamic covalent bond led to a significant decrease in material toughness, indicating synergistic behavior between the two dynamic covalent sacrificial networks (Fig. 34f). The authors were also able to modulate the tensile strength and toughness of the material by tuning the relative abundance of the three different sacrificial networks.
9.2. Mechanophores
Mechanophores are motifs which undergo a structural rearrangement when mechanical force is applied to them, leading to cleavage of at least one covalent bond.185 Such motifs have been identified as potential energy-dissipating units, as the structural rearrangement often reverts to its original state upon removal of the applied force. Recent reviews highlight many such mechanophore moieties that have been explored as mechanochromic units or as energy-dissipating units in materials.168,186,187 Consequently, we discuss here just two illustrative examples of mechanophore chemistry in the context of sacrificial bonds.Otsuka and coworkers have explored difluorenylsuccinonitrile (DFSN)-based crosslinkers as mechanophores in polyacrylates.188,189 When force is applied to DFSN, the central symmetric C–C bond undergoes homolytic fission to form two stable cyanofluorene radicals (Fig. 35a). These radicals are bright pink in color, which enables the authors to visualize the exact point in the matrix where these dynamic bonds have broken under stress (Fig. 35b). The incorporation of DFSNs into a double-network polyacrylate was observed to increase fracture energies by 2.6-fold compared to a mechanophore-free control polymer (Fig. 35b).188 Moreover, the radical nature of the cyanofluorenes allowed the extent of sacrificial bond rupture to be monitored using electron paramagnetic resonance (EPR) spectroscopy, which revealed that increasing mechanical toughness correlated well with an increase in the number of generated radicals. Subsequent work from the same group introduced a non-symmetric mechanophore, cyanofluorenyl–arylbenzofuranone, as a crosslinker between poly(n-hexyl methacrylate) chains.190 Unlike DFSN, this mechanophore generates two different radicals that have distinct photochromic properties and hence shows mixed mechanochromism. While the tensile strength and toughness of this material was markedly improved over a mechanophore-free control, no substantial differences were observed compared to the DFSN-based materials. However, EPR studies showed that the concentration of radicals generated at 300% strain was 10 times higher for materials containing the non-symmetric mechanophore than for the symmetric DFSN-based materials.
 | ||
| Fig. 35 Mechanophores that act as sacrificial bonds to dissipate energy under high stress or strain (a) Schematic representation of how the central bond in DFSN undergoes homolytic cleavage (b) Compared to the control, the material with DFSNs show much higher yield strain and toughness evident in uniaxial tensile tests. Site of elongation turns pink due to formation of cyanofluorene radicals. Adapted with permission from ref. 188. Copyright 2020 American Chemical Society. (c) Spiropyrans in a poly(ethylacrylate) matrix undergo conversion from the SP to the MC form under stress. (d) Decreasing the density of spiropyrans (DNx, where x is the mol% of spiropyran) leads to loss in yield strength but greater stretchability. (e) Application of lateral stress leads to macroscopic color change. (f) Cyclic tensile tests show large hysteresis loops before fracture. Figure adapted from ref. 191. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Spiropyrans are a common mechanochromic motif that have mostly been used to visualize the stress point in polymer materials, due to their readily observable color change upon mechanically-induced cleavage from the colorless spiropyran (SP) form to the pink merocyanine (MC) form.192,193 Cao recently employed spiropyrans as sacrificial motifs in a double-network polymer in which both networks contained poly(ethyl acrylate) crosslinked with spiropyrans (Fig. 35c).191 The resultant material showed high ultimate strength and maximum elongation attributable to the strong, rigid nature of the spiropyran crosslinks. Even with a low concentration of spiropyran crosslinker (0.096 mol%), a maximum fracture energy of 8.4 kJ m−2 and a maximum elongation of 3400% were achieved (Fig. 35d). The rupture of the sacrificial network could be visualized by a macroscopic color change (Fig. 35e), and cyclic tensile loading–unloading experiments showed large hysteresis loops indicative of substantial energy dissipation (Fig. 35f). Spiropyrans have also been explored as visualizable sacrificial toughening agents by Creton and coworkers.194–196
10. Hidden length
Most of the examples discussed so far have employed sacrificial bonds as the connection (crosslinker) between polymer chains. However, if sacrificial bonds are incorporated into a polymer chain such that the sacrificial bond bridges two atoms on the same chain, the formation of the bond produces a macrocyclic portion of the polymer chain that is necessarily not fully extended and, therefore, does not experience strain.197 Consequently, cleavage of the sacrificial bond “reveals” a portion of the polymer backbone—known as hidden length—that can take on strain and reduce the strain experienced by other segments of the polymer chain (Fig. 36a). In many cases, the stretching of this hidden length dissipates much more energy than the enabling bond cleavage event.198 Unsurprisingly, many natural systems employ the combination of sacrificial bonds and hidden length (SBHL) to dissipate energy under external stress in biomaterials, including bone and abalone shells.9,10,14,198 However, examples of synthetic materials that harness SBHL to withstand impact are rare. | ||
| Fig. 36 (a) Mechanism of energy dissipation in systems with sacrificial bonds and hidden length. Reprinted from ref. 198 with permission from Elsevier, copyright 2006. (b) Force-elongation graphs with different sacrificial bond strengths (red) and different loop radii (blue). The energy required to release the hidden length remained constant, but the elongation length increased because of the increased loop radius. The force required to release the hidden length increased with increasing bond strength. Used with permission of American Society of Mechanical Engineers (ASME), from ref. 199; permission conveyed through Copyright Clearance Center, Inc. | ||
Theoretical insights into the design of SBHL materials were gleaned by Deng and coworkers, who employed MD studies to examine how structural parameters affected the mechanical properties of a model SBHL system.199 Polymer fibers were modeled using a simple bead-spring model, due to its similarity to biological systems such as collagen, spider silk, and amyloid. Simulations were performed while varying parameters including loop radii, number of loops, crosslink interaction strength, and fiber stiffness, to establish the nature of the dependence of material toughness on these parameters (Fig. 36b). For example, it was found that toughness increased linearly with the number of loops, because of the increased number of breaking and unfolding events, but toughness exhibited a quadratic dependence on the strength of crosslinks. This study may guide the rational design of SBHL-based materials with tunable toughness and mechanical properties.
One of the few experimental examples of SBHL being deployed in a synthetic material was reported by Goult and coworkers, who realized a shock-absorbing material, based on a modified talin protein, that showed impact resistance from supersonic projectiles.200 Talin is a naturally-occurring mechanosensitive protein implicated in the transduction of mechanical force to the cytoskeleton. In this work, a talin mutant, modified with cysteine residues to permit crosslinking by maleimides, consisting of three rod-like domains (R1, R2, and R3) was used as a mechanosensitive module; these three rod-like domains in the wild-type protein undergo stepwise force-induced unfolding (Fig. 37a).201 To make the talin shock-absorbing material (TSAM), protein monomers were crosslinked by a trivalent maleimide crosslinker (Fig. 37b–f), forming a gel. A control sample prepared by reacting talin monomers with a monovalent maleimide did not show gelation, confirming that only the trivalent crosslinker was successfully polymerizing the talin monomer (Fig. 37e). TSAM and a poly(vinylpyrrolidone) (PVP) hydrogel control were subjected to impact tests with projectiles sized between 20 and 70 microns at a velocity of 1.5 km s−1. Whereas PVP showed no resistance to the projectile, TSAM resisted the projectile and did not allow the projectiles to permeate the bulk material; this compared favorably to a commercial aerogel that permitted particle penetration up to 8 mm. Here, the myriad non-covalent interactions between the amino acid residues of the three domains act as sacrificial bonds, and the polypeptide backbone acts as hidden length (Fig. 37f); by combining these two, protein unfolding leads to extraordinary energy dissipation in these hydrogels.200
 | ||
| Fig. 37 (a) Talin polymers undergo sequential unfolding to reveal hidden length. (b) Maleimide reagents were reacted with (c) cysteine-modified talin monomers to form (d) crosslinked polymers. (e) Trivalent maleimide crosslinkers led to the formation of gels whereas monovalent maleimides did not. (f) Crosslinked talin polymers. (g) Folded talin domains undergo (h) strain-induced unfolding under applied force to form extended polypeptides that can be (i) stretched under further strain to ∼10× their original length. Figure reproduced from ref. 200 with permission from Springer Nature, copyright 2023. | ||
11. Sacrificial conformation
Recently, Wang et al. reported a novel strategy to dissipate energy without cleavage of a sacrificial bond or interaction, instead relying on the flexible conformation of β-cyclodextrin to tolerate strain.202 Their cross-linked polymers (CCN1–3) were prepared by subjecting a cyclodextrin monomer functionalized with two pendent alkene motifs (CM) to photo-induced thiol–ene click chemistry with different amounts of a linear chain monomer (LM) (Fig. 38a). A control material composed entirely of the linear monomers (LCN) was also prepared. The authors found that increasing the CM content increased the Young's modulus of the material but also decreased its maximum elongation. As a result, CCN2 (CD:LM = 1:3) showed the highest toughness of the samples tested, 60-fold higher than that of the control LM (Fig. 38b). CCN2 also exhibited the maximum energy dissipation (Fig. 38c). These results indicate that the conformational flexibility of β-cyclodextrins can be harnessed as an energy dissipation mechanism without breaking any bonds, covalent or non-covalent, signifying a unique sacrificial conformation approach to engineering tough materials.
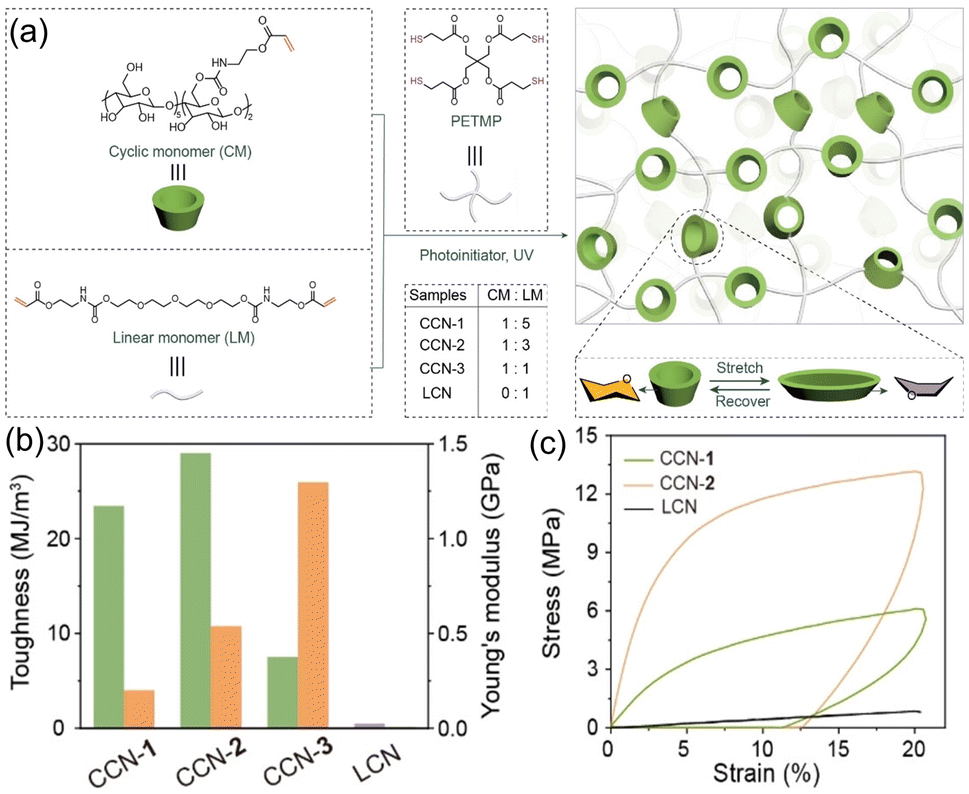 | ||
| Fig. 38 (a) Scheme showing the formation of cross-linked CCN polymers from cyclic monomers (CM) containing β-cyclodextrins and linear monomers (LM). (b) Comparing toughness (green) and Young's modulus (orange) of different polymers with different CM:LM compositions reveals superior mechanical performance by CCN2. (c) Cyclic tensile tests show maximum energy dissipation by CCN2. Figures adapted from ref. 202. Copyright 2024 Wiley-VCH GmbH. | ||
12. Conclusions and outlook
The incorporation of sacrificial bonds into synthetic materials is a powerful strategy to enhance material toughness and strength via the dissipation of energy under applied load. Progress over the past several years has advanced multiple chemical motifs that can serve as sacrificial interactions, ranging from non-covalent interactions such as H-bonding, metal coordination, and the hydrophobic effect to covalent bonds such as dynamic linkages and mechanophores. In concert with experimental studies, the development of new theories to describe energy dissipation in network materials and more widespread simulation of these theories using MD are providing experimentalists with a clearer understanding of how to program energy-dissipating materials with a desired set of mechanical properties, balancing tensile toughness, maximum elongation, and dissipative capacity. The result is a host of new materials with mechanical properties approaching or even surpassing those of natural biomaterials.Since the last comprehensive review on this topic almost a decade ago,17 progress in the field has continued apace and led to several enabling innovations, three of which we highlight here. First, advances in computational and theoretical approaches can now employ fully atomistic MD simulations to reproduce and rationalize experimental observations. Second, the incorporation of multiple sacrificial bonding motifs, whether using a single type of interaction (e.g., HBs using ureas and alcohols) or distinct bonding modalities (e.g., H-bonding and metal coordination), is enabling materials with more sophisticated responses to applied force. Third, the continued expansion of the toolkit of sacrificial bonding motifs, including noncanonical energy dissipation modes such as mechanophores and conformationally-flexible cyclodextrin units, provides ever more chemical handles for the design of new, class-leading materials.
However, as we opined in the Introduction, the most substantial unmet challenges in the field are those that relate to the design of materials with specific properties. Table 1 summarizes some key mechanical properties measured for materials discussed in this review. While the absolute values of many of these metrics are remarkable, the large ranges of strength, strain, and toughness values suggest that structure–property relationships are underdeveloped and warrant rigorous attention. The routine comparison of new materials against well-defined standard reference materials, such as commercial samples of poly(urethane)s and Kevlar, could ease comparison between different studies and help to parse differences arising from sacrificial bond design.
| Sacrificial bond strategy | Ultimate strength | Maximum strain | Tensile toughness | Ref. |
|---|---|---|---|---|
| a Reported value denotes stiffness. | ||||
| Hydrogen bonding (UPy) | 4.5–87.2 MPa | 3%–1285% | 1–386 MJ m−3 | 44, 52, 56, 64 and 67–70 |
| Hydrogen bonding (amides) | 0.35–98.5 MPa | 17%–2160% | 1.6–550 MJ m−3 | 42, 45, 77 and 78–85 |
| Hydrogen bonding (alcohols) | 0.57–83.5 MPa | 2%–1876% | 0.55–1896 MJ m−3 | 89–91, 95 and 96 |
| Metal coordination | 0.20–25.7 MPa | 120%–10000% |
0.0025–96 MJ m−3 | 106, 108–110, 114, 116, 125–127 and 133 |
| Host–guest complexation | 0.09–45.1 MPa | 65%–2400% | 9.63–22 MJ m−3 | 153–155, 158 and 162–164 |
| Electrostatic interactions | 0.25–1.32 MPa | 1100%–6007% | 6.67–8.7 MJ m−3 | 136–138,140 |
| Dynamic covalent bonds | 2.4–24.7 MPa | 988%–4300% | 13.6 MJ m−3 | 180, 183 and 184 |
| Mechanophores | 0.48–3.9 MPa | 921%–3200% | 0.008–16.9 MJ m−3 | 188 and 191 |
| Sacrificial conformation | 538 MPaa |
110% | 30 MJ m−3 | 202 |
Developing comprehensive structure–property relationships will also facilitate the design of next-generation impact-resistant architectures. In the remainder of this section, we highlight several opportunities for continued innovation in this area as the community continues its pursuit of biomimetic load-bearing materials, along with potential strategies to address each opportunity.
Programmable sacrificial bond dynamics. Arguably the most salient challenge is the tendency of synthetic materials to require long relaxation times, on the order of minutes to hours, to fully recover their capacity for energy dissipation. These long relaxation times, which result from the slow re-formation of broken sacrificial bonds, hinder the development of practical materials that tolerate the accumulated damage of frequent, repetitive loads. Incorporating extrinsic sacrificial crosslinkers (cf.Fig. 2c) with mobilities that approach the diffusion limit may be a viable strategy to address this limitation.
Directionality as a design parameter. Most sacrificial bond motifs reported to date rely on directional interactions, such as H-bonding and metal coordination. However, non-directional interactions such as electrostatics or the hydrophobic effect provide substantial advantages, especially with respect to recovery time, and warrant further study. Indeed, the directionality of a sacrificial bond could be viewed as an important design parameter in addition to canonical considerations such as bond strength.
Accumulated damage to covalent networks. Although the presence of sacrificial bonds greatly increases the resistance of materials to strain-induced deformation, prolonged exposure to applied loads will degrade typical soft materials due to damage to the underlying covalent polymer network. Supramolecular polymer networks offer distinct advantages in this regard, as discussed earlier, but the mechanical properties of the pristine materials are often unsatisfactory due to the reliance of the network structure on weaker, non-covalent interactions. Establishing methods to prepare strong and tough structures scaffolded entirely by non-covalent bonds between building blocks may usher in new classes of resilient soft materials.
Synergistic, hierarchical sacrificial networks. More exploration of systems with multiple sacrificial bonds is needed. Though many of the types of sacrificial bond discussed in this review have not been explored for their ability to endow impact resistance, even fewer have been combined to form synergistic sacrificial networks; those materials that do leverage multiple sacrificial bonds, especially those that do so in a hierarchical manner, already show great promise. Achieving such hierarchical bond breakage will require precise control over the number, strength, and spatial distribution of sacrificial bonds, the kinetics of their breaking and re-forming, and the dynamic nature of the covalent polymer network. Chemical strategies to modulate these parameters include the use of electron-withdrawing or -donating groups on bonding motifs to tune the strength of individual sacrificial bonds, employing molecular strain to affect the energetics of bond scission, and leveraging iterative polymerization methods to better control the distribution of sacrificial bonding motifs along covalent polymer backbones. Rational design of such networks, which requires renewed attention from both experimentalists and theoreticians, should produce materials with hitherto unprecedented properties.
Designing heterogeneity across length scales. One of the largest opportunities in materials design, not just for materials with sacrificial bonds, is the ability to design heterogeneity into materials architecture, particularly on the macroscale. This is especially pertinent for impact-resistant materials: consider the crumple zones of a modern car, which direct energy around the passenger cabin and protect the human inhabitants from the most severe damage. How might similar macroscale heterogeneity be programmed into polymer networks? One potential solution emerges from biological systems, which routinely employ hierarchical assembly as an information-efficient means to program structural sophistication over multiple length scales. Designing hierarchically-architected sacrificially-bonded structures may provide similarly heterogeneous materials from synthetic components.
Abbreviations
| 2DPA | 2D polyamide |
| ADPS | 2-Aminophenyl disulfide |
| APBIA | 2-(4-Aminophenyl)-1H-benzimidazol-5-amine |
| ASC | Acylsemicarbazide |
| BF | Bamboo fiber |
| B21C7 | Benzo-21-crown-7 |
| [BMIM][TFSI] | 1-Butyl-3-methylimidazolium-bis((trifluoromethylsulfonyl)imide) |
| BPE | Biobased polyester elastomer |
| C18M | Stearyl methacrylate |
| CD | Cyclodextrin |
| ChNW | Chitin nanowhisker |
| Chol | Cholesterol |
| CNC | Cellulose nanocrystal |
| CCN | Covalently-crosslinked network |
| CNF | Cellulose nanofibril |
| CNT | Carbon nanotube |
| CTAC | Hexadecyltrimethylammonium chloride |
| DABA | 4,4-Diaminobenzanilide |
| DDM | Diamino-diphenylmethane |
| DFB | 1,2-Diformylbenzene |
| DFSN | Difluorenylsuccinonitrile |
| DGEBA | Diglycidyl ether-bisphenol A |
| DPA | 3-Hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid |
| ENR | Epoxidized natural rubber |
| EPR | Electron paramagnetic resonance |
| HAPAA | Hydrophobic-associating poly(acrylic) acid |
| HB | Hydrogen bond |
| HDI | Hexamethylene diisocyanate |
| HEMA | 2-Hydroxyethyl methacrylate |
| HFBA | 2,2,3,4,4,4-Hexafluorobutyl acrylate |
| HGM | Host–guest macromer |
| HMDI | Cyclohexylmethylmethane-4,4′-diisocyanate |
| iGWLC | Inelastic glassy worm-like chain |
| IHP | Impact-hardening polymer |
| IL | Ionic liquid |
| Im | Imidazole |
| IPDI | Isophorone diisocyanate |
| IU | Imidazolidinyl urea |
| LCN | Linearly-crosslinked network |
| MA | Methacrylate |
| MBA | Methylene bis-acrylamide |
| MD | Molecular dynamics |
| MWCNT | Multiwall carbon nanotube |
| NAC | N-Acetyl-L-cysteine |
| NAGA | N-Acryloyl glycinamide |
| PAA | Poly(acrylic acid) |
| PAAm | Poly(acrylamide) |
| PANI | Poly(aniline) |
| PBSA | Poly(butylene succinate-co-butylene adipate) |
| PCL | Poly(caprolactone) |
| pdca | 2,6-Pyridinedicarboxamide |
| PDETAS | Diethylenetriamine-functionalized polysiloxanes |
| PDMS | Poly(dimethylsiloxane) |
| PEG | Poly(ethylene glycol) |
| PEG-DE | Poly(ethylene glycol) diglycidyl ether |
| PESC | Polyelectrolyte-surfactant complexes |
| PPG | Poly(propylene glycol) |
| PTA | Poly(tannic acid) |
| PTMEG | Poly(tetramethylene ether glycol) |
| PU | Polyurethane |
| PVA | Poly(vinyl alcohol) |
| PVP | Poly(vinylpyrrolidone) |
| ROMP | Ring-opening metathesis polymerization |
| RSF | Regenerated silk fibroin |
| SBHL | Sacrificial bond-hidden length |
| SDS | Sodium dodecyl sulfate |
| SF | Silk fibroin |
| SPM | Supramolecular polymeric material |
| SPN | Supramolecular polymeric network |
| (S)SBR | (Solution-polymerized) styrene–butadiene rubber |
| SSG | Shear-stiffening gel |
| STF | Shear-thickening fluid |
| TA | Tannic acid |
| TPY | Terpyridine |
| TSAM | Talin shock-absorbing material |
| TST | Transition state theory |
| WLC | Worm-like chain |
| UPy | Ureidopyrimidinone |
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this submission.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Air Force Office of Scientific Research (AFOSR) through the Young Investigator Program (award FA9550-24-1-0104).References
- M. Gralka and K. Kroy, Inelastic mechanics: A unifying principle in biomechanics, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 3025–3037 CrossRef CAS PubMed.
- S. E. Naleway, M. M. Porter, J. McKittrick and M. A. Meyers, Structural Design Elements in Biological Materials: Application to Bioinspiration, Adv. Mater., 2015, 27, 5455–5476 CrossRef CAS PubMed.
- B. S. Lazarus, A. Velasco-Hogan, T. Gómez-del Río, M. A. Meyers and I. Jasiuk, A review of impact resistant biological and bioinspired materials and structures, J. Mater. Res. Technol., 2020, 9, 15705–15738 CrossRef CAS.
- R. O. Ritchie, The conflicts between strength and toughness, Nat. Mater., 2011, 10, 817–822 CrossRef CAS PubMed.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Bioinspired structural materials, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- M. G. Mazzotta, A. A. Putnam, M. A. North and J. J. Wilker, Weak Bonds in a Biomimetic Adhesive Enhance Toughness and Performance, J. Am. Chem. Soc., 2020, 142, 4762–4768 CrossRef CAS PubMed.
- W.-W. Yu, W.-Z. Xu, Y.-C. Wei, S. Liao and M.-C. Luo, Mechanically Robust Elastomers Enabled by a Facile Interfacial Interactions-Driven Sacrificial Network, Macromol. Rapid Commun., 2021, 42, 2100509 CrossRef CAS PubMed.
- B. L. Smith, T. E. Schäffer, M. Viani, J. B. Thompson, N. A. Frederick, J. Kindt, A. Belcher, G. D. Stucky, D. E. Morse and P. K. Hansma, Molecular mechanistic origin of the toughness of natural adhesives, fibres and composites, Nature, 1999, 399, 761–763 CrossRef CAS.
- J. B. Thompson, J. H. Kindt, B. Drake, H. G. Hansma, D. E. Morse and P. K. Hansma, Bone indentation recovery time correlates with bond reforming time, Nature, 2001, 414, 773–776 CrossRef CAS PubMed.
- P. K. Hansma, G. E. Fantner, J. H. Kindt, P. J. Thurner, G. Schitter, P. J. Turner, S. F. Udwin and M. M. Finch, Sacrificial bonds in the interfibrillar matrix of bone, J. Musculoskeletal Neuronal Interact., 2005, 5, 313–315 CAS.
- L. Osorio, E. Trujillo, A. W. VanVuure, F. Lens, J. Ivens and I. Verpoest, The relationship between the bamboo fibre microstructure and mechanical properties, in Proceedings of the 14th European Conference on Composite Materials.
- H. Rhee, M. F. Horstemeyer, Y. Hwang, H. Lim, H. El Kadiri and W. Trim, A study on the structure and mechanical behavior of the Terrapene carolina carapace: A pathway to design bio-inspired synthetic composites, Mater. Sci. Eng., C, 2009, 29, 2333–2339 CrossRef CAS.
- K. Tushtev, M. Murck and G. Grathwohl, On the nature of the stiffness of nacre, Mater. Sci. Eng., C, 2008, 28, 1164–1172 CrossRef CAS.
- G. E. Fantner, T. Hassenkam, J. H. Kindt, J. C. Weaver, H. Birkedal, L. Pechenik, J. A. Cutroni, G. A. G. Cidade, G. D. Stucky, D. E. Morse and P. K. Hansma, Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture, Nat. Mater., 2005, 4, 612–616 CrossRef CAS PubMed.
- B. Rennekamp, C. Karfusehr, M. Kurth, A. Ünal, D. Monego, K. Riedmiller, G. Gryn'ova, D. M. Hudson and F. Gräter, Collagen breaks at weak sacrificial bonds taming its mechanoradicals, Nat. Commun., 2023, 14, 2075 CrossRef CAS PubMed.
- E. Degtyar, M. J. Harrington, Y. Politi and P. Fratzl, The Mechanical Role of Metal Ions in Biogenic Protein-Based Materials, Angew. Chem., Int. Ed., 2014, 53, 12026–12044 CrossRef CAS PubMed.
- X. Zhou, B. Guo, L. Zhang and G.-H. Hu, Progress in bio-inspired sacrificial bonds in artificial polymeric materials, Chem. Soc. Rev., 2017, 46, 6301–6329 RSC.
- K. Fukao, K. Tanaka, R. Kiyama, T. Nonoyama and J. P. Gong, Hydrogels toughened by biominerals providing energy-dissipative sacrificial bonds, J. Mater. Chem. B, 2020, 8, 5184–5188 RSC.
- J. Yang, K. Li, C. Tang, Z. Liu, J. Fan, G. Qin, W. Cui, L. Zhu and Q. Chen, Recent Progress in Double Network Elastomers: One Plus One is Greater Than Two, Adv. Funct. Mater., 2022, 32, 2110244 Search PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Double-Network Hydrogels with Extremely High Mechanical Strength, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- T. Nonoyama and J. P. Gong, Tough Double Network Hydrogel and Its Biomedical Applications, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 393–410 Search PubMed.
- C. Creton, 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough, Macromolecules, 2017, 50, 8297–8316 Search PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Soft Materials by Design: Unconventional Polymer Networks Give Extreme Properties, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- X. Lin, X. Zhao, C. Xu, L. Wang and Y. Xia, Progress in the mechanical enhancement of hydrogels: Fabrication strategies and underlying mechanisms, J. Polym. Sci., 2022, 60, 2525–2542 CrossRef CAS.
- X. Zhang, J. Xiang, Y. Hong and L. Shen, Recent Advances in Design Strategies of Tough Hydrogels, Macromol. Rapid Commun., 2022, 43, 2200075 Search PubMed.
- X. Kuang, M. O. Arıcan, T. Zhou, X. Zhao and Y. S. Zhang, Functional Tough Hydrogels: Design, Processing, and Biomedical Applications, Acc. Mater. Res., 2023, 4, 101–114 Search PubMed.
- K. Kroy and J. Glaser, The glassy wormlike chain, New J. Phys., 2007, 9, 416–416 Search PubMed.
- L. Wolff, P. Fernandez and K. Kroy, Inelastic mechanics of sticky biopolymer networks, New J. Phys., 2010, 12, 053024 Search PubMed.
- L. Wolff, P. Fernández and K. Kroy, Resolving the Stiffening-Softening Paradox in Cell Mechanics, PLoS One, 2012, 7, e40063 CrossRef CAS PubMed.
- K. Kothari, Y. Hu, S. Gupta and A. Elbanna, Mechanical Response of Two-Dimensional Polymer Networks: Role of Topology, Rate Dependence, and Damage Accumulation, J. Appl. Mech., 2018, 85, 031008 CrossRef.
- W. K. Sun, B. B. Yin and K. M. Liew, Damage-induced energy dissipation in artificial soft tissues, J. Mech. Phys. Solids, 2025, 194, 105933 CrossRef.
- A. Suhail, A. Banerjee and R. Rajesh, Kinetic model description of dissipation and recovery in collagen fibrils under cyclic loading, Phys. Rev. E, 2022, 106, 044407 CrossRef CAS PubMed.
- Q. Chen, W. Huang, L. Zhang, L. Xi and J. Liu, Fully atomistic molecular dynamics simulation of chemically modified natural rubber with hydrogen-bonding network, Polymer, 2023, 284, 126284 CrossRef CAS.
- Q. Chen, W. Huang, L. Zhang, Y. Chen and J. Liu, Impact of Sacrificial Hydrogen Bonds on the Structure and Properties of Rubber Materials: Insights from All-Atom Molecular Dynamics Simulations, Langmuir, 2024, 40, 11470–11480 CrossRef CAS PubMed.
- L. Xie, K. Wu, X. Liang, Z. Song, J. Ding, J. Jin, Y. Yao, L. He and Y. Ni, Toughening by interfacial self-healing processes in bioinspired staggered heterostructures, Int. J. Mech. Sci., 2025, 285, 109847 CrossRef.
- J. Yu, C. Zhai, M. Wang, Z. Cai, J. Yeo, Q. Zhang, C. Zhao and S. Lin, Hybridly double-crosslinked carbon nanotube networks with combined strength and toughness via cooperative energy dissipation, Nanoscale, 2022, 14, 2434–2445 RSC.
- Y. Shen, B. Wang, D. Li, X. Xu, Y. Liu, Y. Huang and Z. Hu, Toughening shape-memory epoxy resins via sacrificial hydrogen bonds, Polym. Chem., 2022, 13, 1130–1139 RSC.
- P. Song and H. Wang, High-Performance Polymeric Materials through Hydrogen-Bond Cross-Linking, Adv. Mater., 2020, 32, 1901244 Search PubMed.
- L. Martikainen, A. Walther, J. Seitsonen, L. Berglund and O. Ikkala, Deoxyguanosine Phosphate Mediated Sacrificial Bonds Promote Synergistic Mechanical Properties in Nacre-Mimetic Nanocomposites, Biomacromolecules, 2013, 14, 2531–2535 Search PubMed.
- B. Zhu, N. Jasinski, A. Benitez, M. Noack, D. Park, A. S. Goldmann, C. Barner-Kowollik and A. Walther, Hierarchical Nacre Mimetics with Synergistic Mechanical Properties by Control of Molecular Interactions in Self-Healing Polymers, Angew. Chem., Int. Ed., 2015, 54, 8653–8657 CrossRef CAS PubMed.
- Z. Zheng, F. Qi, X. Sun, N. Zhao, B. Zhang, F. Qi and X. Ouyang, Synergistic enhancement of mechanical properties and impact resistance of polyurethane elastomers by composite fillers containing quadruple hydrogen bonds and nano-CaCO3, J. Mater. Sci., 2023, 58, 3582–3596 CrossRef CAS.
- D. S. Lee, Y.-S. Choi, J. H. Hwang, J.-H. Lee, W. Lee, S. Ahn, S. Park, J.-H. Lee, Y. S. Kim and D.-G. Kim, Weldable and Reprocessable Biomimetic Polymer Networks Based on a Hydrogen Bonding and Dynamic Covalent Thiourea Motif, ACS Appl. Polym. Mater., 2021, 3, 3714–3720 CrossRef CAS.
- S. Shi, X. Peng, T. Liu, Y.-N. Chen, C. He and H. Wang, Facile preparation of hydrogen-bonded supramolecular polyvinyl alcohol-glycerol gels with excellent thermoplasticity and mechanical properties, Polymer, 2017, 111, 168–176 CrossRef CAS.
- Y. Song, Y. Liu, T. Qi and G. L. Li, Towards Dynamic but Supertough Healable Polymers through Biomimetic Hierarchical Hydrogen-Bonding Interactions, Angew. Chem., Int. Ed., 2018, 57, 13838–13842 CrossRef CAS PubMed.
- J. A. Neal, D. Mozhdehi and Z. Guan, Enhancing Mechanical Performance of a Covalent Self-Healing Material by Sacrificial Noncovalent Bonds, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, Strong Dimerization of Ureidopyrimidones via Quadruple Hydrogen Bonding, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- J. Verjans and R. Hoogenboom, Supramolecular polymer materials based on ureidopyrimidinone quadruple hydrogen bonding units, Prog. Polym. Sci., 2023, 142, 101689 CrossRef CAS.
- I. Agnarsson, M. Kuntner and T. A. Blackledge, Bioprospecting Finds the Toughest Biological Material: Extraordinary Silk from a Giant Riverine Orb Spider, PLoS One, 2010, 5, e11234 Search PubMed.
- S. Tang, J. Li, R. Wang, J. Zhang, Y. Lu, G.-H. Hu, Z. Wang and L. Zhang, Current trends in bio-based elastomer materials, SusMat, 2022, 2, 2–33 Search PubMed.
- B. Guo, Y. Chen, Y. Lei, L. Zhang, W. Y. Zhou, A. B. M. Rabie and J. Zhao, Biobased Poly(propylene sebacate) as Shape Memory Polymer with Tunable Switching Temperature for Potential Biomedical Applications, Biomacromolecules, 2011, 12, 1312–1321 CrossRef CAS PubMed.
- W. Lei, X. Zhou, T. P. Russell, K. Hua, X. Yang, H. Qiao, W. Wang, F. Li, R. Wang and L. Zhang, High performance bio-based elastomers: energy efficient and sustainable materials for tires, J. Mater. Chem. A, 2016, 4, 13058–13062 RSC.
- B. Liu, Z. Tang, Z. Wang, L. Zhang and B. Guo, Integrating transient and sacrificial bonds into biobased elastomers toward mechanical property enhancement and macroscopically responsive property, Polymer, 2019, 184, 121914 CrossRef CAS.
- Y. Xu, G. Lubineau, G. Liao, Q. He and T. Xing, Rate-dependent viscoelasticity of an impact-hardening polymer under oscillatory shear, Mater. Res. Express, 2020, 7, 075701 CrossRef CAS.
- H. Wen, J. Sun, K. Yu, X. Yang, X. Dai and Z. Zhang, A self-healing and energy-dissipating impact-hardening polymer based on a variety of reversible dynamic bonds, Mater. Des., 2023, 231, 112057 CrossRef CAS.
- K. Liu, L. Cheng, N. Zhang, H. Pan, X. Fan, G. Li, Z. Zhang, D. Zhao, J. Zhao, X. Yang, Y. Wang, R. Bai, Y. Liu, Z. Liu, S. Wang, X. Gong, Z. Bao, G. Gu, W. Yu and X. Yan, Biomimetic Impact Protective Supramolecular Polymeric Materials Enabled by Quadruple H-Bonding, J. Am. Chem. Soc., 2021, 143, 1162–1170 CrossRef CAS PubMed.
- T. T. T. Myllymäki, L. Lemetti, Nonappa and O. Ikkala, Hierarchical Supramolecular Cross-Linking of Polymers for Biomimetic Fracture Energy Dissipating Sacrificial Bonds and Defect Tolerance under Mechanical Loading, ACS Macro Lett., 2017, 6, 210–214 CrossRef PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Carbon Nanotubes–the Route Toward Applications, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Carbon Nanotubes: Present and Future Commercial Applications, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- A. Micoli, M. Nieuwenhuizen, M. Koenigs, M. Quintana, R. Sijbesma and M. Prato, Supramolecular Macrostructures of UPy–Functionalized Carbon Nanotubes, Chem. – Eur. J., 2015, 21, 14179–14185 CrossRef CAS PubMed.
- K. Guo, D.-L. Zhang, X.-M. Zhang, J. Zhang, L.-S. Ding, B.-J. Li and S. Zhang, Conductive Elastomers with Autonomic Self-Healing Properties, Angew. Chem., Int. Ed., 2015, 54, 12127–12133 CrossRef CAS PubMed.
- Y. You, J. Yang, Q. Zheng, N. Wu, Z. Lv and Z. Jiang, Ultra-stretchable hydrogels with hierarchical hydrogen bonds, Sci. Rep., 2020, 10, 11727 CrossRef CAS PubMed.
- M. F. Griffin, Y. Premakumar, A. M. Seifalian, M. Szarko and P. E. M. Butler, Biomechanical characterisation of the human nasal cartilages; implications for tissue engineering, J. Mater. Sci.: Mater. Med., 2015, 27, 11 CrossRef PubMed.
- C. Li, G. Guan, R. Reif, Z. Huang and R. K. Wang, Determining elastic properties of skin by measuring surface waves from an impulse mechanical stimulus using phase-sensitive optical coherence tomography, J. R. Soc., Interface, 2011, 9, 831–841 CrossRef PubMed.
- T. Yang, X. Lu, X. Wang, Y. Li, X. Wei, W. Wang and J. Sun, Healable, Recyclable, and Scratch-Resistant Polyurethane Elastomers Cross-Linked with Multiple Hydrogen Bonds, ACS Appl. Polym. Mater., 2023, 5, 2830–2839 CrossRef CAS.
- S. Utrera-Barrios, R. Verdejo, M. A. López-Manchado and M. H. Santana, Evolution of self-healing elastomers, from extrinsic to combined intrinsic mechanisms: a review, Mater. Horiz., 2020, 7, 2882–2902 RSC.
- J. Luo, Z. Demchuk, X. Zhao, T. Saito, M. Tian, A. P. Sokolov and P.-F. Cao, Elastic vitrimers: Beyond thermoplastic and thermoset elastomers, Matter, 2022, 5, 1391–1422 CrossRef CAS.
- Y. Cai, H. Li, C. Li, J. Tan and Q. Zhang, A Strategy of Thiolactone Chemistry to Construct Strong and Tough Self-Healing Supramolecular Polyurethane Elastomers via Hierarchical Hydrogen Bonds and Coordination Bonds, Ind. Eng. Chem. Res., 2023, 62, 6416–6424 CrossRef CAS.
- T. Li, T.-Z. Zheng, Z.-X. Guo, J. Xu and B.-H. Guo, A Well-defined Hierarchical Hydrogen Bonding Strategy to Polyureas with Simultaneously Improved Strength and Toughness, Chin. J. Polym. Sci., 2019, 37, 1257–1266 CrossRef CAS.
- K. Cao and G. Liu, Low-Molecular-Weight, High-Mechanical-Strength, and Solution-Processable Telechelic Poly(ether imide) End-Capped with Ureidopyrimidinone, Macromolecules, 2017, 50, 2016–2023 CrossRef CAS.
- J. Wu, F. Zeng, Z. Fan, S. Xuan, Z. Hua and G. Liu, Hierarchical Hydrogen Bonds Endow Supramolecular Polymers with High Strength, Toughness, and Self-Healing Properties, Adv. Funct. Mater., 2024, 34, 2410518 CrossRef CAS.
- X. Chang, Y. Geng, H. Cao, J. Zhou, Y. Tian, G. Shan, Y. Bao, Z. L. Wu and P. Pan, Dual-Crosslink Physical Hydrogels with High Toughness Based on Synergistic Hydrogen Bonding and Hydrophobic Interactions, Macromol. Rapid Commun., 2018, 39, 1700806 CrossRef PubMed.
- R. E. Hubbard and M. Kamran Haider, in Encyclopedia of Life Sciences, John Wiley & Sons, Ltd, 2010 Search PubMed.
- Y. Li, Y. Jin, W. Fan and R. Zhou, A review on room-temperature self-healing polyurethane: synthesis, self-healing mechanism and application, J. Leather Sci. Eng., 2022, 4, 24 CrossRef CAS.
- C. J. Demott, M. R. Jones, C. D. Chesney, D. J. Yeisley, R. A. Culibrk, M. S. Hahn and M. A. Grunlan, Ultra-High Modulus Hydrogels Mimicking Cartilage of the Human Body, Macromol. Biosci., 2022, 22, e2200283 CrossRef PubMed.
- X. Liu, J. Liu, S. Lin and X. Zhao, Hydrogel machines, Mater. Today, 2020, 36, 102–124 CrossRef CAS.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Weak Hydrogen Bonding Enables Hard, Strong, Tough, and Elastic Hydrogels, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- B. Narayanan Narasimhan, G. Sjoerd Deijs, S. Manuguri, M. S. Hao Ting, M. A. K. Williams and J. Malmström, A comparative study of tough hydrogen bonding dissipating hydrogels made with different network structures, Nanoscale Adv., 2021, 3, 2934–2947 RSC.
- Y. Yao, Z. Xu, B. Liu, M. Xiao, J. Yang and W. Liu, Multiple H-Bonding Chain Extender-Based Ultrastiff Thermoplastic Polyurethanes with Autonomous Self-Healability, Solvent-Free Adhesiveness, and AIE Fluorescence, Adv. Funct. Mater., 2021, 31, 2006944 CrossRef CAS.
- C. Shi, X. Li, X. Zhang and M. Zou, Dual dynamic network structures of recyclable epoxy resins with high strength and toughness via sacrificial hydrogen-bonding clusters and imine bonds: surpassing the strength-toughness trade-off, Chem. Eng. J., 2024, 493, 152361 CrossRef CAS.
- Y. Xing, J. Li, J. Cheng, L. Lu, T. Xue, J. Xu, X. Xu and F. Zhang, 2D Polyamides Enable Self-Healing and Recyclable Elastomers with High Robustness, Toughness, and Crack Resistance via Supramolecular Interactions, Small, 2025, 21, 2411040 CrossRef CAS PubMed.
- T. Guan, X. Wang, Y.-L. Zhu, L. Qian, Z. Lu, Y. Men, J. Li, Y. Wang and J. Sun, Mechanically Robust Skin-like Poly(urethane-urea) Elastomers Cross-Linked with Hydrogen-Bond Arrays and Their Application as High-Performance Ultrastretchable Conductors, Macromolecules, 2022, 55, 5816–5825 CrossRef CAS.
- Q. Zhong, X. Chen, Y. Yang, C. Cui, L. Ma, Z. Li, Q. Zhang, X. Chen, Y. Cheng and Y. Zhang, Hydrogen bond reinforced, transparent polycaprolactone-based degradable polyurethane, Mater. Chem. Front., 2021, 5, 5371–5381 RSC.
- K. Zhou, K. Zhang, L. Wang, Y. Tan, Y. Meng, X. Li and X. Wang, Engineering Ultratough and Impact-Resistant Poly(urethane-urea) Elastomers for Advanced Protective Equipment, ACS Appl. Polym. Mater., 2025, 7, 4038–4049 CrossRef CAS.
- S. Zhan, Y. Bo, H. Liu, R. Yuan, W. Ding, Y. Zhang, D. Zhang, S. Wang and M. Zhang, High-Strength Polyurethane Composite Film Reinforced by Cellulose Nanocrystals, ACS Appl. Polym. Mater., 2024, 6, 1763–1771 CrossRef CAS.
- C. Tian, F. Zhao, N. Yang, Y. Jiang, L. Huang, F. Zhou, D. Yuan and X. Cai, Durable, Super-Resilient, and Ultra-Strong Polyurethane Elastomers Via a Dense Hydrogen Bond Cross-Linking Strategy, Macromolecules, 2025, 58, 2905–2916 CrossRef CAS.
- S. Tommasone, F. Allabush, Y. K. Tagger, J. Norman, M. Köpf, J. H. R. Tucker and P. M. Mendes, The challenges of glycan recognition with natural and artificial receptors, Chem. Soc. Rev., 2019, 48, 5488–5505 RSC.
- C. M. Altaner, L. H. Thomas, A. N. Fernandes and M. C. Jarvis, How Cellulose Stretches: Synergism between Covalent and Hydrogen Bonding, Biomacromolecules, 2014, 15, 791–798 CrossRef CAS PubMed.
- H. Fan, Getting glued in the sea, Polym. J., 2023, 55, 653–664 CrossRef CAS PubMed.
- F. Lin, Z. Wang, J. Chen, B. Lu, L. Tang, X. Chen, C. Lin, B. Huang, H. Zeng and Y. Chen, A bioinspired hydrogen bond crosslink strategy toward toughening ultrastrong and multifunctional nanocomposite hydrogels, J. Mater. Chem. B, 2020, 8, 4002–4015 RSC.
- X. Lin, L. Zhang and B. Duan, Polyphenol-mediated chitin self-assembly for constructing a fully naturally resourced hydrogel with high strength and toughness, Mater. Horiz., 2021, 8, 2503–2512 RSC.
- G. Su, Y. Zhang, X. Zhang, J. Feng, J. Cao, X. Zhang and T. Zhou, Soft yet Tough: a Mechanically and Functionally Tissue-like Organohydrogel for Sensitive Soft Electronics, Chem. Mater., 2022, 34, 1392–1402 CrossRef CAS.
- K. S. Koutroupi and J. C. Barbenel, Mechanical and failure behaviour of the stratum corneum, J. Biomech., 1990, 23, 281–287 CrossRef CAS PubMed.
- N. K. Simha, C. S. Carlson and J. L. Lewis, Evaluation of fracture toughness of cartilage by micropenetration, J. Mater. Sci.: Mater. Med., 2004, 15, 631–639 CrossRef CAS PubMed.
- J. Gao, Y. Zhang, Y. Bi, K. Du, J. Su and S. Zhang, A strong hydrogen bond bridging interface based on tannic acid for improving the performance of high-filled bamboo fibers/poly (butylene succinate-co-butylene adipate) (PBSA) biocomposites, Int. J. Biol. Macromol., 2024, 267, 131611 CrossRef CAS PubMed.
- A. Kar, M. A. Rather, M. Mandal and N. Karak, Elastomeric biodegradable poly(ester amide urethane) as a tough and robust material, Prog. Org. Coat., 2023, 182, 107684 CrossRef CAS.
- L. Luo, F. Zhang, Y. Liu and J. Leng, Super-tough, self-sensing and shape-programmable polymers via topological structure crosslinking networks, Chem. Eng. J., 2023, 457, 141282 CrossRef CAS.
- B. Dereka, Q. Yu, N. H. C. Lewis, W. B. Carpenter, J. M. Bowman and A. Tokmakoff, Crossover from hydrogen to chemical bonding, Science, 2021, 371, 160–164 CrossRef CAS PubMed.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Iron-Clad Fibers: A Metal-Based Biological Strategy for Hard Flexible Coatings, Science, 2010, 328, 216–220 CrossRef CAS PubMed.
- Z. Wei, J. He, T. Liang, H. Oh, J. Athas, Z. Tong, C. Wang and Z. Nie, Autonomous self-healing of poly(acrylic acid) hydrogels induced by the migration of ferric ions, Polym. Chem., 2013, 4, 4601–4605 RSC.
- M. Mauro, Dynamic Metal–Ligand Bonds as Scaffolds for Autonomously Healing Multi-Responsive Materials, Eur. J. Inorg. Chem., 2018, 2018, 2090–2100 CrossRef CAS.
- L. Shi, P. Ding, Y. Wang, Y. Zhang, D. Ossipov and J. Hilborn, Self-Healing Polymeric Hydrogel Formed by Metal–Ligand Coordination Assembly: Design, Fabrication, and Biomedical Applications, Macromol. Rapid Commun., 2019, 40, 1800837 CrossRef PubMed.
- C.-H. Li and J.-L. Zuo, Self-Healing Polymers Based on Coordination Bonds, Adv. Mater., 2020, 32, 1903762 CrossRef CAS PubMed.
- S. Zechel, M. D. Hager, T. Priemel and M. J. Harrington, Healing through Histidine: Bioinspired Pathways to Self-Healing Polymers via Imidazole–Metal Coordination, Biomimetics, 2019, 4, 20 CrossRef CAS PubMed.
- M. Krogsgaard, V. Nue and H. Birkedal, Mussel-Inspired Materials: Self-Healing through Coordination Chemistry, Chem. – Eur. J., 2016, 22, 844–857 CrossRef CAS PubMed.
- F. Byette, A. Laventure, I. Marcotte and C. Pellerin, Metal–Ligand Interactions and Salt Bridges as Sacrificial Bonds in Mussel Byssus-Derived Materials, Biomacromolecules, 2016, 17, 3277–3286 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Control of hierarchical polymer mechanics with bioinspired metal-coordination dynamics, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS PubMed.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. H. Waite, J. N. Israelachvili, B. K. Ahn and M. T. Valentine, Toughening elastomers using mussel-inspired iron-catechol complexes, Science, 2017, 358, 502–505 CrossRef CAS PubMed.
- Y. Na and C. Chen, Catechol-Functionalized Polyolefins, Angew. Chem., Int. Ed., 2020, 59, 7953–7959 CrossRef CAS PubMed.
- J. Chen, S. Wang, J. Huan, Z. Li, X. Yang, X. Li and Y. Tu, Effect of Coordination Sacrificial Bond Strength on Toughening Properties of Polyesters, Macromolecules, 2024, 57, 4054–4061 CrossRef CAS.
- S. Kawahara, H. Nishioka, M. Yamano and Y. Yamamoto, Synthetic Rubber with the Tensile Strength of Natural Rubber, ACS Appl. Polym. Mater., 2022, 4, 2323–2328 CrossRef CAS.
- J. Liu, S. Wang, Z. Tang, J. Huang, B. Guo and G. Huang, Bioinspired Engineering of Two Different Types of Sacrificial Bonds into Chemically Cross-Linked cis-1,4-Polyisoprene toward a High-Performance Elastomer, Macromolecules, 2016, 49, 8593–8604 CrossRef CAS.
- C. Tian, H. Feng, Y. Qiu, G. Zhang, T. Tan and L. Zhang, Facile strategy to incorporate amidoxime groups into elastomers toward self-crosslinking and self-reinforcement, Polym. Chem., 2022, 13, 5368–5379 RSC.
- X. Zhang, J. Huang, Z. Tang, B. Guo and L. Zhang, Iron ion cluster-OH coordination as high-efficiency sacrificial bond for reinforcement of elastomer, Polymer, 2020, 186, 122059 CrossRef CAS.
- N. Domun, H. Hadavinia, T. Zhang, T. Sainsbury, G. H. Liaghat and S. Vahid, Improving the fracture toughness and the strength of epoxy using nanomaterials – a review of the current status, Nanoscale, 2015, 7, 10294–10329 RSC.
- Y. Liu, Z. Tang, D. Wang, S. Wu and B. Guo, Biomimetic design of elastomeric vitrimers with unparalleled mechanical properties, improved creep resistance and retained malleability by metal–ligand coordination, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- Q. Wang, G. He, X. Shen, H. Cong, B. Wang, J. Tian and Y. Xiang, Effect of interfacial sacrificial bonds on the vulcanization and mechanical properties of styrene butadiene rubber/talcum powder composites, J. Appl. Polym. Sci., 2024, 141, e55400 CrossRef CAS.
- Y. Huang, J. Li, M. Qi, G. Si and C. Tan, In Situ Modification to Reinforce Isoprene Rubber by Sacrificial Bonds, ACS Appl. Polym. Mater., 2023, 5, 4080–4087 CrossRef CAS.
- M. Mareliati, L. Tadiello, S. Guerra, L. Giannini, S. Schrettl and C. Weder, Metal–Ligand Complexes as Dynamic Sacrificial Bonds in Elastic Polymers, Macromolecules, 2022, 55, 5164–5175 CrossRef CAS.
- R. Hu, X. Jiang, Y. Chen, J. Wang, Y. Guo, Q. Zheng and Y. Shangguan, Strain hysteresis and Mullins effect of rubber vulcanizates with a reversible sacrificial network, Soft Matter, 2025, 21, 399–410 RSC.
- P. Lin, S. Ma, X. Wang and F. Zhou, Molecularly Engineered Dual-Crosslinked Hydrogel with Ultrahigh Mechanical Strength, Toughness, and Good Self-Recovery, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- J. Yang, F. Xu and C.-R. Han, Metal Ion Mediated Cellulose Nanofibrils Transient Network in Covalently Cross-linked Hydrogels: Mechanistic Insight into Morphology and Dynamics, Biomacromolecules, 2017, 18, 1019–1028 CrossRef CAS PubMed.
- C. Zeng, P. Wu, J. Guo, N. Zhao, C. Ke, G. Liu, F. Zhou and W. Liu, Synergy of Hofmeister effect and ligand crosslinking enabled the facile fabrication of super-strong, pre-stretching-enhanced gelatin-based hydrogels, Soft Matter, 2022, 18, 8675–8686 RSC.
- X. Wang, F. Zhao, B. Pang, X. Qin and S. Feng, Triple network hydrogels (TN gels) prepared by a one-pot, two-step method with high mechanical properties, RSC Adv., 2018, 8, 6789–6797 RSC.
- D. Li, H. Gao, M. Li, G. Chen, L. Guan, M. He, J. Tian and R. Cao, Nanochitin/metal ion dual reinforcement in synthetic polyacrylamide network-based nanocomposite hydrogels, Carbohydr. Polym., 2020, 236, 116061 CrossRef CAS PubMed.
- C. Shao, H. Chang, M. Wang, F. Xu and J. Yang, High-Strength, Tough, and Self-Healing Nanocomposite Physical Hydrogels Based on the Synergistic Effects of Dynamic Hydrogen Bond and Dual Coordination Bonds, ACS Appl. Mater. Interfaces, 2017, 9, 28305–28318 CrossRef CAS PubMed.
- I. Hussain, S. M. Sayed and G. Fu, Facile and cost-effective synthesis of glycogen-based conductive hydrogels with extremely flexible, excellent self-healing and tunable mechanical properties, Int. J. Biol. Macromol., 2018, 118, 1463–1469 CrossRef CAS PubMed.
- X. Guo, H. Zhang, M. Wu, Z. Tian, Y. Chen, R. Bao, J. Hao, X. Cheng and C. Zhou, Silicon-Enhanced PVA Hydrogels in Flexible Sensors: Mechanism, Applications, and Recycling, Gels, 2024, 10, 788 CrossRef CAS PubMed.
- N. Wang, H.-W. Feng, X. Hao, Y. Cao, X.-D. Xu and S. Feng, Dynamic covalent bond and metal coordination bond-cross-linked silicone elastomers with excellent mechanical and aggregation-induced emission properties, Polym. Chem., 2023, 14, 1396–1403 RSC.
- S. Yu, H. Zuo, X. Xu, N. Ning, B. Yu, L. Zhang and M. Tian, Self-Healable Silicone Elastomer Based on the Synergistic Effect of the Coordination and Ionic Bonds, ACS Appl. Polym. Mater., 2021, 3, 2667–2677 CrossRef CAS.
- L. Wang, J. Zhou, L. Li and S. Feng, Poly(β-hydroxyl amine)s: Valuable Building Blocks for Supramolecular Elastomers with Tunable Mechanical Performance and Superior Healing Capacity, Polymers, 2022, 14, 699 CrossRef CAS PubMed.
- Y. Ji, Z. Wen, J. Fan, X. Zeng, X. Zeng, R. Sun and L. Ren, Adaptable thermal conductive, high toughness and compliant Poly(dimethylsiloxane) elastomer composites based on interfacial coordination bonds, Compos. Sci. Technol., 2023, 231, 109840 CrossRef CAS.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, A highly stretchable autonomous self-healing elastomer, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Highly stretchable and tough hydrogels, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- M. Tian, H. Zuo, J. Wang, N. Ning, B. Yu and L. Zhang, A silicone elastomer with optimized and tunable mechanical strength and self-healing ability based on strong and weak coordination bonds, Polym. Chem., 2020, 11, 4047–4057 RSC.
- Z. Jin, T. Chen, Y. Liu, W. Feng, L. Chen and C. Wang, Multivalent Design of Low-Entropy-Penalty Ion–Dipole Interactions for Dynamic Yet Thermostable Supramolecular Networks, J. Am. Chem. Soc., 2023, 145, 3526–3534 CrossRef CAS PubMed.
- C. N. Zhu, S. Y. Zheng, H. N. Qiu, C. Du, M. Du, Z. L. Wu and Q. Zheng, Plastic-Like Supramolecular Hydrogels with Polyelectrolyte/Surfactant Complexes as Physical Cross-links, Macromolecules, 2021, 54, 8052–8066 CrossRef CAS.
- H. N. Qiu, X. P. Hao, L. X. Hou, K. Cui, M. Du, Q. Zheng and Z. L. Wu, Formation and Destruction of Polyelectrolyte/Surfactant Complexes for the Toughening of Hydrogels, Macromolecules, 2023, 56, 8887–8898 CrossRef CAS.
- J. Gao, A. Zeb, H. Li, Y. Xie, Z. Li, J. Zhang, Y. Zhang and S. Zhang, Poly(ionic liquid)s-Based Ionogels for Sensor Applications, ACS Appl. Polym. Mater., 2024, 6, 14260–14272 CrossRef CAS.
- P. Shi, Y. Wang, W. W. Tjiu, C. Zhang and T. Liu, Highly Stretchable, Fast Self-Healing, and Waterproof Fluorinated Copolymer Ionogels with Selectively Enriched Ionic Liquids for Human-Motion Detection, ACS Appl. Mater. Interfaces, 2021, 13, 49358–49368 CrossRef CAS PubMed.
- J. Chen, Y. Wang, L. Li, Y.-E. Miao, X. Zhao, X.-P. Yan, C. Zhang, W. Feng and T. Liu, Visible-Light Transparent, Ultrastretchable, and Self-Healable Semicrystalline Fluorinated Ionogels for Underwater Strain Sensing, ACS Appl. Mater. Interfaces, 2023, 15, 16109–16117 CrossRef CAS PubMed.
- W. Li, L. Li, S. Zheng, Z. Liu, X. Zou, Z. Sun, J. Guo and F. Yan, Recyclable, Healable, and Tough Ionogels Insensitive to Crack Propagation, Adv. Mater., 2022, 34, 2203049 CrossRef CAS PubMed.
- P. Kujawa, A. Audibert-Hayet, J. Selb and F. Candau, Rheological properties of multisticker associative polyelectrolytes in semidilute aqueous solutions, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 1640–1655 CrossRef CAS.
- F. Candau and J. Selb, Hydrophobically-modified polyacrylamides prepared by micellar polymerization1Part of this paper was presented at the conference on ‘Associating Polymer’, Fontevraud, France, November 1997.1, Adv. Colloid Interface Sci., 1999, 79, 149–172 CrossRef CAS.
- Y. Wang, X. Fang, S. Li, H. Pan and J. Sun, Complexation of Sulfonate-Containing Polyurethane and Polyacrylic Acid Enables Fabrication of Self-Healing Hydrogel Membranes with High Mechanical Strength and Excellent Elasticity, ACS Appl. Mater. Interfaces, 2023, 15, 25082–25090 CrossRef CAS PubMed.
- G. Miquelard-Garnier, S. Demoures, C. Creton and D. Hourdet, Synthesis and Rheological Behavior of New Hydrophobically Modified Hydrogels with Tunable Properties, Macromolecules, 2006, 39, 8128–8139 CrossRef CAS.
- A. M. Al-Sabagh, N. G. Kandile, R. A. El-Ghazawy, M. R. Noor El-Din and E. A. El-sharaky, Synthesis and characterization of high molecular weight hydrophobically modified polyacrylamide nanolatexes using novel nonionic polymerizable surfactants, Egypt. J. Pet., 2013, 22, 531–538 CrossRef.
- S. Abdurrahmanoglu, V. Can and O. Okay, Design of high-toughness polyacrylamide hydrogels by hydrophobic modification, Polymer, 2009, 50, 5449–5455 CrossRef CAS.
- D. C. Tuncaboylu, M. Sahin, A. Argun, W. Oppermann and O. Okay, Dynamics and Large Strain Behavior of Self-Healing Hydrogels with and without Surfactants, Macromolecules, 2012, 45, 1991–2000 CrossRef CAS.
- L. Meng, C. Shao, C. Cui, F. Xu, J. Lei and J. Yang, Autonomous Self-Healing Silk Fibroin Injectable Hydrogels Formed via Surfactant-Free Hydrophobic Association, ACS Appl. Mater. Interfaces, 2020, 12, 1628–1639 CrossRef CAS PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Functional Supramolecular Polymeric Networks: The Marriage of Covalent Polymers and Macrocycle-Based Host–Guest Interactions, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed.
- B. Liu, H. Zhou, S. Zhou and J. Yuan, Macromolecules based on recognition between cyclodextrin and guest molecules: Synthesis, properties and functions, Eur. Polym. J., 2015, 65, 63–81 CrossRef CAS.
- B. Yang, Z. Wei, X. Chen, K. Wei and L. Bian, Manipulating the mechanical properties of biomimetic hydrogels with multivalent host–guest interactions, J. Mater. Chem. B, 2019, 7, 1726–1733 RSC.
- X. Huang, M. Zhang, J. Ming, X. Ning and S. Bai, High-Strength and High-Toughness Silk Fibroin Hydrogels: A Strategy Using Dynamic Host–Guest Interactions, ACS Appl. Bio Mater., 2020, 3, 7103–7112 CrossRef CAS PubMed.
- C. Liu, N. Morimoto, L. Jiang, S. Kawahara, T. Noritomi, H. Yokoyama, K. Mayumi and K. Ito, Tough hydrogels with rapid self-reinforcement, Science, 2021, 372, 1078–1081 CrossRef CAS PubMed.
- R. Ikura, J. Park, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Supramolecular Elastomers with Movable Cross-Linkers Showing High Fracture Energy Based on Stress Dispersion, Macromolecules, 2019, 52, 6953–6962 CrossRef CAS.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. Del Barrio and O. A. Scherman, Cucurbituril-Based Molecular Recognition, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, Y. Lan, C. Abell and O. A. Scherman, Biomimetic Supramolecular Polymer Networks Exhibiting both Toughness and Self-Recovery, Adv. Mater., 2017, 29, 1604951 CrossRef PubMed.
- C. S. Y. Tan, G. Agmon, J. Liu, D. Hoogland, E.-R. Janeček, E. A. Appel and O. A. Scherman, Distinguishing relaxation dynamics in transiently crosslinked polymeric networks, Polym. Chem., 2017, 8, 5336–5343 RSC.
- C. S. Y. Tan, J. del Barrio, J. Liu and O. A. Scherman, Supramolecular polymer networks based on cucurbit[8]uril host–guest interactions as aqueous photo-rheological fluids, Polym. Chem., 2015, 6, 7652–7657 RSC.
- C. Li, M. J. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. A. Scherman and D. Liu, Responsive Double Network Hydrogels of Interpenetrating DNA and CB[8] Host–Guest Supramolecular Systems, Adv. Mater., 2015, 27, 3298–3304 CrossRef CAS PubMed.
- R. Bai, H. Zhang, X. Yang, J. Zhao, Y. Wang, Z. Zhang and X. Yan, Supramolecular polymer networks crosslinked by crown ether-based host–guest recognition: dynamic materials with tailored mechanical properties in the bulk, Polym. Chem., 2022, 13, 1253–1259 RSC.
- J. Deng, R. Bai, J. Zhao, G. Liu, Z. Zhang, W. You, W. Yu and X. Yan, Insights into the Correlation of Cross-linking Modes with Mechanical Properties for Dynamic Polymeric Networks, Angew. Chem., 2023, 135, e202309058 CrossRef.
- C.-Y. Shi, Q. Zhang, C.-Y. Yu, S.-J. Rao, S. Yang, H. Tian and D.-H. Qu, An Ultrastrong and Highly Stretchable Polyurethane Elastomer Enabled by a Zipper–Like Ring–Sliding Effect, Adv. Mater., 2020, 32, 2000345 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, 1st edn, 2016 Search PubMed.
- H. Yokochi, R. T. O’Neill, T. Abe, D. Aoki, R. Boulatov and H. Otsuka, Sacrificial Mechanical Bond is as Effective as a Sacrificial Covalent Bond in Increasing Cross-Linked Polymer Toughness, J. Am. Chem. Soc., 2023, 145, 23794–23801 CrossRef CAS PubMed.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: A Molecular Platform for Designing Functions beyond Chemical Recycling and Self-Healing, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- E. M. Lloyd, J. R. Vakil, Y. Yao, N. R. Sottos and S. L. Craig, Covalent Mechanochemistry and Contemporary Polymer Network Chemistry: A Marriage in the Making, J. Am. Chem. Soc., 2023, 145, 751–768 CrossRef CAS PubMed.
- Z. J. Wang and J. P. Gong, Mechanochemistry for On-Demand Polymer Network Materials, Macromolecules, 2025, 58, 4–17 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, A Thermally Re-mendable Cross-Linked Polymeric Material, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- H. M. Colquhoun, D. F. Lewis, A. Ben-Haida and P. Hodge, Ring-Chain Interconversion in High-Performance Polymer Systems. 2. Ring-Opening Polymerization-Copolyetherification in the Synthesis of Aromatic Poly(ether sulfones), Macromolecules, 2003, 36, 3775–3778 CrossRef CAS.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, Poly(thioether) Vitrimers via Transalkylation of Trialkylsulfonium Salts, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS PubMed.
- Y. Chen and K.-H. Chen, Synthesis and reversible photocleavage of novel polyurethanes containing coumarin dimer components, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 613–624 CrossRef CAS.
- I. Nakazawa, S. Suda, M. Masuda, M. Asai and T. Shimizu, pH-dependent reversible polymers formed from cyclic sugar- and aromatic boronic acid-based bolaamphiphiles, Chem. Commun., 2000, 881–882 RSC.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. Jin, H. J. Qi and W. Zhang, Heat- or Water-Driven Malleability in a Highly Recyclable Covalent Network Polymer, Adv. Mater., 2014, 26, 3938–3942 CrossRef CAS PubMed.
- J.-M. Lehn, Dynamers: dynamic molecular and supramolecular polymers, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- J. M. Winne, L. Leibler and F. E. D. Prez, Dynamic covalent chemistry in polymer networks: a mechanistic perspective, Polym. Chem., 2019, 10, 6091–6108 RSC.
- C. Lv, K. Zhao and J. Zheng, A Highly Stretchable Self-Healing Poly(dimethylsiloxane) Elastomer with Reprocessability and Degradability, Macromol. Rapid Commun., 2018, 39, 1700686 CrossRef PubMed.
- P. Wang, Z. Wang, L. Liu, G. Ying, W. Cao and J. Zhu, Self-Healable and Reprocessable Silicon Elastomers Based on Imine–Boroxine Bonds for Flexible Strain Sensor, Molecules, 2023, 28, 6049 CrossRef CAS PubMed.
- Y. Zhou, K. Mai, S. Yang, R. Huang, Q. Zhu, G. Yu, Y. Feng and J. Li, B–O Bond Reinforced Mechanical Stability of Alginate-Based Jammed Emulsion for Enhancing Pesticide Droplet Foliar Deposition, ACS Sustainable Chem. Eng., 2025, 13, 447–460 CrossRef CAS.
- H. Deng, J. Ye, Z. Zu, Z. Lin, H. Huang, L. Zhang, X. Ye and H. Xiang, Repairable, reprocessable and recyclable rigid silicone material enabled by dual dynamic covalent bonds crosslinking side-chain, Chem. Eng. J., 2023, 465, 143038 CrossRef CAS.
- F. Wang, C. Zhang, Y. Han, G. Yi, Z. Mao, J. Li, Z. Li and W. Wang, A hybrid reversible crosslinked polysiloxane elastomer with high toughness and recyclability, J. Polym. Sci., 2024, 62, 2147–2156 Search PubMed.
- S. Garcia-Manyes and A. E. M. Beedle, Steering chemical reactions with force, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Mechanochemical tools for polymer materials, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- S. Jung and H. J. Yoon, Heterocyclic Mechanophores in Polymer Mechanochemistry, Synlett, 2022, 863–874 CAS.
- H. Sakai, D. Aoki, K. Seshimo, K. Mayumi, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, Visualization and Quantitative Evaluation of Toughening Polymer Networks by a Sacrificial Dynamic Cross-Linker with Mechanochromic Properties, ACS Macro Lett., 2020, 9, 1108–1113 CrossRef CAS PubMed.
- T. Watabe, D. Aoki and H. Otsuka, Polymer-Network Toughening and Highly Sensitive Mechanochromism via a Dynamic Covalent Mechanophore and a Multinetwork Strategy, Macromolecules, 2022, 55, 5795–5802 CrossRef CAS.
- K. Yanada, D. Aoki and H. Otsuka, Mechanochromic elastomers with different thermo- and mechano-responsive radical-type mechanophores, Soft Matter, 2022, 18, 3218–3225 RSC.
- Z. Cao, Highly Stretchable Tough Elastomers Crosslinked by Spiropyran Mechanophores for Strain-Induced Colorimetric Sensing, Macromol. Chem. Phys., 2020, 221, 2000190 CrossRef CAS.
- J. W. Kim, Y. Jung, G. W. Coates and M. N. Silberstein, Mechanoactivation of Spiropyran Covalently Linked PMMA: Effect of Temperature, Strain Rate, and Deformation Mode, Macromolecules, 2015, 48, 1335–1342 CrossRef CAS.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, 3D-Printed Mechanochromic Materials, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS PubMed.
- Y. Chen, C. J. Yeh, Y. Qi, R. Long and C. Creton, From force-responsive molecules to quantifying and mapping stresses in soft materials, Sci. Adv., 2020, 6, eaaz5093 CrossRef CAS PubMed.
- Y. Chen, C. J. Yeh, Q. Guo, Y. Qi, R. Long and C. Creton, Fast reversible isomerization of merocyanine as a tool to quantify stress history in elastomers, Chem. Sci., 2021, 12, 1693–1701 RSC.
- Y. Chen, G. Sanoja and C. Creton, Mechanochemistry unveils stress transfer during sacrificial bond fracture of tough multiple network elastomers, Chem. Sci., 2021, 12, 11098–11108 RSC.
- Y. Tian, X. Cao, X. Li, H. Zhang, C.-L. Sun, Y. Xu, W. Weng, W. Zhang and R. Boulatov, A Polymer with Mechanochemically Active Hidden Length, J. Am. Chem. Soc., 2020, 142, 18687–18697 CrossRef CAS PubMed.
- G. E. Fantner, E. Oroudjev, G. Schitter, L. S. Golde, P. Thurner, M. M. Finch, P. Turner, T. Gutsmann, D. E. Morse, H. Hansma and P. K. Hansma, Sacrificial Bonds and Hidden Length: Unraveling Molecular Mesostructures in Tough Materials, Biophys. J., 2006, 90, 1411–1418 CrossRef CAS PubMed.
- Y. Deng and S. W. Cranford, Tunable Toughness of Model Fibers with Bio-Inspired Progressive Uncoiling Via Sacrificial Bonds and Hidden Length, J. Appl. Mech., 2018, 85, 111001 CrossRef.
- J. A. Doolan, L. S. Alesbrook, K. Baker, I. R. Brown, G. T. Williams, K. L. F. Hilton, M. Tabata, P. J. Wozniakiewicz, J. R. Hiscock and B. T. Goult, Next-generation protein-based materials capture and preserve projectiles from supersonic impacts, Nat. Nanotechnol., 2023, 18, 1060–1066 CrossRef CAS PubMed.
- A. Schön, B. R. Clarkson, M. Jaime and E. Freire, Temperature stability of proteins: Analysis of irreversible denaturation using isothermal calorimetry, Proteins: Struct., Funct., Bioinf., 2017, 85, 2009–2016 CrossRef PubMed.
- H. Wang, Z. Wei, Z. Liu, B. Zheng, Z. Zhang, X. Yan, L. He, T. Li and D. Zhao, Energy Dissipation and Toughening of Covalent Networks via a Sacrificial Conformation Approach, Angew. Chem., Int. Ed., 2025, 64, e202416790 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |