
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoluminescence fluctuations in single perovskite nanocrystals: structural, environmental and ligand effect
Hawi N. Nyiera
and
Jing Zhao
 *
*
Department of Chemistry, University of Connecticut, Storrs, CT 06269, USA. E-mail: jing.zhao@uconn.edu
First published on 10th June 2025
Abstract
Perovskite nanocrystals (PNCs) show great promise for optoelectronic devices; yet at the single-particle level, they are susceptible to photoluminescence (PL) fluctuations. Single-particle studies provide key insights into the photophysical processes responsible for these fluctuations. This review discusses both intrinsic factors, such as size and surface defects, and extrinsic factors, including moisture and oxygen, that contribute to PL instability in PNCs. We also highlight recent advancements in surface passivation techniques that effectively reduce or suppress the PL fluctuations, thereby enhancing the stability and optical performance of PNCs. Ultimately, understanding and mitigating PL fluctuations are essential for improving the stability and efficiency of PNC-based devices.
 Hawi N. Nyiera | Hawi N. Nyiera is a PhD candidate in the Department of Chemistry at the University of Connecticut, working under the supervision of Dr Jing Zhao. She earned her bachelor's degree in chemistry from Luther College in 2020. Her doctoral research centers on the surface functionalization of quantum dots for bio-labeling and optical spectroscopy of ensemble and single perovskite nanocrystals. She has been awarded the Masterton–Hurley Teaching Award, the Bobbitt-Chou Fellowship, and the Materials and Interfaces Outstanding Presentations by Young Investigators Award. |
 Jing Zhao | Dr Jing Zhao is a Professor and Associate Department Head of Chemistry at the University of Connecticut. She earned her PhD from Northwestern University and was a postdoc with Nobel Laureate Moungi Bawendi at MIT. She joined UConn in 2012 and became a full professor in 2022. Her research focuses on quantum dots, plasmon–exciton interactions, and nanoparticle applications in catalysis and biosensing. She received an NSF CAREER Award and several recognitions for career excellence. Dr Zhao is a member of the Connecticut Academy of Science and Technology and serves on the editorial board of Communications Chemistry. |
Introduction
Perovskite nanocrystals (PNCs) have the chemical formula ABX3, where A represents an inorganic or organic cation (i.e., Cs+, CH3NH3+ (MA+), CH(NH2)2+ (FA+)), B is a metal cation (i.e., Pb2+, Sn2+, Zn2+), and X is a halide anion (i.e., Cl−, Br−, I−). They have attracted significant attention due to their remarkable properties, including narrow emission line widths, high photoluminescence (PL) quantum yields, and size- and composition-dependent tunable band gaps.1–7 These characteristics make them promising candidates for light-emitting diodes, lasers, and photovoltaic applications.8–16 Nonetheless, similar to traditional II–VI, III–V, and IV–VI semiconductors, at the single-particle level, PNCs exhibit PL fluctuations, where the emission intensity varies among high (ON), intermediate (gray), and low (OFF) levels over time under continuous photoexcitation. The lowering in PL intensity occurs when the photogenerated excitons (electron–hole pairs) undergo non-radiative recombination after excitation such as trapping at a defect site, resulting in a temporary loss of PL emission. When non-radiative processes are not activated, radiative recombination can resume, resulting in the recovery of PL intensity. Fundamentally, these PL fluctuation properties reflect the inherent structural properties of the single emitters and their microenvironment. Specifically, for semiconductor NCs, they are related to their crystal structures, defects, and surfaces.PNCs offer greater defect tolerance compared to conventional semiconductor NCs such as CdSe and GaAs, primarily due to the higher formation energy of ion misplacement in the perovskite structure.4,7,17–20 As a result, the formation of interstitial and anti-site point defects is less likely, which reduces the possibility of creating deep traps (energy levels within the band gap). Nevertheless, due to their ionic nature, PNCs are not entirely free of defects and are susceptible to structural instability.18,21 Ion migration in the PNCs can create vacancies and cause the detachment of weakly bound surface ligands, which affects the stability of the PNCs. These vacancies then act as electron traps, increasing the rate of nonradiative recombination.20,22,23 Additionally, the oleylamine (OLA) and oleic acid (OA) ligands commonly used during PNC synthesis dynamically bind to and dissociate from the NC surface.24–26 The dynamic binding results in an unpassivated NC surface, giving rise to surface defects that act as traps and facilitate nonradiative recombination.22,26 Thus, although PNCs are less prone to deep trap formation due to their defect tolerance property, their ionic nature and dynamic surface chemistry contribute to PL fluctuations in these materials.
Photoluminescence fluctuation models
Different PL intensity fluctuation patterns have been observed in PNCs, known as “blinking” and “flickering”, shown in Fig. 1a and b, respectively. PL intensity “blinking” is characterized by abrupt switching between “ON” and “OFF” states, whereas “flickering” involves gradual intensity fluctuations between multiple intensity states. In addition, fluorescence lifetime intensity distribution (FLID) diagrams have been reported for PNCs. FLID diagrams are two-dimensional plots where the PL intensity and lifetime are determined and plotted, for each time bin (typically tens of milliseconds) of the PL data. These diagrams show the relationship between the PL intensity and PL lifetimes (Fig. 1c–e) and can be related to the different PL fluctuation models. Both the PL intensity time traces and FLIDs vary across the PNCs even when synthesized in the same batch. To date, several models have been proposed to explain the cause of PL fluctuations. Auger recombination has been shown to cause PL fluctuations in many kinds of semiconductor NCs (Fig. 1f).27–30 This phenomenon occurs due to the charging and discharging of PNCs, influenced by deep traps. These deep traps capture the electrons or holes generated after excitation, leaving behind an extra charge. When a new exciton forms, the NC enters a charged state (also known as a “trion”). In this trion state, the exciton can recombine radiatively; or more likely, the exciton undergoes a non-radiative Auger-like process, which often has a much higher rate than the rate of radiative recombination, by transferring energy to the additional charge (Fig. 1f). Compared to the high PL intensity states, the low intensity states caused by the Auger process are characterized by shorter PL lifetimes, as shown by the FLID in Fig. 1c.31,32 In the presence of shallow traps, hot carrier (HC) trapping and nonradiative band-edge carrier (NBC) recombination are proposed to cause PL fluctuations.33 HC trapping occurs before the hot carrier relaxes to the band edge, where the hot charge carrier (e.g. an excited electron) rapidly relaxes through a nonradiative recombination pathway via the traps near the band-edge, before another exciton is generated (Fig. 1g). This type of nonradiative recombination leads to a decrease in PL intensity but is not accompanied by a significant change in PL lifetime.33–36 However, the origin of these traps remains unknown. NBC recombination, on the other hand, is proposed to occur through the activation and deactivation of multiple recombination centers. This may result from the emptying and filling of surface trap states, potentially caused by the dynamic binding of surface ligands. The shallow and short-lived traps capture charge carriers from the band-edge as they relax, resulting in nonradiative recombination (Fig. 1h). As a result, the PL intensity fluctuates gradually due to competition between a fixed radiative rate and varying nonradiative rates in the NCs. The PL fluctuation pattern resulting from NBC recombination is characterized by gradual changes over time, known as flickering, with a gray state occurring more frequently than the ON state (Fig. 1b).33,34 Although FLID diagrams are presented to support the proposed models, more work is needed to clearly explain them, particularly given the complexity when analyzing flickering (as discussed later). Among the few studies that have reported flickering in PNCs, the corresponding FLID diagram typically shows a positive correlation between PL intensity and lifetime (Fig. 1e).37–39 While the proposed models described and shown in Fig. 1f–h represent possible recombination pathways responsible for the observed PL fluctuations in PNCs, it is important to note that a single PNC may exhibit PL fluctuations caused by a combination of these processes rather than by just one.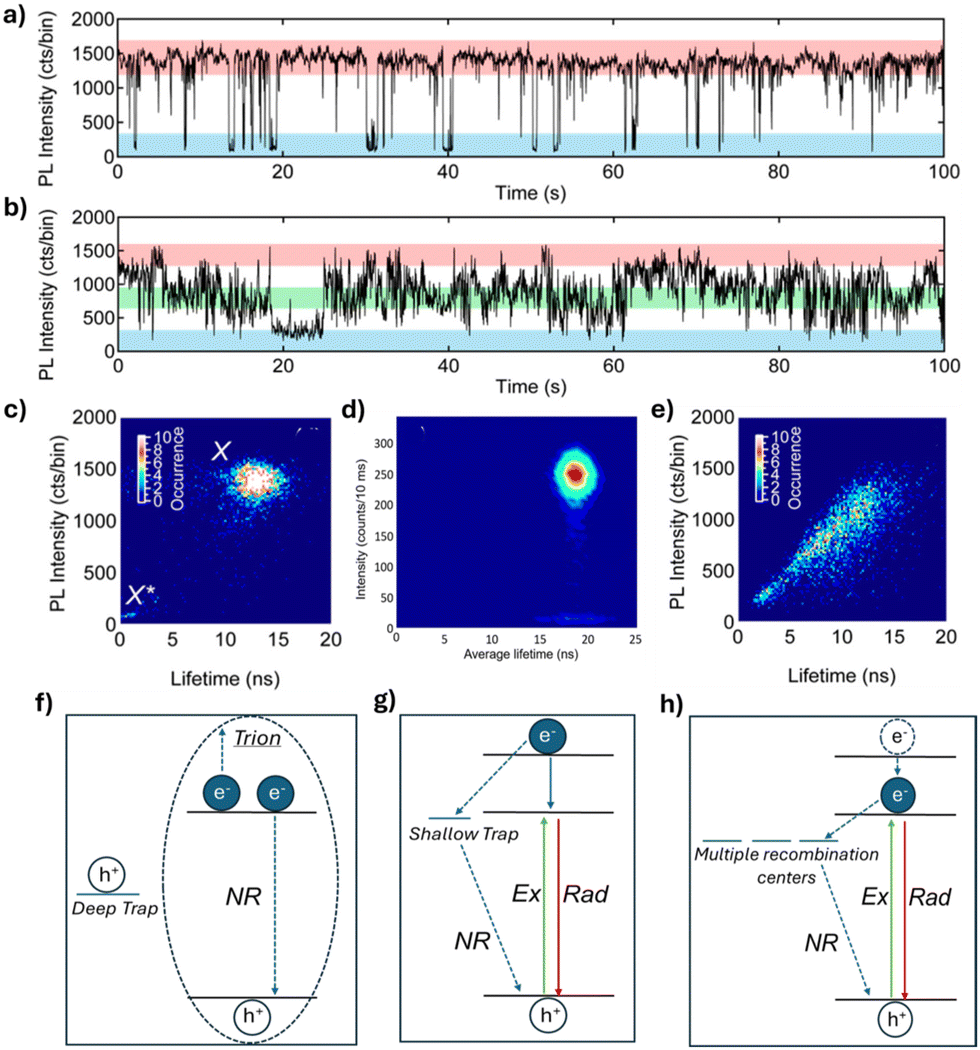 | ||
| Fig. 1 Representative PL intensity traces showing (a) blinking and (b) flickering in FAPbBr3 NCs. Reproduced from ref. 39 with permission from American Chemical Society, copyright 2017. (c–e) FLID diagrams of three FAPbBr3 NCs. Reproduced from ref. 36 with permission from American Chemical Society, copyright 2017, and ref. 39 with permission from Scientific Reports, copyright 2020. (f–h) Schematic illustrations of the PL fluctuation models: (f) Auger recombination; (g) hot carrier (HC) trapping; and (h) non-radiative band-edge exciton recombination via multiple recombination centers. Where “Ex” represents excitation, “Rad” represents radiative recombination, and “NR” represents nonradiative recombination. | ||
While PL fluctuations have been observed in PNCs, further research is needed to fully understand the mechanisms causing the fluctuations in single PNCs. PL fluctuations are more complex in PNCs, exhibiting flickering (Fig. 1b) – a behavior not commonly observed in traditional semiconductor NCs. Additionally, the ionic nature of perovskites makes them sensitive to environmental factors such as oxygen, moisture, high temperatures, and light, which can lead to irreversible phase transformations and decomposition during data acquisition, thereby limiting their experimental durations and complicating data collection and analysis.21,40–42 Despite these challenges, single PNC studies are sensitive to the variations between individual NCs and allow for the analysis of quenching, recombination, trapping, and the impact of external stimuli on exciton dynamics without being averaged by the ensemble. They provide a better understanding of how the various surface defects affect PNCs, which is of great importance to improve the durability and performance of PNC-based applications. This understanding can also give rise to more efficient strategies for material design, surface passivation, and device optimization. The following sections of this review will discuss recent advancements in single-particle PL studies of PNCs and examine the factors that affect their PL fluctuations (illustrated in Fig. 2). It also includes the authors’ perspective on future work in this research area.
 | ||
| Fig. 2 Summary of factors influencing PL fluctuations in PNCs. | ||
Role of polymer matrices during data collection
As previously mentioned, PNCs are highly sensitive to moisture and oxygen. Over time, this sensitivity worsens their optical properties and can lead to the ultimate decomposition of the PNCs, significantly affecting data collection. To overcome this problem, researchers have found that dispersing PNCs in a protective polymer matrix can prevent decomposition and extend the duration of data collection. Rainò et al. showed that the choice of polymer matrix plays a crucial role in this process.43 The polymer used must be soluble in the same apolar solvents as the PNCs and have low autofluorescence. Among the four polymers tested with inorganic CsPbBr3 NCs, i.e. poly(methyl methacrylate) (PMMA), cyclo-olefin copolymer (TOPAS), styrene–ethylene–butylene–styrene block copolymer (SEBS), and polystyrene, polystyrene displayed the best performance by ensuring stable PL emission and suppressing PL blueshift caused by decomposition of the PNCs (Fig. 3a). This is primarily due to its hydrophobic nature, with the aromatic rings in polystyrene showing a stronger interaction with the hydrophobic ligand shell of the CsPbBr3 NCs.43 Although the results suggested that PMMA may not be an ideal polymer matrix for PNCs, Chouhan et al. demonstrated that PMMA effectively maintained stable PL emission of organic–inorganic MAPbI3 NCs (Fig. 3b).44 Additional work has been conducted on tailoring polymer matrices to improve ligand–polymer interactions. For example, an oxygen-scavenging thiol-based polymer, off-stoichiometry thiol–ene (OSTE), was utilized to prevent photodegradation of MAPbX3 NCs caused by oxygen.45 As shown in Fig. 3c, the OSTE polymer encapsulates the NCs, providing a protective layer that prevents PL blueshift caused by decomposition of the NCs.42,45 While polymer matrices offer significant benefits in stabilizing PNCs and extending data collection time, the choice of polymer is critical and must be tailored to the specific type of NC to effectively prevent decomposition and maintain optical performance. | ||
| Fig. 3 (a) Two-dimensional PL spectral maps of CsPbBr3 NCs without polymer protection and embedded in PMMA and polystyrene. Reproduced from ref. 43 with permission from American Chemical Society, Copyright 2019. (b) PL intensity traces of single MAPbI3 NCs in air, argon, PMMA, and displaying a long-lived OFF state in air. Reproduced from ref. 44 with permission from Angewandte Chemie International Edition, Copyright 2019. (c) PL spectra evolution of single MAPbBr3 NCs without polymer protection and with OSTE film coating, under laser exposure for 15 and 30 minutes, respectively. Reproduced from ref. 45 with permission from American Chemical Society, Copyright 2019. | ||
Influence of excitation wavelength and intensity
Extrinsic factors, such as the excitation wavelength and intensity, have been shown to influence the PL fluctuations of PNCs. In a study by Gibson et al., PL intensity traces of single CsPbBr3 NCs were collected at varying excitation intensities.46 The traces were then analyzed by using an intensity threshold to define the “ON” (bright) and “OFF” (dark) states, and the durations (τi) for which the PL intensity remains in the ON and OFF states were then determined. By analyzing the probability distributions of the ON and OFF times, valuable information about the state-to-state kinetics can be obtained. Gibson et al. showed that in CsPbBr3 NCs, the ON-state times can be fitted with a truncated power law, , where the truncation time τc, is the duration at which the power law behavior of the ON-state transitions to exponential decay.46 The ON-state truncation time becomes shorter as the excitation intensity increases from 2.5 to 17.7 W cm−2 until saturation (Fig. 4a). Unlike the ON-state, the OFF-state did not show a truncation time dependence on the excitation intensity. Similar results were reported for organic–inorganic FAPbBr3 and CH3NH3PbBr3 NCs.47,48 In addition to their dependence on excitation intensity, the durations of the ON and OFF states have also been studied at different excitation wavelengths. Since the distribution of OFF times represents the recovery from low to high intensity states, the power-law behavior in the OFF times provides insights into the trapping and de-trapping dynamics of charge carriers within NCs.49,50 Singha et al. investigated the ON and OFF distributions of FaPbBr3 NCs excited at 400 nm and 440 nm, with power densities ranging from 1.4 to 4 W cm−2 (Fig. 4b).33 The results show that according to the truncated power law, an increase in excitation power density leads to a longer OFF-state duration for both 400 nm and 440 nm excitation. The key difference is that the OFF-state durations differ more significantly at 440 nm than at 400 nm, indicating a stronger dependence on the excitation power at higher wavelengths. This suggests that, at longer excitation wavelengths, the competition between HC trapping and Auger processes becomes more pronounced. This is due to the fact that at longer excitation wavelengths, the excitation energy is closer to the band edge, which reduces the likelihood of carriers being trapped in deep traps. This results in faster de-trapping rates and slower trapping rates.33 This phenomenon was also observed by Mandal et al., where the ON fraction (the percentage of time the NC spends in the ON state) of CsPbBr3 NCs increases as the excitation wavelength increases from 405 to 453 and then to 488 nm.51 This change is attributed to an increase in the ratio of carrier de-trapping rate to trapping rate at longer excitation wavelengths.
, where the truncation time τc, is the duration at which the power law behavior of the ON-state transitions to exponential decay.46 The ON-state truncation time becomes shorter as the excitation intensity increases from 2.5 to 17.7 W cm−2 until saturation (Fig. 4a). Unlike the ON-state, the OFF-state did not show a truncation time dependence on the excitation intensity. Similar results were reported for organic–inorganic FAPbBr3 and CH3NH3PbBr3 NCs.47,48 In addition to their dependence on excitation intensity, the durations of the ON and OFF states have also been studied at different excitation wavelengths. Since the distribution of OFF times represents the recovery from low to high intensity states, the power-law behavior in the OFF times provides insights into the trapping and de-trapping dynamics of charge carriers within NCs.49,50 Singha et al. investigated the ON and OFF distributions of FaPbBr3 NCs excited at 400 nm and 440 nm, with power densities ranging from 1.4 to 4 W cm−2 (Fig. 4b).33 The results show that according to the truncated power law, an increase in excitation power density leads to a longer OFF-state duration for both 400 nm and 440 nm excitation. The key difference is that the OFF-state durations differ more significantly at 440 nm than at 400 nm, indicating a stronger dependence on the excitation power at higher wavelengths. This suggests that, at longer excitation wavelengths, the competition between HC trapping and Auger processes becomes more pronounced. This is due to the fact that at longer excitation wavelengths, the excitation energy is closer to the band edge, which reduces the likelihood of carriers being trapped in deep traps. This results in faster de-trapping rates and slower trapping rates.33 This phenomenon was also observed by Mandal et al., where the ON fraction (the percentage of time the NC spends in the ON state) of CsPbBr3 NCs increases as the excitation wavelength increases from 405 to 453 and then to 488 nm.51 This change is attributed to an increase in the ratio of carrier de-trapping rate to trapping rate at longer excitation wavelengths.
 | ||
| Fig. 4 (a) ON-state probability distributions of CsPbBr3 NCs showing truncation at earlier times with increasing excitation intensity. Reproduced from ref. 46 with permission from American Chemical Society, Copyright 2018. (b) ON and OFF-state duration events extracted from PL intensity traces at different excitation wavelengths and intensities. Reproduced from ref. 33 with permission from the Journal of Chemical Physics, Copyright 2024. (c) PL fluctuation trajectories of a relatively small-sized CsPbI3 NC (σ = 1.61 × 10−13 cm2) and a relatively large-sized CsPbI3 NC (σ = 5.11 × 10−13 cm2), along with the corresponding FLID diagrams. Reproduced from ref. 37 with permission from the Journal of Chemical Physics, Copyright 2024. | ||
Dependence on size
The optical properties of PNCs depend not only on their composition but also on their size. In a recent study, Yang et al. determined the absorption cross section of single CsPbI3 NCs by recording the PL intensity of individual NCs at varying excitation powers to generate a PL saturation curve.37 The curve was then fit using the equation, I ∝ 1 − e−σj, where σ is the absorption cross section and j is the excitation photon flux calculated from the laser power, repetition rate, and photon energy. They observed different PL fluctuation patterns at low excitation powers for CsPbI3 NCs with an absorption cross section of 1.61 × 10−13 cm−2 and 5.11 × 10−13 cm2. The smaller NCs exhibit a series of continuously distributed emission states, and were considered to be “flickering” by the authors; while the larger NCs exhibited distinct binary ON and OFF emission states (Fig. 4c). Moreover, the PL intensity and lifetime show a linear correlation in smaller NCs and a non-linear correlation in larger NCs. Smaller NCs differ from larger ones primarily in the degree of overlap between their exciton wave functions and the electronic states associated with surface defects. In smaller NCs with sizes smaller than their exciton Bohr diameter, there is significant overlap between the two, which leads to fast trapping and de-trapping of carriers, resulting in NBC recombination via multiple recombination centers. In contrast, there is less overlap in larger NCs with sizes greater than the exciton Bohr diameter; therefore, trapping and de-trapping of carriers become slower and less efficient. This suggests that Auger recombination is primarily responsible for PL fluctuations in larger-sized CsPbI3 NCs.37 The influence of size in quantum-confined PNCs has also been studied. Paul et al. studied CsPbBr3 NCs with three sizes, 3.80, 4.80, and 5.90 nm.52 These different-sized NCs exhibit similar PL intensity traces. However, the FLID diagrams reveal that in smaller CsPbBr3 NCs, the high PL intensity states are associated with long lifetimes, while the low intensity states display either short or long lifetimes, corresponding to short- and long-lived carrier traps, respectively. In contrast, the larger size NCs showed mostly high-intensity-long-lifetime features. Smaller NCs also exhibit a lower ON-state fraction, indicating a higher carrier trapping rate and suggesting that de-trapping is more difficult in smaller NCs compared to larger ones.52 Similar results were reported in phenethylammonium bromide-treated CsPbBr3 NCs with sizes ranging from 3.6 to 14 nm.53 Overall, the size of PNCs plays a critical role in determining their charge carrier dynamics and PL fluctuations. Among the different-sized PNCs studied so far, smaller NCs exhibit higher trapping rates and more complex PL emission patterns, such as flickering, while larger NCs primarily display distinct ON and OFF states.Effects of external stimuli
Due to their ionic nature, PNCs are highly sensitive to changes in the external stimuli such as oxygen, moisture, heat, and light, which can lead to irreversible phase transitions and decomposition. In particular, elevated temperatures can accelerate ion migration, promoting the formation of non-radiative recombination centers, while humidity can facilitate displacement of surface ligands due to phase transformations affecting the stability of PNCs.40,42,54,55 To investigate this effect, Hong et al. monitored the PL intensity of individual CsPbBr3 NCs under various conditions, including nitrogen gas, dry air, oxygen, and moisture.56 When the CsPbBr3 NCs were exposed to a cycle of dry air, dry air + H2O, and then dry air again, an increase in the PL fluctuation intensity was observed in the presence of H2O (Fig. 5a). Specifically, the PL intensity increased at a humidity level of 40%. The increase in the PL intensity and the reduced OFF durations were attributed to the adsorption of H2O molecules on the PNC surface, which lowers the energetic barrier of midgap halide vacancies. These vacancies act as carrier traps; by lowering their energy barrier, H2O adsorption promotes their transition from an active state, where they capture charge carriers and cause nonradiative recombination, to a passive state, where they no longer trap charge carriers and allow radiative recombination to occur. It is worth noting that when the humidity was raised to 60%, a decrease in PL intensity, followed by complete quenching, was observed, attributed to the effect of aggregated water, which is known to induce PNC decomposition. This study also showed that pure oxygen causes strong PL quenching of the PNCs (Fig. 5b). This is due to the interaction of oxygen with surface defects and traps, which accelerates the degradation of the NCs.56 In a study by Yuan et al., the effect of moisture on the PL of single CsPbI3 PNCs in the presence of light was monitored.42 An increase in the PL intensity of CsPbI3 NCs was observed when exposed to moisture, as previously observed and explained.56 However, under continuous excitation, it was found that the CsPbI3 NCs lost PL emission within 5–10 min, indicating that light accelerates the degradation process. Additionally, a blue shift was observed in the PL spectra of the CsPbI3 NCs when exposed to either moisture or continuous laser excitation, indicating a decrease in the size of the NCs due to decomposition.42 While the aforementioned studies focus on quantum-confined PNCs, similar degradation behavior in the presence of moisture and oxygen has been observed in MAPbI3 single crystals, approximately 800 nm in size, which also exhibit PL fluctuations.57 | ||
| Fig. 5 (a) PL intensity trace of single CsPbBr3 NC monitored under alternating exposure to dry air, air + H2O, and dry air atmosphere. (b) PL intensity trace of single CsPbBr3 NC monitored under alternating exposure to air, oxygen, and air atmosphere. Reproduced from ref. 56 with permission from American Chemical Society, Copyright 2022. (c) Normalized temperature-dependent PL intensity traces and corresponding intensity histograms with logarithmic vertical axis. Reproduced from ref. 59 with permission from Nature Communications, Copyright 2019. | ||
Temperature-dependent PL properties have been observed in CdSe-based NCs and also in PNCs.6,31,58 The PL quantum yield of organo-metal PNCs has been shown to increase at lower temperatures, indicating reduced nonradiative recombination. Gerhard et al. studied the temperature-dependent PL properties of single MAPbI3 NCs and observed a progressive reduction in PL intensity fluctuation upon cooling from 300 K to 77 K (Fig. 5c), with the most pronounced reduction occurring below 200 K.59 This reduction in PL fluctuation is attributed to the nonradiative channels switching from an active to a passive state at lower temperatures. This switching is driven by thermal barriers in the range of 0.2–0.8 eV. At lower temperatures, ion migration is less likely to occur due to the high activation barrier, which leads to reduced PL fluctuation and increased PL intensity. At higher temperatures, ions can easily overcome the activation barrier, leading to ion migration. This migration generates defects such as vacancies or interstitials, which act as traps, resulting in increased non-radiative recombination.59 Similarly, Rainò et al. showed that at low temperatures of 6 K, CsPb(Cl/Br3) shows good photostability without surface passivation.60 In general, the sensitivity of PNCs to environmental factors such as humidity, oxygen, and temperature significantly impacts their PL behavior and stability. Understanding these effects, such as moisture-induced PL enhancement/quenching and oxygen-induced quenching, provides valuable insights into the degradation mechanisms of PNCs and highlights the importance of environmental control for optimizing their optical performance.
Achieving PL fluctuation suppression through surface passivation
Although the atomistic nature of the surface states in PNCs is often unknown, several studies have shown that surface treatment can significantly improve the optical properties of PNCs, primarily by reducing surface defects caused by vacancies and desorption of surface ligands, which create vulnerable sites for moisture and oxygen to react with the PNCs.4 The strategies include encapsulation with an inorganic shell,61,62 surface treatment through the addition of excess halide salts or pseudohalogens to passivate any vacancies,23,34,63–65 and using alternative ligands that have a stronger binding affinity to the NC surface.38,66,67One effective method of surface passivation is through shell growth. The shell can serve as a protective barrier against environmental factors, such as oxygen and moisture, and improve the optical properties of NCs.5,17 Guo et al. demonstrate improved PL properties of Cs4PbBr6 NCs through the encapsulation using alumina. The alumina-coated Cs4PbBr6 NCs show reduced PL intensity fluctuation and improved stability due to the passivation of surface defects. Additionally, delayed emission was observed, resulting from charge trapping, storage, and subsequent recovery to the emissive manifold.61 Tang et al. encapsulated CsPbBr3 NCs with CdS to reduce the occurrence of nonradiative Auger recombination.62 The CsPbBr3/CdS core/shell NCs exhibited an average ON fraction larger than 99% with little to no grey states, indicating reduced deep-trap formation due to the CdS shell.
In addition to shell growth, passivation of surface states can also be achieved through filling the halide vacancies caused by ion migration or the loss of capping ligands.22,68 The halide vacancies formed on the NC surface act as electron traps, increasing the non-radiative rate. Surface passivation can be achieved through the addition of halide salts. Park et al. investigated the PL fluctuation properties of CsPbBr3 NCs modified with ZnBr2. The modified NCs exhibited a low charge trapping rate compared to pristine CsPbBr3 NCs. The excess bromide increased the surface Br− ratio and resulted in improved surface capping and suppressed PL fluctuation.63 Similar observations have been made by Chouhan et al. by adding MABr and MAI during data collection to suppress PL intensity fluctuations of single MAPbX3 NCs (Fig. 6a).23 In addition to halide salts, pseudohalogens, which resemble the chemistry of true halogens, can be used to passivate halide vacancies.34,39 Yarita et al. observed the impact of sodium thiocyanate (NaSCN) on the PL fluctuation of single FaPbBr3 NCs. Of the two types of PL fluctuation patterns observed, blinking and flickering, flickering behavior was completely suppressed while blinking remained unchanged (Fig. 6b), suggesting that surface states or the surrounding environment play an important role in the origin of flickering as opposed to charging/discharging of the NCs.39
 | ||
| Fig. 6 (a) PL intensity trajectories showing fluctuation suppression of MAPbI3 NCs (i) before and (ii) after treatment with a MAI solution. Reproduced from ref. 23 with permission from American Chemical Society, Copyright 2021. (b) Time-integrated PL intensity for the low PL intensity region plotted for untreated and sodium thiocyanate (NaSCN) treated single FAPbBr3 NCs. Reproduced from ref. 39 with permission from American Chemical Society, Copyright 2017. (c) Distribution of the ON percentages for oleic acid/oleylamine (red) and lecithin (blue) capped CsPbBr3 NCs. Reproduced from ref. 66 with permission from American Chemical Society, Copyright 2024. | ||
Due to the dynamic binding of oleylamine (OLA) and oleic acid (OA) ligands that are commonly used in the synthesis of PNCs, the resulting NC surface is prone to disorder and defect formation. Proton transfer is required to transform OA and OLA into their ionic forms, which act as capping ligands for PNCs. While the oleate anion binds strongly to surface lead cations, the oleylammonium cation interacts weakly with surface halides, resulting in a dynamic equilibrium that promotes ligand desorption and leads to an unpassivated surface.69,70 To mitigate this issue, alternative ligands with a stronger binding affinity to the NC surface have been employed. Recent studies have explored amine-free synthesis routes, Praneeth et al. demonstrated that replacing oleylamine with trioctylphosphine as a capping ligand in the synthesis of CsPbBr3 NCs resulted in amine-free PNCs exhibiting stable, non-blinking PL with no long-lived OFF states.71 This is attributed to reduced nonradiative recombination due to more efficient surface passivation by trioctylphosphine compared to oleylamine. Gallagher et al. investigated how changes in ligand equilibrium affect the PL properties of lecithin-capped CsPbBr3 NCs compared to those capped with OA/OLA ligands.66 Lecithin-capped NCs exhibited more stable PL emission, spending on average 68% of the time in the ON state compared to 30% for OA/OLA capped NCs, and showed a higher probability of staying in the ON state (Fig. 6c). This enhanced stability is attributed to the stronger binding affinity of lecithin to the CsPbBr3 NC surface, which better preserves surface integrity. In contrast, OA/OLA ligands are more weakly bound and tend to detach during the dilution process used to prepare single-particle samples, leading to surface degradation and less stable emission.66 Comparable results were reported by Kuang et al., who found that the density of trap states increases as the surface ligands of CsPbBr3 NCs decrease.72 Similarly, alkylthiols such as ethanethiol have also been utilized as an alternative capping ligand for CsPbBr3 NCs. Seth et al. observed both blinking and flickering PL fluctuation behaviors in single CsPbBr3 NCs. Treating these NCs with ethanethiol converts the flickering of some NCs to blinking, but no change was observed for the blinking ones. The reduction of flickering is attributed to the passivation of uncoordinated lead atoms on the NC surface, which serve as shallow electron traps.38 These findings highlight the significant impact of surface treatment and passivation strategies on the optical properties and stability of PNCs. By employing methods such as shell growth, halide salt addition, and alternative ligands, suppressed PL fluctuations and enhanced stability can be achieved.
Conclusions and future perspectives
In summary, single-particle studies have greatly improved our understanding of the stability and degradation mechanisms in PNCs, particularly with regard to photoinduced degradation and ion migration. By monitoring the optical behavior of individual NCs, these studies have provided valuable insights into how external factors like light exposure, temperature, and moisture contribute to the degradation process. Photoinduced degradation, often marked by PL quenching and spectral shifts, has been shown to reduce the photostability of PNCs over time. Ion migration, driven by factors such as temperature and humidity, exacerbates this issue by creating surface defects, which in turn lead to non-radiative recombination and PL fluctuations. These studies also highlight potential solutions, such as surface passivation through encapsulation or ligand modification, which can mitigate the impact of these degradation processes and improve the stability of PNCs. Single PNC studies deepen our understanding of fundamental degradation pathways, thereby enabling the development of more stable and efficient PNC-based materials for a wide range of optoelectronic applications.Experimental challenges arise when considering the impact of environmental factors, such as moisture and oxygen, on the measurements of single PNCs. Although various strategies have been developed to improve the stability of PNCs for single particle studies, there is still a clear demand for methods to achieve long-term stability of PNCs in air or aqueous environments under photoexcitation, enabling their applications in single particle tracking in biological systems. Additionally, low laser power is typically required for long acquisition times in single PNC studies to reduce photodegradation. However, this results in a lower PL signal, reducing the signal-to-noise ratio and photon counts, which can make it difficult challenging to distinguish between different intensity states, a difficulty further compounded by “flickering”. “Flickering” is characterized by continuous variation of PL intensity over time but has not been clearly defined to date. This qualitative description of PL intensity “flickering” makes it harder to identify distinct intensity states and to understand the mechanism causing this behavior.46 To address this, statistical methods such as change point analysis have been attempted.33,46,66,73 However, caution is needed when applying these methods, as the quality of the data can heavily influence the results. Customized statistical analysis tools have also been developed in analyzing single PNCs. For example, Gallagher et al. integrated unsupervised clustering with change point analysis to classify CsPbBr3 NCs based on the number of discrete intensity levels in their PL intensity traces.66 This approach also enabled the classification of ON and OFF states. Simulated PL traces were used for validating the custom change point analysis package, which demonstrated that the method can reliably resolve up to five distinct intensity states. This shows that the method could be confidently used to analyze experimental PL fluctuation data of single PNCs. Beyond statistical analysis tools, machine learning tools have also been developed to track fluorescence trajectories in single molecules.74 The algorithm automatically segments and clusters data without prior assumptions, identifying patterns in complex datasets. Such tools can be utilized in the analysis of PNCs given their complex nature. While statistical methods and machine learning offer promising tools for resolving intensity states of single PNCs, their effectiveness is limited by data quality, amount of data, and low PL signal level, which can lead to underestimation of the number of intensity states or intensity state changes being missed or wrongly identified.73 Purely data-driven machine learning or statistical approaches may lack the interpretability of physical processes in PNCs, while physics-informed models offer mechanistic insights into the exciton lifecycles in PNCs. Thus, integrating these approaches with physics-informed models will be essential to connect single-particle data with fundamental charge carrier dynamics in PNCs.75
In addition to the challenge of analyzing PL intensity fluctuations, the FLID diagrams observed in single PNCs vary from NC-to-NC, even if they exhibit similar PL intensity fluctuations. Fig. 7a and b are examples of FLIDs for “blinking” and “flickering” PNCs and they differ significantly from the ones in Fig. 1c–e.38 Thus, a deeper understanding of the origins of shallow versus deep traps, particularly in relation to surface chemistry and defect types, is crucial for advancing mechanistic insights. Shallow traps are often associated with processes such as dynamic ligand binding or the presence of uncoordinated lead atoms, are believed to cause “flickering”. In contrast, deep traps, which may arise from intrinsic lattice defects such as vacancies or interstitials, are more likely to cause distinct OFF states, characteristic of “blinking”. Surface chemistry, including the presence of moisture, oxygen, and dynamic ligand passivation, can influence the formation of both shallow and deep traps, which can affect the observed PL fluctuation patterns. For example, a halide vacancy that would act as a deep trap in an unpassivated PNC may not lead to trap formation in the presence of strongly binding ligands such as sulfonic or phosphonic acids.76,77 A hybrid approach that considers both the influence of surface-induced shallow traps and intrinsic deep traps may provide a more comprehensive understanding of PL fluctuations in PNCs. To recognize the connections between PL intensity trajectories and FLIDs, advanced statistical methods and computational approaches could be considered and developed.
 | ||
| Fig. 7 FLID diagrams with false color representation of single CsPbBr3 NCs exhibiting (a) blinking and (b) flickering. Reproduced from ref. 38 with permission from American Chemical Society, Copyright 2018. | ||
Ever since single-particle PL fluctuations were observed in single semiconductor NCs, the mechanistic understanding of these phenomena has always been of great interest. Although several models have been proposed to explain PL intermittency in PNCs (Fig. 1f–h), the nature of the traps (such as deep vs. shallow) is still unknown, especially at the atomistic scale. Using correlated multi-modal measurements (e.g., combining PL with electron microscopy), it is possible to link PL emission behavior to structural dynamics at the single-particle level. However, such measurements require complicated and expensive instrumentation and special technical skills. Moreover, the changes in the structure of PNCs upon exposure to electron beams cannot be ignored. Alternatively, atomic-level computations may provide useful information about the defects in PNCs and how they influence the electronic band structures. It remains a challenge to correlate these properties with exciton dynamics in single PNCs. While single-particle studies provide valuable insights that are not detectable at the ensemble level, combining them with ensemble measurements offers additional benefits.78,79 For instance, Yarita et al. employed femtosecond transient-absorption spectroscopy, time-resolved PL spectroscopy to study PNCs at the ensemble level, and second-order photon correlation spectroscopy on individual CsPbBr3 NCs.79 This approach allowed them to gain a deeper understanding of the behaviors of excitons, charged excitons, and biexcitons. Future studies may integrate single-particle and ensemble measurements to gain a more comprehensive understanding of the optical properties of PNCs. Moving beyond isolated conditions, it is highly valuable to probe PNCs in device-like conditions or working devices (e.g., LEDs or solar cells) to obtain the exciton dynamics of PNCs in operando.
Author contributions
H. N. prepared the manuscript under the guidance of J. Z. Both authors have approved the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support from the National Science Foundation (CHE-2203854) for this project.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- H. Zhu, T. Šverko, J. Zhang, D. B. Berkinsky, W. Sun, C. J. Krajewska and M. G. Bawendi, Nano Lett., 2022, 22, 8355–8362 CrossRef CAS PubMed.
- M. C. Brennan, A. Forde, M. Zhukovskyi, A. J. Baublis, Y. V. Morozov, S. Zhang, Z. Zhang, D. S. Kilin and M. Kuno, J. Phys. Chem. Lett., 2020, 11, 4937–4944 CrossRef CAS PubMed.
- S. Seth, T. Ahmed, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 1610–1618 CrossRef CAS.
- T. K. T. Tran, H. N. Nyiera, C. Brea, S. N. Ruiz, C. Wang, H. Tan, S. L. Suib, G. Hu and J. Zhao, ACS Appl. Nano Mater., 2024, 7, 12153–12162 CrossRef.
- H. Utzat, W. Sun, A. E. K. Kaplan, F. Krieg, M. Ginterseder, B. Spokoyny, N. D. Klein, K. E. Shulenberger, C. F. Perkinson, M. V. Kovalenko and M. G. Bawendi, Science, 2019, 363, 1068–1072 CrossRef CAS PubMed.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- C. Y. Huang, C. Zou, C. Mao, K. L. Corp, Y. C. Yao, Y. J. Lee, C. W. Schlenker, A. K. Y. Jen and L. Y. Lin, ACS Photonics, 2017, 4, 2281–2289 CrossRef CAS.
- Q. Zhang, R. Su, W. Du, X. Liu, L. Zhao, S. T. Ha and Q. Xiong, Small Methods, 2017, 1, 1700163 CrossRef.
- J. Tian, Q. Y. Tan, Y. Wang, Y. Yang, G. Yuan, G. Adamo and C. Soci, Nat. Commun., 2023, 14, 1433 CrossRef CAS PubMed.
- M. Hao, S. Ding, S. Gaznaghi, H. Cheng and L. Wang, ACS Energy Lett., 2024, 9, 308–322 CrossRef CAS.
- C. Wang, W. Meng, Y. Li, G. Xu, M. Peng, S. Nie and Z. Deng, Nanoscale, 2023, 15, 1661 RSC.
- Y. Dong, Y.-K. Wang, F. Yuan, A. Johnston, Y. Liu, D. Ma, M.-J. Choi, B. Chen, M. Chekini, S.-W. Baek, L. K. Sagar, J. Fan, Y. Hou, M. Wu, S. Lee, B. Sun, S. Hoogland, R. Quintero-Bermudez, H. Ebe, P. Todorovic, F. Dinic, P. Li, H. K. Kung, M. I. Saidaminov, E. Kumacheva, E. Spiecker, L.-S. Liao, O. Voznyy, Z.-H. Lu and E. H. Sargent, Nat. Nanotechnol., 2020, 15, 668–674 CrossRef CAS PubMed.
- X. Li, K. Zhang, J. Li, J. Chen, Y. Wu, K. Liu, J. Song, H. Zeng, X. Li, K. Zhang, J. Li, J. Chen, Y. Wu, K. Liu, J. Song and H. Zeng, Adv. Mater. Interfaces, 2018, 5, 1800010 CrossRef.
- H. S. Jung and N.-G. Park, Small, 2015, 11, 10–25 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- T. K. T. Tran, H. N. Nyiera and J. Zhao, Nano Res., 2024, 17, 10607–10619 CrossRef CAS.
- I. du Fossé, J. T. Mulder, G. Almeida, A. G. M. Spruit, I. Infante, F. C. Grozema and A. J. Houtepen, J. Am. Chem. Soc., 2022, 144, 11059–11063 CrossRef PubMed.
- D. Meggiolaro, S. G. Motti, E. Mosconi, A. J. Barker, J. Ball, C. Andrea, R. Perini, F. Deschler, A. Petrozza and F. De Angelis, Energy Environ. Sci., 2018, 11, 702–713 RSC.
- H. Jin, E. Debroye, M. Keshavarz, I. G. Scheblykin, M. B. J. Roeffaers, J. Hofkens and J. A. Steele, Mater. Horiz., 2020, 7, 397–410 RSC.
- B. P. Kore, M. Jamshidi and J. M. Gardner, Mater. Adv., 2024, 5, 2200–2217 RSC.
- D. Kim, T. Yun, S. An and C. L. Lee, Nano Converg., 2024, 11, 4 CrossRef CAS PubMed.
- V. Biju, L. Chouhan, S. Ito, E. M. Thomas, Y. Takano, S. Ghimire and H. Miyasaka, ACS Nano, 2021, 15, 2831–2838 CrossRef PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- N. Fiuza-Maneiro, K. Sun, I. López-Fernández, S. Gómez-Graña, P. Müller-Buschbaum and L. Polavarapu, ACS Energy Lett., 2023, 8, 1152–1191 CrossRef CAS.
- J. De Roo, M. Iba, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- R. Vaxenburg, A. Rodina, A. Shabaev, E. Lifshitz and A. L. Efros, Nano Lett., 2015, 15, 2092–2098 CrossRef CAS PubMed.
- A. T. Nguyen, P. Cavanaugh, I. J. La Plante, C. Ippen, R. Ma and D. F. Kelley, J. Phys. Chem. C, 2021, 125, 15405–15414 CrossRef CAS.
- V. I. Klimov, J. Phys. Chem. B, 2006, 110, 16827–16845 CrossRef CAS PubMed.
- M. Nirmal, B. O. Dabbousi, M. G. Bawendi, J. J. Macklin, J. K. Trautman, T. D. Harris and L. E. Brus, Nature, 1996, 383, 802–804 CrossRef CAS.
- A. L. Efros and D. J. Nesbitt, Nat. Nanotechnol., 2016, 11, 661–671 CrossRef CAS PubMed.
- C. Galland, Y. Ghosh, A. Steinbrück, M. Sykora, J. A. Hollingsworth, V. I. Klimov and H. Htoon, Nature, 2011, 479, 203–207 CrossRef CAS PubMed.
- P. K. Singha, T. Mukhopadhyay, E. Tarif, F. Ali and A. Datta, J. Chem. Phys., 2024, 161, 054704 CrossRef CAS PubMed.
- T. Ahmed, S. Seth and A. Samanta, ACS Nano, 2019, 13, 13537–13544 CrossRef CAS PubMed.
- G. Yuan, D. E. Gómez, N. Kirkwood, K. Boldt and P. Mulvaney, ACS Nano, 2018, 12, 3397–3405 CrossRef CAS PubMed.
- C. T. Trinh, D. N. Minh, K. J. Ahn, Y. Kang and K. G. Lee, Sci. Rep., 2020, 10, 2172 CrossRef CAS PubMed.
- C. Yang, G. Zhang, Y. Gao, B. Li, X. Han, J. Li, M. Zhang, Z. Chen, Y. Wei, R. Chen, C. Qin, J. Hu, Z. Yang, G. Zeng, L. Xiao and S. Jia, J. Chem. Phys., 2024, 160, 174505 CrossRef CAS PubMed.
- S. Seth, T. Ahmed and A. Samanta, J. Phys. Chem. Lett., 2018, 9, 7007–7014 CrossRef CAS PubMed.
- N. Yarita, H. Tahara, M. Saruyama, T. Kawawaki, R. Sato, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 6041–6047 CrossRef CAS PubMed.
- S. Cheng and H. Zhong, J. Phys. Chem. Lett., 2022, 13, 2281–2290 CrossRef CAS PubMed.
- T. D. Siegler, W. A. Dunlap-Shohl, Y. Meng, Y. Yang, W. F. Kau, P. P. Sunkari, C. E. Tsai, Z. J. Armstrong, Y. C. Chen, D. A. C. Beck, M. Meilǎ and H. W. Hillhouse, J. Am. Chem. Soc., 2022, 144, 5552–5561 CrossRef CAS PubMed.
- G. Yuan, C. Ritchie, M. Ritter, S. Murphy, D. E. Gómez and P. Mulvaney, J. Phys. Chem. C, 2018, 122, 13407–13415 CrossRef CAS.
- G. Rainò, A. Landuyt, F. Krieg, C. Bernasconi, S. T. Ochsenbein, D. N. Dirin, M. I. Bodnarchuk and M. V. Kovalenko, Nano Lett., 2019, 19, 3648–3653 CrossRef PubMed.
- L. Chouhan, S. Ghimire and V. Biju, Angew. Chem., Int. Ed., 2019, 58, 4875–4879 CrossRef CAS PubMed.
- L. Liu, L. Deng, S. Huang, P. Zhang, J. Linnros, H. Zhong and I. Sychugov, J. Phys. Chem. Lett., 2019, 10, 864–869 CrossRef CAS PubMed.
- N. A. Gibson, B. A. Koscher, A. P. Alivisatos and S. R. Leone, J. Phys. Chem. C, 2018, 122, 12106–12113 CrossRef CAS.
- C. T. Trinh, D. N. Minh, K. J. Ahn, Y. Kang and K.-G. Lee, ACS Photonics, 2018, 5, 4937–4943 CrossRef CAS.
- X. Han, G. Zhang, B. Li, C. Yang, W. Guo, X. Bai, P. Huang, R. Chen, C. Qin, J. Hu, Y. Ma, H. Zhong, L. Xiao and S. Jia, Small, 2020, 16, 2005435 CrossRef CAS PubMed.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2000, 112, 3117–3120 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028–1040 CrossRef CAS.
- S. Mandal, S. Mukherjee, C. K. De, D. Roy, S. Ghosh and P. K. Mandal, J. Phys. Chem. Lett., 2020, 11, 1702–1707 CrossRef CAS PubMed.
- S. Paul, G. Kishore and A. Samanta, J. Phys. Chem. C, 2023, 127, 10207–10214 CrossRef CAS.
- C. Mi, G. C. Gee, C. W. Lander, D. Shin, M. L. Atteberry, N. G. Akhmedov, L. Hidayatova, J. D. DiCenso, W. T. Yip, B. Chen, Y. Shao and Y. Dong, Nat. Commun., 2025, 16, 204 CrossRef CAS PubMed.
- X. Yuan, P. Jing, J. Li, M. Wei, J. Hua, J. Zhao, L. Tian and J. Li, RSC Adv., 2016, 6, 78311–78316 RSC.
- M. H. Miah, M. B. Rahman, M. Nur-E-Alam, M. A. Islam, M. Shahinuzzaman, M. R. Rahman, M. H. Ullah and M. U. Khandaker, RSC Adv., 2025, 15, 628–654 RSC.
- D. Hong, Y. Zhang, S. Pan, H. Liu, W. Mao, Z. Lu and Y. Tian, J. Phys. Chem. Lett., 2022, 13, 10751–10758 CrossRef CAS PubMed.
- A. Halder, N. Pathoor, A. Chowdhury and S. K. Sarkar, J. Phys. Chem. C, 2018, 122, 15133–15139 CrossRef CAS.
- S. A. Empedocles, D. J. Norris and M. G. Bawendi, Phys. Rev. Lett., 1996, 77, 3873–3876 CrossRef CAS PubMed.
- M. Gerhard, B. Louis, R. Camacho, A. Merdasa, J. Li, A. Kiligaridis, A. Dobrovolsky, J. Hofkens and I. G. Scheblykin, Nat. Commun., 2019, 10, 1698 CrossRef PubMed.
- G. Rainò, G. Nedelcu, L. Protesescu, M. I. Bodnarchuk, M. V. Kovalenko, R. F. Mahrt and T. Stöferle, ACS Nano, 2016, 10, 2485–2490 CrossRef PubMed.
- T. Guo, R. Bose, X. Zhou, Y. N. Gartstein, H. Yang, S. Kwon, M. J. Kim, M. Lutfullin, L. Sinatra, I. Gereige, A. Al-Saggaf, O. M. Bakr, O. F. Mohammed and A. V. Malko, J. Phys. Chem. Lett., 2019, 10, 6780–6787 CrossRef CAS PubMed.
- X. Tang, J. Yang, S. Li, Z. Liu, Z. Hu, J. Hao, J. Du, Y. Leng, H. Qin, X. Lin, Y. Lin, Y. Tian, M. Zhou and Q. Xiong, Adv. Sci., 2019, 6, 1900412 CrossRef CAS PubMed.
- J. Park, Y. Kim, S. Ham, J. Y. Woo, T. Kim, S. Jeong and D. Kim, Nanoscale, 2020, 12, 1563–1570 RSC.
- H. Jin, J. A. Steele, R. Cheng, N. Parveen, M. B. J. Roeffaers, J. Hofkens and E. Debroye, Adv. Opt. Mater., 2021, 9, 2002240 CrossRef CAS.
- S. Paul and A. Samanta, J. Phys. Chem. Lett., 2022, 13, 5742–5750 CrossRef CAS PubMed.
- S. Gallagher, J. Kline, F. Jahanbakhshi, J. C. Sadighian, I. Lyons, G. Shen, B. F. Hammel, S. Yazdi, G. Dukovic, A. M. Rappe and D. S. Ginger, ACS Nano, 2024, 18, 19208–19219 CrossRef CAS PubMed.
- C. Mi, M. L. Atteberry, V. Mapara, L. Hidayatova, G. C. Gee, M. Furis, W. T. Yip, B. Weng and Y. Dong, J. Phys. Chem. Lett., 2023, 14, 5466–5474 CrossRef CAS PubMed.
- Y. Wu, C. Wei, X. Li, Y. Li, S. Qiu, W. Shen, B. Cai, Z. Sun, D. Yang, Z. Deng and H. Zeng, ACS Energy Lett., 2018, 3, 2030–2037 CrossRef CAS.
- S. Akhil, S. Biswas, M. Palabathuni, R. Singh and N. Mishra, J. Phys. Chem. Lett., 2022, 13, 9480–9493 CrossRef CAS PubMed.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. Ten Brinck, P. Aellen, A. Süess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C. J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS PubMed.
- N. V. S. Praneeth, S. Akhil, A. Mukherjee, S. Seth, S. Khatua and N. Mishra, Adv. Opt. Mater., 2024, 12, 2303222 CrossRef CAS.
- Y. Kuang, C. Zhu, W. He, X. Wang, Y. He, X. Ran and L. Guo, J. Phys. Chem. C, 2020, 124, 23905–23912 CrossRef CAS.
- I. M. Palstra and A. F. Koenderink, J. Phys. Chem. C, 2021, 125, 12050–12060 CrossRef CAS PubMed.
- D. S. White, M. P. Goldschen-Ohm, R. H. Goldsmith and B. Chanda, eLife, 2020, 9, 53357 CrossRef PubMed.
- Y. Sun and J. Zhao, J. Phys. Chem. C, 2021, 125, 1171–1179 CrossRef CAS.
- N. Fiuza-Maneiro, J. Ye, S. K. Sharma, S. Chakraborty, S. Gómez-Graña, R. L. Z. Hoye and L. Polavarapu, ACS Energy Lett., 2025, 10, 1623–1632 CrossRef CAS PubMed.
- J. Ran, B. Wang, Y. Wu, D. Liu, C. M. Perez, A. S. Vasenko and O. V. Prezhdo, J. Phys. Chem. Lett., 2023, 14, 6028–6036 CrossRef CAS PubMed.
- T. Kim, S. Il Jung, S. Ham, H. Chung and D. Kim, Small, 2019, 15, 1900355 CrossRef PubMed.
- N. Yarita, H. Tahara, T. Ihara, T. Kawawaki, R. Sato, M. Saruyama, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 1413–1418 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |